
Contemporary Issues for Protecting Patients in Cancer Research: Workshop Summary
In the nearly 40 years since implementation of federal regulations governing the protection of human participants in research, the number of clinical studies has grown exponentially. These studies have become more complex, with multisite trials now common, and there is increasing use of archived biospecimens and related data, including genomics data. In addition, growing emphasis on targeted cancer therapies requires greater collaboration and sharing of research data to ensure that rare patient subsets are adequately represented. Electronic records enable more extensive data collection and mining, but also raise concerns about the potential for inappropriate or unauthorized use of data, bringing patient protections into a new landscape. For example, there is growing interest in developing learning health care systems1 (LHCSs) and there are new funding sources for conducting comparative effectiveness research. There are also long-standing concerns about the processes and forms used to obtain informed consent from patients participating in clinical studies. These changes and challenges raise new ethical and practical questions for the oversight of clinical studies,
______________
1 Defined as a health care system in which science, informatics, incentives, and culture are aligned for continuous improvement and innovation, with best practices seamlessly embedded in the care process, patients and families active participants in all elements, and new knowledge captured as an integral by-product of the care experience (IOM, 2013).
and for protecting patients and their health information in an efficient manner that does not compromise the progress of biomedical research.
Recognizing this new landscape, the National Cancer Policy Forum of the Institute of Medicine convened a workshop to explore contemporary issues in human subjects protections2 as they pertain to cancer research, with the goal of identifying potential relevant policy actions. This workshop,3 held in Washington, DC, on February 24 and 25, 2014, brought together clinical researchers, government officials, members of Institutional Review Boards (IRBs), and patient advocates to discuss a wide range of topics, including
- The current regulatory arena and challenges in protecting participants in cancer research
- The patient perspective on current protections for research participants
- New oversight challenges stemming from genetic advances and the use of biospecimens in cancer research
- The evolving context of cancer research and care within LHCSs and multisite trials
- Education and research needs related to improving participant protections in research
This report is a summary of the workshop. A summary of suggestions from individual participants is provided in Box 1. The workshop agenda and statement of task can be found in the Appendix. The speakers’ biographies and presentations (as PDF and audio files) have been archived at http://www.iom.edu/Activities/Disease/NCPF/2014-FEB-24.aspx.
______________
2 “Human subjects protections” is the term used in regulations for the oversight of research involving humans. However, patient advocates at the workshop made an impassioned plea for changing this impersonal terminology. In this report, we often refer to “protections for participants in research.”
3 This workshop was organized by an independent planning committee whose role was limited to the identification of topics and speakers. This workshop summary was prepared by the rapporteurs as a factual summary of the presentations and discussions that took place at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants; are not necessarily endorsed or verified by the Institute of Medicine or the National Cancer Policy Forum; and should not be construed as reflecting any group consensus.
The current oversight of clinical research developed in response to past abuses of participants in clinical trials, such as the Tuskegee trial, in which some patients with syphilis received no treatment for their condition so that doctors could document the natural history of the disease. Awareness of such ethical lapses in the 1960s and 1970s led to the requirement that clinical studies undergo prospective ethical review by an IRB, and that participants give informed consent prior to participating in research.
IRB reviews are tiered according to the perceived risk involved in the study. Detailed review occurs for those studies in which participants would be exposed to the greatest risk, such as the study of experimental cancer drugs with potential serious side effects. Minimal risk studies, such as quality of life studies using questionnaires, can undergo what is known as expedited review, which entails review by the IRB chair or by one or more experienced IRB members designated by the chair, rather than review by the full IRB membership.4 Some studies, such as those involving the study of existing publicly available data or specimens, are considered so low risk that they are exempt from IRB review.5
Regulations governing IRBs for oversight of human research came into effect in 1981 and were further defined by the 1991 Common Rule,6 which specifies a baseline standard of ethics and protections of human research participants that has been adopted by 18 U.S. government agencies that support research. Additional oversight on clinical research was instituted after increasing use of electronic health records (EHRs) led Congress to pass the Health Insurance Portability and Accountability Act (HIPAA) in 1996, with the primary goals of improving the portability of health insurance, facilitating use of EHRs for patients, and protecting the privacy and security of personal health information. The Secretary of the U.S. Department of Health and Human Services (HHS) promulgated the final version of the HIPAA Privacy Rule7 in 2002. The Privacy Rule applies to “covered
______________
4 45 CFR 46.110, Categories 1 through 9.
5 45 CFR 46.101, Categories (b)(1) through (b)(6).
6 45 CFR 46.
7 45 CFR 160, 164.
BOX 1
Suggestions Made by Individual Workshop Participants
Overarching Suggestions to Improve Patient Protections in Cancer Research
- Actively engage patients in setting research priorities and in research oversight.
- Conduct research to better understand what protections patients want in clinical studies, and how best to achieve those protections.
- Clarify regulatory language to facilitate greater consistency in interpretations and implementation.
- Use online participant-centric registries in which participants can select the research options.
- Adopt the integration model of research and practice (i.e., a learning health care system [LHCS]).
- Develop an LHCS that is transparent to the patient community by providing information on studies that are being done and how patients and their medical care are affected by those studies.
- Rely less on informed consent for uses of data in an LHCS that are considered routine, and instead pursue other models of patient engagement to enhance protections for research participants.
- Provide educational opportunities to enhance the health and scientific literacy of the public.
Improving Deidentification Procedures and Privacy Protections
- Employ data protections that are proportional to the potential harm and consider the context of the research.
- Develop a national clearinghouse of models, methods, and evaluations of data deidentification.
- Use data deidentification methods that include both a risk estimation and a risk mitigation procedure.
- Discourage potential misuse of health data through laws prohibiting discrimination in health and life insurance and employment, as well as criminal penalties for hacking or stealing data.
Improving Consent Forms and Processes
- Shorten and simplify consent forms.
- Use health literacy principles and lexicons to simplify the language used in consent documents.
- Include a one-page summary of the consent form.
- Use simplified schemas to describe trial arms and key steps in each study, and use pictorials to show how any ancillary/ correlative studies (and their additional consents) fit into the trial structure.
- Use a teaching approach rather than a persuasive mode of speaking to patients during the consent process.
- Require confirmation of patient comprehension of consent as an entry criterion for studies.
- Develop and evaluate models of the consent process.
- Develop and disseminate evidence-based best practices for informed consent and incorporate feedback loops to foster improvement.
- Offer support for patients in the event that they decide not to participate in the trial offered.
- Clarify language for consent for future research.
- Incorporate interactive education technologies in the consent process.
Improving the Conduct and Oversight of Multisite Studies
- Standardize the operation of central institutional review boards (IRBs).
- Establish metrics to assess the quality of IRB review.
- Enroll patients in screening protocols that allow investigators to molecularly profile a patient’s cancer and then match them to interventional trials.
entities”8 and restricts the use or disclosure of an individual’s Protected Health Information (PHI) unless authorized (given written permission) by the individual, or unless permitted by the Privacy Rule for such things as treatment, payment, and health care operations; public health efforts; law enforcement; product recalls; or judicial or administrative proceedings. No such exemption exists for research (IOM, 2009a).
Alice Leiter, director of Health Information Technology Policy, National Partnership for Women & Families (formerly policy counsel, Health Privacy Project, Center for Democracy & Technology), said that under HIPAA, health care operations include “conducting quality assessment and improvement activities, including outcomes evaluation and development of clinical guidelines, provided that obtaining generalizable knowledge is not the primary purpose of any studies resulting from such activities.” Health care operations also include “population-based activities related to improving health or reducing health care costs and protocol development.” Research, in contrast, is defined by both HIPAA and the Common Rule as a “systematic investigation, including research development, testing, and evaluation, designed to develop or contribute to generalizable knowledge.”
Authorization to use or disclose PHI must specify in writing how, why, and to whom the health information can be used or disclosed. This authorization is distinct from, and in addition to, the traditional and well-established informed consent for research specified in the Common Rule. When patients sign the latter, they give consent to participate in a specific clinical study, after they have been informed of the specific potential risks and benefits of that research. Some informed consents also give permission for future research uses of patients’ health data or biospecimens, such as blood or tissue.
However, Melissa Bianchi, partner at Hogan Lovells US LLP, said that “research authorizations could not authorize future unspecified research.” Future research was initially not considered specific enough to comply with the patient authorization requirements of the Privacy Rule (IOM, 2009a). Consequently, information and biospecimens collected for one research study could not be used in another study without reauthorization from the original participants in the first research study, unless a waiver of authoriza-
______________
8 Defined as health plans, health care clearinghouses, and health care providers who transmit information in electronic form in connection with transactions for which HHS has adopted standards under HIPAA (45 C.F.R. § 164.510).
tion was granted for a subsequent study by an IRB or Privacy Board. The waiver could be granted if the board determined that the new research would pose minimal risk to patient confidentiality and safety, and that the research could not practically be done without the waiver. But no guidance was provided as to what factors should be considered in determining whether those criteria are met (IOM, 2009a).
SHORTCOMINGS OF CURRENT
REGULATIONS AND GUIDANCES
Several workshop participants discussed the perceived shortcomings of current regulations and guidances related to protection of participants in research. Some of that discussion focused on whether there was an appropriate balance when considering the potential risks and benefits of clinical research in oversight. When cancer patients have exhausted all available therapies, a clinical trial may be the best option for some patients, in spite of the uncertainty regarding the potential benefit of an experimental therapy. John Mendelsohn, Director of the Khalifa Institute for Personalized Cancer Therapy at the MD Anderson Cancer Center, said, “Our goal is to avoid unjustifiable risks, but to allow for uncertainty in order to let patients have access to what they need and what they might benefit from in treating their cancer. All of us need to do a better job at this and recognize there is no perfection. Perfection is often the enemy of the excellent.”
Deborah Collyar, founder and president, Patient Advocates in Research was particularly critical of what she called an evolution of patient consent from a document aimed mainly at protecting patients to one aimed at protecting institutions and thus viewing patients as liabilities. “We have to stop saying that informed consents are for patients and then create them for the institutions that are managing risks. It is okay to put in risk management if we need to, but please don’t pretend that is for the patient’s benefit. Risk management of the institution has to be separate from informed consent.”
Collyar also noted that current regulations were developed in reaction to what had happened to research subjects in the past. “It hasn’t been strategic because we wanted to put together a whole research program that made sense. Instead it has been reactive,” she said, noting that the concerns that led to the regulations were valid but “it has created a fearful environment that everybody focuses on because you don’t want to get your hands slapped or don’t want to get shut down. On the other end, people [who participate in research] want individual results and in a way that they can understand
them. They want data shared and the regulations we have actually thwart that process.” Susan Ellenberg, professor of biostatistics and associate dean for clinical research, University of Pennsylvania School of Medicine, added, “it is difficult to pull back on oversight mechanisms because most have been put in place because something awful happened and HHS or Congress had the view that this is never going to be allowed to happen again.” One conferee suggested that patient advocacy groups are in the best position to influence IRBs and regulators “so they have the courage to accept more risk.”
Other participants pointed out that risk varies depending on context, which is not adequately considered in current regulations and guidances. Ruth Faden, director of the Johns Hopkins Berman Institute of Bioethics, noted that health care systems vary tremendously in terms of transparency, accountability, and the extent to which patients are engaged.
Suanna Bruinooge, director of research policy, American Society of Clinical Oncology, added that IRB review should consider the context of the particular disease being studied. Many cancers are deadly and have no effective treatments when they are in advanced stages; as such, cancer research tends to be more integrated into the treatment setting. Such integration makes it difficult to apply current oversight, which makes strong divisions between research and clinical care, several participants noted. (See the “The Changing Context of Research and Care” section.) Patricia Ganz, distinguished university professor at the Fielding School of Public Health and director, Cancer Prevention & Control Research at the Jonsson Comprehensive Cancer Center, University of California, Los Angeles, noted “Often the best treatment for a patient is a clinical trial in oncology because we don’t have enough information about so many different kinds of cancers and the genetic tests we now have to explore tumors. We want to have every patient potentially be a research participant.”
Deidentified information does not qualify as protected health information. Therefore it is not protected under the Privacy Rule and can be disclosed to researchers at any time (IOM, 2009a). The Privacy Rule offers two methods to deidentify personal health information. Under the statistical method, a statistician or person with appropriate training verifies that
enough identifiers have been removed that the risk of identification of the individual is very small. Alternatively, data are considered deidentified if 18 specified personal identifiers are removed from the data (IOM, 2009a) (see Box 2).
However, there has been some confusion as to whether adequate procedures are being used to meet the Privacy Rule’s stipulations for statistical deidentification methods. In 2013, HHS issued a more specific guidance on deidentification that clarifies much of that confusion, Bianchi noted.
However, others at the workshop noted that although deidentification is an important data protection tool, it is not infallible. Brad Malin, associate professor of biomedical informatics and vice chair for research at the Vanderbilt University School of Medicine, said, “Given enough effort, time, and incentives, you can break into any system.” He pointed out several published cases in which researchers had reidentified patients using public databases (Sweeney, 1997). However, he added that data have been deidentified not just as an academic exercise “by a single computer scientist spending lots of time and graduate students’ capabilities,” but by people with little expertise in this area. Despite this, he said, “To the best of my knowledge there are no lawsuits that show deidentified information is leading to actual reidentifications and exploitation of individuals. But absence of proof is not the same as proof of absence.” He also noted that unique identifiers may not be accessible to the public in any known resource, and not all identifiers are unique or reproducible. “You have to recognize that your adversaries usually have incomplete knowledge about the world,” he said.
He also described efforts he and others have made to improve data protections, including automated data scrubbing systems that rely on machine learning and remove or provide substitutions for key identifiers. These systems are 90 to 99 percent effective in protecting health data, he said (Aberdeen et al., 2010; Carrell et al., 2013; Deleger et al., 2013). The data scrubbing should satisfy the HIPAA stipulation that “the risk is very small that the information could be used alone or in combination with other reasonably available information, by the anticipated recipient to identify the subject of the information.”
Malin suggested researchers use a risk-based deidentification model (see Figure 1) in which health data stripped of the 18 identifiers specified by the Privacy Rule is subjected to both a risk estimation and risk mitigation procedure (Benitez et al., 2010; Malin et al., 2011; Xia et al., 2013). Alternatively researchers can employ risk models that assess if the number of identifiers they scrub from their data makes their health information more or less
BOX 2
HIPAA “Safe Harbor” Deidentification Method
The Health Insurance Portability and Accountability Act (HIPAA) “Safe Harbor” Deidentification of Medical Record Information requires that each of the following identifiers of the individual or of relatives, employers, or household members of the individual must be removed from medical record information in order for the records to be considered deidentified:
- Names;
- All geographical subdivisions smaller than a state, including street address, city, county, precinct, ZIP code, and their equivalent geocodes, except for the initial three digits of a ZIP code, if according to the current publicly available data from the Bureau of the Census: (a) the geographic unit formed by combining all ZIP codes with the same three initial digits contains more than 20,000 people; and (b) the initial three digits of a ZIP code for all such geographic units containing 20,000 or fewer people is changed to 000.
- All elements of dates (except year) for dates directly related to an individual, including birth date, admission date, discharge date, date of death; and all ages over 89 and all elements of dates (including year) indicative of such age, except that such
protected than if they scrubbed it of the 18 standard identifiers (Malin et al, 2011). Malin added that the term “Big Data” does not mean the end of privacy and can actually mean “Big Privacy” because most studies showing that privacy breaches are possible assume an extremely strong adversary perusing a small specific subset of subjects. But in the current era of Big Data, cohorts are rarely that small. “In the real world, people have much limited identifiability and you can actually support the types of studies you want to support with almost perfect accuracy and still have the protection guarantees that you anticipated,” he said.
Although deidentification is not a panacea and there is always a risk of reidentification, risk exists in any security setting, Malin pointed out, so one must determine an appropriate level of risk and ensure accountability that this level is achieved. Data use agreements should indicate the potential for that risk of privacy breach, he added, and stressed that risk is proportional
-
ages and elements may be aggregated into a single category of age 90 or older;
- Phone numbers;
- Fax numbers;
- Electronic mail addresses;
- Social Security numbers;
- Medical record numbers;
- Health plan beneficiary numbers;
- Account numbers;
- Certificate/license numbers;
- Vehicle identifiers and serial numbers, including license plate numbers;
- Device identifiers and serial numbers;
- Web Universal Resource Locators;
- Internet Protocol address numbers;
- Biometric identifiers, including finger and voice prints;
- Full-face photographic images and any comparable images; and
- Any other unique identifying number, characteristic, or code (this does not mean the unique code assigned by the investigator to code the data).
SOURCES: 45 C.F.R. § 164.514(b); IOM, 2009a.
to the anticipated recipient’s trustworthiness, with a vetted investigator having more trustworthiness than the public at large. One study he conducted reviewed all actual reidentification attempts through 2010 and found only one case with health data subjected to deidentification of the 18 required identifiers—a reidentification success likelihood of 0.00013 (El Emam et al., 2011). Malin concluded his presentation by noting the need for a national clearinghouse of models, methods, and evaluations of data deidentification. He also said that data protections should be proportional to the potential harm and should consider the context of the research.
Stephen Joffe, Emanuel and Robert Hart associate professor and director, Penn Fellowship in Advanced Biomedical Ethics in the Department of Medical Ethics and Health Policy at the University of Pennsylvania Perelman School of Medicine, pointed out the necessity for having dates in patient data for cancer research in order to assess how much a specific treat-
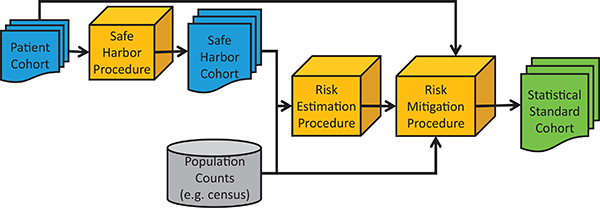
FIGURE 1 A risk-based deidentification model. Health data stripped of the 18 identifiers specified by the HIPAA Privacy Rule are subjected to both a risk estimation and risk mitigation procedure.
SOURCES: Benitez et al., 2010; Malin et al., 2011; Xia et al., 2013. Reprinted with permission from the Journal of the American Medical Informatics Association.
ment delays the progression of cancer and other key indicators. He asked how Malin’s data scrubbing systems addressed this need for dates. Malin responded that in a manner acceptable to HIPAA, he substituted random dates for when a patient received cancer treatment and when their cancer progressed that still preserved the amount of time between the two. “This way you can look at what happens to a patient over time, but you cannot do an aligned epidemiological study across the entire population. That’s why it depends on what you are using the information for. You need to know this before you redact some dates because you want to make sure you keep the data in a useful form. If what you are doing is an epidemiologic study, dates become extremely important and you want to retain them,” Malin said.
Malin also pointed out that the 18 identifiers specified in the Privacy Rule for deidentification were based on what could be used to reidentify data in the 1990s. “Those 18 were probably sufficient at that time, but whether it will be 5 years from now I do not know. The data are not just getting bigger, but the dimensionality of the data is increasing. As more dimensions are added, the data become more identifiable. But as more people are added, the data become less identifiable. If the rate of the dimensions is growing at a faster pace than the number of people in your studies, you might have a problem,” Malin stressed.
Collyar noted that many patients actually wish to have their data reidentified in order to take advantage of innovative treatments indicated by new molecular signatures that could be assessed in their stored biospecimens or data. “Deidentification is great for discovery research, but when the new
molecular signatures come out, patients want to be able to use their sample or data to find out if they have [relevant markers],” she said.
Impediment to Quality Improvement and Learning
Several participants were critical of the distinction that current regulations make between quality improvement activities and research. Leiter pointed out that two quality improvement efforts using the same data and addressing the same questions will be treated as health care operations if the institutions only use the results internally, but as research if the institutions share the results with others so that learning may occur. “It creates a real roadblock to the learning health care system because you have these disincentives, while not increasing the protection to the patient or to a human subject by setting up this distinction,” she stressed. She noted that the Health IT [Information Technology] Policy Committee9 suggested that rules be aligned better so that even if the intent is to share results for generalizable knowledge, use of clinical data to evaluate safety, quality, and efficacy should be treated like health care operations as long as the provider entity maintains oversight and control over data use decisions (Comments on the ANPRM, 2012).
Richard Schilsky, chief medical officer for the American Society of Clinical Oncology, noted that if one is collecting data for health care operations and has no prespecified hypotheses or research plan, but rather is just observing trends that emerge in a dataset collected for another purpose, most people would not consider that to be a systematic investigation that falls under the research definition of HIPAA and the Common Rule. Leiter agreed but noted that some clinicians are still not 100 percent sure that such investigations are not systematic research. As such, they tend to err on the side of caution and either forgo the investigation or seek patient consent and IRB review of it. “There is a fear climate that you are going to be wrong given that it is so easy to make the argument on the side that you are doing systematic research,” she said.
______________
9 The Health IT Policy Committee makes recommendations to the National Coordinator for Health IT on a policy framework for the development and adoption of a nationwide health information infrastructure, including standards for the exchange of patient medical information. The American Recovery and Reinvestment Act of 2009 (ARRA) provides that the Health IT Policy Committee shall at least make recommendations on the areas in which standards, implementation specifications, and certifications criteria are needed in eight specific areas (see http://www.healthit.gov/FACAS/health-it-policy-committee) (accessed June 16, 2014).
Several patient advocates at the workshop also pointed out that current regulations thwart the data sharing that patients favor, to enable learning from their own health care experiences.
Collyar pointed out another problem with current regulations and guidances; they contain vague language that is interpreted differently by IRBs. Greater consistency in interpretations and implementation is needed, she suggested. “People don’t have equal access [to clinical trials] across the country, based on what their IRBs allow. How is it ethical for somebody in Nebraska not to be able to have the same thing that is available to them in Houston?” Collyar asked.
Others argued that regulations and guidances are not sufficient if they conflict with the existing incentives in the system. Regulations and incentives should ideally be aligned to encourage the most ethical conduct. Speaking in regard to the oversight of patient consent and authorization, Jeffrey Botkin, associate vice president for research integrity at the University of Utah, and chair of the Secretary’s Advisory Committee on Human Research Protections, stressed that “Incentives are too strong in favor of greater complexity [of consent forms]. I am skeptical that fundamental change will happen without changing those incentives. Significant progress will only be made if incentives change. New regulations or guidance from HHS will be essential to change incentives.” He suggested that an Office for Human Research Protections (OHRP) guidance could require ensuring that patients understand what has been said to them as part of the consent process, as it is for the consent process specified by the 2000 World Medical Association Declaration of Helsinki. Such a guidance might be an appropriate way to leverage changes in IRB standards for informed consent. “Often the IRBs are afraid of negative findings by OHRP so they will pay close attention to OHRP guidances,” he said.
Lack of Harmonization with International Standards
Some participants called for harmonization of U.S. regulations and guidances with international standards because of the increasing interna-
tional scope of biomedical research. “When coming up with standards, we need to include our international colleagues,” said Jeffrey Peppercorn, associate professor of medicine in the Division of Medical Oncology at Duke University Medical Center. Ellen Wright Clayton noted that universal standards will be difficult to devise, given the wide range of views on patient privacy just within Europe. “It is going to be a real challenge because there are different regulatory regimes and different cultural settings. We also need to realize that what the European Union laws say and what some European countries do couldn’t be more different,” she said.
A large portion of the workshop was devoted to discussing the current shortcomings in patient informed consent for participation in clinical studies. Many workshop participants spoke about long-standing concerns that most patient consent forms for research are too long and complex, with formatting that is too dense, and filled with technical terms patients do not understand. Consequently, many patients do not gain the essential information necessary for informed consent.
One study found that 63 percent of individuals tested did not recognize the potential for incremental risk posed by a clinical trial, and 70 percent did not understand the unproven nature of the treatment being evaluated in the study (Joffe, 2001). Another study concluded that oncology consent documents are “often so long that the average patient is unlikely to read the document and/or are written in language that is likely to be too complex for them to understand” (Sharp, 2004, p. 573). The Agency for Healthcare Research and Quality (AHRQ) has stated that “[Informed consent] documents are long and written at a reading level beyond the capacity of most potential subjects” (AHRQ, 2009, p. 1). Despite the awareness that consent forms are too long, the median number of pages for informed consent forms increased to 11 between 2000 and 2005 (Beardsley et al., 2007). A National Cancer Institute (NCI) audit found that the median page number for the consent forms used in 97 NCI-sponsored Phase III cancer clinical trials was 16, reported Mary McCabe, chair of the Ethics Committee and director of the Survivorship Program at Memorial Sloan Kettering Cancer Center.
Leiter pointed out that patient authorization forms tend to either be so short and understandable that they are too general and do not provide meaningful protections, “or they are incredibly meaningful because they explain absolutely everything, but then you get the 22-page, 10-point font
authorization form that gets put immediately in the trash after it is signed, or it is just signed without being read because it is too long.”
McCabe and Botkin offered several reasons for the length and complexity of consent forms, including
- Research sponsors, investigators, and IRBs want to provide comprehensive medical information and avoid legal liability. They all seek to be accurate by using technical language.
- A longer and more complex form is easier to write than a shorter, easier to read form, which requires communications expertise. “Simply barking at investigators to do a better job writing the consent forms is not an effective solution and instead institutions should provide people who can help individuals write in a more appropriate way,” Botkin said.
- There are no regulatory requirements that forms be shorter and simpler or that require key information conveyed is understood by participants.
- IRBs add too many unnecessary details to forms to cover institution-specific information. The institutional boilerplates they use to provide that information is usually written in an incomprehensible “legalese.”
- A lack of comprehension does not appear to reduce recruitment to trials. Patients are embarrassed to ask questions and reveal what they do not know, and often instead trust their practitioners to make the decisions about research participation that will be in their best interest.
Recognizing the shortcomings of consent forms, NCI started an initiative in 2011 to write an improved informed consent template that was first used in NCI-sponsored clinical trials in 2013. The new template has several changes over the older template provided by the Institute, including
- Use of a study title that a lay person can understand.
- Risks of the study are written from the patient perspective. They are listed according to how common or serious they are, and presented in an easy-to-read table format. Instead of describing risks as percentages, the new format describes them as “X out of 100.”
- Length limits stated for each section. Anything that is optional, such as correlative studies, is not described in the main body of the consent document.
- Information included about mandatory specimen collection and optional studies, including future studies in which patients can participate.
- Standard of care is described prior to describing the care that would be received in the clinical trial.
But Laura Cleveland, patient advocate, Alliance for Clinical Trials in Oncology, and member of the NCI Central Institutional Review Board (NCI CIRB), noted that despite those improvements, the new NCI template is still too long and overwhelming for patients, and has a reading level of a college sophomore according to her Flesch-Kincaid analysis. The average U.S. reading level in English is about eighth or ninth grade, said Michael Paasche-Orlow, associate professor of medicine, Boston University, with only about half the population being health literate. The elderly, minorities, immigrants, and those with chronic diseases tend to be less health literate, his research found (Paasche-Orlow et al., 2005).
“There are very large ethnic, racial, and economic gaps in literacy. The whole conversation we are having here really has another whole social justice lens to it, which is about what does it mean that we have a 20-page consent form—what is that going to do in society,” he said. One study mentioned by Paasche-Orlow found that African Americans received less information about clinical trials than whites (Penner et al., 2013). Another participant added that clinicians are often less inclined to discuss a clinical trial with minority, low-income participants with a minimum of education because they assume they will not understand the consent form. “It’s a factor that plays into health disparities and a real disparity in access to trials,” she said.
Another study by Paasche-Orlow found that after HIPAA was instituted, the average Flesch-Kincaid reading level of academic institutions’ template consent forms for research was greater than 11th grade, and that among institutions that had grade-level reading standards, only 6 percent of their templates met that standard, with the mean being 4.2 grade levels above the standard (Paasche-Orlow et al., 2013). He strongly suggested shortening and simplifying consent forms, noting that the people reading them “are physically and emotionally not at their best.”
Cleveland suggested using health lexicons to simplify the language used in consent documents and determining their reading levels by using the tools offered by various word processing programs. She also suggested including a one-page summary of the consent form for a potential cancer clinical trial participant. This summary could concisely state what a clinical trial is; what the specifics of the trial are under consideration; why it is being
done; and how it will be different from receiving usual care, as well as the costs involved. Practical helpful information could also be provided, such as what participants should bring to appointments and where they should park when being treated.
Several speakers noted that the consent form is only one component of informed consent, and that the consent process is equally if not more important. Botkin pointed out that the current consent process does not usually ascertain whether participants understand key information elements before a decision is made. “Rather than looking at whether the form is signed and has the right date on it, shouldn’t we also look and see if the institution is assessing understanding of the form?” McCabe added.
Paasche-Orlow noted a frequent lack of shared meaning between researcher and patient. For example, though disclosure of investigators’ ties to the drug industry is considered one of the main mechanisms to reveal potential conflicts of interest, he found patients’ often were impressed by such disclosures, with some responding with such comments as, “I did not realize he was such a big shot. I should be in this study because he is such a big shot.” Paasche-Orlow stressed that “If you do not check, you do not know what the person understands.”
Terrence Albrecht, associate center director, Barbara Ann Karmanos Cancer Institute, concurred, noting that her research on clinical interactions among patients, family members, and oncologists has shown that the participants tend to agree on the topics discussed, but disagree about the details. One of her studies found that patients often have misconceptions about whether they were offered the opportunity to participate in a clinical trial. Thirty-nine percent of patients who only discussed a trial with their practitioners, but were not formally offered participation, erroneously said they had been offered participation in a trial, whereas 14 percent of patients clearly offered the opportunity to participate in a trial reported they were not offered that option, including some who consented to the trial.
Cleveland added, “To many patients, consent for participation in a clinical study means ‘If I want a medical procedure I have to sign this form.’” She stressed, “We have to protect people in a way that they are going to be able to understand and process information to make a decision that is right for them. We need our health care professionals who are talking to people about clinical trials to have good communication skills.”
Paasche-Orlow suggested that clinicians use a teaching approach instead of using a persuasive mode of speaking to patients during the consent process (see Box 3). “We need to flip the default toward taking responsibility to confirm comprehension,” he said. Albrecht added that the consenting process often tends to be more of a monologue than a dialogue. She noted a conversation occurs when all participants have equal rights to speak, but stressed that often the balance of a consenting conversation is
Teach to goal is a learning mechanism to confirm comprehension. It involves teaching followed by assessing the person’s comprehension and then continued focused teaching until the person exhibits mastery of what is being taught. When this approach is taken in the consenting process, the medical professional starts with phrases such as “I want to make sure we have the same understanding about this research,” or “It’s my job to explain things clearly. To make sure I did this I would like to hear your understanding of the research project.”
The purpose of teach to goal is to check comprehension, not memory of the educational material. Consequently the medical professional should allow the potential research subject to consult the consent document when answering questions, which are aimed at confirming understanding of all the important elements of the study. The medical professional should also listen for simple parroting, and if a potential subject uses technical terms, ask them to explain further. Paasche-Orlow suggested asking open-ended questions such as “Tell me in your own words about the goal of this research and what will happen to you if you agree to be in this study” or “What do you expect to gain by taking part in this research?”
Any misinformation should be corrected until potential research subjects indicate that they have understood by correctly answering all the questions. Medical professionals need to make clear that the need to repeat information is due to their failure to clearly convey the information rather than the “fault” of the potential subject. For example, one could say, “Let’s talk about the purpose of the study again because I think I have not explained the project clearly.”
SOURCE: Paasche-Orlow presentation on February 24, 2014.
tipped to the rights of patients to be informed about research with little opportunity for them to engage in a dialogue with their practitioners.
Paasche-Orlow agreed and pointed out that the current standard is that if the patients want to know more, they have to ask questions, but usually such questions are not entertained until after a 45-minute monologue on the part of the clinician. “That is not an honest play at interaction,” he said. He suggested asking patients open-ended questions, such as “Tell me in your own words, what risks would you be taking and what do you expect to gain by participating in this study?” and then correcting any misinformation they have. “You have to make the consent discussion an interactive experience,” he said and suggested requiring confirmation of comprehension as entry criteria for studies.
Cleveland also suggested checking for patient understanding by asking a simple question, such as “Tell me what you understand about this,” which could be part of the clinical trial protocol template. Taylor noted that another advantage of asking patients such questions after a consent discussion is that they give practitioners feedback as to how well they are conducting the discussion, which could lead to improved consent discussions in the future. Paasche-Orlow concurred, noting in his presentation that “Right now people get very little feedback about their own skill level—what they are doing well, and what they are not doing well and need to improve.”
Several speakers said that there is a need to enhance practitioner knowledge, skills, and behavior in consent processes because many do not have the appropriate skills and expertise, or do not take the time to do it properly, and may be insensitive to the stressful circumstances in health care environments. “No matter how cool a customer, no matter how deep their education and how much they have studied their disease, they come in emotionally impaired—they are thinking about their families, themselves, and if they are going to leave a legacy. We have to address them as such,” Cleveland said. She noted that “the sickest people tend to be the most vulnerable,” and make privacy of their health information a low priority compared to using it to find a cure for their disease. “They want you to take everything and do anything with it in order to help them,” she said. “So there does need to be something in place to protect people when they are most vulnerable. The challenge is to put protections in place that are not paternalistic and are designed with the patient’s engagement,” she added.
Patients also often have little experience with research, Botkin noted, and the assumption that if you give people information they will make logical decisions in their own best interest “is a shallow, inaccurate description of how people live their lives, particularly in the context of serious illness.”
Cancer and other life-threatening ailments can provoke fear and anxiety that can make patients more passive and more dependent on and trusting of their care providers to make their medical decisions, including whether to enroll in a trial, Botkin said.
Albrecht suggested that in addition to adhering to a standard consent checklist of what to discuss during the consent process, there should be adherence to discussion points that offer support for patients if they decide not to participate in the trial offered. Such support would include reassuring patients that their doctor will continue to see them, that they will have support to address side effects, and that if they are not doing well on the study, their care will be reevaluated and a new care plan drawn up.
Although several speakers made suggestions for improving both consent forms and the consent process, Botkin noted that which ones might work best is unknown because insufficient research has been done to address this question. “It is not evidence-based as it is currently conducted,” he said. But more important, according to Botkin, is changing the incentives of stakeholders in the consent process. New ideas and data will not change these incentives, he said, and instead suggested new regulations or guidances from HHS that will change those incentives that have made consent forms and processes too complicated. In that regard, the Secretary’s Advisory Committee on Human Research Protections (SACHRP) made specific recommendations in 2013 for simplifying informed consent by reducing the number of required elements and increasing the number of optional elements (SACHRP, 2013) (see Table 1). Botkin also suggested HHS consider requiring that part of informed consent include ensuring subjects have understood the information in order to leverage some of the changes needed in the process. “My hope is to encourage a SACHRP initiative in this particular domain and we have had support from OHRP and the Assistant Secretary of Health for making this effort,” Botkin said.
Speaking from the patient perspective, Collyar stressed that whatever changes are made to the consenting process, a key component should be partnership with and respect and trust of patients. She also stressed that patients just want to have the essential information they need to make their decisions. “They don’t want to have to learn a whole new field themselves. No patient says ‘yes, I’m excited about becoming health literate.’”
Collyar agreed with the SACHRP recommendations and also suggested building multiple models of the consent process and evaluating and learning from them, as well as incorporating feedback loops so the consent process continues to improve. She also suggested that consent processes should continually adapt to new technologies as they become available.
TABLE 1 Elements of Informed Consent
| Current Elements of Informed Consent (45 CFR 46.116) | Revisions Proposed by SACHRP (2013) |
Required Elements
|
Required Elements
|
Optional Elements
|
Optional Elements
|
NOTE: SACHRP = Secretary’s Advisory Committee on Human Research Protections. SOURCES: HHS, 2013; Botkin presentation on February 25, 2014; http://www.hhs.gov/ohrp/sachrp/commsec/attachmentd-sec.letter19.pdf (accessed June 16, 2014).
Several participants suggested that a number of new technologies could improve the consent process, including interactive computer programs and audiovisuals that educate the patient about the trial; tablets that patients can use to fill out forms in the waiting room; and greater involvement of
research nurses during the entire consenting process. Paasche-Orlow demonstrated a computer program he developed that uses embodied interactive conversational agents to emulate face-to-face communication. In the example he showed, a cartoon version of a person appears on the computer screen and talks about how consent forms can be long and complicated “and my job is to help you understand as much as possible.” The conversational agent then asks the patient questions, such as if they have ever been in a research study before, and responds with empathy when needed. For example, if the patient says their previous experience with a research study was bad, the agent responds, “I’m sorry to hear that. If you choose to be in any more research studies, I certainly hope you have a better experience.”
The computer avatar educates the patient by explaining potential side effects with a visual that makes it easy to see what portion of participants are likely to experience those side effects. It also acts as an advocate for the potential participant, stating, “You mentioned before that you had been in an earlier study and were not satisfied. You know if you choose to join this study and then found you were not happy, you could withdraw. If you tell the research team, they will help you leave the study in a way that is safe for you.”
Paasche-Orlow noted that even the elderly who have little experience with such computer programs respond favorably, waving back at the avatar and easily manipulating the touch screen provided for their responses. He said his studies indicate that in some ways, the computer avatar performs better, on average, than the physicians discussing consent with patients because it remembers the personal information the potential participant provides and restates that information to personalize the process. In the real world, “these are often hour-long consent conversations and the research staff frequently follows a script, and although they have many opportunities to personalize and empathize, they choose not to exercise those options,” he said. For example, when taking a family history, the physician may note that the patient’s father died of cancer without making any empathetic comment about it. “It is very strange and has something to do with the culture of medicine in general. I think we can do better,” he said. He noted that the degree of empathy expressed by the computer conversational agent can vary. His studies show that patients with depression or lower literacy spend more time asking more questions if the computer program is altered so that the avatar expresses more empathy.
Michaele Christian, former associate director of the Cancer Therapy Evaluation Program in the Division of Cancer Treatment and Diagnosis at
NCI, suggested using audiovisuals to reduce the amount of time potential participants need to spend reading materials related to the study. Albrecht responded that some investigators, such as Neal Meropol at Cleveland Clinic, are experimenting with videos that patients can watch prior to meeting with their physician to discuss a clinical trial. “It is a preparatory aid tailored around the basic concerns and values of the patient,” she said.
Sharon Terry, president and chief executive officer of the Genetic Alliance, suggested using computer tablets on which potential research participants fill out their family and personal medical history in the waiting room. That information could be reviewed by a computer program that finds appropriate clinical trial matches for them. The patients could indicate their initial interest in a study on the tablet so that a clinician or clinical trial recruiter can then speak with the patient about that option. A similar tool for alerting physicians to inherited disorders and greater risk of preterm birth in their obstetric patients is currently undergoing testing. “This type of tool could aid community-based hospitals and clinics with more limited resources than major academic medical centers and so could be particularly important for increasing the diversity of the participants in clinical trials,” Terry said.
Albrecht noted a pilot study that she conducted in which the research nurse screens potential participants before they meet with the doctor, but then continues to be present when the doctor meets with the patient to discuss the clinical trial. “They sit in the background, but they have the imprimatur of the physician,” she noted, and they provide more details about the study once the physician leaves. This shortens the amount of time the physicians spend consenting patients and “that research nurse becomes a lifeline for that patient going forward throughout the rest of the decision process and into treatment.” But she noted that more incentives and system-wide changes need to be provided for such a supportive consenting process. Collyar suggested the development of a smartphone app that would make it easier to find and reconsent patients because cell phone numbers tend to stay constant even if they move, unlike landlines.
As McCabe said, “There is a whole new landscape out there and we really ought to move beyond that paper consent form and thinking about it in the usual way.”
Leiter pointed out that although informed consent is important, it alone cannot ensure adequate protection of participants in clinical
research. She suggested relying less on consent, especially for uses of data in an LHCS, and instead pursuing other models of patient engagement, such as seeking patient input into research, having greater transparency on the research uses of data, and imposing requirements to share results with patients. Terry concurred, saying, “Consent cannot bear the burden of the research industry. It is only part of the picture and we need to go to Fair Information Practice Principles more robustly” (see Box 4).
Even if they want their health data shared, Clayton cautioned that “people should be protected from unwarranted deidentification of all research data. The [key consideration] is not so much what they give consent to, but rather what is our oversight, accountability, transparency, limitations on downstream uses, etc. We should be paying much more attention to that.” Leiter pointed out that federated, decentralized research, in which data remain within the originating institution, offers more data protections than if data are collected in a big centralized location. “If we think about ways to protect data, we provide some way to incentivize keeping the data where it is to have the least exposure possible,” she said. “There are lots of other ways besides consent to layer on protection and lots of different tools to employ, and they too often get ignored in policy and legal discussions about data protection.”
The Health Information Technology for Economic and Clinical Health (HITECH) Act of 2009 called for numerous changes to the Privacy Rule, and in 2013, HHS announced a number of changes to the Rule and guidance (HHS, 2013). HHS specified that a HIPAA authorization can permit future research if the authorization adequately describes the future research such that it would be reasonable for an individual to expect that his or her protected health information could be used or disclosed for that purpose. Compound authorizations for research purposes are also now permitted as long as certain conditions are met. For example, the patient could simultaneously authorize the use of his or her health information for a specific current research project as well as for a biospecimen bank that distributes tissue samples and/or data for future research projects.
“This has made a big difference in terms of our ability to go forward and create forms that allow future research, but it is hard to know how to write these authorizations so that they adequately describe the future research so it would be reasonable for an individual to expect that their
BOX 4
Fair Information Practice Principles
The Fair Information Practice Principles (FIPPs) are a widely accepted framework at the core of the Privacy Act of 1975. They have become the basis of related laws and regulations adopted by many federal and state agencies. The FIPPs are not precise legal requirements, but principles for balancing the need for privacy with other interests in an era of computerized information. The FIPPS were first articulated in a comprehensive manner in the former U.S. Department of Health, Education, and Welfare’s 1973 report titled “Records, Computers, and the Rights of Citizens” (HEW, 1973). The eight FIPPs, as stated in the Nationwide Privacy and Security Framework for Electronic Exchange of Individually Identifiable Health Information, are
Individual Access: Individuals should be provided with a simple and timely means to access and obtain their individually identifiable health information in a readable form and format.
Correction: Individuals should be provided with a timely means to dispute the accuracy or integrity of their individually identifiable health information, and to have erroneous information corrected or to have a dispute documented if their requests are denied.
Openness and Transparency: There should be openness and transparency about policies, procedures, and technologies that directly affect individuals and/or their individually identifiable health information.
information is going to be used for it,” Bianchi said. “It has removed some of the roadblocks to researchers, but it is not clear how far it is going to get us,” she added, noting that it is not yet clear if current ongoing studies can be grandfathered because they described potential future research in the informed consent process.
Another issue that has affected biomedical research is the question regarding what constitutes sale of PHI, which is not allowed under HIPAA. Past language addressing this issue was quite vague, and researchers were concerned that payments they received to cover the cost of data transfer
Individual Choice: Individuals should be provided a reasonable opportunity and capability to make informed decisions about the collection, use, and disclosure of their individually identifiable health information.
Collection, Use, and Disclosure Limitation: Individually identifiable health information should be collected, used, and/or disclosed only to the extent necessary to accomplish a specified purpose(s) and never to discriminate inappropriately.
Data Quality and Integrity: Persons and entities should take reasonable steps to ensure that individually identifiable health information is complete, accurate, and up-to-date to the extent necessary for the person’s or entity’s intended purposes and has not been altered or destroyed in an unauthorized manner.
Safeguards: Individually identifiable health information should be protected with reasonable administrative, technical, and physical safeguards to ensure its confidentiality, integrity, and availability and to prevent unauthorized or inappropriate access, use, or disclosure.
Accountability: These principles should be implemented and adherence ensured through appropriate monitoring and other means, and methods should be in place to report and mitigate non-adherence breaches.
SOURCES: HEW, 1973; ONCHIT, 2008; http://itlaw.wikia.com/wiki/Fair_Information_Practice_Principles (accessed June 16, 2014).
would be interpreted as sale of PHI. HHS now specifies that a reasonable fee to cover the cost to prepare and transmit data is allowable.
ADVANCED NOTICE OF PROPOSED RULEMAKING
Holly Taylor, associate professor of health policy and management, Johns Hopkins Bloomberg School of Public Health and core faculty, Johns Hopkins Berman Institute of Bioethics, provided an overview of the recent Advanced Notice of Proposed Rulemaking (ANPRM) for the Common
Rule that HHS put forth in July 2011, with the aim of clarifying regulatory ambiguity and relieving perceived impediments to clinical research. Subtitled “Human Subjects Research Protections: Enhancing Protections for Research Subjects and Reducing Burden, Delay, and Ambiguity for Investigators,” this document made several suggestions related to protecting research subjects from informational risk, aligning IRB reviews to degree of risk, using one IRB for multisite studies, harmonizing human subject protections regulations, and improving consent forms and processes.
The ANPRM proposed that the HIPAA regulations be adopted as the universal standard for privacy protection in research, and that collection of data for research purposes, even if it does not have identifiers, should require patient consent. The ANPRM proposed requiring broad consent for future research use of archived biospecimens. This would be a change from current rules, which allows research without consent when biospecimens are deidentified. The proposed broad consent would enable research on archived biospecimens, even with identifiable samples, without the time and expense of reconsenting patients for additional studies. But Ellen Wright Clayton, Craig Weaver professor of pediatrics and professor of law, Vanderbilt Center for Biomedical Ethics and Society, noted that such consent for future research does not apply if research results will be returned to participants. In that case, patients would first need to be recontacted and reconsented for that particular study.
The lack of clarity in distinguishing research and quality improvement activities was also recognized by the ANPRM, which states “The Common Rule has been criticized for inappropriately being applied to and inhibiting research in certain activities including quality improvement, public health activities, program evaluation studies regarding quality improvement. These activities are in many instances conducted by health care and other organizations under clear authority to change internal operative procedures to increase safety or otherwise improve performance, often without the consent of staff or clients, followed by monitoring or evaluation of the effects,” Menikoff reported. He added, “It is far from clear that the Common Rule was intended to apply to such activities nor that having it apply produces any meaningful benefits to the public. It might have a chilling effect on the ability to learn from and conduct important types of innovation.”
Taylor said that other proposed changes would aim to better calibrate IRB review to the level of risk. For example, one suggestion was to revise and simplify the review approach for expedited review by eliminating the requirement for continuing review. The ANPRM also suggested replacing
“exempt research” with “excused research.” This category of research would require Principal Investigators to report to an IRB, which would audit such requests, with the assumption that as soon as a report is filed, the investigator could move forward with the research. Another key ANPRM proposal was the use of a single IRB of record for domestic multisite studies. This was in response to the recognition that multiple IRB reviews fail to improve human subject research protections, while lengthening the time of review and diverting valuable resources.
The ANPRM also called for improving consent forms and the consent process. Jerry Menikoff, director of the HHS Office for Human Research Protections, noted, “We’re all recognizing that consent forms have reached a point where they’re just getting longer and have more boilerplate [content] so you end up burying a lot of the key details that a person should actually be thinking about when they enroll in a research study. One of the purposes of ANPRM was to change the rules so that there would be greater authority to go from a 20-page consent form to a 3-page consent form that puts the boilerplate [material] somewhere else and is more to the point on what’s going to happen in the clinical trial and what the patient should be thinking about in terms of his or her best interests in enrolling.” (See the section on consent forms.)
Taylor said another ANPRM stipulation is that there should be an improved and more systematic approach for the collection and analysis of data on unanticipated problems and adverse events. The ANPRM also called for improved harmonization of regulations and related agency guidances related to human subjects protections. Finally, the ANPRM proposed that federal regulatory protections be extended to all research with human participants regardless of funding source, as long as this research is conducted at institutions in the United States that receive some federal funding from an agency that has adopted the Common Rule.
The ANPRM generated more than 1,000 comments. A joint response by the American Association for Cancer Research, the American Society of Clinical Oncology, and the Association of American Cancer Institutes indicated general support of the ANPRM, but requested better delineation of the responsibilities of the IRB of record, and reconsideration of the consent requirements for deidentified data. These organizations disagreed with the adoption of HIPAA as the universal standard for privacy protection. A committee of the National Research Council’s Board on Behavioral, Cognitive, and Sensory Sciences also concluded that HIPAA should not be adopted as the universal standard, and stated that the proposed requirement for con-
sent for use of deidentified data should be reconsidered (NRC, 2014). This committee also called for criteria to define what is and is not considered human subjects research. They also endorsed the new category of excused research, but noted the need to operationalize some of the procedures for that type of research.
The ANPRM has been closed for public commentary since October 2011. If revised regulations are ultimately developed and implemented, they would have to be endorsed by all the federal agencies that currently use the Common Rule. Taylor noted that it took 10 years to get such approval for the current Common Rule.
PATIENT PERSPECTIVES ON RESEARCH PROTECTIONS
Patients have a wide range of views on sharing their data for health research, Terry noted. One study found that about one quarter of survey respondents who expressed an opinion would be willing to share their data without consent if their identity is not revealed and the study is supervised by an IRB (20 percent of those surveyed said they were not sure). About half of respondents said they would want each study seeking to use their data to contact them in advance and seek specific consent each time (IOM, 2009a). “There’s really a wide spectrum of patient views on this. One size does not fit all,” Terry noted.
Clayton agreed, noting that a systematic review found substantial variability in participants’ stated desires for control over research use of their health information and their willingness to accept broad consent for use of their biospecimens in studies (Brothers et al., 2011). She also pointed out, “A lot of this research is affected by how you ask the question. If you ask them, are they worried about their information not being private and the research being risky, then you get the answer that they think this research is risky. If you ask them whether they think this research is worthwhile and a biobank is good for an institution to have, the answer is emphatically yes,” she said.
Clayton noted that 88 percent of patients surveyed at Vanderbilt about sharing their deidentified health data were either neutral or stated that data sharing made them more amenable to participating in research. A similar percentage of patients in Group Health Cooperative reconsented to data sharing. In a study Peppercorn conducted, about three quarters of participants agreed to having their DNA and biospecimens stored for potential use in future unspecified studies related to cancer or other diseases (Ludman
et al., 2010). “Patients basically are altruistic and want to help in clinical trials,” Cleveland stressed.
Taylor pointed out that many patients are willingly providing their health information on Facebook and other social media sites, which could provide investigators with a public resource for study recruitment. “There is some interesting debate in the literature about what is an ethical use of something like Facebook to try and track or retain [research] subjects,” she said. Leiter noted the increasing willingness of the younger generation to share personal information, including health information, on Internet forums, but noted a “fundamental lack of knowledge as to how unprotected those data are.” Health data uploaded onto the Internet have no legal protections, she said. “With the evolving tolerance for sharing information, there needs to be an understanding or something formulated so that health data are not as exposed as they are right now, even with people more willing to share it,” she added.
Bianchi agreed there is a lack of protection for health data in settings outside of those stipulated by HIPAA, including health data that patients upload to the Internet. She noted that the European Union has a “right to be forgotten” regulation that requires Internet servers and websites to erase individuals’ personal information that has been made public without legal justification (Data Guidance: Global Guidance in One, 2013). “Don’t we have a right to be forgotten too?” she asked. Malin added that studies by Alessandro Acquisti at Carnegie Mellon show that when people eagerly embrace new technologies or platforms, such as Facebook, they tend to make their personal information public using them, but then at a later point in time, “there is a lot of remorse that kicks in and they want to take that information offline. Just because people right now may feel comfortable putting information up online, they may not later when they try to get a job.” Gail Jarvik, head and professor, Division of Medical Genetics, University of Washington School of Medicine, noted that the elderly tend not to be as concerned about the privacy of their health information, studies show. “By the time you are 90, you have gotten the diseases you are going to get and there is not a secret in your genome hiding anymore so there is less of a concern,” she said.
Terry suggested using online participant-centric registries with which participants can interact and change the research options that can be pursued with their data or biospecimens. She said many registries are currently centered on investigators, institutions, research sponsors, etc., rather than on the patient. She presented a model for such a registry called Platform for
Engaging Everyone Responsibly or PEER (see Box 5). According to Terry, PEER empowers participants to create and manage access to all of their information by moving participant contact details into the registry and giving them the ability to manage the right to access their health data (see Figure 2). “This is our vision of the future—that individuals will always be in the center of their data,” Terry said. She added that instead of individuals having the onerous task of finding a trial in which they can participate through clinicaltrials.gov, the trial can find the individuals, as long as they have provided enough of their health data on PEER.
Terry said that PEER not only meets the privacy standards of HIPAA, but exceeds them. By providing participants with easy access to information about clinical trials that are investigating treatment alternatives, PEER is doing the case management that is included in the HIPAA definition of “health care operations” that enables use or disclosure of protected health information. PEER affords individual participants a level of privacy protection that is beyond HIPAA, according to Terry, because it holds no personal information other than that which the participant or their representative explicitly provides, or requests that a third party provide, and the participant maintains continuous control over the use of his or her information. PEER has been deployed by a number of organizations and institutions, including various disease organizations and universities that are part of the National Patient-Centered Clinical Research Network of the Patient-focused Drug Development Initiative of the Food and Drug Administration.
Botkin suggested polling a focus group of some study participants about their willingness to share their data in the newer study instead of reconsenting all people for future research projects. He suspected that the majority of people in the focus group would want to have their data reused and such a finding could be leverage to have the study approved by an IRB.
Peppercorn suggested that potential misuse of health data could be discouraged through laws prohibiting discrimination in health and life insurance and employment, as well as by imposing criminal penalties for hacking or stealing data. He noted that the law has had trouble in the past keeping up with technological advances in cyberspace. “Let’s get ahead of it and prohibit things we don’t want done,” he said. Collyar concurred and emphasized the criminal penalty for inappropriate use of data because we cannot keep people from illicitly collecting data.
BOX 5
Platform for Engaging Everyone Responsibly (PEER)
PEER is an online participant-centric registry with which participants can interact and change the research options that can be pursued with their data or biospecimens. A number of access controls on PEER enable participants to specify, in a granular manner, medical researchers, organizations, data analysis platforms, and others that can and cannot use their health information and for what purpose. These controls, which can be operated independently of each other, include:
- Discovery, which controls who can discover their information • Contact information, which determines who can access their contact information
- Export, which controls who can export deidentified information from PEER
Participants have the option to edit their responses to these controls from a computer or smartphone, as well as to indicate if they want more information about a specific study or organization before deciding how it can use their health data. They can also access knowledgeable and trusted guides from their particular community, including patient advocates and activists, who describe in videos the different privacy settings and their ideas about sharing information, and also make recommendations on how to choose privacy settings based on the degree of privacy participants might want. The platform also sends participants notifications of data-sharing opportunities for which they can consent, deny, or delay making a decision.
PEER can be embedded in any website, such as those provided by disease foundations or cancer centers, and can be easily customized. This helps build trust for the platform on the local level at which participants would interact, according to Sharon Terry. One group that used PEER on their website, for example, changed the working language to make it more relevant to their community.
SOURCES: Terry presentation on February 25, 2014; Genetic Alliance (http://www.geneticalliance.org/programs/biotrust/peer; accessed June 16, 2014).
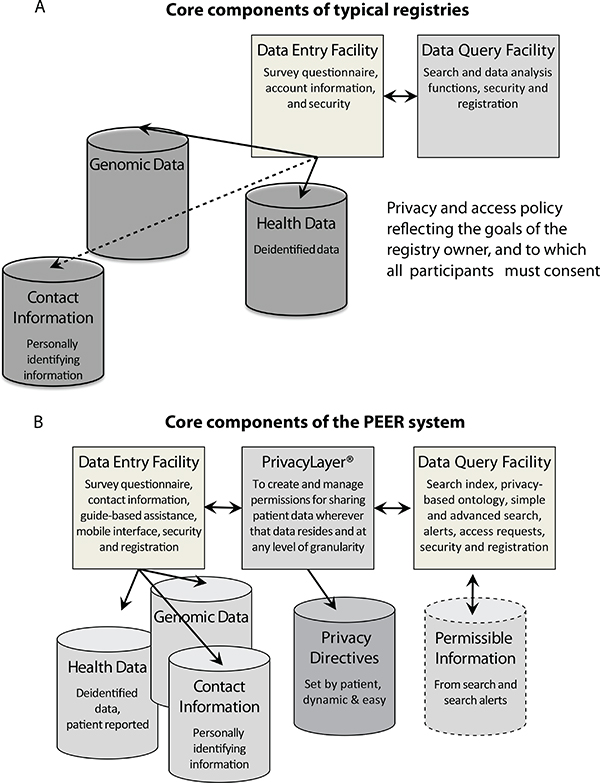
FIGURE 2 A. Typical registry architecture. Usual core components of registries include: functionality for data entry, a database of deidentified data, and facility for inquiry and/ or analysis of that data based on the privacy and access policy of the registry owner, to which all participants must consent in advance. Contact information for individual participants—if available—is most often loosely coupled, and outside the registry. B. Core component of the Platform for Engaging Everyone Responsibly (PEER). PEER is participant-centric with privacy controls from Private Access that empower participants to create and manage access to all of their information. The right to access contact details in the registry is separately managed by each participant.
SOURCE: Terry presentation on February 24, 2014.
ETHICAL CHALLENGES OF GENETIC ADVANCES
Angela Bradbury, assistant professor of medicine, Perelman School of Medicine at the University of Pennsylvania, opened the session on ethical challenges of genome-based cancer research by noting that recent technological advances enable researchers to easily and inexpensively determine the entire sequence of a person’s DNA (genome). In this era of the “$1,000 genome,” there is renewed interest in developing precision medicine (also called personalized medicine), whereby clinicians can use genetic findings in a person’s blood or tumor sample to attempt to determine the most suitable prevention efforts and/or treatments for certain cancers. Variants of BRCA, HER2, BRAF, and other genes are already being used in clinical oncology to select treatment, she noted (Burke and Psaty, 2007; Schwartz et al., 2008).
However, Collyar noted that while medicine is moving in this direction, it is premature to discuss this as if it is a reality in all cases. Despite technological advances, interpreting genetic findings remains challenging. For example, results from the numerous genetic sequencing platforms may vary, and there is a lack of information on genetic differences that can be reliably translated to appropriate clinical action. Much genetic information does not provide “yes” or “no” answers akin to that given for many infectious disease tests, but rather probabilities of developing a disease or of the disease being aggressive. This introduces uncertainty into discussions about genetic findings “and there are questions about how much uncertainty patients are willing to tolerate,” Bradbury said. Another challenge is what to do with the secondary incidental findings in genomic test results. Genetic tests are usually done to determine possible prevention or treatment avenues for a specific disease, but they may also reveal clinically relevant information about other disorders, some of which may not yet be apparent.
The ethical issues discussed in more detail at the workshop were those linked to patient consent for using archived biospecimens, and the return of test results. Speakers also explored liability issues in genomics research and the unique privacy issues raised by having genetic information in public databases. Workshop participants reported a wide range of perspectives on all these issues.
Patient Consent for Use of Archived Biospecimens
Researchers are increasingly using archived biospecimens, such as blood and tumor tissues, and related health data in genetic studies that could have
clinical implications. This research occurs subsequent to and has different objectives than the purpose for which the biospecimens were originally collected and for which participants consented to use of their tissues.
Clayton pointed out that the ANPRM would require consent for collection and use of all biospecimens on grounds that DNA can reveal a participants’ identity, but it fails to address the fact that clinical information can often be more identifiable and easier to obtain than genetic information. “If I were on an airplane and I had a choice between leaving my deidentified medical record on the plane or leaving my DNA on the plane, the medical record is the one I would worry about,” she said. She added that to be consistent with this reasoning, there should be consent for using all clinical data. “This would be a dramatic change from current practice and would threaten epidemiology, which has conferred great contributions to our clinical knowledge,” she said, adding, “This recommendation of the ANPRM is based on unwarranted and unfounded genetic exceptionalism. This response to the low risk of reidentification of genetic information pays more attention to creating systems of oversight and control, instead of just saying we are going to do informed consent.”
Peppercorn added that requiring reconsent for use of biospecimens for future unanticipated studies would violate the trust of the participants who donated those biospecimens with the anticipation that they would help advance science and help future patients. “This issue of public trust that we worry about, in terms of violating informed consent, really cuts both ways. Just as we want to be careful about passing the smell test if the public learned about the data we are sharing from archived biospecimens, what would the public think if they learned we didn’t use this information to try to address the diseases that still plague us?” he asked.
Peppercorn described how he weighed the pros and cons of sharing the genetic data gathered in an NCI-sponsored clinical trial of treatment for early-stage breast cancer, through a genetics database called dbGaP.10 This database, which is housed by the National Center for Biotechnology Information, aims to advance science and health by providing access to biomedical and genomic information. The trial was initiated long before the database was established. The informed consent document used in the trial discussed future research in general terms that the participants could opt to participate in, as well as the uncertainty regarding details of those future research questions. But it did not use the terms “genetics” or “genetic
______________
10 See http://www.ncbi.nlm.nih.gov/gap (accessed June 16, 2014).
information,” nor did it mention sharing the participants’ genetic or clinical data with a national database. However, the informed consent document did mention privacy risks and the potential for recontact by researchers.
Peppercorn and his colleagues weighed several factors in determining the appropriateness of sharing genetics data from the biospecimens without undergoing the expensive and likely unfeasible process of reconsenting the patients. These factors included whether there were
- Potential societal benefit from sharing data;
- Informed consent for future research;
- Uncertainty of risks from future unspecified research explained in the consent form; and
- Data sharing consistent with the goals and terms of the initial consent form, even though the language used was not perfect by modern standards.
He noted the low bars to overcome were the first two on the list. “If there was no societal benefit from sharing the data or at least informed consent for future research, you have a big problem,” he said. Peppercorn added that the research team also considered whether reconsenting was feasible and what the expressed interests of the participants had been. “Whether the expressed interests of the participants are better honored through data sharing or withholding the data is really the ultimate question to address in each case,” he said.
Peppercorn considered similar factors when deciding if archived tumor samples originally collected for a study of two different treatments for gastric cancer could be shared with The Cancer Genome Atlas (TCGA), an NCI-supported effort to molecularly characterize various cancers, including gastric cancers. These samples were thought to be a unique collection, especially useful for TCGA endeavors, and because the study was conducted at 500 sites without a central IRB and most of the patients from whom the samples were collected had died, reconsenting was not a feasible option.
Peppercorn also considered TCGA guidelines which specify that samples used in their project should have informed consent documents that
- Have some description of genetic or genomic research.
- Explain the concept of data sharing, in broad terms as well as how data might be used by individuals other than the principal investigator for the original project.
- Indicate the possibility of future research, including research beyond cancer research, as well as research that may result in commercial products.
- Explain the risk of loss of privacy and confidentiality and describe measures taken to reduce risks.
Peppercorn decided that sharing the biospecimens with TCGA was appropriate for participants in the trial who had opted on their consent form to let their tissue or blood be kept for future unknown use in research to learn about, prevent, treat, or cure cancer or for research about other health problems. About three-quarters of the participants had opted in for this future research.
Peppercorn concluded his presentation by noting, “We are not going to be able to predict the questions we are going to want to ask in the future or what the technologies will be.” Consequently, he said that the informed consent process is going to be imperfect for future research and thus should be accompanied by “a transparent and multidisciplinary process that our participants would be happy with for reviewing the use of their archived biospecimens and deciding what we can or can’t do with them.” He also suggested prospectively improving informed consent for future unspecified research. Such consents should be broad, given the broad scope of science, and should explain the uncertainty of what that future research will be.
Jarvik described the principles and recommendations about return of individual research results11 developed by a consortium of members of eMERGE (Electronic Medical Records and Genomics) and CSER (Clinical Sequencing Exploratory Research). In a consensus statement, these genetics experts concluded that research, even in a clinical setting, differs from clinical care in both its goals and its procedures; as a result, the minimal and maximal information that should be returned to participants may differ. Another principle stipulated was that resources for research should be directed primarily at scientific discovery; thus, researchers do not have a duty to look for actionable genomic findings beyond those uncovered in the normal process of their investigations.
______________
11 This does not pertain to general results summaries that may be created for trial participants and other interested stakeholders.
The consortium agreed that analytically and clinically valid information of an important and actionable medical nature that is identified as part of the research process should be offered to research subjects, but that participants should have the right to refuse any results that may be offered. However, parents’ right to refuse results of their children should be trumped when the result is deemed medically important enough during childhood that it would be in the best interest of the child to act on it at that time. It should also be clear at enrollment what kinds of results will be returned to participants, the consortium concluded. When studies do not allow participants to opt out of potentially receiving results, this should be clearly addressed in the consent form, which should also clarify when participants may be contacted in the future.
Bradbury presented both sides of the debate as to whether to return to individual patients the research results of large studies using archived DNA. The reasons she cited for not returning research results were to
- Maintain the distinction between clinical care and research. Research seeks to increase generalizable knowledge while clinical care focuses on advancing the care of individual patients.
- Avoid overinterpretation and misunderstandings or inaccuracies on the clinical significance of the findings. If the genetic analysis is done in research laboratories that are not certified under the Clinical Laboratory Improvement Amendments (CLIA),12 the reliability of the findings may be less certain.
- Maintain health information privacy and a patient’s right to not know.
Return of research results could also incur prohibitive costs on research teams, biobanks, and the health care system in general, as well as potentially have negative impacts on research progress, Bradbury said. “We need to be very cognizant of the impact of returning results on research. If we are going to do it at all, we need to be very narrow about it, talk about it at the front end, and be very cautious about how we proceed,” Clayton said.
______________
12 Passed by Congress in 1988, CLIA established quality standards for all non-research laboratory testing performed on specimens derived from humans for the purpose of providing information for the diagnosis, prevention, and/or treatment of disease, or impairment of or assessment of health. CLIA established these standards to ensure the accuracy, reliability, and timeliness of patient test results regardless of where the test is performed.
Bradbury cited the following reasons to return research results:
- Increase the clinical benefits to patients, especially findings that would change their clinical management.
- Show respect and autonomy for the patients’ efforts to give their time and biospecimens to the research endeavor and to reciprocate those efforts.
- Recognize that research and clinical care are often intertwined to the benefit of both, especially in the cancer arena. Bradbury noted that “Oncology providers are often thinking about research as part of that armamentarium that we discuss with our patients and patients are often coming in looking for trials.”
Clinically Actionable Findings
According to Bradbury, currently most researchers and ethicists find it acceptable to not return research findings to individual patients unless there appears to be a consensus that select results are clearly actionable clinically, could change the course of clinical care for a patient, and have been confirmed in a CLIA-certified laboratory. But there is no nationwide consensus among genetic experts and organizations on what those clinically actionable genetic findings are, several speakers pointed out.
Bradbury noted that a number of genetic tests are entering the clinical arena without prior studies showing their clinical usefulness, so defining actionable genetic findings as those coming from commercial tests may not be appropriate. In addition, some findings may not currently alter clinical care, but that does not preclude them from being actionable in the future. Bradbury pointed out there is some research to suggest that participants want to see the results of their clinical tests, even when no clinical interventions can be taken at the time. However, little is known about the outcomes of returning individual results to participants. Often, researchers are unable to return results to more than half of participants due to a lack of current contact information, Bradbury said.
However, evidence does show that clinically actionable findings are relatively rare at present, according to Jarvik and Clayton. “It is actually quite rare to stumble across something that is not the primary goal of your research,” Jarvik said. In her extensive review, these actionable findings comprised only about 2 to 3 percent of all genetic findings (Dorschner et al., 2013). But she added, “Even though it is not something that happens
commonly,… I would much rather know if the person wants to know something.”
Clayton said that doing genetic tests and maintaining databases may increase liability if inaccurate results are returned to participants, but that risk is still low. She found that even in the clinical setting, few courts have held that physicians can be liable for failing to identify or act on incidental findings. For such liability, legal counselors have to show a breach in the standard of care, but in many cases it was decided that a failure to identify or disclose a genetic finding did not breach standard of care, Clayton reported (Clayton et al., 2013). “Anyone can walk into a courthouse, but the basis for liability in the context of litigation for failing to return individual research results is really probably pretty low, although that may change,” she said.
Bradbury, Clayton, and Jarvik noted there appears to be general agreement in the field that consent forms should address the return of individual results to participants who are currently enrolling in research studies, and that researchers have no duty to hunt for actionable genetic findings beyond the aims of their research. This perspective was advanced in a recent report, Anticipate and Communicate: Ethical Management of Incidental and Secondary Findings in the Clinical, Research, and Direct-to-Consumer Context, by the Presidential Commission for the Study of Bioethical Issues (2013), as well as by the consensus statement jointly authored by eMERGE and the CSER Return of Results committees. Both of these specifically reject expanding the clinical recommendations from the American College of Medical Genetics and Genomics (Green et al., 2013) to the research setting, Clayton noted. Bradbury added, “There are still a lot of questions and challenges to think through” in this regard.
Context may be important in deciding when to return individual research results, Bradbury noted. “If the research participant is engaged with a clinical team, is that a context where there is an obligation to be returning the results, as opposed to a research participant who participated in a registry and maybe has no ongoing contact?” she asked. As Clayton noted, most people would agree there is a duty for an investigator to return research results to their current patients, but it is “questionable whether a downstream investigator has the same level of duty to a patient participant as the treating clinician does.” But it can be hard to draw the line between clinical care and research, Jarvik noted. “We find that even though we try
very hard to do this and tell participants ‘this is your research visit, this is your clinical visit, this is your clinical doctor, this is your research team,’ that they are calling the researchers for their clinical questions because they don’t really understand the distinction, and they seem to know us better because they have seen more of us.”
Another ethical conundrum is whether to return genetic findings in children that indicate susceptibility to diseases with onset in adulthood. Jarvik noted that some experts believe an adult-onset finding should never be returned in a pediatric case, but there is no consensus on this issue. The eMERGE and CSER consortium decided that adult-onset findings that were not previously known in the family could be offered to the families of pediatric patients, she reported.
Bradbury raised the question about who should pay for the confirmatory testing and counseling linked to the return of incidental findings. This question was echoed by Mendelsohn. He noted that in many cases, the genetic information will be relevant to a disorder that does not fall under the domain or expertise of the physician managing the patient. “Who should be relaying this complex information to patients?” he asked. Clayton responded, “If you are the one who returns the information and you don’t know how to interpret it, then you have to refer to someone who can. That duty to refer is absolutely implicit in the physician–patient relationship.” Clayton added that genetic counselors have the expertise to interpret genetic findings, but such counselors are in short supply.
Another conferee pointed out that even if the treating physician may know how to interpret the genetic data, they may be poorly trained in how to manage an issue that could affect other family members. Jarvik concurred, noting that “the education of general physicians is woefully inadequate” with regard to interpreting genetic findings. “We are putting whole genome reports in the medical record and lots of physicians are looking at these who don’t really know what it all means. They need some support for how to interpret it,” Jarvik said. As for who should pay for such genetic consults or additional tests, she pointed out that insurance companies sometimes are willing to cover the costs, especially for findings on a mutation that is highly likely to cause disease.
Biospecimens from Deceased Participants
Medical information that can be captured from stored biospecimens and related health data from research participants can be useful for a long time, even after those patients have died. For example, studies to identify biomarkers that indicate which cancers are the most aggressive and thus need more aggressive therapy can be done effectively using the biospecimens and health records of research participants many years after they have died. As Ganz noted, “Clearly these deceased individuals have nothing to lose. They have already lost their lives and these annotated biospecimens would have important valuable data that could be retrieved from them.” However, Bradbury asked, “If these deceased participants had requested the return of research results in their consent forms, should the results be returned to the next of kin?”
The HIPAA Privacy Rule is silent on whether authorization is required from the personal representative or next of kin of deceased individuals to conduct research using their personal health information. Some organizations interpret the Privacy Rule to require researchers to obtain authorization from next of kin or a waiver of authorization from an IRB or Privacy Board to access the personal health information of those research participants who have died (Ness, 2007). State laws may also affect the use of biospecimens in research from deceased participants. Jarvik noted that deceased people are considered human subjects of research by Washington state law.
Peppercorn said when deciding the appropriateness of using such biospecimens in research, one has to consider how broad the consent form is that the participants signed. “If they consented to broad future use, then it is fine to use them, but if you just happened to have their biospecimens archived and had limited informed consent, even though the patient is deceased, I think my gut response is that it would be a large violation of the trust they placed in us to use them,” he said.
THE CHANGING CONTEXT OF RESEARCH AND CARE
Ethical and oversight issues in research depend on context, especially whether applied to a traditional system in which research is clearly delineated from routine patient care, or to a “learning health care system,” in which the transition from patient care to research is seamless. Consequently, when discussing appropriate protections for research participants, there was
a debate about whether to keep research separate from patient care, or to integrate it with practice as in an LHCS.
Faden noted that ethical abuses of some patients participating in research in the 1960s and 1970s led to the current regulations aimed at separating research from clinical practice, which she called the separation model. “The separation model was and continues to be terribly important for protecting people from potentially harmful and abusive research and for more broad violations of human rights,” she said.
But Faden added that a major downside to this model that has emerged over the decades is that it overprotects people from low-risk research activities. The separation model also underprotects patients, according to Faden, because it is not conducive to addressing the more common and significant harms to patients, which are due to medical errors and patients receiving inappropriate care due to lack of evidence about what the right care is for specific patient groups. “The current regulatory structure makes it more difficult than ethically it should be to generate the evidence to find the solutions that will allow us to address medical errors and inappropriate care,” Faden said.
To counter these shortcomings of the separation model of care, Faden suggested adopting the integration model of research and practice, which is, essentially, a learning health care system. In such a system, “knowledge is so embedded into the core of the practice of medicine that it is a natural outgrowth and product of the health care delivery process and leads to continual improvement in care,” as described in a previous IOM workshop summary (IOM, 2007).
One feature of an LHCS is that knowledge is generated from every clinical encounter, so every opportunity for patient care also becomes an opportunity for systematic learning (IOM, 2007, 2010). This learning is then translated into improved care in the same settings in which the knowledge was generated. “The two twin commitments of a learning health care system are delivering care and learning systematically and rigorously from that care,” Faden stressed.
The landscape of learning that can occur in a health care system is large and varied, Faden, Joffe, and Menikoff all noted. This learning derives from clinical trials of previously untested treatments; studies of surgical, procedural, or other innovations in treatments currently given; quality improvement studies; and comparative effectiveness studies. Another category of study that is increasingly being used is called the pragmatic trial, which is aimed at discovering whether an intervention is effective under real-world
conditions. Joffe noted that all these activities overlap with each other and occur against a background of clinical care. “Although it looks like these activities are sharply delimited from ordinary clinical care, in the real world, we all know these boundaries are blurred,” he said (see Figure 3). He added that many of the learning activities involve low to no incremental risk over and above the risks that participants would be exposed to in the course of their ordinary clinical care.
Faden pointed out that the main two types of knowledge generated by a learning health care system are focused on improving patient safety; quality improvement, and determining which existing clinical options work best for patients (that is, comparative effectiveness research). The latter has been defined as “the generation and synthesis of evidence that compares the benefits and harms of alternative methods to prevent, diagnose, treat, and monitor a clinical condition or to improve the delivery of care” (IOM, 2009b, p. 29). Most comparative effectiveness research compares existing approved and widely used low-risk interventions in routine care to assess which are safer, more effective, or more cost-effective.
FIGURE 3 Landscape of learning activities. All these activities overlap with each other and occur against a background of clinical care. Many of the learning activities involve low to no incremental risk over and above the risks that participants would be exposed to in the course of their ordinary clinical care.
SOURCE: Joffe Presentation on February 25, 2014.
Comparative effectiveness trials can be observational or randomized. If they are observational, “no aspect of clinical care or experience that a patient has is altered or changed by the research. The physician and the other clinicians interact with the patient and make the decisions they would otherwise already make. The research questions are answered using data that are routinely collected for general clinical purposes, sometimes augmented by additional data as well,” Faden said. In contrast, with randomized comparative effectiveness studies, some element of the patient’s care is determined by a protocol.
No fully operational LHCS currently exists, Faden said. “I tend to think of learning health care systems a little bit like human rights—we have progressive realization toward the goal of a learning health care system. We’re on our way there but we’re not there yet,” she said. She added that “the learning health care system confronts the fact that the way in which we attempt to generate new knowledge now, while incredibly important and successful at giving us all kinds of wonderful new treatments and advances in health care, has, in many respects, failed us in not being able to give us as much information as quickly as we need it in order to make really good decisions in everyday experience of being a patient or a doctor.”
But Faden added, “It is not known yet whether a fully realized learning health care system can actually be implemented in an ethically acceptable way and will realize the benefits it’s intended to realize.” Menikoff noted that learning health care systems are not likely to eliminate ethical issues tied to research, including when informed consents are necessary. “Lines will have to be drawn and these difficult issues will not necessarily go away with a learning health care system,” he said.
Oversight in a Learning Health Care System
A few speakers discussed appropriate oversight in an LHCS, including transparency and disclosure, and data monitoring. Faden suggested that in a learning health care system, patients should be actively engaged in setting comparative effectiveness and other research priorities and in the ethics and oversight of the studies that are conducted, including disclosure and consent practices. She also suggested that an LHCS should be completely transparent to the patient community, and use multiple modalities so patients can find out what studies are being done, what types of patients are affected by those studies, and the effects of those studies on medical care. Accountability to the patient community it serves is a key feature of an LHCS, Faden
stressed. “It has to be able to explain to the patients what has been learned from the research conducted in the system and whether anything changed in that setting due to the learning that took place, and if nothing changed due to that learning activity, why not?”
Ganz added, “Many patients want their experience to be captured, if not for themselves, then for other people who might benefit.” Ganz pointed out that patient-centered care depends in part on a learning health care system. She noted that “when you do not have a standard of care that is effective or there may be something more promising, you could see how you could just transparently move into a clinical trial as part of patient engagement. To build a better quality cancer care delivery system, these clinical trials need to be part of the seamless care that we deliver,” she said. Collyar added that it would be optimal to have every patient in a health care system sign up for a long-term outcomes registry that would also inform them of potential research in which they could participate.
Chris Daugherty, professor of medicine, and chair, Biological Sciences Division, Institutional Review Board, University of Chicago, raised the question of how effective disclosure would be in an LHCS and whether participants would fully understand how their clinical care and data would be affected by the learning activities that occur. Faden responded that in any health care system, including an LHCS, patients should be informed about what studies are being done and be given the opportunity to opt in or out of them. But the LHCS could address ethical disclosure failures in many more traditional medical centers. “If I want to know today what research is going on at Johns Hopkins Hospital as a patient, it’s not so easy to find out and that is something that needs to be fixed,” she said.
Leiter noted that patients are increasingly sharing their health information themselves, and such patient engagement might substantially lower the inherent risk and potential for violation of patient expectations. She suggested less reliance on informed consent for uses of data in a learning health care system that would be considered “routine,” and that we should instead pursue other models of patient engagement to enhance protections for research participants. Such models include patient input into the research done, greater transparency with regard to research uses of data, and requirements to share research results with patients.
Several speakers noted current regulatory roadblocks to an LHCS. Leiter said many health care systems want to conduct and publish the results of their quality improvement activities or other low-risk research conducted in their care settings. However, because the HIPAA regulations are vague,
they are not sure that their research will fall under the “health care operations” category and be exempt from IRB review. “It tends to lead to a ‘better safe than sorry’ approach,” in which the work is subjected to IRB review, and “jumping through these hoops can be unbelievably time consuming and expensive,” she said.
Menikoff added that there are comparative effectiveness studies that are probably not particularly risky, and these could meet the criteria for being eligible for IRB waivers of patient consent under the current regulatory system. “But the difficulty is figuring out which ones those trials are,” he said. He added that even a study that compares two different standards of care that are already in practice “does not necessarily turn it into a minimal risk trial.” Taylor noted that even defining standard of care can be tricky.
Ellenberg gave several examples of how ethical considerations vary according to the context of the research and its endpoints. She noted if a hospital doesn’t need to get IRB review when deciding to switch brands of antibacterial soap, why should they need IRB review if they want to do a study comparing two brands of antibacterial soap, or two types of dispensers? Hospitals often arbitrarily adopt a new diagnostic assay as a routine procedure or change the duration of a routine procedure, such as dialysis, without any oversight, but when they wish to study the effectiveness of those measures, should their studies undergo IRB review? There is still great debate on answers to those questions, Ellenberg noted.
Ellenberg explained the distinction between explanatory and pragmatic trials. She said the purpose of explanatory trials is to answer a scientific question very precisely by controlling heterogeneity; the aim is to isolate a treatment effect by making everything as homogeneous as possible. Explanatory trials are used in the regulatory setting to evaluate new drugs and other kinds of new medical treatments. She said the purpose of a pragmatic trial is to answer a practical question about routine clinical practice.
Oversight of pragmatic trials includes not only ensuring protections for study participants, Ellenberg said, but also ensuring adequate data quality and monitoring of study conduct. “If a study is not conducted well, it might produce results that are wrong that people believe are right, so there are ethical considerations there,” she said. She suggested most data quality can be monitored centrally, with onsite visits when central review indicates potential quality problems. Ellenberg noted that data entry errors
in pragmatic trials should be minimal and random and thus not likely to create systematic bias.
Randomized controlled trials with significant implications for patient outcomes or with anticipated safety issues typically are overseen by a data monitoring committee composed of outside experts not involved in the trial or without a stake in the trial’s outcome. Such monitoring may or may not be needed for pragmatic trials, according to Ellenberg, but if it is done, the same principles and practices of data monitoring would be applicable to pragmatic trials. She speculated that “most pragmatic trials will require this monitoring because they are going to be addressing public health questions and will probably be fairly large.”
Because pragmatic trials are likely to be conducted on a more heterogeneous population than explanatory trials, researchers need to be more cautious about interpreting a “no difference” finding in their pragmatic comparative effectiveness studies. Such a finding could be due to outcomes that were too variable to detect an effect, according to Ellenberg. More stringent criteria for stopping a study early in pragmatic trials may also be needed, she added, because practitioners may be comfortable with their long-held standard practices and will be reluctant to adopt a new approach unless the evidence is especially strong. In addition, futility analyses that indicate there will not be any difference shown in a study will probably not be relevant for pragmatic trials because if the two treatments are nearly the same, more data points are needed to narrow the confidence interval around the possible difference, Ellenberg said.
Researchers are increasingly using cluster randomized trial (CRT) designs for their pragmatic studies. In a CRT, instead of randomizing individual participants to a particular treatment, clusters of patients undergo randomization. For example, researchers may randomize hospitals, clinics, or hospital units to different approaches to infection control. For these trials, statistical power depends more on the number of clusters or units than on the number of individuals per cluster. CRTs pose many challenges, including finding enough researchers experienced in how to conduct or monitor them. In addition, because more participants are required for a CRT than for a standard randomized trial, potentially more participants will be exposed to an inferior intervention, Ellenberg noted. Keeping investigators blinded to interim results is also difficult when everyone at the site receives the same treatment. This may raise concerns about interim changes in study conduct. Another special concern with pragmatic CRTs is that it is hard to assess if enough sites are entered into the study to give a statistically
significant result because the number needed can vary depending on the heterogeneity of sites.
MULTISITE STUDIES AND IRB REVIEW
Cancer clinical trials are increasingly multisite and international in scope, which may necessitate different oversight mechanisms in order to conduct these trials effectively and efficiently. Several deficiencies in oversight of multisite studies were pointed out by speakers at the workshop. These deficiencies included clinical trial launch delays and inefficiencies due to multiple IRB reviews, conflicting IRB reviews and no standards for a high-quality IRB review, increased complexity and length of consent forms reviewed by multiple IRBs, and the impracticality of IRB reviews of clinical studies on rare diseases.
Schilsky noted that multiple IRB reviews can decrease the efficiency of study launch and leads to a redundancy of review even though few changes will ultimately be permitted in the protocol. “When the study sponsor puts a protocol and consent form out there to 200 to 800 sites, you get a lot of questions that you have an obligation to respond to. Then you need to track what the variation is across all the consent forms and know exactly what each participant in the study consented to, and biospecimens cannot be distributed for any research purpose unless you’re sure that it’s been appropriately consented for that specific research purpose. It becomes incumbent on the sponsor to have mechanisms in place to track all those things,” Schilsky said.
David Parda, chair of the Department of Radiation Oncology at West Penn Allegheny Health System, noted an article in which the authors estimated the cost of regulations per life-year gained through phase I cancer clinical trials as $2.7 million.13 This article also asserted that regulatory delays in development of effective therapies result in tens to hundreds of thousands of life-years lost, but save very few lives in return. The authors concluded that this imbalance between potential life-years lost versus saved
______________
13 To calculate the cost per life-year saved by the regulations, the authors estimated that 30 percent of the per-patient costs of clinical trials (or approximately $8,000 per patient) are due to the increasingly complex regulatory environment, estimated that the toxic death rate in phase I cancer clinical trials decreased by 0.3 percent from 1979 to 2002 and that all of that decrease in toxic deaths is due to enhanced safety engendered by more stringent regulations, and assumed that patients with cancer on phase I trials have a life expectancy of 12 months.
renders the regulatory burden potentially unethical (Stewart et al., 2010). On a more personal note, Parda recounted that one of the members of his institution’s local IRB developed kidney cancer. At the time there were significant IRB-induced delays in starting a clinical trial of an experimental drug for kidney cancer, and the ailing IRB member commented at a meeting, “By the time you guys activate this trial, I’m going to be dead.”
Multiple IRB reviews can delay a trial’s onset, which is especially problematic for time-sensitive studies. Barbara Bierer, senior vice president of research, Brigham and Women’s Hospital, and program director, Harvard Catalyst Regulatory Knowledge and Support Program, described such delays as “hours of ping ponging between the IRB and the investigator.” Schilsky added that these delays are tied to a failure to complete enrollment, which he said “is a huge waste of resources.” Furthermore, if a clinical trial is not completed and published, participants are exposed to risk without the potential for benefit to be fully realized. He noted that there is almost a linear relationship between time to the first patient enrolled and the probability of success in completing accrual (Cheng and Dilts, 2011), “largely because the science moves on while everybody’s waiting to get the study enrolled,” Schilsky said. Daugherty countered that studies indicate that IRB reviews are not the only reason for delays in study onset, nor are they necessarily the “rate-limiting step” for launching a clinical trial. Schilsky agreed, but stressed that IRB review can be a significant contributor to delays, which could be substantially reduced through greater use of central IRBs (CIRBs, see section on “Central IRBs”).
Another frequent problem with multiple IRB reviews is their inconsistency. “If you have three different sites and one IRB is very conservative and one IRB less conservative, it can grind things very quickly to a halt,” said Leiter. “On the other hand, the institutions feel very strongly that their IRBs are a representation of their values and of their patient values and it’s hard to say any one of them is wrong,” she added. Schilsky noted that the changes local IRBs make to consent forms can introduce errors or confusion, and substantive problems pointed out by a local IRB with the consent form might not be communicated broadly to all the sites participating in the study. Often the IRBs are not aware of the experiences or concerns raised at other sites participating in the study. In addition, “if you give a group of people a job they are going to feel obligated to justify their time and effort by doing something, so local IRBs tend to make lots of unnecessary changes,” Schilsky said. Currently, there are no standard metrics to define what a high-quality IRB review is, “so it’s really difficult to know, looking
across multiple sites, whether an IRB is doing a good job,” Schilsky added. The time, effort, and cost of multiple IRB reviews are substantial, he added.
From a patient perspective, the variable quality and extent of local IRB reviews can be unethical, Collyar pointed out. Multiple IRB reviews typically make the consent form longer and more complex, Schilsky added, which might increase risk to patients. The highly vetted NCI model consent form is almost never adopted wholesale by local institutions, he said. Schilsky cited one review article on IRB review of multisite trials that found there was an average of 5.2 changes made in the protocol or consent form per site, most of which were to the latter (Ravina et al., 2010). Another study found 16 of 18 IRBs in a multisite study made changes to the informed consent form (Stark et al., 2010). A third study of 25 sites participating in a multisite trial found a median of 46.5 changes made to the consent forms, of which 82 percent changed the wording without changing the meaning of the informed consent document (Burman et al., 2003). Another study of 16 sites participating in a multisite trial found that the consent forms in only 3 of these sites contained all of the required elements, and the reading levels ranged from grades 8 to 13 (Silverman et al., 2001).
“It’s not clear that local IRB review is actually improving the transmission of information to the research participants,” Schilsky stressed and added, “Sometimes we would find that certain required elements of the consent form were dropped out by the local IRB, which gave us considerable concern.” He added that patients might miss an opportunity to participate in a study if their particular hospital or clinical site declines to participate due to local IRB concerns.
Multiple IRB reviews also diffuse responsibility and potentially lead to more superficial review because no IRB feels like they have true responsibility for the research and are empowered to change the protocol, according to one study (Menikoff, 2010). “If there are substantive problems with the protocol that are identified by an IRB, they may not be communicated because the site just chooses not to participate in the study instead of raising their concerns with the study sponsor,” Schilsky said. Bierer added, “There is enormous administrative confusion. The IRB offices don’t know who they should contact for what and whether it’s their responsibility to inform the sponsor or the regulatory bodies. There’s also increased administrative burden in IRB offices. We have one person fully assigned to simply track which IRBs are going where.”
Multiple local IRB reviews at sites for the occasional patients who qualify for genetic studies is also impractical, Schilsky pointed out. “And
does every local IRB have the expertise necessary to review these increasingly complex biomarker-driven studies?” he asked. This is especially problematic in oncology with the growing recognition that each cancer has multiple genetic subtypes. “Every cancer that we deal with increasingly is becoming a group of rare cancers,” Schilsky said. “If you want to do a clinical trial in which you match a drug treatment to a particular genotype in the tumor, you may have to screen 10,000 or more patients for the one or two patients at each site who might be eligible to participate in the trial. This is not feasible and it’s fraught with peril.”
Instead, he suggested enrolling patients in a screening protocol. The informed consent for this protocol would allow investigators to molecularly profile a patient’s cancer and follow their outcomes, but would not specify any interventions. This would provide a pool of profiled patients who then could be offered participation in specific intervention trials. Only the patients who have the particular genetic biomarker of interest would be offered participation in an interventional trial. This is in contrast to the traditional model of offering patients at specific institutions whatever trials they currently have in their portfolio for which they are qualified to participate. “We need to figure out how to deliver the trial to the patient, not the patient to the trial, because these patients are going to be rare as hen’s teeth and scattered all over the globe,” Schilsky said. With this approach, individual IRB reviews would be impractical and costly for the small number of trial participants expected at each site, Schilsky noted, saying, “We have to be able to get the trial with an associated IRB approval to wherever the patient is. This will require more central IRB oversight.”
He concluded his presentation by stating, “It seems to me that increasingly little is gained from individual site review in multisite studies, and there is potential for multisite reviews to actually increase risk and diminish the quality of information conveyed to research participants. Recruitment in the genomic era is only practical with use of CIRBs. We need a regulatory framework that will encourage site acceptance of CIRB review as well as additional research on the appropriate metrics to define what a high-quality IRB review is.”
Parda agreed with many of the disadvantages of local IRB reviews cited by others at the workshop and added a few additional ones. He noted that local IRB reviewers tend to have limited reviewer expertise, and it can be a challenge to recruit and retain members and fund their work. Consequently many local IRBs are overwhelmed with the volume of review requests, which can cause delays in reviewing protocols. On the other hand, Parda
also listed the advantages of having local IRBs review study protocols, including their knowledge and responsiveness to local investigators, culture, and values, and their ability to address state and local laws.
In 2001, NCI instituted two central IRBs, one for adult cancers and one for pediatric cancers, which can be used for all clinical trials supported by NCI. The NCI CIRBs were formed to help reduce the administrative burden on local IRBs and investigators, while promoting a high level of protection for research participants. One study found that use of an NCI CIRB was linked to faster reviews, less staff effort, and reduced cost, Schilsky reported (Wagner et al., 2010). Parda pointed out that the average number of days it took for his multisite health care system to approve an NCI-sponsored Phase III clinical trial was 116 days in 2006. After the adoption of the NCI CIRB, that dropped to 15 days in 2010.
Schilsky noted increasing and widespread adoption of the NCI CIRBs in cancer clinical trials. That increase is likely due to several advantages that CIRBs offer. In his presentation, Parda listed these advantages, including more disease-specific expertise; greater efficiency, which reduces time delays; reduced variation on consent forms; increased safety due to expedient reviews of adverse events; and greater cost-effectiveness. The disadvantages he cited for CIRBs included the perception that their highest priority is activating the trial, rather than protecting research participants. There also are concerns that a CIRB cannot review protocols for local content, laws, and compliance as effectively as local IRBs, and that there might be increased potential for institutional liability through loss of control of the IRB review.
Parda noted that many institutions will not rely on “outside” review, whether for lack of familiarity, liability concerns, or preference for control. This results in a significant increase in workload and resource commitment without a clear benefit, as well as delays in approval and initiation of clinical trials, Parda pointed out. In addition, multiple local IRBs can lower the scientific integrity and introduce risk in a multisite trial that often requires the same protocol and consent form to be used at each institution to reduce bias. If a significant concern is raised by a local IRB, often the only option is to not participate rather than addressing that concern through all IRBs for the study, Parda said, echoing Schilsky and Bierer.
Since 2013, NCI’s CIRBs have operated on an independent model for review of NCI-sponsored studies, rather than doing facilitated reviews.14 In the independent model, the CIRB is the sole IRB of Record responsible for study review as well as review of local context considerations for enrolled institutions. The CIRB also reviews locally developed recruitment or educational materials, locally occurring unanticipated problems, or serious or continuing non-compliance, and responds to investigator or institution questions. The institution is still responsible for monitoring conduct of research and reporting any concerns to the CIRB, Parda reported. Bierer stressed that “Local oversight will still be needed to ensure quality care in quality human subjects research.”
The NCI CIRBs have oncology-specific, multidisciplinary members with wide representation from surgical, medical, and radiation oncology; pharmacy; biostatistics; and patient advocates. “That is very hard to reproduce in every local environment,” Parda noted. “This improves the quality, experience, and cost for patients and health care professionals, and improves professionalism and a scholarly approach to medical practice that most physicians embrace.” He added that the CIRB eliminates the back- and-forth between investigator and local IRBs to gain study approval, and also eliminates frequent submissions to IRBs for amendments, continuing reviews, adverse events, etc. With NCI’s CIRBs, it is also no longer necessary to complete multiple IRB application and submission packets, and unlike local IRBs, the CIRB has some leverage to make a substantive change to the protocol, if necessary.
But Parda added that there are still challenges involved in using NCI’s CIRBs, including balancing local and central leadership and cultures. However, he also noted that NCI’s CIRBs enable more clinical trial participation in community settings where expertise, time, and money to run IRBs are limited. Another challenge with using the NCI CIRBs is compatibility of the computerized systems used for information transfer, but the recent development of a Web-based system should help with that. The CIRBs require full-time operational and regulatory experts and support.
______________
14 Facilitated review relies on IRB Authorization Agreements. These are written agreements between two institutions that allow one institution to act as the “IRB of Record” for another institution (the “Named IRB”). The bulk of the human subject protection considerations are made by the IRB of Record, with secondary review being done by the Named IRB using the information obtained from the IRB of Record.
New England Reliance Agreement
Other institutions have also set up central IRBs to review multisite studies, including a group of more than 20 institutions in the Boston area that use the New England Reliance Agreement,15 which was described by Bierer in her presentation. The key component of this system is a master common reliance agreement that creates a framework for the reviewing IRB and a relying institution. Once the institution relies on (cedes IRB review to) another, then that IRB is the IRB of record. Ability to accept or reject IRB review reliance is on a case-by-case basis. That decision is made by the site’s primary investigators before the protocol is written in final form. Duplicative IRB reviews are eliminated entirely once the reliance agreement is accepted.
This system, which is flexible and scalable, promotes collaboration and reduces administrative burden and the costs for the IRBs and study teams, Bierer noted. From its launch in 2009 to 2013, about 90 percent of institutions opted to cede IRB review to another institution, with the reviewing IRB accepting review for at least one additional site. One-quarter of reliance applications involve three sites using the same IRB for review, and eight has been the maximum number that can apply to the same IRB for review of a study. To avoid “IRB shopping,” the default is that the primary investigator institution is the IRB of record unless this institution chooses to rely on another because of the expertise of the other IRB or other issues.
Certain responsibilities remain with the relying institution, including ensuring compliance with state laws, assessing investigator and team competency and education, and conducting an initial conflict of interest review and sending that report for the reviewing IRB to improve, agree, or disagree.
Relying institutions need to also make sure administrative functions are aligned with those of the reviewing IRB, including the grants and contracts office, and to oversee non-compliance reporting. “We’ve made a number of educational and service tools available on the Web to help with this so that we can really start to reduce the duplicative effort at training and moving this all to a better quality platform,” Bierer said.
A key to making such collaborative IRB review systems work is defining the roles and responsibilities of the reviewing IRB and the relying institution and setting up other ways to address additional roles as needed, according to Bierer.
______________
15 Formerly the Harvard Catalyst Reliance Agreement.
Building trust between institutions is also essential. “We all trust ourselves, and we don’t trust the IRB next door to us,” she noted. “The IRBs need to get familiar with each other. Initially when we started this, no one wanted to rely on another institution for IRB review and wanted everything based on a person-to-person conversation they could have with someone at their own institution. But a year after we set up the system, they got comfortable with it and said, ‘Why do we have to make the telephone call—can’t we just automate it?’” Bierer said. She added that established channels of communication are needed among institutions to clarify who should be contacted when issues arise. She also recommended having a template for informed consent language on which all the IRBs in the system agree.
Bierer suggested providing some incentives for increasing single-site IRB review for multisite research. “There’s an advantage to having many different central IRBs,” she said. She also suggested standardizing the operation of central IRBs “because we’ve got too many models out there and ways of doing business and we need some clarity of roles and responsibilities.”
But Daugherty noted that although the New England Alliance Agreement is a good model, “90 percent of institutions are not going to be able to use it,” he said, because they are small providers of community care and “do not have the resources to do things well or do them perhaps at all compared to more traditional academic institutions.”
Daugherty voiced strong opposition to relying on central IRBs. In his presentation, he asserted that the loss of local IRB attention to multisite trials would be a loss of a valuable local resource, as most IRBs see themselves as a service organization providing valuable guidance and advice to clinical researchers. In a CIRB system, this resource will be lost, he said. “There will be no in-house support for your unanticipated problems—you will have to contact the central IRB to figure out whether that death on the study needs to be reported, but sometimes people like someone local to talk to,” he said.
Daugherty also pointed out that when CIRB reviews are done, local institutions will still need to support protocol and consent tracking, and monitoring of conflict of interest. With a CIRB review, the cost of local oversight will go down, but it isn’t going to go down to zero, he said. There will still be a significant amount of administrative burden that will have to be borne by the local institution. Daugherty also noted that the NCI’s
CIRB is outsourced to the Emmes Corporation. “The central IRB members are paid and at the local level this has been contrary to longstanding local IRB models,” he said, adding that there might be conflict of interest issues that need to be addressed with NCI’s CIRB.
Daugherty countered the statements other speakers made about IRB reviews causing undue delays in trial launchings. He noted that IRB review time generally only comprises 2 to 3 percent of the time between the development of a trial concept and enrollment of the first participant, and there are other steps necessary to trial launch that occur during that time span, such as assigning and training study nurses.
He added that some of the delays attributed to IRB review might be due to the time it takes for a primary investigator and cancer center administrator to respond to IRB review comments, which can take as long as a month, he found at the University of Chicago, where he chairs the local IRB. “The complaint is that local IRB review results in too much time consumed, but are there equal and more meaningful times to consider?” he asked.
He suggested that undue time is often spent between initial protocol development and submission of the protocol to the local IRB, or the time between IRB approval and enrollment of the first participant. “We see protocols come in and their dates are calendar years that precede that of the submission date,” he said. He also stressed that the only time a CIRB can save is time spent on the initial IRB review. Botkin added, “What’s the marginal improvement in getting the IRB centralized when the rest of the reviewing committees to date have not been centralized and perhaps can’t be? In our institution the IRB is not the rate-limiting step.”
Daugherty suggested some CIRB review times are too short. He described an anecdotal experience in which he said the NCI CIRB spent less than 5 minutes discussing an annual review of two large Cooperative Group trials, encompassing more than 2,000 subjects, and thousands of adverse event and toxicity reports from dozens of local institutions. He noted that in the absence of local IRB review, there would be no other review of such ongoing research. “At best, the central IRB continuing review process is a view from 30,000 feet. It may be adequate, but this relative lack of more granular review needs to be acknowledged. There is no intuitive reason to believe that human subjects risks would lessen or protections increase in this context,” he said. “Time alone cannot be used as a measure of the quality of IRB review.” Daugherty supported his position with a 1969 quote from Han Jonas: “A slower progress in the conquest of disease would not threaten
society, grievous as it is to those who have to deplore that their particular disease be not yet conquered, but that society would indeed be threatened by the erosion of those moral values whose loss, possibly caused by the ruthless pursuit of scientific progress, would make its most dazzling triumphs not worth having.”
However, Monica Bertagnolli, chief, Division of Surgical Oncology, Brigham and Women’s Hospital, professor of surgery, Harvard Medical School, countered that all large cancer clinical trials are subject to extensive oversight by NCI’s Data Safety Monitoring Board, which provides a thorough, detailed review of trials in addition to the IRB reviews. Daugherty conceded that this board and NCI’s CIRB would be able to act more quickly than multiple local IRBs to terminate experimental therapies shown to be too risky, but he suggested it would be beneficial to have local IRBs shape the communications to patients about why these trials or certain arms of the trials were terminated.
Steven Piantadosi, Phase One Foundation chair and director, Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, asked if a downside to using a CIRB would be that it would cause atrophy in the expertise and ability of local investigators in state-of-the-art research. Schilsky responded that institutions will still conduct scientific reviews of every clinical trial they offer, even if it undergoes CIRB review, so faculty members at those institutions will have the opportunity to serve on such review committees. Also, the adoption of the NCI CIRB for certain kinds of research studies does not require disbanding of the local IRB. It just requires that the local IRB focus on other types of research, he said.
Parda added that sometimes local IRBs inappropriately do both a scientific and regulatory review of studies and “in some ways, outsourcing the IRBs gives more time and opportunity for more rigorous scientific review locally.” Bierer agreed with both Schilsky and Parda and added that “it will give the local IRB more time to think about phase 1 and 2A and investigator-initiated trials.” But Piantadosi stressed, “I’ve seen thousands of trials in my career, and it was most valuable to see their methodology and become acquainted with their questions, even though I wouldn’t be one of those investigators learning in the Cooperative Group setting you outlined.” But Joffe countered that the CIRB can fulfill an educational and modeling function because it has not only scientific and clinical expertise, but also ethics and methodological expertise. “The central IRB can both bring in people from the broader community who can learn and bring their experience back to their home institutions. These central IRBs can become
models for improving IRB practice both with respect to central review, but also with respect to local review and oversight,” he said.
Several speakers noted educational deficits regarding protections for research participants, for both practitioners and the public. “Physicians tell me they have little training in how to present consent forms to their patients,” Albrecht said. Paasche-Orlow suggested doing a national survey of the training, supervision, and documentation approaches that are already being used in the consent process and using best practices, especially those rooted in the “teach-to-goal” approach to develop a model training program with teacher guides that can be applied across organizations (see Box 3).
Daugherty added that there should be more extensive training of IRB members and investigators on human subjects protections. “For the most part, that training is either non-existent or superfluous—you go on a website, click through three or four modules and slides and answer questions. Most of us can answer the questions without having read the content before the question,” he said. Bierer agreed and noted there are organizations that do this to some degree, such as Public Responsibility in Medicine and Research, whose objective is to communicate best practices and create a culture and home for IRB administrative efforts.
Several workshop participants also recommended educating the public about the value of research and biospecimens. One suggested using community advisory boards or approaches that go outside of the informed consent process to increase the public’s scientific literacy more generally. Albrecht responded to this suggestion and noted that she conducted a pilot program aimed at informing older, underserved African American residents of the greater Detroit metropolitan area about biospecimen donation, collection, and banking. She worked with a series of engaged focus groups and conducted interviews to develop a pilot workshop and video. Stakeholders and community advisors were involved at all stages of the planning and research process.
Laura Cleveland suggested beginning such educational efforts earlier so that school children are aware of what clinical trials are. “I think education about human participation in cancer clinical trials should be done at younger ages,” she said. Botkin said it would be helpful if institutions did a better job at branding themselves as research institutions. “Folks who walk through the door do not have a basic understanding that [research] is a big
part of what we do. We want to raise the awareness of the importance of research in general as a whole part of the process as opposed to making it something extra and special,” he said.
Several speakers noted a lack of empirical evidence in a number of areas related to protections for research participants, including whether changes to the HIPAA Privacy Rule and guidances have improved patients’ trust, what consent forms and processes are most effective, what the public thinks about sharing biospecimens and linked data, outcomes of returning individual genetic results to research participants, and the impact of CIRBs.
Botkin noted that the informed consent process was not evidence based when it was first instilled within the regulations and it is still not evidence based as it is currently conducted. He suggested conducting research to address that evidence deficit. Leiter added, “There needs to be some explicit studies done on the efficacy of whatever we are trying out to build patient trust. If we are trying all these things and then there is no measurement, it is going to be very hard to determine whether or not we are making any progress or just putting into place another system that is imperfect in ways that are potentially detrimental to our end goal.”
Mendelsohn pointed out that there needs to be more data on what research participants want in terms of oversight and protections and whether they would be willing to trade a modestly increased risk to their privacy for greater use of their biospecimens and health information, with the aim of facilitating research that improves clinical care, for example. Terry noted that she has gone to various organizations and institutions and told them that patients want greater flexibility in how they interpret certain HIPAA restrictions so more research can be done more easily on their disorders, but without data on what patients want, it is hard to present a convincing argument that changes need to be made. Peppercorn added, “We clearly need research in this area because there isn’t a lot in the scientific literature about what the public thinks. People donate their biospecimens and consent to future unspecified research. Now when the concepts of data sharing or greater use of their biospecimens are posed, which side would they like the scientific community to be erring on and what are the contextual factors that influence that?” Clayton noted that the National Institutes of Health (NIH) recently funded 10 groups as part of the eMERGE consortium to study the acceptability of broad data sharing, including doing a survey of
16,000 people about the acceptability of broad consent for data sharing and broad research use.
Little is also known about the outcomes of returning individual research results to participants. One of the recommendations in a white paper by eMERGE and CSER is that researchers measure the benefits and harms of returning genomic information to research participants and evaluate and determine the best practices for conveying this information in both research and clinical settings, Jarvik reported.
Schilsky noted that there is little empirical research about the impact of CIRBs. One extensive literature review could only identify 11 empirical studies on IRB review of multisite trials (Ravina et al., 2010). He and Daugherty also suggested that metrics be established to assess the quality of IRB review. “We need more precise and meaningful metrics than IRB review times to know whether we’re doing it right,” Daugherty said. Taylor reported that the NIH has a new funding opportunity for empirical research on the ethical issues related to CIRBs and consent for research using clinical records and data.
At the end of the workshop, Bradbury highlighted key points from the discussions. She noted a major theme was the need to balance risk and uncertainty with trust, and “to understand there’s risk in everything that we do, but we cannot be so risk averse that we cripple the system and hamper the advance of cancer research. A regulatory structure that’s based on legalistic fears of rare events may not benefit the mission to prevent and cure cancer and as we heard from our patient advocates, it will not help our patients.”
She added that a recurring theme throughout the workshop was to encourage a culture change of risk-based oversight that may need to consider innovative ways to approach informed consent. “We heard from our patients and advocates that they are concerned with what we don’t do with the gifts and resources that they provide more so than information on other risks that we sometimes outline in multipage consents.” She added that is not to say there are no risks in clinical research, but to recognize that the degree of those risks varies depending on what type of study is being done and in what type of context.
Another major theme, according to Bradbury, was the need to consider the process of informed consent, sharing data, and returning research
results, rather than focusing just on the regulations and forms. More thought also needs to be given to the process for defining research activities, the oversight of various trial designs, and the promise of learning health care systems, she added. “We’ll need change not just at the government regulation level but also at the institutional level,” she said.
She concluded by saying that the workshop was not intended to develop specific recommendations for change, but rather to frame and discuss the issues and to encourage action. She added that “progress does start here and we can and must all be agents of change. We heard many opportunities throughout the workshop, including continuing individual involvement in the ANPRM process, and continuing to persistently push the agenda at all levels and in parallel efforts at the regulatory, institutional, and individual encounter levels. Your examples at an individual level do not go unnoticed by those you mentor and over time can instill the culture change that I think we have all been aspiring to.”
Aberdeen, J., B., S., Yeniterzi, B. Wellner, C. Clark, D. Hanauer, B. Malin, and L. Hirschman. 2010. The mitre identification scrubber toolkit: Design, training, and assessment. International Journal of Medical Informatics 79(12):849-859.
AHRQ (Agency for Healthcare Research and Quality). 2009. Informed consent and authorization toolkit for minimal risk research. Rockville, MD.
Beardsley, E., M. Jefford, and L. Mileshkin. 2007. Longer consent forms for clinical trials compromise patient understanding: So why are they lengthening? Journal of Clinical Oncology 25(9):e13-e14.
Benitez, K., G. Loukides, and B. Malin. 2010. Beyond safe harbor: Automatic discovery of health information de-identification policy alternatives. In Proceedings of the ACM International Health Informatics Symposium. Pp. 163-172.
Brothers, K., D. Morrison, and E. Clayton. 2011. Two large-scale surveys on community attitudes toward an opt-out biobank. American Journal of Medical Genetics Part A 155(12):2982-2990.
Burke, W., and B. M. Psaty. 2007. Personalized medicine in the era of genomics. Journal of the American Medical Association 298:1682-1684.
Burman, W., P. Breese, and S. Weis. 2003. The effects of local review on informed consent documents from a multicenter clinical trials consortium. Controlled Clinical Trials 24(3):245-255.
Carrell, D., B. Malin, J. Aberdeen, S. Bayer, C. Clark, B. Wellner, and L. Hirschman. 2013. Hiding in plain sight: Use of realistic surrogates to reduce exposure of protected health information in clinical text. Journal of the American Medical Informatics Association 20(2):342-348.
Cheng, S. K., and D. M. Dilts. 2011. Predicting accrual achievement: Monitoring accrual milestones of NCI-CTEP–sponsored clinical trials. Clinical Cancer Research 17(7):1947-1955.
Clayton, E. W., S. Haga, P. Kuszler, E. Bane, K. Shutske, and W. Burke. 2013. Managing incidental genomic findings: Legal obligations of clinicians. Genetics in Medicine 15(8):624-629.
Data Guidance: Global Guidance in One. 2013. EU: Right to be forgotten now the right to erasure. Cecile Park Publishing Ltd. http://www.dataguidance.com/dataguidance_privacy_this_week.asp?id=2119 (accessed May 29, 2014).
Deleger, L., H. Brodzinski, H. Zhai, Q. Li, T. Lingren, E. Kirkendall, E. Alessandrini, and I. Solti. 2013. Developing and evaluating an automated appendicitis risk stratification algorithm for pediatric patients in the emergency department. Journal of the American Medical Informatics Association 20(e2):e212-e220.
Dorschner, M., L. Amendola, and G. Jarvik. 2013. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. American Journal of Human Genetics 93(4):631-640.
El Emam, K., E. Jonker, L. Arbuckle, and B. Malin. 2011. A systematic review of reidentification attacks on health data. PLoS ONE 6(12) e28071.
Green, R., J. Berg, W. Grody, and S. Kalia. 2013. American College of Medical Genetics and Genomics (ACMG) recommendations for reporting of incidental findings in clinical exome and genome sequencing. https://www.acmg.net/docs/IF_Statement_Final_7.24.13.pdf (accessed April 25, 2014).
HEW (Department of Health, Education, and Welfare). 1973. Records, computers and the rights of citizens: Report of the Secretary’s Advisory Committee on Automated Personal Data Systems. http://www.justice.gov/opcl/docs/rec-com-rights.pdf (accessed May 21, 2014).
HHS (Department of Health & Human Services). 2013. New rule protects patient privacy, secures health information. http://www.hhs.gov/news/press/2013pres/01/20130117b.html (accessed May 21, 2014).
IOM (Institute of Medicine). 2007. The learning healthcare system. Washington, DC: The National Academies Press.
IOM. 2009a. Beyond the HIPAA Privacy Rule: Enhancing privacy, improving health through research. Washington, DC: The National Academies Press.
IOM. 2009b. Initial national priorities for comparative effectiveness research. Washington, DC: The National Academies Press.
IOM. 2010. A foundation for evidence-driven practice: A rapid learning system for cancer care. Washington, DC: The National Academies Press.
IOM. 2013. Best care at lower cost: The path to continuously learning health care in America. Washington, DC: The National Academies Press.
Joffe, S., E. F. Cook, P. D. Cleary, J. W. Clark, and J. C. Weeks. 2001. Quality of informed consent in cancer clinical trials: A cross-sectional survey. Lancet 358(9295):1772-1777.
Ludman, E., S. Fullerton, L. Spangler, S. Trinidad, M. Fuji, G. Jarvik, E. Larson, and W. Burke. 2010. Glad you asked: Participants’ opinions of re-consent for dbGaP data submission. Journal of Empirical Research on Human Research Ethics 5(3):9-16.
Malin, B., K. Benitez, and D. Masys. 2011. Never too old for anonymity: A statistical standard for demographic data sharing via the HIPAA Privacy Rule. Journal of the American Medical Informatics Association 18(1):3-10.
Menikoff, J. 2010. The paradoxical problem with multiple-IRB review. New England Journal of Medicine 363(17):1591-1593.
Ness, R. 2007. Influence of the HIPAA Privacy Rule on health research. Journal of the American Medical Association 298(18):2164-2170.
ONCHIT (Office of the National Coordinator for Health Information Technology). 2008. Nationwide privacy and security framework for electronic exchange of individually identifiable health information. http://www.healthit.gov/sites/default/files/nationwideps-framework-5.pdf (accessed April 24, 2014).
Paasche-Orlow, M., R. Parker, J. Gazmararian, L. Nielsen-Bohlman, and R. Rudd. 2005. The prevalence of limited health literacy. Journal of General Internal Medicine 20(2):175-184.
Paasche-Orlow, M., F. Brancati, H. Taylor, S. Jain, A. Pandit, and M. Wolf. 2013. Readability of consent form templates: A second look. Institutional Review Board 35(4):12-19.
Penner, L., S. Eggly, J. Griggs, W. Underwood, III, H. Orom, T. Albrecht. 2012. Life-threatening disparities: The treatment of black and white cancer patients. Journal of Social Issues 25(2):328-357.
Presidential Commission for the Study of Bioethical Issues. 2013. Anticipate and communicate: Ethical management of incidental and secondary findings in the clinical, research, and direct-to-consumer context. Washington, DC: PCSBI.
Ravina, B., L. Deuel, and E. Dorsey. 2010. Local Institutional Review Board (IRB) review of a multicenter trial: Local costs without local context. Annals of Neurology 67(2):258-260.
SACHRP (Secretary’s Advisory Committee on Human Research Protections). 2013. SACHRP comment regarding the June 4, 2013, FDA Request for Comment relating to the availability of masked and de-identified non-summary safety and efficacy data. http://www.hhs.gov/ohrp/sachrp/commsec/attachmentasecretarialletter21.pdf (accessed May 29, 2014).
Schwartz, G. F., K. Hughes, H. Lynch, C. Fabian, I. Fentiman, M. Robson, S. Domchek, L. Hartmann, R. Holland, and D. Winchester. 2008. Proceedings of the International Consensus Conference on Breast Cancer Risk, Genetics, & Risk Management, April 2007. Cancer 113(10):2627-2637.
Sharp, S. 2004. Consent documents for oncology trials: Does anybody read these things? American Journal of Clinical Oncology 27(6):570-575.
Silverman, H., S. C. Hull, and J. Sugarman. 2001. Variability among Institutional Review Boards’ decisions within the context of a multicenter trial. Critical Care Medicine 29(2):235-241.
Stark, A. R., J. E. Tyson, and P. L. Hibberd. 2010. Variation among Institutional Review Boards in evaluating the design of a multicenter randomized trial. Journal of Perinatology 30(3):163-169.
Stewart, D. J., S. N. Whitney, and R. Kurzrock. 2010. Equipose lost: Ethics, costs, and the regulation of cancer clinical research. Journal of Clinical Oncology 28(17):2925-2935.
Sweeney, L. 1997. Weaving technology and policy together to maintain confidentiality. Journal of Law, Medicine & Ethics 25(2):98-110.
Wagner, T. H., C. Murray, J. Goldberg, J. Adler, and J. Abrams. 2010. Costs and benefits of the National Cancer Institute Central Institutional Review Board. Journal of Clinical Oncology 28:662-666.
Xia, W., R. Heatherly, X. Ding, J. Li, and B. Malin. 2013. Efficient discovery of deidentification policies through a risk-utility frontier. In Proceedings of the 3rd ACM Conference on Data and Applications Privacy and Security (CODASPY). Pp. 59-70.
This page intentionally left blank.


































































