
DOUBLING UP
How The Genetic Code Replicates Itself
by Anne Simon Moffat
This year marks the forty-first anniversary of one of the most important scientific discoveries ever made: the finding that the universal genetic material is made of deoxyribonucleic acid, commonly called DNA. Although it had been known for hundreds of years that plants and animals had some sort of genetic material that carried information from generation to generation, what it was and how it worked remained unknown until 1953, when the American James Watson and the Englishman Francis Crick made their announcement from Cambridge, England. Their description of DNA as a twisted, ladder-like structure with rungs of complementary pairs of simple chemical bases offered a basic architecture for the genetic code. And because this physical structure involved a template where information from one DNA strand could be carried to another, it also hinted at how information could be passed from one generation to another with great fidelity. What happens is that the bases on each of the parent strands become paired with their complements on the newly
formed daughter strands. Indeed, in a classic example of British—and scientific—understatement, Watson and Crick wrote in their landmark Nature paper that, ''It has not escaped our notice that the specific pairings (of bases in DNA) we have postulated immediately suggests a possible copying mechanism for the genetic material."
This discovery was the culmination of decades of work by scores of researchers. Yet it marked the beginning, not the end, of a new era of biological research. It triggered a desire to know more about the bricks, mortar, and embellishments that filled out the basic architecture of DNA described by Watson and Crick and the precise building mechanisms used to construct it. Specific issues that were once beyond the scope of experiments—such as when does a cell know when to duplicate its genetic material and how does it do it?—could now be tackled. Moreover, broad problems—such as the way DNA duplication fits into the total cell cycle, the ongoing process of cell growth and division—could be addressed (see Box beginning on p. 100).
Over the past 40 years, the incentives for pursuing such research have been many and diverse. On one level there is the sheer intellectual excitement of getting to know the ways of life on an intimate basis. On another, such studies offer the opportunity to use new information about the structure and behavior of DNA for useful purposes: for example, improved understanding of DNA replication provides a means for controlling the unrestrained proliferation of DNA found in cancer cells (see Box beginning on p. 106).
And, over the years, experimental approaches to the study of genetic material have been equally diverse. Some researchers look at the simple genomes of viruses, which are often considered to be in a biological netherworld since they can't live on their own, while others look at the slightly larger, circular genomes of bacteria. The genetic material of yeasts, which are the simplest organisms to have DNA in organized chromosomes (and are used to make bread and beer), are the focus of still other labs. Finally, some researchers accept the challenge of looking at cells taken from advanced organisms, such as rats, frogs, plants, and man.
As with the study of most complex phenomena, the early results from studies of DNA replication and gene expression in these various organisms were murky and incomplete. It was not always clear that the piecemeal results could be synthesized into a coherent picture of what
|
The Big Picture: The Cell Cycle DNA replication, although important and time consuming (many cells spend one-half their existence copying their genome), is only one stage in the cell cycle, the complex process of cell growth and division that leads to the birth of new cells.* Cells also spend considerable time simply growing in size or gathering up enough nutrients to build and maintain their subcellular apparatus, such as the energy-producing mitochondria and, in plants, the light-gathering chloroplasts. In fact, the myriad activities that must be started, completed, and integrated with others during the cell cycle would rival those found in a car assembly plant. And like the action that takes place on an assembly line, the cell cycle has an elaborate scheme of feedback controls, where the activities of one stage determine what follows. For a better sense of the role of DNA replication in the larger scheme of cellular matters, here is an overview of the key stages of the cell cycle. The G1 Phase The cell cycle is defined as starting at the G1 (for first gap) phase, which lies between the previous nuclear division and the start of DNA synthesis. Once cells reach a critical size in G1, a decision is made whether to commit |
|
the cell to further involvement in the subsequent S or synthetic phase, leading to cell division. Some researchers refer to this part of the G1 phase as "Start." Cells that exit from the cell cycle typically do so here, and enter the so-called G0 (or resting) phase. The S Phase (DNA synthesis) DNA replication takes place during the synthetic or S phase. Although there is great activity in the genome during this stage of the life cycle, the chromosomes cannot be seen as distinct structures under the light microscope. The complete cell cycle in a mammalian cell needs about 16 hours, and the S phase can take almost half that. The time devoted to DNA synthesis is even more striking in E. coli. These bacteria can divide every 30 minutes or less, and all but about 1 minute is often needed to complete one round of DNA synthesis. The G2 Phase The second gap phase is placed between DNA duplication and actual nuclear division. During this stage, cells can be very busy making proteins, lipids and carbohydrates, and other substances essential to the support of life. Also, during this period, any DNA that was damaged during the S phase is repaired. M or Mitotic Phase During the brief but very busy period called mitosis (Figure 4.1), new daughter chromosomes separate and the cell divides. Its various stages are divided into prophase, metaphase, anaphase, telophase, and interphase |
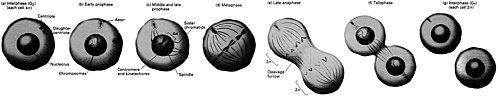
FIGURE 4.1 All phases of mitosis. (From Molecular Cell Biology by J. Darnell, H. Lodish, and D. Baltimore. Copyright © 1990 by Scientific American Books, Inc. Reprinted with permission of W.H. Freeman and Company.)
Pulling It All Together Although much is known about what happens in the individual stages of the cell cycle, one of the biggest challenges facing cell biology is to learn |
|
more about how the signals that guide cells from one stage of the cycle into another control events such as DNA replication. "Feedback controls" are known to exist, since in most cells inhibiting a downstream event can arrest programming of the cell cycle. For example, stopping DNA duplication keeps cells from entering mitosis. Although early studies of the cell cycle focused on external extracellular signals, such as hormones and other growth factors, in recent years many clues have emerged about regulatory signals that come from within the cell. Many of the key regulatory proteins have been positively identified. For example, from the study of yeast mutants that are blocked through the normal progression of the cell cycle and of frog oocytes and their development into early embryos, it has become clear that one protein, called cyclin, is critical to the smooth progress of the cell cycle. Such molecules build up during interphase and are lost during mitosis. In fact, the degradation of cyclin appears essential for exiting mitosis. Cyclin was discovered as part of a large protein complex, called the maturation-promoting factor (MPF), which also includes a critical kinase enzyme, called the cdc 2 (for cell division cycle) kinase. This enzyme is positively regulated by cyclin to fuel the mitotic process. There appear to be complex regulatory schemes acting between this kinase and cyclin, and, indeed, cyclin-cdc 2 complexes also control the commitment to cell division at Start in the G1 phase of the cell cycle. Another likely player in the regulation of the cell cycle is a protein, ubiquitin, whose name suggests its ever present nature. It appears to have some role in the degradation of cyclin. One theory suggests that MPF stimulates an activity that makes cyclin a better substrate for ubiquitination and consequent destruction. Interestingly, there is one other known example of a protein having regulated degradation by ubiquitin, the light-activated phytochrome in plants. It is no surprise that there are many layers of control, involving many different compounds, on the cell cycle. The challenge remains, however, to describe all such substances and how they work together to trigger cells into a smooth transition from one phase of the cell cycle to another. |
goes on in living cells as they grow and multiply. But, eventually, these various experimental approaches converged on some key findings. It was recognized that DNA is the universal genetic code: evolution had fashioned a system for transferring biological information from one generation to another that works equally well for the simple genomes of viruses and bacteria as for the more complex genomes of worms, monkeys, and humans. But, equally important, it became clear that while the double helical architecture of DNA is relatively simple and straightforward, the mechanisms for building it and expressing it are not. DNA needs a large cast of supporting characters to help it get its job done. In recent years some of the most significant advances in DNA studies have been the discovery of these adjunct players—enzymes, other proteins, and various cofactors—and their roles in DNA synthesis and expression.
STARTING AT THE VERY BEGINNING
One approach to the study of DNA and its supporting players is to look at the very first events associated with DNA's replication. About 30 years ago, using a prime analytical tool of the day, the microscope, some researchers noticed large bits of chromosome that appeared to balloon up at predictable and specific times in a cell's life cycle. These so-called replication bubbles were identified in the chromosomes of animal, plant, and bacterial viruses as well as those of more advanced organisms, such as yeast and some mammals. Other researchers fed microbial and animal cells labeled isotopes at the start of DNA synthesis, with normal isotopes offered later in the same round of DNA synthesis. In both animal and yeast experiments it was found that some sections of the genetic material were preferentially synthesized before the bulk of the genome. In a variation on this research, other experimenters tagged DNA with a fluorescent compound that is similar to one of the bases of the genetic code. By looking through a microscope, with suitable light filters, small amounts of fluorescence could be seen in certain regions of the nucleus. This work offered further evidence that not all DNA copies itself at the same time and that certain bits of the DNA were preferentially copied early on in the replicative process.
At about the same time that these researchers were using microscopic techniques to examine DNA replication visually, other labs were developing biochemical strategies for examining this event. For example, cell fusion experiments done in the early 1970s showed that diffusible factors were needed for DNA synthesis. When cells in the "resting phase," the so-called G1 phase immediately prior to the stage
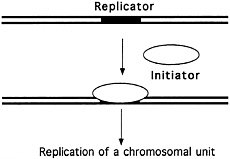
FIGURE 4.2 The "replicon" model. (Courtesy of Bruce Stillman, Cold Spring Harbor Laboratory.)
when DNA synthesis takes place, were fused with cells in the DNA synthetic phase, the so-called S phase, the S-phase cells did something to the G1 cells to trigger them into the S phase. Some substance present in the S phase appeared to coax the onset of DNA replication in the G1 nucleus. Moreover, this unknown trigger was shown to be measurable in the following experiment. When three or more cells were fused, the higher the ratio of S-phase to G1-phase cells in the fusion, the faster the G1 cells entered the S phase. With very high S:G1 ratios, entry into the S phase by the G1 cells was almost instantaneous.
All these experiments supported the so-called Replicon model of DNA duplication (see Figure 4.2), proposed in 1963 by François Jacob and Sydney Brenner, which suggested that DNA replication is controlled by a biochemical regulator, known as the initiator, which triggered the start of DNA duplication by acting on a specific collection of DNA bases, called the replicator, or replication origin. But this work did not offer definitive proof of the existence of the postulated replicator and its helpmate, the initiator. Almost a decade would pass before such proof was in hand.
To tackle this problem in mammalian cells, researchers chose to work with a tumor-inducing monkey virus known as SV40, which has a very small genome, only 1.5 microns (0.0000015 of a meter) long, and contains only about 5000 based pairs of DNA. The reasoning was that if a possible replicator origin was present, it would be easier to find it on a virus's small genome than on the genome of, for example, a human, which has about 3 billion base pairs and is about 1 meter long. The search initially involved studies of various replication intermediates taken from cells infected with SV40 to identify where in the small chromosome DNA replication started. Once this origin of DNA replication was found, researchers hunted for a specific protein, the initiator, that would bind DNA at the replication origin. The search for this protein, among millions of proteins in the cellular soup, was a nontrivial task. After many years of fractionations and purifications, where cellular
|
How Some Cancer Drugs Might Work Cancer, in the broadest sense, is uncontrolled cell growth. DNA replicates and cells proliferate wildly, depriving more organized growth of needed resources. Tumorous growths can appear as amorphous, undifferentiated, and ungovernable masses. Yet despite giving the appearance of complete disarray, cancer can be the end result of just a few molecular events gone wrong. Too much, or too little, of one of the many proteins that control cell division and DNA replication can disrupt normal cell growth and division, leading to cancer. Until fairly recently, most anticancer drugs were developed using empirical techniques, that is, by trial and error. Within the last year or two, this has begun to change. Now that some details are known about the various proteins needed to guide DNA replication, cell growth, and division in normal cells, efforts can be made to design drugs that correct some of the biochemical imbalances in cancer cells. By adding or deleting key proteins, such anticancer drugs may inject a level of management and structure that is missing in cancer cells. Numerous new biotechnology companies, many large, established pharmaceutical houses, and a number of independent research organizations worldwide are developing anticancer drugs based on this strategy. To be sure, not all cancers will be amenable to this sort of treatment, and it is unlikely that such therapies will be clinically available before the turn of the century. Still, says Bruce Stillman, the design of drugs that target key proteins and add a measure of control into a system that has run awry "represents a front line clinical approach to the treatment of cancer." One class of nuclear proteins that has received much attention from some cancer specialists are the topoisomerases, enzymes that are required for DNA replication and expression by affecting the overall topology of strands of DNA (see pp. 120–121). One research stratagem suggests that by poisoning the activity of these essential enzymes through the use of various antitopoisomerase drugs, the uncontrolled DNA replication and cell division characteristic of cancer cells could be curtailed. In fact, Paul J. Smith, of the Medical Research Council's Clinical Oncology and Radiotherapeutics Unit in Cambridge, England, has suggested that, "Tumor cells carry the seeds of their own destruction in the form of these enzymes [and] it is up to the chemotherapist to take advantage of this unique opportunity." In the last 5 to 6 years two discoveries were made that suggested that drugs that functioned as antitopoisomerases might work as anticancer drugs. First, newly replicated DNA was found to be a preferential target for antitopoisomerase II drugs. Second, several classes of known anticancer drugs were found to work by poisoning the topoisomerase enzymes. These drugs trap the topoisomerases on DNA, thus impairing the normal replication process. Topoisomerase II "poisons'' include various drugs, |
|
such as the acridines (e.g., Amsacrine); anthracyclines (e.g., Adriamycin); anthraquinones (e.g., Mitoxantrone); and the epipodophyllotoxins (e.g., VP-16). But because only a few of these agents, such as the epipodophylloxins, trap topoisomerases in a nonproductive and stable form, the list of candidate anticancer drugs is narrowed. So, as a first step to further development of antitopoisomerase anticancer drugs, researchers are focusing on how these drugs tie up the enzymes, the character of the enzyme/drug complex, and why it is toxic to actively growing cancer cells. One theory suggests that topoisomerase poisons trap the enzyme in a useless state, resulting in the loss of an enzyme critical to normal DNA replication. Another posits that the complex of the topoisomerase plus poison itself damages the system. But from the chemotherapist's view, the when and where of topoisomerase inactivation is at least as important as how it is done. Some evidence suggests that a collision of a replication fork with a trapped topoisomerase is the damaging event. There is also the possibility that the ability of an impaired topoisomerase to upset normal replication depends on where its sits on the chromosome. Certain sequences on the chromosome may be more vulnerable to the action of a topoisomerase poison. Another complication that might affect chemotherapeutic strategies is the finding that inhibiting RNA or protein synthesis protects cells from the cell-killing action of the topoisomerase poison, Amsacrine. So now there is a search for factors that could inhibit, or enhance, the impact of topoisomerase poisons. "The hope [is] that such factors can be manipulated to the chemotherapist's advantage," says Smith. Still another possibility for enhancing the effect of topoisomerase poisons as anticancer drugs is to coax cells to increase their production of topoisomerases and, hence, their vulnerability to this enzyme's poisons. This is not as farfetched as it sounds. For example, in laboratory studies with human breast cancer cells, it was found that the manipulation of estrogen could stimulate the expression of topoisomerases at a particular point in time and, consequently, the effectiveness of well-timed doses of the topoisomerase poisons, VP-16 and Amsacrine. The end result is the increased killing of cancer cells. This therapeutic strategy will soon begin clinical trials. Smith says that in the past, "Not knowing the full nature of the cytotoxic interaction between an antitumor agent and its target cell has always undermined our ability to design more effective drugs, to target treatment modalities and to gain some therapeutic advantage by manipulating the biology of neoplastic cells in situ. This situation has now changed for several classes of potent antitumor agents which poison DNA topoisomerases." And there are signs that nuclear proteins other than the topoisomerases will soon be targets for therapeutic agents designed to fight cancer. |
proteins were repeatedly divided and tested for activity as an initiator, Robert Tjian, then at the Cold Spring Harbor Laboratory (and now at the University of California, Berkeley) announced in 1978 discovery of the SV40T antigen as the initiator that specifically bound to the virus origin of DNA replication. This protein was first found with antisera from animals that had tumors produced by SV40: it was therefore called tumor antigen, or "T" antigen. When the virus gene encoding T antigen is introduced into cells that are not dividing, expression of T antigen induces the infected cells into the S phase, causing replication of the host cell's DNA and allowing virus DNA replication. In some cell types, T antigen will also transform the cells so that they now cause tumors in animals. Thus, T antigen is an initiator protein that binds to the virus origin of DNA replication and is a potent onco-protein, a cancer-causing agent.
How this T antigen initiator bound to the genome was found by examining both natural mutants and artificially mutated specimens of SV40. These and other experiments revealed that the T antigen was a relatively large protein of 708 amino acids and that 114 of these bound the origin (replicator) sequences on the DNA. It was also shown that only 65 base pairs from the whole virus genome were the target upon which the T antigen acted to trigger DNA replication. Flanking this essential core were other DNA sequences in the virus genome that stimulated DNA replication.
A key issue, of course, was what the initiator protein actually did to trigger DNA duplication. Work by several groups in the 1980s revealed that this T antigen, in the presence of other compounds that could supply energy, would unwind eight base pairs of DNA and, once unwound, the single strands of DNA could serve as templates for copying, thus starting the sequence of events that would ultimately lead to complete duplication of the DNA.
It was also realized that T antigen alone is not able to replicate the virus genome but requires proteins from the host cell. It was expected that these proteins would be the very same ones that are responsible for replicating the cell chromosomes in each S phase of the cell cycle. A key breakthrough toward demonstrating this was the development, by Joachim Li and Thomas Kelly, of the Johns Hopkins University School of Medicine, of cell-free protein extracts from monkey or human cells that would support the replication of SV40 DNA in the test tube, when T antigen was added (Figure 4.3). Subsequent work in Kelly's laboratory, as well as work by Jerard Hurtwitz, of the Sloan Kettering Cancer Center, and Bruce Stillman, of the Cold Spring Harbor Laboratory,
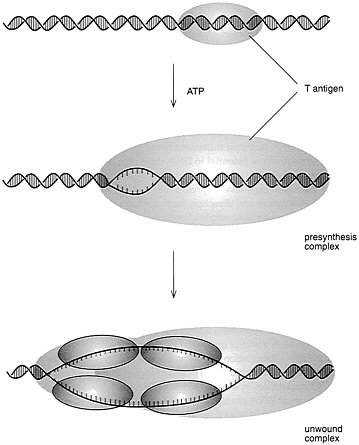
FIGURE 4.3 Initiation of SV40 DNA replication. (Courtesy of Bruce Stillman, Cold Spring Harbor Laboratory.)
identified these key cell proteins and revealed that they were required for replication of virus as well as for cellular DNA.
TACKLING THE NEXT STAGE OF COMPLEXITY
A second key issue was whether other organisms, especially those more complex than viruses, have replication origins and used proteins like T antigen to get the process going. Was the system used by viruses,
which, after all, are not genuine living organisms, an interesting anomaly? Or was this system relevant to all biology? To help answer that question, many researchers turned to the study of bacteria, the next most advanced group of organisms. In particular, they looked at Escherichia coli (E. coli), a common intestinal bacterium with a genome of about 4 million base pairs, about 1 millimeter in length. Here, too, researchers at first sidestepped the fact of not knowing exactly where DNA synthesis began, and they focused on a protein that seemed critical for the start of DNA replication, a protein that is the product of a gene called dna A. It is a scarce protein but appeared critical for cell viability, since E. coli rarely survived mutations to the gene. Knowing that this protein was somehow essential to DNA duplication but not knowing its details, researchers cloned the gene for dna A in recombinant DNA vectors that allow high-level expression in bacteria by taking over their metabolic machinery and directing them to churn out proteins. This gave scientists reasonably ample quantities of this usually scarce protein to work with. (It should be noted that developing such production schemes can be tough. Duplicating in vivo production schemes in alternate hosts can require considerable tinkering to get the desired results.) It was found that this protein binds to a particular site on the bacterial chromosome, in between two genes known to code for two key enzymes. The initiator protein dna A binds tightly to a small number of such nucleotide sequences, "seeding" the cooperative binding of 20 to 40 more copies of dna A, to form a large nucleoprotein complex. This complex serves as a sort of molecular pliers, pulling apart the double strand of DNA. Replication begins at this site and proceeds in both directions along the strand. This origin of replication site in bacteria is about 245 nucleotides in length. Moreover, research has shown that within this short DNA stretch, two-thirds of such nucleotides occur at the same position in at least five bacteria studied, including the distant relative Vibrio harveyi, a marine bacterium. This information suggests that these genomic sites for initiating DNA synthesis have been conserved during evolution. Such conservation among species hints that these nucleotides are important for biological function and may well interact with initiator proteins that are also conserved among the species. A key challenge, then, was to demonstrate that this general strategy for starting duplication of DNA—having a protein identify a specific origin sequence, among millions of others on the double helix, and then having it uncoil the DNA—also worked for the more advanced organisms known as eukaryotes, organisms that have a nucleus and have the genome divided among multiple chromosomes.
SNARING KEY EVIDENCE
But proof was not in hand until 1992, when Bruce Stillman of the Cold Spring Harbor Laboratory in Cold Spring, New York, and a speaker at the 1993 "Frontiers of Science" Symposium, together with his colleague Stephen Bell, captured an initiator protein from yeast, the simplest of the eukaryotes, which, like humans, have true chromosomes enclosed in an organized nucleus. It seemed a reasonable hypothesis that yeast, plants, monkeys, and man, all eukaryotes, would share a general scheme for duplicating DNA that involved replication origins on their chromosomes and initiator proteins, like the viral T antigen or E. coli's dna A protein. But there were also signs that, because of the size of their genomes, eukaryotic systems had complexities that are unknown in viruses and bacteria. For this reason scientists could not safely conclude that the replication scheme that worked so well in lower prokaryotic organisms would work the same way at the next level up, in the eukaryotes. For example, in human cells the complete cell cycle can take about 24 hours, with about one-third of that time devoted to duplicating the cell's DNA. With about 3 billion bases to replicate in 8 hours, such cells need more than one replication origin to get the job done in time. "The way nature solves this problem," says Stillman, "is to plump a lot of these initiator sequences along the chromosome [see Figure 4.4]. Then
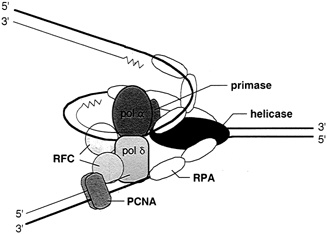
FIGURE 4.4 The multiprotein complex of the replisome keeps DNA synthesis progressing smoothly. In eukaryotes it includes two polymerases, alpha and delta; primase; helicase (T antigen in the case of SV40 virus DNA replication); replication protein A (a single-stranded binding protein); replication factor c; and the proliferating cell nuclear antigen. (Courtesy of Bruce Stillman, Cold Spring Harbor Laboratory.)
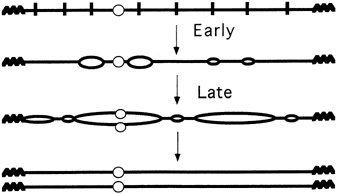
FIGURE 4.5 Multiple replication origins in chromosomes. (Courtesy of Bruce Stillman, Cold Spring Harbor Laboratory.)
you initiate from all of them and you'll replicate the genome." Like a parallel-processing supercomputer, and very unlike a simple virus, eukaryotic cells have hundreds, thousands, or even tens of thousands of replication processes going on simultaneously (see Figure 4.5). Indeed, it is now known that in the particular experimental yeast favored by researchers, called Saccharomyces cerevisiae, at least 400 replication origins exist in its 16 chromosomes. Moreover, the fruit fly Drosophila melanogaster can have 6000 replication segments at one time, just in its largest chromosome.
Thus, while it seemed that there are some strong parallels between DNA replication in higher and lower organisms, there also seemed to be some key differences. The challenge was to find out just what they are.
Early clues as to the nature of DNA replication in yeast came in the late 1970s when general studies of this organism revealed that yeast chromosomes need several distinct structures to duplicate, including special DNA sequences known as autonomously replicating sequences (ARs). In 1979 Ron Davis and his colleagues at Stanford University Medical School and John Carbon and his colleagues at the University of California, Santa Barbara, showed that select DNA sequences could confer a high rate of stability when reintroduced into yeast cells. A subset of these ARs was subsequently shown to act as origins of replication in chromosomes. Deletion analysis, which involves mutating yeast chromosomes by selectively removing some bases in the DNA, had shown that at least 100 bases are needed for these ARs sequences to work efficiently. By fine tuning their studies of the ARs, researchers also
discovered that a minimal 11-base-pair sequence is essential for origin activity. Known as the "A" element, which appears common to all origins from Saccharomyces, the 11-base-pair essential element is always accompanied by "B" elements that stimulate origin activity. Last year, after an intense search by many labs for this protein, Stillman and Bell, also of the Cold Spring Harbor Laboratory, announced discovery of a multiprotein complex in yeast that could act as an initiator by binding the essential 11-base-pair element of the ARs.
The search required arduous testing of many proteins present in yeast cell extracts. What Stillman calls "molecular shrapnel" was shot at the yeast DNA, splitting it into random pieces. However, if the initiator protein is bound over the origin, it protects the DNA from being cut. With this information, a "footprint" of the initiator protein binding to a particular set of DNA sequences was pieced together, and, using special biochemical techniques, the protein was extracted and purified from the complex. This protein complex, it was found, bound to all of the yeast's replication origins. Moreover, although this initiation protein complex binds strongly to natural, wild-type ARs sequences, it does not bind to inactive DNA elements whose ARs have been tinkered with, and destroyed, by mutations.
Like the initiator protein T antigen, but unlike most other DNA binding proteins of viruses, this newly discovered protein complex needs the addition of energy from the compound adenosine triphosphate (ATP) to work properly. In yeast the initiator protein appears to bind the DNA by wrapping the DNA around itself "and is reminiscent of the pattern observed when the E. coli initiation protein dna A binds to its cognate origin," says Stillman. But the development of new reagents, such as antibodies against particular protein subunits and cloning of the genes encoding the multiple protein subunits, "will help us understand its many roles in initiating DNA replication," Stillman adds.
IMPLICATIONS FOR THE FUTURE
Discovery of a yeast initiator protein complex is significant for many reasons, as has been recognized by the editors of several leading scientific publications, who rushed commentaries on this finding into print. First, the presence of at least six protein subunits suggests that the triggering of DNA replication may be controlled in a complicated way. In fact, Stillman has noted that there is precedent for the use of multiple proteins to recognize an origin of DNA replication. In 1991 it was discovered by Michael Botchan and his colleagues at the University of
California, Berkeley, that the bovine papilloma virus initiator proteins, E1 and E2, recognize their origin of replication cooperatively.
Second, Stillman suggests there may be important parallels between the mechanisms for initiating duplication in DNA and the mechanisms for controlling expression of the replicated DNA. Both may use an array of multipartite switches, in which a critical number are turned on before the system is set in operation. In particular, Stillman "proposes that an origin of DNA replication in yeast, element 'A,' provides an initiator function which, like the TATA box of a eukaryotic promoter, is essentially inactive unless stimulated by the other 'B' elements, called activator elements."
Discovery of the origin recognition complex will also allow other experiments that may reveal why the initiation of DNA replication at each origin is limited to one round only per cell cycle. It has been suggested that the initiator protein function is destroyed following each initiation event or that a newly replicated DNA strand is made insensitive to the presence of the initiator. With an initiator protein complex in hand, these ideas can be tested. In fact, Joachim Li of the University of California, San Francisco, and Bruce Alberts, formerly at that institution and now president of the National Academy of Sciences, have noted that, "By following the fate of the Saccharomyces cerevisiae initiator protein during the replication reaction and throughout the cell cycle, we can expect to learn how the cell prepares itself for a new round of initiation and how it prevents those preparations from occurring prematurely."
Another possibility is to use information about the yeast origin recognition protein to fill gaps in our understanding of the cell cycle (see Figure 4.6). For example, much attention has been given to the study of the various biochemical events of the G1 phase immediately prior to the synthesis of DNA. It is known that certain genes that are active during this phase produce enzymes known as kinases, which are important in the energy transfer reactions known as phosphorylations. Using the initiation protein complex as a starting point, it may be possible to work backwards to define a relationship between this complex and the important kinases produced during the preceding G1 phase. Perhaps some of the products produced in the G1 phase help with the synthesis or activity of one of the proteins that make up the origin recognition complex.
Still another strategy for learning more about the many proteins needed for eukaryotic DNA duplication is to work backwards from the yeast origin recognition complex to the genes that code for it. Joel A. Huberman, of the Roswell Park Cancer Institute, Buffalo, New York, has
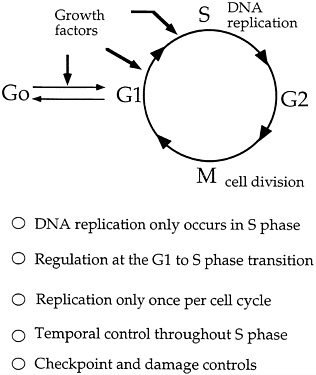
FIGURE 4.6 Cell cycle and regulation of DNA replication. (Courtesy of Bruce Stillman, Cold Spring Harbor Laboratory.)
noted that, because Bell and Stillman have already succeeded in getting pure preparations of the origin recognition complex (ORC), "We won't have long to wait until the individual polypeptides are purified and their genes cloned." Then, Huberman adds, "It will … be possible to compare the predicted amino acid sequences of the ORC polypeptides to those of known proteins; any similarities that turn up will suggest roles for the individual polypeptides in the initiation process." And, add Li and Alberts, "The discovery of homologues to the ORC, and analysis of their DNA binding sites, could provide a shortcut to defining replication origins in [other] higher eukaryotes."
Finally, identification of the yeast ORC may permit a scheme for replicating eukaryotic DNA in the test tube, a procedure that would
greatly ease its study. ''Learning how to unleash them [the ORC] in vitro could therefore reveal how this protein is regulated in vivo," say Li and Alberts.
In short, discovery of the multiprotein initiation factor in yeast provides a long sought after key for unlocking the secrets of the genomes of advanced organisms. For the past 40 years necessity often demanded that most research on the molecular details of DNA replication and expression be done on relatively simple, prokaryotic organisms. Now, a new era of biological studies could begin, one that puts more emphasis on the workings of the genome in advanced organisms, including humans.
EPILOGUE
Just as the discovery of the T antigen, dna A protein, and yeast origin recognition complex have yielded important clues about how DNA replication starts, the discovery of other proteins has revealed other details about the later stages of DNA replication. In most cases such information has been gained by analyses of mutant microbes that fail to replicate properly and are defective in a single protein. But the search can be very difficult since some proteins are only partially active in isolation and may have detectable activity only if found with large and, often elusive, protein complexes. Still, despite such difficulties, knowledge of the large number of proteins needed to complete DNA replication in E. coli is quite detailed, and there has been reasonable, but slow, progress in defining those proteins that have a role in the replication of eukaryotic DNA.
Here is a rundown of the key accessory proteins—sometimes called the protein machine—that help DNA to copy itself. Although the polypeptide components of the machinery may differ in detail from organism to organism, enough is known about them to conclude that some key polypeptides fill certain roles in DNA replication in all organisms. The proteins described here go into action after the initiator proteins described earlier.
Proteins That Keep Replication in Order
Once DNA is unraveled and replication begins, the active site of replication advances in both directions along the DNA strand until it meets an advancing replicating segment from a neighboring origin or the entire chromosome is copied. The spot where the newly synthesized
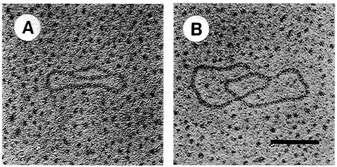
FIGURE 4.7 Electron microscopy of in vitro replication intermediates. DNA was mounted for electron microscopy by the method of Davis et al. The extent of replication of molecule A is approximately 20 percent and that of molecule B is approximately 90 percent. Bar = 0.2 μm. (Reprinted by permission from Li and Kelly, 1984.)
strands meet the original unreplicated region is called a replication fork (see Figure 4.7). At each growing fork, one new DNA strand, called the leading strand, grows continuously in the same direction of the fork; the second strand, the so-called lagging strand, grows in the opposite direction (see Figure 4.8). At the critical juncture of the growing fork, a large collection of accessory proteins is needed to keep the complex process in order.
Helicases and Single-Stranded Binding Proteins
Among the first proteins to go into action are the helicases, which unwind the DNA and prepare the template strand for copying. These enzymes are found at every replication fork. A related protein, known as the single-stranded DNA binding protein or the helix destabilizing protein, also comes into action early on in replication. It binds to a site where the double helix has unwound but is not yet copied. It is thought to work by preventing the reformation of double helix, single-stranded tangles, or odd structures, such as hairpin loops, all of which would upset replication. The single-stranded DNA binding protein speedily dissociates as a different enzyme, DNA polymerase, approaches.
The Polymerases
The DNA polymerases were first recognized for their ability to add deoxynucleotides into DNA. All DNA polymerases discovered so far can only elongate preexisting chains: they can't start them. These enzymes were first purified from E. coli, where at least three different
polymerases have been described, and others have since been extracted from more advanced organisms, too. In fact, many years after DNA polymerases were purified from E. coli, four were identified from mammalian cells. Some of their roles are known, although they remain to be as well described as their cousins in E. coli.
In E. coli one complex polymerase, known as the DNA polymerase III holoenzyme, is responsible for the elongation of DNA chains at the growing fork. It is made up of two multiunit polymers, and it speeds along the parent chain, residue by residue, assisting with the addition of nucleotides, one by one. One segment of the complex, known as the alpha subunit, seems to catalyze the addition of individual nucleotides, and it does so most efficiently, at a rate inside the cell of about 1000 nucleotides per second. This is one of the fastest polymerization reactions known, in vivo. One factor that assists DNA polymerase III is its tenacity. It is believed that the giant asymmetric polymerase III clamps on to the growing DNA chain throughout the polymerization, instead of dissociating and binding randomly to other growing chains.
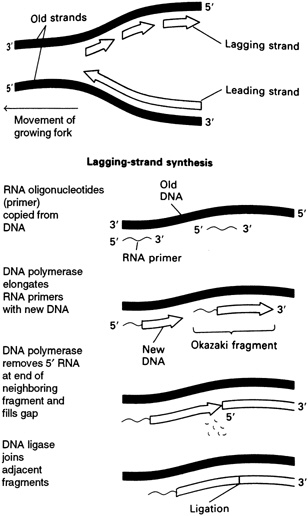
Exonucleases, Polymerases, and Ligases That Piece Together the Leading and Lagging Strands
As mentioned earlier, two different types of DNA strands are made at the replication fork: the leading strand builds in the direction of the replication fork, and the other, the lagging strand, builds in the opposite direction. These two strands are made by different means, requiring different enzymes.
The primer for synthesis of the leading strand is thought to be a loose end from a newly made strand at the origin of DNA replication, which then grows continuously. In contrast, synthesis of the lagging strand is carried out in a discontinuous manner. Each DNA fragment on the lagging strand, known as an Okazaki fragment, is begun with a ribonucleic acid (RNA) primer. Many such fragments are attached to primers extending along the lagging strand template and, eventually, the RNA primers are cut off by enzymes known as exonucleases. Then what comes into play is a particular type of DNA polymerase, known as DNA polymerase I, that fills in the gaps left by the removal of the RNA primers by adding deoxynucleotides. Finally, the various pieces of the lagging strand are cleanly sealed with the aid of another enzyme, DNA ligase.
The Replisome Protein Complex Keeps All in Order
Keeping synthesis of the leading and lagging strands in phase is nontrivial, and it is thought that a special protein complex, called the replisome, is needed to orchestrate fast and clean synthesis. It is made of a large collection of accessory proteins: Two DNA polymerase III holoenzymes, one for the leading strand and another for the lagging strand; a primosome complex, which includes the enzyme primase (the enzyme that makes the short RNA primer on the Okazaki fragments); dna B helicase, a second helicase (sometimes called a rep protein), the single-stranded DNA binding protein; and others. The concerted action of these various accessory proteins keeps synthesis of the leading and lagging strands clean flowing.
The Topoisomerases Adjust Swivel to DNA
The protein apparatus of the replisome appears to prevent tangles at the outset of synthesis, but another set of proteins, the topoisomerases, probably help resolve tangles that are created later in synthesis and exist in newly replicated DNA: they seem essential to the successful conclusion of replication and the separation of finished chromosomal copies.
It is well established that DNA has an unusual twisted, double helical structure. The tightness of its coiled form is described in terms of the linking number, which describes the number of times one strand of a helix winds around another; twisting, which refers to the periodicity of the winding; and writhe, the reciprocal of twist. Typically, the linking number equals twist plus writhe. Underwinding yields negative supercoils, and overwinding yields positive supercoils. Once it was realized that DNA replication and its subsequent packaging into chromosomes depended on supercoiling, a search began for those enzymes that could affect DNA topology. They are the topoisomerases.
The first of these to be discovered was the omega protein in E. coli , which can remove negative supercoils. This type I topoisomerase does this by breaking a single strand of DNA, passing the intact strand through the gap, and then resealing the gap, thus changing the extent of strand interwinding. If this process is carried out repeatedly, supercoiled DNA can be relaxed. The so-called E. coli topo I is less active on positively supercoiled than on negatively supercoiled molecules. However, those topo I enzymes found in eukaryotes are more versatile: They relax both positive and negative supercoils. Bacteria and yeast with mutant topo I enzymes suffer a slower rate of growth, but the mutant is
not lethal. These enzymes also seem involved in the transcription or "reading" of DNA.
Another group of topoisomerases, found in all cells, is called Type II or, in bacteria, gyrase. They can change the linking number by cutting and then resealing a split double helix. There appear to be two forms of the Topo II, alpha and beta, which differ in a number of biochemical properties. The alpha variety seems to bind better to DNA segments that are rich in the bases adenine and thymine; and the beta variety prefers DNA sequences that are rich in the bases guanine and cytosine. Most important, the topoisomerases appear essential for segregating the replicated daughter chromosomes, and they may also be needed to resolve complicated, interlocked, or knotted DNA strands.
Using Protein Editors to Correct Errors
An unusual feature of replicating DNA that requires the help of yet more accessory proteins is its ability to correct errors. DNA is the only cellular macromolecule that can be self-repaired, probably because the cost of damaged DNA outweighs the costs of repair to the cell. If accumulated errors, both those made during the act of replication and those brought on by environmental factors, such as thermal stress and chemical mutagens, are left uncorrected, growing cells might accumulate so much genetic misinformation that they could no longer function. Faulty proteins would be produced at an unacceptable rate, barring normal cellular activities. Moreover, DNA in sex cells might be so damaged as to prevent the production of viable offspring. So a system is needed to identify errors in DNA and to make corrections.
What happens in E. coli, for example, is that one segment of the DNA polymerase III holoenzyme, the ε subunit, proofreads newly synthesized DNA by recognizing the distortions produced by an incorrectly paired base. This is probably done by some physical means where the enzyme detects a malformation in the three-dimensional structure of the DNA. For example, if ultraviolet radiation, which can disrupt thymine bases by causing the dimerization of their pyrimidine structures, skews a nascent DNA strand, this can be "felt" by a probing enzyme. The damaged bit is then cut out by a special enzyme, known as an endonuclease, before extension of the strand is complete. Repairs are then made by the now familiar enzymes, DNA polymerase I and DNA ligase. Such inspections and repairs are made with exceptional efficiency, assuring that DNA is replicated with great fidelity. For example, a wrong base is added by the holoenzyme to the growing chain about once every 10,000 steps, and the
correction activity itself errs about once every 1000 steps. This gives an overall error rate of 10−7, one of the lowest rates for any enzyme.
DNA repair in more advanced organisms is less well understood. But studies of humans with xeroderma pigmentosum, a malady characterized by easily pigmented skin and vulnerability to skin cancer, has suggested that they lack the ability to repair DNA that has been damaged by ultraviolet radiation. Some other diseases, such as the leukemias, Franconi's anemia and Bloom's syndrome, are associated with heightened sensitivity to agents that can damage DNA.
Additional progress in learning about DNA repair in eukaryotes has come from studies of mutant yeast and Chinese hamster cells. For example, yeast mutants sensitive to damage by ultraviolet light have been used to locate several radiation-sensitive genes, including one that directs production of a helicase, an enzyme that unwinds DNA and is essential to the normal replication. Moreover, ultraviolet-sensitive Chinese hamster cells have been manipulated to reveal a human gene that is involved in DNA repair.
Both these lines of study may reveal more molecular details about DNA repair in advanced organisms, including humans. But they could also offer an explanation of how uncorrected damage to DNA leads to the uncontrolled proliferation of DNA and cell growth characteristic of cancer. Such research offers a clear link between the erudite aspects of DNA synthesis described earlier and the more general goal of using basic science to reduce human suffering.
BIBLIOGRAPHY
Bell, S. P., and B. Stillman. 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357(May 14):128–134.
Diffley, J. F. X., and B. Stillman. Dec. 1990. The initiation of chromosomal DNA replication in eukaryotes. Trends in Genetics 6(12):427–432.
Darnell, J., H. Lodish, and D. Baltimore. 1990. Molecular Cell Biology, Chapter 12. Scientific American Books. pp. 449–487.
Fangman, W. L., and B. J. Brewer. 1992. A question of time: Replication origins of eukaryotic chromosomes. Cell 71(Oct. 30):363–366.
Glotzer, M., A. W. Murray, and M. W. Kirschner. 1991. Cyclin is degraded by the ubiquitin pathway. Nature 349(Jan. 10):132–138.
Huberman, J. A. 1992. Quest's end and question's beginning. Current Biology 2(7):351–352.
Li, J. J., and B. M. Alberts. 1992. Eukaryotic initiation rites. Nature 357(May 14):114–155.
Li, J. J., and T. J. Kelly. 1984. Proceedings of the National Academy of Sciences USA 81:6976.
Marahrens, Y., and B. Stillman. 1992. A yeast chromosomal origin of DNA defined by multiple functional elements. Science 255(Feb. 14):817–823.
Murray, A. W. 1992. Creative blocks: Cell-cycle checkpoints and feedback controls. Nature 359(Oct. 15):599–604.
Smith, P. J. 1990. DNA Topoisomerase dysfunction: A new goal for antitumor chemotherapy. BioEssays 12(4).
Tsurimoto, T., T. Melendy, and B. Stillman. 1990. Nature 346(Aug. 9):534–539.
RECOMMENDED READING
Murray, A. W., and M. W. Kirschner. 1991. What controls the cell cycle. Scientific American (March):56–63.
Stillman, B. 1989. Initiation of eukaryotic DNA replication in vitro. In Annual Reviews of Cell Biology 5:197–245.


























