
“The problem is that most antimicrobial agents don’t conform to [Lipinski’s] rules.”
Karen Shaw
“There are three, and only three, methods that I can think of that have a chance of reviving the discovery of new natural antibacterial natural products.”
Chaitan Khosla
Two speakers addressed the challenges of identifying potential new antibiotics capable of overcoming resistance. Karen Shaw, Vice President of Biology at Cubist Pharmaceuticals, presented some advice on how to conduct screening and described some of the pitfalls based on knowledge accumulated in the pharmaceutical industry. Chaitan Khosla, Professor of Chemistry and Chemical Engineering at Stanford University and Director of the Stanford Institute of Chemical Biology, spoke about the approaches that he believes have a chance of reviving the discovery of new natural antibacterial agents.
CHALLENGES IN DISCOVERING NEW ANTIBIOTICS THROUGH SCREENING
In the 1990s, genomics promised a wealth of new targets for antibiotic discovery, but that promise has gone unfilled, said Karen Shaw. “The bottom line is that genomics has not revealed new targets that gave us new drugs.” The 1990s also saw the failure of screening and optimization paradigms for both synthetic molecule collections and natural product libraries, she said. To illustrate the failure of both approaches, she recounted the results of a seven-year effort at GlaxoSmithKline that examined 300 genes that were conserved among bacteria, did not have a human homolog, and were essential to bacterial survival. The company conducted 70 high-throughput screening campaigns, producing 16 hits that resulted in five leads, two of which were optimized and none of which progressed to human clinical trials. “So why aren’t we finding new inhibitors?” asked Shaw.
One answer is that big pharmaceutical libraries were designed largely to follow Lipinsky’s rules1 that have guided the search for drugs in other therapeutic areas. “The problem is that most antimicrobial agents don’t conform to these rules,” said Shaw. Even with the advent of new tools such as genomics, researchers in numerous laboratories kept finding the same compounds repeatedly, as was the case with the compound actinonin, a peptide deformylase inhibitor, that was discovered simultaneously by many laboratories. Efforts to use combinatorial chemistry to create new libraries did not generate significant new leads, a failure that Shaw pinned on the fact that the design of the combinatorial libraries being tested was usually driven by the needs of other therapeutic areas. Natural product screening produced many positive hits, but there was a problem prioritizing which compounds to pursue and how to synthesize what were often complex molecular structures.
Shaw noted that too often, screens for antimicrobial activity and enzyme activity are not concordant. “Those two things are often not linked, more often than one would like to see,” said Shaw. Off-target toxicity is often the real source of antimicrobial activity, even with compounds generated in chemical optimization programs. Cytotoxicity assays can also provide false positives or even rule out potential compounds too early if mammalian cell toxicity results from a functional group that would have had the potential to be eliminated in optimization work. On the other hand, molecules that appear “clean,” that is, they are not toxic to mammalian cells, might be nonspecifically absorbed by proteins such as albumin and therefore at lower effective concentrations in the assay medium.
_________________
1 Lipinsky’s rules are a set of five criteria for the design of orally available drugs.
Another problem in screening arises from the reliance on model microbial organisms. “We’ve known for quite a long time, especially in the genomics era, that testing in one species is not sufficient,” said Shaw. She added that some species have bypass pathways that are absent in other species, while there are duplications of genes in some species and not others. She reiterated Silver’s earlier remark that the field must start focusing on dual targeting to avoid and surmount resistance.
Cell penetrance is an issue in gram-negative bacteria given that any compound has to get past two different cell membranes to gain entry to the cell. Shaw noted that one strategy for addressing that issue is to create a charged, water-soluble molecule that can pass through the outer membrane via embedded porins. Then, once the compound reaches the periplasm between the two membranes it assumes an uncharged, hydrophobic state that enables it to pass through the cytoplasmic membrane and bypass resistance-associated efflux pumps. This ability to change charge given the different environmental conditions is what makes the fluoroquinolones effective against gram-negative bacteria, she explained.
Natural products have been the subject of intense screening over many decades, yielding large numbers of active compounds but few novel ones with suitable biological mechanisms. Most of the hits identified in natural products screens, said Shaw, are generally toxic. “Potency is not a key parameter, mechanism is,” she said. Too often, investigators pursue compounds with the best results in terms of minimum inhibitory concentration (MIC), but these compounds tend to be generally toxic. “What matters is having a molecule in hand, even if it’s a weak hit, and a target that it binds to,” Shaw explained, since that then gives medicinal chemists a molecule that they can try to optimize to improve its bacterial toxicity.
This last idea, she said, brings up an important philosophical question: “Are you looking for a drug, or are you looking for a hit? If you’re after a hit, you start with the most susceptible organism you can find with the idea that you’ll identify many compounds that you can then sort through later,” she said. “If you want to find a drug, you start out with the most resistant organism and screen your library with the hope that you’ll find a molecule that you need to modify a little bit.”
Shaw then discussed synergy screening to look for molecules that inhibit β-lactamase or that are cell wall synergists. The key is to conduct large-scale searches for targets that have the potential to interact in vivo. Another approach, one that is quite old, is to screen pathways to identify inhibitors of bacterial macromolecule synthesis. More recently, protein-driven screens have looked at enzyme activity, binding properties, and the ability to inhibit protein–protein interactions. RNA targets have also become a focus of assays in the era of genomics.
Discussing the work that her group at Trius Therapeutics (which was purchased by her current employer, Cubist Pharmaceuticals, in July 2013) conducted to screen marine natural products, she said the philosophy driving the project was to identify a validated hit-target pair that could then become the focus of structure-based drug design to optimize target affinity, specificity, antibacterial activity, and in vivo properties. Based on the results of screening around 10,000 mixtures isolated from marine sources, it turned out to be easier to find compounds that were active versus gram-positive organisms and rare to find gram-negative activity against wild-type strains. The only way that she and her colleagues identified compounds active against gram-negative bacteria was to use an E. coli permeability mutant, and most of the hits identified using this mutant were also effective against gram-positive organisms.
After describing some of the details of how she and her colleagues screened the resulting hits for macromolecular synthesis inhibition activity, she noted that this type of assay is capable of distinguishing inhibition of pathways specific for DNA synthesis, RNA synthesis, protein synthesis, and cell wall assembly. Compounds that inhibited multiple pathways were discarded, while those that were selective for one pathway became the subject of further study. She added that it was possible to screen crude extracts using these assays rather than needing to isolate the individual components of the extracts (Figure 3-1). Using this approach, her group was able to screen 2,000 fractions in roughly nine months, which she characterized as good throughput, and follow those initial results with resistance studies to quickly prioritize potential hits for further study. For nucleic acid hits, subsequent screens looking at specific mechanisms of action, such as inhibitors of DNA polymerase or ligase, and at general nucleic acid binding or DNA intercalation were able to quickly and efficiently separate good leads from bad.
One interesting profile that emerged during this effort was what Shaw called the “flatline” profile. This profile was seen with compounds that had antimicrobial activity but no inhibition of any of the macromolecular pathways. Mixtures displaying this profile often led to new targets that could be the subject of further study.
Shaw then turned to the subject of antisense assays, which look for nucleic acid sequences that selectively attenuate gene expression. For essential genes, attenuation can result in growth inhibition. These assays are run by introducing plasmids that produce antisense RNA sequences under the control of a regulatable promoter. The point of this approach is that moderate levels of antisense induction can be used to titrate growth rate and that cells become hypersensitized to any further insult that is specific to the attenuated target. Her group’s experience with this type of assay showed that screening for growth inhibition is only the first step and that not all antisense clones are equally susceptible to drugs act-

FIGURE 3-1 An example of macromolecular synthesis inhibition assays using fractions and crude extracts.
SOURCE: Karen Shaw (2013).
ing on the target of interest, for reasons that are still unclear. In addition, it turns out that not all targets can be assayed using antisense approaches, again for reasons that are not clear.
After briefly describing how these assays are run, she showed how researchers at Merck used this approach to identify both known and new inhibitors of synthases FabH and FabF from libraries of natural products. In another set of experiments, Shaw and her colleagues ran an antisense assay looking at cell wall targets. These experiments demonstrated synergy between known cell wall inhibitors and inhibition of several early steps in cell wall biosynthesis. She noted that a benefit of conducting antisense screening is that it is a focused approach that provides strains optimized for target-specific hypersensitivity. Another benefit is that antisense screening quickly identifies antibacterial agents that act by defined mechanisms. Antisense assays can enable screening for on-target activity during structure-based drug design optimization and they can identify compounds that act synergistically with those targets. For example, antisense assays showed that Mur ligase pathway enzyme inhibitors are likely to be synergistic with penicillin binding protein inhibitors.
Summarizing the results of screening 22,000 crude samples and fractions from marine sources, Shaw said that about seven percent demonstrated antimicrobial activity. Macromolecular synthesis inhibition eliminated 80 percent of those hits as being nonspecific inhibitors. About half of the remaining samples were eliminated through cross-resistance studies, DNA intercalation assays, and other rapid screens. Those samples remaining then needed to go through refermentation, fractionation, and structural elucidation, which she said are always going to be the rate limiting steps in natural products screening. In the end, only a very few compounds were judged worthy of entering a structure-based drug design program.
Before discussing new approaches and sources of compounds, Shaw said that one of the lessons learned from screening efforts is that an enzyme hit might not translate to how the cell is being killed and that improvements in antimicrobial activity are often not associated with target-based inhibition. “It is important to prove that initially and consistently through optimization,” she said. Hits are a plentiful, so the real issue is prioritizing which hits are worth pursuing. Hits from combination screens, such as those looking for β-lactamase inhibitors, need to be confirmed to be working via a multiples mechanisms, and it is important to do resistance studies early and often. Finally, it is important to get the biology right and to beware of functional and nonfunctional homologs in different species.
Another new approach that she encountered recently uses mass spectrometry to create “molecular fingerprints” that can then be used to track molecular networking across an entire organism. The challenge with this approach, which is being pursued by a company called Siernas, is data analysis, but it is being used to identify novel compounds that then can be put through antibacterial screens. In one analysis of a marine sponge, this approach yielded 20 pure compounds for further testing.
Shaw concluded her talk by briefly reviewing the work that her team did looking for inhibitors of topoisomerase IV,
also known as ParE, and DNA gyrase B (GyrB) that would not display cross-resistance with fluoroquinolone antibiotics. This project had its origins in the 1950s with the discovery of novobiocin, a natural product that showed some promise against gram-positive bacteria but that was not dual targeting and was judged to be prone to resistance. Using crystal structures, Shaw and her colleagues were able to map the sequence diversity of GyrB and ParE in gram-negative and gram-positive bacteria and use that information to develop highly potent, broad spectrum antibiotics that are undergoing further development. She noted that of the four compounds that initially were most promising, macromolecular synthesis assays quickly showed that two of the compounds were working by nonspecific mechanisms.
NEW WAYS OF LOOKING AT OLD ANTIBIOTICS AND THEIR TARGETS
“There are three, and only three, methods that I can think of that have a chance of reviving the discovery of new natural antibacterial natural products,” said Chaitan Khosla. The first, highlighted in the presentations by Silver and Shaw, is the tried and true method of conventional activity-based screening, though today driven by new methodologies. The second involves mining orphan secondary metabolic pathways, and the third engineers known antibiotic pathways to make new molecules. To illustrate these approaches, Khosla discussed how they are being used with a class of antibiotics derived from an assembly-line process involving polyketide synthases (Figure 3-2). The best-known member of this family of antibiotics is erythromycin, but it also includes the 14- to 16-membered macrolide antibiotics, rifamycin, mupirocin, tiacumicin, and the streptogamins. He noted, too, that of all the antibiotic synthetic pathways explored so far, the polyketide assembly line is perhaps the most mature in terms of foundational mechanistic knowledge and the availability of associated technologies for engineering this pathway.
As an example of the first approach, screening against new targets using modern technologies, Khosla discussed the type three secretion system (TTSS) that many gram-negative bacteria use to inject bacterial effector proteins directly into the host cell cytoplasm, bypassing the cell’s membrane-bound defense mechanisms. The TTSS has emerged as a potential target only within the past decade, he explained, in part because of the idea that it is not essential for bacterial survival or reproduction and therefore may not be subject to the selective pressures that generate resistance, though he added that this idea has not yet been tested rigorously. When the TTSS was first characterized, medicinal chemists screened large numbers of existing medicinal chemistry libraries with little success until the discovery in 2008 by a group in Japan of a new type of antibiotic that “doesn’t resemble anything that we’ve seen before through traditional screening because they were specifically screening for compounds that block this new target,” said Khosla. Working with the group in Japan, Khosla and his colleagues have now characterized the assembly-line process that creates this new class of antibiotics, known as the guadinomines.
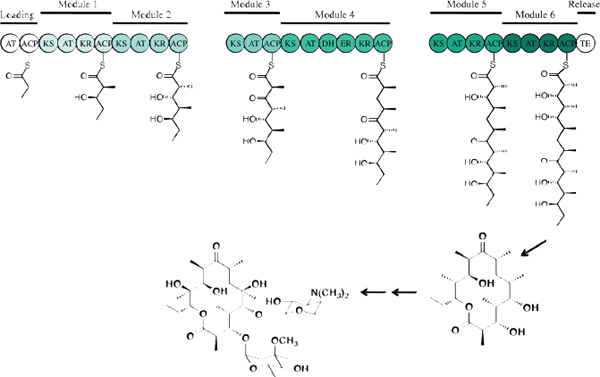
FIGURE 3-2 An example of mining orphan metabolic pathways. Assembly-line polyketide antibiotic biosynthesis.
SOURCE: Chaitan Khosla (2013).
He explained that this is an important advance given the difficulty in producing these molecules synthetically or isolating them from natural sources in quantities sufficient for further development.
Other examples of new classes of molecules discovered by looking at novel targets using modern screening technologies include kibdelomycin, which was identified from soil samples by a team at Merck using the type of antisense-induced sensitivity screen that Shaw discussed. Khosla noted that making this molecule is likely beyond the capabilities of synthetic chemists and that his group is working with Merck to characterize the biosynthetic pathway used to make it and various derivatives. Other approaches include conducting single-molecule biophysical studies of important targets, such as DNA gyrase, to identify key molecular features involved in target binding, and using reconstituted lipid metabolism systems to look for inhibitors of yet another aspect of macromolecule synthesis. “These kinds of approaches can be very resource-efficient ways to discover new species-specific [narrow-spectrum] antibiotics,” said Khosla.
Turning to the second method of identifying new antibiotics—mining orphan secondary metabolic pathways— Khosla explained that his group has used whole-genome sequencing and other technologies to identify close to 900 distinct assembly-line polyketide synthases, of which fewer than 20 percent have well-characterized substrates and products. To his knowledge, there is no ongoing large-scale effort to mine this family of polyketide assembly lines despite the fact that more than a dozen commercially important antibiotics come from the 20 percent of assembly lines that are well characterized and that make known molecules. His group has been developing techniques for refactoring these assembly-line pathways in heterologous hosts to produce novel compounds from glucose and propionic acid. In one project using such a system, Khosla’s team and collaborators from Kosan Biosciences were able to produce commercial quantities of the anticancer agent epothilone D within three years after this compound’s discovery.
Given the success of this and a few other similar projects, Khosla said that industry is interested in this approach but that there are a number of challenges that have to be overcome for it to gain wider use. Assembling the DNA constructs needed to create one of these assembly lines is feasible, but still too costly. “This will cease to be a relevant problem once we get to the stage where we can assemble DNA at less than five cents a base,” he said. A number of research groups, including his, are developing promising methods of expressing the very large proteins that make these assembly lines, another challenge facing the field, but more work is needed to identify methods of tailoring enzymes to create novel structures and to address the supply of all but the simplest precursor molecules to feed into these assembly lines. There is also the need for continued development of analytical methods to more rapidly elucidate molecular structures, though using heterologous systems simplifies this problem if one knows the precursors and enzymes involved that limit the structural possibilities for the products. This challenge may become simpler still with the successful development of reconstituted in vitro systems rather than heterologous systems using whole organisms.
Looking to the future, Khosla said that it will one day be possible to engineer these assembly lines to make entirely new molecules, the third approach to antibiotic discovery. His group and others have developed a number of methods of engineering assembly-line polyketide biosynthetic pathways, but more work is needed to truly realize the promise of this approach. The main challenge to address is conceptual rather than technical, he explained. “We do not yet understand fully how these assembly lines work, and we certainly do not yet understand the structural basis for the modularity of these assembly lines,” he said. “Until we do, it is premature to predict where this approach will take us.”
Over the past 15 years, research in his laboratory has shown that protein–protein interactions through the assembly lines are critical to moving reactive intermediates through the assembly line in a directional manner. His group is now exploring methods of intentionally designing new pathways using protein–protein interaction principles, but he characterized this work as “slow science” that requires a design, build, and test paradigm.
One point raised during the discussion period was that when organisms are screened for the purpose of antibiotic discovery, they are often grown under benign conditions rather than in circumstances in which they might turn on orphan pathway or metabolic defense mechanisms and produce novel compounds. Shaw and others in the audience noted that it is becoming more common to add either various stimulatory compounds, such as lipopolysaccharide, or other organisms to the culturing system, but this has yet to be done in a sufficiently organized way to determine if that approach makes a difference in terms of the antibacterial compounds the organisms produce. Regarding this last point, Silver argued that bacteria have been waging war against each other for millions of years and that it is unlikely that experiments of this sort would yield anything that has not been identified already in natural product screens from soil and other bacterial environments.
Khosla noted during the discussion period that some of the products of the polyketide assembly lines are further modified by oxidases, oxygenases, and transferases. In most cases, the biochemistry and genomic locations of these adjunct “tailoring” enzymes have been described in the literature. In general, the genes coding for these enzymes are clustered with the genes coding for the assembly lines.
While tailoring may be necessary to produce a molecule with maximum antibacterial activity, the unmodified molecules typically have enough activity to show up as hits in screening assays. He added, too, that with the new in vitro systems it is possible to examine intermediate compounds that are rarely seen in the natural or heterologous systems for biological activity.
Cortés, J., S. F. Haydock, G. A. Roberts, D. J. Bevitt, and P. F. Leadlay. 1990. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348(6297):176-178.
Donadio, S. M. J. Staver, J. B. McAlpine, S. J. Swanson, and K. Katz. 1991. Modular organization of genes required for complex polyketide biosynthesis. Science 252(5006):675-679.
Haste, N. M., V. R. Perera, K. N. Maloney, D. N. Tran, P. Jensen, W. Fenical, V. Nizet, and M. E. Hensler. 2010. Activity of the streptogamin antibiotic etamycin against methicillin-resistant Staphylococcus aureus. Journal of Antibiotics (Tokyo) 63(5):219-224.
Holmes, T. C., A. E. May, K. Zaleta-Rivera, J. G. Ruby, P. Skewes-Cox, M. A. Fischbach, J. L. DeRisi, M. Iwatsuki, S. Ōmura, and Chaitan Khosla. 2012. Molecular insights into the biosynthesis of guadinomine: a type III secretion system inhibitor. Journal of the American Chemical Society 134(42):17797-17806.
Iwatsuki, M., R. Uchida, H. Yoshijima, H. Ui, K. Shiomi, A. Matsumoto, Y. Takahashi, A. Abe, H. Tomoda, and S. Omura. 2008. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. I: taxonomy, fermentation, isolation and biological properties. Journal of Antibiotics (Tokyo) 61(4):222-229.
Iwatsuki, M., R. Uchida, H. Yoshijima, H. Ui, K. Shiomi, Y. P. Kim, T. Hirose, T. Sunazuka, A. Abe, H. Tomoda, and S. Omura. 2008. Guadinomines, Type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. II: physico-chemical properties and structure elucidation. Journal of Antibiotics (Tokyo) 61(4):230-236.
Jacobson, J. R., C. R. Hutchinson, D. E. Can, and C. Khosla. 1997. Precursor-directed biosynthesis of erythromycin analogs by an engineered polyketide synthase. Science 277(5324)367-369.
Kapur, S., B. Lowry, S. Yuzawa, S. Kenthirapalan, A. Y. Chen, D. E. Cane, and C. Khosla. 2012. Reprogramming a module of the 6-deoxyerthythronolide B synthase for iterative chain elongation. Proceedings of the National Academy of Sciences 109(11):4110-4115.
Manchester, J. L., E. T. Buurman, G. S. Bisacchi, and R. E. McLaughlin. 2012. Molecular determinants of AcrB-mediated bacterial efflux implications for drug discovery. Journal of Medicinal Chemistry 55(6):2532-2537.
Nikaido, H., and D. G. Thanassi. 1993. Penetration of lipophilic agents with multiple protonation sites into bacterial cells: tetracyclines and fluoroquinolones as examples. Antimicrobial Agents and Chemotherapy 37(7):1393-1399.
Phillips, J. W., M. A. Goetz, S. K. Smith, D. L. Zink, J. Polishook, R. Onishi, S. Salowe, J. Wiltsie, J. Allocco, J. Sigmund, K. Dorso, S. Lee, S. Skwish, M. de la Cruz, J. Martín, F. Vicente, O. Genilloud, J. Lu, R. E. Painter, K. Young, K. Overbye, R. G. Donald, and S. B. Singh. 2011. Discovery of kibdelomycin, a potent new class of bacterial type II topoisomerase inhibitor by chemical-genetic profiling in Staphylococcus aureus. Chemistry and Biology 18(8):955-965.
Tari, L. W., M. Trzoss, D. C. Bensen, X. Li, Z. Chen, T. Lam, J. Zhang, C. J. Creighton, M. L. Cunningham, B. Kwan, M. Stidham, K. J. Shaw, F. C. Lightstone, S. E. Wong, T. B. Nguyen, J. Nix, and J. Finn. 2013. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part I: Structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorganic and Medicinal Chemistry Letters 23(5):1529-1536.
Young, K., H. Jayasuriya, J. G. Ondeyka, K. Herath, C. Zhang, S. Kodali, A. Galgoci, R. Painter, V. Brown-Driver, R. Yamamoto, L. L. Silver, Y. Zheng, J. I. Ventura, J. Sigmund, S. Ha, A. Basilio, F. Vicente, J. R. Tormo, F. Pelaez, P. Youngman, D. Cully, J. F. Barrett, D. Schmatz, S. B. Singh, and J. Wang. 2006. Discovery of FabH/FabF inhibitors from natural products. Antimicrobial Agents and Chemotherapy 50(2):519-526.






