
Psaty BM, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73.
Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature.2010;464:59.
R. D. C. Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2005
Scupham AJ, et al. Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microbiol. 2006;72:793.
Seow CH, et al. Novel anti-glycan antibodies related to inflammatory bowel disease diagnosis and phenotype. Am J Gastroenterol. 2009;104:1426.
Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978.
Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162.
Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8:31.
Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228.
Willing BP, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844.
METAGENOMICS AND PERSONALIZED MEDICINE87
Herbert W. Virgin88,*and John A. Todd89,*
The microbiome is a complex community of Bacteria, Archaea, Eukarya, and viruses that infect humans and live in our tissues. It contributes the majority of genetic information to our metagenome and, consequently, influences our resistance and susceptibility to diseases, especially common inflammatory diseases, such as type 1 diabetes, ulcerative colitis, and Crohn’s disease. Here we discuss how host–gene–microbial interactions are major determinants for the development of these multifactorial chronic disorders
________________
87 Reprinted from Cell 147(1), Virgin, H. V., J.A. Todd. 2011. Metagenomics and personalized medicine, pages 44-56, Copyright 2011, with permission from Elsevier.
88 Department of Pathology and Immunology, Department of Molecular Microbiology, and Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research, Washington University School of Medicine, St. Louis, MO, 63110, USA.
89 Juvenile Diabetes Research Foundation/Wellcome Trust Diabetes and Inflammation Laboratory, Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 0XY, UK.
* Correspondence: virgin@wustl.edu (H.W.V.), john.todd@cimr.cam.ac.uk (J.A.T.).
and, thus, for the relationship between genotype and phenotype. We also explore how genome-wide association studies (GWAS) on autoimmune and inflammatory diseases are uncovering mechanism-based subtypes for these disorders. Applying these emerging concepts will permit a more complete understanding of the etiologies of complex diseases and underpin the development of both next-generation animal models and new therapeutic strategies for targeting personalized disease phenotypes.
Recent advances in diverse areas of science and technology make this a unique time to study the genetics and pathogenesis of complex diseases, such type 1 diabetes (T1D) and inflammatory bowel disease (IBD), which includes Crohn’s disease (CD) and ulcerative colitis (UC). These distinct diseases are now understood to share important common characteristics and aspects of their disease mechanisms. In all three diseases, the immune system damages tissues: T1D is likely an autoimmune disease, whereas CD and UC are likely caused by inappropriate inflammatory responses to components of our microbiome (see Box A19-1 for definition of key terms). Many genetic loci regulate the risk for each disease. Although a threshold dose of these susceptibility alleles provides the foundation for developing the disease, these alleles are not sufficient to cause the disease.
It has been obvious for decades that complex gene–gene and gene–environment interactions govern these diseases, but not surprisingly, untangling this web of interactions has been extremely difficult (Figure A19-1). Despite the failure to identify single causal agents for each disease, there is strong evidence that microbes contribute to pathogenesis. Furthermore, genomewide association studies (GWAS), which use large study populations and careful replication of results, have effectively identified many important loci in the host that increase one’s risk for the disease, and these results have fundamentally altered how we conceptualize these diseases (Stappenbeck et al., 2011; Khor et al., 2011; Anderson et al., 2011; Franke et al., 2010; Todd, 2010). Correlation of GWAS data with genome-wide gene expression analyses (eQTLs), in combination with protein–protein interaction data, is greatly assisting the identification of candidate causal genes within these loci (Anderson et al., 2011; Franke et al., 2010; Cotsapas et al., 2011; Rossin et al., 2011; Fehrmann et al., 2011). Recently, numerous approaches have been developed to start defining mechanisms for complex inflammatory diseases by using leads from GWAS and analyses of the microbiome. These promising approaches include the following: the introduction of mutations in GWAS-identified loci into the mouse genome (Cadwell et al., 2010; Bloom et al., 2011); the creation of induced pluripotent stem cells (iPSCs) from patients and their differentiation into relevant cell types (e.g., Rashid et al., 2010); and humanized mouse models in which the murine immune system is replaced by transplantation (e.g., Brehm et al., 2010; Esplugues et al., 2011) or human microbial communities are transplanted into formerly germ-free mice (Goodman et al., 2011). Currently the great challenges in this field are to (1) understand how
Dysbiosis: Most commonly refers to a disruption in the normal homeostatic and beneficial relationship between microbes and their host, including disruptions in microbial community structure and function. Alterations in microbial community structure, involving Bacteria, Archaea, and/or Eukarya, can occur in any body habitat but have been best described in the gut where they have been associated with a number of disease states including, for example, inflammatory bowel disease.
Familial clustering: If a family member is diagnosed with a disease such as type 1 diabetes, ulcerative colitis, or Crohn’s disease, then the risk of other first-degree family members is much greater (perhaps as much as 50-fold for some multiactorial disorders) than that for a person taken at random from the general population. Familial clustering is caused by a combination of inherited genetic variants from the parents to the children and shared environmental factors within the families. Susceptibility variants are being discovered rapidly by GWAS, but the environmental factors remain unknown, although numerous candidates are recognized, most particularly a role for the microbiome and infections.
GWAS: Analysis of common alleles (mostly single-nucleotide polymorphisms, SNPs) in a population that associates genetic loci with disease susceptibility. These loci contain “candidate” disease genes.
Metagenetics: Approaching genetic and genomic studies by considering all of the genes in the metagenome as opposed to considering, in isolation, host genes or genes that confer particular properties (e.g., virulence or commensalism) upon an individual microbe. Importantly, the history of microbial inputs into the metagenomic profile of an individual is important for identifying the causes of complex disease, requiring expensive but essential longitudinal studies, including information from maternal and gestational exposures and phenotypes.
Metagenome: As used here, metagenome is the sum of all genes and genetic elements and their modifications in the somatic and germ cells of a host plus all genes and genetic elements in all microorganisms that live on or in that host at a given time. The metagenome has transient elements (e.g., during infection with a pathogen) and more persistent elements (e.g., infection with latent eukaryotic virus; presence of commensal bacteria).
Microbiome: As used here, the microbiome is the sum of all microbial organisms that live in or on the host at a given time. The microbiome includes members of Bacteria, Archaea, Eukarya, and the viruses of these organisms. In other articles this term may be used to refer to the genes of these organisms.
Virome: The sum of all viruses living in the tissues of the host or infecting organisms in the microbiome. These viruses maybe further divided into viruses that infect members of each of the three domains of life (e.g., bacterial virome or bacterial phages or the eukaryotic virome).
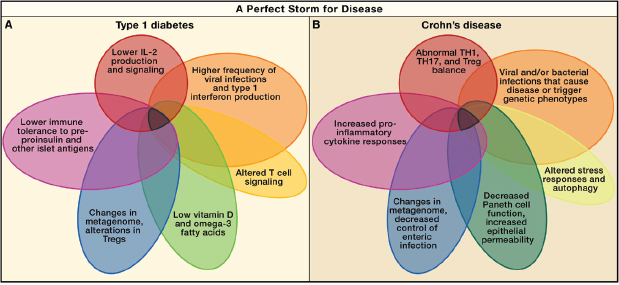
FIGURE A19-1 Perfect storms for developing Crohn’s disease and type 1 diabetes. A series of overlapping events and phenotypes driven by metagenetic and environmental processes that, in sum, contribute to the development and pathogenesis of type 1 diabetes (A) and Crohn’s disease (B).
both microbiome and GWAS-identified genes contribute to disease; (2) elucidate the molecular mechanisms by which causal genes act during pathogenesis; and (3) validate biomarkers and druggable pathways via genotype- phenotype studies (e.g., Dendrou et al., 2009; Bloom et al., 2011; Cadwell et al., 2010).
By peering through the lens of recent studies on CD, UC, and T1D, this review seeks to delineate emerging concepts in research on complex inflammatory diseases and to comment on the implications of these concepts for the interpretation of genetic and pathogenetic data. Two concepts are emphasized and integrated herein: (1) that single disease diagnoses are unlikely to be single phenotypes and may instead be the sum of multiple mechanism-based disease subsets, and (2) that the interactions of individual microorganisms and their genomes with specific host genes or pathways underpin the relationship between genotype and phenotype in these complex diseases. In this view, disease genetics may be combinatorial with different host–gene–microbial interactions, contributing to the pathogenesis of disease in subsets of patients. These two interrelated concepts, therefore, define T1D, CD, and UC as metagenetic (Box A19-1), rather than simply “genetic,” diseases. These concepts will guide the design and interpretation of future experiments that seek to dissect the pathophysiologic mechanisms underlying a number of complex diseases and to identify more effective approaches for their treatment and prevention.
Host Genetic Grist for the Metagenetic Mill
Recently, meta-analyses of GWAS of large cohorts of patients of European descent with UC or CD have been performed (Franke et al., 2010; Anderson et al., 2011; Khor et
al., 2011). These studies identified 98 loci, and candidate genes within these loci, that have a putative role in IBD. Similar studies of T1D identified 53 disease susceptibility loci (Barrett et al., 2009) (http://www.t1dbase.org). Importantly, many disease susceptibility loci are shared among common autoimmune and inflammatory diseases, including T1D, Graves’ disease, celiac disease, CD, UC, psoriasis, rheumatoid arthritis, alopecia areata, multiple sclerosis, and systemic lupus erythematosus (Cotsapas et al., 2011; Khor et al., 2011). It is striking that T1D and CD share 13/52 (25%) risk loci outside the human leukocyte antigen (HLA) gene complex despite the fact that these diseases are neither thought to be related diseases nor reported to be shared within families more often than expected by chance (http://www.t1dbase.org). Notably, the candidate causal genes in these 13 susceptibility loci regulate immunity. These include (Khor et al., 2011) PTPN22, which is involved in T and B cell signaling; IL10, encoding a powerful cytokine that suppresses inflammatory responses (including in specialized T regulatory cells in the gut) (Maloy and Powrie, 2011); BACH2, which regulates B cell gene expression and possibly IgA production; TAGAP, which is involved in T cell activation; IKZF1, which negatively regulates B cells; IL2RA, which controls T regulatory lymphocyte development and function; GSDMB/GSDMA/ORMDL3, which is involved in stress responses; FUT2, which controls microbial susceptibility (Smyth et al., 2011; Franke et al., 2010; McGovern et al., 2010); and IL27, which suppresses inflammatory responses and regulates IL-10 signaling (Imielinski et al., 2009; Barrett et al., 2009). This is a remarkable concordance of involved genes for two unrelated diseases, indicating that different diseases can have common mechanistic components and that the immune system is key for both diseases.
However, not withstanding all insights into disease mechanisms that the GWAS approach has already provided, the inheritance and the strong clustering of these multifactorial diseases within families (Box A19-1), which encompass both inherited genetic variants and intrafamilial environmental factors, remain only partially explained. Assuming a simple statistical model of gene interaction (Clayton, 2009), the numerous identified loci account for not more than 25% of the familial clustering of CD and UC (Anderson et al., 2011; Franke et al., 2010). This contrasts with T1D, in which the HLA effect is uniquely large and, together with 52 non-HLA loci, can account for almost all of the familial clustering (Clayton, 2009). For T1D, the massive effect of the HLA region, owing to functional polymorphisms in the HLA class II and class I genes, contributes almost 50% of familial clustering on its own (Clayton, 2009; Todd, 2010). There are, however, probably hundreds of non-HLA loci affecting the risk of CD, UC, and T1D that remain unmapped owing to their very small effect sizes (Barrett et al., 2009; Anderson et al., 2011; Franke et al., 2010). These putative loci will be difficult to map unless they contain rare mutations of higher penetrance, an occurrence that is just beginning to yield informative findings (Nejentsev et al., 2009; Rivas et al., 2011) and holds continued promise with the rapid use of highthroughput next-generation sequencing.
In humans, the HLA locus contains a large number of genes encoding the major histocompatibility complex (MHC) molecules (which are responsible for presenting antigens to cells of the immune system), along with a number of other genes that modulate immune responses. The remarkable contribution of HLA variations (Todd, 2010) to T1D risk is an unusual feature of a common disease. Nevertheless, HLA genotypes that greatly predispose individuals to T1D are not sufficient to cause the disease because only ~5% of high-risk HLA carriers develop T1D. HLAs are expressed by antigen-presenting cells (APCs), such as macrophages, B lymphocytes, and dendritic cells (DCs). DCs are highly potent APCs that reside in the pancreas and its islets (i.e., collections of insulin-producing beta cells and other endocrine cells) and could initiate the autoimmune destruction of beta cells by T cells (Calderon et al., 2011a, 2011b). Interestingly, the pancreatic lymph nodes, where DC priming of T cells for the induction of T1D may occur, also drain parts of the intestine, providing a site where the microbiome might influence the genesis of T1D (Turley et al., 2005; Wen et al., 2008). Because the insulin gene is one of the strongest non-HLA T1D susceptibility loci in the genome (Todd, 2010) (http://www.t1dbase.org), insulin and its precursors are likely primary autoantigens. These very strong associations with both HLA and this autoantigen gene are not a feature of CD or UC, in which no particular antigen is known to be targeted, hence their classification as inflammatory rather than autoimmune diseases. GWAS point to several other immunologic components of T1D etiology, including IL-2 production and receptor signaling (IL-2 gene, IL-2 receptors IL2RA [CD25] and IL2RB [CD132]; Todd, 2010), immune tolerance and T cell receptor signaling (PTPN2 [Long et al., 2011], PTPN22 [Arechiga et al., 2009; Bottini et al., 2006]), and recently, the immune response to viral infections and the type 1 interferon responses (IFIH1 [encoding MDA5], GPR183 [EBI2] [Heinig et al., 2010], TLR7 and TLR8, and FUT2 [Smyth et al., 2011]).
Twenty-eight loci (28/71, 39%) of CD risk loci are shared with UC, indicating that a set of core mechanisms participate in these diseases (Figure A19-2) (Khor et al., 2011). These diseases genes implicate numerous processes in both CD and UC, including T cell differentiation and function, autophagy, endoplasmic reticulum stress, oxidative stress, and mucosal immune defenses, among others. There are important gene–gene and pathway–pathway interactions within this core set of processes. For example, the CD risk gene NOD2 links to autophagy through interactions with the Nod2 protein and with Atg16L1, induction of proinflammatory cytokines, control of bacterial infection, and sensing of pathogen-associated molecular patterns (Levine et al., 2011). Particularly notable are pathways involving the cytokines IL-23 and IL-12, which regulate the development of TH1 and TH17 CD4 T cells, and IL-10, which is essential in the function of certain regulatory T cells (Tregs) via its anti-inflammatory activity. Rare mutations in genes encoding IL-10 receptors confer susceptibility to early-onset IBD (Glocker et al., 2009). These genetic clues point to a key role for regulating
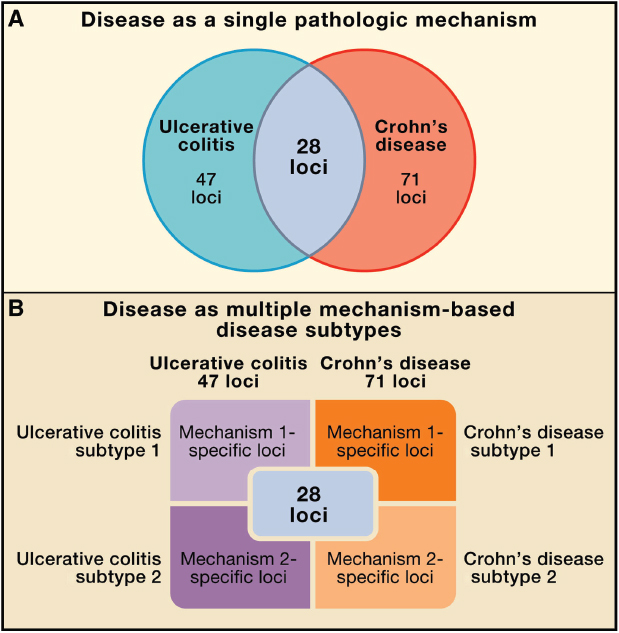
FIGURE A19-2 Refining the relationship between genotype and phenotype in complex inflammatory diseases. (A) Traditionally, a disease is considered as a single phenotype, with genes or loci conferring risk to two diseases shown as overlapping in a Venn diagram. (B) We propose a new view of the genotype–phenotype relationship in which different sets of loci are responsible for mechanistically distinct subtypes of diseases, and the sum of these subtypes constitutes the overall diagnosis. Here two disease subtypes are indicated for simplicity, but many such subtypes may exist, and sets of overlapping risk loci may be associated with these multiple mechanistically distinct disease phenotypes.
the balance between pro- and anti-inflammatory T cells in CD and UC. The regulation of T cell differentiation is also a key target for host-gene-microbial interactions, as discussed below.
Mechanism-Based Disease Subtypes
GWAS have revealed a wealth of genes potentially involved in T1D, UC, and CD, but no single gene or set of genes is prognostic. How can we interpret this observation? Here, we argue for an important contributor to this observation—the concept that “diagnosis” does not equal “single phenotype.” Without a distinct phenotype, genetic results are often difficult to interpret. This basic principle comes into sharp focus as one considers current genetic and pathogenesis studies of CD, UC, and T1D.
Why is a diagnosed “disease” an imprecise phenotype? It is not because patients have been misdiagnosed—the diagnoses of UC, CD, or T1D have stood the test of time to predict patient prognosis. However, we believe that there are many pathways to the same diagnosis. A diagnosis may be “clinically” precise but “mechanistically” imprecise. Thus, clinical diagnoses are poor phenotypes for genetic studies unless a single mechanism is responsible for the diagnosis, as in the case of a rare gene mutation in a monogenic disease. The complexity of GWAS results is consistent with the existence of multiple disease subtypes within T1D, UC, or CD, each based on a specific mechanism (Figure A19-2). Support for this idea comes from the observation that subsets of IBD patients respond differentially to mechanistically distinct interventions (Melmed and Targan, 2010).
Why do diagnostic categories group different mechanistic processes under the same moniker? Over many decades, pathologists have lumped patients with similar but nonidentical clinical and pathological signs and symptoms into diagnostic categories that predict outcome and complications. Indeed, this has enormous value clinically, but it emphasizes similarities between patients in outcome rather than differences in pathways that lead to a common endpoint. Complex diseases are diagnosed by summing up multiple factors that may be causes or mere consequences of the disease process. Disease “diagnosis” does not require the presence in the tissue of all of the abnormalities that may be “classically” seen in a given disease (Gianani et al., 2010; Odze, 2003). For example, at the polar extreme, CD is easily distinguished from UC by its classical ileal involvement (i.e., involvement of tissue at the end of the small intestine), fissures, granulomas, transmural inflammation (i.e., inflammation through the entire intestinal wall), fat wrapping of the intestine, patchy pathology, skip lesions, and patient presentation with bowel strictures or percutaneous fistulae. However, like UC, CD can be restricted to the colon, and the inflammatory infiltrates of CD and UC overlap. UC can be patchy, and the patient presentations of the two diseases can overlap extensively. Similarly, the genetics, pathology, and pathogenesis of IBD may differ between young and old patients with the same diagnosis (Imielinski et al.,
2009; Odze, 2003). Even when all classical aspects of a disease are present, the mechanism responsible for the pathology observed may differ from one person to another. Based on these considerations, it is no surprise that the genetics of T1D, CD, and UC are complex because different phenotypes may have been grouped into a single analysis.
This putative mechanistic heterogeneity is reflected in sometimes subtle, but quantifiable, characteristics of the disease process and pathology. Taking such differences into account can be used to identify disease subtypes that are more recognizable as molecularly defined pathological conditions and that more closely relate to specific pathogenetic mechanisms underpinned by distinct sets of genetic risk loci (Figure A19-3). For example, variations in the ATG16L1 gene (i.e., hypomorphic expression in the mouse and homozygosity for the T300A variant in humans) result in abnormalities in Paneth cell granules and secretion (Cadwell et al., 2008, 2010). Paneth cells are innate immune epithelial cells positioned at the base of small intestinal crypts, where they secrete antimicrobial peptides and other factors that help shape the configuration of the intestine’s bacterial community. Abnormalities in Paneth cells are observed in the subset of CD patients homozygous for the T300A allele, thus defining a pathologic subtype of CD (Figure A19-2B). If one used criteria including Paneth cell abnormalities in CD diagnosis, the frequency of the ATG16L1 T300A allele would be higher in patients with the “Paneth cell subtype” of CD than in the CD population as a whole (Figure A19-3). If multiple risk loci contribute to such Paneth cell changes, one might be able to detect gene-gene interactions in this subset of patients compared to other subsets.
A similar situation exists in T1D. Biopsy specimens of the pancreas are virtually impossible to obtain. Therefore, T1D is defined clinically by the downstream consequences of destroying the insulin-secreting b cells of the pancreatic islets, namely, high blood glucose and absolute insulin dependence, rather than by the mechanisms for their destruction. It is, therefore, possible that several different pathologic processes result in this disease. T1D patients diagnosed under age 10 years frequently exhibit islet inflammation or insulitis, whereas patients diagnosed over age 10 years exhibit insulitis less frequently. More recently, this histopathological heterogeneity has become even more evident (Gianani et al., 2010), thanks to the Juvenile Diabetes Research Foundation nPOD project (http://www.jdrfnpod.org). As for CD, the diagnosis of T1D may reflect the presence of more than one pathogenetic mechanism and, thus, represent more than one disease subtype, although in T1D the HLA effects are an essential common pathway.
The concept that disease diagnoses include mechanism-based disease subtypes has many implications for interpreting human genetic studies and for understanding the relationship between the microbiome and genetic susceptibility, as discussed below. Including disease subtypes within a single diagnosis would decrease the power to define causal alleles and to detect gene-gene interactions that contribute to a single disease subtype. In this view, the difficulty of interpreting
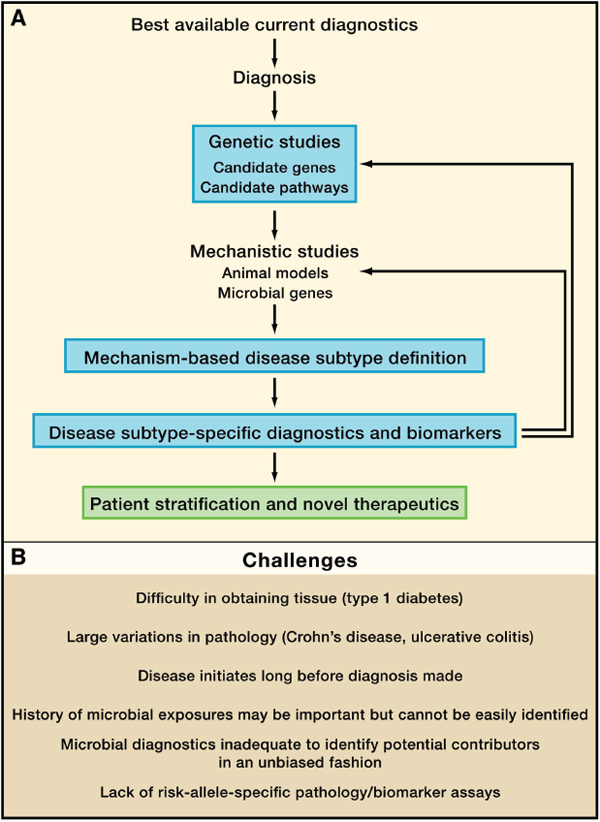
FIGURE A19-3 The iterative redefinition of mechanism-based disease subtypes. Here we present a conceptual workflow for breaking a broad disease diagnosis into its component subtypes by the iterative application of genetics and mechanistic studies. One output would be therapeutics based on disease subtype and patient stratification into groups more likely to respond to a given therapy or preventive strategy (A). (B) shows specific challenges for this process for type 1 diabetes and Crohn’s disease.
how multiple small genetic effects sum to predispose an individual to a clinical diagnosis may partly reflect insufficient precision in selection of specific phenotypes to study.
It is important to recognize that it is the power and informativeness of GWAS themselves that drive the concept of mechanism-based disease subtypes (Figure A19-2). In the absence of candidate genetic mechanisms for defining disease subtypes, there is limited clinical utility in focusing on low-frequency characteristics or subtypes within a larger diagnostic category that predicts patient outcome. We, therefore, argue for iterative high-precision phenotyping of patients into mechanism-based subtypes in future studies; this will allow more accurate interpretation of genetic, pathogenesis, outcome, and therapeutic studies (Figure A19-3). Such definitions must be iteratively reassessed as risk alleles are defined and disease mechanisms are delineated so that the field is not limited by inflexible definitions of disease that may obscure mechanistic heterogeneity. This type of approach is a necessary presage to so-called stratified or personalized medicine. The genetic and pathological complexity of T1D, CD, and UC is particularly well suited for testing whether iteratively redefining disease diagnoses can enhance the value of genetic and pathogenesis studies. Importantly, precision in disease categorization would make defining the impact of host-gene-microbial interactions on disease processes more robust.
Host-Gene-Microbial Interactions in Metagenetics
Metazoan organisms are complex communities that include a core organism in combination with a veritable zoo of other organisms that live on or in the body—our microbiome. The microbiome includes eukaryotic viruses, Eukarya, bacteria viruses, Bacteria, Archaea, and, for many, helminths (Virgin et al., 2009; Kau et al., 2011; Garrett et al., 2010b; Spor et al., 2011). The importance of understanding the microbiome has been repeatedly emphasized, giving rise to a large number of international human microbiome projects (e.g., https://commonfund.nih.gov/hmp/, http://www.metahit.eu/) that have focused initially on the bacterial component of the microbiome. The host plus non-host genes of this polyglot and interactive community constitute our metagenome (Box A19-1). A critical emerging concept is that bacterial and viral interactions in the pathogenesis of inflammatory disease occur in a host gene-specific fashion (see below; Virgin et al., 2009; Cadwell et al., 2010; Bloom et al., 2011; Elinav et al., 2011). Understanding the metagenome is, therefore, highly relevant to understanding T1D, UC, CD, and other common multifactorial diseases.
Intestinal bacteria play a role in driving IBD, and emerging data support a similar view for T1D (Wen et al., 2008; Giongo et al., 2011; Roesch et al., 2009). The evidence that bacteria play a role in IBD includes two major observations: that surgical diversion of the fecal stream ameliorates inflammation (Sartor, 2008), and that antibiotics help some patients. In mouse models of colitis, viruses,
bacteria, or both acting together can contribute to the pathology via signaling through innate immune sensors and regulation of pro- and anti-inflammatory cytokines (Levine et al., 2011; Maloy and Powrie, 2011; Khor et al., 2011). For many years, enterovirus infection has been associated with T1D (e.g., Yeung et al., 2011; Oikarinen et al., 2011; Stene et al., 2010), and the major sensor for enterovirus RNA is the T1D susceptibility gene IFIH1, encoding MDA5 (Nejentsev et al., 2009; McCartney et al., 2011). The mechanisms for these associations between components of the microbiome and T1D, CD, or UC have proven elusive. The lack of integration among scientific disciplines and among training programs, together with limitations in technology, has substantially limited the understanding of metagenomic contributions to disease.
Adult mammals are permanently infected by many viruses, and they are populated by large site-specific bacterial and phage communities without overt negative effects (Virgin et al., 2009; Foxman and Iwasaki, 2011; Spor et al., 2011). Thus, the bacterial microbiota (and their phages) and the eukaryotic virome are two major (but not the only) contributors to the metagenome. The intestinal microbiome plays a critical role in mammalian physiology by synthesizing vitamins and harvesting energy from food (Spor et al., 2011; Kau et al., 2011). Further, the normal function of the innate immune system, which is critically involved in the pathogenesis of T1D, UC, and CD, is regulated by both chronic viral infections and resident bacterial communities (Barton et al., 2007; White et al., 2010; Virgin et al., 2009; Spor et al., 2011; Kau et al., 2011). The microbiome and metagenome vary from person to person based on host genetics, diet, exposure, geography (including westernization as approximated by the gross national product of a country), socioeconomic status, mode of delivery, gestational age at birth, breast feeding, antibiotic use, and additional factors (Virgin et al., 2009; Benson et al., 2010; Spor et al., 2011; Kau et al., 2011; Penders et al., 2006). Such variations could certainly provide environmental inputs that contribute to the incidence of T1D, UC, and CD, within the genetic foundation revealed by GWAS (Bach, 2002; Vehik and Dabelea, 2011; Ehlers and Kaufmann, 2010).
There are extensive interactions between host and non-host genes within the metagenome, and bacteria and eukaryotic viruses alter our physiology and fitness (Spor et al., 2011; Virgin et al., 2009; Hansen et al., 2010). These genetic interactions within the metagenome create a complex and poorly understood host-genemicrobial interaction matrix that can define phenotype. Host genes, such as those involved in innate and adaptive immunity (e.g., NOD2, NLRP6, HLA, TLR2, and MYD88), shape the bacterial microbiota (Spor et al., 2011; Elinav et al., 2011; Wen et al., 2008). Forward genetic screens in mice suggest that resistance to individual viruses involves hundreds of genes (Virgin et al., 2009), making it likely that many host genes regulate the microbiome (and thus the metagenome). Importantly, key interactions between members of the microbiome have been and are increasingly being reported. For example, murine norovirus can trigger an intestinal inflammatory process in mice with a mutation in Atg16L1. This process
can be treated with antibiotics and is thus presumed to be bacteria dependent (Cadwell et al., 2010) (Figure A19-4). The existence of such interactions indicates that it will be important to consider that the disease contributions of microbiome members (e.g., helminths and bacteria) are potentially dependent on each other.
Metagenetic influences on disease could occur in various ways. Familial disease clustering may reflect intrafamilial behavioral and dietary factors that define the metagenome. A major influence on the human metagenome may be vertical
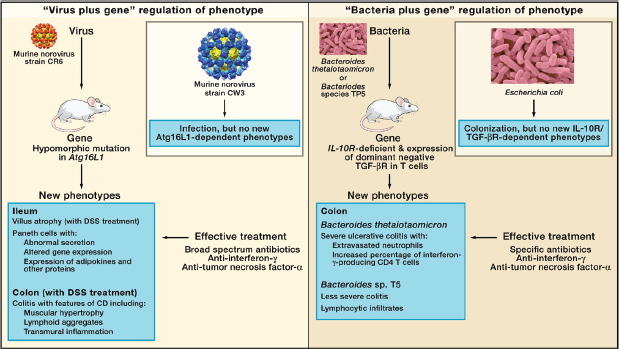
FIGURE A19-4 Microbe plus gene interactions determine inflammatory bowel disease phenotypes. (A) Two recent studies analyzed the capacity of two different strains of murine norovirus, MNV strain CR6 versus MNV strain CW3, to trigger phenotypes when orally inoculated into mice with a mutation in the Crohn’s disease risk gene Atg16L1 (Cadwell et al., 2008, 2010). This mutation results in decreased expression of Atg16L1 protein (hypomorphic, Atg16L1HM). Even though MNV CW3 and MNV CR6 are closely related, they have different effects on intestinal pathology in Atg16L1HM mice. Some of these interactions are observed only when mice are fed the chemical dextran sodium sulfate (DSS). (B) Two other studies analyzed the capacity of two different species of Bacteroides to trigger phenotypes in combination with mutations in the IL-10 receptor and T cell expression of a dominant-negative form of the TGF-β receptor (dnKO mice) (Bloom et al., 2011; Kang et al., 2008). dnKO mice are cured of their spontaneous colitis by treatment with antibiotics, but oral feeding of “cured” mice with fecal contents or specific bacteria reinduces disease. Even though Bacteroides thetaiotaomicron and Bacteroides sp. TP5 are closely related, they induce different forms of inflammation when fed to antibiotic-cured dnKO mice. In the same studies, dysbiosis (Box A19-1) with increases in the numbers of Enterobacteriaceae was noted in dnKO mice prior to curing the mice with antibiotics. However, E. coli inoculation did not trigger the pathologies seen with either Bacteroides species.
transmission of the maternal microbiome. In the controlled environment of mouse colonies, the bacterial microbiota is clearly maternally inherited. Furthermore, this microbiome can have profound pathological effects in mice carrying specific mutations, such that studies of host-gene functions must now consider contributions of the metagenome (see below).
The situation in humans is more complex (Hansen et al., 2011; Benson et al., 2010), although the importance of early environmental exposures has been well documented, including studies with mono- and dizygotic twins (Turnbaugh et al., 2009). Children delivered vaginally initially acquire a distinctly different intestinal bacterial microbiota than those born by caesarian section (Penders et al., 2006; Dominguez-Bello et al., 2010). In a meta-analysis, delivery by caesarean section increases the risk of T1D by 20% (Cardwell et al., 2008). An association with increased risk for T1D has also been reported for higher birth weight (Cardwell et al., 2010) and early infant diet (Pflüger et al., 2010) (Figure A19-5). Furthermore, changing microbial exposures and infections likely has a major
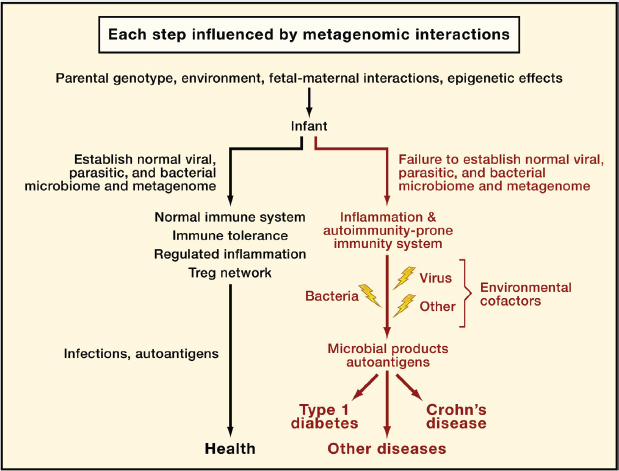
FIGURE A19-5 A metagenetic view of developing normal and pathological immune responses. This flowchart depicts stages in the development of normal immune responses or autoimmune and inflammatory diseases at which metagenetic interactions (i.e., gene-gene and gene-microbe interactions) might play a determining role. ‘‘Microbial products’’ refers to molecules that interact with host innate immune sensors and initiate inflammation.
influence on the incidence of other diseases. The dramatic rise in the incidence of allergy, asthma, and T1D in the last 60 years correlates with vast improvements in health care and sanitation (Bach, 2002; Vehik and Dabelea, 2011; Ehlers and Kaufmann, 2010). For example, severe rhinovirus infection before the age of 3 years coupled to an asthma-predisposing inherited host genome has been associated with increased risk of asthma (Foxman and Iwasaki, 2011). Thus, the metagenome could contribute to disease susceptibility and potentially explain a proportion of the familial clustering, the so-called “missing heritability” of multifactorial diseases.
Metagenetic Effects on Immunity and Autoimmunity
GWAS point to a fundamental role for the immune system in the pathogenesis of T1D, UC, and CD. An emerging concept is that bacterial and viral interactions contribute to both normal immune physiologies and abnormal pathologic responses that occur in a host gene-specific fashion (Virgin et al., 2009; Cadwell et al., 2010; Bloom et al., 2011; Garrett et al., 2007; Elinav et al., 2011). The microbiome has significant effects on the development of the immune system (Lee et al., 2011; Mazmanian et al., 2008; Sartor, 2008; Ivanov et al., 2009) and on physiology, including susceptibilities to obesity and the metabolic syndrome (Kau et al., 2011). Serum IgE responses to antigenic challenge are lower in mice colonized with Clostridium, confirming that bacteria can have profound effects on systemic immune responses involved in allergy (Atarashi et al., 2011). Central nervous system (CNS) inflammation induced by autoantigen is limited in germfree mice, but it can be restored by colonization with specific bacteria (Lee et al., 2011). In the non-obese diabetic (NOD) mouse model mouse model, autoimmune diabetes is regulated by a MyD88-dependent interaction of intestinal microbes with the innate immune system (Wen et al., 2008). Germ-free K/BxN T cell receptor transgenic mice are resistant to arthritis caused by autoantibodies to the self-antigen glucose-6-phosphate isomerase. When these animals are colonized with segmented filamentous bacteria (SFB), they regenerate TH17 responses in the small intestine, autoantibody production, and arthritis (Wu et al., 2010). Furthermore, the normal intestinal microbiome is essential for effective resistance to oral inoculation with Toxoplasma gondii and for generating appropriate CD8+ T cell responses to influenza (Ichinohe et al., 2011; Benson et al., 2009).
The mechanisms responsible for these observations are under intensive investigation. One recent study shows that intestinal bacteria induce Tregs in an antigen-specific and T cell receptor-dependent fashion (Lathrop et al., 2011). This is a key observation because it provides a mechanism, in addition to thymic exposure to self-antigens, for how regulatory responses can be generated to blunt inflammation. Given the continuous presence of the stimulating antigens for these Tregs in the normal intestinal microbiome, such cells could have profound effects on both intestinal and systemic immune responses, including responses
to self-antigens. This is particularly important because it has been reported that T lymphocytes migrate to the intestine to accept differentiation signals regulating autoimmune responses (Esplugues et al., 2011). It was also shown that injection of Staphylococcus aureus or its superantigen S. aureus enterotoxin B (SEB) was able to induce these intestinal regulatory TH17 cells, which is consistent with SEB injection being immune tolerogenic (Esplugues et al., 2011). These studies suggest that variation in the metagenome between individual humans, between mice in different research facilities, or even between animals from different cages within the same facility could have profound effects on many aspects of the immune response. This concept has key implications for the interpretation of mouse studies. The microbiome is maternally inherited in mice, but it can differ among research facilities; there may even be significant microenvironmental variation between cages of mice or between mice born of different dams. Given that the microbiome influences immunity so extensively, experiments must control for these factors. Currently, this is neither consistently performed nor required by peer reviewers.
Host-Gene-Metagenome Interactions in UC and CD
Correlations between communities of intestinal bacteria and CD or UC have led to the concept of dysbiosis (Box A19-1) as a contributor to these diseases (e.g., Sartor, 2008). This important hypothesis emphasizes the potential role that changes in the bacterial microbiota have on disease. However, now this hypothesis needs to expand and include both nonbacterial components of the metagenome and highly specific interactions between individual bacteria or viruses and host genes, which have recently been identified as contributors to disease pathogenesis. The relative contribution of dysbiosis versus the contribution of single organisms within the microbiome to the etiology of complex inflammatory diseases is unresolved. A confounding element has been the reliance on antibiotic treatment to assess bacteria as causes for intestinal disease. Because antibiotics can treat enteric inflammatory disease triggered by viruses (Figure A19-4) (Cadwell et al., 2010), a broader approach—including proof that specific bacteria or viruses are both necessary and sufficient for a phenotype—will be required to understand metagenetics of disease. Specific risk alleles for CD or UC could affect IBD by altering bacterial populations or individual bacterial types (Maloy and Powrie, 2011; Garrett et al., 2010b; Spor et al., 2011). Data from numerous mouse models of transmissible colitis confirm this point and are discussed below. The complexity of these reciprocal interactions between host and non-host genes within the metagenome underlines the critical need for new concepts and methodologies in computational and systems biology that can deal with individual host-gene microbial interactions in the broader context of the metagenome.
IBD in humans and mice is associated with alterations in the balance between TH1, TH17, and Treg cells, and this balance is dependent on the metagenome
(Garrett et al., 2010b; Maloy and Powrie, 2011). The relevance of these studies to human CD and UC is strongly supported by the identification of genes regulating these pathways in GWAS on IBD (see above). The role of specific bacteria and helminths in regulating these T cell responses in both the small and large intestine is highly relevant to understanding the genetics and pathogenesis of IBD. Polysaccharide A synthesized by the common colonic commensal Bacteroides fragilis induces Tregs that secrete IL-10 and inhibit intestinal inflammation (Round and Mazmanian, 2010; Mazmanian et al., 2008). Similarly, a protein antigen secreted by the intestinal helminth Heligmosomoides polygyrus induces Foxp3+ Treg cells in vitro and in vivo in mice (Grainger et al., 2010). Furthermore, enteric carriage of a community of Clostridium species induces IL-10-secreting Foxp3+ Tregs in the colon, likely via induction of TGF-β (Atarashi et al., 2011). These findings are interesting in light of the ubiquity of Bacteroides and Clostridia as commensal organisms in human and mouse, and differences in human carriage of helminths across the world.
In mice, the presence of distant relatives of Clostridia, called SFB, drives resistance to the enteric pathogen Citrobacter rodentium and the induction of CD4+ TH17 cells in the lamina propria of the small intestine (Ivanov et al., 2009; Gaboriau-Routhiau et al., 2009). The discovery that SFB influence CD4+ T cell differentiation was made when investigators noticed differences in intestinal TH17 cell numbers between mice of the same strain purchased from different vendors, followed by the demonstration that co-housing of these mice resulted in induction of TH17 cells (Ivanov et al., 2009). SFB are highly evolved for their commensal relationship with the mouse intestine (Sczesnak et al., 2011). Similar organisms have not yet been reported in humans, but it seems likely that similarly coevolved organisms will play a role in human intestinal biology and immunoregulation. The discovery of the role for SFB in CD4+ T cell responses is similar to the discovery of a virus-plus-gene trigger for an intestinal disease in mice with symptoms similar to those in CD (Cadwell et al., 2010). This finding occurred by comparing intestinal phenotypes in one strain of mice bred in two different facilities. Both of these findings underline the critical importance of directly analyzing the contributions of the entire microbiome, rather than individual components, in animal models of diseases.
Transmissible Colitis and Host-Gene-Metagenome Interactions
Recent studies have made the striking observation that genetically determined colitis is transmissible, revealing a key role for host genes in defining the microbiome and for metagenomic contributions to enteric disease. Mice lacking both Rag2 and the transcription factor T-bet develop colitis that can be transmitted from a mutant mother to wild-type fosterling mice (Garrett et al., 2007, 2010a). Although there are expansions of specific bacterial types in these mice, including Klebsiella pneumoniae and Proteus mirabilis, another cofactor, in addition to
these bacteria, is required to generate the colitis phenotype. This cofactor is not yet identified. Similarly, mice deficient in NLRP6, caspase- 1, IL-18, or ASC (all proteins that regulate the expression of proinflammatory cytokines such as IL-18) develop colitis that is transmissible to co-housed wild-type mice (Elinav et al., 2011). Recent studies in another mouse model of transmissible colitis, which has similarities to UC, provide an example of the specificity of host-gene-bacterial relationships and IBD (Figure A19-4) (Bloom et al., 2011; Kang et al., 2008). Mice lacking the IL-10 receptor and expressing a dominant-negative form of the TGF-β receptor in T lymphocytes develop IFN-γ- and TNF-α-dependent colitis (Kang et al., 2008). The disease is cured by antibiotic treatment and reinduced by co-housing diseased and cured animals or by simply feeding cured mice the common commensal bacteria Bacteroides thetaiotaomicron (B. theta) (Bloom et al., 2011). In the same mice, the related Bacteroides sp. TP5 induced a lymphocytic inflammatory infiltrate different from that induced by B. theta, indicating the remarkable specificity of host-gene-bacterial interactions (Figure A19-4). The authors noted dysbiosis in diseased animals with increased numbers of Enterobacteriaceae, but these bacteria did not induce disease despite being present in higher numbers in sick mice. This study shows that a single bacterial type can cause IBD-like pathology in the proper genetic setting, a bacteria-plus-gene interaction that triggers intestinal inflammation. Importantly, the observation that two closely related bacteria induce different pathologies in the same genetically susceptible host provides support for the concept that genes present in the non-host metagenome can determine a host phenotype.
A similar observation, in this case of a virus-plus-gene interaction that triggers IBD-like pathology, has been described in mice mutant for the CD risk gene Atg16L1 (Figure A19-4) (Cadwell et al., 2008, 2010). Abnormal Paneth cells were observed in humans carrying the ATG16L1 T300A allele and mice hypomorphic for expression of Atg16L1 raised in a conventional clean barrier (Cadwell et al., 2008). Importantly, the phenotype of the mice varied between different facilities and could be induced in mutant mice, but not wild-type mice, by inoculation with a specific strain of murine norovirus (Karst et al., 2003; Thackray et al., 2007). When these mice were challenged with dextran sodium sulfate (DSS), they developed inflammatory phenotypes specific to the combination of Atg16L1 mutation and an individual norovirus strain (Figure A19-4). Virus-triggered pathologies could be treated by blocking TNF-α or IFN-γ or by treatment with antibiotics. Interestingly, infection with murine norovirus enhances signaling through Nod1 and Nod2 via the induction of type 1 IFN, potentially providing a direct link between enteric viral infection and NOD signaling pathways implicated in IBD risk (Kim et al., 2011). These data raise the possibility that patterns of viral infection and specific components of the bacterial metagenome act together to influence the penetrance of UC and CD susceptibility risk alleles in humans. Furthermore, these data show that closely related viruses can have quite different effects on the phenotype of a host genetically prone to a disease process. This finding further
supports the concept that genes in the non-host metagenome can determine host phenotypes.
Host-Gene-Metagenome Interactions in T1D
For T1D, recent observations fit with a “perfect storm” scenario in which numerous events combine to increase susceptibility to disease development in early childhood (Figure A19-1). These events include susceptibility alleles in HLA class II genes and INS that cause increased autoreactivity against insulin, its precursors, and other islet antigens; lowered IL-2, IL-10, and IL-27 production and signaling; altered T cell receptor signaling and regulation (via, for example, susceptibility alleles in PTPN2, PTPN22, CTLA4, and IL2RA); and increased type 1 IFN production and responsiveness (Todd, 2010; Robinson et al., 2011; Bluestone et al., 2010). The “perfect T1D storm” is generated when these factors combine with a permissive, modern environment of widespread vitamin D deficiency (Cooper et al., 2011) and other still unidentified environmental factors (Figure A19-5). In particular, the T1D susceptibility genes and candidates IFIH1 (Nejentsev et al., 2009), GPR183 (EBI2) (Heinig et al., 2010), TLR7, TLR8 (Barrett et al., 2009), and FUT2 (Smyth et al., 2011) strongly suggest an etiological role for virus-induced, type 1 interferon production. A common knockout mutation of FUT2 in several populations causes the nonsecretor status (i.e., a lack of shedding of the A and B blood group antigens into saliva and intestinal secretions). This T1D-predisposing FUT2 genotype is also associated with increased risk of CD (McGovern et al., 2010; Franke et al., 2010), providing another direct mechanistic link between these two diseases and microbial infections. The FUT2 nonsecretor genotype is associated with resistance to certain strains of norovirus and Helicobacter pylori (Smyth et al., 2011). Investigations of the mechanisms involved in the FUT2 associations with chronic and infectious disease are urgently required, as is the case for many of the newly identified GWAS candidate genes.
Defining the Metagenome Now and in the Future
Technologies for analyzing human loci involved in complex diseases have, until recently, outstripped technologies for analyzing the metagenome. For example, single-nucleotide polymorphism (SNP)-based GWAS cover the entire human genome, although at low resolution, whereas most common tools and methods applied to the non-host metagenome focus on only one component, such as a particular bacteria, viruses, or phage. The non-host metagenome is so complex that researchers have focused on DNA sequencing, even though many organisms relevant to disease—including enteroviruses that have been linked to T1D and viruses that cause intestinal disease—have RNA genomes. Although our knowledge of the human gut metagenome is in its infancy, this metagenome can
now be explored in detail by deep, next-generation sequencing of both RNA and DNA, then stratified by host genotype, disease risk, or disease status. Investigators are increasingly using shotgun sequencing of RNA + DNA, which theoretically can detect any organism (e.g., Finkbeiner et al., 2008). However, studies to date have often relied on the DNA sequencing of 16S rRNA genes of bacteria. This standard and reliable method has identified dysbiosis in IBD and T1D (Wen et al., 2008; Roesch et al., 2009; Giongo et al., 2011; Sartor, 2008; Garrett et al., 2010b). Whether these changes are causal or secondary to disease is unclear.
An outstanding example of consequences of relying on the analysis of only a subset of the metagenome is the recent appreciation that bacterial phage viruses are a major and dynamic part of the intestinal microbiome (Reyes et al., 2010). This adds an enteric bacterial “virome” to the eukaryotic virome that lives in our tissues (Virgin et al., 2009). Bacteria are not the only cells, in addition to host cells, that can be infected by viruses with consequent changes in biology. For example, an RNA virus infects the eukaryotic pathogen Leishmania and regulates the host inflammatory responses during parasite infection (Ives et al., 2011). Thus, like bacteria and their phages, all Eukarya in the microbiome are candidates for viral infection that might alter biological processes.
The tools to detect and quantify the entire non-host metagenome at a reasonable cost will undoubtedly develop rapidly as metagenomic sequencing technologies and computational approaches to phylogeny and microbe detection are developed and applied. Similarly, sequencing the entire host genome is becoming more cost efficient and practical. This wealth of data will set the stage for metagenetics, but meaningful and robust analyses of the complex interactions within the metagenome will require new computational tools and new conceptualizations of gene-gene and gene-microbe interactions.
Conclusion: The Metagenetics of Mechanism-Based Disease Subtypes
Here we have argued that two factors need to be considered as key contributors to the genetics and pathogenesis of complex inflammatory diseases, such as T1D, CD, and UC: specific host-gene-microbial interactions and the mechanistic heterogeneity of phenotypes that constitute complex diseases. Although we have used the lens of T1D, CD, and UC research to support these concepts, it is clear that these ideas may apply to a broader array of diseases as well. The striking effects of the microbiome on systemic immunity and on diseases that affect both visceral and mucosal tissues suggest that any physiologic process may be altered by the microbiome and gene-specific interactions of the microbiome with the host. At a minimum, the diverse diseases that have been revealed by GWAS to share risk alleles are strong candidates for considering the metagenome, rather than only the host genome, as contributing to health or disease.
The concepts of mechanism-defined disease subtypes and host-gene-microbial interactions cooperate in important ways. For example, if the single
diagnosis of CD or T1D includes multiple mechanistic phenotypes (Figure A19-2 and Figure A19-3), a specific host-gene-microbial interaction (Figure A19-4) might contribute to only one of these phenotypes. In this setting, the impact of interactions between genes in the metagenome, of either microbial or host origin, would be obscured. This could, for example, obscure the role of a single microbe in causing one mechanism-based disease subtype rather than causing all cases of a disease. Failure to identify such an agent would prevent the use of approaches that treat or vaccinate against the agent (Figure A19-2 and Figure A19-5). It is logical and anticipated that stratifying patients for treatment with pathway-specific drugs will improve outcomes and success of phase II and III clinical trials (Figure A19-3). This paradigm is highly effective and increasingly used in the treatment of cancer, but it also seems likely to benefit those with germline-based predisposition to disease as well.
Deconvoluting the complex matrix of interactions within the metagenome that contribute to disease will require more complete analyses of the metagenome. It also requires an iterative redefinition of disease subtypes using markers that distinguish between patients based on the mechanism responsible for injury rather than the presence of tissue injury per se. This ambitious goal is daunting to consider, but data discussed herein from human studies, animal studies, and analyses of the microbiome lead us to the inescapable conclusion that complex interactions within the metagenome control phenotypes. We must face this complexity head-on to solve the puzzle of the etiology and pathogenesis of complex diseases.
We, therefore, argue for the inclusion of the metagenome in human genetic studies for these diseases. We view complex diseases as “metagenetic,” reflecting the contributions of both host and non-host genes within the metagenome. The nonhost genes in the metagenome that are relevant to a disease might be viral, bacterial, or derived from additional members of the microbiome, which are still largely uncharacterized. Parasites likely play a critical role in some populations. These metagenetic interactions probably contribute to the development of disease at two levels (Figure A19-5). First, we envision the normal immune system developing via harmonious relationships within the metagenome. For example, the level of innate immunity in mice is regulated by chronic herpesvirus infection (Barton et al., 2007; White et al., 2010), and therefore acquisition of a specific chronic virus might predispose the host to either helpful or harmful responses to other components of the microbiome. It will be important to develop quantitative and robust ways to identify such a “normal” immune system. Second, once a poorly balanced immune system is generated, host-gene interactions, with either other host genes or the non-host metagenome, likely synergize to generate inappropriate levels of inflammation in response to microbial products (e.g., CD and UC) or to set the stage for development of HLA-dependent autoimmunity (T1D). Understanding this level of biological complexity will require the involvement of statisticians, computational biologists, geneticists, pathogenesis experts, virologists, bacteriologists, and parasitologists in an integrated fashion to identify
mechanistically important interactions. Such an integrated approach can then perhaps make sense of the metagenetics of complex diseases, to the advantage of us all.
Acknowledgments
The authors would like to acknowledge helpful conversations with Thad Stappenbeck, Ramnik Xavier, Emil Unanue, Jeff Gordon, Balfour Sartor, Adolfo Garcia-Sastre, and Dermot McGovern. We thank Tom Smith for providing the images of noroviruses used in Figure A19-4. H.W.V. is supported by the NCI, NIAID, NCRR, the Crohn’s and Colitis Foundation of America, and the Broad Medical Foundation. J.A.T. is supported by the NIDDK, NIHR, the Wellcome Trust, the Juvenile Diabetes Research Foundation International, and the European Union.
References
Anderson, C.A., Boucher, G., Lees, C.W., Franke, A., D’Amato, M., Taylor,K.D., Lee, J.C., Goyette, P., Imielinski, M., Latiano, A., et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252.
Arechiga, A.F., Habib, T., He, Y., Zhang, X., Zhang, Z.Y., Funk, A., and Buckner, J.H. (2009). Cutting edge: the PTPN22 allelic variant associated with autoimmunity impairs B cell signaling. J. Immunol. 182, 3343–3347.
Atarashi, K., Tanoue, T., Shima, T., Imaoka, A., Kuwahara, T., Momose, Y., Cheng, G., Yamasaki, S., Saito, T., Ohba, Y., et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341.
Bach, J.F. (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920.
Barrett, J.C., Clayton, D.G., Concannon, P., Akolkar, B., Cooper, J.D., Erlich, H.A., Julier, C., Morahan, G., Nerup, J., Nierras, C., et al; Type 1 Diabetes Genetics Consortium. (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41, 703–707.
Barton, E.S., White, D.W., Cathelyn, J.S., Brett-McClellan, K.A., Engle, M., Diamond, M.S., Miller, V.L., and Virgin, H.W., 4th. (2007). Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447, 326–329.
Benson, A., Pifer, R., Behrendt, C.L., Hooper, L.V., and Yarovinsky, F. (2009). Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6, 187–196.
Benson, A.K., Kelly, S.A., Legge, R., Ma, F., Low, S.J., Kim, J., Zhang, M., Oh, P.L., Nehrenberg, D., Hua, K., et al. (2010). Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 107, 18933–18938.
Bloom, S.M., Bijanki, V.N., Nava, G.M., Sun, L., Malvin, N.P., Donermeyer, D.L., Dunne, W.M., Jr., Allen, P.M., and Stappenbeck, T.S. (2011). Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9, 390–403.
Bluestone, J.A., Herold, K., and Eisenbarth, G. (2010). Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464, 1293–1300.
Bottini, N., Vang, T., Cucca, F., and Mustelin, T. (2006). Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin. Immunol. 18, 207–213.
Brehm, M.A., Bortell, R., Diiorio, P., Leif, J., Laning, J., Cuthbert, A., Yang, C., Herlihy, M., Burzenski, L., Gott, B., et al. (2010). Human immune system development and rejection of human islet allografts in spontaneously diabetic NODRag1null IL2rgammanull Ins2Akita mice. Diabetes 59, 2265–2270.
Cadwell, K., Liu, J.Y., Brown, S.L., Miyoshi, H., Loh, J., Lennerz, J.K., Kishi, C., Kc, W., Carrero, J.A., Hunt, S., et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263.
Cadwell, K., Patel, K.K., Maloney, N.S., Liu, T.C., Ng, A.C., Storer, C.E., Head, R.D., Xavier, R., Stappenbeck, T.S., and Virgin, H.W. (2010). Virus-plussusceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145.
Calderon, B., Carrero, J.A., Miller, M.J., and Unanue, E.R. (2011a). Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc. Natl. Acad. Sci. USA 108, 1561–1566.
Calderon, B., Carrero, J.A., Miller, M.J., and Unanue, E.R. (2011b). Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc. Natl. Acad. Sci. USA 108, 1567–1572.
Cardwell, C.R., Stene, L.C., Joner, G., Cinek, O., Svensson, J., Goldacre, M.J., Parslow, R.C., Pozzilli, P., Brigis, G., Stoyanov, D., et al. (2008). Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia 51, 726–735.
Cardwell, C.R., Stene, L.C., Joner, G., Davis, E.A., Cinek, O., Rosenbauer, J., Ludvigsson, J., Castell, C., Svensson, J., Goldacre, M.J., et al. (2010). Birthweight and the risk of childhood-onset type 1 diabetes: a meta-analysis of observational studies using individual patient data. Diabetologia 53, 641–651.
Clayton, D.G. (2009). Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 5, e1000540.
Cooper, J.D., Smyth, D.J., Walker, N.M., Stevens, H., Burren, O.S., Wallace, C., Greissl, C., Ramos-Lopez, E., Hyppönen, E., Dunger, D.B., et al. (2011). Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes 60, 1624–1631.
Cotsapas, C., Voight, B.F., Rossin, E., Lage, K., Neale, B.M., Wallace, C., Abecasis, G.R., Barrett, J.C., Behrens, T., Cho, J., et al; on behalf of the FOCiS Network of Consortia. (2011). Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 7, e1002254.
Dendrou, C.A., Plagnol, V., Fung, E., Yang, J.H., Downes, K., Cooper, J.D., Nutland, S., Coleman, G., Himsworth, M., Hardy, M., et al. (2009). Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat. Genet. 41, 1011–1015.
Dominguez-Bello, M.G., Costello, E.K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., and Knight, R. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 107, 11971–11975.
Ehlers, S., and Kaufmann, S.H.; Participants of the 99(th) Dahlem Conference. (2010). Infection, inflammation, and chronic diseases: consequences of a modern lifestyle. Trends Immunol. 31, 184–190.
Elinav, E., Strowig, T., Kau, A.L., Henao-Mejia, J., Thaiss, C.A., Booth, C.J., Peaper, D.R., Bertin, J., Eisenbarth, S.C., Gordon, J.I., and Flavell, R.A. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757.
Esplugues, E., Huber, S., Gagliani, N., Hauser, A.E., Town, T.,Wan, Y.Y., O’Connor, W., Jr., Rongvaux, A., Van Rooijen, N., Haberman, A.M., et al. (2011). Control of TH17 cells occurs in the small intestine. Nature 475, 514–518.
Fehrmann, R.S.N., Jansen, R.C., Veldink, J.H., Westra, H.J., Arends, D., Bonder, M.J., Fu, J., Deelen, P., Groen, H.J.M., Smolonska, A., et al. (2011). Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 7, e1002197.
Finkbeiner, S.R., Allred, A.F., Tarr, P.I., Klein, E.J., Kirkwood, C.D., and Wang, D. (2008). Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog. 4, e1000011.
Foxman, E.F., and Iwasaki, A. (2011). Genome-virome interactions: examining the role of common viral infections in complex disease. Nat. Rev. Microbiol. 9, 254–264.
Franke, A., McGovern, D.P., Barrett, J.C., Wang, K., Radford-Smith, G.L., Ahmad, T., Lees, C.W., Balschun, T., Lee, J., Roberts, R., et al. (2010). Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 42, 1118–1125.
Gaboriau-Routhiau, V., Rakotobe, S., Lécuyer, E., Mulder, I., Lan, A., Bridonneau, C., Rochet, V., Pisi, A., De Paepe, M., Brandi, G., et al. (2009). The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689.
Garrett, W.S., Lord, G.M., Punit, S., Lugo-Villarino, G., Mazmanian, S.K., Ito, S., Glickman, J.N., and Glimcher, L.H. (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45.
Garrett, W.S., Gallini, C.A., Yatsunenko, T., Michaud, M., DuBois, A., Delaney, M.L., Punit, S., Karlsson, M., Bry, L., Glickman, J.N., et al. (2010a). Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300.
Garrett, W.S., Gordon, J.I., and Glimcher, L.H. (2010b). Homeostasis and inflammation in the intestine. Cell 140, 859–870.
Gianani, R., Campbell-Thompson, M., Sarkar, S.A., Wasserfall, C., Pugliese, A., Solis, J.M., Kent, S.C., Hering, B.J., West, E., Steck, A., et al. (2010). Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia 53, 690–698.
Giongo, A., Gano, K.A., Crabb, D.B., Mukherjee, N., Novelo, L.L., Casella, G., Drew, J.C., Ilonen, J., Knip, M., Hyöty, H., et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 5, 82–91.
Glocker, E.O., Kotlarz, D., Boztug, K., Gertz, E.M., Schäffer, A.A., Noyan, F., Perro, M., Diestelhorst, J., Allroth, A., Murugan, D., et al. (2009). Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045.
Goodman, A.L., Kallstrom, G., Faith, J.J., Reyes, A., Moore, A., Dantas, G., and Gordon, J.I. (2011). Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 108, 6252–6257.
Grainger, J.R., Smith, K.A., Hewitson, J.P., McSorley, H.J., Harcus, Y., Filbey, K.J., Finney, C.A., Greenwood, E.J., Knox, D.P., Wilson, M.S., et al. (2010). Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-β pathway. J. Exp. Med. 207, 2331–2341.
Hansen, E.E., Lozupone, C.A., Rey, F.E., Wu, M., Guruge, J.L., Narra, A., Goodfellow, J., Zaneveld, J.R., McDonald, D.T., Goodrich, J.A., et al. (2011). Pan-genome of the dominant human gutassociated archaeon Methanobrevibacter smithii, studied in twins. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4599–4606.
Hansen, J., Gulati, A., and Sartor, R.B. (2010). The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol. 26, 564–571.
Heinig, M., Petretto, E., Wallace, C., Bottolo, L., Rotival, M., Lu, H., Li, Y., Sarwar, R., Langley, S.R., Bauerfeind, A., et al; Cardiogenics Consortium. (2010). A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 467, 460–464.
Ichinohe, T., Pang, I.K., Kumamoto, Y., Peaper, D.R., Ho, J.H., Murray, T.S., and Iwasaki, A. (2011). Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 108, 5354–5359.
Imielinski, M., Baldassano, R.N., Griffiths, A., Russell, R.K., Annese, V., Dubinsky, M., Kugathasan, S., Bradfield, J.P., Walters, T.D., Sleiman, P., et al; Western Regional Alliance for Pediatric IBD; International IBD Genetics Consortium; NIDDK IBD Genetics Consortium; Belgian-French IBD Consortium; Wellcome Trust Case Control Consortium. (2009). Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 41, 1335–1340.
Ivanov, I.I., Atarashi, K., Manel, N., Brodie, E.L., Shima, T., Karaoz, U., Wei, D., Goldfarb, K.C., Santee, C.A., Lynch, S.V., et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498.
Ives, A., Ronet, C., Prevel, F., Ruzzante, G., Fuertes-Marraco, S., Schutz, F., Zangger, H., Revaz-Breton, M., Lye, L.F., Hickerson, S.M., et al. (2011). Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331, 775–778.
Kang, S.S., Bloom, S.M., Norian, L.A., Geske, M.J., Flavell, R.A., Stappenbeck, T.S., and Allen, P.M. (2008). An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 5, e41.
Karst, S.M., Wobus, C.E., Lay, M., Davidson, J., and Virgin, H.W., 4th. (2003). STAT1-dependent innate immunity to a Norwalk-like virus. Science 299, 1575–1578.
Kau, A.L., Ahern, P.P., Griffin, N.W., Goodman, A.L., and Gordon, J.I. (2011). Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336.
Khor, B., Gardet, A., and Xavier, R.J. (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317.
Kim, Y.G., Park, J.H., Reimer, T., Baker, D.P., Kawai, T., Kumar, H., Akira, S., Wobus, C., and Núρez, G. (2011). Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe 9, 496–507.
Lathrop, S.K., Bloom, S.M., Rao, S.M., Nutsch, K., Lio, C.W., Santacruz, N., Peterson, D.A., Stappenbeck, T., and Hsieh, C.S. (2011). Peripheral education of the immune system by colonic commensal microbiota. Nature 10.1038/nature10434.
Lee, Y.K., Menezes, J.S., Umesaki, Y., and Mazmanian, S.K. (2011). Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4615–4622.
Levine, B., Mizushima, N., and Virgin, H.W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335.
Long, S.A., Cerosaletti, K., Wan, J.Y., Ho, J.C., Tatum, M., Wei, S., Shilling, H.G., and Buckner, J.H. (2011). An autoimmune-associated variant in PTPN2 reveals an impairment of IL-2R signaling in CD4(+) T cells. Genes Immun. 12, 116–125.
Maloy, K.J., and Powrie, F. (2011). Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306.
Mazmanian, S.K., Round, J.L., and Kasper, D.L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625.
McCartney, S.A., Vermi, W., Lonardi, S., Rossini, C., Otero, K., Calderon, B., Gilfillan, S., Diamond, M.S., Unanue, E.R., and Colonna, M. (2011). RNA sensor-induced type I IFN prevents diabetes caused by a b cell-tropic virus in mice. J. Clin. Invest. 121, 1497–1507.
McGovern, D.P., Jones, M.R., Taylor, K.D., Marciante, K., Yan, X., Dubinsky, M., Ippoliti, A., Vasiliauskas, E., Berel, D., Derkowski, C., et al; International IBD Genetics Consortium. (2010). Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum. Mol. Genet. 19, 3468–3476.
Melmed, G.Y., and Targan, S.R. (2010). Future biologic targets for IBD: potentials and pitfalls. Nat. Rev. Gastroenterol. Hepatol. 7, 110–117.
Nejentsev, S., Walker, N., Riches, D., Egholm, M., and Todd, J.A. (2009). Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387–389.
Odze, R. (2003). Diagnostic problems and advances in inflammatory bowel disease. Mod. Pathol. 16, 347–358.
Oikarinen, S., Martiskainen, M., Tauriainen, S., Huhtala, H., Ilonen, J., Veijola, R., Simell, O., Knip, M., and Hyo ¨ ty, H. (2011). Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 60, 276–279.
Penders, J., Thijs, C., Vink, C., Stelma, F.F., Snijders, B., Kummeling, I., van den Brandt, P.A., and Stobberingh, E.E. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521.
Pflüger, M., Winkler, C., Hummel, S., and Ziegler, A.G. (2010). Early infant diet in children at high risk for type 1 diabetes. Horm. Metab. Res. 42, 143–148.
Rashid, S.T., Corbineau, S., Hannan, N., Marciniak, S.J., Miranda, E., Alexander, G., Huang-Doran, I., Griffin, J., Ahrlund-Richter, L., Skepper, J., et al. (2010). Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest. 120, 3127–3136.
Reyes, A., Haynes, M., Hanson, N., Angly, F.E., Heath, A.C., Rohwer, F., and Gordon, J.I. (2010). Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338.
Rivas, M.A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C.K., Boucher, G., Ripke, S., Ellinghaus, D., Burtt, N., et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 10.1038/ng.952.
Robinson, T., Kariuki, S.N., Franek, B.S., Kumabe, M., Kumar, A.A., Badaracco, M., Mikolaitis, R.A., Guerrero, G., Utset, T.O., Drevlow, B.E., et al. (2011). Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J. Immunol. 187, 1298–1303.
Roesch, L.F., Lorca, G.L., Casella, G., Giongo, A., Naranjo, A., Pionzio, A.M., Li, N., Mai, V., Wasserfall, C.H., Schatz, D., et al. (2009). Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 3, 536–548.
Rossin, E.J., Lage, K., Raychaudhuri, S., Xavier, R.J., Tatar, D., Benita, Y., Cotsapas, C., and Daly, M.J.; International Inflammatory Bowel Disease Genetics Constortium. (2011). Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 7, e1001273.
Round, J.L., and Mazmanian, S.K. (2010). Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 107, 12204–12209.
Sartor, R.B. (2008). Microbial influences in inflammatory bowel diseases. Gastroenterology 134, 577–594.
Sczesnak, A., Segata, N., Qin, X., Gevers, D., Petrosion, J.F., Huttenhower, C., Littman, D.R., and Ivanov, I.I. (2011). The genome of Th17 cell-inducing segmented filamentous bacteria reveals extensive auxotrophy and adaptations to the intestinal environment. Cell Host Microbe 10, 260–272.
Smyth, D.J., Cooper, J.D., Howson, J.M.M., Clarke, P., Downes, K., Mistry, T., Stevens, H., Walker, N.M., and Todd, J.A. (2011). FUT2 non-secretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes. Published online October 24, 2011. 10.2337/db11-0638.
Spor, A., Koren, O., and Ley, R. (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 9, 279–290.
Stappenbeck, T.S., Rioux, J.D., Mizoguchi, A., Saitoh, T., Huett, A., Darfeuille-Michaud, A., Wileman, T., Mizushima, N., Carding, S., Akira, S., et al. (2011). Crohn disease: a current perspective on genetics, autophagy and immunity. Autophagy 7, 355–374.
Stene, L.C., Oikarinen, S., Hyöty, H., Barriga, K.J., Norris, J.M., Klingensmith, G., Hutton, J.C., Erlich, H.A., Eisenbarth, G.S., and Rewers, M. (2010). Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 59, 3174–3180.
Thackray, L.B., Wobus, C.E., Chachu, K.A., Liu, B., Alegre, E.R., Henderson, K.S., Kelley, S.T., and Virgin, H.W., 4th. (2007). Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J. Virol. 81, 10460–10473.
Todd, J.A. (2010). Etiology of type 1 diabetes. Immunity 32, 457–467. Turley, S.J., Lee, J.W., Dutton-Swain, N., Mathis, D., and Benoist, C. (2005). Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc. Natl. Acad. Sci. USA 102, 17729–17733.


























