
5
Genomic and Evolutionary Challenges for Biodemography
Kenneth M. Weiss
The world is experiencing a flood of new knowledge about the evolution and nature of genomic causation. New technologies and data have, in general, supported prior clearly predictable and expectations from the best of 20th century biology (Weiss, 1992; Weiss and Terwilliger, 2000). Inherited factors with strong effect on biodemographic traits promise opportunities for major clinical application (e.g., Gordon et al., 2014). However, relevant causation is more often so subtle and complex as to defy simple solutions and, instead, to raise serious epistemological challenges.
The challenges go beyond increased data collection. What is needed is better theoretical understanding of genomic causation. This applies particularly to behavior traits that are hard to define and affected by experience. Improved ability to relate genetic effects to age patterns, and hence life history, reflects causal mechanisms and could be a fruitful area of research. However, an important question is when, where, or even whether a genomic approach is justified. In what follows, I will try to provide a picture of the state of the science, briefly highlighting some of the many issues, of which I have described my views elsewhere in more detail (Buchanan et al., 2009; Weiss and Buchanan, 2009a, 2009b, 2011; Weiss, 2010; Weiss et al., 2011).
EVOLUTION AND DEVELOPMENT: NESTED GENOMIC SYMMETRIES
Each person is the product of development, constrained by the products of evolution. Evolution among populations and development among
cells both generate trees of nested local descent, as shown schematically in Figure 5-1. Both processes depend on differential proliferation among cells that inherit their progenitors’ genomes.
In a descent lineage among individuals or from a fertilized egg during the lifetime of one individual, the DNA sequence will experience mutational changes. These arise randomly and are essentially irreversible. But in addition to a tree of sequence variation, each individual is a tree of DNA usage variation that is altered during development and life history by controlled, context-dependent mechanisms that are reversed by similar context-dependent mechanisms.
The phenotype in an individual is the net result of its inherited constitutive (germ line) genotype and the distribution of somatically generated cellular phenotypes. The trait distribution in a population jointly reflects these two independent sources of variation.
These biodemographic phenomena generate life history-based nested descent trees, but DNA sequence samples from the population may not be a very reliable indicator of the integrated genome of each individual. The effect of somatic mutation, overlaid on inherited genotype, results in a net

FIGURE 5-1 Symmetry between evolutionary and somatic genomic variation trees.
NOTE: Population (left) and embryological growth yield many localized variants. In the case of populations, recent variants are newly arisen mutations that have not had time to rise in frequency or spread spatially. In the case of somatic mutations, recent ones have arisen later in development and are localized to more specific tissues, based on the trees of related tissues as they arise embryologically. The arrows identify the sequences that are sampled in typical genomic studies—individuals from populations sampled for their inherited genotype (but each individual (right) is not typically sampled for his/her tree of somatically arisen genotypes).
SOURCE: Modification of a base drawing courtesy of A.G. Clark.
phenotype that includes hidden somatic variation, presenting a precision problem for genome-wide trait mapping aimed at finding causal variants of traits of interest. In a similar way, the inherited genomes as a set vary among populations. Both in evolution and development, variants that arise early in a descent tree, whether it be that within an individual, among individuals in a population, or among populations, are generally more common and widely dispersed than more recent arrivals, and the same may be generally said about the effects they may have on phenotypes.
What are typically sampled in mapping studies are collections of individual constitutive genotypes in different individuals (arrow in left panel, Figure 5-1), which correspond to the single genotype of each individual (arrow on right). Unfortunately, no real theory exists about the degree to which this multiply-nested variation matters. Instead of explicit theoretical modeling, genomic causal inference typically has rested on inductive, exhaustively reductionist, retrospective data fitting, rather than rigorous prediction. The approaches rest essentially on generic statistical association and regression models, which are mechanism-free approaches that, in the presence of the kinds of complex causation I will try to outline, can lead to conclusions so indirect as to provide weak predictive power and hamper understanding of causal mechanisms at the genetic or evolutionary level. This matters because a major public promise has been personalized prediction. There are subtleties here that some in the profession understand: This may be possible in some situations, but the drive for individual prediction and the belief in strong genomic causation are persistent and quite widespread. Many newly documented genomic phenomena are clearly not well treated by these approaches.
THE DISSOLVING IDENTITY OF “GENES” AND THEIR FUNCTION
Genomes achieve complex functions of many kinds beyond protein coding, many of which are not yet understood. These functions are differently active within each cell’s context and concealed from other cells except through intercellular communication. The scientific community has very incomplete knowledge of these mechanisms, especially from quantitative (dose-response or age-related) predictive perspectives.
Context Is Everything
DNA is basically an inert molecule that functions only by its interactions with other factors in a cell. Remarkably, despite enormous amounts of data and knowledge, there is still heated controversy over how much of the genome is “functional” at all. As a reductio ad absurdum shows, ATG is the DNA code for the amino acid methionine, but not all ATGs
are involved in coding (roughly 1/64 of all adjacent nucleotide triplets are ATGs but only a tiny percent are protein-coding). So what are they doing and why are they there?
DNA sequences function only in the context of overlapping sequence-based multiway interactions (often modeled as “networks”) of factors. In fact, most genes are primarily used to regulate or process other genes. In this sense, life is a “Boolean” (combinatorial) phenomenon that functions via the context-specific arrangement and spatiotemporal combination of factors, which, as Figure 5-2 suggests, occurs not necessarily in the location where the interacting factors are produced. A gene activated in one cell that leads it to secret a signal may have its effect in distant cells that monitor their external environment for signals. Though generated in diverse other locations, combinations of such signals in the latter location may be responsible for the local effect. This is rather abstract, again nested, causation relative to the usual linear model of DNA as a long necklace of discrete causal elements.
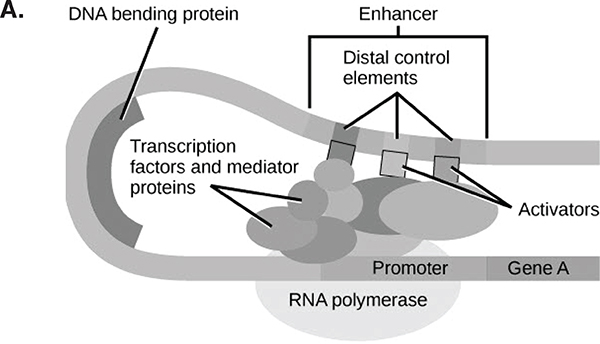
FIGURE 5-2 The Boolean nature of genomic function.
Panel A. Combinations of regulatory factors are needed to express a given gene. Many short DNA sequences near a gene are bound by proteins (themselves coded for by genes elsewhere in the genome, with their own comparable structure). The combination of these proteins binding the regulatory sequences and with each other affects the amount of transcription of the gene.
SOURCE: Mahr (2014).
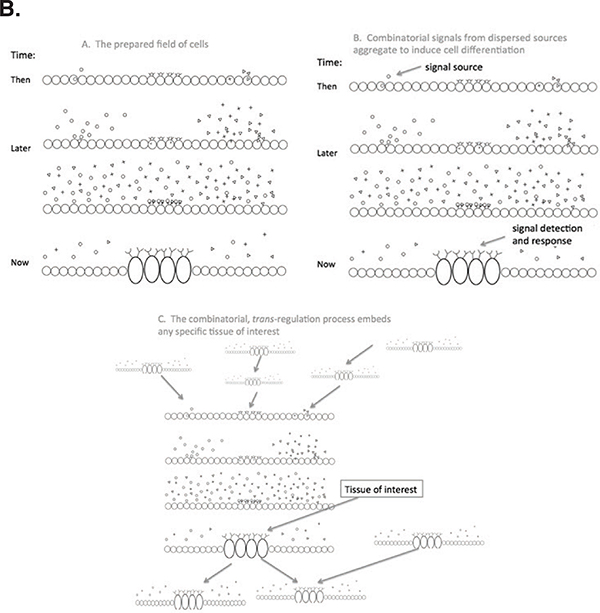
FIGURE 5-2 The Boolean nature of genomic function.
Panel B. Stages in tissue differentiation: Left to right, a bed of cells shown, in which some cells produce signal molecules and others (at the center here) have receptors for specific signals; next, local cell differentiation activated over time by the receipt of appropriate combinations of signals and receptors, coded from various genes in various cellular sources elsewhere. The lower panel shows that this is a multiple step, hierarchical phenomenon without a beginning or end: a trait is studied by measuring some chosen set of cells at some time (or by ignoring all of this and assuming causation can be inferred from the inherited genome alone).
Genomic Functions Are Not Easy to Define
Many parts of the genome are transcribed into RNA. But while there are hundreds of thousands of these RNA transcription sites (Mattick et al., 2010; Kells et al., 2014), only a fraction of the resulting RNA has known function. But what is known already is enough that the very definition of biological “function” is contentious (Doolittle, 2013; Eddy, 2013; Graur et al., 2013).
Humans are the collective product of the population processes of evolution, and if one holds the strong adaptationist view that almost everything that is functional has been made so by natural selection, then an obvious criterion for function is DNA sequence conservation: Functional sequence should not vary much compared to parts of DNA with no or little apparent function. The nominal Darwinian explanation is that over billions of years, what works has been favored and thence kept in place by “purifying” selection against new mutations, which in the face of this adaptive history are likely harmful if they have any effect. Even variants that are strongly adaptive are rather quickly fixed in the population and thereafter maintained by purifying selection. By contrast, mutational variation that arises in a functionless sequence element will therefore not be screened for fitness and will accumulate among individuals in the population or among species.
However, sequence conservation can also arise from other processes. Close phylogenetic relationship means general sequence conservation, since divergence is largely due to the slow, stochastic, clock-like accumulation of mutational differences. Your and your parents’ sequences are very nearly identical. And about half of humans’ DNA is comprised of short “repeat” sequence elements that disseminate from place to place and time to time within the genome by inserting copies of their sequence elsewhere in the genome. The resulting dispersed elements retain a sequence identity even though gradually diverging because of mutations that arise slowly over the generations.
Thus, sequence conservation itself is not a definitive criterion for locating evolutionary function in the genome and generally must be assessed by means such as calibration in comparative context. That is, sequence differences that are “important” must be statistically judged relative to what would be expected based on differences accumulated by phylogenetic relationships alone. Even though a newly documented DNA sequence can be annotated to identify many likely functions by recognizing conserved canonical sequence structures, even this can be problematic, and much or most of the sequence at present remains unannotatable. For example, function may reside in the length of a sequence element, serving as a spacer, rather than its exact sequence. Such facts reduce the a priori utility of con-
servation for extrapolating genomic function from one species to another, as in comparative biodemographic analysis.
Even more problematic is that, though not generally recognized even by evolutionary geneticists, there are reasons to think that conservation is not a necessary prerequisite for biological function. The reasons relate to complex, or “polygenic,” control, and will be discussed below. But first, I wish to identify a few rather strange phenomena that frustrate attempts to identify function even when mapping has statistically implicated a specific location in the genome.
STRANGE ENCOUNTERS OF THE N-DIMENSIONAL KIND
The general theory of “the” gene has not changed much for many decades. It is basically a linear-regression theory of point-causation by cis-coding: that is, by discrete elements located along a string of nucleotides. With the discovery of sequence used for local gene regulation (binding by transcription factors) and transcribed RNAs of various directly functional types other than protein-coding, research has only gone slightly beyond this classical linear-causation view. That in part explains why the basic methodologies for finding genomic causality have not fundamentally changed.
Trans Functionality
It has long been known that the usage (expression) of “gene” X is regulated by the binding of multiple proteins to DNA recognition sites chromosomally near to the regulated gene (shown earlier in Figure 5-2). Typically tens of such regulatory proteins form a local complex by interacting with the local DNA and with each other. This local control phenomenon is known as cis-regulation. But the binding of cis-regulatory sites by proteins coded elsewhere in the genome clearly shows that local gene function is also a trans phenomenon that depends on the context of things elsewhere in the genome, and cannot be inferred just from local mapping hits.
The idea has been that cis-regulation involves the regulatory proteins finding chemically attractive DNA binding sites. But far more subtle trans effects are being identified. There appears, in fact, to be 4-dimensional arrangement of genomes in cell nuclei that are 3-dimensional in space but also time-dependent as cells change their gene usage depending on their contextual circumstances (Dekker et al., 2013). Specific areas within and between chromosomes juxtapose in the nucleus in clustered subregions in which active transcription—for a given cell’s given context—is taking place. There is at least some stochastic cell-specific variation (Nagano et al., 2013). This is at least to some extent DNA-DNA rather than DNA-protein
affiliation, and at this early stage, no theory exists for this aggregation and how it occurs.
Given this, gene mapping may identify a region whose variation has a statistical association with a trait. However, the reason for the effect may be elusive, because it is not clear a priori how such hits reflect the 4D genomic usage phenomenon: A local map hit may relate to effect(s) elsewhere—but why, where, and how? If the result of mapping is strong and consistent, it may possible to follow the networks involved (e.g., Chen et al., 2008; Emilsson et al., 2008; Keller et al., 2008), but, to date, results are weak and not highly transferrable across species.
Dynamic Feedback
Lamarckian inheritance has a bad name. Jean Lamarck held that inherited factors were themselves changed by an organism’s behavior in ways that directly (and adaptively) reflected that behavior. The famously comical example (only mentioned in passing by Lamarck himself) is that a giraffe successfully stretching its neck to reach high leaves and hence to survive will produce long-neck-causing elements and transmit them to its offspring.
As far as DNA sequence changes go, such ideas of the inheritance of acquired traits seem totally incorrect. But it has been shown that context-specific “epigenetic” chemical changes do modify DNA molecules in ways that affects gene usage, without changing their nucleotide sequence. Epigenetic changes can be inherited and thus mimic Mendelian familial correlations (Flanagan et al., 2006; Petronis, 2010). There is a long history, going back to early 20th century genetics, of study of variation in genetically identical (inbred) organisms showing no evidence of inherited nonclassically genetic variation. However, more direct evidence for at least short-term inheritance of epigenetic variation is being found in at least some instances, as documented elsewhere in this volume, so the degree to which this is a current technologically driven fashion or an important source of heritable variation is as yet unclear.
In any case, epigenetic DNA modification is driven by context-specific, genome-encoded processes that respond to various conditions in the cell. But variation in epigenetic patterns can be affected not just by environmental conditions, but also by either cis or trans genomic sequence variation. This is because an epigenetic modification involves both the modified sequence and the modifying enzymes, coded by genes located elsewhere. Understanding epigenetics is challenging, because the effects are tissue- and context-specific, not directly assessable from constitutive DNA sequence alone. There are some sequence-based methods for identifying potential epigenetic sites in mapped genome regions, but showing that epigenetic change is actually responsible for a studied phenotype may require directly testing the right cells at the right time.
Monoallelic Expression
Another manifestation of sophisticated genome-wide trans communication within the nucleus of a cell is monoallelic expression. Mendelian theory for diploids is that the two alleles carried at any gene location are both expressed, in their appropriate cellular context. The phenotype of the individual is the net result. That is the basis of classical (and I think persistent but rather dated) Mendelian concepts like “dominant” and “recessive” causation. But there are thousands of instances already known in which only one allele is used. Given these known cases, it seems inevitable that there are others (Chess, 2012).
The first-known example was X-inactivation. Most X-linked genes are inactivated in one of the two X chromosomes in a female. Several genes in the immune system undergo local rearrangement on one copy and inactivation of the other. Genes are differentially modified epigenetically in sperm and egg, so the early embryo is using only one of them. Olfactory reception is a most extreme instance. There are about 1,000 olfactory receptor (OR) genes (2,000 in humans’ diploid set) that code for cell-membrane proteins that bind odorant molecules leading to a signal pulse to the brain. These genes are located in multigene clusters found among most of the chromosomes. But in each olfactory neuron, only one OR gene from one cluster, and not its homolog, is chosen for expression. The rest are inactivated: even if there turns out to be some “leaky” multi-OR expression, this pattern reflects genome-wide regulatory interactions.
These and other well-documented monoallelic phenomena occur at appropriate developmental stages but are thence inherited in the cell’s somatic lineage. They produce mosaic tissue organization, unique in each individual. The known examples are ancient (have been around for millions of years) and produced by active genome-encoded mechanisms. Precedent would suggest that many more are to be discovered; indeed, some enticing new evidence suggests monoallelic expression may be stochastically ubiquitous. At the very least, such effects can be expected to reduce statistical signal strength in mapping studies.
To Thine Own Self Be … What?
The vertical transmission of somatic mutations generates a hierarchical descent tree of variation among the tissues as part of each person’s life history. This has long been recognized and most clearly documented in regard to cancer, where there has been some clinical progress but where the picture is clearly complex (Vogelstein et al., 2013). However, there is also horizontal transmission. Viruses can sometimes be incorporated into the genome and join the somatic tree in any branch and at any point. More recently
documented is the potential importance of parallel hierarchies of infecting microbes, collectively called the microbiome, which introduces a complex ecosystem of tissue- and context-related colonization. Though typically transferred horizontally from the environment, its elements interact with each other and with the genomes, evolving unique descent patterns with each individual. Genome mapping alone may generate hits whose effects are not to be seen within the genome itself. There are clear pathological interactions, and the microbiome may have major contributor to aging patterns (Gordon et al., 2014). However, at present, little more than very generic theory for these phenomena has been developed.
THE HUMAN GENOME DOESN’T EXIST, AND NEITHER DOES YOURS!
“The” human genome sequence available online in GenBank is often treated as a Platonic ideal. But though the sequence is real, it is not that of any individual. It is a wholly arbitrary composite reference sequence. People neither carry two imperfect instances of “the” human genome nor “copies” of it. Each person carries instances of human genomes, landmarked against an arbitrarily chosen referent. That is all!
The reference sequence is from person(s) unaffected by identified diseases at the time. This does not make it “normal,” and abnormal genetic traits are not reliably to be found by differences from this reference. Indeed, as discussed above, individuals don’t carry even two instances, because their billions of cells each have unique sequences, closely related but not identical to what was inherited. From this perspective, it is a little like science fiction to attempt to map genomic causation from single (or pairs of) constitutive sequences, sometimes assessing function by comparison with an arbitrary reference. This is a strange definition of “normal” since the donors of “the” sequence will eventually incur a variety of age-related disorders that may be affected by their particular genotype.
Implications for Comparative Strategies
The picture is comparably dicey in relation to comparative analysis. There is no such thing as “the” mouse or dog genome. There are different inbred strains, which can be ordered off the shelf from various suppliers or obtained from the kennel club. But none of these are “normal”! Most have been bred under controlled, protective conditions, often for some prespecified trait, whether a healthy or pathogenic one.
The purpose of inbreeding is to have identical, replicable genomes to work with on the assumption that the resulting genome causes the desired trait. There is indeed much variation among inbred animals, but I think
it is generally assumed to be of environmental, epigenomic, or stochastic origin (Martin, 2009, 2012; Smith, 2011). But even forgetting (or, rather, choosing to ignore) somatic mutation in each individual, inbred mice are genomically variable, if less so than people (or real mice). There are not many in-depth tests of this that I am aware of, but a simple computer simulation of genomes of strains, and crosses between strains, generates considerable allelic variation comparable to what is empirically seen (work not shown).
The arbitrary reference nature of species-specific genome sequences may seem obvious, but is subtly ignored every day in many ways. For example, researchers routinely engage in this sort of Platonism when inserting a single transgene to demonstrate its effect in “the” mouse. That this is a kind of Platonic fiction is clearly shown by the high fraction of phenotypic effects among strains into which the same transgene is engineered. This clearly shows that genomic “background” is not just incidental noise. The literature is rife with careless claims or reports from animal experiments (Couzin-Frankel, 2013). These facts have important implications for comparative studies, between natural and even inbred animals, especially for complex biodemographic traits whose genomic basis one wishes to understand. But these well-known facts of unkown, because usually untested, importance are typically and conveniently ignored.
Genomic Causation Is Typically Polygenic
From a genomic point of view, many traits important in behavior and aging are clearly polygenic: The traits are each affected by “genes” often numbering in the tens or even hundreds. The term is in quotes because it can involve protein-coding genes as well as regions serving regulatory or other functions. Variation among genomic sites contributes (along with environmental factors) to the trait’s variation. Even in lucky situations, it is often clear that major easily found variants in individual genes are responsible for only a small subset of the total trait variation. Most sites identified by genome-wide mapping methods, such as comparing variation in cases with controls, contribute only trivially small individual effects, such that sample structure and arbitrary significance criteria are more important than biology in whether they are identified. A long-standing argument, or I would say defense of (or excuse for) continued mapping in the face of this reality is that it can identify important “druggable” pathways. But the history to date does not provide sufficient reason to persist in that hope.
The sequence variants at each of the contributing sites have some frequency in a sample or population. Each person represents a sampling from the “pool” of these variants in the population. The result is that to a good approximation, each individual’s relevant genotype is unique.
One thing learned from genome sequences of randomly sampled individuals is the importance of context, involving the genomic background and environmental experience of each individual. People are walking happily around with numerous mutationally defunct genes. Even studies of genes known to be associated with given conditions are mutated in damaging ways in unaffected people. This might be attributed to environmental exposure differences and the clearly genetic strain-specific transgenic results in animals raised in identical conditions, but models in which there are very different responses to the same transgenic manipulation show that genomic context is important.
The major implication of polygenic causation is, first, phenogenetic equivalence: Many different genotypes can generate the same trait value (here, even ignoring environmental interactions). Secondly, every individual has a different genotype for the implicated set of variants, even ignoring somatic variation. The upshot is that the Detectance, the Pr[Genotype | phenotype], is often quite small: that is, the genotype from the trait cannot be reliably inferred. Perhaps worse is that the Predictance, the Pr [Phenotype | Genotype], is also small and unreliable. Thus even retrospective model-fitting may not yield high Predictance in new samples. This is a widely known fact, though, in my view, persistent hope often overrides facing up to the reality.
If the relevant genotype of each person in a population is unique, the frequencies and even presence of given variants will vary from sample to sample. Over time, as individual variants change frequency or are lost by failure to be transmitted, or new variants arise by random mutation, isolated populations will evolve increasing differences in both the identity and frequency of relevant variants. This makes the tactic of metaanalysis to increase sample size problematic. For common variants—those that preceded human expansion into the several groups included in a meta-analysis—replication can be found, and there are cases in which this has been the case (Hindorff et al., 2009). But this is to me a somewhat selective optimism rather than the rule, in particular because it is difficult to interpret that lack of confirmation that is commonly found. For the same reason cross-species comparative analysis presents fundamental challenges. The challenge is greater if genomic action is interactive with lifestyles and yields gradual effects on age-related life history functions.
Genomic Effects Are Often Essentially Age-Specific
Age is an ultimate rate-dependent measure, so that genetics is at the heart of much biodemography in long-lived species. Complex traits not obvious at birth depend on the amount, timing, and quantitative level of interacting genetic and environmental effects. They can affect the amount
and tempo of responses to environmental changes, and hence the rate at which they accumulate with age. Despite their fundamental importance, age-specific effects are usually tested only very generally, such as by contrasting “early” vs. “late” onset. An obvious problem is that age-matched unaffected controls can later become cases. It is difficult to do better with a reliance on statistical sampling in the absence of solid predictive theory. Compounding the problem is that life history events vary greatly even among closely related species—including preferred comparative model animals. Age effects seem highly stochastic within species, with high variance in onset among related individuals, usually raised in an essentially arbitrary controlled, standardized environment. Yet there are major characteristic species differences in hazard functions. A long-standing question is why mice, with biology very similar to humans and fewer at-risk cells, have cancer hazards that increase over 30 months compared to humans’ 80 years. But cancer is a “mechanical” disease at the cell level, largely due to genomic mutations, and one must expect much greater hazard-function related questions regarding species-specific behaviors that involve learning, if inferring genomic causation is the objective.
There are two other factors to be considered. Model animals have been bred for specific traits, including behavior, and hence the genomic results may not accurately reflect the more complex mechanisms in naturally evolved populations. The selection is generally strong and teleological, fundamentally different from natural selection. So the genomic consequences in a model may not be easily transferrable to wild animals … like humans. In addition, while model species’ behavior is often context-dependent, humans respond to environments culturally in confounding ways, such as changing exposures because of knowledge of their effects. When I read that eating eggs is good for you, I eat them … until I read that they’re bad. There is no good theory for this!
SOME RELEVANT EPISTEMOLOGICAL ISSUES
As I noted at the outset, most models of genomic causation are essentially sample-based, theory-free internal statistical comparisons, such as cases vs. controls, rather than tests of a formal theory. Or “theory” is nothing more than the assumption that with “appropriate” samples, contrasts between the comparison groups will be detectable by some inherently subjective “significance” or other criterion. Without adequate formal theory for what to expect to find, this means finding strong effects will perforce ignore smaller ones that may be present. Significance cutoffs are important and have been explicitly used to avoid having to follow up a sea of false positives, but they force the apparent fact that many or even the bulk of causal effects are too small to detect with chosen cutoff criteria.
Individually this may make little practical difference; in aggregate, perhaps not so.
When there is strong point-like causation, experimental confirmation and understanding is achieved, which can be followed up experimentally. Examples abound. This is how, by careful choice of traits, Mendel opened the door to the discovery of genetics in the first place. Indeed, such a result in one species can sometimes be applied to others, which is why mouse models are often useful. But no formal evolutionary or genomic theory predicts that “sometimes.”
High-penetrance “Mendelian” variants are an addictive lure, relative to the more general pattern. What is now known about genomes and causal complexity stretches the ability of current explanations and methods to account adequately for is seen, with implications for both experimental and comparative research. Indeed, what is known affects basic epistemological criteria. These implications are easy to see, but it’s far more difficult to understand how to respond.
Life in a Nonclocklike Universe
If life were a purely Newtonian clockwork phenomenon, then with complete information the entire future could be predicted and the entire past retrodicted. “Extrapolation” might not even be a word in the lexicon. But this is not the reality of the world. Time’s arrow in this cosmos does have a direction. It is due to the probably inherent probabilistic nature of fundamental causation.
As a result, a fundamental challenge is that genomic risks are necessarily estimated retrospectively. The past is over and to some extent can be measured, but the future is yet to come. The past can be retrofitted if the right things are measured, but that isn’t the same as retrodiction and certainly not prediction. It’s often a huge and sometimes inescapable problem that the relevant risk factors were unknown, much less measured.
But what about the future? Genetic risk factors are typically treated as inherent, present at conception. Based on this is the widespread promise to predict future outcomes. However, although environmental factors predominate even in strongly “genomic” traits (where heritability is still usually far less than 1.0), future environmental exposures cannot even in principle be predicted. This also applies to stochastic effects, especially without an accurate determination about what is environmental and what is stochastic. In general, these considerations make genome-based prediction epistemically inaccurate to an unknown degree.
Time’s cosmic arrow is based on the accumulation of entropy. The corresponding biodemographic arrow is of the accumulation of irreversible effects of age and the one-way tree of population history. The degree
of probabilism in genomic causation is unclear. Genome mapping suggests that countless small contributions are responsible for complex traits. Each contributes so small an amount that any given element’s long-term survival cannot be predicted; they will often be lost to drift, especially since most are rare in the population (of people, or of cells). Thus, among populations and species, and over time, a fluid flow of causal contributors is to be expected.
This relates to sequence conservation, which I mentioned earlier is a commonly invoked criterion for biological function. If polygenic contributors have only weak individual effects, these effects may be ephemeral even in the presence of natural selection. In turn, their sequence will not be highly conserved relative to expectations of drift alone. This applies both between and even within populations, even within species.
Yet, perplexingly, in sequence conservation patterns, known functional elements of the genome are relatively highly conserved. Likewise, experimental evidence, in animals and other model species, and in the evolution of domestication in plants and animals, suggests an expectation to find a much higher level of determinism. Inbred animals are approximate replicates of a same genome, with generally specifiable traits (e.g., body size, susceptibility to a given type of cancer), yet despite this increased genomic determinism, even in standardized environments there is variation (Gartner, 1990). In any case, it is clear that environmental context is an important part of variation in traits, but phenogenetic equivalence is also fundamental.
Replication
Modern life science is derived based on Enlightenment-era principles of replicable cause and effect. That is essentially the basis of statistical inference. But if everyone is unique, then epistemic limitations are rarely taken seriously if even recognized. Lifetime or age-specific risk effects are based on statistical sampling designs and are probabilistic, usually in unclear ways. Often huge numbers of measured genetic and other factors are included in survey samples, inference depending on the user’s choice of sample, which is affected by facts already known, what is practicable, and other factors, which are difficult to account for or even to be aware of in study design or analysis. The issue here is individual vs. group predictability. This might be said to be the difference between medicine and public health. Probabilistic causation can mean that individual prediction is weak but group prediction stronger, but the issues of unknowable future factors that I mentioned above can vitiate even that to an unknown extent. And I think it is true that currently there is a pressure to extrapolate group mean values to individuals (e.g., in policy or “personalized” medicine).
Because of the essential nature of variation in evolution, which is, after all, a population process, no two individual organisms are alike. Indeed,
a key aspect of polygenic traits is phenogenetic equivalence. There is no specific theory for how or, except generically, how much two individuals vary relative to the genomic causal basis of a trait. As a result, the bedrock criterion of replicability is not a reliable one for genomic studies. As noted above, people are all walking around with many entirely defunct genes, which should affect health but don’t, and the same will surely be extended to the other functional elements of the genome.
There are legitimate reasons for this kind of approximate approach, and statistical sampling is a core element of the research toolbox. But in important ways, inferences are based on the methods rather than the biology. There are often major multiple-testing problems that are hard to correct for accurately, despite extensive and very sophisticated statistical corrections that are routinely applied, because so many issues are being examined by so many investigators in so many studies. Still, the issue is taken seriously and carefully (Yang et al., 2012; Hemani et al., 2014). But to me, most importantly is that such corrections explicitly trade conservative inference, to escape masses of false positives as noted earlier, for dismissal of small effects even if they are numerous and large in aggregate.
Also as noted earlier, over time evolution generates genomic divergence both within and between populations. For this reason, meta-analysis relies on detecting older variants still found in populations descendant from a common founding population. The variant’s effects must not depend too heavily on the genomic background, which will have diverged, and must have similar effects in the diverse environments. They are older if they are shared among populations and that makes them relatively more common than the typical genomic variant. Replicable results may occasionally follow, but they will typically be for strong-effect alleles that do not really require replication studies. But because humans are a very large population with a recent history of rapid expansion, most genetic variants are new or recently arisen in some place in the world. This same property must apply to those variants that contribute to traits of interest, normal or otherwise. Rare variants may be discoverable in families if they have very strong effects, but not in statistical association studies.
Replication has its central place in science and is useful, of course, and there are clear examples where it works as expected. But because of evolution, it is not a guaranteed criterion. Even when the same factor, perhaps a given locus genotype, is present and relevant to an outcome of interest, the strength of effect may not be replicated, and a serious level general theory about the extent of these issues is lacking.
Falsification
If replication is not a reliable criterion because failure to replicate may mean simply that samples have different characteristics for reasons including evolutionary divergence, what about the oft-invoked Popperian criterion of falsification? For many reasons, that is not a very useful criterion. Superficially, failure to replicate may be due to measurement or laboratory error. But the evolutionary reason is that two samples are not necessarily expected to have replicate causal distributions. The alleles and genes involved may depend on variation that affects their frequency (and hence detectability) or even their actual relevance to a trait.
A gene may contribute, even fundamentally, to a trait but simply not vary in a given sample. Methods based on sampling variation cannot detect the presence of such effects. The problem may apply especially to inbred crosses, but variation in a given natural population may simply be low or only have weak effects. The latter could be because the gene is too important for the studied trait to withstand serious mutations. Not finding a mapping hit in the gene is nonreplication but is not falsification of the idea that the gene is involved. It is to be expected with many or even most non-obvious genomic contributions to traits. To the contrary, falsification is in a sense reinforcement of the correctness of an understanding of evolution.
Parsimony
Physical theory is often taken to imply that nature follows the simplest path between two points and that the simplest explanation of a phenomenon is the “most likely” (or, said in a more neutral way, to be preferred). But if one accepts the many-paths view of physics, not even light rays travel in the simplest direct path.
Evolution is a local, highly stochastic process. There is no reason to think that the simplest way to evolve a causal regime is taken, nor is it required to be similar in different lineages. There are tens of different paths to vision and many other traits for which there are data. Polygenic control shows that there are countless ways to the same height, body mass index, psychopathology, or blood pressure. Even if some elements are conserved, there is no “simplest” way.
While it can be an occasional general guide, Occam’s famous razor can cut in many directions. It is simply a weak guide to choosing among hypotheses. One might say that, for statistical data, a likelihood or Bayesian approach addresses this issue. But I (though no statistician) am not convinced: These methods only optimize among a prespecified range of options.
Descriptive statistical modeling is in a sense an invocation of parsimony, in that it detects strongest effects and dismisses lesser ones. The
results of multiple studies may not generate a specific mechanistic theory, but can provide general guides about pattern. There are many quasiregular patterns found in various aspects of genomes and their usage (Koonin, 2011a, 2011b). Figure 5-3 provides a generic description of the way things look (in my view).
Rather than a Darwinism-Mendelian duality in which individual genetic variants are the causally deterministic units of life, fixed by an equally deterministic force like Darwinian natural selection, life has evolved to be a multidimensional web, with at least a generic statistical spectral structure of effects. The “shape” shown is schematic and the details case-specific, but
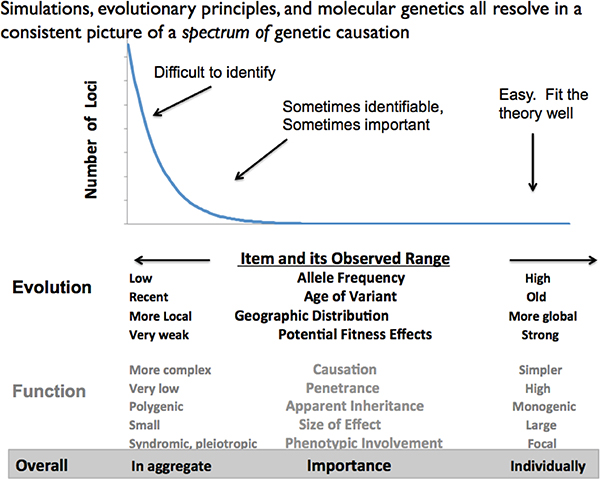
FIGURE 5-3 The causal and inferential spectrum of life.
NOTE: The relative distribution of effect strengths, and their manifestation, are generally found as suggested here. Evolutionarily, most effects are rare, recent, and hence geographically localized. Common variants are older and geographically widespread, but are rarer. Corresponding to this, functionally most variants have weak effect are of low frequency in the population and have their effects jointly. Strong individual effects are relatively rare, but they are the ones easy to detect. The bottom line suggests that the numerous, local, weak effects generally have their effects in aggregate while strong effects more clearly act individually on their own. However, this is schematic and empirical, because there is no formal theory for the shape of the distribution.
what can be said is that a minority of cases follow simple classical ideas, perhaps seducing researchers to think that everything else will, if only they have adequate samples and enumeration. The middle ground presents with some predictive power, but much—often the bulk—of instances are of many-to-many, essentially unenumerable causal factors. It is they that present the challenge, and, of course, when dealing with age effects the causation and requisite study scope are much more problematic. Between species, knowledge is still largely in never-never land a priori understanding is sought.
SOME STRATEGIC THOUGHTS
The aspects of genomic causation that I have outlined are substantial and relevant to biodemography. While they do not present problems with “solutions,” they can affect research strategies. Here are some thoughts about what that might involve.
Where Is Genetics Really Relevant?
I am a geneticist, but to me the first strategy might be to understand whether a question of importance really requires a genetic answer. If relevant genetic causation is complex and polygenic, perhaps one should concentrate on the empirical trait itself, rather than attempting to enumerate its genomic causation. This would be especially apt if the trait is common in humans or is a reflection of cultural attributes, or is clearly shared between species and their ecology. When I look at the roster of biodemographic topics considered in this volume, most seem to be sufficiently tractable (or challenging!) at the trait level itself. At least, given the nature of genomic causal complexity, one should be clear what serious biodemographic information would justify the time and cost of attempting to reduce to genomic terms.
Isolating Age Effects and Developing Useful Theory
Where it seems appropriate, even for basic knowledge, there is still much to be learned about the basic mechanisms by which genomic variation affects age-specific risk or outcomes. An obvious tactic would be to take a more carefully designed rather than incidental age-stratified approach to data, to increase contrasts such as comparing very early to very late onset of traits. Comparison of age-stratified results among different populations (rather than pooling them in meta-analysis), or animal strains, searching for correlates of signal strength, without relying on significance cutoffs, could provide clues.
I must add, however, that tail-based mapping comparisons have not found very much, either within the normal or pathogenic range, in part because the tails are affected by sets of rare, strong-effect variants. Indeed, while there are important exceptions, long, focused, large, and very costly attempts to understand traits like cancer or cardiovascular disease have generated only rather generic ideas like the gradual accumulation of various sorts of cell or tissue damage whose implicated genetic basis remains unstable and largely elusive.
Still, mapping studies have rarely made serious attempts to fit hazard functions to genomic effects, site-by-site or in aggregate. One might adjust current regression approaches to do that. I don’t think it is just nostalgia to suggest that resuscitating serious hazard and competing-cause modeling of these effects, in light of what is known about genomes, especially where these can be based on at least an approximation to relevant mechanism.
But what causal model would one use, if one really wished to reflect the nested population and somatic processes of age-related change? One might try modifying current aggregate genome-wide risk estimation methods. But since relevant phenotypes are affected by so many genes that each person is unique, innovative methods would have to be devised. It is possible that plant or even animal breeders using mapping methods have relevant experience.
The Value of Controlled Crosses
Mapping in strain crosses as in dogs, mice, and perhaps some other husbanded species could reveal aspects of mechanism. Crosses between strains or breeds with widely differing biodemographically relevant traits could be particularly revealing, and here I would suggest that dogs are a good model because they have been bred to have specific traits that vary widely among breeds. Much good work is actively being done on dog genomic causation. But dogs are large animals, slow growing with long generations, and expensive to study and often problematic to interpret in human terms. Some relevant mouse social or behavioral traits could be substituted if the traits are carefully chosen.
Identifying Noncoding Functional Processes
Mapping generally results in hits in many areas of the genome, even where no known, or known-relevant, functional elements are there. Expedient whole-exome mapping is not suitable for this. However, comparative mapping in different species or strains for clearly homologous traits could at least confirm that mapped function-unknown elements really are involved.
Facing the Somatic-Mutation Realities
Somatic variation is vitally important in some traits, and there is no reason why this should not also affect behavior, even within a “normal” (nonpathological) range. There may be value in sampling different embryological tissue lineages (on separate developmental branches, or left vs. right side) to identify somatic allelic differences and then see if they have phenotype correlates (probably not useful for behavioral traits).
Small Data Rather Than Big Data?
Even when mapping is felt to be important, I personally think one should stick to smallish samples, which will at least detect major effects with less statistical noise and greater useful replicability than huge (and hugely expensive) samples.
Somewhat more positively, as major “Big Data” databases come online, if multiple trustworthy measures of relevant traits at different ages are included, the data can be retrospectively mined, at low additional cost, to identify genomic (or environmental) contributing factors, in ways I have suggested above. It should be kept in mind that there can only be analysis on what has been measured, and how, at the time.
Overall
Overall, I believe that what is most needed is better mechanistic theory for biodemographic processes if they are to be understood in genomic (including evolutionary) terms. To me, that means moving away from the very indirect linear models that assume replicable phenomena or at least coming to better terms with their ephemeral nature in the face of the great amount already known about genomic variation and the evolutionary processes that generate it—rather than the relentless pursuit of ever more data of the same kind.
CONCLUSIONS: CAN THE CAUSAL WEB BE UNWOVEN?
Evolution involves a complex local, ad hoc web of natural selection, migration, and chance. The sieving of genomic effects, slowly over eons of time and space, is responsible for the causal complexity found. In essence, causal effects that natural selection cannot “detect” (directly screen) will be as hard to detect as causal factors in contemporary mapping studies. Slow evolution generates, or at least tolerates, complex variation. This is what is found even for abnormal or unusual traits, or indeed even in relatively low variation model systems like mice or even flies—or yeast (Bloom et al., 2013).
Obviously, when it comes to genetic explanation, I am less sanguine about the current state of the science than are most, including other authors here. To me, the allure of known simple Mendelian answers engenders a simplistic Darwinian-Mendelian way of thinking. The perplexing complexity of genomic causation being revealed is thus often treated as surprising or even disappointing. But it’s actually a positive reinforcement of theoretical expectations present (but often not recognized) for nearly a century. This shows an understanding of important general aspects of genomic causation.
An important difference in perspectives is that between individual “personalized” and group-based patterns, risks, and predictability. Most papers and most authors recognize the difficulties, but too often, in my view, in caveats or small print, and too often persist in the same costly studies knowing the landscape. Individual prediction works when there are strong enumerable factors that are relatively unaffected by environments. But from common diseases and patterns related to aging, these seem to be the exception. A population perspective, in which interventions can be applied without knowing the specific genotypes that might be most affected, seems most promising. Nonetheless, I believe that researchers should by now know, given the rather massive secular changes in the most common disorders (and traits like stature), that even population prediction based on genotypes can be problematic. Without a better formal theory, I think that the extent this is so is not known, and, of course, it will differ with each trait of interest.
Perhaps there is no new theory to be found that will reduce this generic complexity to a desired kind of Mendelian predictability except in those instances, many already known, where it does happen to work. If so, it is correct that evolution is an ad hoc, largely stochastic process of divergence and diversity. Researchers will just have to soldier on enumerating variation and using subjective statistical criteria for assessing causation.
Alternatively, the current reliance on readily available and essentially standard statistical survey approaches may impede fully acknowledging the lack of a better way of understanding. Whatever the answer, this is a time of discovery by enumerative induction, enamored with mass data collection for various venal, bureaucratic, as well as scientifically legitimate reasons, but which has, to date, yielded only limited ability to generalize or extrapolate.
In my view, there should be major investment in innovative attempts to develop better theory for well-posed questions, rather than mega-projects of standard types, essentially to refine statistical evidence or association that may not point very directly to genomically causal elements of importance. And perhaps more effort should be expended on understanding what is “normal”—or even the meaning of a “norm” in considering both evolution and contemporary causation.
Both during evolution and today, competing causes, population distribution, and age-specific effects are core topics that are given rather scant attention. They present sampling and analytic challenges, even in the limited experimental settings available. Cross-species comparisons are interesting and potentially informative, but their limitations in the current theoretical environment need to be recognized.
Such a focus raises societal challenges of its own, given the politics and economics of modern science, the competition for resources, and, it must be said, the appeal and immediacy of technology and “Big Data” in which thinking is, often explicitly, deferred. Yet there is the accompanying promise of biomedical miracles.
Insightful creative genius cannot be ordered up just because there are serious problems with the current “paradigm.” But this exculpation only can legitimately go so far. Incremental success, technology, and scale provide convenient excuses for not having to slow down and think about a problem. Known challenges exist, and they should be taken seriously.
ACKNOWLEDGMENTS
Thanks to the editors, reviewers, and Anne Buchanan for very helpful comments on this manuscript. I have tried to respond to their comments, but they may not agree with my views, and are not responsible for any errors that may remain. The references cited are very selectively chosen to illustrate main points, from a literature of countless thousands that deal with this broad topic.
I maintain a blog that regularly deals with these sorts of topics: The Mermaid’s Tale (a blog: ecodevoevo.blogspot.com)
REFERENCES
Bloom, J.S., Ehrenreich, I.M., Loo, W.T., Lite, T.L., and Kruglyak, L. (2013). Finding the sources of missing heritability in a yeast cross. Nature, 494(7436), 234-237.
Buchanan, A.V., Sholtis, S., Richtsmeier, J., and Weiss, K.M. (2009). What are genes “for” or where are traits “from”? What is the question? BioEssays: News and Reviews in Molecular, Cellular and Developmental Biology, 31(2), 198-208.
Chen, Y., Zhu, J., Lum, P.Y., Yang, X., Pinto, S., MacNeil, D.J., Zhang, C., Lamb, J., et al. (2008). Variations in DNA elucidate molecular networks that cause disease. Nature, 452(7186), 429-435.
Chess, A. (2012). Mechanisms and consequences of widespread random monoallelic expression. Nature Reviews Genetics, 13(6), 421-428.
Cousin-Frankel, J. (2013). When mice mislead. Science, 342(6161), 922-923, 925.
Dekker, J., Marti-Renom, M.A., and Mirny, L.A. (2013). Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nature Reviews Genetics, 14(6), 390-403.
Doolittle, W.F. (2013). Is junk DNA bunk? A critique of ENCODE. Proceedings of the National Academy of Sciences of the United States of America, 110(14), 5294-5300.
Eddy, S.R. (2013). The ENCODE project: Missteps overshadowing a success. Current Biology, 23(7), R259-R261.
Emilsson, V., Thorleifsson, G., Zhang, B., Leonardson, A.S., Zink, F., Zhu, J., Carlson, S., Helgason, A., et al. (2008). Genetics of gene expression and its effect on disease. Nature, 452(7186), 423-428.
Flanagan, J.M., Popendikyte, V., Pozdniakovaite, N., Sobolev, M., Assadzadeh, A., Schumacher, A., Zangeneh, M., Lau, L., et al. (2006). Intra- and interindividual epigenetic variation in human germ cells. American Journal of Human Genetics, 79(1), 67-84.
Gartner, K. (1990). A third component causing random variability beside environment and genotype. A reason for the limited success of a 30 year long effort to standardize laboratory animals? Laboratory Animals, 24(1), 71-77.
Gordon, L.B., Rothman, F.G., Lopez-Otin, C., and Mistelli, T. (2014). Progeria: A paradigm for translational medicine. Cell, 156, 400-406.
Graur, D., Zheng, Y., Price, N., Azevedo, R.B., Zufall, R.A., and Elhaik, E. (2013). On the immortality of television sets: “Function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biology and Evolution, 5(3), 578-590.
Hemani, G., Shakhbazov, K., Westra, H.J., Esko, T., Henders, A.K., McRae, A.F., Yang, J., Gilbon, G., Martin, N.G., Metspalu, A., Franke, L., Montgomery, G.W., Visscher, P.M., and Powell, J.E. (2014). Detection and replication of epistasis influencing transcription in humans. Nature, 508(7495), 249-253.
Hindorff, L.A., Sethupathy, P., Junkins, H.A., Ramos, E.M., Mehta, J.P., Collins, F.S., and Manolio, T.A. (2009). Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America, 106(23), 9362-9367.
Keller, M.P., Choi, Y., Wang, P., Belt Davis, D., Rabaglia, M.E., Oler, A. T., Stapleton, D.S., Argmann, C., et al. (2008). A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Research, 18(5), 706-716.
Kells, M., Wold, B., Snyder, M., et al. (2014). Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences of the United States of America. doi: 10.1073/pnas.1318948111. Available: wsww.pnas.org/11/17/6131/supple/ DC Supplemental. [July 2014].
Koonin, E.V. (2011a). Are there laws of genome evolution? PLOS Computational Biology, 7(8), e1002173.
Koonin, E.V. (2011b). The Logic of Chance. Upper Saddle River, NJ: FT Press.
Mahr, J. (2014). Gene Expression—Eukaryotic Transcriptional Regulation (GPC). Available: http://cnx.org/content/m49660/1.2/ [March 2014].
Martin, G.M. (2009). Epigenetic gambling and epigenetic drift as an antagonistic pleiotropic mechanism of aging. Aging Cell, 8(6), 761-764.
Martin, G.M. (2012). Stochastic modulations of the pace and patterns of ageing: impacts on quasi-stochastic distributions of multiple geriatric pathologies. Mechanisms of Ageing and Development, 133(4), 107-111.
Mattick, J.S., Taft, R.J., and Faulkner, G.J. (2010). A global view of genomic information—moving beyond the gene and the master regulator. Trends in Genetics, 26(1), 21-28.
Nagano, T., Lubling, Y., Stevens, T. J., Schoenfelder, S., Yaffe, E., Dean, W., Laue, E.D., Tanay, A., and Fraser, P. (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature, 502(7469), 59-64.
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature, 465(7299), 721-727.
Smith, G.D. (2011). Epidemiology, epigenetics and the ‘Gloomy Prospect’: embracing randomness in population health research and practice. International Journal of Epidemiology, 40(3), 537-562.
Vogelstein, B., Papadopoulos, N., Velculescu, V.E., Zhou, S., Diaz, L.A., Jr., and Kinzler, K.W. (2013). Cancer genome landscapes. Science, 339(6127), 1546-1558.
Weiss, K.M. (1992). Genetic Variation and Human Disease: Principles and Evolutionary Approaches. Cambridge, England: Cambridge University Press.
Weiss, K.M. (2010). Seeing through the gene-trees. Evolutionary Anthropology, 19, 210-221.
Weiss, K.M., and Buchanan, A.V. (2009a). The cooperative genome: Organisms as social contracts. The International Journal of Developmental Biology, 53(5-6), 753-763.
Weiss, K.M., and Buchanan, A.V. (2009b). The Mermaid’s Tale: Four Billion Years of Cooperation in the Making of Living Things. Cambridge, MA: Harvard University Press.
Weiss, K.M., and Buchanan, A.V. (2011). Is life law-like? Genetics, 188(4), 761-771.
Weiss, K.M., Buchanan, A.V., and Lambert, B.W. (2011). The red queen and her king: Cooperation at all levels of life. American Journal of Physical Anthropology, 146(Suppl. 53), 3-18.
Weiss, K.M., and Terwilliger, J.D. (2000). How many diseases does it take to map a gene with SNPs? Nature Genetics, 26(2), 151-157.
Yang, J., Loos, R.J., Powell, J.E., Medland, S.E., Speliotes, E.K., Chasman, D.I., et al. (2012). FTO genotype is associated with phenotypic variability of body mass index. Nature, 490(7419), 267-272.


























