
Microbial Forensics and Clinical and Public Health Considerations: Commonalities and Differences
This chapter explores the commonalities and differences between the needs for microbial forensics on the one hand and for clinical diagnostics and public health protection on the other in terms of purposes, procedures, technologies, and best practices. The answers to these questions touch on key goals and objectives, sample type, level of characterization required, interpretation and reporting of differences, and differences in governance and associated requirements.
Dr. Stephen Morse of the U.S. CDC outlined what public health needs to know in the event of an infectious disease outbreak due to either natural causes or a biological attack:
- The identity of the agent,
- Whether it has antimicrobial susceptibilities,
- The method of dispersal or dissemination,
- The type of preparation (was it weaponized?) and potential for reaerosolization,
- Who was potentially exposed,
- The area involved and the extent of contamination,
- Whether to advise those in the affected area to evacuate or shelter in place, and
- Whether other factors (e.g., safety of food and water supplies) need to be addressed.
Professor Alemka Markotić, University Hospital for Infectious Diseases, Zagreb, an associate member of the Croatian Academy of Sciences and Arts, also emphasized that the distinction between natural and deliberate infectious disease outbreaks is very important. The Biological Weapons Convention (BWC) does not apply to natural outbreaks of disease. Therefore, it is essential to determine as quickly as possible if the origins of an outbreak are natural or deliberate. Any State Party to the BWC may request assistance when a suspicious outbreak of disease has occurred (see Chapter 1). Various evaluation criteria have been developed to assist in assessing whether an outbreak is suspicious (see, e.g., Box 3-1). An interesting paper by Grunow and Finke (2002) applied a model using many of these criteria to an epidemic of tularemia in Kosovo in 1999 and 2000. Grunow and Finke determined that the outbreak was the result of natural causes. But the procedure used was retrospective and would not have been in time for an intervention to prevent further spread of the disease if the cause had been due to a deliberate act.
Dr. Dana Kadavy of Signature Science, LLC, noted some general differences between the procedures used for clinical diagnostics and public health versus microbial forensics:
- Clinical diagnostic analyses are designed to identify rapidly the species and possibly the strain of an infectious organism to inform effective treatment strategies. Microbial forensics laboratories generate more fine-grained analyses of evidentiary samples, looking not only at species and strains but also perhaps at isolates of strains that possess unique genetic markers. Such in-depth analyses are likely to take much more time and effort (Figure 3-1).
- For microbial forensics, data must hold up to the scrutiny of judges and juries in a court of law as well as political figures, the media, and the public. Standards are defined for human DNA analysis, and diagnostic data are generated in conformity with Certified Authorization Professional (CAP), Clinical Laboratory Improvement Amendments (CLIA), or other certification standards. However, such guidance is less well defined for microbial forensics.
- In the clinical and public health sectors in the United States, governance lies with the American Medical Association and the Food and Drug Administration. For microbial forensics, governance lies in law enforcement, the judicial system, and perhaps the U.S. Department of Defense.
Microbial forensics may encompass evidence from environmental background, epidemiological data, and other forensic findings. Collec-
BOX 3-1
Epidemiological Clues That May Signal a Biological or Chemical Terrorist Attack
- Single case of disease caused by an uncommon agent (e.g., glanders, smallpox, viral hemorrhagic fever, inhalation, or cutaneous anthrax) without adequate epidemiological explanation;
- Unusual, atypical, genetically engineered, or antiquated strain of agent (or antibiotic resistance pattern);
- Higher morbidity and mortality in association with a common disease or syndrome or failure of such patients to respond to usual therapy;
- Unusual disease presentation (e.g., inhalation anthrax or pneumonic plague);
- Disease with an unusual geographic or seasonal distribution (e.g., plague in a nonendemic area, influenza in the summer);
- Stable endemic disease with an unexplained increase in incidence (e.g., tularemia, plague);
- Atypical disease transmission through aerosols, food, or water in a mode suggesting sabotage (i.e., no other possible physical explanation);
- No illness in persons who are not exposed to common ventilation systems (have separate closed ventilation systems) when illness is seen in persons in close proximity who have a common ventilation system;
- Several unusual or unexplained diseases coexisting in the same patient without any other explanation;
- Unusual illness that affects a large, disparate population (e.g., respiratory disease in a large heterogeneous population may suggest exposure to an inhaled pathogen or chemical agent);
- Illness that is unusual (or atypical) for a given population or age group (e.g., outbreak of measles-like rash in adults);
- Unusual pattern of death or illness among animals (which may be unexplained or attributed to an agent of bioterrorism) that precedes or accompanies illness or death in humans;
- Unusual pattern of death or illness in humans that precedes or accompanies illness or death in animals (which may be unexplained or attributed to an agent of bioterrorism);
- Significant number of ill persons who seek treatment at about the same time (point source with compressed epidemic curve);
- Similar genetic type among agents isolated from temporally or spatially distinct sources;
- Simultaneous clusters of similar illness in noncontiguous areas, domestic or foreign;
- Large numbers of cases or unexplained diseases or deaths;
SOURCE: Reprinted from Khan and Pesik (2011). Copyright 2011, with permission from Elsevier.
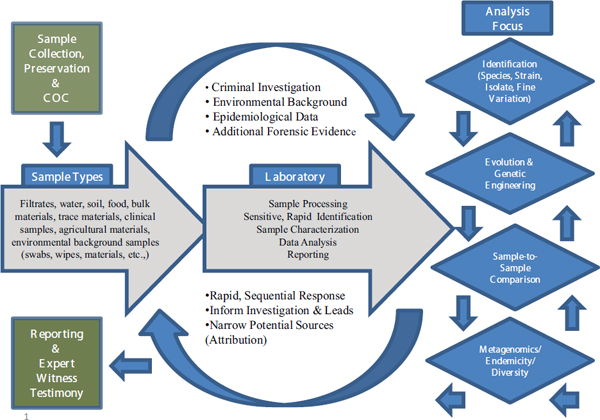
FIGURE 3-1 The microbial forensic process.
SOURCE: Budowle et al., 2013.
tively these aspects serve to help frame the forensics work required, and provide understanding of, and goals for, the types of laboratory analyses to be performed. The range of potential sample types that may be encountered in microbial forensics is vast. Clinical samples also can be highly complex, but the sample types generally are known and their matrices are well characterized. In microbial forensics, samples may come not only from clinical sources, but also environmental, food, and other matrices or be a completely unexpected target, as in a hoax. Sample types range from the simple to the complex, and across a clinical, clinical/forensic, and forensic continuum. They may comprise pure isolates, a prominent isolate in a minor mixture, a trace isolate in a mix of hundreds of organisms, and every other permutation. Samples also may contain copious amounts of a target organism or just a trace of the sought-for (or unknown) organism, which represents an analytical target against a background of sample “noise.” These challenges necessitate alternative sample-processing strategies.
Laboratory analyses for microbial forensics are intended to (1) serve as rapid screening mechanisms, (2) provide information for investigative leads, and (3) narrow the potential sources for a particular microorganism
in order to link it to a particular origin or activity for attribution. Microbial forensic methods need the capability to drill deeply and with very fine variances to fully assign attribution. Analysis extends to strain, subtype, and/or isolate; type and abundance of organism present in simple to complex samples; presence of antibiotic resistance or virulence genes; evidence of genetic engineering and/or isolate evolution (is it endemic, wild type, or a cultured strain repeatedly passed around labs?); and in-depth, sample-to-sample comparison that may be informed by SNPs and other genetic markers. Some of these are also major concerns for public health, for example, the presence of antibiotic resistance or virulence genes, which may dictate differences in treatment options. Reporting requirements for characterization also are specific for the microbial forensics space, and some of these are shared with clinical diagnostic practices and food analysis systems. In such cases, the sectors should leverage one another. The data generated may inform investigative leads and requests for additional samples, and will be used in reporting and for expert testimony. Documentation and reporting must be in place at each step.
The following case studies will illustrate in practical terms how some analytical approaches are shared between clinical medicine or public health and the use of microbial forensics to deal with biocrime.
THE RAJNEESHEE CULT IN OREGON, E. COLI O104, AND KLEBSIELLA PNEUMONIAE: THREE CASE STUDIES OF RESPONSES TO INFECTIOUS DISEASE OUTBREAKS
In 1984, the Rajneeshee cult deliberately contaminated restaurant salad bars in The Dalles, Oregon, with Salmonella enterica. The motivation for this biocrime was to influence the outcome of a local election by incapacitating the local populace to ensure that the cult’s candidates would win the county elections. More than 750 people were sickened, but fortunately there were no fatalities. But the deliberate nature of the attack was not confirmed until nearly a year later.
In this instance, the agent and context of the attack mimicked a naturally occurring event that plays out several times a year across the United States, with large, multifocal outbreaks such as the one that plagued the tomato and pepper supplies in 2008-2009, causing disease in more than 1400 people. (Skowronski and Lipkin, 2011:174)
This example of a biocrime (a more frequent category of intentional use of biological agents than bioterrorism; see Carus, 2001) highlights several important points. First, it can be a major challenge to differentiate among natural, accidental, or deliberately caused disease outbreaks. Second, such an outbreak requires similar responses to protect public
health regardless of the cause. Finally, modern technology makes it possible to determine the identity and characteristics of the organism being dealt with much more rapidly, as the next two cases dealing with natural outbreaks illustrate. The first case is one that also originated in the consumption of food from a salad bar, this time in Germany in 2011.
Escherichia coli, or E. coli, is usually a harmless bacterium that grows naturally in the intestinal tracts of humans and other animals. However, there are many different strains of E. coli, just as there are different strains of B. anthracis and Y. pestis. Some of these strains are highly pathogenic, such as the infamous O157:H7 and O104:H4 strains. The letter O in the name refers to a marker on the bacterium’s surface, which presents in hundreds of different immunogenic forms, in the latter instance, form number 104. The H describes another immunologic marker found on the bacterium’s flagellum. Both of these markers are easily detected by specific antibodies and can be used to identify strains. Pathogenic E. coli must be able to attach to or invade the cells of the intestinal lining, where they disrupt the normal function of the intestine by producing toxins, such as the Shiga-like toxin produced by the O157:H7 strain (AAM, 2011b). This toxin is very similar to the toxin produced by Shigella dysenteriae and is able to kill host cells. Both S. dysenteriae and E. coli that produce Shiga-like toxin are able to cause hemolytic uremic syndrome (HUS) in infected individuals. Some pathogenic E. coli may have extra genes not found in the normal intestinal strains that confer these abilities.
Dr. Dag Harmsen of University Hospital, Munster, Germany, described the recent E. coli O104:H4 outbreak in Germany and a Klebsiella pneumoniae outbreak in the Netherlands to illustrate the usefulness of next-generation gene sequencing (NGS) in public health responses. NGS is becoming both an essential component of microbial forensic analyses and the new preferred standard in epidemiology. For example, the E. coli O104:H4 outbreak has become a textbook case on technology development and microbial surveillance (Lipkin, 2013). It ushered in a new kind of epidemiological investigation and was directly instrumental in the development and evaluation of several new bioinformatics tools. It also provided proof of principle for the capability of smaller sequencing machines, which are reasonably affordable and, perhaps more importantly, allow rapid turnaround times.
As can be seen in Figure 3-2, which provides the timeline of the case, the O104 E. coli outbreak began in early May 2011 in the Hamburg area of Germany. In contrast with previous outbreaks, the incubation period was long, approximately 10 days. The outbreak was not declared in Germany until May 20th. Two days later, the European Centre for Disease Prevention and Control issued a warning for Europe. Labs were instructed to send isolates to the University of Münster, and the first samples arrived
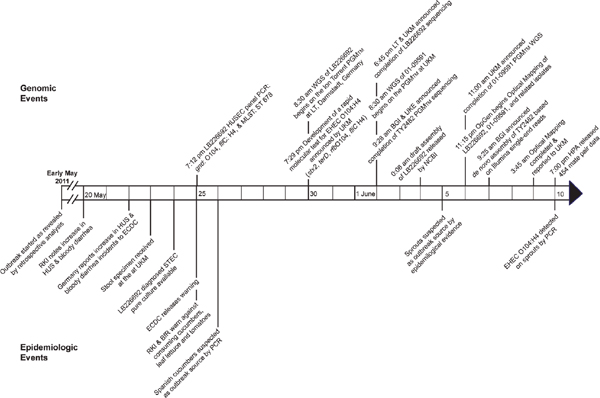
FIGURE 3-2 Events timeline of enterohemorrhagic E. coli O104:H4 outbreak. BfR, Bundesinstitut für Risikobewertung (Federal Institute of Risk Assessment, Germany); BGI, Beijing Genomics Institute (People’s Republic of China); EDC, European Centre for Disease Prevention and Control (Sweden); HPA, Health Protection Agency (United Kingdom); HUS, hemolytic uremic syndrome; LT, Life Technologies Group; PGM™, Ion Torrent Personal Genome Machine™; RKI, Robert Koch Institute (Germany); UKM, University Hospital Muenster (Germany).
SOURCE: Mellmann et al. (2011).
on May 22. After growing a pure culture, the organism was characterized and a screening test was created. This screening test, based on Sanger sequencing, was published on the Internet on May 30, and 15 days later in Lancet Infectious Disease. Eventually several other groups sequenced the E. coli so that its genome was eventually produced on all available NGS platforms. Several peer-reviewed papers published the sequence very quickly.
Phylogenetic analysis based on Sanger multilocus sequence typing (MLST) showed that the closest related genome was enteroaggregative E. coli, which had previously been sequenced by a French group (Mellmann et al., 2011). Harmsen pointed out that routine surveillance is very important. The HUSECO41 E. coli strain was identified in a 2008 publication (see Mellmann et al., 2008), yet little notice was taken, perhaps because this organism rarely causes infections. Using the sequence data, the MLST technique showed convincingly that this previous isolate is indeed most closely related to E. coli O104:H4. Although Harmsen’s group and others have proposed differing hypotheses about E. coli O104:H4’s evolutionary descent, there is as yet no resolution of this question.
Although there had already been an outbreak in Japan related to bean sprouts, it was not until June 5 that sprouts were suspected as the source. Unfortunately, most people remember eating a salad but will not remember all of its ingredients. This deficiency provides a very strong argument for changing the way epidemiological investigations are conducted, because the goal is to elucidate and contain the outbreak. It takes considerable time to identify a single contaminated component of a mixed salad and there are many false leads because microorganisms and/or toxins can be transferred from one component to another. There are numerous examples where one component of a salad was alleged to be the culprit only to find out later that something else was responsible. In the current case, two clusters of affected people had eaten at the same restaurants, which proved to be the epidemiological breakthrough. Based on this information, a very detailed food supply chain analysis was performed, and that is how the sprouts were finally implicated. Although no isolate had been recovered from sprouts in the German case as of October 2013, the epidemiological investigation confirmed convincingly that sprouts were involved. There was also a smaller outbreak in Marseilles, France, linked to sprouts. In the German case, once the sprouts were identified, the outbreak was essentially over.
Hospitals need fast, sensitive, and specific screening tests. In hospital surveillance terms, the ability to exclude pathogens is very important owing to response and cost. If a hospital can exclude the presence of an outbreak, it can avoid closing wards and putting patients into isolation, which are very costly measures.
According to Harmsen, while the laboratories and treatment provid-
ers were well prepared to confront the outbreak, communication with the public was not handled as well. For example, an accusation made early in the outbreak implicating Spanish cucumbers came from a local food agency in Hamburg. This agency used screening tests that were not specific for the O104 strain and the Spanish cucumbers were wrongly implicated as the source. Spain sustained substantial economic losses and sought compensation. Communication among the national, state, and local authorities was not always consistent, nor was there agreement between public health and food communications groups. Intense media attention is common in such situations and is not always helpful. Harmsen noted that he himself was misquoted during interviews. He suggests that there should be centralization of information for these kinds of communication.
The E. coli case demonstrated that rapid NGS can be used almost in real time during outbreaks, and Harmsen described a second example that proved the usefulness of NGS for diagnostics during an outbreak. Only a month after the German E. coli case in June 2011, there was an outbreak of K. pneumoniae at Dutch Maasstad Hospital in Rotterdam. The strain was multidrug-resistant K. pneumoniae OXA-48. The outbreak did not receive the global attention that the German outbreak had, but it was an enormous public health issue in the Netherlands.
The Dutch National Institute for Public Health and the Environment (RIVM) sent K. pneumoniae strains to the University of Münster to sequence. A draft genome of the K. pneumoniae OXA-48 outbreak strain was developed and compared with other publicly available Klebsiella genomes. Scientists identified 36 candidate regions to use in developing a strain-specific multiplex PCR test. They enlisted the help of the Wellcome Trust Sanger Institute in Cambridge in the United Kingdom, which was conducting a global surveillance of Klebsiella. By comparing the candidate signature sequences against Sanger’s additional 200 Klebsiella genomes, they identified two candidate regions that were specific for the Dutch outbreak (Netherlands National Institute for Public Health and the Environment, 2013). This information was given to RIVM, and a multiplex molecular diagnostic test assay that targeted one of the two signatures as well as antibiotic resistance genes was developed. This test assay was supplied to every Dutch hospital and is still used today for screening patients, mainly for exclusion purposes. The case is an excellent example of the important role of genomics in diagnostics as well as microbial forensics.
As detailed in Box 3-2, clinical microbiologists see many potential applications for NGS that can be broadly organized under these categories: (1) ad hoc epidemiology; (2) diagnostics; (3) therapeutics; and (4) global surveillance, early warning, and outbreak detection. Benchtop NGS is a democratizing force, enabling small- and medium-sized labora-
BOX 3-2
Rapid NGS Diagnostic Applications and
Public Health Microbiology
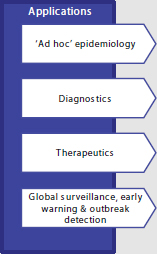 |
Details |
|
■ Introduction of benchtop Next Generation Sequencing (NGS) machines, enables small- and medium-sized laboratories (‘democratizing of NGS’) to perform ‘ad hoc’ genomic prospective epidemiology ■ Speciation / identification & pathogenicity profiling ■ Molecular diagnostic screening tests ■ Ultra-deep sequencing for pathogen discovery from human tissues (e.g., hemorrhagic viruses) ■ Susceptibility profiling ■ Vaccine preventability ■ Reverse vaccinology (rationale vaccine design) ■ Non-targeted new drug detection ■ Standardized Whole Genome Shotgun [WGS] NGS for detection of transmission between individuals ■ Outbreak detection, i.e., establishing the spread of particular strains locally or regionally ■ Longer-term and evolutionary studies to identify the emergence of particularly pathogenic or virulent variants |
SOURCE: Harmsen presentation, 2013.
tories to acquire these powerful new diagnostics, and some believe this technology can “leapfrog” into developing countries. NGS can be used for diagnostic and screening tests, and in therapeutics it has been used for susceptibility profiling. Some of the very early genome sequencing was performed for reverse vaccinology for the design of new vaccines, and NGS is now being used for drug development. It seems likely that many of the problems with sequencing will eventually be overcome, and its use could become fairly routine in clinical diagnostics. If this is the case, stored clinical data can also be analyzed for microbial forensics purposes if mechanisms for forensic analysts to access the clinical data can be devised.
Clinical microbiologists are particularly interested in global surveillance for use in detecting early outbreaks. To maximize surveillance data, a kind of molecular “Rosetta stone” is needed—a nomenclature that will enable global comparisons of data.
Harmsen also suggested that another ultimate goal should be to develop plain-language reporting that is understandable to physicians and epidemiologists. Translating “there is a SNP at position 4,000,000” to

FIGURE 3-3 Four approaches to using whole-genome sequencing. Pros (green), cons (red).
SOURCE: Adapted from Harmsen presentation, 2013.
“this confers resistance to tetracycline” greatly improves usefulness. For certain bacteria, plain-language reporting could be easily achieved, and several groups are already working to accomplish this.
Figure 3-3 summarizes Harmsen’s thoughts on four approaches for using whole-genome nomenclature. Genome-wide SNPs work especially well for monomorphic organisms, but Harmsen does not think it is the best choice. He agrees with Maiden et al. (2013) in advocating genome-wide, gene-by-gene allele typing. Although MLST is not popular for use with monomorphic organisms owing to its limited discriminatory power, it is still useful for other organisms and is influential from an intellectual point of view. Harmsen suggests using core-genome 1 MLST, or using MLST+, which enables analysis of the seven “housekeeping” genes2 as
_______________
1 The core genome is the set of genes that are present in all members of a species, suggesting that they are required for essential cellular functions.
2 Housekeeping genes are constitutive genes that are required for the maintenance of basic cellular function.
well as hundreds or thousands of other genes, which will provide the discriminatory power needed for outbreak investigation for most organisms (Jolley et al., 2012). There are also the benefits of it being additive, expandable, and nomenclature friendly.
NGS is a technology for analyzing all bacteria, whether performing evolutionary or phylogenetic analysis. It could enable standardized hierarchical microbial typing, surveillance and outbreak investigation, evolutionary analysis, and resistome/toxome3 analysis. Harmsen ranked the discriminatory power of microbial typing approaches in Figure 3-4. He believes that MLST will endure for backwards compatibility; it has a legacy of 15 years of publication and data generation and it can be extracted easily from NGS data. Canonical SNPs offer the same range, if not better, of discriminatory powers. Martin Maiden recently proposed a system called ribosomal MLST (rMLST) (Jolley et al., 2012) to implement a combined taxonomic and typing approach for the whole domain of bacteria. Harmsen believes that MLST+ ranks high for discriminatory power and that it can be standardized. Using SNPs and alleles would offer even more discriminatory power (Köser et al., 2012), but standardizing the method will be challenging.
Harmsen also sees a need for quality assurance and quality control guidelines for microbiology and microbial forensics. Published guidelines are primarily for use in human genetics. There is an initiative called Global Microbial Identifiers4 that is trying to address this and harmonize, but it is not well funded. He believes that a One Health approach,5 which links the communities concerned with food safety, veterinary medicine, clinical medicine, and microbial forensics, should be implemented, both within and among countries. Many nations may assume such collaborations are already in place, but during an actual event, difficulties will arise.
From Harmsen’s perspective, the future of NGS looks bright. Phenotyping based on genotyping should become a reality. Organisms such as tuberculosis and methicillin-resistant Staphylococcus aureus (MRSA) would be particularly appropriate for this analysis. These capabilities should be coupled with early-warning and geographic information systems.
_______________
3 The “resistome” is the collection of antibiotic resistance genes and their precursors in bacteria (Wright, 2007). The “toxome” is the collection of toxicity pathways, most of which are only partly known (Hartung, 2011).
4 For more information, see http://www.globalmicrobialidentifier.org/; accessed October 24, 2013.
5 The One Health Initiative is a movement to forge all inclusive collaborations between physicians, veterinarians, nurses, and other scientific-health and environmentally related disciplines. More information is available at http://www.onehealthinitiative.com/.
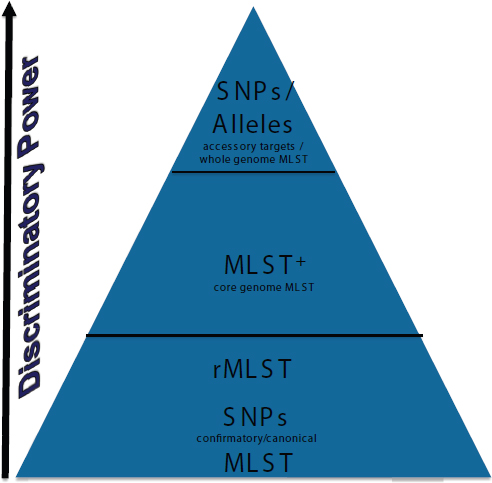
FIGURE 3-4 A ranking of the discriminatory power of microbial typing approaches, from bottom to top, with increasing discriminatory power. MLST, multilocus sequence typing; MLST+, core-genome MLST; rMLST, ribosomal MLST. SOURCE: Harmsen presentation, 2013.
HEPATITIS C IN SPAIN: A PUBLIC HEALTH PROBLEM THAT BECAME A LAW ENFORCEMENT ISSUE
Dr. Fernando González-Candelas, University of Valencia, Spain, described how molecular technology and phylogenetics were used successfully in court to convict an anesthetist of a biocrime involving the infection of multiple patients with hepatitis C virus (HCV). González-Candelas described how phylogenetic inference and coalescent theory
were employed to establish association between the presumed source and those that were infected.
González-Candelas explained that by early 1998, the Spanish public health authorities had noted a steady rise in HCV cases in Valencia since 1994. The cases appeared to be unrelated. Because intravenous (IV) drug use was on the rise and viruses are transmitted easily by blood, the increase was at first attributed to drug use. But in early 1998, a physician reported that four of his patients who had been sent to a private hospital for minor surgery had tested positive for HCV a few weeks afterward. The hospital stay seemed to be the only common denominator. Public health officials launched an epidemiological investigation, analyzing 66,000 surgical patients in two hospitals. They concluded that an anesthetist working at the private hospital was the likely common source. A search for other potentially infected patients revealed a sizeable outbreak of HCV that appeared to be clearly linked to this one medical professional. The anesthetist was ultimately convicted for being responsible for the infection of 275 of patients with HCV.
Both the hospital and the anesthetist were sued. Public health authorities and the judge heading the respective epidemiological and judicial investigations requested that González-Candelas and his colleagues use their expertise in evolutionary biology to specifically ascertain
- Was there an outbreak?
- What was the source?
- Can other sources be excluded?
- Which patients were included in the outbreak and which were not?
- When did it start and how long did it last?
- When was each patient infected?
- When was the index case infected? (González-Candelas et al., 2013)
The question “when was each patient infected?” was highly relevant to the judge and multiple insurance companies who had provided malpractice insurance coverage to the anesthetist during his working years; the time of transmission could determine liability. In addition, the anesthetist contended that he was a simply another victim.
Many people who are infected with HCV remain without symptoms and are unaware of their infection yet still can transmit the virus. Of those infected, 15-30 percent experience spontaneous clearance of the virus, and 70-85 percent become chronic carriers, of whom 25 percent do not experience a progression to disease. But HCV can be silent for many years. Of those in whom illness progresses, a process that can take 10-30 years, out-
comes can include cirrhosis, end-stage liver disease, and hepatocellular carcinoma (Bowen and Walker, 2005).
The HCV genome is organized as a positive sense single-stranded RNA virus and has a high mutation rate, similar to that of HIV. It evolves quickly—one million times faster than human DNA—but variability is not equally distributed across the genome, and some areas evolve much faster than others. The rapid evolution is due to high mutation rates, high virus production (approximately 1010 viral particles/day in an infected patient), and a short generation time. Cell-infection cycles occur within a few hours (Penin et al., 2004).
The background prevalence of HCV in Spain is about 2.5 percent, which is relatively high, and many of those infected are unaware that they are. Evolution can occur quickly within an infected individual, and over years of infection, the number of viruses that accumulate in that individual can be substantial. In addition, there are reports of compartmentalization of HCV in individuals; various organs and tissues in the same individual can be infected by slightly different and divergent populations of the virus (Di Liberto et al., 2006). For this reason, one cannot expect to find the same virus at the same time throughout the host. In addition, although transmission between infected patients is horizontal (i.e., from person A to person B), the viruses are related through vertical processes. Whenever there is an infection, there is a strong “bottlenecking” of the infections passed on, such that patients infected by the same source may receive slightly different samples of the virus. Therefore one cannot expect to see exactly the same viral genome in the source and all patients infected by the source.
González-Candelas and his team sequenced about 134 clones for each patient who agreed to supply a sample. This limited sampling is why consideration of compartmentalization is so important. He gave an example of patients co-infected with two HCV genotypes, who, depending on the day of sampling, showed only one of the two genotypes, alternating from one day to another. The dynamics of viruses within an individual’s blood are largely unknown, but are more complicated than one simple representation of a viral population. There are many populations, and the longer a person is infected the more opportunity exists for minor differences. Most differences appearing in the widely divergent clade shared by the patients in the Valencia HCV outbreak probably represented sampling from the same source at different times over 10 years. The source would transmit a slightly different virus, but one still recognizable as a sample of the source’s viral diversity; it was still possible to determine the common ancestor. González-Candelas proposed that if one can find the common ancestor of a sample, compared with the common ancestor of
the reference population, one is in a good position to make all additional inferences.
For the court case, phylogenetic inference and coalescent theory6 were used to analyze the outbreak. González-Candelas believes that these approaches should be incorporated into microbial forensics.
In 1998, González-Candelas and colleagues were using gel-based Sanger sequencing. They decided to use two approaches to analyze the clinical viral samples: (1) direct sequencing of PCR products generated with primers directed to a relatively slow-evolving polymerase region of the HCV genome (NS5B), and (2) sequencing of PCR products derived from the faster-evolving regions (E1 and E2). For the latter, they analyzed several clones from multiple victims to estimate viral diversity in each individual (González-Candelas et al., 2013). They also cloned sample sequences from local population controls, which were not related to the case based on epidemiological determinations. In analyzing 300+ samples, it was also possible that some were positive for HCV but were not related to the source despite the fact that they had either been anesthetized by the alleged source or there were other circumstances that could have made him the presumed source of their infection.
Analyzing a single gene from the virus samples, however, did not provide all the necessary information. The graph in Figure 3-5 shows the distribution of differences when compared with the sequence in the NS5B region (229 nucleotide [nt]) of each patient isolate with that from the presumed source. There were many identical sequences, which appeared to be the “smoking gun” that linked about 150 patients to the anesthetist. The scientists, however, wanted to know how they should characterize possible victims who show a single nucleotide difference between their
_______________
6 Coalescent theory and phylogenetic inference: Phylogenetic inference uses the genetic variance between members of a population to infer evolutionary relationships; at the level of specific genes, alleles are used to create trees to represent how members split into separate branches, converge, or become extinct. Coalescent theory, on the other hand, seeks to analyze all known alleles of a specific gene to identify the most recent common ancestor (MRCA). In its simplest form, coalescent theory does not consider evolutionary pressure, recombination, or other interactions with the environment. If one follows the ancestry of two haploid organisms, eventually a single organism (the MRCA) will be identified that gave rise to the two lineages and the two organisms will have coalesced. These analyses can be used to predict the time of appearance of the alleles, as a sort of molecular clock, by converting the degree of change in a sequence to a specific interval of time. Gonzalez-Candelas and his colleagues were the first to use these two approaches in understanding origin and relatedness in the context of a criminal trial. Problems remain with juror perception of the validity of these kinds of data, in view of the complexity of the concepts and the tendency to accept all DNA-based evidence as incontrovertible; validation of the approach for use in criminal court remains to be performed. Nevertheless, the Valencia case highlights the value of using phylogenetics in conjunction with traditional epidemiological data when building a case (Vandamme and Pybus, 2013).
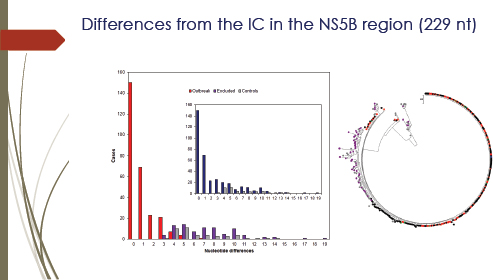
FIGURE 3-5 Differences from the Index Case in the NS5B region (229 nt). The bar graph (left) shows the distribution of differences in the NS5B region. Sequences were compared with a 229-nt fragment of the NS5B gene derived from the presumed source. The graph shows the distribution of nucleotide differences (Hamming’s distance) for sequences derived from patients included in the outbreak (red bars), from patients excluded (dark purple) from the outbreak, and from local controls (gray). The bar graph inset shows the same distribution for putative outbreak samples (dark blue) and local controls (gray) before the former were divided into included and excluded from the outbreak. The neighbor-joining tree (right) was obtained with the NS5B-region sequences of hepatitis C virus (HCV)1a samples analyzed. Color codes: outbreak sequences are in black, red, and green, excluded from the outbreak are in dark purple, and local unrelated controls are in gray. The presumed source (PS) sequence is shown in blue. No clade was found with bootstrap support higher than 70 percent.
SOURCE: González-Candelas et al. (2013).
virus and that of the presumed source—a circumstance that occurs commonly in virology. The court decided that those showing one difference could also be considered victims, which increased the number of potential victims to 200+. But, the scientists asked, if one difference is accepted, why not two differences? They had controls with as few as three differences from the source. Being concerned about false negatives, they asked whether they should use this number as a threshold defining those with three or more differences as not part of the outbreak.
The second problem with NS5B phylogeny is illustrated by the
neighbor-joining tree analysis in Figure 3.5 (right), which failed to group all of the control samples in a monophyletic group. Furthermore, none of the nodes in this tree receive bootstrap support higher than 70 percent by either method. Analysis with the slowly evolving NS5B region was unable to discriminate among the three epidemiological groups: local controls, patients infected by a common source, and patients infected by alternative sources. Therefore the emphasis was shifted to the more rapidly evolving E1-E2 regions.
Sequencing of the E1-E2 region was carried out for a total of 4,184 cloned PCR fragments, representing 134 clones sequenced from the presumed source, 321 samples initially considered part of the outbreak, and 42 local controls; the average number of clones from each sample (excluding the presumed source7) was 10.77. Multiple alignments were performed with the 4,184 sequences to derive neighbor-joining and maximum likelihood phylogenetic trees for the 4,184 cloned sequences, shown in Figure 3-6. The conclusion of this approach was that sequences from 274 patients were grouped with the sequences from the presumed source, while the second group included all the sequences derived from the local controls and sequences from 47 patients initially considered to belong to the outbreak. Thus the separation between the two groups became clear.
However, while they could show the global analysis of the phylogeny, the data could not represent a forensic conclusion, because forensics normally requires an individual analysis. This requirement had not previously been applied to an epidemiological analysis. They adapted a statistical framework for forensic analysis to molecular epidemiology (González-Candelas et al., 2013). They used Evett and Weir’s (1998) method for applying genetic analyses to forensic settings, using a Bayesian framework. Evett and Weir propose that scientific experts limit their contributions to forensic analyses of genetic information to the evaluation of available genetic data in light of two competing hypotheses, those of the prosecution and the defense. The forensic expert should present the likelihood ratio: what is the likelihood of the genetic information seen in light of two mutually exclusive hypotheses combined with other types of evidence? The police or other investigators would provide additional independent evidence.
It is up to the judge or jury to evaluate and integrate this information with the information provided by other types of evidence, ideally in a numerical way. Molecular phylogenetics provides a logical way to provide likelihood ratios or probabilities. In this case, there were two large groups—the outbreak and the non-outbreak. Based on what was
_______________
7 Under Spanish law, only a single sample is required to be provided by a suspect.
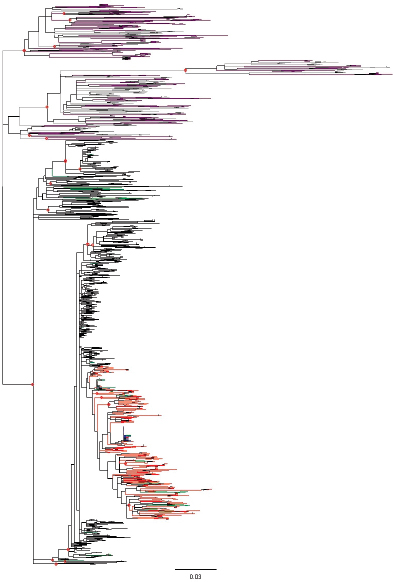
FIGURE 3-6 Maximum likelihood tree for cloned sequences in the E1-E2 region. The tree includes 4,184 sequences from a 406-nt fragment of the E1-E2 region including hypervariable region (HVR)1 and HVR2. Sequences were obtained from patients included in the outbreak (274 patients, 3,038 sequences), patients excluded from the outbreak (47 and 559, dark purple), local controls (42 and 453, gray), and the presumed source (PS, 134 sequences, dark blue). Sequences and branches in the monophyletic clade defined by all the cloned sequences from the PS are labeled in red. Sequences from polyphyletic samples with some representatives in the clade delimited by the PS sequences and others outside it are labeled in green. Relevant nodes with bootstrap support larger than 90 percent are indicated by red dots.
SOURCE: González-Candelas et al. (2013).
being tested, the results were either tests of the defense or the prosecution hypothesis.
Eventually, González-Candelas and his team answered all of the questions posed by the court. There was an outbreak, and the source was a practicing anesthetist. Other potential sources could be discarded; molecular surveillance of HCV has never turned up another case with samples from the clade of the outbreak samples. No other common source of infection was found. They identified 275 individuals in the outbreak and excluded 47. The outbreak began at the end of 1988, and ran until 1998, intensifying from 1996 onward. The presumed source had been infected for 10 years, and this infection was prior to the time of his patients’ infection. Two-thirds of the estimates for the time of infection coincided with those offered by the prosecution. The anesthetist was convicted of professional malpractice and was sentenced to a lengthy jail term.
For moving forward, González-Candelas’ conclusions are
- Molecular phylogenetics and coalescent theory are essential for microbial forensics.
- A meaningful statistical treatment (maximum likelihood, Bayesian inference) is mandatory.
- In particular, this case shows that recent developments in evolutionary biology can be used to estimate dates and places of relevant events.
- A good sample of a reference population is absolutely essential to draw any conclusion about the origin of a set of samples. This may change from one case to another.
- The forensic expert is one among others: his/her conclusions have to be evaluated in the appropriate context.
- Although current sequencing methodologies allow us to work with complete genomic information, a lot of work remains to be done to standardize and control laboratory and analytical procedures.




















