
1
Introduction
The past 20 years have seen a rapid increase in our understanding of the biology of cancer. The fundamental concept guiding research in this field has been the recognition of cancer as a genetic disease in the sense that cells within a tumor inherit malignant characteristics from a transformed progenitor cell. This research has led to general acceptance of the view that cancer is the end result of a series of acquired or inherited mutations in the structure of certain genes contained within the DNA of the cell. These mutations are passed from the original transformed progenitor cell to its clonal descendants, which compose the cells of the tumor. It is the abnormal function of specific mutated genes that ultimately leads to the uncontrolled cell growth and behavior that characterize all cancers.
Advances in understanding the genetics of cancer are beginning to have an impact on the clinical management of malignant disease. Many of the genetic changes that underlie malignant transformation of cells and/or distinguish malignant clones can be used as markers to diagnose, monitor, or characterize various forms of cancer. The purpose of this report is to assess the current status of genetic testing in cancer management both from the standpoint of those tests and genetic markers that are presently available and from the perspective of genetic approaches to cancer testing that are likely to have an impact on cancer management in the near future.
Outline of Molecular Genetics
Many features of gene structure and action are now well understood. Genetic information is stored within the nuclear DNA of each cell in the body. DNA consists of two intertwined strands—the DNA double helix—with each strand consisting of many linked nucleotides. There are four different nucleotides in DNA: adenosine (A), guanosine (G), thymidine (T), and cytidine (C). The two strands of DNA are held together by the hydrogen bonds between nucleotides; specifically, A nucleotides in one strand bond with T nucleotides in the other, and G's bond with C's. An A nucleotide bonded to a T or a G bonded to a C is sometimes referred to as a base pair. Two stretches of single-stranded DNA having nucleotide sequences such that each A in one strand can bond with a T in the other strand and each G can bond with a C are said to be complementary.
DNA replication occurs by separation of the two strands in a DNA molecule, followed by synthesis of two new daughter strands having nucleotide sequences based on the template provided by the two parent strands. The result is a doubling of the DNA, with each molecule containing a parental strand and a new daughter strand complementary to the parent strand.
The total nuclear DNA content of a cell (referred to as the genome of the cell) is divided into chromosomes. Each chromosome consists of a very long, single DNA molecule complexed with a variety of proteins. There are more than six billion nucleotide pairs in the normal diploid human genome in nondividing cells. To pack this amount of DNA into chromosomes, the DNA must be extensively coiled and looped about the central axis of the chromosome. The degree of packing changes with the cell cycle. DNA is most tightly packed—and consequently the chromosomes are most condensed—during cell division, thereby facilitating the orderly and equitable distribution of DNA to the two progeny cells.
The vast majority of DNA in human cells has no known function. Genetic information for the synthesis of proteins constitutes only a few percent of the genome. The genes exist as defined stretches of nucleotide sequence scattered throughout the genome. Each gene is associated with several regions of nucleotide sequence responsible for regulating protein synthesis. In addition, the portion of the gene that determines the amino acid sequence of the encoded protein is often divided into pieces called exons, which are separated by noncoding sequences of nucleotides referred to as introns. Introns vary in length from dozens to several hundred thousand base pairs.
Synthesis of protein is basically a two-step process. The initial step in protein synthesis is the building of a so-called messenger RNA (mRNA) molecule from information in one strand of the DNA making up the gene, a process called transcription. These messenger RNAs are single-stranded
polymers of nucleotides, closely resembling DNA in structure except for an extra oxygen atom in each nucleotide. The sequence of nucleotides in the mRNA is dictated by that of the template DNA strand and is complementary to that template. Before passing out of the nucleus, the intronic sequences are spliced from the RNA to produce a continuous sequence of nucleotides encoding information for the polymerization of amino acids into protein. This step is accomplished in the cytoplasm while the RNA is bound to ribosomes. At this site, successive triplets of nucleotides are read by the protein-synthesizing apparatus, each triplet corresponding to a specific amino acid. The amino acids are sequentially covalently bonded to each other, resulting in a newly synthesized protein polypeptide.
Proteins synthesized from mRNA control directly or indirectly all functions of the cell, including cell division. Different genes are active in different tissues and at different stages of development. Abnormalities in gene structure (i.e., mutations) may affect a cell in either subtle or drastic ways, depending on the normal function of the protein and the degree to which the structure or activity of that protein has been changed.
Recombinant DNA technology primarily involves the use of methods to isolate discrete double-stranded fragments of DNA and to ligate these fragments into vector DNA molecules, which can then be propagated after insertion of the vector into a host bacterial cell. Bacteria carrying the vector can be grown in culture, allowing subsequent isolation of large amounts of vector, together with any recombinant DNA sequence that has been ligated into it. The propagation of isolated fragments by this method is often referred to as DNA cloning. Abundant, pure DNA fragments generated by cloning have been invaluable for the structural and functional analyses of nucleotide sequences in DNA and for the production of reagents used in diagnosis.
Brief Overview of Cancer Genetics
Recognition of the basic role of mutation in the genesis of cancer required the identification of unique genetic changes associated with particular tumors. Important early insights came from investigation of the chromosomes in normal and malignant cells, an area of study known as cytogenetics. By 1970, it had become possible by using improved staining techniques to identify individually each of the human chromosomes. This advance allowed the demonstration of recurrent cytogenetic abnormalities associated with specific kinds of tumors. Often, these abnormalities are translocations—a rearrangement of two or more chromosomes that leads to the formation of a new fusion or chimeric chromosome (Figure 1-1). Other tumors have been shown to contain deletions, consisting of the loss of an entire chromosome or a portion of a chromosome, inversions (which in-
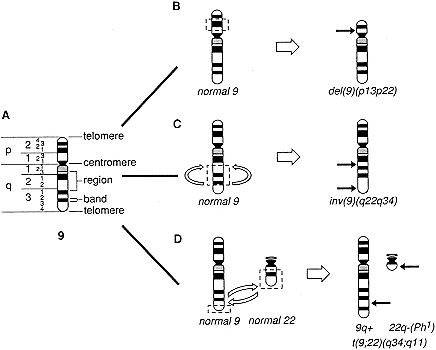
Figure 1-1 Schematic diagram illustrating a normal chromosome and three chromosomal abnormalities observed in human neoplasms. A. Diagram of the banding pattern of a normal chromosome 9. The chromosome arms (p, short arm; q, long arm), regions, and band numbers are indicated on the left of the chromosome: specific chromosome structures are indicated on the right of the chromosome. B. Diagram of the mechanism of an interstitial deletion of the short arm of chromosome 9, a common abnormality in acute lymphoblastic leukemia. Chromosome breaks occur in bands 9p13 and 9p22, and the intervening chromosomal segment (band 9p21 and parts of bands 9p13 and 9p22) is lost [del(9)(pl3p22)]. C. Diagram of the mechanism of a paracentric inversion. Chromosome breaks occur in two bands within a single chromosome arm, in this case, within 9q22 and 9q34; the intervening segment is inverted and the chromosome breaks are repaired [inv(9)(q22q34)]. D. Diagram of the mechanism of the reciprocal translocation involving chromosomes 9 and 22, t(9;22)(q34;q11), which gives rise to the Philadelphia (Ph1) chromosome in the malignant cells of patients with chronic myelogenous leukemia. Breaks occur in bands q34 and q11 of chromosomes 9 and 22, respectively, followed by a reciprocal exchange of chromosomal material. This rearrangement results in the translocation of the ABL oncogene, normally located at 9q34, adjacent to the BCR gene on chromosome 22, giving rise to a chimeric BCR-ABL gene, whose protein product plays a role in the transformation of myeloid cells.
volve flipping a segment of chromosome at a particular location), or cytogenetic evidence of segmental chromosome amplification. Significantly, identical chromosomal changes were often observed in every malignant cell within a tumor. This led to two major conclusions: (1) that the observed chromosomal anomalies likely played a part in the genesis of the tumor and (2) that the population of cells comprising a tumor arose as a clonal expansion from an original progenitor cell containing the aberrant chromosome or chromosomes.
The concept of cancer as a clonal disorder has important practical ramifications for diagnosis. In most tumors, genetic changes specific to the progenitor cell are faithfully passed down to all progeny. While genetic heterogeneity due to the appearance of subclones during growth and progression of the tumor may complicate this picture, those changes that are directly responsible for the malignant phenotype are likely to be maintained throughout the entire population of cells. Such changes constitute a specific marker that can be exploited to distinguish malignant from normal cells. Furthermore, a mutation need not be directly related to the transformation event to be diagnostically useful, as long as that mutation is specific for the malignant clone. This principle has been exploited extensively in the diagnosis of lymphocytic neoplasms, which is discussed below.
The genes that are mutated in various forms of cancer are still being discovered and characterized, but they appear to fall into two major categories: proto-oncogenes and tumor suppressor genes. Proto-oncogenes encode proteins that play an important role in normal cells in controlling growth, differentiation, and expression of other genes. Many of these proteins function as growth factors or play a part in formation of the receptor apparatus that allows a cell to respond to external stimuli such as growth factors. In some cases, genes of this class code for enzymes called protein kinases that alter the function of target proteins by catalyzing the addition of a phosphate group to serine, threonine, or tyrosine residues. Other proto-oncogenes act as transcription factors, proteins that regulate the amount of other proteins within the cell by altering the transcription of DNA into RNA.
In general, mutations that convert proto-oncogenes into oncogenes lead to increased activity of the encoded protein. In some cases, the mutation involves the coding sequence of the gene and results in an altered protein with abnormal activity. In other instances, the mutation does not change protein structure, but produces its effect by causing the protein to be synthesized in increased amounts or at inappropriate times during cell differentiation. Proto-oncogene activation frequently occurs as a consequence of chromosomal translocations but may also be produced by point mutation, a change in a single base pair, or gene amplification, a process that leads to an increase in the number of copies of the gene per cell. Detection of
activated proto-oncogenes at the breakpoints of translocations has validated the idea that genes that play a direct role in cancer pathogenesis are likely to be found at sites involved in recurrent, clonal chromosomal aberrations.
In contrast to proto-oncogenes, tumor suppressor genes encode proteins that normally have a negative influence on cell proliferation. The precise manner in which this occurs is still being defined, but many of these genes seem to regulate the entrance of resting cells into the cell cycle, an event necessary for cell proliferation. For this reason, mutations involving tumor suppressor genes usually abolish the function of the protein and result in increased proliferative potential. This often occurs through gene deletion, physical loss of the gene from the cell, but may also be produced by point mutations.
A line of investigation that has recently converged with the study of tumor suppressor genes concerns the mechanism of inherited predisposition to cancer. It has been known for some time that the tendency to develop certain cancers appears to be inherited within particular families in a Mendelian fashion. Research carried out over the last few years on several forms of pediatric cancer and now more recently on kindreds with high frequencies of multiple types of adult tumors has shown that the critical factor governing the predisposition to cancer among affected families is the inheritance of a defective copy of a tumor suppressor gene. In some instances, an acquired mutation inactivating the second copy of the tumor suppressor gene appears to be the event that initiates actual tumor development in an individual.
Many different sources of information have confirmed the conclusion that malignant transformation is often a multistep process requiring the accumulation of a series of mutations before a cell is rendered fully malignant. Thus, any single mutation associated with a given form of cancer may be necessary but not sufficient for the malignant behavior of that cancer. In addition, tumor cells may characteristically develop other mutations after they have already undergone malignant transformation. These mutations may not contribute directly to the malignancy of these cells per se but can have an important effect on other features of the cancer, such as rate of growth, invasion, or metastatic potential.
Infections by certain viruses have recently been implicated in the etiology of several types of human cancer. In these cancers, viral genomes are found in virtually every tumor cell making up the cancer, and evidence favors the notion that the genomes of these viruses harbor some gene or genes that act as an oncogene to promote malignant transformation. Because infection of an individual usually precedes the appearance of the cancer by many years, it appears that the acquisition of additional mutations is necessary before an infected cell becomes overtly malignant. An alternative interpretation, which may not be entirely different from this in prin-
ciple, is that the presence of the viral genomes in the cell acts as a cofactor in malignant transformation by stimulating the cell to divide, and that more rapid replication of cellular DNA predisposes the cell to mutation. Regardless of the mechanism of oncogenesis, the presence of viral genomes and virus-specific proteins produces changes in the genotype and phenotype of the host cell, and in some circumstances, these changes can serve as tumor-specific markers.







