
2
Challenges In Overcoming Antibiotic Resistance
“The last novel class [of antibiotics] to be licensed was discovered in 1987.”
Lynn Silver
“If you are able to reverse the resistant phenotype, then you are rescuing drugs that have become obsolescent.”
Shahriar Mobashery
The predominant focus of this workshop was to understand the barriers to developing new antibiotics that can overcome or bypass resistance to existing therapies. Two speakers addressed these challenges. Lynn Silver, a consultant at LL Silver Consulting, LLC, discussed the need to select drug targets that are not subject to rapid resistance selection, and Shahriar Mobashery, Professor of Life Sciences at Notre Dame University, spoke about the mechanisms that bacteria use to neutralize many of the most potent antibiotics.
SELECTING ANTIBACTERIAL TARGETS TO AVOID RESISTANCE SELECTION
What plagues the antibiotic field today, said Lynn Silver, is that the field is suffering from a discovery void, a gap of over 30 years when efforts to discover novel classes of antibiotics largely failed. “If you look at when things were discovered, we stopped discovering novel antibiotics in 1987,” she stated, noting that every new antibiotic that has been approved for human use since then is a member of a chemical class discovered before 1987. For example, retapamulin, which was approved by the FDA in April 2007 and the European Medicines Agency in May 2007 for the treatment of bacterial skin infection, is a member of a class of compounds discovered in 1950. Newer antibiotics such as fourth-generation fluoroquinolones derive from nalidixic acid, which was discovered in 1962. By itself, that “innovation gap” would not be that big of an issue, but the problem is that resistance arose to most classes of antibiotics soon after their introduction, and there was little in the pipeline (a discovery void) in terms of antibiotics with novel mechanisms of action that could overcome resistance. One important exception has been vancomycin, which was discovered in 1953 and approved in 1958, but now even vancomycin-resistant strains of Enterococcus and Staphylococcus are becoming increasingly prevalent (Figure 2-1).
With each parry by bacteria, medicinal chemists developed new drugs that were not just molecules copied from others but derivatives of existing antibiotic classes with better pharmacological properties or modifications that overcame specific resistance mechanisms. For example, when cases of methicillin-resistant Staphylococcus aureus (MRSA) started appearing, medicinal chemists responded with compounds that block the action of the extended-spectrum β-lactamases that are responsible for resistance. Some of these compounds, which should also combat CRE, are now in clinical trials. “This is real science and it makes very good drugs,” said Silver, “but the problem is that resistance keeps happening.”
By and large, however, the discovery of novel compounds, especially in new classes toward new targets, that can be developed has so far failed, and Silver listed several possible reasons for this failure. One possibility is that research has focused on only a few bacterial targets, and to remedy this problem the drug discovery community has turned to genomics, crystallography, and bioinformatics to identify new targets. These approaches, however, have not been particularly successful. Another possibility is that medicinal chemists have not screened enough compounds, and in response to that possibility the field turned to high-throughput screening of large chemical libraries and natural product isolates. High-throughput screening has yielded few leads, and naturally occurring antibiotics have largely proven to be so difficult to isolate or have such poor pharmacological properties that the field has for the most part largely abandoned that avenue of discovery. The problem with all of these efforts, said Silver, is that antibiotic developers generally keep applying new technologies to address the discovery gap without understanding why these approaches were not succeeding in the first place.
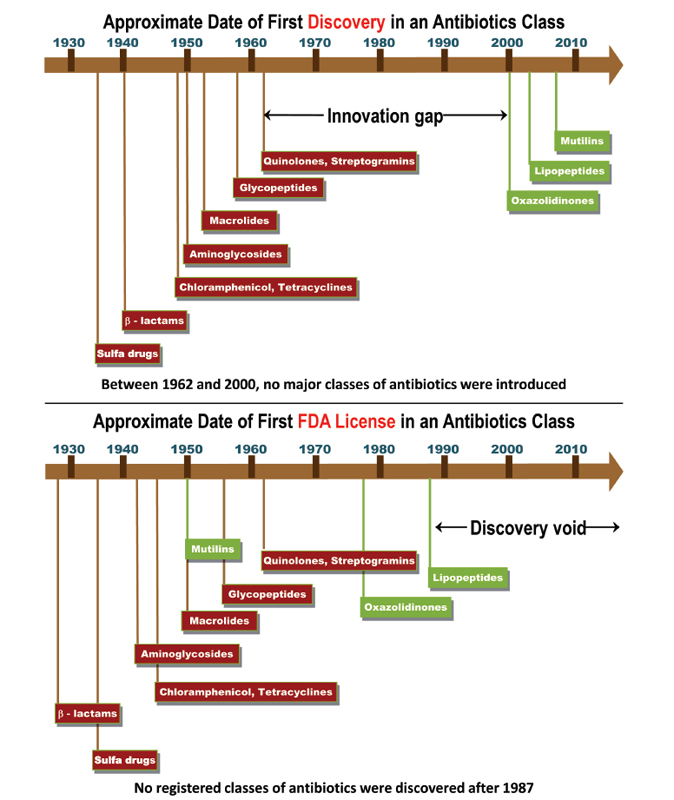
FIGURE 2-1 The innovation gap and discovery void in antibiotics discovery and development. A comparison of this first discovery in an antibiotic class and the first FDA licensure.
SOURCES: Top: Fischbach, M. A., and C. T. Walsh (2009). Antibiotics for emerging pathogens. Science 325(5944):1089-1093 with permission from AAAS. Bottom: Lynn L. Silver.
In her mind, there are two rate-limiting steps in antibiotic discovery. The first is the selection of targets that are not subject to rapid selection of resistance, and the second is the use of chemistries that are appropriate for antibacterial discovery. “Antibacterial agents have certain physiochemical parameters that are different from those of other human health drugs,” said Silver, and as a result the chemical libraries that have been screened for antibacterial activity were unlikely to generate suitable leads for further development. In addition, there is no complete set of general rules or an agreed upon rational approach to getting drugs into gram-negative bacteria.
What makes a good antibacterial target? Silver explained that research has largely focused on attributes such as the
target not having a human homolog, that it is present in a range of bacteria, that it is essential, that it is druggable in the sense that it is possible to create molecules that interfere specifically with the activity of the target, and that the target has a low potential for cross-resistance with existing antibiotics. She believes, though, that this list should include two other largely ignored attributes: location of the target on or in bacteria and a low frequency of resistance to new compounds. She then noted that when she and her colleagues in industry looked at the targets of successful antibiotics, they found that these compounds fell into one of these categories. Either they had multiple targets or targets encoded by multiple genes, in which case high-level, target-based resistance to these compounds does not occur by single-step mutations, or they had single enzyme targets that were subject to single-step target-based resistance.
Based on this evaluation, Silver concluded that successful monotherapeutic antibacterials are not subject to single mutations for high-level resistance because they are multitargeted, and that current drugs that inhibit single enzymes, because they are subject to single-step mutation, are generally used in combinations and when organismal load is low. This conclusion, she said, leads to the hypothesis that multiple targets are preferable for systemic monotherapy, and that the null hypothesis for single targeted agents is that they will select rapidly for resistance. In fact, she said, this is what has happened with tuberculosis and HIV, where single-agent therapy led to the rapid development of resistance but multi-drug therapy has proven to be highly successful. She also cited several examples of single-target monotherapies that entered clinical trials but failed because resistance developed rapidly. As an example, she said that one promising agent that targeted leucyl tRNA synthetase demonstrated excellent activity in vitro against a broad spectrum of gram-negative bacteria, but that resistance occurred in four of 34 patients after just one day of therapy during clinical trials. When isolated, these mutants were highly fit and grew as fast as wild-type bacteria. Silver noted that these findings were replicated in an in vitro system called the hollow fiber resistance model that mimics the pharmacokinetics of drug dosing and cell growth under conditions of cyclic dosage of antibiotics.
One way to move forward when the resistance frequency to new lead compounds is high is to optimize the chemistry, probably through an iterative approach, to reduce the resistance frequency. As an example of this approach, Silver cited a case in which researchers developed an analog of trimethoprim, which is an inhibitor of dihydrofolate reductase, which has higher affinity to its target. This compound demonstrated efficacy in clinical trials but has not yet been approved for human use. The one caveat to this approach is that as molecules are designed to have more than one interaction with specific enzymes, they may be effective against a smaller set of organisms, which could make the business case for the development of these agents less attractive.
Another path forward is to discover more multitargeted inhibitors, which Silver admitted is a more difficult approach. Potential targets in this class include multiple cell wall enzymes; DNA gyrase and topoisomerase IV; and other enzymes sharing active sites, such as DNA polymerases PolC and DnaE. Compounds that inhibit protein synthesis by binding to multiple targets in ribosomal RNA or that inhibit lipid formation might be good candidates, too, since these processes all involve multi-protein complexes. Compounds that damage the integrity of the bacterial cell wall, such as daptomycin or amphotericin B, have proven useful in treating resistant gram-positive bacteria and fungi, respectively, with little incidence of resistance. Silver noted that the field needs to explore additional pathways with similar active sites or ligands, such as the tRNA synthetases, the purine synthesis pathway, or cofactor synthesis pathways. In silico analyses of known ligand–target interactions combined with chemi- and bioinformatics could prove useful for identifying families of targets by the interaction with similar ligands.
Other targets for antibiotic development could fall into what she called the adjunctive category of therapies. These would include inhibitors and dispersers of biofilms, permeability enhancers, and efflux pump inhibitors. Virulence targets and antitoxins have been touted as resistance-proof since they do not kill the organism, but that idea awaits proof of concept. Metabolic targets are possible, but most metabolic targets identified so far have been single enzymes, and while regulatory targets are interesting scientifically, the general consensus within the antibiotic development community is that these pathways could be bypassed or be highly prone to resistance selection.
There are also a number of clinical approaches that could be taken, Silver explained. The simplest approach would be to dose patients with drug levels above the mutation prevention concentration, the concentration above which single-step mutations to resistance are not selected. However, dosing levels used today are generally geared toward efficacy, not resistance avoidance, and the mutation prevention concentration may be incompatible with toxicity and pharmacokinetic parameters. More promising are efforts to identify combinations of drugs that work by different or even synergistic mechanisms. A major challenge with this approach is to match pharmacokinetic properties so that the levels of each drug are high enough to overcome resistance. It may also be difficult, or even unethical, to conduct the necessary clinical trials that can demonstrate decreased resistance with combinations of drugs that by themselves result in resistance. Following a discussion of the myriad approaches to overcoming antibiotic resistance, Silver explained that there is some need for prioritization on which approaches are best suited for further development.
Two of the most challenging infections to treat are those caused by gram-positive MRSA and gram-negative carbapenem-resistant Enterobacteriaceae (CRE). In his presentation, Shahriar Mobashery discussed the work that his group has done in understanding the resistance mechanisms employed by these bacteria and developing approaches for overcoming resistance in those organisms. He noted that these resistance mechanisms are complex, multistep processes, but each step along the way toward manifestation of resistance offers the opportunity for intervention to reverse the resistant phenotype. “If you are able to reverse the resistant phenotype, then you are rescuing drugs that have become obsolescent,” he explained.
Staphylococcus aureus was susceptible to β-lactam antibiotics such as penicillin well into the 1960s, when the β-lactamases were first discovered. Medicinal chemists countered that development by altering the structures of these drugs to overcome the catalytic function of those enzymes. However, within four years of the introduction of these second-generation penicillins, MRSA had not only appeared but had spread worldwide. Health officials expressed concern that vancomycin-resistant strains of MRSA would appear, and indeed they have. Mobashery said that by his count, 13 variants of vancomycin-resistant MRSA have been identified.
The mechanism that produces vancomycin resistance is complex, he explained, involving a set of genes whose origins were not from Staphylococcus aureus. The first such gene codes for a signal transduction protein, BlaR1, that binds β-lactams irreversibly as a result of a decarboxylation reaction triggered by β-lactam binding that in turn produces a conformational change in the protein that spans the bacterial cell membrane (Figure 2-2). This conformational change is observable using Fourier transform infrared spectroscopy. Once binding occurs, it activates a cytoplasmic protease
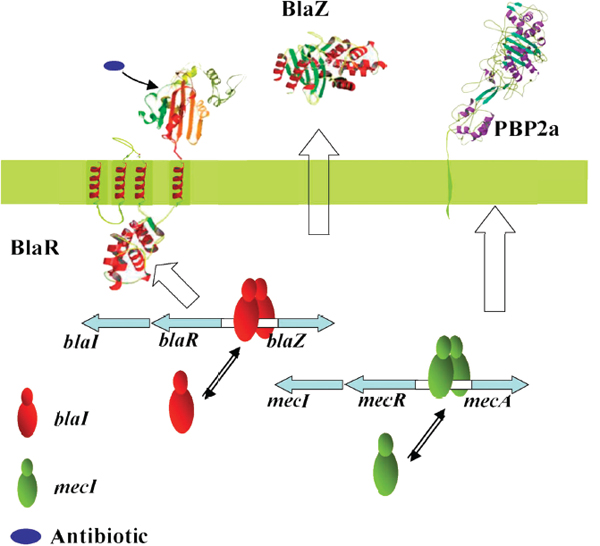
FIGURE 2-2 An example of antibiotic resistance. Sensing of β-lactam antibiotics requires a conformational change for transduction of information to the cytoplasmic side.
SOURCE: Shahriar Mobashery (2011).
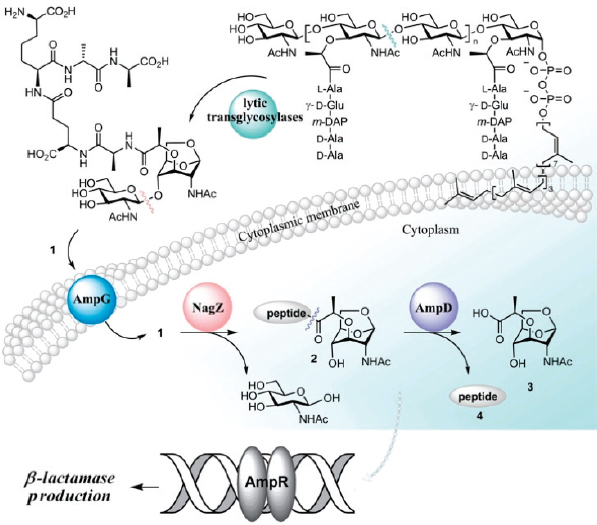
FIGURE 2-3 The link between β-lactamase production and cell wall recycling is mediated by several enzymes, including ligase AmpD.
SOURCE: Shahriar Mobashery (2011).
domain that degrades several gene repressors. Upon degradation of these repressors, full-blown resistance develops within minutes to tens of minutes of exposure as a result of the BlaZ gene expressing β-lactamase and the MecA gene expressing the penicillin-binding protein 2A (PBP2a). Work in Mobashery’s laboratory has shown that this is a reversible process when antibiotic is no longer present, suggesting that PBP2a could be an interesting target for drugs that could block the development of resistance.
One of the functions of PBP2a is to crosslink peptides in the bacterial cell wall, a critical function for viability of the cell wall, and in nonresistant organisms the β-lactam antibiotics are able to inactivate this enzyme through an irreversible acylation reaction. From the results of x-ray crystallography experiments, it appears that allosteric control is involved in the activity of this protein and that it is possible to impact that control with certain cephalosporins. Indeed, ceftaroline, a fifth-generation cephalosporin that is active against MRSA, appears to overcome resistance by binding to mutated PBP2a at the allosteric site. Binding to the allosteric site induces a conformational change in this enzyme that opens its active site and enables a second molecule of ceftaroline molecule to bind there and inhibit PBP2a’s ability to act as a transpeptidase in cell wall synthesis.
Briefly turning to the subject of gram-negative bacteria, Mobashery discussed his group’s work synthesizing a number of small carbohydrates that mimic small glycans released when the bacterial cell wall is damaged. These glycans serve as signaling compounds that activate lytic transglycosylases that are involved in the development of resistance in gram-negative bacteria (Figure 2-3). Activation of one enzyme in particular, ligase AmpD, triggers the production of a β-lactamase involved in resistance. His group has been conducting crystallographic studies in an attempt to better understand the function of the AmpD protein and to determine if there are sites in the protein that might be good targets for drug development.
In response to a question about the use of nanotechnology to develop novel antibiotics that might overcome resistance, Silver said that this is an avenue worth pursuing, but that she worried about toxicity. She noted that nanoparticles may help with getting compounds into bacteria, particularly gram-negative species. Mobashery said his concern with nanoparticles was with how they are cleared from and metabolized by the human body.
Silver asked Mobashery if it might be worth looking at existing libraries of natural products to see if any of them could induce the type of conformational changes his research has identified. He thought this could be a productive exercise and that it may be possible to find compounds that are not β-lactams that would bind to the allosteric site and synergize with a β-lactam.
Carrasco-López, C., A. Rojas-Altuve, W. Zhang, D. Hesek, M. Lee, S. Barbe, I. André, P. Ferrer, N. Silva-Martin, G. R. Castro, M. Martínez-Ripoll, S. Mobashery, and J. A. Hermoso. 2011. Crystal structures of bacterial peptidoglycan amidase AmpD and an unprecedented activation mechanism. Journal of Biological Chemistry 286(36):31714-31722.
Fischbach, M.A., and C. T. Walsh, 2009. Antibiotics for emerging pathogens. Science 325(5944):1089-1093.
Hernandez, V., T. Crépin, A. Palencia, S. Cusack, T. Akama, S. J. Baker, W. Bu, L. Feng, Y. R. Freund, L. Liu, M. Meewan, M. Mohan, W. Mao, F. L. Rock, H. Sexton. A. Sheoran, Y. Zhang, Y.-K. Zhang, Y. Zhou, J. A. Nieman, M. R. Anugula, E. M. Keramane, K. Savariraj, D. S. Reddy, R. Sharma, R. Subedi, R. Singh, A. O’Leary, N. L. Simon, P. L. De Marsh, S. Mushtaq, M. Warner, D. M. Livermore, M. R. K. Alley, and J. J. Plattner. 2013. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrobial Agents and Chemotherapy 57(3):1394-1403.
Llarrull, L. I., M. Toth, M. M. Champion, and S. Mobashery. 2011. Activation of BlaR1 protein of methicillin-resistant Staphylococcus aureus, its proteolytic processing, and recovery from induction of resistance. Journal of Biological Chemistry 286(44):38148-38158.
Otero, L. H., A. Rojas-Altuve, L. I. Llarrull, C. Carrasco-López, M. Kumarasiri, E. Lastochkin, J. Fishovitz, M. Dawley, D. Hesek, M. Lee, J. W. Johnson, J. F. Fisher, M. Chang, S. Mobashery, and J. A. Hermoso. 2013. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proceedings of the National Academy of Sciences 110(42):16808-16813.
Silver, L. L. 2007. Multi-targeting by monotherapeutic antibacterials. Nature Review: Drug Discovery 6(1):41-52.
Silver, L., and K. Bostian. 1990. Screening of natural products for antimicrobial agents. European Journal of Clinical Microbiology and Infectious Diseases 9(7):455-461.
Silver, L. L., and K. A. Bostian. 1993 Discovery and development of new antibiotics: the problem of antibiotic resistance. Antimicrobial Agents and Chemotherapy 37(3):377-383.
VanScoy, B. D., C. C. Bulik, C. Moseley, N. E. Scangarella-Oman, L. A. Miller, S. M. Bhavnani, and P. G. Ambrose. 2013. Hollow fiber infection model (HFIM) mimics both the time-to-resistance emergence and magnitude of E. coli resistance to GSK052 occurring in a phase 2b clinical study. Poster A-016, 53rd ICAAC Meeting, Denver. http://www.icaaconline.com/php/icaac2013abstracts/data/papers/2013/A/2013_A-016.htm (accessed October 30, 2013).
Villegas-Estrada, A., M. Lee, D. Hesek, S. B. Vakulenko, and S. Mobashery. 2008. Co-opting the cell wall in fighting methicillin-resistant Staphylococcus aureus: potent inhibition of PBP2a by two anti-MRSA β-lactam antibiotics. Journal of the American Chemical Society 130(29):9212-9213.






