
THE ROLE OF BIOINFORMATICS IN MICROBIAL FORENSICS
In a 2009 report, the American Academy of Microbiology stated that the application of computer analysis to molecular biology, otherwise known as “bioinformatics,” is “a fundamental corollary to biodefense research” (AAM, 2009b). The report also pointed out a theme very much consistent with the presentations at the Zagreb workshop—that advancements in biodefense and bioinformatics research, although meant to lead to national security improvements, will also reverberate across almost all biological disciplines. Specific fields that will benefit include
- Understanding genetic diversity,
- Epidemiology,
- Vaccinology,
- Global health,
- Metabolic reconstruction,
- Systems biology, and
- Personalized medicine.
As most of the workshop speakers noted, WGS has made possible giant steps forward for epidemiology and microbial forensics. The ability to examine and compare the genetic sequences of bacteria, archaea, viruses, and microbial eukaryotes has revolutionized microbiology and provided researchers with insights “into the processes microbes carry out, their pathogenic traits, and new ways to use microorganisms in medi-
cine and manufacturing” (AAM, 2009b). Gene sequencing, WGS, and the techniques these make possible are moving microbiology forward more rapidly than ever before. They also have given rise to new tools—genomics, proteomics, metabolomics, transcriptomics, molecular phylogeny, and others—that now are available to scientists studying the relationships among microbes, their pathogenicity mechanisms, and their metabolic potential (AAM, 2009b). The AAM report went on, however, to note that a “major effort in functional annotation to understand the DNA sequence we already have” is needed (2009b:16). “Annotation is a critical part of making genome sequences into resources, but it represents a huge bottleneck.” It also called for the implementation of a set of standard annotation platforms and a central annotation resource “with defined methods and standards, and guidelines for people to use the resource” (2009b:16).
THE INTERPLAY BETWEEN SEQUENCING AND BIOINFORMATICS
Dr. Jongsik Chun of Seoul National University in South Korea believes that WGS provides the ultimate information for both epidemiology and microbial forensics. As an illustration of the power of WGS, he pointed to its use in tracking the transmission of carbapenem-resistant K. pneumoniae among individuals during the NIH Clinical Center’s 2011 outbreak (Snitkin et al., 2012). WGS is superior to all other identification methods if cost, time, and bioinformatics are not issues. However, using genomic information is the area of microbial forensics in which the application of bioinformatics is most needed. Chun sought to provide an understanding of the scope of microbial knowledge that is lacking and the uses to which the data that are available could be applied with better tools. He reviewed the challenges faced in assembling and curating databases, as well as in developing bioinformatics to efficiently and effectively exploit the data.
Chun, who is a trained taxonomist, reminded the workshop participants that the traditional concept of species is “groups of actually or potentially interbreeding natural populations that are reproductively isolated from other such groups” (Mayr, 1942). But in the bacterial world, this definition falls short because there is no “sex” or “breeding,” per se. Instead, bacteria may exchange genetic information through lateral transmission both within species and among different species, allowing for high genetic diversity. For bacteria the fundamental concept of a species is actually based on how similarity between strains is measured. Bacte-
rial classification and identification should be based on the comparison between type strain1 and isolate.
Chun believes that the indirect DNA-DNA hybridization method—the previous “gold standard” for determining the genetic distance between two species—is not a reliable methodology. The 16S rRNA sequencing is revolutionizing taxonomy and now WGS is available. A new index has been developed called Average Nucleotide Identity (ANI). In DNA-DNA hybridization, the DNA would be fragmented and hybridized in solution. The same general analysis now can be done digitally. The DNA of one strain is fragmented, the sequences are compared using BLASTN or MUMmer programs, the similarity of each fragment is established, and the findings are averaged. The method has been demonstrated to be very reliable (Goris et al., 2007).
NGS and ANI have provided for the first time an accurate, objective, and reproducible method for bacterial classification and identification. Sequencing may be expensive but capacity is not a problem. High-throughput NGS sequencers have a capacity equal to tens of thousands of conventional Sanger sequencers. However, Chun noted that big gaps exist between the ability to sequence and the cost of accessing the necessary hardware and bioinformatics. Genomics in microbial forensics presents a typical big data problem: high-volume, high-velocity, and high-variety, coupled with a need for quick answers.
A very conservative estimate puts the number of prokaryotic species at 1 million, of which only approximately 11,000 species have valid names. The routine use of 16S rRNA sequencing is accelerating the description of new species, and in 2012, 721 new species were described. However, Chun pointed out that there is little or no taxonomy funding in many countries, except perhaps China. Chinese microbiologists are now responsible for the description of over 30 percent of new species of bacteria and archaea.
There has been a huge jump in genome releases since the onset of NGS. Of the roughly 11,000 bacteria and archaea species with valid names, 99 percent have 16S rRNA gene sequences in public databases. There are genome sequences, however, for only 24 percent of these prokaryotic species and only 16 percent of prokaryotic species have been sequenced for type strain. Thus about 85 percent of known species have no genome sequences for their type strains. Applying genome sequencing to species-level identification is limited because reference genomes do not exist. Accurate identification based on genome data is possible with ANI, but more type strains should be sequenced.
_______________
1 Usually the first strain of a new species of bacteria to be described is known as the “type strain.” A bacterial species is a collection of strains that share many common features (Black, 2002).
Chun pointed out that sampling for genomics is biased toward clinically important strains, such as E. coli, Staphyllococcus aureus, S. enterica, and Enterococcus faecalis. There are big gaps in genome-sequenced taxa. If there is bacterial contamination in soil, for example, how much background can be identified? Only about 5 percent of detectable soil bacteria can be identified as species with valid names, while 95 percent cannot. This is because most soil bacteria are not cultivable. Perhaps single-cell genomics can solve this problem, but the currently available technology is not suitable for large-scale surveys.
Creating a database is easy, but maintaining a large and rapidly growing database is very difficult. If someone adds 2,000 E. coli strains to a database, it can take a year to curate. Many databases have been retired or destroyed, likely owing to the inability to cope with the quantity of new data and the lack of the funding to maintain existing databases. Databases Chun considers to be up to date and stably funded include the following:
- NIH’s National Center for Biotechnology Information (NCBI) maintains several databases: GenBank, RefSeq, Microbial Genomes Resources.
- Integrated Microbial Genomes and Metagenomes (IMG) supports the annotation, analysis, and distribution of microbial genome and metagenome datasets sequenced at DOE’s JGI.
- The European Bioinformatics Institute’s (EBI) Ensembl Genomes.
- EzGenome.2
Chun explained that his lab created EzGenome because it wished to provide a prokaryotic genome database that was manually curated, taxonomically correct, and useful for microbial systematics as well as ecology and microbial forensics. In the GenBank database, for example, genomes are identified to the species level in some cases. EzGenome contains about 12,000 prokaryotic genomes. All genome projects have complete hierarchical taxonomic information (from phylum to species) according to the EzTaxon taxonomic system. Some genome projects are identified at the genus level owing to a lack of 16S rRNA sequence and adequate reference genome sequences to which to compare them. ANI-based dendrograms are produced for all genera and families.
Chun agreed that GenBank is a good primary database, but its entries may be incorrectly labeled and include contaminants, likely due to contributor’s error, which is a common occurrence in databases. A survey by
_______________
2 Information is available at http://ezgenome.ezbiocloud.net/; accessed November 23, 2013.
Chun’s lab of GenBank revealed many errors, and such errors can lead to problems—for example, when taxon-specific primers are designed.
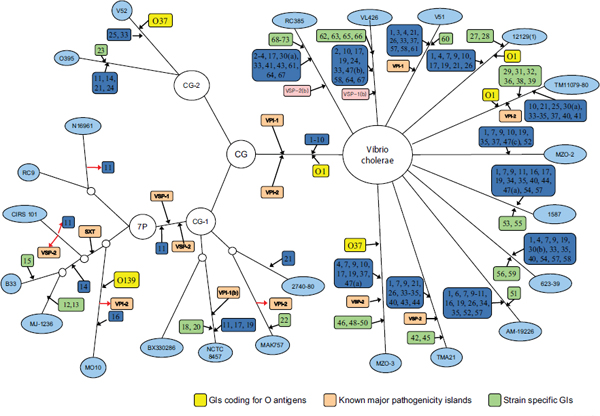
Chun used work his laboratory has done with pandemic strains of Vibrio cholerae as an example of the power, and limitations, of current technology and bioinformatics. Cholera researchers continually encounter newly emerging and reemerging pathogenic clones with diverse combinations of phenotypic and genotypic properties, and this hinders control of the disease (Chun et al., 2009). The seven pandemic V. cholerae strains belong to O1 or O139 serotypes and have almost identical genome backbones, including only about 100 SNPs. However, they show highly variable gene content in some regions of the genome (Cho et al., 2010). Short-term evolution is caused by a combination of SNPs and lateral gene transfer, but it is possible to trace the gene transfer events. Chun’s laboratory performed SNP analysis on a variety of V. cholerae strains, mapped these findings, and created the phylogenetic tree in Figure 7-1. It portrays the genomic islands in closely related strains. They discovered that sequence variations among orthologs were more common than SNP variations and that SNP variations appear to be rare among highly conserved genes. They also determined that the degree of genome variation appears to be species specific; for example, there is more variation within V. cholerae than within Streptococcus agalactiae. The tree appearing in Figure 7-1, however, was based on only 26 genomes and took 6 months to create. How long would it take to perform analysis on 1,000 E. coli? Better bioinformatics are needed.
Examples of two targets in microbial forensics are the pure culture sample or a single microorganism or virus, and the metagenomic sample, which can comprise a mixture of organisms (e.g., human, bacteria, viruses). Metagenomic sample analysis involves detecting target-specific information against a large background by using single-gene-based community analysis (e.g., 16S rRNA-based microbiome) or shotgun metagenomic DNA sequencing. Issues, methods, and bioinformatic analyses differ for pure cultures versus metagenomic samples, but Chun emphasized that properly curated genome databases are necessary for both.
Pure culture sample analysis involves the assembly of genomes, taxonomic identification, identification of variation, such as SNPs, gene content, tracing of gene transfer, and matching against a database. Metagenomic sample analysis involves assembly of a metagenome, taxonomic analysis of community composition, and matching against a database. Chun stressed that the technical difficulty of a metagenomic sample analysis is 1,000 times greater than that of a pure culture sample. Clinicians continually ask him, “Can you do quantification using NGS sequencing?” He believes the answer is no, and a method for quantification needs to
be developed. The components of a bioinformatics program for microbial forensics should include
- Curated, regularly updated databases;
- Data mining tools for large-scale databases;
- Real-time analytical capability;
- Adequate reporting functions; and
- Effective data visualization—it is crucial to import user experience into this process because people (e.g., judges, decision makers, first responders) must understand the outcome of an analysis.
Challenges that Chun sees in microbial forensic bioinformatics include
- Integrating data generated by different methods, such as WGS, multilocus sequence typing (MLST), and pulsed-field gel electrophoresis. The answer lies in bioinformatics and manual curation.
- Integrated real-time sharing of NGS data. Labs A, B, C, and D should be able to share in real time. We can even upload raw data to the cloud servers while sequencing. This is doable (e.g., via BaseSpace genomics cloud computing),3 but poses a policy problem.
- Storage capacity is not an issue, but processing secondary data for microbes is.
For efficient sharing, standardization must be developed for
- Metadata (data about data) and database format/schema,
- Sequencing instruments,
- Protocols for sequencing-library preparation,
- Minimum sequencing depths (for each species),
- Assembly software and/or pipeline,
- Annotation software and/or pipeline, and
- Other bioinformatics tools.
There are challenges to achieving analysis repeatability. A bioinformatics pipeline typically involves many software tools, parameters, and hidden “know-how.” For this reason it is difficult to reproduce most of the large-scale genomics papers (e.g., genome assembly, metagenome comparison). Two solutions he suggests are
_______________
3 A cloud platform directly integrated into the industry’s leading sequencing platforms. Built by Illumina on Amazon’s AWS cloud infrastructure. For more information, see https://basespace.illumina.com/home/sequence; accessed November 24, 2013.
- Virtualization. A model could be the ENCODE Virtual Machine and Cloud Resource for human DNA.4 Complex bioinformatics pipelines and parameters are packaged as an image and can be plugged into any computer.
- Cloud computing. If a lab does not have adequate information technology (IT) infrastructure, software can be applied using cloud computing. The cloud service comes with IT.
There is a need for bioinformatics and databases for the purposes of detecting antibiotic resistance and genetic engineering. A retrospective study revealed the potential for rapid WGS to reduce the time it takes to diagnose a patient with multidrug-resistant tuberculosis from weeks to days (Köser et al., 2013). In this study, the patient’s first sputum sample became culture-positive after 3 days in the mycobacterial growth indicator tube (MGIT) culture system. The DNA was extracted directly from the MGIT tube and sequenced using the Illumina MiSeq platform. Antibiotic susceptibility was determined in less than 2 days. For this purpose, a database of SNPs responsible for antibiotic resistance would be enormously helpful. Similarly, the ability to genetically engineer microbes would be greatly empowered by assembling databases of sequence data to compare. This could be particularly useful for analyzing viruses. A great number of kits exist—for example, for PCR and microarrays—and if new virus sequences were to be added to the dataset, one could mine the database to update primers, probes, and other tools.
Chun believes that bioinformatics for metagenomics faces serious hurdles. The computational challenges are (1) the databases are ever-growing, (2) software and hardware are needed to improve searching/matching, and (3) improved data visualization capabilities are needed.
Cloud computing offers a way to share metadata, and to send out streams of data that can enable accurate identification, provide global epidemiology and treatment information, and inform genetic engineering. Overall, Chun believes that cloud computing technology and bioinformatics will find a way. But he sees real barriers in policy. Two key questions are
- Who owns the data? Only 23 B. anthracis sequences are in public databases (versus more than 1,000 E. coli). So it is likely that many B. anthracis genome sequences have not been shared.
- Who is responsible if something goes wrong? If there were to be a problem with a certain software that was used in a court case,
_______________
4 For more information, see http://www.nature.com/encode; accessed November 24, 2013.
for example, would the developers be liable? This can be a problem, especially because most popular bioinformatics software is developed in academic institutions.
One possible solution to the information-sharing dilemma, since the majority of genome data for biothreats may not be publically available, is defining and establishing two data tracks, one that can be shared and one that cannot. Ideally, academia and governments can at least share standards for data, if not the data itself. In this way, the data can be easily integrated when possible.
In a discussion following Dr. Chun’s presentation, Dr. Paul Keim noted that technology development today differs from that of a decade ago. Technologies then were not forward compatible, which limited their power. But WGS is forward compatible. If one investigator has a genome sequence using Life Technologies’ PGM sequencer and another using an Illumina MiSeq sequencer, the data can be easily compared, put in a database, and will be forward compatible with any technology that comes along. He agrees that a committee will never decide anything, but the genome is the genome and once we have it, we can use it.
Dr. Munirul Alam of the International Center for Diarrhoeal Diseases Research in Bangladesh suggested that individual committees be formed to guide what typing tools, for example, should be used for each pathogen. Scientists are sporadically doing everything, but not in a collaborative way leading to consensus. The findings could be used in microbial forensics. Dr. Dag Harmsen agreed it would be desirable to have an international committee to agree on typing methods, but thinks that such a thing is unrealistic—committees cannot even agree on names. In addition, needs differ between developing and developed countries, so there will never be one assay method that fits all needs. It is ultimately the users who decide what is going to be used and will declare a de facto standard. Also, technology continues to evolve, and novel and even more powerful analytical tools likely will arise in the near future.
MANAGING LARGE DATASETS
Dr. Aaron Darling reviewed changes that have already occurred and are still under way in the development of hardware, software, and systems for collaborative computing. Enormous quantities of data are being generated, and there are challenges to handling, storing, and analyzing them in reproducible ways that can be validated. He discussed the pros and cons of cloud computing and “software as a service” in a scientific arena. He touched on issues regarding the funding of and responsibility
for microbial forensics dataset management. He offered possible solutions for some of the obstacles he sees.
Looking backward, he noted that when there were few genomes to analyze, researchers performed analyses and then disagreed about the meaning of the analyses because there is a great deal of observational bias when data are limited. Now that a great deal of data exists, however, the taxonomic biases within databases should be considered, in particular, the lack of commensals.
In addition to the uncertainties regarding which organisms to sequence, organisms are naturally and constantly changing. The sequences added to databases represent a moving target because they come from living systems that evolve. Is the population evolving, or is one observing “standing” variation?5 Moreover, the databases used for microbial forensics are likely to be based on public databases. The standards for contributions to public databases are unlikely to meet the rigor one would expect for admissibility in court. Can these data be trusted?
Ten years ago, a major challenge was how to sequence a genome. Today a major challenge is how to deal with the immense amount of sequencing data being generated. This issue is becoming a major problem, both in isolate genomics and metagenomics. Two years ago, the sequence read archive at NCBI reached 100 terabases, 11 percent of which was metagenomic (Kodama et al., 2012). The MG-RAST6 resource offers 38 terabases of metagenomic data for download.
Sending large amounts of data over the Internet is not a problem because the aggregate bandwidth of the network is enormous. Moving data is possible using available, well-engineered, peer-to-peer (e.g., Bit-Torrent and Coral Content Distribution Network) and multicast solutions. However, although the aggregate bandwidth of the network is substantial, the endpoint bandwidth (e.g., at the source and the destination) may be limited, which could be of concern for truly large data volumes. The nature of how data arrives and how analysis is performed is also changing. The trend is away from data arriving in batches and toward delivery of continuous data streams. Three examples of initiatives to release data streams as they are being generated by public health labs and other sources are (1) the Global Microbial Identifier (a multiorganizational international effort), (2) the 100K Foodborne Pathogen Genome Project (University of California, Davis, School of Veterinary Medicine), and (3) the Genome Trakr Network (FDA). The data-stream approach is changing the nature of how analyses are performed. The previous paradigm was that data were collected from various sources, computational analysis was
_______________
5 Standing variation is genetic variation for fitness traits in natural populations.
6 For more information, see http://metagenomics.anl.gov/; accessed November 24, 2013.
performed, and the results were published. This approach does not adapt well to streaming data sources and may not be viable going forward. Unfortunately, most of the existing algorithms were engineered with the assumption that all data would be available from the start. Bioinformaticians need to start developing algorithms that scale to arbitrarily large datasets.
Algorithms in bioinformatics already exist that fit those two criteria. They include multiple sequence alignment with profile-hidden Markov models, phylogenetic placement on reference trees, and bloom filters. The nature of the data has changed, and more methods like these must be developed. Once it is possible to analyze all the data, researchers will have some basis from which to make a decision about whether data are useful or can be deleted from the data that are archived for future analysis.
The computing infrastructure used to analyze data also has changed greatly. The approach is shifting from individuals doing analyses on PCs or within a computing cluster to people performing analyses within cloud systems. The Amazon Web Services Elastic Computing Cloud is the best-known of these systems, and there are others, such as OpenStack, a cloud infrastructure that virtually everyone can download and set up on his or her own computer hardware. In Australia, the government set up a national research cloud named NeCTAR. Those at Australian research institutions are issued free allocations for use; in instances when large computing resources and many computing hours are needed, users can make special requests.
Other paradigms for using cloud computing have emerged, including the HTCondor project, which essentially leverages idle computing time; it is a cycle-scavenging system. The Condor can launch virtual machine images on idle computers. If an institution has a large number of computers that sit idle after everyone goes home at 5:00 p.m., that overnight time becomes a large computing resource.
The cloud-computing model is attractive because it enables one to rent time on a large, commercial, professionally managed computing facility, and to pay only for what is used. It enables a researcher to quickly scale computing resources up or down. One does not have to worry about how to dispose of all of the outdated computers in 5 years because the cloud provider will manage that. A challenge, however, is that data motion and storage in the cloud can become very expensive and will increase the funding researchers need from their sponsors. Researchers who wish to perform large dataset analyses in clouds must factor in how to most efficiently move the data around and store it. One possible solution is to use third-party providers for data storage.
Hardware architectures have changed also, largely owing to the fact that central processing unit (CPU) clock speed reached its limits about
10 years ago. A CPU cannot be pushed much further than 2.5 gigahertz without overheating. Speedups are largely attributable to doing more work in the same clock cycles. In particular, the graphics processing paradigm has emerged; these systems are constructed to enable thousands of arithmetic operations in parallel during each clock cycle. Today there are a number of graphics processor–enabled bioinformatics applications. For sequence alignment, there is the Short Oligonucleotide Analysis Package (SOAP); for sequence indexing and generic sequence analysis algorithms, the SeqAn library; and for phylogenetic analysis, graphics processing unit–enabled versions of MrBayes and Bayesian evolutionary analysis by sampling trees, which use the BEAGLE Library (beagle-lib).
Another innovation in computing hardware that is very relevant for large-data processing is solid-state disk (SSD) architectures. SSDs are similar to “thumb drives” but are scaled-up higher performance versions. They use essentially the same technology except that these are used as a normal disk drive in a computer; their random read/write performance is much higher than the classic hard-platter disk drive. When coupled with the classic disk in a unified storage system, these provide both high capacity and the ability to do a very fast random-access and reading and writing of data, which is a very useful feature when performing large genome analyses.
Computing costs have begun to exceed sequencing costs (Loman et al., 2012; Sboner et al., 2011). The major costs are the development of novel algorithms and software and training and maintaining skilled personnel to perform the analyses. When computation results in a public good, one must determine who will pay for it. Darling believes that costs can be divided into three steps:
- Development of novel algorithms and software (likely by academia);
- Integration of new software with existing software for usable systems (engineering); and
- Maintenance, support, and operation of computing.
Creating software is by nature very different from developing a physical technology. Once software is developed, it can be easily copied and disseminated. The cost for distributing it widely is negligible. The prevailing model in bioinformatics, and perhaps scientific software in general, is that through its funding institutions and scientific organizations, the public pays for step 1 and perhaps a portion of step 2. In Darling’s opinion, industry generally supports steps 2 and 3. This model appears to be reasonably workable because if a software system breaks, someone should be accountable, and private industry appears to be in a better posi-
tion to handle this responsibility. A complication, however, is intellectual property considerations. Much of the intellectual property is generated during the first step, and recognition of this must be conveyed to the parties who operate and maintain the software. One of the most common approaches is to make the software open source. The open sourcing of software is actually advantageous for industry because it can avoid investing in the development step. It is attractive for the bioinformatics industry to pick up these open-source tools and operationalize and validate them for users.
In microbial forensics analysis, absolute reproducibility may be an unachievable goal. So a challenge becomes, how reproducible should it be? In theory, computers should make repeatability and reproducibility very easy. Many scientists who practice data analysis, however, lack training in fundamental aspects of computing and software engineering that would enable them to undertake reproducible computing. A recent trend toward educating this community can be seen in the series of workshops called Software Carpentry.7 These workshops offer training in basic computing skills, such as version control, as well as literate programming, so scientists can generate workflows that are reproducible on different systems and engineered according to standards generally accepted in software engineering. In addition, there are a number of efforts to generate point-and-click visual systems that will enable users to generate reproducible workflows. These include the Galaxy, Taverna, Knime, and Kepler systems. Users can access a large number of small bioinformatics components, which they can connect and reconnect for arbitrarily different and unique workflows, and which they can then share with others. When users perform data analysis, the systems will track and record every step applied to data, and the users can share analysis metadata.
In terms of the operational aspects of repeatability and reproducibility, Darling believes that the ability to examine the software unit should be taken into account. With closed-source software, the best one can achieve is repeatability and copying the closed-source software to another computer; one cannot know how or why it works, nor can the computation being performed by it be independently reproduced. With open-source software, it is possible to examine the nuts and bolts of why it does what it does. Often when software is developed, and a manuscript is published about it, there are inconsistencies, and frequently large discrepancies, between what the software is actually doing and what is described in the manuscript. This can be attributed to the fact that when software is engineered and implemented, the developer must make approximations
_______________
7 More information is available at http://software-carpentry.org/; accessed November 24, 2013.
to the model described in the manuscript for basic engineering issues because computers have finite resources. The trend toward cloud computing has largely been advantageous, but an important disadvantage exists. In the old model of computing, software developers sold copies of their software and users installed it on their computers. The problem was that the software was difficult to maintain, and it was very difficult for software developers to distribute bug fixes. The new model is that the user does not even receive a copy of the software. Instead, the software is purchased as a Web-based service. The software runs on a server that is managed by the entity that developed it, and the user pays for access. It simplifies software deployment and maintenance; updates are managed centrally from the software developer servers; and piracy issues are eliminated. The disadvantage, however, especially when systems must be validated, is that the user has no control over software versioning. Therefore, reproducible research is not possible. The ever-present fear is that when the service provider decides to fix a bug in its software system, it could have unintended consequences for the analysis a researcher has already completed. Similarly, doing the same analysis on different days may yield very different answers.
Darling thinks that there is probably a way to generate reproducible results and still obtain careful version control of analyses using cloud systems, but it should be something that the service provider builds into the system at the very basic level. This needs to be thought about and discussed with the providers who develop the necessary software.
Chun suggested that there currently are too many software choices, and what is needed is a trusted body to evaluate this software. He added that bioinformatics is engineering and he believes all bioinformatics issues are solvable. Darling agreed, noting that software reviewers don’t always run the software or confirm that the source code is correct. A peer-review avenue for software systems is a good idea. The AAM (2009b) report also noted that there is a lack of statistical rigor for bioinformatics algorithms that needs to be addressed.
Some typing methods offer more sensitivity with less variability, and others offer more variability with less sensitivity. What can be relied on in the meantime? Darling said that he cannot comment on what should be relied on, but that genome-based phylogenies and their applicability are based on the type of organism one examines. Different lineages evolve in very different ways. Some recombine heavily, and if one builds phylogenies on these, a different result will emerge than if one builds a genome phylogeny on B. anthracis. For E. coli, he estimates it would be necessary to examine at least 200–400 kilobases of the genome in a concatenate alignment before getting a consistent answer about the phylogeny (Didelot et al., 2012).













