
Validation and Reference Materials for Microbial Forensics
In the introduction to their chapter on validation of microbial forensics, Murch and Bahr (2011:649) state the following:
The use of effective, robust, and properly validated methods for the collection, preservation, transport, analysis, interpretation, and communication of probative evidence is a linchpin of reliability and confidence-building measures that contribute to the acceptance, use, and understanding of science by investigators, judges, attorneys, juries, the media, and lay public. Stakeholders expect that forensic methods, protocols, and techniques have been validated properly. All science proposed and admitted to court is subject to discovery and scrutiny under U.S. case law and prescribed legal procedures. In recent years, as courts and the media have become more aware of the value, power, risks, and uncertainties of forensic science, whether or not methods have been validated properly is receiving increasing attention.
Microbial forensics is a relatively new discipline and the science and technologies it uses are evolving rapidly. Because of the demands to produce and apply reliable and robust capabilities, validation measures, requirements, and protocols are essential. Dr. Bruce Budowle, one of the early developers of microbial forensics, and his colleagues Steven Schutzer, Roger Breeze, Paul Keim, and Stephen Morse produced a Microbial Forensics textbook, now in its second edition (Budowle et al., 2011), which is used as an informative resource regarding many aspects of microbial forensics. In an authoritative presentation at the Zagreb workshop, Dr. Budowle reviewed established and recommended measures and
approaches for validating components of each stage of microbial forensics processes. He provided insight into gaps in validation coverage and the challenges of designing protocols in a system encountering substantial and often undefined diversity and sample variation.
Dr. Budowle began by stressing that a microbial forensics investigation does not have to produce a “smoking gun” solution to be informative. It needs only to provide a conclusion to the level necessary for producing a useful piece of a puzzle. The goal is attribution, which is
1.) To establish the exclusion or possible inclusion of the source of a sample. The level of attribution can be determining the species or strain of a microbe to the individualization of a particular sample, that is, coming from a particular isolate or test tube. The latter is not readily achievable with currently validated genetic typing capabilities; and
2.) To integrate the microbial forensic findings with other forensic and investigative evidence to address the ultimate question of guilt or lack of guilt.
He outlined four undesirable scenarios for the application of a methodology:
1. Applying a bad method and doing it poorly. Operators are often unaware their methods and performance are poor. The information generated lacks a firm basis and is frequently incorrect.
2. Applying a bad method well. Operators believe the data are good because they followed standard operating protocols carefully, but the data may be meaningless or, worse, erroneous.
3. Applying a good method poorly. Operators produce content for which the criteria or limitations for use are undefined or compromised or, worse, erroneous.
4. Applying a good method well, but the science community or stakeholders do not accept the data. The operator may, for example, have validated the method without documenting the validation, so there is no confidence in the system, or there have been documented problems with the particular laboratory and there is a lack of confidence even though the procedure(s) were carried out properly.
The goal, of course, is to perform a good method well. Budowle noted that a great deal of focus is placed on technology. Empowerment by using sophisticated technology can be deceptive if its use is not validated. Moreover, while validation is necessary, perceptions of what constitutes validation vary. In practice, the basis for validation may be as tenuous as “because I’ve used it for so long it must be the right thing,” or “based on my experience of doing this three times” or “it is a community validated method so my use is valid.” Because there are no absolute criteria, and
each application may have unique requirements, validation is a challenge. The goals for validation in microbial forensics are to
- Promote development of a program that is scientifically valid and rigorous.
- Define criteria for development and validation of methods that will support attribution for criminal investigations.
- Establish national/international working guidelines for quality assurance and quality control.
There are two general types of validation for microbial forensics: developmental and internal. Both essentially have the same goal: to define limitations of use and/or interpretation.
- Developmental validation refers to the acquisition of test data and the determination of conditions and limitations by the developers of the method. Measures or determinants of developmental validation would include specificity, sensitivity, reproducibility, bias, precision, false positives, false negatives, determining appropriate controls, and the choice and/or quality of reference databases (Murch and Bahr, 2011).
- Internal validation refers to accumulation of test data within the laboratory that intends to use the method to demonstrate that established methods perform as expected. Methods must be tested in one’s own laboratory to ensure that they perform as expected and that limitations are understood, so that interpretations do not cross the boundaries of the abilities of the methodology.
Because microbial forensics may be applied in exigent circumstances, “preliminary validation” may be considered. For example, during the U.S. anthrax letters case when a research tool had to be applied to an investigation, Paul Keim’s multiple-locus VNTR analysis (MLVA) was used to determine that the B. anthracis strain was Ames. In the midst of such an event, a result was needed in a short time to protect human health, and it was unreasonable to wait until the laboratory can validate a procedure to the degree normally desired. Response time does not allow for such a lengthy process. The criteria recommended for considering “preliminary validation” are (1) exigent circumstances, (2) an inability to wait for some lengthy time period to completely validate the method, and (3) the acquisition of limited test data that will enable an evaluation of a method that provides support to investigate a biocrime or bioterrorism event.
One caution raised was being so tied to a protocol that the scientist does not think about the assay and its result and operates more or less
as an automaton. Standard operating protocols (SOPs) are for routine work. A danger of SOPs is that they can restrict analytical thinking and disregard both inculpatory and exculpatory evidence. The ideal balance to strike is one that enables thinking “out of the box” while still applying boundaries on use.
The minimal criteria that should be addressed for validation are
- Specificity,
- Reproducibility,
- Precision,
- Accuracy,
- Robustness,
- Analysis of specified samples commensurate with the intended application of the assay (e.g., reference panels and mock or nonprobative materials), and
- Limit of detection.
In his experience, Budowle has encountered scientists who contend that there is no need for a quantitative element in validation when the test result “is a yes/no answer.” He stressed that this view is absolutely incorrect because one must quantify the limits of detection in order to draw inferences about the meaning of what one is trying to assess. An assay will require some level of quantification.
Validation measures should be applied from the very first stage of the investigation—sample collection—and through all subsequent stages of the process, including shipping and storage; extraction, which greatly impacts results; analysis; and finally, interpretation. It is critical that validation be applied to the interpretation stage. Interpretation is dependent on, for example, which databases and procedures are employed for drawing inferences; a value can change based on the database or inference framework used.
If evidence is not collected, it cannot be tested. If it is not collected correctly, results may be suspect or destroyed. The “proper” collection of evidence is not a new concept; clearly, mixing evidence items in a bag is unacceptable. Storing evidence improperly also will be problematic as crucial targets may be destroyed.
Microbial forensics relies not only on microbial evidence but also on additional forensic evidence, and the collection of microbial evidence can sometimes be incompatible with other types of forensic evidence. Given a choice between a fingerprint and a spore, the level of individualization offered by the fingerprint could make it the more desirable piece of evidence to preserve. Swabbing may obliterate the fingerprint and
destroy valuable evidence. Triaging strategies must be incorporated into the sampling process.
A challenge is to develop investigative protocols that optimize the choice and priority of methods. Among factors to consider are the amount of sample available, evidence preservation/conservation, and trade-offs between speed and accuracy and/or precision. Given the limitations of wearing protective equipment speed may become a requisite over accuracy and chain of custody. Making priority decisions given collection constraints should be determined ahead of time and methods developed to minimize the loss of sample and less than desired collection and documentation. Simple strategies, such as pre-labeling tubes before beginning sampling due to difficult manipulation of labels when wearing protective gear, is an example of a simple consideration.
Collection validation must address
- Recovery,
- Stability,
- Integrity,
- Target—organism or analyte (e.g., DNA, RNA, protein, toxin, agar), and
- Influence of sample matrix.
As an illustration, Budowle pointed out that the ideal swab is the one that adsorbs the sample well. But the worst swab for extraction of the target is the one that adsorbs well. A swab that adsorbs well may be better for recovery from surfaces at a crime scene, but the swab matrix holds the target analyte such that it is not efficiently removed from the swab during extraction. The result can be low yield of a sample even though sufficient material was collected at the crime scene. Selection of collection tools often requires that a balance be struck. Samplers must work out these details and ensure that the application aligns with final objectives. Collecting an isolate from a victim is quite different from going into an air duct to collect trace samples. Good procedures exist for collecting, preserving, and shipping blood, but they do not exist for handling a flask of liquid that no one wants to open during collection. It may be necessary to make an educated guess about the target in the sample or the nature of the sample may dictate the best option for preserving the sample. Options to preserve the contents of a flask can vary from maintaining it as a liquid or frozen. There may be presumptive testing that can be performed at the scene or intelligence to guide this determination—rightly or wrongly. If there is a substantial amount of material, it may be desirable to store it multiple ways to increase the odds of preserving the target of interest. Procedures must continue to be developed.
Extraction efficiency depends on the collection medium. Different extraction procedures can be productive or counterproductive in obtaining the particular target medium. When choosing an extraction procedure, analysts must consider
- Specific target—virus, bacteria, fungal, toxin;
- Spore vs. vegetative cell;
- Active vs. inactive (culture plan, amplification plan);
- Analyte that will be assayed—DNA, RNA, protein, lipid, stabilizers, media, fatty acid, etc.;
- Stability of analyte;
- Matrix effect—substrate, other co-extracted analytes, materials such as soil; and
- Downstream assay impact.
Budowle emphasized that it is essential to consider what effect extraction methods will have on downstream analysis. There is no point in extracting material with a particular method if co-purifying compounds impedes the function of an assay downstream.
The basic criteria for validation during the analytical stage appear in Box 6-1A, and additional criteria may be warranted. The amount of target that can be analyzed by an assay should be defined. It is not enough to implement an assay; it is essential to define the acceptable range. A low amount may be acceptable, but associated stochastic effects may affect interpretation. The critical reagents should be defined. Often assays are developed to perform in optimum ranges and performance criteria. Optimization can be misleading as it implies maximum performance. However, a window of performance criterion is established for robust methodologies. It is usually undesirable to design an assay that is at the edge of a window of performance because any small perturbation in some part of the process could cause it to fail. Windows can be set for criteria, for example, as simple as the temperature for annealing primers during PCR. If the highest temperature for successful primer annealing is 63°C it would not be best for a robust assay to set the temperature at 63°C. Given that thermocyclers have some variation, it may be desirable to set the temperature slightly lower so that the temperature in the tube is favorable for primer annealing. It is best to find the range for the performance criterion, and use a condition somewhere within the performance range of the analysis but not at the edge—so it will perform robustly.
Another point is to consider a real-time PCR assay with a linear range between 20 and 30 cycles, and the user observes a positive result somewhere between 40 to 50 cycles. Since the result is no longer in the linear range, one cannot predict quantity of the target, but there is a posi-
BOX 6-1
Validation for the Analysis Stage
| A. Basic Criteria | B. Broader Criteria |
|
|
SOURCE: Budowle presentation, 2013.
tive result. The challenge becomes in determining the meaning of a positive result that may be due to a single molecule. Highly sensitive assays may detect a signal but the reliability or confidence in the result may be suspect.
An important issue that Budowle believes receives too little attention is the process for handling conflicting results or considering alternative hypotheses. Conflicting results will occur. Good scientists should always question results. But in the face of conflicting results, the action to follow could be to verify with orthogonal testing, if available. Alternatively, reports should contain language about results that may be questionable or that alternative hypotheses may be supported.
Budowle and colleagues developed additional criteria (Box 6-1B) for the analysis-stage validation to supplement basic criteria. Their intent was to provide test/data recipients, funders, or stakeholders with a more comprehensive list of criteria. Being more informed, one could ask if appropriate validation criteria were addressed with the development of a method. It is the responsibility of the developers and users to demonstrate why they chose the criteria they did to evaluate and validate the method and, just as importantly, explain why they rejected what they thought unim-
portant. Thus, more accountability and documentation will be associated with “validated” methods. Not all criteria listed would apply in every situation. While testing, for example, specificity across a range of species may be necessary to validate a hand-held assay; once implemented the assay would not require a comprehensive database to effect interpretation. It may, however, apply to determining whether or not the Ames strain is common or rare in a particular circumstance.
With a new technology or method, the results often are compared with the results of an existing technology—for example, comparing MLVA and canonical SNPs to determine if they correlate and where they do not. One would expect that at some point, one technology may resolve better than the other and that observation should not be considered discordant data in itself. If assessing an entirely new technology for assaying a target and generating results with no comparable existing data, one must define the criteria by which to test it, develop the test, and validate the performance. Two approaches to evaluating new technology are portrayed in Figure 6-1.
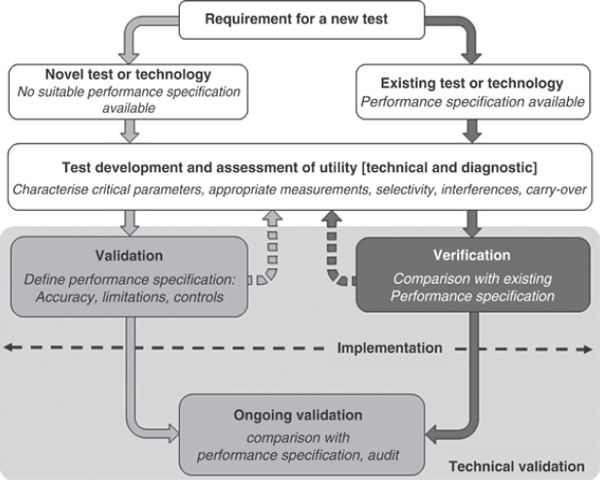
FIGURE 6-1 Process of implementing a test for diagnostic use.
SOURCE: Mattocks et al. (2010).
SOPs must be created, and once an entire system is created, another validation must be performed. The SOPs should contain sufficient detail, including, where appropriate,
- All steps in the procedure,
- Proper controls,
- All reagents and preparations,
- Calibration,
- Criteria for analysis of results, and
- Interpretation of results.
Analysis and interpretation are two different things. Analysis could be “I have a positive result.” Interpretation would be, “It is ten times more likely to observe this result if X compared with if Y.” Budowle emphasized that there are feedback validation mechanisms in both approaches.
Validation is an ongoing process; it does not stop once the method is up and running. To illustrate its importance, he described an incident in which a lot of commercially available human DNA identification kits were sold to customers, and subsequently the manufacturer discovered that the deoxynucleotide triphosphates (dNTPs) could degrade while kits sat on the shelf. The company sent letters to crime laboratories apprising them of this possibility, but some laboratories continued using the kits. The signal of results was dropping (i.e., a loss of sensitivity of detection) and in actuality could have presented negative results. This outcome is a very serious problem when working with small amounts of DNA. The positive control supplied with the kit also was diminishing in signal intensity. Yet some users ignored it or were not cognizant of the signal loss. Instead, as long as they had a qualitative call that was consistent with the profile of the positive control, the users were satisfied that quality control was acceptable. Both qualitative and quantitative signals should have been monitored. Had the users done so, it would have been evident early on that the system was not performing appropriately and the assay could have been halted rather than consuming valuable evidence.
Error is an important consideration because error is ubiquitous. Different types of error are shown in Figure 6-2. There is, for example, systematic error, which includes measurement error. There also is bias, both in methods and in the individuals interpreting the data. It is human nature to want to accommodate data to scenarios that make sense to us, regardless of alternative hypotheses or explanations. This human nature affects how we perceive false positives and false negatives. There is no absolute point where the line can be drawn between the two false categories for acceptable performance. As the line shifts in one direction or the other, more false positives will be obtained at the expense of false nega-
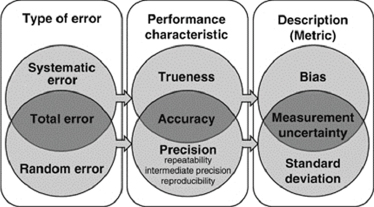
FIGURE 6-2 Performance characteristics, error types, and measurement metrics.
SOURCE: Adapted from Mattocks et al. (2010).
tives, or vice versa (see Mattocks et al., 2010). The reality is that a decision is made based on an evaluation of the necessary risks related to the application at hand with available resources, and these must be defined.
The sample analysis flowchart in Figure 6-3 provides an idea of the variety of microbial analyses one might consider. Investigators might perform classic taxonomic analysis, for example, and analyze for weaponization engineering. They also might examine spore density. If a sample
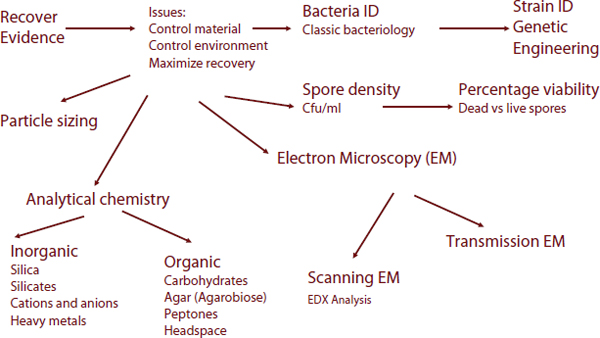
FIGURE 6-3 Sample analysis flowchart.
SOURCE: FBI Quantico laboratory.
is collected from a host, this analysis might not be very helpful, but if it is collected from a package found in a train station, it might be useful.
The range of possible analyses in the flowchart conveys the challenge encountered with microbial forensic evidence. The field comprises a number of subdisciplines, each requiring substantial expertise. Moreover, it illustrates why next-generation sequencing (NGS, also known as massively parallel sequencing), is so appealing. Many other techniques have been designed to identify a specific target or targets; but they require knowledge about the target to be able to design the assay. But with NGS, the technology can target all bacterial and viral select agents using the same basic approach. NGS also provides high throughput, which can translate into a high sensitivity of detection and resolution. Higher throughput can result in greater depth of coverage, potentially allowing for detection of low-abundance or trace-level targets. Therefore, NGS will likely become one of the primary tools for microbial forensics. To use the technology, or for that matter any of the microbe identification methodologies, a panel of isolates is required to validate the assays. Much thought should go into the appropriate panel to test/validate the tool. The panel should be broader than two samples, but narrower than all possible samples, because the latter would be impractical. The challenges will be to define the panel breadth, how to obtain the materials, and who will make them available.
Many procedures for collection exist but have not been compiled, some have never been disseminated, and some have not been validated. These protocols, however, should be leveraged. Budowle believes that the literature, particularly older literature, should not be ignored. Access to older literature is needed because some perpetrators may not have the same resources that advanced laboratories have for routine microbiological research. It may be delusional thinking to assume that someone will go into a BSL-3 facility to produce a weapon when that someone may actually be in a garage using a washing machine as a centrifuge. If we are not familiar with the old procedures, we may miss telltale signs that a weapon is being developed, miss invaluable leads, and may not collect informative evidence. Digital archives could be created to preserve older literature. Workshop participant Jens Kuhn of the National Institutes of Health (NIH) agreed that access to older literature is very important. Moreover, he believes that researchers who rely solely on PubMed for research and database creation may be unaware that there is a bias in this resource. The journals of many countries and even the literature on specific diseases are underrepresented in PubMed. PubMed is, for example, of little value if you seek case representations on hantaviruses, whereas some countries produce much literature on the subject. Another problem is that younger scientists may mistakenly believe that older literature has little value. It
is important that libraries receive funding, and that libraries retain their old relevant journals, which even the NIH library finds difficult to do.
Budowle believes that interpretation is perhaps the most critical step requiring validation because there is a tendency to become unthinkingly trusting once a protocol is running. For example, in a real-time PCR assay, if a signal is obtained at 48 cycles, it may not have much value, or it may have some lead value. With enough amplification cycles, it is likely everything will become positive. There is a need to have an interpretation of “inconclusive.” He pointed to a situation in 2005 in which a partial signal was indicative of the presence of Francisella tularenis in the area of the National Mall in Washington, DC. Four positive amplicons were considered sufficient for identification of F. tularensis; however, only two were positive, and there was no clear interpretation for a partial profile and what actions to take.
Budowle believes the concept of “missing data” is also an important factor to be considered when interpreting results and should be part of the validation equation. As an example, he recounted an anecdote about the process the Allies in World War II followed to protect bombers from being shot down during bombing runs in Germany. This anecdote may or may not be true, but either way, it is instructive. Returning planes were examined, the location of bullet holes mapped, and vulnerable areas were reinforced with shielding based on the location of the bullet holes. Much later, it was pointed out that the planes that returned were not necessarily informative. The planes that did not return should have been the focus. The investigators were missing key data. Budowle sees this as an ongoing challenge in microbial forensics because a lot of data is missing. Interpretations must be made using limited data, and biology is mutable. The sample an investigator tests today may be linked to something ten passages back, or may have traveled through a host and mutated. We must accept that we lack data, yet build around this to make the process work as well possible. While we can never have absolutes, we must still produce conclusions, but they should be tempered appropriately.
Considerations to help guide this interpretation process are listed in Box 6-2. Putting a confidence limit on a conclusion can be very difficult. In some cases, a qualitative statement may be all that is possible and may be appropriate, as it was in the Amerithrax case in which there was no accompanying statement of a percent confidence that the microbe was the Ames strain. Limitations are a reality. An answer with limitations is not a wrong answer; it simply reflects what we know and can do. Even with a qualitative statement, some verbal constraints are necessary on the strength of the evidence or extant data support. In addition, saying, “I am 92 percent confident of X” may not convey the right information for the circumstances. Instead of quantitative or qualitative statements, we may
BOX 6-2
Interpretation of Results
- Statements—qualitative, quantitative, semiquantitative
- Database—type, relevance, representative, quality
- Background data—normal values, reference range, endemicity
- Does a result require follow-up or further analysis—temporal/spatial analysis, effect of passage
- Limits of interpretation
- Statistical approach—match, similarity, most recent common ancestor, identical
- Thresholds
- Software
- Alternative explanations
SOURCE: Budowle presentation, 2013.
need qualitative statements with extra information to guide the end user or other scientists who may review the analysis.
Databases also have limitations. Our databases comprise samples of organisms that infected something. The organisms that did not infect anything may be just as prevalent but have lacked the opportunity or ability to infect. This reality should be considered during the investigation process. We do not know what is endemic, and this can be an issue in the overall interpretation process. If we identify a “match” or a “similarity” or the most common ancestor, the significance of these associations must be supported by something defensible.
Thresholds are another challenge. Thresholds are critical not just for the analytical phase but also for the bioinformatics phase of NGS. Budowle stressed that there always can be alternative explanations and that good scientists should confront them, position them appropriately, and through good hypothesis testing determine whether they have low probability or high probability of support. Especially in forensics it is crucial to follow the scientific method, which is to attempt to disprove one’s hypothesis rather than to prove it.
Some of the major questions that might be asked during a source exclusion, association, and attribution analysis appear in Box 6-3. Perhaps instead of asking whether samples are the same or different, we should be asking “how different are they?” What conclusions can be made, and how do they associate in an evolutionary context? In contrast, one might argue that the question to ask about the evidence in the Amerithax case—the
BOX 6-3
Challenges for Any Target:
Source Exclusion, Association, and Attribution
- Are these isolates the same or different? What are the discrimination criteria?
- How dependent is the conclusion on samples taken, method used, and interpretive approach?
- What would an exclusion, association, or “match” really mean? How precise can or should one be?
- Do the methods and interpretation account for variation, evolutionary change (genome dynamics), and influences imparted by environment and ecology?
- How should significance to a comparison be assigned? What are the confidence limits? Can a probability or likelihood be assigned?
SOURCE: Budowle presentation, 2013.
flask marked RMR 1029—might not have been an evolutionary question since the flask’s contents comprised a mixture of samples. A more appropriate question may have been “How probable would it be to observe these variants if they arose from a single source versus from mixtures?” A great quantity of epidemiological data exists that can be consulted to help us understand associations.
Forensic questions include “What is it, and was the release intentional or natural?” How the threat agent was made can be a critical piece of evidence for an investigation, which in turn can feed back to help answer other questions and also may provide a lead to the perpetrator’s identity.
Forensic questions based on genetics-based analyses appear in Box 6-4. Investigators want to know, “Is the information provided with genetic markers probative? Is it meaningful?” For example, in a foodborne pathogen investigation, one might ask if there is an expectation that E. coli will be in a room besides what we carry in our bodies? The probability of E. coli being in a room occupied by humans (and animals) is high. What the appropriate level is may be unknown but it could affect the confidence of our conclusions.
Richard Vipond of Public Health England, Porton Down, offered an observation based on his experience of trying to take PGM (Ion Torrent Personal Genome Machine™) through validation for metagenomics work. His lab wanted to “freeze” the technology in time to assess performance and result quality. Because the manufacturers are in competition with each other and the technologies are evolving rapidly, every couple of months there is a release of a new version—for example, a chemistry
BOX 6-4
Microbial Forensics Questions: Genetics
- What might be deduced concerning the nature and source of the evidentiary sample?
- Is the pathogen detected of endemic origin or introduced?
- Do the genetic markers provide a significant amount of probative information?
- Does the choice of markers allow effective comparison of samples from known and questioned sources?
- If such a comparison can be made, how definitively and confidently can a conclusion be reached?
- Are the genetic differences too few to conclude that the samples are not from different sources (or lineages)?
- Are these differences sufficiently robust to consider that the samples are from different sources?
- Is it possible that the two samples have a recent common ancestor or how long ago was there a common ancestor?
- Can any samples be excluded as contaminants or be recent sources of the isolate?
- Are there alternative explanations for the results that were obtained?
SOURCE: Reprinted from Hunt et al. (2009) with permission from Elsevier.
or server upgrade—and this can dramatically change performance. The Porton Down laboratory has seen error rate, critical to any decision it makes, decrease with quality enhancements, and then increase with the introduction of a novel step. It is quite difficult to freeze assessment. The lab cannot even go back and rerun tests using the same reagents and software because these materials will not be available 6 months later. However, the rapidly evolving technology environment must be embraced or there will be a risk of reducing capabilities.
Deep sequencing generates substantial data, and sequencing capabilities are enabling scientists to develop elaborate databases tailored specifically to the demands of a case. This capacity demands novel ways to handle and analyze data that are now routinely obtained in terabyte amounts. Given the immense quantity of data, bioinformatics is an absolute necessity. It is unlikely (at least in the United Kingdom), however, that bioinformaticians will be on staff in application-oriented laboratories, so investigators will need to validate the bioinformatics pipelines and there will be a demand that the pipelines be sufficiently robust.
Factors to consider that affect data interpretation and quality include
- Quality metrics of sequence data;
- Sequence errors and uncertainties;
- Reliable standards for genomic data representation;
- Uncertainty with databases used;
- Inferences based on available data, including metadata;
- Formulation of well-defined hypothesis/hypotheses: testing methods for assessing the weight of microbial forensics evidence;
- Criteria for comparisons: match, similar, different, inconclusive;
- Rigor of reasoning by the expert; and
- Software.
Quality metrics are another important issue. What criteria should be used? Budowle pointed out that while some analysts use Q20 (a quality metric score associated with a base call) for base calling, others use Q30 (a higher quality score by an order of magnitude than Q20). He suggested that neither by itself may be sufficient, and validation is necessary to guide the user on how best to use quality metrics. For example, if the user were to encounter 500 reads with a Q9 score for one base in a homo-polymeric stretch in all these reads, despite the low Q score, there may be high confidence associated in this single base with complete representation because the low score might be the result of a chemistry artifact. The PGM system is more refractory than the MiSeq with sequencing of homo-polymers but with so many reads (or fragments) that the data may still be reliable because of the quantitative representation. Such considerations will be necessary to validate so that data can be used as effectively as possible. It is very difficult for one lab to replicate all essential details of any bioinformatics pipeline used by another lab because there are so many factors that affect outcomes. Attention to documentation will be requisite.
The extraction method one uses can cause variation of the result, as can the amplification method, including enrichment, PCR, and primer selection. The primers may seem to be an obvious consideration, but the enrichment process or capture method may not.
There are also differences in sequencing techniques. One technique will show a gap in a sequence whereas another will show a base. We must consider how to resolve such differences. Budowle’s lab runs analyses on both MiSeq and PGM so he can exploit orthogonal chemistries to help determine what is reliable and valid. Neither system is immune to problems that need to be addressed. This testing enables one to improve both systems. Standards and controls must be created to better assess such concerns.
Box 6-5 lists the components of a “bioinformatics genetic toolbox” for microbial forensics. Budowle noted that there are processes in which alignment is used, and processes in which it is not; the processes are
BOX 6-5
A Bioinformatics Toolbox for Microbial Forensics
- Assemblers;
- Aligners;
- Phylogenetic algorithm(s) for clonal and sexually inherited markers, recombination, gene conversion, and horizontal gene transfer;
- Capability to identify informative markers and their power to address specific forensic issues;
- Better understanding of mutation rates and the effects of environment and host on these rates;
- Discrimination and match criteria to quantitatively interpret results with confidence bounds;
- Endemicity;
- Capability to relate diversity to function;
- Capability for comparative and functional genomics;
- Ability to contain or access curated (genetic marker) databases on pathogens and near neighbors and their background occurrence with epidemiological history, when available; and
- Data management with the capability to access and process large amounts of diverse genetic data and to communicate data rapidly with stringent informational security (i.e., fully functioning information interoperability).
SOURCE: Budowle et al. (2005).
different and must be validated. Assembly may be sought for simple samples, but currently would be extremely challenging for metagenomic samples. Phylogenetic algorithms differ and also must be validated. Validation also will be an issue in data management. An enormous amount of data is being generated, and there is a recommendation or trend in the greater science community toward deleting raw data and simply saving the data at the fast level. This approach may be practical and economically appealing, but concordance testing may require that the biological material initially used for sequencing be archived. Otherwise, there may not be proper comparisons made as modifications or novel approaches are developed. Note that the large amounts of data common today require new bioinformatics algorithms because old tools (e.g., BLAST) will not scale to handle large data needs.
There are inference and error validation concerns, as well (Box 6-6). The base error rate of the particular sequencing protocol used must be defined. Moreover, there are different kinds of error. Sequencing errors, which vary from site to site, may occur as a result of chemistry and soft-
BOX 6-6
Inference and Error Issues
- Forensic analysis of whole-genome sequence data often will compare two or more sequences, for example, an evidence sample profile with that of a reference sample that may be considered a direct link or have a common ancestor.
- Sequencing error and other factors will most likely inflate the dissimilarity between samples, creating a degree of “uncertainty” to some extent.
- Defining and quantifying the error rates associated with each platform/chemistry is critically important and includes extraction, amplification, library preparation, software.
SOURCE: Budowle presentation, 2013.
ware. Alignments can cause a great deal of noise, and there is other noise that cannot be identified solely by simulation, for example, with a metagenomic sample analysis in which chimeras have been created.
When scientists perform 16S rRNA diversity studies, everything that differs from what has been previously seen is considered “new”—another organism—yet Budowle notes that this might simply be junk. The diversity of what has been identified may be overstated or understated because of stochastic effects in the process.
Validation of materials is essential to a successful microbial forensics program, both for cross-comparing data and for running routine tests. Controls—reference samples, panels, and reagents—are required and must be accessible. The magnitude of the problem of developing material standards is illustrated by the wide variety of targets, which include but are not limited to genes, proteins, morphology, physiology, and biochemistry. Reference materials are needed for all developers and analysts. Moreover, standards will change. The Ames strain has become a sort of standard for B. anthracis only because it was used as a weapon. But how we define or select a standard for each species should be made more judiciously. A single strain may never suffice as a standard, but a standard must be established at some level because using 50 standards in a run with one sample may not be practical and currently would be costly. Establishing standards becomes a process in itself.
The number of technologies is wide and expanding. Among them are NGS, other DNA/RNA assays, traditional morphological and biochemical identification approaches, SEM/TEM, micro-Raman spectrometry, atomic force microscopy, isotopes, secondary ion mass spectroscopy, particle-induced X-ray emission, and field immunoassays. A single method cannot
address all targets and all evidentiary materials that will be encountered. Nor can one validation method or standard address all technologies; each technology must be validated individually.
There are two types of standards: performance standards and material standards.1 Most often, users focus on material standards, but performance is equally important. Traceability, a documentation of measurements to some standard, is needed. In the United States, standards developed by the National Institute of Standards and Technology (NIST) are typically used, but the available standards do not meet all needs. We need to decide who will make the standards to bridge the gaps in what is available. Some of the gaps in standard reference materials appear in Box 6-7. A major concern is how to identify a good standard versus “just a standard.” For example, surrogates may or may not suffice; they are often, but not always, suitable (Anderson et al., 2005). Near neighbors will be required to describe the sensitivity and specificity of an assay. Budowle also posed the question of who will prepare these standard reference materials and who will maintain them? Is it government’s, industry’s, or an individual laboratory’s responsibility? Likely the responsibility will vary based on the need.
Whole-genome sequencing of a metagenomics sample will generate sequences that span the genomes of the microorganisms that reside within the sample. In metagenomic samples, species and/or strains can be represented in widely varying abundance. The limited depth of coverage and amplification-bias stochastic effects on portions of individual genomes might affect representation of critical sequences that define species, causing them to be missed. Part of a genome will likely be represented, but phylogenetically and genetically the parts that are detected may not be able to resolve at the species level (most importantly near neighbors) or even at higher taxonomic levels. An analytical assay may perform perfectly well but may be uninformative on the presence of a target even if it is truly in the sample. So, we have to consider this process of resolving at the near-neighbor level and understanding which sequence reads are informative for taxonomic resolution or classification. One should rightly pose the question, whether only one, two, three, or more reads are sufficient to render an interpretation of identification.
Primer development and the quality of primer synthesis require validation as well. Primer design programs do not validate primers, and the need for such validation is underappreciated. Similarly, bar coding or
_______________
1 A performance standard specifies what is to be accomplished but does not dictate the particular method or material to be used as long as the desired end is achieved. A materials standard specifically directs that a certain material or method be used to accomplish the desired end.
BOX 6-7
Gaps in Standard Reference Materials (SRMs)
- Traceability of controls. At this point, most reference materials lack SOPs for routine typing. Preanalytical, assay, and interpretation: reaction mode
- Sensitivity, specificity, contamination, technical issues: framework for investigators.
- How to identify a good standard?
- Supporting materials, for example,
- nucleic acid
■ extraction method,
■ quantification method, and
■ integrity, purity.
- nucleic acid
- NIST-traceable model.
- Criteria to qualify a reference sample: What is the reference?
- Standards for preparation of SRM:
- process
- appropriate analyte of interest
- Validation—also acquire samples other than reference: range (and the logic behind that)
- Can surrogates suffice?
- Are near neighbors required? Or should this be an individual researcher responsibility?
SOURCE: Budowle presentation, 2013.
indexing for NGS may require reliability testing. Some reads may have uninformative bar codes,2 likely because the index is low quality, because there may be synthesis errors in the generation of the barcodes. Perhaps adequate differentiation between/among indices should be implemented so that an error does not place sequencing content in the wrong sample. Computationally it should be feasible to define barcodes, but depending on how they are used may limit the numbers of samples that can be indexed.
New reference materials must be generated, but old ones must be maintained, or there may be no good way to compare back with existing or former methods going forward. It will be necessary to select the best methods for generating databases. Concerns that should be addressed
_______________
2 Barcodes or indexes are short unique sequence tags added to every fragment of a sample during library preparation to “tag” the fragments unique to a sample. Thereby, different samples can be pooled (or multiplexed) and data separated (i.e., demultiplexed) bioinformatically after sequencing, based on the unique tagged sequences.
appear in Box 6-8. For years, there has been discussion about the concept of (1) a centralized database or (2) a decentralized virtual centralized database. There are challenges to creating these databases. One is how to convince and incentivize people to share. Potential solutions might be to make funding dependent on sharing, or to provide database access in return for sharing. There will be patent and intellectual property issues. There should be, at a minimum, some centralized knowledge database of what is held by governments and the private sector.
Any centralized entity or group of entities that produces reference materials should have access to a sufficiently comprehensive collection, understand the rationale for isolates, and have sufficient long-term support. Such an entity would need to maintain the highest possible QA/QC, using accepted, standardized methods. It should be responsive to research and development (R&D) and support and incorporate R&D that updates extant knowledge of diversity. Finally, it should be governed as a community resource, while balancing this access against security concerns.
Budowle does not foresee development of an “ideal” reference resource because the characteristics of such a resource will continually change. Instead he foresees the creation of multiple reference resources, some of which may be set up in real time during events. Another limitation to achieving an ideal reference resource is the inability to capture the full diversity of the microbial world and all the permutations neces-
BOX 6-8
Databases and Resources
- The Select Agent Registry system has created a database, but it has limitations—no uniformity, and decentralized holdings hamper the speed, accuracy, efficiency, and reliability.
- Databases could refer to physical materials and/or related data; microbes, toxins, nucleic acids; and metadata; genomic, proteomic, transcriptomic, and metabolomic data.
- A centralized, comprehensive physical archive of reference materials would facilitate
- Implementation of a standardized characterization system,
- Uniform QA/QC,
- Development of standard typing techniques,
- Standardization of new techniques and analytical methods,
- Reference samples for high-resolution genomic comparisons, and
- Duplication.
SOURCE: Budowle presentation, 2013.
sary for the multitude of analytical methods. For archival purposes, it is reasonable to maintain representatives of pathogens that may be used in biocrimes or bioterrorism. Centralized collections are likely to be inadequate for most investigations, but suitable for basic research. Culturing strains from an archive will be done on a limited basis owing to the cost of maintaining a comprehensive set that is continually cultured and tested over time to ensure viability.
Microbial forensics investigators need increased access to reference resources, yet there is a real conflict: One position may demand that access be restricted for security reasons while another position is that tests and countermeasures cannot be developed without access to the materials. To make any progress, we must consider and understand both positions. Budowle’s suggestions for immediately moving forward are to
- Identify the experts now. There should be a global consortium of experts who have agreed to be available for, at a minimum, consultation should an event occur.
- Review the current (and past) state-of-the-art technologies.
- Establish scientific working groups and guidelines.
- Establish standards and standardization.
- Better define—and encourage—validation and peer review of the science.
- Share information and capabilities within the law enforcement and intelligence communities.
- Foster partnerships.
- Develop ways for greater access to genomes or microorganisms to facilitate validation.
Budowle emphasized that we must begin an interactive and committed process to address issues now. Scrambling to prepare as an event occurs is not a desirable scenario.
Cindi Corbett of Canada’s National Microbiology Laboratory agreed that the idea of microbial working groups is a good one. Large problems exist, but they can be attacked one step at a time by the typing experts, database experts, and so forth. She would like to see Round Robins because investigators do things differently. Given the same set of data, would we all come up with an acceptable answer? Initially such an exercise could use just data, instead of an actual sample. Many opportunities exist for collaboration, interacting with the community, and for employing experts in various areas.
REFERENCE COLLECTIONS AND DATABASES
As noted in numerous places in this report, there is widespread agreement that more reference collections and databases that are properly curated and maintained are needed. Reference collections house actual organisms while databases comprise only genomic or other information about microorganisms.
The American Type Culture Collection (ATCC) is one of the premier sources for microbial reference strains. It contains the world’s largest collection of bacteria, viruses, yeast, fungi, protozoa, nucleic acids, and molecular tools. The ATCC Bacteriology Collection contains more than 18,000 strains in over 750 genera as well as more than 3,600 type cultures of validly described species and nearly 500 bacteriophages. For viruses, ATCC houses a wide assortment of cultures for use in research related to pathogenesis, epidemiology, molecular assay development, and vaccine discovery and production. ATCC offers an extensive array of infectious disease organisms intended to promote research leading to novel methods of detecting, minimizing, and treating infectious diseases. It is an entity that could be studied as a model for a reference collection devoted to microbial forensics.
Dr. Juncai Ma, a member of the organizing committee for the Zagreb workshop, is the Director of WFCC-MIRCEN World Data Center for Microorganisms (WDCM) of the Institute of Microbiology at the Chinese Academy of Sciences and an executive of the World Federation of Culture Collections. He discussed the WDCM,3 which is a user-friendly international database resource for the compilation of data on the location and function of culture collections of microorganisms, cultured cell lines, and genetic elements. The WDCM also provides access to the data, serving as an online gateway to international databases on microbial diversity, culture collection catalogs, services, and molecular data relating to microorganisms.
The WDCM was developed under the auspices of the World Federation of Culture Collections and UNESCO’s Microbial Resources Centres (WFCC-MIRCEN). The WFCC is a Multidisciplinary Commission of the International Union of Biological Sciences (IUBS) and a federation within the International Union of Microbiological Societies (IUMS). The WDCM is now located at the Institute of Microbiology, Chinese Academy of Sciences (IMCAS) in Beijing. Brief descriptions of the WDCM databases and resources appear in Box 6-9.
The Information Center of IMCAS is collaborating with the interna-
_______________
3 More information is available at http://www.wdcm.org/; accessed November 23, 2013.
BOX 6-9
Databases and Resources:
The World Data Centre for Microorganisms (WDCM)
1. World Directory of Culture Collections (CCINFO).
Directory of all registered culture collections: 652 collections from 70 countries/regions.
Microorganisms: 2,304,840
Bacteria: 966,143
Fungi: 689,946
Viruses: 36,807
Cell lines: 31,178
Can be browsed by country, region, or acronym. Can be searched by collection, strains, or keyword. Drill-down information and statistics on culture collections—including key collection personnel, subject coverage (e.g., agriculture, biotechnology), preservation methods, culture availability criteria, services, and entry and update information.
2. CCINFO Strains.
List of holdings of registered culture collections.
3. Reference Strain Catalogue.
Provides access to the reference strains listed by the ISO TC 34 SC 9 Joint Working Group 5 and by the Working Party on Culture Media of the International Committee on Food Microbiology and Hygiene (ICFMH-WPCM) from its Handbook of Culture Media for Food and Water Microbiology.1
4. Analyzer of Bio-resource Citations (ABC).
A platform to support researchers in checking literature citations. Data-mines 3,005 journals; years 1953-2012. Can search by paper, patent, and strain number. Offers drill-down statistics on most-referenced strains. Users can upload their own papers.
tional Barcode of Life (iBOL) project,4 and with the China Central DNA Barcode of Life program. WDCM collaborates with multiple collections throughout the world to advance the field and to organize educational workshops and symposia. WDCM also cooperates with the International Organization for Standardization (ISO) to develop the WDCM Reference Strain Database for all microbial resources in conformance with ISO standards, and all of this information is available to the public.
Ma believes that the WDCM can make contributions to microbial forensics and perhaps serve as another model for information on culture
_______________
4 More information is available at http://ibol.org/; accessed November 23, 2013.
5. Global Catalogue of Microorganisms (GCM).
A free information-service platform to help culture collections to manage, disseminate, and share information related to their holdings. Fifty-two different collections from 25 countries. By year end 2014, there will be 100 collections. Small collections will be offered support in creating their own linking homepages. Includes detailed strain information (e.g., patents, sequences, bioinformatics analysis), related citations, isolation sources, geographic origin, phylogenetic analysis, species identification, and access to online exchange of data. Drill-down information available for countries. Searches can be refined by collection, temperature, organism type, and isolation origin, and can be displayed in multiple formats. Android version available; iPad version soon to be released
6. Statistics on Organism Patents.
Via collaboration with World Intellectual Property Organization, information and statistics on patents associated with culture collections.
7. Resources:
Nomenclature—National Center for Biolotechnology Information, Species 2000, List of Prokaryotic Names with Standing in Nomenclature.
Metagenome—Joint Genome Institute (JGI, U.S. Department of Energy). Metagenome portal, European Bioinformatics Institute Metagenomics, metagenome submission guide.
8. The World Directory of Culture Collections.
Book. Sixth version will be released in 2014; 191 collections updated their information in 2013.
________________
1 WDCM Reference Strain Catalogue, available at http://refs.wdcm.org/home.htm.
SOURCE: Ma presentation, 2013.
collections and databases. Issues that should be addressed by all culture collections before sharing would include
- Data standards: What do we need? What can we share? Minimum datasets (MDS) and recommended datasets (RDS). Range of microorganisms and software development.
- Data policy, software availability for data sharing, data club (should it be member only?). The data in WDCM’s Global Catalogue are currently open to all.
Ma proposed development of a Global Catalogue for Microbial Forensics, as outlined in Box 6-10. He noted that a Global Microbial Forensics Catalogue and its knowledge base would make the discipline more vis-
BOX 6-10
Proposed Global Catalogue for Microbial Forensics
| Proposed Content: | Proposed Functions: |
|
|
SOURCE: Ma presentation, 2013.
ible, and pointed out that it is much easier to begin cooperative efforts among stakeholders when working in the information field. Working groups could be organized to address information issues.


























