
4
Information Related to Biologic Plausibility
The committee reviewed all relevant experimental studies of 2,4-dichlorophenoxyacetic acid (2,4-D), 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), 4-amino-3,5,6-trichloropicolinic acid (picloram), dimethylarsinic acid (DMA, also called cacodylic acid), and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) that have been published since Update 2010 (IOM, 2011) and has incorporated the findings into this chapter when it is appropriate and into the biologic-plausibility sections of Chapters 7–13 when they are of consequence for particular health outcomes. For each substance, this chapter includes a review of toxicokinetic properties, a brief summary of the toxic outcomes investigated in animal experiments, and a discussion of underlying mechanisms of action as illuminated by in vitro studies. The final section of this chapter presents two newly emerging subjects of molecular and biologic science that provide novel insight into potential mechanisms of xenobiotic-induced disease and that may increase the biologic plausibility of toxic actions of the herbicides sprayed in Vietnam.
Establishment of biologic plausibility through laboratory studies strengthens the evidence of a cause–effect relationship between herbicide exposure and health effects reported in epidemiologic studies and thus supports the existence of the less stringent relationship of association, which is the target of this committee’s work. Experimental studies of laboratory animals or cultured cells allow observation of effects of herbicide exposure under highly controlled conditions, which is difficult or impossible to achieve in epidemiologic studies. Such conditions include genetic differences between people, and frequency and magnitude of exposure, exposure to other chemicals, and pre-existing health conditions, all of which can be controlled in a laboratory animal study.
Once a chemical contacts the body, it becomes subject to the processes of
absorption, distribution, metabolism, and excretion. The combination of those four biologic processes determines the concentration of the chemicals in the body and how long each organ is exposed to it and thus influences its toxic or pharmacologic activity.
Absorption of a substance in an organism normally takes place by uptake into the bloodstream from mucous surfaces, such as the intestinal walls of the digestive tract during ingestion. Low solubility, chemical instability in the stomach, and inability to permeate the intestinal wall can all reduce the extent to which a substance is absorbed after being ingested. The solubility of a chemical in fat and its hydrophobicity influence the pathways by which it is absorbed, its relative potential to be metabolized (structurally transformed), and ultimately whether it persists in the body or is excreted. Absorption is a critical determinant of a chemical’s bioavailability, that is, the fraction of it that reaches the systemic circulation. In addition to ingestion, routes of exposure experienced by humans are inhalation (entry via the airways) and dermal exposure (entry via the skin). Animal studies may involve additional routes of exposure that are not ordinarily encountered by humans, such as intravenous or intraperitoneal injection, in which a chemical is injected into the bloodstream or abdominal cavity, respectively.
Distribution refers to the movement of a substance from the site of entry to the tissues and organs where it may have its ultimate effect or be sequestered. Distribution takes place most commonly via the bloodstream.
Metabolism is the breaking down that all substances begin to experience as soon as they enter the body. For many foreign substances, the breakdown to carbon dioxide and water is not complete and is more precisely referred to as biotransformation. Most of this process takes place in the liver by the action of enzymes, including cytochrome P450s, which catalyze the oxidative metabolism of many chemicals. As metabolism occurs, the parent chemical is converted to new chemicals called metabolites, which are often more water-soluble (polar) and thus more readily excreted. When the resulting metabolites are pharmacologically or toxicologically inert, metabolism has deactivated the administered dose of the parent chemical and thus reduced its effects on the body. Metabolism may activate a chemical to a metabolite that is more potent or more toxic than it is.
Excretion is the removal of substances or their metabolites from the body, most commonly in urine or feces, whereas elimination applies to the disappearance of the parent molecule from the bloodstream. The rate of excretion of a chemical from the body is often limited by the rate of metabolism of the parent chemical into more water-soluble, readily excreted metabolites. Elimination is often incomplete, especially in the case of chemicals that resist biotransformation, and incomplete excretion results in the accumulation of foreign substances that can adversely affect biologic functions. Elimination is first-order when its rate is directly proportional to the amount of chemical then in the body, which would also mean that the chemical’s half-life is independent of dose.
The routes and rates of absorption, distribution, biotransformation or me-
tabolism, and excretion of a toxic substance collectively are termed toxicokinetics (or pharmacokinetics for chemicals used as pharmaceutical agents). Those processes determine the amount of a particular substance or metabolite that reaches specific organs or cells and that persists in the body. Understanding the toxicokinetics of a chemical is useful for valid reconstruction of exposure of humans, but it is most important for assessing the risk of effects from exposure to a chemical by determining the concentration of the active chemical in target tissues. The principles involved in toxicokinetics are similar among chemicals, although the degree to which different processes influence distribution depends on the structure and other inherent properties of the chemicals. Thus, the lipophilicity or hydrophobicity of a chemical and its structure influence the pathways by which it is metabolized and whether it persists in the body or is excreted. The degree to which different toxicokinetic processes influence the toxic potential of a chemical depends on metabolic pathways, which often differ among species. For that reason, attempts at extrapolation from experimental animal studies to human exposures must be done extremely carefully.
Many chemicals were used by the US armed forces in Vietnam. The nature of the substances themselves was discussed in more detail in Chapter 6 of the original Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam (VAO) report (IOM, 1994). Four herbicides documented in military records were of particular concern and are examined here: 2,4-D, 2,4,5-T, picloram, and cacodylic acid. This chapter also examines TCDD, the most toxic congener of the tetrachlorodibenzo-p-dioxins (tetraCDDs), also commonly referred to as dioxin—which is a contaminant of 2,4,5-T—because its potential toxicity is of concern. Considerably more information is available on TCDD than on the herbicides themselves. Other contaminants present in 2,4-D and 2,4,5-T are of less concern. Except as noted, the laboratory studies of the chemicals of concern used pure compounds or formulations; the epidemiologic studies discussed in later chapters often tracked exposures to mixtures.
Chemistry
Picloram (Chemical Abstracts Service Number [CAS No.] 1918-02-1; see chemical structure in Figure 4-1) was used with 2,4-D in the herbicide formulation Agent White, which was sprayed in Vietnam. It is also used commonly in Australia in a formulation that has the trade name Tordon 75D®. Tordon 75D contains several chemicals, including 2,4-D, picloram, a surfactant diethyleneglycolmonoethyl ether, and a silicone defoamer. A number of studies of picloram used such mixtures as Tordon formulations or other mixtures of 2,4-D and picloram that are similar to Agent White.
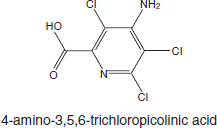
FIGURE 4-1 Structure of picloram.
Toxicokinetics
The original VAO committee reviewed studies of the toxicokinetics of picloram. Studies of animals showed rapid absorption through the gastrointestinal tract and rapid elimination of picloram as the unaltered parent chemical in urine. Nolan et al. (1984) examined the toxicokinetics of picloram in six healthy male volunteers who were given single oral doses of 0.5 or 5.0 mg/kg and a dermal dose of 2.0 mg/kg. Picloram was rapidly absorbed in the oral study and rapidly excreted unchanged in urine. More than 75% of the dose was excreted within 6 hours, and the remainder with an average half-life of 27 hours. On the basis of the quantity of picloram excreted in urine in the skin study, the authors noted that only 0.2% of the picloram applied to the skin was absorbed. Because of its rapid excretion, picloram has low potential to accumulate in humans.
In general, the literature on picloram toxicity continues to be sparse. Studies of humans and animals indicate that picloram is rapidly eliminated as the parent chemical. Studies of animals indicate that picloram is sparingly toxic at high doses.
Toxicity Profile
The original VAO committee reviewed studies of the carcinogenicity, genotoxicity, acute toxicity, chronic systemic toxicity, reproductive and developmental toxicity, and immunotoxicity of picloram. In general, there is some evidence on cancer in some rodent models but not in other species (NCI, 1978). Because of some concern that contaminants in the picloram (in particular, hexachlorobenzene) might be responsible for the carcinogenicity, picloram itself has not been established as a chemical carcinogen.
Studies conducted by the Environmental Protection Agency (EPA) (1988c) yielded no evidence that picloram is a genotoxic agent. Picloram is considered a mild irritant; it has produced erythema in rabbits only at high doses. The available information on the acute toxicity of picloram is paltry. Some neurologic effects—including hyperactivity, ataxia, and tremors—were reported in pregnant rats exposed to picloram at 750 or 1,000 mg/kg (Thompson et al., 1972).
Chronic Systemic Toxicity
Several studies have reported various effects of technical-grade picloram on the livers of rats. In the carcinogenicity bioassay conducted by Stott et al. (1990), treatment-related hepatomegaly, hepatocellular swelling, and altered tinctorial properties in the central regions of the liver lobules were noted in the groups exposed at 60 and 200 mg/kg per day. Males and females exposed at the 200 mg/kg dose had higher liver weights than controls. The no-observed-effect level (NOEL) was 20 mg/kg per day, and the lowest observed-effect level was 60 mg/kg per day for histologic changes in centrilobular hepatocellular tissues. According to the EPA (1988c), hexachlorobenzene (a contaminant of technical grade picloram at 197 ppm) was probably not responsible for the hepatic effects. Gorzinski and colleagues (1987) also reported a dose-related increase in liver weights, hepatocellular hypertrophy, and changes in centrilobular tinctorial properties in male and female F344 rats exposed to picloram at 150 mg/kg per day and higher in the diet for 13 weeks. In a 90-day study, cloudy swelling in the liver cells and bile duct epithelium occurred in male and female F344 rats given 0.3% or 1.0% technical picloram in the diet (EPA, 1988c). Hepatic effects have also been reported in dogs exposed to picloram: increased liver weights were reported in beagles that received 35 mg/kg per day or more in the diet for 6 months (EPA, 1988c). No other effects of chronic exposure to picloram have been reported.
Reproductive and Developmental Toxicity
The reproductive toxicity of picloram was evaluated in a two-generation study; however, few animals were evaluated, and no toxicity was detected at the highest dose tested, 150 mg/kg per day (EPA, 1988c). Some developmental toxicity was produced in rabbits exposed to picloram by gavage at 400 mg/kg per day on days 6–18 of gestation. Fetal abnormalities were forelimb flexure, fused ribs, hypoplastic tail, and omphalocele, each occurring in a single litter (John-Greene et al., 1985). Some maternal toxicity was observed at that dose, however, and EPA concluded on the basis of the sporadic findings that the malformations were not treatment-related (EPA, 1988c). No teratogenic effects were produced in the offspring of rats given picloram by gavage at up to 1,000 mg/kg per day on days 6–15 of gestation, but the occurrence of bilateral accessory ribs was significantly increased (Thompson et al., 1972).
Immunotoxicity
Studies of the potential immunotoxicity of picloram included dermal sensitization in humans and rodent immunoassays. In one study, 53 volunteers received nine 24-hour applications of 0.5 mL of a 2% potassium picloram solution on the skin of both upper arms. Each volunteer received challenge doses 17–24 days
later. The formulation of picloram (its potassium salt) was not a skin sensitizer or an irritant (EPA, 1988c). In a similar study, a 5% solution of picloram (M-2439, Tordon 101 formulation) produced slight dermal irritation and a sensitization response in six of the 69 volunteers exposed. When the individual components of M-2439—picloram, triisopropanolamine (TIPA) salt, and 2,4-D TIPA salt—were tested separately, no sensitization reaction occurred (EPA, 1988c). Tordon K+, but not technical-grade picloram, was also found to be a skin sensitizer in guinea pigs (EPA, 1988c). CD1 mice exposed to Tordon 202C (94% 2,4-D and 6% picloram) had no consistent adverse effects on antibody responses (Blakley, 1997), but the lack of a consistent response may be due to the fact that CD1 mice are outbred.
Mechanisms
No well-characterized mechanisms of toxicity for picloram are known.
Chemistry
Arsenic (As) is a naturally occurring element that exists in a trivalent form (As+3 or AsIII) and a pentavalent form (As+5 or AsV). See Figure 4-2 for chemical structures of selected arsenic-containing compounds—sodium arsenite, which contains AsIII, is generally considered to be the most toxic of these arsenic compounds. Arsenic is commonly present in drinking-water sources that are associated with volcanic soils and can reach high concentrations (over 50 ppb). Numerous human health effects have been attributed to drinking-water exposure, particularly bladder, skin, and lung cancers and vascular diseases.
Arsenic exists in both inorganic and organic (methylated) forms and is readily metabolized in humans and other species. Inorganic arsenic can be converted to organic forms. Although organic forms can be converted into inorganic forms by microorganisms in the soil, there is no evidence that this can occur in humans or other vertebrate species (Cohen et al., 2006). The arsenic in cacodylic acid (CAS No. 75-60-5) has a valence of +5. Cacodylic acid (also known as dimethylarsinic acid [DMAV] by its more standard chemical name), disodium methanearsonate, and monosodium methanearsonate are herbicides that EPA approved for use in the United States, where they are occasionally applied on golf courses and large open spaces. Cacodylic acid was the form of arsenic used in Agent Blue, one of the mixtures used for defoliation in Vietnam; DMAV made up about 30% of Agent Blue. Agent Blue was chemically and toxicologically unrelated to Agent Orange, which consisted of phenoxy herbicides contaminated with dioxin-like compounds. As shown in Figure 4-3, DMAIII and DMAV, as well as monomethyl arsonous acid (MMAIII) and monomethyl arsonic acid (MMAV), are metabolic
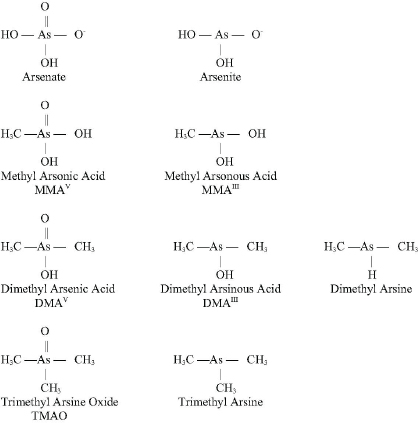
FIGURE 4-2 Structures of selected arsenic-containing compounds.
products of exposure to inorganic arsenic. Methylation of inorganic arsenic used to be considered a detoxification process associated with increased excretion (Vahter and Concha, 2001). However, some of the methylated metabolic intermediates, especially MMAIII, have been found to be more toxic than the parent sodium arsenite (Aposhian et al., 2000). The methylation pathway of inorganic arsenic results in the formation of DMAV and DMAIII.
The committee contemplated the relevance of animal data collected after exposure to inorganic arsenic, leading to DMAV being formed endogenously, vs data collected after direct exposure to exogenous DMAV, which is the form of arsenic and manner of potential exposure applicable to Vietnam veterans. It has not been established—nor can it be inferred—that the observed effects of exposure to inorganic arsenic are caused by endogenous formation of DMAV. Furthermore, results of recent studies suggest that there is an increased incidence of cancer in people who generate less DMAV endogenously (Huang SK et al., 2008), which would imply that complete methylation of inorganic arsenic to DMAV is associated with reduced risk of inorganic arsenic–induced adverse health outcomes
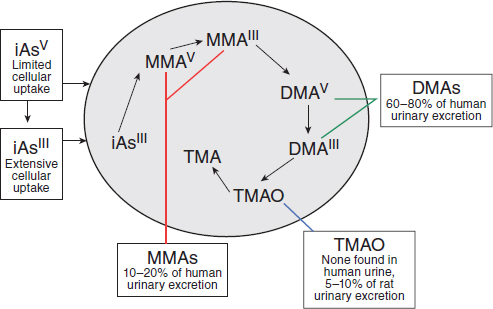
FIGURE 4-3 General pathways of arsenic metabolism after exposure to inorganic arsenic (iAs).
SOURCE: Adapted with permission from Cohen et al., 2006.
(Hall and Gamble, 2012). Finally, because there is no evidence that DMA is demethylated to inorganic arsenic in humans or other animals (Cohen et al., 2006), the committee chose not to consider the literature on inorganic arsenic for this report. The reader is referred to Arsenic in Drinking Water (NRC, 1999a) and Arsenic in Drinking Water: 2001 Update (NRC, 2001). The committee considered and reviewed only those toxicologic studies in which animals were directly exposed to DMAV.
Toxicokinetics
The metabolism and disposition of DMAV has recently been reviewed (Cohen et al., 2006; Suzuki et al., 2010). In general, it is rapidly excreted mostly unchanged in the urine of most animal species after systemic exposure. However, rats differ from most other mammals (including humans) in that a larger percentage (10%) of DMAV binds to hemoglobin in red blood cells, and that leads to a considerably longer half-life in blood (Cui et al., 2004; Suzuki et al., 2004). The binding of DMAV to hemoglobin is 10 times higher in rats than in humans (Lu et al., 2004). Chronic exposure of normal rat hepatocytes to DMAV resulted in decreased uptake and increased excretion, so that over time they developed resistance to its cytotoxic effects (Kojima et al., 2006); the tolerance was mediated
by induction of glutathione-S-transferase activity and of multiple-drug–resistant protein expression. Adair et al. (2007) examined tissue distribution of DMA in rats after dietary exposure for 14 days and found that it was extensively metabolized to trimethylated forms that may play a role in toxicity.
A physiologically based pharmacokinetic (PBPK) model of intravenous and ingested DMAV has been developed on the basis of mouse data (Evans et al., 2008). Similar models have been developed for humans on the basis of exposure to inorganic arsenic (El-Masri and Kenyon, 2008), but these models have limited utility for considering the toxicity of DMAV exposures that are relevant to Vietnam veterans.
Toxicity Profile
This section discusses the toxicity associated with organic forms of arsenic, most notably DMAV because it is the active ingredient in Agent Blue. The toxicity of inorganic arsenic is not considered relevant to veteran exposures to Agent Blue.
Neurotoxicity
Kruger et al. (2006) found that DMAIII and DMAV significantly attenuated neuronal ion currents through N-methyl-D–aspartate receptor ion channels whereas only DMAV inhibited ion currents through α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. The data suggest that those methylated forms of arsenic may have neurotoxic potential.
Immunotoxicity
Previous studies have shown that a low concentration of DMAV (10–7 M) could increase proliferation of human peripheral blood monocytes after their stimulation with phytohemagglutinin whereas it took a high concentration (10–4 M) to inhibit release of interferon-γ. This suggested that various immunomodulatory effects of DMAV have their own concentration specificity (Di Giampaolo et al., 2004).
Skin Toxicity
In a recent evaluation of effects of topical exposure of pregnant mice to DMA (valence not stated) on the skin of the dams and offspring (Kim et al., 2012), no effects were observed in offspring, but exposure increased skin thickness in the area of application and altered expression of apoptosis-related genes (Bcl-2, Bad, caspase-12). The results suggested that transient DMA exposure can be a skin irritant and produce dermatitis.
Genotoxicity and Carcinogenicity
DMAIII and DMAV are genotoxic and increase oxidative stress and cause DNA damage. Gómez et al. (2005) demonstrated that DMAIII induced a dose-related increase in DNA damage and oxidative stress in Jurkat cells. DMAIII was considerably more potent than DMAV in inducing DNA damage in Chinese hamster ovary cells (Dopp et al., 2004), and this was associated with a greater uptake of DMAIII into the cells. An additional study showed that DMAV is poorly membrane-permeable, but when forced into cells by electroporation it can induce DNA damage (Dopp et al., 2005). Similarly, analysis of arsenical dimethylated metabolites in human bladder cancer cells found dimethylmonothioarsinic acid (DMMTAV) and DMAIII to be the most toxic and DMAV to be less toxic in terms of DNA damage (Naranmandura et al., 2011). DNA damage from DMMTAV was shown to be related to accumulation of reactive oxygen species and down-regulation of p53 and p21 (DNA repair proteins); these processes were mediated in part through intracellular conversion to DMAV and DMAIII (Naranmandura et al., 2011). Thus, although extracellular DMAV has little toxic effect in cells because of its low uptake, intracellular DMAV can be highly toxic. Gene-expression profiling of bladder urothelium after chronic exposure to DMAV in drinking water showed significant increases in genes that regulate oxidative stress (Sen et al., 2005), whereas hepatic gene-expression profiling showed that DMAV exposure induced changes consistent with oxidative stress (Xie et al., 2004). In vivo, DMAV-induced proliferation of the urinary bladder epithelium could be attenuated with the antioxidant N-acetylcysteine (Wei et al., 2005).
DMAIII and DMAV are carcinogenic. Cancer has been induced in the urinary bladder, kidneys, liver, thyroid glands, and lungs of laboratory animals exposed to high concentrations of DMA. In a 2-year bioassay, rats fed 40 or 100 ppm DMAV developed epithelial carcinomas and papillomas in the urinary bladder and nonneoplastic changes in the kidneys (Arnold et al., 2006). Similarly, Wang et al. (2009) found that exposure of F344 rats to DMAV in drinking water at 1, 4, 40, or 100 ppm resulted in a change in the urinary bladder epithelium, but there were no changes in DNA repair capacity. In another study, Cohen et al. (2007a) exposed F344 rats to DMAV in the diet for 2 years and found an increase in bladder tumors in those receiving 100 ppm; they postulated that trimethylated forms of arsenic may be responsible for bladder cancer in rats. Direct bladder exposure of rats to 90 mg/kg DMAV induced infiltration and proliferation of urothelial epithelium mediated through oxidative stress pathways and extracellular signal-regulated kinase signaling (Takahashi et al., 2011). It is noteworthy that co-treatment with an antioxidant, N-acetylcysteine, worsened the DMAV-induced bladder injury, rather than ameliorating it as expected. In the mouse lung, DMAV acted as a tumor initiator (Yamanaka et al., 2009) and as a tumor promoter (Mizoi et al., 2005). DMAV can also act as a complete carcinogen, inducing lung tumors in susceptible strains of mice, including those with deficient DNA-repair activity (Hayashi et
al., 1998; Kinoshita et al., 2007). Yamanaka et al. (2009) suggest that DMAIII can act as a tumor promoter through the formation of a DMAIII radical after reduction of DMAV. Recent studies have also found that oral exposure of adult mice to 200 ppm DMAV in addition to fetal arsenic exposure can act as a promoter of renal and hepatocellular carcinoma, markedly increasing tumor incidence beyond that produced by fetal arsenic exposure alone (Tokar et al., 2012). Those findings emphasize how multiple life events can contribute to an adverse health outcome in which adult DMAV exposure triggered an otherwise dormant disease.
Mechanisms
Oxidative stress is a common theme that runs through the literature on the mechanisms of action of arsenic, particularly with regard to cancer in animals, although some studies have suggested that methylated arsenicals (MMAIII and DMAIII) can induce mutations in mammalian cells at concentrations below those required to produce oxidative stress after in vitro exposure (Klein et al., 2008). Recent studies have shown that mice that are deficient in enzymes associated with repair of oxidative DNA damage are highly susceptible to induction of tumors, particularly lung tumors, by DMAV (Kinoshita et al., 2007). The chemical reaction of arsenicals with thiol groups in sensitive target tissues, such as red blood cells and kidneys, may also be a mechanism of action of organic arsenicals (Naranmandura and Suzuki, 2008).
The variation in the susceptibility of various animal species to tumor formation caused by inorganic and organic arsenic is thought to depend heavily on differences in metabolism and distribution. Thus, genetic differences may play an important role. Numerous investigators are examining potential human susceptibility factors and gene polymorphisms that may increase a person’s risk of cancer and other diseases induced by arsenicals (Aposhian and Aposhian, 2006; Hernandez et al., 2008; Huang SK et al., 2008; Huang YK et al., 2008; McCarty et al., 2007; Meza et al., 2007; Steinmaus et al., 2007, 2010), but it is not yet possible to identify polymorphisms that may contribute to a person’s susceptibility to DMA-induced cancer or tissue injury.
PHENOXY HERBICIDES 2,4-DICHLOROPHENOXY ACID AND 2,4,5-TRICHLOROPHENOXYACETIC ACID
Chemistry
2,4-D (CAS No. 94-75-7) is an odorless and, when pure, white crystalline powder (Figure 4-4); it may appear yellow when phenolic impurities are present. The melting point of 2,4-D is 138°C, and the free acid is corrosive to metals. It is soluble in water and in a variety of organic solvents (such as acetone, alcohols, ketones, ether, and toluene). 2,4,5-T (CAS No. 93-76-5) is an odorless, white

FIGURE 4-4 Structures of 2,4-D and 2,4,5-T.
to light-tan solid with a melting point of 158°C. 2,4,5-T is noncorrosive and is soluble in alcohol and water. It reacts with organic and inorganic bases to form salts and with alcohols to form esters.
Uses of 2,4-D and 2,4,5-T
2,4-D has been used commercially in the United States since World War II to control the growth of broadleaf plants and weeds on range lands, lawns, golf courses, forests, roadways, parks, and agricultural land; it remains a widely used herbicide approved for use by the European Union and EPA. Formulations include 2,4-D amine and alkali salts and esters, which are mobile in soil and readily absorbed through the leaves and roots of many plants. Like 2,4-D, 2,4,5-T was developed and marketed as a herbicide during World War II. However, the registration for 2,4,5-T was canceled by EPA in 1978 when it became clear that it was contaminated with TCDD during the manufacturing process. It is recognized that the production of 2,4-D also involves the generation of some dioxin contaminants, even some with dioxin-like activity, but the fraction of TCDD is comparatively very small, as illustrated in Chapter 4.
The herbicidal properties of 2,4-D and 2,4,5-T are related to their ability to mimic the plant growth hormone indole acetic acid. They are selective herbicides in that they affect the growth of only broadleaf dicots (which include most weeds) and do not affect monocots, such as wheat, corn, and rice.
Toxicokinetics
Several studies have examined the absorption, distribution, metabolism, and excretion of 2,4-D and 2,4,5-T in animals and humans. Data on both compounds are consistent among species and support the conclusion that absorption of oral or inhaled doses is rapid and complete. Absorption through the skin is much lower but may be increased with the use of sunscreens or alcohol (Brand et al., 2002; Pont et al., 2004). After absorption, 2,4-D and 2,4,5-T are distributed widely in
the body but are eliminated quickly, predominantly in unmetabolized form in urine (Sauerhoff et al., 1977), but 2,4,5-trichlorophenol and 2,4-dichlorophenol have been identified as trace metabolites in urine. The half-life of single doses of 2,4-D or 2,4,5-T in humans has been estimated to be about 18–23 hours (Gehring et al., 1973; Kohli et al., 1974; Sauerhoff et al., 1977; WHO, 1984). Hines et al. (2003) found that concentrations of 2,4-D and its metabolites in the urine of herbicide applicators were consistent with 2,4-D urinary half-life estimates of 13–40 hours in humans.
Toxicity Profile
The toxicity database on 2,4-D is extensive (http://toxnet.nlm.nih.gov/ search on “2,4-D;” accessed April 23, 2013), whereas the available data on the toxicity of purified 2,4,5-T, independent of its contamination by TCDD, are sparse. TCDD is much more toxic than 2,4,5-T, and much of the toxicity attributed to 2,4,5-T in early studies was later shown to be caused by the TCDD contaminant. The following summary therefore focuses on 2,4-D toxicity, and information on pure 2,4,5-T is added when it is available.
After a single oral dose, 2,4-D is considered to produce moderate acute toxicity with an LD50 (dose lethal to 50% of exposed animals) of 375 mg/kg in rats, 370 mg/kg in mice, and from less than 320 to 1,000 mg/kg in guinea pigs. Rats and rabbits have dermal LD50s of 1,500 mg/kg and 1,400 mg/kg, respectively. 2,4,5-T itself also produces moderate acute toxicity, with oral LD50s of 389 mg/kg in mice and 500 mg/kg in rats. Death from acute poisoning with 2,4-D or 2,4,5-T has been attributed to the ability of the chemicals to uncouple oxidative phosphorylation, a vital process used by almost all cells in the body as the primary means of generating energy. After exposure to high doses, death due to multiple organ failure can occur rapidly. Studies in rats, cats, and dogs indicate that the central nervous system is the principal target organ for acute 2,4-D toxicity in mammals and suggest that the primary site of action is the cerebral cortex or the reticular formation (Arnold et al., 1991; Dési et al., 1962a,b). Neurotoxicity in humans is the predominant effect of acute inhalation and oral exposure to 2,4-D; symptoms include stiffness of arms and legs, incoordination, lethargy, anorexia, stupor, and coma. 2,4-D is also an irritant of the gastrointestinal tract, causing nausea, vomiting, and diarrhea.
Chronic exposure to 2,4-D at relatively high concentrations has been shown to produce a variety of toxic effects, including hepatic and renal toxicity, neurotoxicity, and hematologic changes. A NOEL of 2,4-D of 1 mg/kg was identified for renal toxicity in rats (Hazleton Laboratories America, 1987). Exposure to 2,4-D was associated with reduced survival and decreased growth rates of offspring of mothers fed high doses during pregnancy that were associated with maternal toxicity. However, even at high exposures, 2,4-D did not affect fertility and did not produce teratogenic effects in the offspring (Charles et al., 2001; Munro
et al., 1992). A recent study suggests that chronic exposure of adult male rats to 2,4-D results in a reduction in the weight of the testes, prostate, epidydimes, and seminal vesicles and a reduction in sperm density (Joshi et al., 2012). The purity of 2,4,5-T has been shown to influence its reproductive toxicity: TCDD contamination increases its fetotoxic effects and induces teratogenic effects. Immunotoxicity of 2,4-D has been reported in a small number of studies. At high doses that produced clinical toxicity, suppression of the antibody response was observed, whereas other measures of immune function were normal. The immunotoxicity of 2,4,5-T has not been evaluated in laboratory animals.
The carcinogenicity of 2,4-D and 2,4,5-T has been studied in rats, mice, and dogs after exposure in their food, direct placement in their stomachs, or exposure of their skin. All the studies had negative results except one that found an increased incidence of brain tumors in male rats—but not female rats—that received the highest dose of 2,4-D. The occurrence of malignant lymphoma in dogs kept as pets was reported to be higher when owners reported that they used 2,4-D on their lawns than when they did not (Hayes et al., 1991, 1995), but detailed reanalysis did not confirm this finding (Kaneene and Miller, 1999). A controlled study that used dogs exposed to 2,4-D in the laboratory had negative results. Timchalk (2004) suggested that dogs are not relevant for comparative evaluation of human health risk attributable to 2,4-D exposure because they excrete 2,4-D less efficiently than rats or humans do. 2,4-D is not metabolized to reactive intermediates capable of interacting with DNA, and the evidence supports the conclusion that 2,4-D is not a genotoxic carcinogen. However, a recent study shows that lymphocytes from smokers show genotoxic damage after exposure to 2,4-D, whereas lymphocytes from nonsmokers do not (Sandal and Yilmaz, 2011); this suggests that although 2,4-D may not be a carcinogen, it may influence the activity of known carcinogens.
2,3,7,8-TETRACHLORODIBENZO-P-DIOXIN
Chemistry
TCDDs are polychlorinated dibenzo-p-dioxins that have a triple-ring structure consisting of two benzene rings connected by an oxygenated ring with four attached chlorine atoms; in the case of the dioxin congener of greatest concern, 2,3,7,8-TCDD (commonly called simply TCDD), the chlorine atoms are attached at the 2, 3, 7, and 8 positions of the benzene rings (see Figure 4-5). The chemical properties of TCDD include a molecular weight of 322, a melting point of 305–306°C, a boiling point of 445.5°C, and a log octanol–water partition coefficient of 6.8 (National Toxicology Program substance profile). It is very lipophilic, or fat soluble, is virtually insoluble in water (19.3 ng/L), and is soluble in organic solvents, such as benzene and acetone. It has been suggested that volatilization of dioxin from water may be an important mechanism of transfer from the aqueous
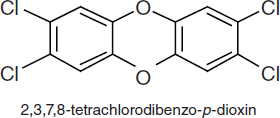
FIGURE 4-5 Chemical structure of TCDD.
to the atmospheric phase (EPA, 2004); however, because of its very low water solubility, most TCDD is bound to sediments and particulate matter.
Toxicokinetics
The absorption, distribution, biotransformation, and excretion of TCDD have been extensively studied in humans and a number of other animal models in the last 25 years. Given the plethora of data, this section highlights and summarizes only key findings. A more exhaustive review may be found at http://www.epa.gov/ncea/pdfs/dioxin/nas-review.
TCDD is absorbed into the body rapidly but is eliminated slowly. Because it is very lipophilic, resistant to biotransformation, and slowly eliminated, the concentration of TCDD in the lipid fraction of blood serum is thought to be in dynamic equilibrium with that in the lipid fraction in other tissue compartments. Thus, the lipid-adjusted blood serum concentration of TCDD is used to estimate total body burdens; at high TCDD concentrations, however, the liver sequesters some of the dioxin, so lipid adjustment that ignores the hepatic fraction would underestimate the total body burden. Exposure of humans to TCDD is thought to occur primarily via the mouth, skin, and lungs. In laboratory animals, oral administration of TCDD has been shown to result in absorption of 50–93% of the administered dose (Nolan et al., 1979; Rose et al., 1976). Similarly, a study performed in a 42-year-old man found that 87% of the oral dose was absorbed (Poiger and Schlatter, 1986). Dermal absorption appears to be dose-dependent: lower absorption occurs at higher doses (Banks and Birnbaum, 1991). Studies performed in vitro with tissues isolated from humans indicate that human skin may be more resistant to absorption (Weber et al., 1991). The varied and complex environmental matrices make environmental exposures difficult to quantify. Animal studies have demonstrated that the presence of soil or lipophilic agents dramatically reduces dermal absorption of TCDD: application in an activated carbon–water paste essentially eliminates absorption in contrast with absorption of pure compound dissolved in solvents. Oral bioavailability of TCDD and related compounds also depends on the matrix: contaminated breast milk and food
products have much higher bioavailability than soil-bound or sediment-bound TCDD, and activated carbon essentially blocks oral bioavailability (Olson, 2012).
After ingestion and gastrointestinal absorption, TCDD associates primarily with the lipoprotein fraction of the blood and later partitions into the cellular membranes and tissues (Henderson and Patterson, 1988). TCDD is distributed to all compartments of the body; the amounts differ from organ to organ, but most studies indicate that the primary disposition of TCDD is in the liver and adipose tissues. For example, in a human volunteer, it was found that 135 days after ingestion 90% of TCDD was in fat (Poiger and Schlatter, 1986), and TCDD persists in adipose tissue in the rhesus monkey (Bowman et al., 1989). The disposition and elimination of TCDD depend on the tissue examined, the time that has elapsed since exposure, total exposure, and other factors. For example, the concentration of cytochrome P450 1A2 (CYP1A2) (Poland et al., 1989) in the liver is increased by TCDD. Direct binding of TCDD to CYP1A2 is thought to result in sequestration of TCDD in the liver and to inhibit its distribution to other tissues. The importance of CYP1A2 concentrations for the toxic actions of TCDD has also been demonstrated in several laboratory situations; for instance, CYP1A2-knock-out mice were more susceptible than wild-type mice to TCDD immunotoxicity (Smialowicz et al., 2008), and maternal hepatic CYP1A2 was found to sequester TCDD and protect mouse fetuses against TCDD-induced teratogenesis (Dragin et al., 2006). In addition, distribution of TCDD is age-dependent, as shown by studies in which young animals displayed the highest concentration of TCDD in the liver and older animals the highest concentrations in kidneys, skin, and muscle (Pegram et al., 1995). Finally, the rate of elimination of TCDD, particularly after low exposures, depends heavily on the amount of adipose tissue mass (Aylward et al., 2005a; Emond et al., 2005, 2006).
In laboratory animals, TCDD is metabolized slowly. It is eliminated primarily in feces as both the parent chemical and its more polar metabolites. However, elimination appears to be dose-dependent: at low doses, about 35% of the administered dose of TCDD was detected in the feces; at higher doses, about 46% was observed (Diliberto et al., 2001). The dose-dependent occurrence of TCDD metabolites in the feces is thought to be due to increased expression of metabolizing enzymes at higher doses and to hepatic sequestration, which makes dioxins more available for metabolism. A measure of elimination is half-life, which is defined as the time required for the plasma concentration or the amount of a chemical in the body to be reduced by half. The half-life of TCDD in humans varies with body-mass index (BMI), age, sex, and concentration in the body and has been found to vary from 0.4 to more than 10 years (see Table 4-1).
Milbrath et al. (2009) conducted a comprehensive review of studies that reported the congener-specific elimination rates of TCDD and related compounds and analyzed the relationships between the apparent half-lives of the compounds as a function of age, body fat, smoking status, and breastfeeding. In infants (under 2 years old), the compounds have a reported half-life of 0.4 year (Leung et al.,
TABLE 4-1 Estimates of TCDD Half-Life in Humans and Animals
| Reference | Half-Lifea | Confidence Interval | Comment |
| Human studies: | |||
| Leung et al., 2006 | 0.4 year | Breastfed infants, 0–1 year after exposure | |
| Aylward et al., 2005a | < 3 years | Toxicokenetic model estimates for exposures > 10,000 pg/g of serum lipid | |
| Emond et al., 2005 | > 10 years | < 50 pg/g of serum lipid PBPK model based on 10 Operation Ranch Hand veterans 40,000 pg/g of serum lipid 138 pg/g of serum lipid | |
| Weeks > 10 years | |||
| Flesch-Janys et al., 1996 | 7.2 years | Adult males, Boehringer cohort | |
| Geusau et al., 2002 | 0–3 years after exposure: | ||
| 1.7 yearsb | Adult female 1, 144,000 pg/g of serum lipid | ||
| 3.4 yearsb | Adult female 2, 26,000 pg/g of serum lipid | ||
| Kumagai and Koda, 2005 | Adult male, incinerator workers, | ||
| 1.1–2.3 years | 0–1.3 years after exposure | ||
| Michalek et al., 2002 | 0.34 yearb | Adult males, Seveso cohort, | |
| 0–3 months after exposure | |||
| 6.9 years | 3–16 years after exposure | ||
| 9.8 years | Adult females, Seveso cohort, | ||
| 3–16 years after exposure | |||
| 7.5 years | Adult males, Operation Ranch | ||
| Hand veterans, | |||
| 9–33 years after exposure | |||
| Needham et al., 1994 | 7.8 years | 7.2–9.7 years | Adults, Seveso cohort |
| Pirkle et al., 1989 | 7.1 years | 5.8–9.6 years | Adult males, Operation Ranch Hand veterans, |
| 9–23 years after exposure | |||
| Milbrath et al., 2009 | 7.2 years | Reference half-life for 48.7-year-old | |
| Sorg et al., 2009 | 15.4 months | Victor Yushchenko: TCDD at 108,000 ppt lipid | |
| Animal studies: | Monkeys | ||
| Neubert et al., 1990 | 73.7 days | 60.9–93.8 days | single injection |
| Mice | |||
| DeVito and Birnbaum, 1995 | 15 days | female B6C3F1 | |
| Gasiewicz et al., 1983 | 11 daysc | C5BL/6J | |
| 24.4 daysc | DBA/2J | ||
| 12.6 daysc | B6D2F1/J | ||
| Koshakji et al., 1984 | 20 days | male ICR/Ha Swiss | |
| Rats | |||
| Reference | Half-Lifea | Confidence Interval | Comment |
| Emond et al., 2006 | Inducible elimination PBPK model estimates, | ||
| 10 days | 103 μg/kg acute treatment | ||
| 75 days | 10–3 μg/kg acute treatment | ||
| Hurst et al., 1998 | 8 days | Long-Evans, excretion from liver | |
| Pohjanvirta and Tuomisto, 1990 | 21.9 days | male Han/Wistar, resistant strain | |
| Viluksela et al., 1996 | 20.2 days | Long-Evans, TurkuAB strain | |
| 28.9 daysd | Long-Evans, Charles River strain | ||
| Weber et al., 1993 | 16.3 ± 3.0 days | male Sprague-Dawley | |
aHalf-lives of TCDD in humans based on measurement of TCDD in serum samples.
bShorter half-lives measured in humans during first months after exposure or in severely contaminated persons consistent with nonlinear elimination predicted by physiologically based pharmacokinetic (PBPK) models (for example, Carrier et al., 1995). Greater half-life in females attributed to greater BMI index.
cTotal cumulative excretion of 3H-TCDD-derived radioactivity.
dAttributed to differences in dilution due to different growth rates.
2006), and in adults, a half-life of 7.2 years (Milbrath et al., 2009). As people age, the growth results in a dilution effect and shorter half-lives. Aging also results in an increase in and redistribution of body fat and lipophilic chemicals that alter their rate of elimination (Van der Molen et al., 1996). Human studies of the Operation Ranch Hand cohort have consistently found a similar relationship between increasing half-life of TCDD and increasing BMI (Michalek and Tripathi, 1999; Michalek et al., 1992, 1996). Smoking and breastfeeding are associated with promoting elimination of TCDD and, in the case of breastfeeding, exposing infants through breast milk. Polycyclic aromatic hydrocarbons (PAHs) in cigarette smoke are capable of inducing CYP1A1, 1A2, and 1B1, which in turn may increase the rate of metabolism and later elimination of TCDD. A 30% decrease in TCDD half-life has been associated with smoking (Flesch-Janys et al., 1996).
Special Case of the Poisoning of Victor Yushchenko
In 2004, Victor Yushchenko, a candidate for the presidency of the Ukraine, was poisoned with TCDD. It led to severe chloracne and a blood serum TCDD concentration of 108,000 ppt (pg/g lipid wt), which is about 50,000 times as great as that in the general population at the time. The incident provided an opportunity to assess the toxicokinetics of TCDD after what was apparently a single large exposure. Serum and fat analysis of TCDD supports the first-order elimination half-life of 15.4 months in Yushchenko, and the similar decay curves confirmed
that TCDD was in equilibrium between serum lipids and subcutaneous fat (Sorg et al., 2009). That is much shorter than the 7.2-year reference half-life reported by Milbrath et al. (2009) and supports the dose-dependent elimination of TCDD, which is associated with induction of potential TCDD-metabolizing enzymes (CYP1A1, 1A2, and 1B1) in very high TCDD exposures. Two metabolites of TCDD (2,3,7-trichloro-8-hydroxydibenzo-p-dioxin and 1,3,7,8-tetrachloro-2-hydroxydibenzo-p-dioxin) were detected in feces, serum, and urine but not in fat and skin. Over a 12-month period, about 38% of the TCDD-derived material was eliminated as metabolites (95% in feces, 5% in urine) and 62% as parent chemical. The metabolite:TCDD ratio in the blood serum was about one-fiftieth of that in the feces; this supports the conclusion that the metabolites were not originally ingested with TCDD. The very slow metabolism of TCDD has been previously reported in laboratory animal models (Gasiewicz et al., 1983; Olson, 1986; Olson et al., 1980; Poiger and Schlatter, 1979), but this is the first report of metabolism in humans. It is also noteworthy that the structures of the human metabolites are the same as previously reported in the rat and dog (Poiger et al., 1982; Sawahata et al., 1982).
In light of the variables discussed above and the effect of differences in physiologic states and metabolic processes, which can affect the mobilization of lipids and possibly of compounds stored in them, complex PBPK models have been developed to integrate exposure dose with organ mass, blood flow, metabolism, and lipid content to predict the movement of toxicants into and out of each organ. A number of recent modeling studies have been performed in an effort to understand the relevance of animal experimental studies to exposures that occur in human populations (Aylward et al., 2005a,b; Beaudouin et al., 2010; Emond et al., 2005).
Toxicity Profile
Effects on Tissues and Organs of Laboratory Animals
The effects of TCDD in laboratory animals have been observed in a number of species (rats, mice, guinea pigs, hamsters, monkeys, cows, and rabbits) after the administration of a variety of doses and after periods that represent acute exposures (less than 24 hours), subchronic exposures (1 day–3 months), and chronic exposure (more than 3 months). Some differences have been observed between species, particularly with respect to degree of sensitivity, but in general the effects observed are qualitatively similar. Relatively high exposures of TCDD affect a variety of organs and result in organ dysfunction and death. Animal species vary widely in lethal toxicity of TCDD; the oral LD50 of the chemical varies from 1 μg/kg (in guinea pigs) to 5,000 μg/kg (in hamsters). There is up to a 5,000-fold interspecies variability in the acute lethal potency of TCDD in mature guinea pigs, rats, and hamsters. The developing fetus, however, is especially vulnerable
to TCDD exposure, and there is only about a 10-fold variability in fetal lethal potency among these species (Kransler et al., 2007; Peterson et al., 1993; Poland and Knutson, 1982). A characteristic of TCDD exposure is a wasting syndrome that includes loss of adipose and muscle tissue and severe weight loss, but the specific mechanisms of lethality remain unknown. In most rodents, exposure to TCDD leads to hepatic enlargement, the presence of hepatic lesions, and impaired hepatic function. The thymus is also sensitive. Finally, in both humans and nonhuman primates, TCDD exposure results in chloracne and associated dermatologic changes. As will be discussed in more detail in Chapters 6–13, studies performed in animal models have indicated that exposure to TCDD adversely affects the heart, the skin, and the immune, endocrine, and reproductive systems and increases the incidence of cancers of the liver, skin, thyroid, adrenal cortex, hard palate, nasal turbinates, tongue, and respiratory and lymphatic systems (ATSDR, 1998; Barouki et al., 2012; Birnbaum, 1994; Huff et al., 1994; Knerr and Schrenk, 2006). When TCDD has been administered to pregnant animals, birth defects—such as cleft palate, malformations of the reproductive organs of male and female progeny, and abnormalities in the cardiovascular, pulmonary, and nervous systems—have been observed.
Effects on Enzymes, Hormones, and Receptors in Laboratory Animals and Cultured Cells
In addition to adversely affecting the ability of specific organs to fulfill their normal physiologic roles, TCDD has been found to alter the function and expression of essential proteins, particularly a number of enzymes. The metabolism of foreign chemicals often changes their biologic properties, increasing their polarity (water solubility) and thus promoting the elimination of the metabolites. The enzymes that are most affected by TCDD are ones that act on or metabolize xenobiotics and hormones. Among the enzymes affected by TCDD, the best-studied is CYP1A1, which metabolizes xenobiotics. In laboratory animals, exposure to TCDD commonly results in an increase in CYP1A1 in most tissues; CYP1A1 therefore is often used as a marker of TCDD exposure. Related enzymes, which are also increased with TCDD exposure, include CYP1B1 and CYP1A2, which with CYP1A1 are capable of biotransforming some procarcinogens to potentially mutagenic and carcinogenic metabolites.
Other enzymes that are affected by TCDD metabolize hormones, such as thyroid hormones, retinoic acid, testosterone, estrogens, and adrenal steroids. Those hormones transmit their signals by interacting with specific proteins called receptors and in this manner initiate a chain of events in many tissues of the body. For example, binding of the primary female sex hormone, estrogen, to the estrogen receptor promotes the formation of breasts and the thickening of the endometrium and regulates the menstrual cycle. Exposure to TCDD can increase the metabolism of estrogen and thus lead to a decrease in the amount of estrogen available
for binding and activating the estrogen receptor. The ultimate effect of TCDD is an interference with all the bodily functions that are regulated by estrogens. Similarly, the actions of TCDD on the adrenal steroids can adversely affect their ability to regulate glucose tolerance, insulin sensitivity, lipid metabolism, obesity, vascular function, and cardiac remodeling. In addition to changing the amount of hormone present, TCDD has been found to interfere with the ability of receptors to fulfill their role in transmitting hormone signals. Animal models have shown that exposure to TCDD can increase the amounts of enzymes in the body and interfere with the ability of hormones to activate their specific hormone receptors. Those actions of TCDD on enzymes and hormone receptors are thought to underlie, in part, observed developmental and reproductive effects and cancers that are hormone-responsive.
Effects on Paths of Cellular Differentiation
The broad spectrum of TCDD effects on hormone and growth factor systems, cytokines, and other signal-transducer pathways indicates that TCDD is an extremely powerful growth dysregulator (Birnbaum, 1994). Research performed primarily in cultured cells has shown that TCDD can affect the ability of cells to undergo such processes as proliferation, differentiation, and apoptosis. During the proliferative process, cells grow and divide. When cells are differentiating, they are undergoing a change from less specialized to more specialized. Cellular differentiation is essential for an organism to mature from a fetal to an adult state. In the adult, proper differentiation is required for normal functions of the body, for example, in maintaining a normally responsive immune system. Processes of controlled cell death, such as apoptosis, are similarly important during development of the fetus and are necessary for normal physiologic functions in the adult. Apoptosis is a way for the body to eliminate damaged or unnecessary cells. The ability of a cell to undergo proliferation, differentiation, and apoptosis is tightly controlled by an intricate network of signaling molecules that allows the body to maintain the appropriate size and number of all the specialized cells that form the fabric of complex tissues and organs. Disruption of that network that alters the delicate balance of cell fate can have severe consequences, including impairment of the function of the organ because of the absence of specialized cells. Alternatively, the presence of an excess of some kinds of cells can result in the formation and development of tumors. Thus, the ability of TCDD to disrupt the normal course of a specific cell to proliferate, differentiate, or undergo apoptosis is thought to underlie (at least in part) its adverse effects on the immune system and the developing fetus and its ability to promote the formation of some cancers.
Mechanisms
TCDD binds and activates the aryl hydrocarbon receptor (AHR) in the cells of virtually every tissue in the body. The ability of TCDD to bind to the AHR with high affinity is considered to be necessary—but not sufficient—to produce the wide array of adverse effects associated with TCDD exposure. The pathologic responses associated with exposure to TCDD are thought to be due to binding to and activation of the AHR and later alterations in the expression of TCDD-regulated genes and to altered signaling of biologic pathways that interact with the AHR signal-transduction mechanism (Poland and Knutson, 1982; Safe, 1990; Schmidt and Bradfield, 1996; Whitlock, 1990).
The AHR functions as a ligand-activated nuclear transcription factor. On binding of agonists (ligands), such as TCDD, the AHR forms a heterodimer with a structurally related protein called AHR nuclear translocator (ARNT). The dimeric complex binds to core DNA sequences called xenobiotic-responsive elements (XREs) or dioxin-responsive elements (DREs) in the promotor region of responsive genes and enhances the transcription of those genes. Many of the AHR–regulated genes encode drug-metabolizing enzymes, such as CYP1A1, CYP1A2, CYP1B1, and a variety of phase II conjugating enzymes. Although the up-regulation of these enzymes is a sensitive biomarker of exposure to TCDD and in part contributes mechanistically to some of the adverse effects of TCDD, the tissue-, species-, time-, and dose-specific modulation (increase or decrease) of many genes is thought to contribute to the wide array of toxic responses to TCDD exposure (Black et al., 2012; Boverhof et al., 2006; Ovando et al., 2006, 2010; Perdew, 2008; Puga et al., 2009).
AHR Signaling Pathways
The primary and most intensely studied pathway by which TCDD elicits biologic responses is depicted in Figure 4-6. In the absence of bound ligand, the inactive AHR is retained in the cytoplasm of the cell in a complex consisting of two molecules of the heat-shock protein hsp90, one molecule of prostaglandin E synthase 3 (p23) (Kazlauskas et al., 1999), and one molecule of the immunophilin-like protein hepatitis B virus X-associated protein 2 (XAP2) (Petrulis et al., 2003), previously identified as either AHR–interacting protein (AIP; Ma and Whitlock, 1997) or AHR–associated protein 9 (ARA9; Carver and Bradfield, 1997). The hsp90 dimer–p23 complex plays multiple roles in the protection of the AHR from proteolysis, maintaining it in a conformation that makes it accessible to ligand binding at the same time that it prevents the premature binding of ARNT (Carver et al., 1994; Pongratz et al., 1992; Whitelaw et al., 1993). XAP2 interacts with the carboxyl terminus of hsp90 and with the AHR nuclear-localization signal (NLS), a short amino acid domain that targets the receptor for interaction with
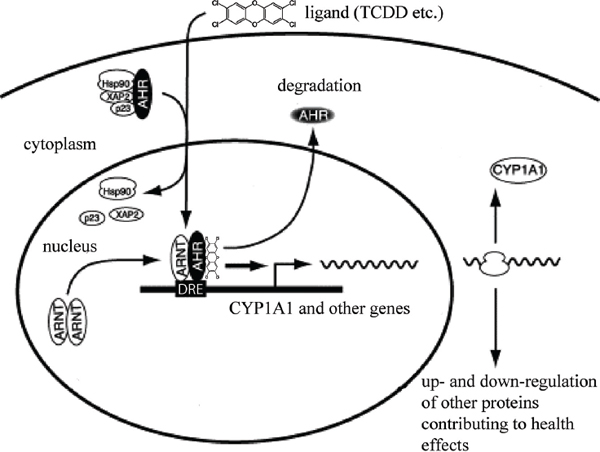
FIGURE 4-6 Mechanism of gene induction and repression after AHR activation by TCDD.
nuclear-transport proteins. Binding of XAP2 blocks such interaction, preventing the inappropriate trafficking of the receptor into the nucleus (Petrulis et al., 2003).
Binding of ligand (such as TCDD) induces the release of XAP2 and the exposure of the NLS and leads to the binding of nuclear-import proteins and translocation of the cytosolic complex into the nucleus (Davarinos and Pollenz, 1999; Song and Pollenz, 2002). Once in the nucleus, hsp90, p23, and XAP2 dissociate from the AHR, and this allows the binding of ARNT (Hoffman et al., 1991; Probst et al., 1993). The activated AHR–ARNT heterodimeric complex is then capable of directly or indirectly interacting with DNA by binding to recognition sequences in the regulatory region of responsive genes (Dolwick et al., 1993; Probst et al., 1993).
The canonical DNA recognition motif of the AHR–ARNT complex is referred to as the AHR–responsive element (AHRE, also referred to as the DRE or the XRE, for dioxin- or xenobiotic-responsive element, respectively). This element is found in the promoter region of AHR–responsive genes and contains the core sequence 5’-GCGTG-3’ (Shen and Whitlock, 1992), which is part of a more extensive consensus-binding sequence, 5’-T/GNGCGTGA/CG/CA-3’ (Lusska
et al., 1993; Yao and Denison, 1992). The AHR-ARNT complex binds to the AHRE core sequence in such a manner that ARNT binds to 5’-GTG-3’ and AHR binds to 5’-TC/TGC-3’ (Bacsi et al., 1995; Swanson et al., 1995). A second type of element, termed AHRE-II, 5’-CATG(N6)C[T/A]TG-3’, has been shown to be capable of acting indirectly with the AHR-ARNT complex (Boutros et al., 2004; Sogawa et al., 2004). The end result of the process is the recruitment of the transcriptional machinery associated with RNA polymerase II and the initiation of differential changes in the expression of the genes bearing the AHR-ARNT recognition motif. Many of the genes code for proteins responsible for detoxification reactions directed at the elimination of the ligand. Research suggests that post-translational modifications in histone proteins may modify the response (Hestermann and Brown, 2003; Schnekenburger et al., 2007).
In addition to the widely accepted view that the actions of TCDD are mediated by binding of the activated AHR-ARNT dimer to AHREs on DNA, which results in altered gene expression (Figure 4-6), more recent studies suggest that a “nongenomic” pathway within the cytoplasm also contributes to the toxic effects of TCDD, as reviewed by Matsumura (2009). The TCDD-mediated activation of AHR within the cytoplasm does not involve binding to ARNT or DNA and appears to contribute to rapid inflammatory responses associated with TCDD (Sciullo et al., 2008). In several cell lines, activation of protein kinase C (PKC) and the later activation of the serine phosphorylated form of cytosolic phospholipase A2 (cPLA2) takes place within 15 min of TCDD exposure (Dong and Matsumura, 2008; Park et al., 2007). It is proposed that within the cytoplasm, TCDD-mediated activation of AHR leads to a rapid increase in intracellular Ca2+, plus activation of cPLA2, protein kinases, and pro-inflammatory proteins, such as cyclooxygenase (COX-2) (Matsumura, 2009). This pathway and other alternative mechanisms of TCDD-mediated AHR activation have also been reviewed by Denison et al. (2011) and Perdew (2008).
AHR Physiology
The vertebrate AHR is presumed to have evolved from its counterpart in invertebrates, in which it serves a ligand-independent role in normal development processes. The ancestral function of the AHR appears to be the regulation of specific aspects of embryonic development, it having acquired the ability to bind xenobiotic compounds only during vertebrate evolution (Hahn, 2001). The invertebrate AHR also functions as a transcription factor and binds to the same dimerization partner (ARNT) and DNA-response elements as the vertebrate protein, but it does not respond to any of the environmental ligands recognized by the vertebrate receptor. Instead, it regulates diverse developmental processes that are independent of exogenous ligand exposure, such as neuronal differentiation during worm development in Caenorhabditis elegans (Huang et al., 2004; Qin and Powell-Coffman, 2004) or normal morphogenesis of legs, antennae, and
bristles in Drosophila melanogaster (Adachi-Yamada et al., 2005; Céspedes et al., 2010). In developing vertebrates, the AHR seems to play a role in cellular proliferation and differentiation and, in keeping with this role in invertebrates, also has a developmental role in craniofacial, renal, and cardiovascular morphogenesis (Birnbaum et al., 1989; Fernandez-Salguero et al., 1997; Lahvis et al., 2005). Other potential functional roles of the AHR include reproduction, innate immunity, tumor suppression, and blood-pressure regulation (Fujii-Kuriyama and Kawajiri, 2010).
The clearest adaptive physiologic response to AHR activation is the induction of xenobiotic-metabolizing enzymes involved in detoxification of toxic ligands. Evidence of that response, which was described above, was first observed in conjunction with the induction of Cyplal, which resulted from exposure to PAHs or TCDD and was directly related to activation of the AHR signaling pathway (Israel and Whitlock, 1983, 1984). Because of the presence of the AHRE motif in their gene promoters, other metabolizing genes were tested and found to be induced by AHR ligands, and this led to the identification of a so-called AHR gene battery of phase I and phase II detoxification genes that code for the drug-metabolizing enzymes CYP1A1, CYP1A2, CYP1B1, NQO1, ALHD3A1, UGT1A2, and GSTA1 (Nebert et al., 2000). Presumably, vertebrates have evolved those enzymes to detect a wide array of foreign, potentially toxic chemicals, represented in the wide variety of substrates that the AHR is able to bind to and whose biotransformation and elimination it is able to facilitate.
A potential complication of the adaptive responses elicited by AHR activation is the induction of a toxic response. Toxicity may result from the adaptive response itself if the induction of metabolizing enzymes results in the production of toxic metabolites. For example, the PAH benzo[a]pyrene (B[a]P), an AHR ligand, induces its own metabolism and detoxification by the AHR-dependent signaling mechanism described earlier but paradoxically becomes bioactivated to a toxic metabolite in several tissues by metabolism that depends on CYP1A1 and CYP1B1 activity (Harrigan et al., 2004). A second potential source of AHR-mediated toxicity may be aberrant changes in global gene expression beyond those observed in the AHR gene battery. The global changes in gene expression may lead to deleterious changes in cellular processes and physiology. Microarray analysis has proved invaluable in understanding and characterizing that response (Boverhof et al., 2006; Martinez et al., 2002; Ovando et al., 2006, 2010; Puga et al., 2000, 2004; Takeda et al., 2012; Vezina et al., 2004).
It is clear that the AHR is an essential component of the toxicity of dioxin and of dioxin-like chemicals (DLCs). Homozygous deletion of the AHR in mice leads to a phenotype that is resistant to the toxic effects of TCDD and to the carcinogenic effects of B[a]P (Fernandez-Salguero et al., 1996; Lahvis and Bradfield, 1998; Schmidt et al., 1996). AHR knockout mice, however, have other phenotypic effects, including reduced liver size, hepatic fibrosis, and cardiovascular abnormalities. Hence, it is likely that dioxin has effects that are due to disruption
of endogenous AHR functions and that are unrelated to the intrinsic toxicity of some of its ligands.
Definition of Dioxin-Like Compounds, Toxic Equivalence Factor, and Toxic Equivalents
TCDD has the highest affinity for the AHR, but many other chemicals have dioxin-like properties: they have similar chemical structures, have similar physiochemical properties, and cause a common battery of toxic responses because of their relatively high affinity for the AHR. Because of their hydrophobic nature and resistance to metabolism, these chemicals persist and bioaccumulate in fatty tissues of animals and humans. Although there are several hundred polychlorinated, polybrominated, and mixed polychlorinated-polybrominated dibenzo-p-dioxins, dibenzofurans, and biphenyls, only a relatively small number of congeners of these chemical classes display dioxin-like activity. Only 17 polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans with chlorine at the 2, 3, 7, and 8 positions and a few of the coplanar polychlorinated biphenyls that are often measured in environmental samples are recognized as being DLCs.
In the context of risk assessment, these polychlorinated-polybrominated dibenzo-p-dioxin, polychlorinated dibenzofuran, and biphenyl DLCs are commonly found as complex mixtures when detected in environmental media and biologic tissues or when measured as environmental releases from specific sources. That complicates the human health risk assessment that may be associated with exposures to varied mixtures of DLCs. To address the problem, the concept of toxic equivalence has been adopted by the scientific community, and the toxic equivalence factor (TEF) has been developed and introduced to facilitate risk assessment of exposure to those chemical mixtures. On the most basic level, TEFs compare the potential toxicity of each DLC found in a mixture with the toxicity of TCDD, the most toxic member of the group. The procedure involves assigning individual TEFs to the DLCs on the basis of in vivo and in vitro potency relative to TCDD, which is assigned a TEF of 1.0. The DLCs have been assigned TEFs ranging from 0.00001 to 1.0 by the World Health Organization (WHO) (van den Berg et al., 2006, as summarized in Table 4-2). Interim TEF values have been established for brominated congeners by the most recent (2011) joint WHO-UNEP (UN Environment Programme) meeting to evaluate the WHO Toxicity Equivalency Factor scheme. The recommendation is to use the TEF of the corresponding chlorinated congener as an interim TEF value for brominated congeners for human risk assessment (van den Berg et al., 2013).
When several chemicals are present in a mixture, the toxicity of the mixture is estimated by multiplying the TEF of each DLC in the mixture by its mass concentration and summing the products to yield the TCDD toxic equivalents (TEQs) of the mixture. In that approach to assessing dioxin-like activity of a complex real-world mixture of DLCs, an environmental or biologic specimen
| Chemical | TEF |
| Chlorinated dibenzo-p-dioxins | |
| 2,3,7,8-TCDD | 1.0 |
| 1,2,3,7,8-PeCDD | 1.0 |
| 1,2,3,4,7,8-HxCDD | 0.1 |
| 1,2,3,6,7,8-HxCDD | 0.1 |
| 1,2,3,7,8,9-HxCDD | 0.1 |
| 1,2,3,4,6,7,8-HpCDD | 0.01 |
| OctoCDD | 0.0003 |
| Chlorinated dibenzofurans | |
| 2,3,7,8-TCDF | 0.1 |
| 1,2,3,7,8-PeCDF | 0.03 |
| 2,3,4,7,8-PeCDF | 0.3 |
| 1,2,3,4,7,8-HxCDF | 0.1 |
| 1,2,3,6,7,8-HxCDF | 0.1 |
| 1,2,3,7,8,9-HxCDF | 0.1 |
| 2,3,4,7,8,9-HxCDF | 0.1 |
| 1,2,3,4,6,7,8-HpCDF | 0.01 |
| 1,2,3,4,7,8,9-HpCDF | 0.01 |
| OctoCDF | 0.0003 |
| Non-ortho-substituted PCBs | |
| PCB 77—3,3′,4,4′-tetraCB | 0.0001 |
| PCB 81—3,4,4′,5-tetraCB | 0.0003 |
| PCB 126—3,3′,4,4′,5-pentaCB | 0.1 |
| PCB 169—3,3′,4,4′,5,5′-hexaCB | 0.03 |
| Mono-ortho-substituted PCBs | |
| PCB 105—2,3,3′,4,4′-pentaCB | 0.00003 |
| PCB 114—2,3,4,4′,5-pentaCB | 0.00003 |
| PCB 118—2,3′,4,4′,5-pentaCB | 0.00003 |
| PCB 123—2′,3,4,4′,5-pentaCB | 0.00003 |
| PCB 156—2,3,3′,4,4′,5-hexaCB | 0.00003 |
| PCB 157—2,3,3′,4,4′,5′-hexaCB | 0.00003 |
| PCB 167—2,3′,4,4′,5,5′-hexaCB | 0.00003 |
| PCB 189—2,3,3′,4,4′,5,5′-heptaCB | 0.00003 |
NOTE: CB, chlorinated biphenyl; CDD, chlorinated dibenzo-p-dioxin; CDF, chlorinated dibenzofuran; PCB, polychlorinated biphenyl; TEF, toxicity equivalency factor.
SOURCE: Adapted from: van den Berg et al. (2006).
with a 100-ppt (100-pg/g) TEQ is toxicologically equivalent to 100-ppt TCDD. There are two accepted specialized methods for assessing the DLCs in a complex biologic or environmental specimen: one involves analytic chemistry that quantifies specific DLCs (high-resolution gas chromatography-mass spectroscopy), and the other a reporter-gene biologic screen that assesses dioxin-like activity due to binding to the AHR in a transformed cell line (CALUX, EPA method 4435).
Epidemiologic studies discussed in this and other updates assess exposure by reporting the specific concentration of TCDD in a specimen or by expressing dioxin-like activity in a complex mixture in units of TEQs.
Carcinogenic Classification
EPA and the International Agency for Research on Cancer (IARC), a branch of WHO, have defined criteria to classify the potential carcinogenicity of chemicals on the basis of the weight of scientific evidence from animal, human, epidemiologic, mechanistic, and mode-of-action studies. EPA classified TCDD as a “probable human carcinogen” in 1985 and as “carcinogenic to humans” in a 2003 reassessment. In 1998, the IARC panel of experts concluded that the weight of scientific evidence supported the classification of dioxin as a class I carcinogen, that is, as “carcinogenic to humans.” Four years later, the US National Toxicology Program upgraded its classification to “known to be a human carcinogen.” In 2006, a panel of experts convened by the National Research Council to evaluate the EPA reassessment concluded that TCDD was “likely to be carcinogenic to humans;” this designation reflected the revised EPA Guidelines for Carcinogen Risk Assessment made public in 2005.
Genotoxicity
Genotoxicity describes a deleterious action that affects the integrity of a cell’s DNA. Genotoxic substances are known to be potentially mutagenic or carcinogenic. Although TCDD is carcinogenic in humans and laboratory animals, it is generally classified as nongenotoxic and nonmutagenic (Wassom et al., 1977). There is no evidence of covalent binding of TCDD or its metabolites to DNA (Poland and Glover, 1979). TCDD does interact with DNA through a receptor-mediated pathway that involves the initial binding of TCDD to the AHR, binding of the activated receptor complex to DREs on DNA and later alterations in expression of TCDD-regulated genes, and altered signaling of biologic pathways that interact with the AHR signal-transduction mechanism (Poland and Knutson, 1982; Safe, 1990; Schmidt and Bradfield, 1996; Whitlock, 1990). TCDD, 2,4,5-T, and 2,4-D were not mutagenic in Salmonella typhimurium with or without the addition of liver metabolic-activation enzymes (Blevins, 1991; Mortelmans et al., 1984). TCDD-induced cytogenetic damage in laboratory mice showed no increase in the frequencies of sister-chromatid exchanges, chromosomal aberrations, or micronuclei in bone marrow cells of either C57Bl/6J or DBA/2J mice after administration of a single high dose of TCDD—up to 150 μg/kg (Meyne et al., 1985). TCDD did not alter the frequency or spectrum of mutations in male and female Big Blue transgenic rats (Thornton et al., 2001). There is one report of a positive result with TCDD in a test that measured induction of chromosomal
deletions resulting from intrachromosomal recombination in mouse embryos in vivo (Schiestl et al., 1997).
In summary, the vast majority of studies did not detect mutagenic activity of TCDD in a variety of in vitro and in vivo short-term tests.
Other Toxic Outcomes
Chloracne is a signature effect of high exposure to TCDD and DLCs in some species and in humans who are sensitive.
There is an extensive body of evidence from experimental studies in animal-model systems that TCDD, other dioxins, and several DLCs are immunotoxic (Kerkvliet, 2009). Although the available evidence on dioxin immunotoxicity in humans is scant, mechanistic considerations support the notion that chemical alterations of immune function would cause adverse health outcomes because of the critical role that the immune system plays in general protection—fighting off infection and eliminating cancer cells at early stages. Because of those considerations, the chemicals are potential immunotoxicants.
Similarly, reproduction and embryonic development clearly are targets of TCDD, other dioxins, and DLCs; it is found consistently that the adverse effects are more prevalent during fetal development than in the adult. Although data on those effects in humans are practically nonexistent, some good data are now emerging on the developmental effects of DLCs in humans (Mocarelli et al., 2008). Human and animal studies have revealed other potential health outcomes, including cardiovascular disease, hepatic disease, thyroid dysfunction, lipid disorders, neurotoxicity, and metabolic disorders, such as diabetes.
A number of effects of TCDD exposure in vitro appear to be independent of AHR-mediated transcription and in at least one instance perhaps independent of AHR itself. Guo et al. (2004) showed that TCDD induced expression of transforming growth factor-α and other genes involved in extracellular matrix deposition in cells from mice that had homozygous ablation of the Ahr gene. Studies have shown that TCDD can mobilize calcium from intracellular sources and increase calcium imported from the culture medium (Puga et al., 1995). Mitochondrial oxidative stress has been shown to be induced when calcium is mobilized (Senft et al., 2002). Calcium mobilization by TCDD may have an important effect on signal-transduction mechanisms that control gene expression, inasmuch as several proto-oncogenes, such as c-fos, are activated by calcium changes.
Summary of Biologic Plausibility That TCDD Induces Adverse Effects in Humans
Mechanistic studies in vitro and in laboratory animals have characterized the biochemical pathways and types of biologic events that contribute to adverse effects of exposure to TCDD. For example, much evidence indicates that TCDD,
acting via the AHR in partnership with ARNT, alters gene expression. Receptor binding may result in release of other cytoplasmic proteins that alter the expression or activity of other cell-regulatory proteins. Mechanistic studies also indicate that many other cellular-component proteins contribute to the gene-regulatory effect and that the response to TCDD exposure involves a complex interplay between genetic and environmental factors. Comparative data from animal and human cells in vitro and from tissues suggest a strong qualitative similarity among species in response to TCDD, and this further supports the applicability to humans of the generalized model of initial events in response to dioxin exposure. Several studies indicate, however, that there may be substantial quantitative differences in qualitatively similar responses among species, with humans generally being less sensitive than rodents.
Biochemical and biologic responses to TCDD exposure are considered adaptive or simply reflective of exposure and not adverse in themselves if they take place within the normal homeostatic ranges of an organism. However, they may exceed normal physiologic boundaries or constitute early events in a pathway that leads to damage in sensitive members of the population. In the latter case, the response is toxic and would be expected to cause an adverse health effect. Those generalizations set the ground rules for the concept of biologic plausibility, which relies on extrapolation from animal studies to human risks, and for the precautionary principle, which bases decision making on minimizing exposure if the precise nature or magnitude of the potential damage that a substance may cause in humans is uncertain.
LIMITATIONS OF EXTRAPOLATING RESULTS OF LABORATORY STUDIES TO HUMAN RESPONSES
In some instances, toxic responses identified in laboratory-animal and cell-culture studies are not detected in epidemiologic studies after human exposure to the same chemicals. Although animal and cell-culture studies provide important links to understanding of biochemical and molecular mechanisms associated with toxicity induced by xenobiotics, many factors must be considered in extrapolating their results to human disease and disease progression. The following are key factors that might limit the ability of laboratory studies to predict human responses completely and accurately.
• Magnitude and duration of exposure. In many instances, animal and cell-culture studies are conducted at higher exposures and for shorter durations than are typical in human exposures. For example, the concentrations of TCDD used in animal studies can be many times higher than in the TCDD exposures of Vietnam veterans during their military service. In addition, TCDD is a persistent organic pollutant, and this results in human exposure that occurs over a lifetime, whereas animal studies seldom
examine chronic low-level exposure that occurs over a period of many months or years. Animal studies that establish a measurement of body burden over a specific period provide the best potential for extrapolation to humans.
• Toxicokinetics. The toxicokinetics—absorption, distribution, metabolism, and excretion—of xenobiotics can vary widely between laboratory animals and humans. As shown in Table 4-1, the biologic half-life of TCDD varies from 8-29 days in rats and mice to about 7 years in humans even though drug-metabolizing enzymes—including cytochrome P450 1A1, 1A2, and 1B1—are up-regulated or induced via TCDD-mediated activation of the AHR in both rat and human livers (Black et al., 2012).
• Timing of exposure. Many organ systems are more susceptible to xenobiotic exposure during critical stages of development, differentiation, or function—such as during gestation or in the face of another external challenge (for example, antigens, smoking, dietary salt, and fat)—than at other times. Therefore, the response of some systems (such as immune or cardiovascular systems) may depend on the timing of exposure relative to the other challenges.
• Exposure composition. Most animal and cell-culture studies involve exposure to single chemicals or a well-defined mixture, but most human exposures are to complex mixtures from multiple sources.
• Difference in AHR affinity. The binding affinity of AHR for TCDD differs between species (discussed in Okey et al., 2005). Many strains of mice used for toxicologic study harbor a high-affinity AHR allele (AHRb) and exhibit greater sensitivity to hepatic CYP1A induction, immunosuppression, birth defects, and other responses than do strains that carry the low-affinity allele (AHRd). That simple allelic difference in AHR affinity has not been observed in humans, and the TCDD-binding affinity of the AHR found in most humans more closely resembles the low-affinity mouse AHRd allele. Nonetheless, Nebert et al. (2004) reported that some people have a TCDD-binding affinity that is 12 times higher than that in others. Thus, although humans are generally considered less sensitive on the basis of an AHR that has a low TCDD-binding affinity, this assumption may not apply to everyone.
• Complex disease etiology. The etiology of human diseases is highly influenced by genetics, environmental factors, and gene-environment interactions; these factors can be protective as well as deleterious. In addition to the chemical of interest, environmental factors commonly influencing human responses include diet, prescription and over-the-counter pharmaceuticals, cigarette smoking, alcohol consumption, and stress. Stress produced via known or unknown sources is a well-known modifier of human disease responses (for example, immune and cardiovascular responses). Furthermore, stress is an ever-present variable that is difficult to assess or
control for in epidemiologic studies because there is substantial individual variation in response to it (Cohen et al., 2007b). In contrast, laboratory studies are often conducted with inbred strains of animals under tightly controlled experimental conditions and thus may underestimate or overestimate the potential contribution of a single chemical exposure to disease development.
• Sex differences. There are well-known differences in susceptibility to xenobiotic exposures between male and female animals, some of which are modified by sex steroids. For example, female Sprague Dawley rats are significantly more responsive to the hepatotoxic (neoplastic and non-neoplastic) effects of TCDD than are males of the same strain (Kociba et al., 1978).
Epigenetics is the term used to describe mechanisms that regulate gene expression and genomic stability and are independent of changes in DNA sequence and mitotically stable; that is, they will be replicated when a cell divides (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010). The epigenetic marks on DNA are maintained every time a cell divides and are needed to maintain the identity and function of the cell type.
The history of epigenetics began in the 1940s when Conrad Waddington coined the term epigenetics to describe gene-environment interactions that alter biologic traits (Waddington, 1940, 1953, 1956). It was not until the 1970s that the first molecular epigenetic factor was described: DNA methylation, the chemical addition of a methyl group to DNA (Holliday and Pugh, 1975). In the 1980s, the role of DNA methylation in modifying gene expression—turning genes on and off—was established (Chen and Riggs, 2005). In the 1990s, the chemical modification of histone proteins associated with DNA also was shown to modify gene expression, thus establishing a second molecular epigenetic mechanism (Turner, 1998). In the early 2000s, various small noncoding RNA molecules were shown to regulate DNA activity (Sato et al., 2011). Around 2005, the first mapping of genome-wide epigenetic marks (epigenomes) was conducted (Pokholok et al., 2005). The marks act together in an exquisitely choreographed fashion to control the cellular ability to interact with, process, and initiate events and to respond to the signals and needs of the individual and local tissue environment.
Today, the processes recognized as epigenetic mechanisms are DNA methylation (Chen and Riggs, 2005; Holliday and Pugh, 1975), histone modification (Turner, 1998), alterations in chromatin structure (Murr, 2010), and modulation of expression by some small RNA molecules (Valeri et al., 2009). DNA methylation is the addition of a methyl group onto specific nucleotides. In mammals, it occurs at cytosine nucleotides that are adjacent to guanine nucleotides. Methylation of DNA can alter the expression of the adjacent gene, particularly if it occurs in the
promoter of the gene. Other modulations of DNA include hydroxymethylation (which is prominent in stem cells) and adenylation. Histones are the proteins that bind and form complex structures with DNA called nucleosomes. Chemical modifications of histones, such as methylation and acetylation, can alter histone structure and modify gene expression, particularly if the modification occurs in the promoter of the gene (Turner, 1998). The coiling or twisting of the DNA-histone complexes creates a structure called chromatin, and the structure of the chromatin can alter gene expression. The most recently recognized epigenetic factor consists of small noncoding RNA molecules that can associate with mRNA and regulate gene expression.
The interaction of all those epigenetic processes creates the epigenome, and the epigenome has a critical role in regulating gene expression independently of changes in DNA sequence (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010). The variation that is possible in the epigenome is startling: the histone proteins that control chromatin configurations have many dozens of possible modifications, and there are upwards of 50 million nucleotides in the DNA where methylation can occur and participate in regulating the cellular state. That implies that trillions of configurations of the epigenome are possible.
Environmental epigenetics involves the ability of environmental factors—such as nutrition, toxicants, and stress—to alter epigenetic programming. Thus, epigenetics provides a molecular mechanism by which environmental factors can influence disease etiology (Jirtle and Skinner, 2007; Szyf, 2007). The role of epigenetics in disease etiology has been shown for cancer and a number of other diseases (Christensen and Marsit, 2011; Cortessis et al., 2012; Skinner et al., 2010). In addition, exposure to environmental factors at critical times of development has the ability to alter epigenetic programming and to cause changes in gene expression (Skinner et al., 2010). Hence, immune responses, fetal development, and reproductive health are important examples of how epigenetic mechanisms play crucial roles in normal physiology.
New investigative tools and a more refined understanding of the epigenetic process have given rise to active research on the nature of the relationship between environmental exposure to epigenetically active agents and the occurrence of diverse disease states, including cancer, reproductive developmental conditions, and immune dysregulation. The committee sought to review data on the potential relationship of the exposures of interest with adverse epigenetic effects in the directly exposed veterans in an attempt to find evidence linking the exposures to disease processes that might have been mediated epigenetically. We also sought to review relevant data on female veterans and male veterans separately inasmuch as the epigenetic consequences of exposures could be different, particularly in the case of adverse reproductive outcomes.
A relevant example is that in vitro treatment of preimplantation embryos with TCDD alters the DNA methylation of imprinted genes (Wu et al., 2004). That in turn affects the functions and development of cells, tissues, and biologic systems.
It is well established that early-life exposures or environmental influences are associated with the onset of disease much later in life (Barker et al., 2010). Thus, an early developmental alteration in the epigenome provides a molecular mechanism whereby adult diseases can have a developmental basis.
Most epigenetic modifications occur in somatic cells and are not heritable, but do have the potential to generate effects in the exposed the individual. There is, however, the possibility of epigenetic transgenerational inheritance, which involves the ability of the environment to promote a permanent alteration in the germ line that is transmitted to later generations (Skinner et al., 2010). Manikkam et al. (2012) have shown that exposure of pregnant female rats to TCDD at critical times of development of the germ line (when epigenetic programming is being established) can lead to abnormalities in the third-generation offspring, including kidney disease and changes in the ovaries and sperm of the offspring. There are few data on possible male-mediated heritable effects of TCDD or other environmental compounds. The sparse data include results of studies of lead, a known developmental toxicant. Lead exposure can alter semen quality in males (Alexander et al., 1996) and has been shown to induce paternally mediated developmental toxicity in rats (Anjum et al., 2011); this demonstrates that male-mediated effects on reproduction can be induced by reproductive toxicants. However, there is a serious need for additional study of the question, particularly of the effects of the compounds of interest to this committee.
In summary, the ability of epigenetic mechanisms to regulate gene expression might underlie the ability of xenobiotic exposure to contribute to disease development and the potential for offspring to inherit effects of the disrupted epigenetic processes.
A second emerging field in biologic science that may provide insight into the mechanism of xenobiotic-induced disease is the disruption of the developing immune system by xenobiotic exposure, developmental immunotoxicity (DIT). The developing immune system is among the most sensitive physiologic targets of prenatal and childhood environmental insult. The sensitivity is due, in part, to the novel processes of gene rearrangement, somatic-cell selection, and immune-cell distribution that are required to produce a security system that can effectively protect not only the child but also the aging adult against external challenges without itself producing immune-mediated chronic disease. To produce that security system, the maturation of the immune system needs a critical series of steps that produce highly specialized immune cells that are capable of self vs nonself recognition and are tailored to the specialized functional environments of different tissues and organs (such as brain, lungs, skin, liver, gastrointestinal tract, and reproductive tract). A disruption of immune development can place the integrity of the organism at risk.
Among the known risk factors for DIT are such chemicals as heavy metals, some pesticides, industrial solvents such as trichloroethylene, and polychlorinated biphenyls. The adverse outcomes of DIT may become apparent soon after exposure or can emerge much later in life. Often, childhood or adult infections can trigger the appearance of DIT-associated immune problems that were established earlier in life (Dietert, 2009). DIT-induced alterations can also contribute to myriad health problems related to dysfunction or pathologic conditions in virtually any tissue or organ. Chemicals, drugs, infectious agents, and physical and emotional stressors can act synergistically and increase the risk of DIT. Not everyone is at identical risk for DIT. People who have particular genotypes may be at increased risk for specific chemical-induced DIT on the basis of heritable factors that affect metabolism or immune vulnerability.
The heightened sensitivity of the developing immune system is due to the existence of critical developmental windows of vulnerability during which environmental interference with key steps of immune maturation can change the entire course of immune development and result in later-life immune dysfunction and increased risk of disease. The events programmed for these critical developmental windows have several basic features:
• They are necessary, usually one-time events of early development, with no equivalents in adults.
• They lock in building blocks on which additional maturational events rely.
• If they do not occur both on time and efficiently, the ramifications are usually profound, prolonged, and irreversible.
Examples of critical windows of immune vulnerability and the chemicals that can cause disruptions have been described in several reviews (Dietert and Dietert, 2008; Dietert and Piepenbrink, 2006; Dietert et al., 2000; Holsapple et al., 2003; Landreth, 2002) and include
• The process of the seeding of immune cells in tissues where they grow into resident populations.
• The selection process of thymocytes in the thymus to distinguish nonself from self during the development of acquired immunity.
• The maturation of macrophage populations in the lung, in the brain, and elsewhere.
• The maturation of dendritic cells to provide balanced immune responses.
• The initial development, expansion, and seeding to the periphery of t-regulatory cell populations.
The increased sensitivity of the fetal, neonatal, and juvenile immune systems compared with the immune system of an adult can be manifested as a sensitivity to lower doses of chemical exposure than doses that affect the adult; a greater
persistence of the immune problems that follow exposure than are seen in adults; a broader array of immune problems than are experienced by adults; and greater likelihood that a second later-life chemical exposure or environmental stressor will trigger an unexpected immune problem.
It is important to note that disruption of immune maturation is not the only route for DIT. Early-life chemical exposure may affect the status of genes (the epigenome) in such a way that their pattern of expression in later life is affected and thereby alters immune functional capacity. Such changes in gene status that affect immune status could occur in the exposed generation (for people exposed in utero or during childhood), or they could carry through one or more additional generations as a result of true epigenetic alterations.
Adachi-Yamada T, Harumoto T, Sakurai K, Ueda R, Saigo K, O’Connor MB, Nakato H. 2005. Wing-to-leg homeosis by spineless causes apoptosis regulated by fish-lips, a novel leucine-rich repeat transmembrane protein. Molecular and Cellular Biology 25(8):3140–3150.
Adair BM, Moore T, Conklin SD, Creed JT, Wolf DC, Thomas DJ. 2007. Tissue distribution and urinary excretion of dimethylated arsenic and its metabolites in dimethylarsinic acid-or arsenate-treated rats. Toxicology and Applied Pharmacology 222(2):235–242.
Alexander BH, Checkoway H, van Netten C, Muller CH, Ewers TG, Kaufman JD, Mueller BA, Vaughan TL, Faustman EM. 1996. Semen quality of men employed at a lead smelter. Occupational and Environmental Medicine 53:411–416.
Anjum MR, Sainath SB, Suneetha Y, Reddy PS. 2011. Lead acetate induced reproductive and paternal mediated developmental toxicity in rats. Ecotoxicology and Environmental Safety 74(4):793–799.
Aposhian HV, Aposhian MM. 2006. Arsenic toxicology: Five questions. Chemical Research in Toxicolology 19(1):1–15.
Aposhian HV, Gurzau ES, Le XC, Gurzau A, Healy SM, Lu X, Ma M, Yip L, Zakharyan RA, Maiorino RM, Dart RC, Tircus MG, Gonzalez-Ramirez D, Morgan DL, Avram D, Aposhian MM. 2000. Occurrence of monomethylarsonous acid in urine of humans exposed to inorganic arsenic. Chemical Research in Toxicology 13:393–397.
Arnold EK, Beasley VR, Parker AJ, Stedelin JR. 1991. 2,4-D toxicosis II: A pilot study of clinical pathologic and electroencephalographic effects and residues of 2,4-D in orally dosed dogs. Veterinary and Human Toxicology 33:446–449.
Arnold LL, Eldan M, Nyska A, van Gemert M, Cohen SM. 2006. Dimethylarsinic acid: Results of chronic toxicity/oncogenicity studies in F344 rats and in B6C3F1 mice. Toxicology 223(1-2):82–100.
ATSDR (Agency for Toxic Substances and Disease Registry). 1998. Toxicological profile for chlorinated dibenzo-p-dioxins. US Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry.
___________________
1Throughout this report, the same alphabetic indicator after year of publication is used consistently for a given reference when there are multiple citations by the same first author in a given year. The convention of assigning the alphabetic indicators in order of citation in a given chapter is not followed.
Aylward LL, Brunet RC, Carrier G, Hays SM, Cushing CA, Needham LL, Patterson DG Jr, Gerthoux PM, Brambilla P, Mocarelli P. 2005a. Concentration-dependent TCDD elimination kinetics in humans: Toxicokinetic modeling for moderately to highly exposed adults from Seveso, Italy, and Vienna, Austria, and impact on dose estimates for the NIOSH cohort. Journal of Exposure Analysis and Environment Epidemiology 15(1):51–65.
Aylward LL, Brunet RC, Starr TB, Carrier G, Delzell E, Cheng H, Beall C. 2005b. Exposure reconstruction for the TCDD-exposed NIOSH cohort using a concentration- and age-dependent model of elimination. Risk Analysis 25(4):945–956.
Bacsi SG, Reisz-Porszasz S, Hankinson O. 1995. Orientation of the heterodimeric aryl hydrocarbon (dioxin) receptor complex on its asymmetric DNA recognition sequence. Molecular Pharmacology 47(3):432–438.
Banks YB, Birnbaum LS. 1991. Absorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) after low dose dermal exposure. Toxicology and Applied Pharmacology 107(2):302–310.
Barker DJ, Gelow J, Thornburg K, Osmond C, Kajantie E, Eriksson JG. 2010. The early origins of chronic heart failure: Impaired placental growth and initiation of insulin resistance in childhood. European Journal of Heart Failure 12(8):819–825.
Barouki R, Aggerbeck M, Aggerbeck, Coumoul X. 2012. The aryl hydrocarbon receptor system. Drug Metabolism and Drug Interactions 27(1):3–8.
Beaudouin R, Micallef S, Brochot C. 2010. A stochastic whole-body physiologically based pharmacokinetic model to assess the impact of inter-individual variability on tissue dosimetry over the human lifespan. Regulatory Toxicology and Pharmacology 57(1):103–116.
Birnbaum LS. 1994. Evidence for the role of the Ah receptor in response to dioxin. Progress in Clinical and Biological Research 387:139–154.
Birnbaum L, Harris M, Stocking L, Clark A, Morrissey R. 1989. Retinoic acid and 2,3,7,8-tetra-chlorodibenzo-p-dioxin selectively enhance teratogenesis in C57BL/6N mice. Toxicology and Applied Pharmacology 98:487–500.
Black MB, Budinsky RA, Dombkowski A, Cukovic D, LeCluyse EL, Ferguson SS, Thomas RS, Rowlands JC. 2012. Cross-species comparisons of transcriptomic alterations in human and rat primary hepatocytes exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicological Sciences 127(1):199–215.
Blakley BR. 1997. Effect of Roundup and Tordon 202C herbicides on antibody production in mice. Veterinary and Human Toxicology 39(4):204–206.
Blevins RD. 1991. 2,3,7,8-tetrachlorodibenzodioxin in fish from the Pigeon River of eastern Tennessee, USA: Its toxicity and mutagenicity as revealed by the Ames Salmonella assay. Archives of Environmental Contamination and Toxicology 20:366–370.
Boutros P, Moffat ID, Franc MA, Tijet N, Tuomisto J, Pohjanvirta R, Okey AB. 2004. Dioxin-responsive AHRE-II gene battery: Identification by phylogenetic footprinting. Biochemical and Biophysical Research Communications 321(3):707–715.
Boverhof DR, Burgoon LD, Tashiro C, Sharratt B, Chittim B, Harkema JR, Mendrick DL, Zacha-rewski TR. 2006. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicological Sciences 94(2):398–416.
Bowman RE, Schantz SL, Weerasinghe NCA, Gross ML, Barsotti DA. 1989. Chronic dietary intake of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) at 5 and 25 parts per trillion in the monkey: TCDD kinetics and dose-effect estimate of reproductive toxicity. Chemosphere 18:243–252.
Brand RM, Spalding M, Mueller C. 2002. Sunscreens can increase dermal penetration of 2,4-dichlo-rophenoxyacetic acid. Journal of Toxicology—Clinical Toxicology 40(7):827–832.
Carrier G, Brunet RC, Brodeur J. 1995. Modeling of the toxicokinetics of polychlorinated dibenzo-p-dioxins and dibenzofurans in mammalians, including humans. II. Kinetics of absorption and disposition of PCDDs/PCDFs. Toxicology and Applied Pharmacology 131(2):267–276.
Carver L, Bradfield C. 1997. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. Journal of Biological Chemistry 272:11452–11456.
Carver L, Jackiw V, Bradfield C. 1994. The 90-kDa heat shock protein is essential for Ah receptor signaling in a yeast expression system. Journal of Biological Chemistry 269:30109–30112.
Céspedes MA, Galindo MI, Couso JP. 2010. Dioxin toxicity in vivo results from an increase in the dioxin-independent transcriptional activity of the aryl hydrocarbon receptor. PLoS ONE 5(11):e15382.
Charles JM, Hanley TR, Wilson RD, van Ravenzwaay B, Bus JS. 2001. Developmental toxicity studies in rats and rabbits on 2,4-dichlorophenoxyacetic acid and its forms. Toxicological Sciences 60:121–131.
Chen ZX, Riggs AD. 2005. Maintenance and regulation of DNA methylation patterns in mammals. Biochemistry and Cell Biology 83(4):438–448.
Christensen BC, Marsit CJ. 2011. Epigenomics in environmental health. Frontiers in Genetics 2:84.
Cohen SM, Arnold LL, Eldan M, Lewis AS, Beck BD. 2006. Methylated arsenicals: The implications of metabolism and carcinogenicity studies in rodents to human risk assessment. Critical Reviews in Toxicology 36(2):99–133.
Cohen SM, Ohnishi T, Arnold LL, Le XC. 2007a. Arsenic-induced bladder cancer in an animal model. Toxicology and Applied Pharmacology 222(3):258–263.
Cohen SM, Janicki-Deverts D, Miller GE. 2007b. Psychological stress and disease. Journal of the American Medical Association 298(14):1685–1687.
Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, Haile RW, Laird PW. 2012. Environmental epigenetics: Prospects for studying epigenetic mediation of exposure-response relationships. Human Genetics 131:1565–1589.
Cui X, Kobayashi Y, Hayakawa T, Hirano S. 2004. Arsenic speciation in bile and urine following oral and intravenous exposure to inorganic and organic arsenics in rats. Toxicological Sciences 82(2):478–487.
Davarinos N, Pollenz R. 1999. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. Journal of Biological Chemistry 274:28708–28715.
Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. 2011. Exactly the same but different: Promiscuity and molecular mechanism of action of the aryl hydrocarbon (dioxin) receptor. Toxicological Sciences 124(1):1–22.
Dési I, Sos J, Nikolits I. 1962a. New evidence concerning the nervous site of action of a chemical herbicide causing professional intoxication. Acta Physiologica Academiae Scientiarum Hungaricae 22:73–80.
Dési I, Sos J, Olasz J, Sule F, Markus V. 1962b. Nervous system effects of a chemical herbicide. Archives of Environmental Health 4:95–102.
DeVito M, Birnbaum L. 1995. The importance of pharmacokinetics in determining the relative potency of 2,3,7,8-tetrachlorodibenzo-p-dioxin and 2,3,7,8-tetrachlorodibenzofuran. Fundamental and Applied Toxicology 24:145–148.
Dietert R. 2009. Distinguishing environmental causes of immune dysfunction from pediatric triggers of disease. The Open Pediatric Medicine Journal 3:38–44.
Dietert R, Dietert J. 2008. Potential for early-life immune insult including developmental immunotoxicity in autism and autism spectrum disorders: Focus on critical windows of immune vulnerability. Journal of Toxicology and Environmental Health—Part B: Critical Reviews 11(8):660–680.
Dietert R, Piepenbrink M. 2006. Perinatal immunotoxicity: Why adult exposure assessment fails to predict risk. Environmental Health Perspectives 114(4):477–483.
Dietert R, Etzel R, Chen D, Halonen M, Holladay S, Jarabek A, Landreth K, Peden D, Pinkerton K, Smailowicz R, Zoetis T. 2000. Workshop to identify critical windows of exposure for children’s health: Immune and respiratory systems work group summary. Environmental Health Perspectives 108(Suppl 3):483–490.
Di Giampaolo L, Di Gioacchino M, Qiao N, Travaglini P, D’Intino A, Kouri M, Ponti J, Castellani ML, Reale M, Gabriele E, Boscolo P. 2004. “In vitro” effects of different arsenic compounds on PBMC (preliminary study). Giornale Italiano di Medicina del Lavoro Ed Ergonomia 26(3):183–186.
Diliberto JJ, DeVito MJ, Ross DG, Birnbaum LS. 2001. Subchronic exposure of [3H]-2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD) in female B6C3F1 mice: Relationship of steady-state levels to disposition and metabolism. Toxicological Sciences 61:241–255.
Dolwick KM, Swanson HI, Bradfield CA. 1993. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proceedings of the National Academy of Sciences 90:8566–8570.
Dong B, Matsumura F. 2008. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Molecular Pharmacology 74:255–263.
Dopp E, Hartmann LM, Florea AM, von Recklinghausen U, Pieper R, Shokouhi B, Rettenmeier AW, Hirner AV, Obe G. 2004. Uptake of inorganic and organic derivatives of arsenic associated with induced cytotoxic and genotoxic effects in Chinese hamster ovary (CHO) cells. Toxicology and Applied Pharmacology 201(2):156–165.
Dopp E, Hartmann LM, von Recklinghausen U, Florea AM, Rabieh S, Zimmermann U, Shokouhi B, Yadav S, Hirner AV, Rettenmeier AW. 2005. Forced uptake of trivalent and pentavalent methylated and inorganic arsenic and its cyto-/genotoxicity in fibroblasts and hepatoma cells. Toxicological Sciences 87(1):46–56.
Dragin N, Dalton TP, Miller ML, Shertzer HG, Nebert DW. 2006. For dioxin-induced birth defects, mouse or human CYP1A2 in maternal liver protects whereas mouse CYP1A1 and CYP1B1 are inconsequential. Journal of Biological Chemistry 281(27):18591–18600.
El-Masri HA, Kenyon EM. 2008. Development of a human physiologically based pharmacokinetic (PBPK) model for inorganic arsenic and its mono-and di-methylated metabolites. Journal of Pharmacokinetics and Pharmacodynamics 35(1):31–68.
Emond C, Michalek JE, Birnbaum LS, DeVito MJ. 2005. Comparison of the use of physiologically based pharmacokinetic model and a classical pharmacokinetic model for dioxin exposure assessments. Environmental Health Perspectives 113(12):1666–1668.
Emond C, Birnbaum LS, DeVito MJ. 2006. Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environmental Health Perspectives 114(9):1394–1400.
EPA (US Environmental Protection Agency). 1988c. Guidance for the Reregistration of Pesticide Products Containing Picloram as the Active Ingredient. Washington, DC: EPA, Office of Pesticide Programs.
EPA. 2004. Exposure and Human Health Reassessment of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) and Related Compounds—National Academy Sciences (NAS) Review Draft. Washington, DC. National Center for Environmental Assessment.
Evans BR, Karchner SI, Allan LL, Pollenz RS, Tanguay RL, Jenny MJ, Sherr DH, Hahn ME. 2008. Repression of aryl hydrocarbon receptor (AHR) signaling by AHR repressor: Role of DNA binding and competition for AHR nuclear translocator. Molecular Pharmacology 73(2):387–398.
Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. 1996. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicology and Applied Pharmacology 140:173–179.
Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. 1997. Lesions of aryl-hydrocarbon receptor-deficient mice. Veterinary Pathology 34(6):605–614.
Flesch-Janys D, Becher H, Gurn P, Jung D, Konietzko J, Manz A, Papke O. 1996. Elimination of polychlorinated dibenzo-p-dioxins and dibenzofurans in occupationally exposed persons. Journal of Toxicology and Environmental Health 47(4):363–378.
Fujii-Kuriyama Y, Kawajiri K. 2010. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proceedings of the Japan Academy—Series B: Physical and Biological Sciences 86(1):40–53.
Gasiewicz T, Geiger L, Rucci G, Neal R. 1983. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metabolism and Disposition 11:397–403.
Gehring PJ, Kramer CG, Schwetz BA, Rose JQ, Rowe VK. 1973. The fate of 2,4,5-trichlorophen-oxyacetic acid (2,4,5-T) following oral administration to man. Toxicology and Applied Pharmacology 26:352–361.
Geusau A, Schmaldienst S, Derfler K, Päpke O, Abraham K. 2002. Severe 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) intoxication: Kinetics and trials to enhance elimination in two patients. Archives of Toxicology 76:316–325.
Gómez SE, Del Razo LM, Muñoz Sanchez JL. 2005. Induction of DNA damage by free radicals generated either by organic or inorganic arsenic (AsIII, MMAIII, and DMAIII) in cultures of B and T lymphocytes. Biological Trace Element Research 108(1-3):115–126.
Gorzinski SJ, Johnson KA, Campbell RA, Landry TD. 1987. Dietary toxicity of picloram herbicide in rats. Journal of Toxicology and Environmental Health 20:367–377.
Guo J, Sartor M, Karyala S, Medvedovic M, Kann S, Puga A, Ryan P, Tomlinson CR. 2004. Expression of genes in the TGF-beta signaling pathway is significantly deregulated in smooth muscle cells from aorta of aryl hydrocarbon receptor knockout mice. Toxicology and Applied Pharmacology 194(1):79–89.
Hahn ME. 2001. Dioxin toxicology and the aryl hydrocarbon receptor: Insights from fish and other non-traditional models. Marine Biotechnology (New York, NY) 3(Suppl 1):S224-S238.
Hall MN, Gamble MV. 2012. Nutritional manipulation of one-carbon metabolism: Effects on arsenic methylation and toxicity. Journal of Toxicology, article ID 595307, 11 pp.
Harrigan JA, Vezina CM, McGarrigle BP, Ersing N, Box HC, Maccubbin AE, Olson JR. 2004. DNA adduct formation in precision-cut rat liver and lung slices exposed to benzo[a]pyrene. Toxicological Sciences 77(2):307–314.
Hayashi H, Kanisawa M, Yamanaka K, Ito T, Udaka N, Ohji H, Okudela K, Okada S, Kitamura H. 1998. Dimethylarsinic acid, a main metabolite of inorganic arsenics, has tumorigenicity and progression effects in the pulmonary tumors of A/J mice. Cancer Letters 125(1-2):83–88.
Hayes HM, Tarone RE, Cantor KP, Jessen CR, McCurnin DM, Richardson RC. 1991. Case-control study of canine malignant lymphoma: Positive association with dog owner’s use of 2,4-D herbicides. Journal of the National Cancer Institute 83(17):1226–1231.
Hayes HM, Tarone RE, Cantor KP. 1995. On the association between canine malignant lymphoma and opportunity for exposure to 2,4-dichlorophenoxyacetic acid. Environmental Research 70(2):119–125.
Hazleton Laboratories America. 1987. Oncogenicity Study in Mice with 2,4-Dichlorophenoxyacetic Acid (2,4-D). Final Report. Prepared for the Industry Task Force on 2,4-D Research Data.
Henderson L, Patterson DJ. 1988. Distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin in human whole blood and its association with, and extractability from, lipoproteins. Bulletin of Environmental Contamination and Toxicology 40(4):604–611.
Hernandez A, Xamena N, Surralles J, Sekaran C, Tokunaga H, Quinteros D, Creus A, Marcos R. 2008. Role of the Met(287)Thr polymorphism in the AS3MT gene on the metabolic arsenic profile. Mutation Research 637(1-2):80–92.
Hestermann E, Brown M. 2003. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Molecular and Cellular Biology 23(21):7920–7925.
Hines CJ, Deddens JA, Striley CA, Biagini RE, Shoemaker DA, Brown KK, Mackenzie BA, Hull RD. 2003. Biological monitoring for selected herbicide biomarkers in the urine of exposed custom applicators: Application of mixed-effect models. Annals of Occupational Hygiene 47(6):503–517.
Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. 1991. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 252:954–958.
Holliday R, Pugh JE. 1975. DNA modification mechanisms and gene activity during development. Science 187(4173):226–232.
Holsapple M, West L, Landreth K. 2003. Species comparison of anatomical and functional immune system development. Birth Defects Research-Part B 68:321–334.
Huang SK, Chiu AWH, Pu YS, Huang YK, Chung CJ, Tsai HJ, Yang MH, Chen CJ, Hsueh YM. 2008. Arsenic methylation capability, heme oxygenase-1 and NADPH quinone oxidoreductase-1 genetic polymorphisms and the stage and grade of urothelial carcinomas. Urologia Internationalis 80(4):405–412.
Huang X, Powell-Coffman JA, Jin Y. 2004. The AHR-1 aryl hydrocarbon receptor and its co-factor the AHA-1 aryl hydrocarbon receptor nuclear translocator specify GABAergic neuron cell fate in C. elegans. Development 131(4):819–828.
Huang YK, Pu YS, Chung CJ, Shiue HS, Yang MH, Chen CJ, Hsueh YM. 2008. Plasma folate level, urinary arsenic methylation profiles, and urothelial carcinoma susceptibility. Food and Chemical Toxicology 46(3):929–938.
Huff J, Lucier G, Tritscher A. 1994. Carcinogenicity of TCDD: Experimental, mechanistic, and epidemiologic evidence. Annual Review of Pharmacology and Toxicology 34:343–372.
Hurst CH, Abbott BD, DeVito MJ, Birnbaum LS. 1998. 2,3,7,8-tetrachlorodibenzo-p-dioxin in pregnant Long Evans rats: Disposition to maternal and embryo/fetal tissues. Toxicological Sciences 45(2):129–136.
IOM (Institute of Medicine). 1994. Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. Washington, DC: National Academy Press.
IOM. 2011. Veterans and Agent Orange: Update 2010. Washington, DC: The National Academies Press.
Israel DI, Whitlock JP Jr. 1983. Induction of mRNA specific for cytochrome P1-450 in wild type and variant mouse hepatoma cells. Journal of Biological Chemistry 258:10390–10394.
Israel DI, Whitlock JP Jr. 1984. Regulation of cytochrome P1-450 gene transcription by 2,3,7,8-tetrachlorodibenzo-p-dioxin in wild type and variant mouse hepatoma cells. Journal of Biological Chemistry 259:5400–5402.
Jirtle RL, Skinner MK. 2007. Environmental epigenomics and disease susceptibility. Nature Review Genetics 8(4):253–262.
John-Greene JA, Ouellette JH, Jeffries TK, Johnson KA, Rao KS. 1985. Teratological evaluation of picloram potassium salt in rabbits. Journal of Food and Chemical Toxicology 23:753–756.
Joshi SC, Tibrewal P, Sharma A, Sharma P. 2012. Evaluation of toxic effects of 2,4-D (2,4-dichlorophenoxyacetic acid) on fertility and biochemical parameters of male reproductive system of albino rats. International Journal of Pharmacy and Pharmaceutical Sciences 4(3):338–342.
Kaneene JB, Miller R. 1999. Re-analysis of 2,4-D use and the occurrence of canine malignant lymphoma. Veterinary and Human Toxicology 41(3):164–170.
Kazlauskas A, Poellinger L, Pongratz I. 1999. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (aryl hydrocarbon) receptor. Journal of Biological Chemistry 274:13519–13524.
Kerkvliet NI. 2009. AHR-mediated immunomodulation: The role of altered gene transcription. Biochemical Pharmacology 77:746–760.
Kim E, Kim M, Sung K, Hyun J, Jang J, Kim K. 2012. Effect of topical dimethylarsinic acid on the expression of apoptosis-related proteins in mouse skin. Bulletin of Environmental Contamination and Toxicology 88:672–677.
Kinoshita A, Wanibuchi H, Wei M, Yunoki T, Fukushima S. 2007. Elevation of 8-hydroxydeoxy-guanosine and cell proliferation via generation of oxidative stress by organic arsenicals contributes to their carcinogenicity in the rat liver and bladder. Toxicology and Applied Pharmacology 221(3):295–305.
Klein OD, Kostiner DR, Weisiger K, Moffatt E, Lindeman N, Goodman S, Tuchman M, Packman S. 2008. Acute fatal presentation of ornithine transcarbamylase deficiency in a previously healthy male. Hepatology International 2(3):390–394.
Knerr S, Schrenk D. 2006. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Molecular Nutrition and Food Research 50(10):897–907.
Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnard SD, Hummel RA, Humiston CG. 1978. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicology and Applied Pharmacology 46(2):279–303.
Kohli JD, Khanna RN, Gupta BN, Dhar MM, Tandon JS, Sircar KP. 1974. Absorption and excretion of 2,4,5-trichlorophenoxy acetic acid in man. Archives Internationales de Pharmacodynamie et de Therapie 210:250–255.
Kojima C, Qu W, Waalkes MP, Himeno S, Sakurai T. 2006. Chronic exposure to methylated arsenicals stimulates arsenic excretion pathways and induces arsenic tolerance in rat liver cells. Toxicological Sciences 91(1):70–81.
Koshakji RP, Harbison RD, Bush MT. 1984. Studies on the metabolic fate of [14C] 2,3,7,8-tetrachlo-rodibenzo-p-dioxin (TCDD) in the mouse. Toxicology and Applied Pharmacology 73:69–77.
Kransler KM, McGarrigle BP, Olson JR. 2007. Comparative developmental toxicity of 2,3,7,8-tetra-chlorodibenzo-p-dioxin in the hamster, rat and guinea pig. Toxicology 229:214–225.
Kruger K, Gruner J, Madeja M, Hartmann LM, Hirner AV, Binding N, Muhoff U. 2006. Blockade and enhancement of glutamate receptor responses in Xenopus oocytes by methylated arsenicals. Archives of Toxicology 80(8):492–501.
Kumagai S, Koda S. 2005. Polychlorinated dibenzo-p-dioxin and dibenzofuran concentrations in serum samples of workers at an infectious waste incineration plant in Japan. Journal of Occupational and Environmental Hygiene 2(2):120–125.
Lahvis G, Bradfield C. 1998. Ahr null alleles: Distinctive or different? Biochemical Pharmacology 56(7):781–787.
Lahvis GP, Pyzalski RW, Glover E, Pitot HC, McElwee MK, Bradfield CA. 2005. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Molecular Pharmacology 67(3):714–720.
Landreth K. 2002. Critical windows in development of the rodent immune system. Human and Experimental Toxicology 21:493–498.
Leung HW, Kerger BD, Paustenbach DJ. 2006. Elimination half-lives of selected polychlorinated dibenzodioxins and dibenzofurans in breast-fed human infants. Journal of Toxicology and Environmental Health-Part A 69(6):437–443.
Lu M, Wang H, Li XF, Lu X, Cullen WR, Arnold LL, Cohen SM, Le XC. 2004. Evidence of hemoglobin binding to arsenic as a basis for the accumulation of arsenic in rat blood. Chemical Research in Toxicology 17(12):1733–1742.
Lusska A, Shen E, Whitlock JP Jr. 1993. Protein-DNA interactions at a dioxin-responsive enhancer. Journal of Biological Chemistry 268:6575–6580.
Ma Q, Whitlock J Jr. 1997. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlo-rodibenzo-p-dioxin. Journal of Biological Chemistry 272(14):8878–8884.
Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK. 2012. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS ONE 7(2):e31901.
Martinez JM, Afshari CA, Bushel PR, Masuda A, Takahashi T, Walker NJ. 2002. Differential toxicogenomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin in malignant and nonmalignant human airway epithelial cells. Toxicological Sciences 69(2):409–423.
Matsumura F. 2009. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochemical Pharmacology 77:608–626.
McCarty KM, Chen YC, Quamruzzaman Q, Rahman M, Mahiuddin G, Hsueh YM, Su L, Smith T, Ryan L, Christiani DC. 2007. Arsenic methylation, GSTT1, GSTM1, GSTP1 polymorphisms, and skin lesions. Environmental Health Perspectives 115(3):341–345.
Meyne J, Allison DC, Bose K, Jordan SW, Ridolpho PF, Smith J. 1985. Hepatotoxic doses of dioxin do not damage mouse bone marrow chromosomes. Mutation Research 157:63–69.
Meza M, Gandolfi AJ, Klimecki WT. 2007. Developmental and genetic modulation of arsenic bio-transformation: A gene by environment interaction? Toxicology and Applied Pharmacology 222(3):381–387.
Michalek JE, Tripathi RC. 1999. Pharmacokinetics of TCDD in veterans of Operation Ranch Hand: 15-year follow-up. Journal of Toxicology and Environmental Health 57(6):369–378.
Michalek JE, Tripathi RC, Caudill SP, Pirkle JL. 1992. Investigation of TCDD half-life heterogenicity in veterans of Operation Ranch Hand. Journal of Toxicology and Environmental Health 35:29–38.
Michalek JE, Pirkle JL, Caudill SP, Tripathi RC, Patterson DG Jr, Needham LL. 1996. Pharmacokinetics of TCDD in veterans of Operation Ranch Hand: 10-year follow-up. Journal of Toxicology and Environmental Health 47:209–220.
Michalek JE, Pirkle JL, Needham LL, Patterson DG Jr, Caudill SP, Tripathi RC, Mocarelli P. 2002. Pharmacokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Seveso adults and veterans of Operation Ranch Hand. Journal of Exposure Analysis and Environmental Epidemiology 12(1):44–53.
Milbrath MO, Wenger Y, Chang CW, Emond C, Garabrant D, Gillespie BW, Jolliet O. 2009. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environmental Health Perspectives 117(3):417–425.
Mizoi M, Takabayashi F, Nakano M, An Y, Sagesaka Y, Kato K, Okada S, Yamanaka K. 2005. The role of trivalent dimethylated arsenic in dimethylarsinic acid-promoted skin and lung tumorigenesis in mice: Tumor-promoting action through the induction of oxidative stress. Toxicology Letters 158(2):87–94.
Mocarelli P, Gerthoux PM, Patterson DG, Milani S, Limonta G, Bertona M, Signorini S, Tramacere P, Colombo L, Crespi C, Brambilla P, Sarto C, Carreri V, Sampson EJ, Terner WE, Needham LL. 2008. Dioxin exposure, from infancy through puberty, produces endocrine disruption and affects human semen quality. Environmental Health Perspectives 116(1):70–77.
Mortelmans K, Haworth S, Speck W, Zeiger E. 1984. Mutagenicity testing of Agent Orange components and related chemicals. Toxicology and Applied Pharmacology 75:137–146.
Munro IC, Carlo GL, Orr JC, Sund KG, Wilson RD, Kennepohl E, Lynch BS, Jablinske M, Lee NL. 1992. A comprehensive integrated review and evaluation of the scientific evidence relating to the safety of the herbicide 2,4-D. American College of Toxicology 11:559–664.
Murr R. 2010. Interplay between different epigenetic modifications and mechanisms. Advances in Genetics 70:101–141.
Naranmandura H, Suzuki KT. 2008. Formation of dimethylthioarsenicals in red blood cells. Toxicology and Applied Pharmacology 227(3):390–399.
Naranmandura H, Carew MW, Xu S, Lee J, Leslie EM, Weinfeld M, Le XC. 2011. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells. Chemical Research in Toxicology 24:1586–1596.
NCI (National Cancer Institute). 1978. Bioassay of Picloram for Possible Carcinogenicity. Report Series No. 23. Bethesda, MD: NCI.
Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. 2000. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochemical Pharmacology 59(1):65–85.
Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. 2004. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. Journal of Biological Chemistry 279(23):23847–23850.
Needham L, Gerthoux P, Patterson D, Brambilla P, Pirkle J, Tramacere P, Turner W, Beretta C, Sampson E, Mocarelli P. 1994. Half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in serum of Seveso adults: Interim report. Organohalogen Compounds 21:81–85.
Neubert DW, Wiesmuller T, Abraham K, Krowke R, Hagenmaier H. 1990. Persistence of various polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDDs and PCDFs) in hepatic and adipose tissue of marmoset monkeys. Archives of Toxicology 64:431–442.
Nolan RJ, Freshour NL, Kastl PE, Saunders JH. 1984. Pharmacokinetics of picloram in male volunteers. Toxicology and Applied Pharmacology 76(2):264–269.
Nolan R, Smith F, Hefner J. 1979. Elimination and tissue distribution of 2,3,7,8-tetrachlorodibenzo-para-dixion (TCDD) in female guinea-pigs following a single oral dose. Toxicology and Applied Pharmacology 48(1):A162.
NRC (National Research Council). 1999a. Arsenic in Drinking Water. Washington, DC: National Academy Press.
NRC. 2001. Arsenic in Drinking Water: Update 2001. Washington, DC: National Academy Press.
Okey AB, Franc MA, Moffat ID, Tijet N, Boutros PC, Korkalainen M, Tuomisto J, Pohjanvirta R. 2005. Toxicological implications of polymorphisms in receptors for xenobiotic chemicals: The case of the aryl hydrocarbon receptor. Toxicology and Applied Pharmacology 207(Suppl 2):43–51.
Olson JR. 2012. Pharmacokinetics of dioxins and related compounds. In: Dioxins and Health, Third Edition, A. Schecter, ed., John Wiley & Sons, Inc., pps 109–169.
Olson JR, Gasiewicz TA, Neal RA. 1980. Tissue distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the golden syrian hamster. Toxicology and Applied Pharmacology 56:78–85.
Olson LJ. 1986. The immune system and pesticides. The Journal of Pesticide Reform 6(2):20–25.
Ovando BJ, Vezina CM, McGarrigle BP, Olson JR. 2006. Hepatic gene downregulation following acute and subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicological Sciences 94(2):428–438.
Ovando BJ, Ellison CA, Vezina CM, Olson JR. 2010. Toxicogenomic analysis of exposure to TCDD, PCB126 and PCB153: Identification of genomic biomarkers of exposure to AhR ligands. BMC Genomics 11:583–598.
Park S, Dong B, Matsumura F. 2007. Rapid activation of c-Src kinase by dioxin is mediated by the Cdc37-HSP90 complex as part of Ah receptor signaling in MCF10A cells. Biochemistry 46:899–908.
Pegram RA, Diliberto JJ, Moore TC, Gao P, Birnbaum LS. 1995. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) distribution and cytochrome P4501A induction in young adult and senescent male mice. Toxicology Letters 76:119–126.
Perdew GH. 2008. Ah receptor binding to its cognate response element is required for dioxin-mediated toxicity. Toxicological Sciences 106(2):301–303.
Peterson RE, Theobald HM, Kimmel GL. 1993. Developmental and reproductive toxicity of dioxins and related compounds: Cross-species comparisons. Critical Reviews in Toxicology 23:283–335.
Petrulis J, Kusnadi A, Ramadoss P, Hollingshead B, Perdew G. 2003. The hsp90 co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. Journal of Biological Chemistry 278:2677–2685.
Pirkle J, Wolfe W, Patterson D, Needham L, Michalek J, Miner J, Peterson M, Phillips D. 1989. Estimates of the half-life of 2,3,7,8-tetrachlorodibenzo-p-dioxin in Vietnam veterans of Operation Ranch Hand. Journal of Toxicology and Environmental Health 27:165–171.
Pohjanvirta R, Tuomisto J. 1990. Remarkable residual alterations in responses to feeding regulatory challenges in Han/Wistar rats after recovery from the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Food and Chemical Toxicology 28:677–686.
Poiger H, Schlatter C. 1979. Biological degradation of TCDD in rats. Nature 281(5733):706–707.
Poiger H, Schlatter C. 1986. Pharmacokinetics of 2,3,7,8-TCDD in man. Chemosphere 15:1489–1494.
Poiger H, Buser HR, Weber H, Zweifel U, Schlatter C. 1982. Structure elucidation of mammalian TCDD-metabolites. Experientia 38:484–486.
Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122(4):517–527.
Poland A, Glover E. 1979. An estimate of the maximum in vivo covalent binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to rat liver protein, ribosomal RNA and DNA. Cancer Research 39(9):3341–3344.
Poland A, Knutson JC. 1982. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annual Reviews of Pharmacology and Toxicology 22:517–554.
Poland A, Teitelbaum P, Glover E. 1989. [125I]2-iodo-3,7,8-trichlorodibenzo-p-dioxin-binding species in mouse liver induced by agonists for the Ah receptor: Characterization and identification. Molecular Pharmacology 36(1):113–120.
Pongratz I, Mason GGF, Poellinger L. 1992. Dual roles of the 90-kDa heat shock protein hsp90 in modulating functional activities of the dioxin receptor. Journal of Biological Chemistry 267:13728–13734.
Pont AR, Charron AR, Brand RM. 2004. Active ingredients in sunscreens act as topical penetration enhancers for the herbicide 2,4-dichlorophenoxyacetic acid. Toxicology and Applied Pharmacology 195:348–354.
Probst MR, Reisz-Porszasz S, Agbunag RV, Ong MS, Hankinson O. 1993. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Molecular Pharmacology 44:511–518.
Puga A, Bohm J, Hoffer A, Leikauf G, Shertzer H, Zhou S. 1995. Dioxin alters calcium homeostasis and the regulation of arachidonate metabolism in mouse hepatoma cells. Proceeding of the 15th International Symposium on Chlorinated Dioxins and Related Compounds 25:381–386.
Puga A, Maier A, Medvedovic M. 2000. The transcriptional signature of dioxin in human hepatoma HepG2 cells. Biochemical Pharmacology 60(8):1129–1142.
Puga A, Sartor MA, Huang MY, Kerzee JK, Wei YD, Tomlinson CR, Baxter CS, Medvedovic M. 2004. Gene expression profiles of mouse aorta and cultured vascular smooth muscle cells differ widely, yet show common responses to dioxin exposure. Cardiovascular Toxicology 4(4):385–404.
Puga A, Ma C, Marlowe J. 2009. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochemical Pharmacology 77(4):713–722.
Qin H, Powell-Coffman JA. 2004. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Developmental Biology 270(1):64–75.
Rose JQ, Ransy JC, Wentzler TH, Hummel RA, Gehring PJ. 1976. The fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin following single and repeated oral doses to the rat. Toxicology and Applied Pharmacology 36:209–226.
Safe SH. 1990. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs). Critical Reviews of Toxicology 21:51–88.
Sandal S, Yilmaz B. 2011. Genotoxic effects of chlorpyrifos, cypermethrin, endosulfan and 2,4-D on human peripheral lymphocytes cultured from smokers and nonsmokers. Environmental Toxicology 26:433–442.
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. 2011. MicroRNAs and epigenetics. FEBS Journal 278:1598–1609.
Sauerhoff MW, Braun WH, Blau GE, Gehring PJ. 1977. The fate of 2,4-dichlorophenoxyacetic acid (2,4-D) following oral administration to man. Toxicology 8:3–11.
Sawahata T, Olson JR, Neal RA. 1982. Identification of metabolites of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) formed on incubation with isolated rat hepatocytes. Biochemical and Biophysical Research Communications 105:341–346.
Schiestl R, Aubrecht J, Yap WY, Kandikonda S, Sidhom S. 1997. Polychlorinated biphenyls and 2,3,7,8-tetrachlorodibenzo-p-dioxin induce intrachromosomal recombination in vitro and in vivo. Cancer Research 57:4378–4383.
Schmidt JV, Bradfield CA. 1996. Ah receptor signaling pathways. Annual Review of Cell Development-Biology 12:55–89.
Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. 1996. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proceedings of the National Academy of Sciences 93(13):6731–6736.
Schnekenburger M, Peng L, Puga A. 2007. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochimica et Biophysica ACTA/General Subjects 1769:569–578.
Sciullo EM, Vogel FA, Li W. 2008. Initial and extended inflammatory messages of the nongenomic signaling pathway of the TCDD-activated Ah receptor in U937 macrophages. Archives of Biochemistry and Biophysics 480(2):143–155.
Sen B, Wang A, Hester SD, Robertson JL, Wolf DC. 2005. Gene expression profiling of responses to dimethylarsinic acid in female F344 rat urothelium. Toxicology 215(3):214–226.
Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. 2002. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radical Biology and Medicine 33(9):1268–1278.
Shen ES, Whitlock JP. 1992. Protein-DNA interactions at a dioxin responsive enhancer: Mutational analysis of the DNA binding site for the liganded Ah receptor. Journal of Biological Chemistry 267:6815–6819.
Skinner MK, Manikkam M, Guerrero-Bosagna C. 2010. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinology and Metabolism 21(4):214–222.
Smialowicz RJ, DeVito MJ, Williams WC, Birnbaum LS. 2008. Relative potency based on hepatic enzyme induction predicts immunosuppressive effects of a mixture of PCDDs/PCDFs and PCBs. Toxicology and Applied Toxicology 227:477–484.
Sogawa K, Numayama-Tsuruta K, Takahashi T, Matsushita N, Miura C, Nikawa J, Gotoh O, Kikuchi Y, Fujii-Kuriyama Y. 2004. A novel induction mechanism of the rat CYP1A2 gene mediated by Ah receptor-ARNT heterodimer. Biochemical and Biophysical Research Communications 318(3):746–755.
Song Z, Pollenz RS. 2002. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Molecular Pharmacology 62(4):806–816.
Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V, Saurat JH. 2009. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 374(9696):1179–1185.
Steinmaus C, Moore LE, Shipp M, Kalman D, Rey OA, Biggs ML, Hopenhayn C, Bates MN, Zheng S, Wiencke JK, Smith AH. 2007. Genetic polymorphisms in MTHFR 677 and 1298, GSTM1 and T1, and metabolism of arsenic. Journal of Toxicology and Environmental Health-Part A 70(2):159–170.
Steinmaus C, Yuan Y, Kalman D, Rey OA, Skibola CF, Dauphine D, Basu A, Porter KE, Hubbard A, Bates MN, Smith MT, Smith AH. 2010. Individual differences in arsenic metabolism and lung cancer in a case-control study in Cordoba, Argentina. Toxicology and Applied Pharmacology 247(2):138–145.
Stott WT, Johnson KA, Landry TD, Gorzinski SJ, Cieszlak FS. 1990. Chronic toxicity and oncogenicity of picloram in Fischer 344 rats. Journal of Toxicology and Environmental Health 30:91–104.
Suzuki KT, Katagiri A, Sakuma Y, Ogra Y, Ohmichi M. 2004. Distributions and chemical forms of arsenic after intravenous administration of dimethylarsinic and monomethylarsonic acids to rats. Toxicology and Applied Pharmacology 198(3):336–344.
Suzuki S, Arnold LL, Pennington KL, Chen B, Naranmandura H, Le XC, Cohen SM. 2010. Dietary administration of sodium arsenite to rats: Relations between dose and urinary concentrations of methylated and thio-metabolites and effects on the rat urinary bladder epithelium. Toxicology and Applied Pharmacology 244(2):99–105.
Swanson HI, Chan WK, Bradfield CA. 1995. DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. Journal of Biological Chemistry 270(44):26292–26302.
Szyf M. 2007. The dynamic epigenome and its implications in toxicology. Toxicological Sciences 100(1):7–23.
Takahashi N, Yoshida T, Ohnuma A, Horiuchi H, Ishitsuka K, Kashimoto Y, Kuwahara M, Nakashima N, Harada T. 2011. The enhancing effect of the antioxidant N-acetylcysteine on urinary bladder injury induced by dimethylarsinic acid. Toxicologic Pathology 39:1107–1114.
Takeda T, Fujii M, Taura J, Ishii Y, Yamada H. 2012. Dioxin silences gonadotropin expression in perinatal pups by inducing histone deacetylases: A new insight into the mechanism for the imprinting of sexual immaturity by dioxin. Journal of Biological Chemistry 287(22):18440–18450.
Thompson DJ, Emerson JL, Strebing RJ, Gerbig CG, Robinson VB. 1972. Teratology and postnatal studies on 4-amino-3,5,6-trichloropicolinic acid (picloram) in the rat. Food and Cosmetics Toxicology 10:797–803.
Thornton AS, Oda Y, Stuart GR, Glickman BW, de Boer JG. 2001. Mutagenicity of TCDD in Big Blue transgenic rats. Mutation Research 478:45–50.
Timchalk C. 2004. Comparative inter-species pharmacokinetics of phenoxyacetic acid herbicides and related organic acids. Evidence that the dog is not a relevant species for evaluation of human health risk. Toxicology 200(1):1–19.
Tokar EJ, Diwan BA, Waalkes MP. 2012. Renal, hepatic, pulmonary and adrenal tumors induced by prenatal inorganic arsenic followed by dimethylarsinic acid in adulthood in CD1 mice. Toxicology Letters 209:179–185.
Turner BM. 1998. Histone acetylation as an epigenetic determinant of long-term transcriptional competence. Cellular and Molecular Life Sciences 54(1):21–31.
Vahter M, Concha G. 2001. Role of metabolism in arsenic toxicity. Pharmacology and Toxicology 89(1):1–5.
Valeri N, Vannini I, Fanini F, Calore F, Adair B, Fabbri M. 2009. Epigenetics, miRNAs, and human cancer: A new chapter in human gene regulation. Mammalian Genome 20:573–580.
van den Berg M, Birnbaum L, Denison MS, DeVito M, Farland W, Feeley M, Fiedler H, Hakansson H, Hanberg A, Haws L, Rose M, Safe S, Schrenk D, Tohyama C, Tritscher A, Tuomisto J, Tysklind M, Walker N, Peterson R. 2006. The 2005 World Health Organization re-evaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicological Sciences 93(2):223–241.
van den Berg M, Denison MS, Birnbaum L, DeVito M, Fiedler H, Falandysz J, Rose M, Schrenk D, Safe S, Tohyama C, Tritscher A, Tysklind M, Peterson RE. 2013. Polybrominated dibenzo-p-dioxins, dibenzofurans and biphenyls: Inclusion in the toxicity equivalency factor concept for dioxin-like compounds. Toxicological Sciences 133(2):197–208.
Van der Molen GW, Kooijman SALM, Slob W. 1996. A generic toxicokinetic model for persistent lipophilic compounds in humans: An application to TCDD. Toxicological Sciences 31(1):83–84.
Vezina CM, Walker NJ, Olson JR. 2004. Subchronic exposure to TCDD, PeCDF, PCB126, and PCB153: Effect on hepatic gene expression. Environmental Health Perspectives 112(16):1636–1644.
Viluksela M, Duong TV, Stahl BU, Li X, Tuomisto J, Rozman KK. 1996. Toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in two substrains of male Long-Evans rats after intravenous injection. Fundamental and Applied Toxicology 31:184–191.
Waddington CH. 1940. Organisers and Genes. United Kingdon: Cambridge University Press.
Waddington CH. 1953. Gene assimilation of an acquired character. Evolution 7:118–126.
Waddington CH. 1956. Principles of Embryology. United Kingdom: George Allen and Unwin Ltd.
Wang A, Wolf DC, Sen B, Knapp GW, Holladay SD, Huckle WR, Caceci T, Robertson JL. 2009. Dimethylarsinic acid in drinking water changed the morphology of urinary bladder but not the expression of DNA repair genes of bladder transitional epithelium in f344 rats. Toxicologic Pathology 37(4):425–437.
Wassom JS, Huff JE, Loprieno N. 1977. A review of the genetic toxicology of chlorinated dibenzo-p-dioxins. Mutation Research 47:141–160.
Weber LW, Zesch A, Rozman K. 1991. Penetration, distribution and kinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin in human skin in vitro. Archives of Toxicology 65:421–428.
Weber LW, Ernst SW, Stahl BU, Rozman K. 1993. Tissue distribution and toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats after intravenous injection. Fundamental and Applied Toxicology 21:523–534.
Wei M, Arnold L, Cano M, Cohen SM. 2005. Effects of co-administration of antioxidants and arsenicals on the rat urinary bladder epithelium. Toxicological Sciences 83(2):237–245.
Whitelaw M, Pongratz I, Wilhelmsson A, Gustafsson JA, Poellinger L. 1993. Ligand-dependent recruitment of the ARNT coregulatory determines DNA recognition by the dioxin receptor. Molecular and Cellular Biology 13:2504–2514.
Whitlock JP Jr. 1990. Genetic and molecular aspects of 2,3,7,8-tetrachlorodibenzo-p-dioxin action. Annual Review of Pharmacology and Toxicology 30:251–277.
WHO (World Health Organization). 1984. 2,4-Dichlorophenoxyacetic Acid (2,4-D). Environmental Health Criteria 29. Geneva: WHO.
Wu Q, Ohsako S, Ishimura R, Suzuki JS, Tohyama C. 2004. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biology of Reproduction 70(6):1790–1797.
Xie Y, Trouba KJ, Liu J, Waalkes MP, Germolec DR. 2004. Biokinetics and subchronic toxic effects of oral arsenite, arsenate, monomethylarsonic acid, and dimethylarsinic acid in v-Ha-ras transgenic (Tg.AC) mice. Environmental Health Perspectives 112(12):1255–1263.
Yamanaka K, Kato K, Mizoi M, An Y, Nakanao M, Hoshino M, Okada S. 2009. Dimethylarsine likely acts as a mouse-pulmonary tumor initiator via the production of dimethylarsine radical and/or its peroxy radical. Life Sciences 84(17-18):627–633.
Yao E, Denison M. 1992. DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry 31:5060–5067.
















































