
4
Toxicology
In this chapter, the results of experiments in which animals were exposed to the substances of concern and observed for particular effects are reviewed, to provide a basis for evaluating the biologic plausibility of the epidemiologic evidence associating exposures and effects described in Chapters 8-11. Assessing the biologic plausibility of the outcomes reported in epidemiologic studies would strengthen any evidence for an association between exposures and effects.
Although there is evidence that multiple chemicals were used for various purposes in Vietnam, the use of four herbicides has been documented in military records; therefore, toxicologic assessment was limited to the compounds 2,4-dichlorophenoxyacetic acid (2,4-D), 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), picloram, and cacodylic acid (Figure 4-1). In addition, the toxicologic properties of a 2,4,5-T contaminant that has caused a great deal of controversy, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), are described. The emphasis of the chapter is on the effects of TCDD, because there is considerably more information available on TCDD than on the herbicides.
The chapter begins with an overview that describes toxicology data on TCDD and the four herbicides in nontechnical terms. The overview is followed by complete toxicity profiles of each of the five substances considered. In reading these profiles, several characteristics of animal studies should be borne in mind. First, animals are exposed to various levels of a compound through multiple routes of exposure. In addition, animals may be exposed once to a very high dose of a compound or multiple times to lower doses. Thus, an effect observed in animals may not necessarily occur
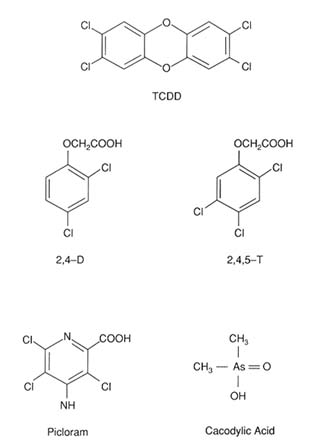
FIGURE 4-1 Chemical structures of the herbicides 2,4-D, 2,4,5-T, picloram, and cacodylic acid, and of the contaminant TCDD.
in humans because of differences in dose, route, and timing of exposure. Second, for the most part, animals are exposed to a single agent and are generally healthy when exposure occurs. Although most of the people exposed to TCDD who are of interest in this report were healthy, they were certainly not exposed solely to TCDD. Third, the toxicity of a given compound varies widely depending on the health status (as determined by nutrition, age, infection, etc.) of the animal examined. When data are available, the contribution of nutrition, age, and other possible factors to the toxicity of the compounds is discussed. Fourth, there is a wide variability in the toxicity of TCDD depending on the species of animal tested. These differences are exemplified in the dose of TCDD required to kill 50 percent of the animals exposed (LD50) (Table 4-1).
In the guinea pig (the most sensitive species to acute lethality by TCDD), the LD50 is 0.6-2.1 µg/kg. On the other hand, the LD50 of TCDD for the hamster (the least sensitive species examined) is 1,157-5,051 µg/kg. It is currently unknown where on this spectrum humans lie; however, studies are under way to determine the sensitivity of humans to a number of effects of TCDD. Lastly, individuals within a species may vary widely in their sensitivity to the effects of a chemical. For example, two strains of mice, C57Bl/6 (sensitive) and DBA/2 (resistant), are very different in their sensitivity to the acute toxicity of TCDD. Studies involving congenic mice (mice that are identical at all genetic sites except one) suggest that for many of the toxicologic
TABLE 4-1 Acute Lethality of TCDD to Various Species and Substrains
|
Species/Strain/Sex |
Route |
LD50 (µg/kg) |
References |
|
Guinea pig/Hartley (male) |
Oral |
0.6-2.1 |
McConnell et al., 1978a; Schwetz et al., 1973 |
|
Mink/not reported (male) |
Oral |
4.2 |
Hochstein et al., 1988 |
|
Chicken/not reported |
Oral |
8 25 |
Greig et al., 1973 |
|
Monkey/rhesus (female) |
Oral |
~ 70 |
McConnell et al., 1978b |
|
Rat/L-E (male) |
Intraperitoneal |
~ 10 |
Tuomisto and Pohjanvirta, 1987 |
|
Rat/Sherman, Spartan |
Oral |
|
Schwetz et al., 1973 |
|
male |
|
22 |
|
|
female |
|
13-43 |
|
|
Rat/Sprague-Dawley |
Intraperitoneal |
|
Beatty et al., 1978 |
|
male |
|
60 |
|
|
female |
|
25 |
|
|
weaning male |
|
25 |
|
|
Rat/Fischer Harian (male) |
Oral |
340 |
Walden and Schiller, 1985 |
|
Rat/H/W/ (male) |
Intraperitoneal |
> 3,000 |
Pohjanvirta and Tuomisto, 1987; Pohjanvirta et al., 1988a |
|
Mouse/B6 (male) |
Oral |
182 |
Chapman and Schiller, 1985 |
|
D2A/2J (male) |
|
2,570 |
|
|
B6D2F1 (male) |
|
296 |
|
|
Mouse/B6 |
Intraperitoneal |
132 |
Neal et al., 1982 |
|
Mouse/D2 |
|
620 |
|
|
Mouse/B6D2F1 |
|
300 |
|
|
Rabbit/New Zealand white (male and female) |
Oral |
115 |
Schwetz et al., 1973 |
|
|
Dermal |
275 |
|
|
Rabbit/New Zealand white (male and female) |
Intraperitoneal |
~50 |
Brewster et al., 1988 |
|
Hamster/golden Syrian (male and female) |
Oral |
1,157-5,051 |
Henck et al., 1981 |
|
Hamster/golden Syrian (male and female) |
Intraperitoneal |
> 3,000 |
Olson et al., 1980b |
|
SOURCE: U.S. EPA, 1992. |
|||
effects described below, the differences in the sensitivity of these two strains are due to differences in the affinity of an intracellular protein—referred to as the Ah receptor—for TCDD. Although all of these considerations have implications for the interpretation of the data described below, it should be kept in mind that the primary purpose of this review is to contribute to a consideration of the biologic plausibility of the associations observed in epidemiologic studies that are relevant to herbicide exposure in Vietnam, not to resolve the continuing scientific and regulatory concerns about TCDD.
OVERVIEW
Information from tests in laboratory animals and other nonhuman systems is useful because it can be combined with information obtained from humans exposed to the herbicides (described in Chapters 6 and 7) to determine the biologic plausibility for health effects observed in humans (described in Chapters 8-11). Establishing the biologic plausibility of effects due to herbicide exposure in the laboratory strengthens the evidence for any effects of the herbicides that are suspected to occur in humans.
The herbicides that were used in the greatest quantities in Vietnam were 2,4-D, 2,4,5-T, picloram, and cacodylic acid. Agent Orange was a one-to-one mixture of 2,4-D and 2,4,5-T. A contaminant of 2,4,5-T, 2,3,7,8-tetrachlorodibenzo-p-dioxin (commonly called TCDD or dioxin), was found at varying levels in different batches of Agents Orange, Pink, Purple, and Green.
Chemistry
TCDD forms as a by-product during the manufacture of 2,4,5-T. TCDD molecules contain carbon, hydrogen, oxygen, and chlorine. TCDD dissolves easily in fats and oils but not in water, and is persistent in the environment. The primary source of TCDD in the environment is combustion and industrial processes, but the primary source of human exposure is through food.
2,4-D and 2,4,5-T are called chlorophenoxy acids and are also made up of carbon, hydrogen, oxygen, and chlorine. They both dissolve in water and are very similar in structure to a natural plant hormone called auxin. As a result of this similarity, 2,4-D and 2,4,5-T can mimic the action of auxin in some plants, and this activity is thought to be the reason these chemicals are herbicidal.
Cacodylic acid contains carbon, hydrogen, oxygen, and arsenic and was called Agent Blue. Picloram contains carbon, hydrogen, oxygen, chlorine,
and nitrogen, and was combined with 2,4-D to become Agent White. Both compounds dissolve in water.
Exposure and Metabolism
When exposure to a chemical occurs, its effects on the body depend on a number of factors: it can be absorbed into the body, it can be distributed to different organs in the body, it can be metabolized by enzymes that change its chemical structure, and it can be eliminated from the body. A chemical's effects ultimately depend on the rate and extent to which all of these activities occur.
When TCDD is ingested by animals (e.g., through contaminated food), more than 50 percent is absorbed into the body through the gastrointestinal tract. Most of the TCDD breathed in the air is thought to be absorbed through the lungs, but this route of exposure is not well-studied. In contrast, TCDD is not absorbed well through the skin. The same pattern of absorption holds true for 2,4-D and 2,4,5-T, and probably for picloram and cacodylic acid, although much less information is available for them.
After a chemical is absorbed into the body, it can be transported to different organs through the blood or lymph system. TCDD is transported by both systems of circulation, and is distributed primarily to the liver and to body fat. Following single doses of TCDD to rats, a dose-related increase occurred in the proportion of the dose that distributed to the liver as compared to the fat. This observation may be due to increased binding of TCDD to liver cells as the doses increased, as well as to the loss of body fat that occurs in rats as doses of TCDD increase. The amount of time that TCDD remains in the liver or fat is different for different species: in rats, TCDD remains in fat longer than in the liver; in mice, it stays in both for about the same time; and in monkeys, it stays in fat for a very long time. Mice and rats eliminate TCDD from the body in both urine and feces, whereas all other species studied eliminate TCDD primarily through feces.
2,4-D and 2,4,5-T are distributed widely in the body and are eliminated quickly, mostly in the urine. The distribution patterns of picloram and cacodylic acid are not known, although they are eliminated rapidly from the body, mostly in urine. Some of the cacodylic acid that is absorbed is bound to red blood cells, however, and is eliminated when the red blood cells to which it is bound die naturally. Although cacodylic acid binds readily to rat red blood cells, it does not bind readily to human red blood cells.
TCDD is removed slowly from the body; as discussed later in Chapter 6, it takes more than 10 years for half of the body burden of TCDD to be removed. TCDD is metabolized by enzymes in the liver to form derivatives that can dissolve in water and thus be more easily eliminated from the body than TCDD itself, which does not dissolve in water. Water-soluble derivatives
of TCDD are thought to be much less toxic to animals than TCDD itself, although at present, no significant correlations have been made between the distribution, metabolism, and elimination of TCDD and its toxicity in different species.
2,4-D, 2,4,5-T, and cacodylic acid are not metabolized to any significant extent in the body. It is not known whether picloram is metabolized.
Carcinogenicity: TCDD
The ability of TCDD to cause cancer in animals has been studied using rats, mice, and hamsters exposed to TCDD for between one and two years. In these studies, TCDD was fed to animals, applied to their skin, injected under their skin, or injected into their abdominal cavities. Table 4-2 summarizes the results of the different studies that have been performed in animals to evaluate the ability of TCDD to cause cancer.
As the table shows, increased tumor rates have been reported to occur at several different sites in the body in different studies, although the liver was consistently a site of tumor formation in different studies and different species. In studies in which liver cancer occurred, other toxic changes in the liver also occurred. Other organs in which increased cancer rates were observed in animals exposed to TCDD include the thyroid and adrenal glands, the skin, and the lung. Organs in which decreased cancer rates were seen in animals exposed to TCDD include the uterus, pancreas, and the pituitary, mammary, and adrenal glands.
In addition to increasing cancer rates in animals by itself, TCDD can increase tumor formation by other chemicals. For example, when a single dose of a known carcinogen is applied to the skin of mice and that dose is followed by multiple doses of TCDD over a period of several months, more skin tumors are seen than would be expected from the single dose of carcinogen alone. Similar results are obtained in rat livers when a single dose of a liver carcinogen is followed by multiple doses of TCDD.
In rats, liver tumor formation associated with TCDD exposure is dependent on the presence of ovaries; in other words, only female rats that have not had their ovaries removed can develop liver tumors when they are exposed to TCDD. This observation indicates that complex hormonal interactions are likely to be involved in TCDD-induced carcinogenesis.
Mechanism of Action
TCDD has a wide range of effects on growth regulation, hormone systems, and other factors associated with the regulation of activities in normal cells. TCDD may thus play a number of different roles that could affect tumor formation. Understanding how TCDD affects tumor formation in
TABLE 4-2 Summary of Carcinogenicity Bioassays of TCDD
|
Reference |
Species/Strain/Sex |
Protocol |
Results |
|
Van Miller et al., 1977 |
Sprague-Dawley rats, male, 10/group |
0.001-1,000 ppb (0.0003-500 µg/kg/wk) in feed for 78 weeks; observed for 17 weeks |
High mortality, poor reporting; total tumors increased in all but lowest dose group; possible increase in lung tumors and liver tumors; no tumors in controls |
|
Kociba et al., 1978 |
Sprague-Dawley rats, male and female, 86/control group, 50/treated groups |
21-2,200 ppt (0.001-0.1 µg/kg/day) in feed for 2 years |
Males: increased tumors of tongue, nose/palate; females: increased tumors of lung, liver, nose/palate |
|
Toth et al., 1979 |
Swiss mice, male, 100/control group, 45/treated groups |
0.007-7.0 µg/kg/wk by gavage for 1 year; observed for life spans |
Liver tumors in 0.7 group; none in 0.007 group; higher dose died |
|
NTP, 1982a |
Osborne-Mendel rats, male and female, 75/control group, 50/treated groups |
0.0014-0.071 µg/kg/day by gavage for 2 years |
Males: increased tumors of thyroid and skin; females: increased tumors of skin, liver, and adrenal gland |
|
NTP, 1982a |
B6C3F1 mice, male and female, 75/control group, 50/treated groups |
Males: 0.0014-0.071 µg/kg/day; females: 0.0057-0.29 µg/kg/day; by gavage for 2 years |
Males: increased tumors of lung and liver; females: increased lymphoma and tumors of liver, thyroid gland, skin |
|
NTP, 1982b |
Swiss-Webster mice, male and female, 45/control group, 30/treated groups |
0.001-0.005 mg/dermal application, 3 times weekly for 2 years |
Males: no effect; females: increased skin fibrosarcomas |
|
Della Porta et al., 1987 |
B6C3F1 mice, male and female, 42-50/group |
2.5-5.0 µg/kg/week by gavage for 52 weeks; observed until 78 weeks |
Both sexes: increased hepatocellular carcinoma |
|
|
B6C3F1 and B6CF1 mice, male and female, 89-106/group |
1-30 µg/kg/week by intraperitoneal injection for 5 weeks; observed until 78 weeks |
All: increased lymphoma; B6C3F1 males: increased hepatocellular adenomas and carcinomas |
|
Rao et al., 1988 |
Syrian golden hamsters, male |
100 µg/kg by intraperitoneal injection; 2-6 treatments over a 4-week period; observed until 12-13 months |
Increased squamous cell carcinoma of facial skin |
|
|
|
50-100 µg/kg by subcutaneous injection; 2-6 treatments over a 4-week period; observed until 12-13 months |
Increased squamous cell carcinoma of facial skin |
|
SOURCE: Adapted from Huff, 1992. |
|||
laboratory animals may help us understand whether TCDD would affect tumor formation in humans. For example, when a chemical's ability to induce tumors in animals is tested, it is administered at doses much higher than those to which humans are normally exposed in the environment. High doses of chemicals can cause toxic effects in animals that may increase their sensitivity to carcinogenesis; in other words, cancer can occur at high doses because of effects that would not occur at low doses (Cohen and Ellwein, 1990). In this case, it would not be appropriate to conclude that a chemical that caused cancer in laboratory animals would do so in humans. Understanding how a chemical causes cancer is thus a very important consideration when using information obtained in the laboratory to evaluate effects in humans.
A normal cell can be transformed into a cancer cell when the information that is coded into the DNA of the cell is changed in critical places. Such changes are called mutations and may result from the direct interaction of a chemical with DNA. TCDD is not considered toxic to DNA; that is, tests of its ability to alter the structure of DNA have been negative.
Another way that a normal cell can be transformed into a cancer cell is when changes occur in the regulation of the manner in which the information encoded in DNA is expressed, and incorrect information is received by the cell. Regulation of DNA is performed by proteins called receptors, which interact both with other molecules and with specific sites on DNA. There is a receptor in liver cells (and probably other cells as well), called the Ah receptor, that can interact with TCDD and then with sites on DNA. Binding of TCDD and the Ah receptor to each other and then to DNA results in a number of biologic effects such as increasing the activity of certain enzymes and affecting the levels of hormones and of molecules that control tissue growth. For example, TCDD treatment can increase the rate at which liver cells multiply; both this effect and TCDD-induced liver tumor formation are dependent on the presence of ovaries. It is thus possible that TCDD, together with the Ah receptor, could alter the information obtained from DNA in such a way that a normal liver cell is transformed into a cancerous liver cell, although direct proof of this possibility has not been obtained.
Carcinogenicity: Herbicides
Several studies of the carcinogenicity of 2,4-D, 2,4,5-T, picloram, and cacodylic acid have been performed in laboratory animals. In general they have produced negative results, although some were not performed using rigorous criteria for the study of cancer in animals, and some produced equivocal results that could be interpreted as either positive or negative. The studies and their results are summarized in Table 4-3.
2,4-D was administered to rats, mice, and dogs in their food, by injecting it under their skin, or by placing it directly into their stomachs. All the results were negative, except for one study that found an increased rate of brain tumors in male rats, but not female rats, receiving the highest dose. These tumors also occurred in the control group and might have occurred spontaneously and not as a result of 2,4-D exposure, however. In another study, the occurrence of cancer of the lymph system (malignant lymphoma) among dogs kept as pets was found to occur more frequently when owners used 2,4-D on their lawns than when they did not (although this test had limitations). These dogs were exposed to other chemicals in addition to 2,4-D, however. Another test using dogs exposed to 2,4-D in the laboratory produced negative results, so it is not clear whether 2,4-D was responsible for the lymphomas in dogs.
2,4,5-T has been tested in rats and mice in their food, in their drinking water, by injecting it under their skin, or by placing it directly into their stomachs. Cacodylic acid has been tested in a very limited study in mice both in their food and by placing it directly into their stomachs. Picloram has been tested in rats and mice in their food. Results of all of these studies were uniformly negative, with the exception of one study using picloram in which liver tumors appeared but were attributed to the presence of hexachlorobenzene as a contaminant.
Mechanism of Action
In the absence of any compelling evidence that the herbicides used in Vietnam are carcinogens in animals, it is difficult to draw conclusions regarding their mechanisms of action as such. The mechanisms of action of the herbicides have not been studied to the same extent as TCDD. Neither 2,4-D nor 2,4,5-T is considered toxic to DNA; that is, they do not interact directly with or change the structure of DNA. Tests on cacodylic acid indicate that it is toxic to DNA only at very high doses, and tests with picloram are extremely limited, but suggest that it is not toxic. None of these compounds is metabolized to reactive intermediates. They do not accumulate in the body. Thus there is as yet no convincing evidence of, or mechanistic basis for, the carcinogenicity in animals of any of the herbicides used in Vietnam.
Immunotoxicity: TCDD
The immune system is a complex network of cells and molecules that play an important role in the maintenance of health and resistance to infection. Suppressing the activity of the immune system could lead to an increase in the incidence and severity of infectious disease and an increase in
TABLE 4-3 Summary of Carcinogenicity Bioassays of Herbicides Used in Vietnam
|
Reference |
Species/Strain/Sex |
Protocol |
Results |
|
Bionetics, 1968a; Innes et al., 1969 |
Strain (C57BL/6×C3H/Anf)F1 and (C57BL/6×AKR)F1 mice, male and female, 18/group |
46.4 mg 2,4-D/kg by gavage at 7 days of age, the same amount unadjusted for body weight daily until 28 days of age, then 149 mg/kg diet until 78 weeks of age |
No effect |
|
|
Strain (C57BL/6×C3H/Anf)F1 and (C57BL/6×AKR)F1 mice, male and female, 18/group |
21.5 mg 2,4,5-T/kg by gavage at 7 days of age, the same amount unadjusted for body weight daily until 28 days of age, then 60 mg/kg diet until 78 weeks of age |
No effect |
|
|
Strain (C57BL/6×AKR)F1, male and female, 18/group |
100 mg 2,4-D/kg by gavage at 7 days of age, the same amount unadjusted for body weight daily until 28 days of age, then 323 mg/kg diet until 78 weeks of age |
No effect |
|
|
Strain (C57BL/6×C3H/Anf)F1 and (C57BL/6×AKR)F1, male and female, 18/group |
Single dose of 215 mg 2,4-D or 2,4,5-T/kg by gavage or subcutaneously on day 28 of age |
No effect |
|
Hansen et al., 1971 |
Osborne-Mendel rats, male and female, 25/group |
0, 5, 25, 125, 625, or 1250 ppm 2,4-D in the diet for 2 years |
No effect |
|
Hazleton, 1986 |
Fischer 344 rats, male and female, 60/group |
0, 1, 5, 15, or 45 mg 2,4-D/kg in the diet for 2 years |
Females: no effect; males: increased astrocytomas at high dose only |
|
|
B6C3F1 mice, male and female, 60/group |
0, 1, 15, or 45 mg 2,4-D/kg in the diet for 2 years |
No effect |
|
Hayes et al., 1991 |
Dogs kept as pets |
Case-control study, information from questionnaires and telephone interviews, no exposure data |
Household with dogs developing malignant lymphoma used 2,4-D more frequently than those that did not; odds ratio = 1.3 |
|
Hansen et al., 1971 |
Beagle dogs, male and female, 3/group |
0, 10, 50, 100, or 500 ppm 2,4-D in the diet for 2 years |
No effect |
|
Muranyi-Kovacs et al., 1976 |
XVII/G mice, 20 male and 19 female; C3Hf mice, 22 male and 25 female |
100 mg 2,4,5-T/l drinking water for 2 months, followed by 80 mg/kg diet for their life spans |
No effect |
|
Kociba et al., 1979 |
Sprague-Dawley rats, male and female, 60/group |
0, 3, 10, or 30 mg 2,4,5-T/kg/d in the diet for 2 years |
No effect |
|
Innes et al., 1969 |
Unspecified strain mice, male and female |
46.4 mg cacodylic acid/kg on day 7 of age, same amount unadjusted for body weight daily until day 28 of age, then 121 ppm (about 18 mg/kg/d) in the diet for 18 months |
No effect |
|
Stott et al., 1990 |
Fischer rats, male and female, 50/group |
0, 20, 60, or 200 mg picloram/kg/d in the diet for 2 years |
No effect |
|
NCI, 1978 |
Osborne-Mendel rats, male and female |
0, 10,000, or 20,000 ppm picloram (0, 500, or 1,000 mg/kg/d) in the diet for 39 weeks, then 0, 5,000, or 10,000 ppm for 41 weeks; observed for additional 33 weeks |
Increase in liver tumors attributed to contamination of picloram by hexachlorobenzene |
|
|
B6C3F1 mice, male and female |
0, 2,500, or 5,000 ppm picloram (0, 357, or 714 mg/kg/d) in the diet for 79 weeks; recovered for additional 10 weeks |
No effect |
some types of cancer. Increasing the activity of the immune system could result in the development of allergies and of autoimmune diseases. TCDD has been shown to have a number of effects on the immune systems of laboratory animals.
Studies in mice, rats, guinea pigs, and monkeys indicate that TCDD suppresses the function of certain components of the immune system in a dose-related manner; that is, as the dose of TCDD increases, its ability to suppress immune function increases. TCDD suppresses the function of cells of the immune system such as lymphocytes (cell-mediated immune response), as well as the generation of antibodies by B cells (humoral immune response). Increased susceptibility to infectious disease has been reported following TCDD administration. In addition, TCDD increased the number of tumors that formed when mice were injected with tumor cells.
The effects of TCDD on the immune system appear to vary among species, although most studies used different treatments and are not completely comparable. Studies indicate, however, that some species are more sensitive to the effects of TCDD on the immune system than others. It is not known whether humans would be more or less sensitive than laboratory animals.
Mechanism of Action
Studies of the mechanism of TCDD-mediated effects on the immune system are conflicting. Most studies indicate that the presence of the Ah receptor is required for TCDD-induced immunotoxicity, but other studies indicate that it is not. It is possible that the Ah receptor could play a role in some types of immunotoxicity and not in others. Additional studies indicate that an animal's hormonal status may contribute to its sensitivity to immunotoxicity. There is not enough information available on the mechanisms of TCDD-mediated immunotoxicity in laboratory animals to be able to predict whether it would be immunotoxic in humans, but the fact that TCDD induces such a wide variety of effects in animals suggests that it is likely to have some effect in humans as well.
Immunotoxicity: Herbicides
The potential immunotoxicity of the herbicides used in Vietnam has been studied to a very limited extent. Effects on the immune system of mice have been reported for 2,4-D administered at doses that were high enough to produce clinical toxicity, but these effects did not occur at low doses. The potential for picloram to act as a contact sensitizer (produces an allergic response on the skin) was tested, but other aspects of immunotoxicology
were not examined. The immunotoxicity of 2,4,5-T and cacodylic acid has not been evaluated in laboratory animals.
Reproductive and Developmental Toxicity: TCDD
TCDD has been reported to have a number of effects on the reproductive and developmental functions of laboratory animals. Reproductive toxicity is defined as the occurrence of adverse effects on the male or female reproductive system, whereas developmental toxicity is defined as the occurrence of adverse effects on the developing animal. Developmental toxicity can occur any time during the lifetime of the animal as a result of either parent's exposure to a toxic agent prior to conception, during the development of the fetus, or after birth until the time of puberty.
For example, administration of TCDD to male rats, mice, guinea pigs, marmosets, monkeys, and chickens can elicit reproductive toxicity by affecting testicular function, decreasing fertility, and decreasing the rate of sperm production. TCDD has also been found to decrease the levels of hormones such as testosterone in rats. These effects generally occur only at doses that are high enough to produce clinical toxicity, however, and are much less common at low doses. The reproductive systems of adult male laboratory animals are considered to be relatively insensitive to TCDD because high doses are required to elicit effects. Potential developmental toxicity following exposure of male animals to TCDD has not been studied.
Studies in female animals are limited but demonstrate reduced fertility, decreased ability to remain pregnant throughout gestation, decreased litter size, increased fetal death, impaired ovary function, decreased levels of hormones such as estradiol and progesterone, and increased rates of fetal abnormalities. Most of these effects may have occurred as a result of TCDD's general toxicity to the pregnant animal, however, and not as a result of a TCDD-specific mechanism that acted directly on the reproductive system.
Mechanism of Action
Little information is available on the cellular and molecular mechanisms of action that mediate TCDD's reproductive and developmental effects in laboratory animals. Evidence from mice indicates that the Ah receptor may play a role: mice with Ah receptors that have a relatively high affinity for TCDD respond to lower doses than mice with a relatively low affinity. Other as yet unidentified factors also play a role, however, and it is possible that these effects occur only secondarily to TCDD-induced general toxicity. Extrapolating these results to humans is not straightforward because
of the many factors that determine susceptibility to reproductive and developmental effects among species.
Reproductive and Developmental Toxicity: Herbicides
Several studies have evaluated the reproductive and developmental toxicity of herbicides in laboratory animals. Results indicate that 2,4-D does not affect male or female fertility and does not produce fetal abnormalities, but it did reduce the rate of growth of offspring and increase their rate of mortality when pregnant rats or mice were exposed. Very high doses were required to elicit these effects, however. The reproductive toxicity of 2,4,5-T has not been evaluated, although it was toxic to fetuses when administered to pregnant rats, mice, and hamsters. Studies of the reproductive toxicity of cacodylic acid are too limited to draw conclusions. Studies of its developmental toxicity indicate that it is toxic to rat, mouse, and hamster fetuses at high doses that are also toxic to the pregnant mother. Very limited data indicate that picloram is not a reproductive toxicant, although it may produce fetal abnormalities in rabbits at doses that are also toxic to the pregnant animal.
Studies of the reproductive toxicity of the herbicides are thus too limited to draw conclusions about their effects on male or female fertility. Studies of the developmental toxicity of the herbicides suggest that they can be toxic to developing animals, but high doses are required.
Other Toxicity: TCDD
TCDD has been reported to elicit several other kinds of toxicity in laboratory animals besides those described above. For example, the liver is a target organ for TCDD-induced toxicity in sensitive species. Sensitivity to TCDD-induced liver toxicity is dependent on the presence of Ah receptors with a high affinity for TCDD. Effects of TCDD on the liver include increasing the rate at which liver cells multiply, increasing the rate of liver cell death, increasing fat levels in liver cells, decreasing bile flow, and increasing the levels of protein and of substances that are precursors to heme synthesis. TCDD also increases the levels of certain enzymes in the liver, but this effect is not considered toxic. Mice and rats are susceptible to TCDD-induced liver toxicity, but guinea pigs and hamsters are not. It is possible that liver toxicity is associated with susceptibility to liver cancer.
Other toxic effects of TCDD that have been reported in laboratory animals include reduced blood glucose levels and starvation, increased rates at which cells in the gastrointestinal tract multiply, and changes in skin cells.
Other Toxicity: Herbicides
The herbicides used in Vietnam have also been reported to elicit adverse effects in a number of organs in laboratory animals. The liver is a target organ for toxicity induced by 2,4-D, 2,4,5-T, and picloram, with changes reportedly similar to those induced by TCDD. Some kidney toxicity has been seen in animals exposed to 2,4-D and to cacodylic acid. Exposure to 2,4-D has also been associated with effects on blood, such as reduced levels of heme and of red blood cells.
TOXICITY PROFILE OF TETRACHLORODIBENZO-p-DIOXIN
Introduction
''Dioxin" is a general term used to describe a subset of halogenated aromatic hydrocarbons, as listed in Table 4-4. The chemical structure of some of these compounds is shown in Figure 4-2. These chemicals are usually considered together because (1) their chemical structures are similar; and (2) they produce similar patterns of toxicity (although they differ in potency; Poland and Knutson, 1982).
As will be discussed further below, the greatest biologic potency is associated with halogenation at three or more lateral positions that gave the molecule a relatively planar configuration (Safe, 1986). Although there are
TABLE 4-4 Hierarchical Tree of Selected Halogenated Aromatic Hydrocarbons: Relationship of TCDD to Other Compounds
|
• Nonchlorinated • Chlorinated |
||||
|
|
• Polychlorinated dibenzofurans (PCDFs; furan) • Polychlorinated biphenyls (PCBs; biphenyl) • Polychlorinated dibenzodioxins (PCDDs; dioxin)a |
|||
|
|
|
• Dioxins with other than four chlorinesb • Tetrachlorinated dibenzodioxinc |
||
|
|
|
|
• Dioxins with four chlorines other than at the 2,3,7,8-positions • 2,3,7,8-Tetrachlorodibenzo-p-dioxind |
|
|
a Theoretically, 75 possible PCDDs differing only in the number of chlorine atoms and their location on the dioxin nucleus. b OCDD, for example, would refer to a molecule with eight chlorines (O=octa-) on the ring structure. c TCDD may exist as 22 different isomers, but the agent generally referred to as "TCDD" is the 2,3,7,8-isomer. d Numbering system refers to the position of the chlorines on the aromatic rings. SOURCE: Fishbein, 1987. |
||||
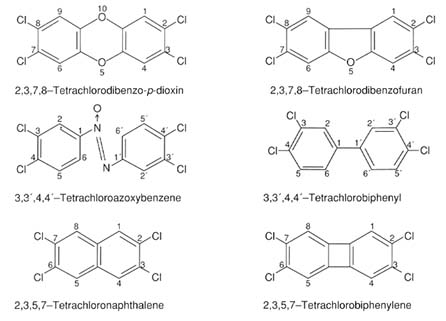
FIGURE 4-2 Chemical structures of some of the halogenated aromatic hydrocarbon compounds.
75 possible chlorine-substituted dibenzo-p-dioxin isomers, the data described below in the sections on toxicology and health effects concern the measured exposure to one dioxin isomer, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). In addition to TCDD, commercial formulations of chlorophenoxy herbicides contain a series of other polychlorinated dibenzodioxins (PCDDs) and dibenzofurans. Although studies have been conducted on many of these structurally similar molecules, such as polyhalogenated dibenzofurans and polyhalogenated biphenyls, these studies are not covered in this report because of the extensive literature base.
Some of the halogenated aromatic hydrocarbons are manufactured as commercial products, but others, like TCDD, occur as contaminants in commercial products. TCDD is formed as a contaminant in the synthesis of 2,4,5-trichlorophenol, which is used to manufacture 2,4,5-T (one of the components of several of the herbicides used in defoliation and crop destruction during the Vietnam war) and hexachlorophene. The degree of TCDD contamination is dependent on the temperature and pressure of the reaction conditions (Lilienfeld and Gallo, 1989). Young and colleagues (1978) reported on the levels of TCDD found in more than 450 samples of Agent Orange and one sample of Agent Purple (Table 4-5).
TABLE 4-5 Concentrations of TCDD in Samples of Agents Orange and Purple
|
|
Number of Samples |
Concentration of TCDD (µg/g) |
||
|
Source of Samples |
Orange |
Purple |
Range |
Mean |
|
Johnston Atoll inventory, 1972a |
200 |
(4)b |
0.05-47 |
1.91 |
|
Johnston Atoll inventory, 1974 |
10 |
|
0.07-5.3 |
1.68 |
|
NCBC, Gulfport inventory, 1972c |
42 |
|
0.05-13.3 |
1.77 |
|
NCBC, Gulfport inventory, 1975 |
238 |
|
0.02-15 |
2.11 |
|
Eglin AFB archived sampled |
|
1 |
— |
45.00 |
|
Eglin AFB inventory, 1972 |
2 |
|
— |
0.04 |
|
a Surplus Agent Orange was shipped from South Vietnam to Johnston Atoll (near Hawaii) for storage in April 1972. b Four of 200 samples may have been Agent Purple. c The Naval Construction Battalion Center (NCBC), Gulfport, Mississippi, served as a storage site for surplus Agent Orange from 1969 to 1977. d Agent Purple was used extensively in the evaluation of aerial spray equipment on Test Area C-52, Eglin Air Force Base (AFB) Reservation, Florida, 1962-1964. SOURCE: Young et al., 1978. |
||||
Using various analytical methods, other investigators determined that TCDD was the dominant compound of its group in Agent Orange (IARC, 1986). Due to their chemical stability and lipophilicity, the chemicals are persistent in the environment and are magnified in the food chain. The primary source of dioxins for human exposure is the food supply (Travis et al., 1989). The main ultimate sources of dioxins are industrial processes and combustion. As stated, the syntheses of some organic chemicals are known to yield dioxins (U.S. EPA, 1980). The use of products contaminated with dioxins and waste disposal from these production processes are two major sources of dioxin exposure (U.S. EPA, 1985). Since 1980, the practices that led to the dispersal of dioxins have been greatly reduced.
TCDD is a molecule that forms colorless needles with a melting point of 295-306°C. It is insoluble in water, but is soluble in many organic solvents (e.g., acetone, alcohol, and benzene) and oils.
Exposure and Pharmacokinetics
The fate of experimentally administered TCDD has been studied in a variety of animal species (reviewed: Neal et al., 1982; Gasiewicz et al., 1983; Olson et al., 1983; Birnbaum, 1985). Drug disposition studies such as these provide important information in developing models that can predict the biodistribution and elimination of TCDD following human exposure.
Ultimately, the disposition of TCDD, like all agents, is influenced by many factors—including the rate of drug absorption, distribution, metabolism, and elimination, and its sequestration and storage in various tissues—all of which have the potential for having an impact on the magnitude of toxicity produced.
Bioavailability Following Various Routes of Exposure
The amount of bioavailable TCDD (i.e., that which is capable of reaching tissue sites sensitive to TCDD-mediated alterations) is dependent on the route of chemical entry. In animal studies, the oral exposure route is most significant because it is believed to be the primary route for human exposure. Although the actual percentage of the total orally administered TCDD dose that undergoes gastrointestinal absorption following oral administration is found to vary among mammalian species, in virtually all cases absorption from either oil vehicles or dietary supplementation is greater than 50 percent. In Sprague-Dawley rats, 84 percent was the mean absorption of a single oral dose of 14C-TCDD (1.0 µg/kg) in an acetone corn oil vehicle (1:25 ratio by volume) (Rose et al., 1976). A similar percentage of the total dose was absorbed when rats were repeatedly administered low doses of TCDD (0.1-1.0 µg/kg) via the oral route, 5 days per week for 7 weeks (Rose et al., 1976). Following multiple oral administrations of high TCDD doses (50 µg/kg) in the rat, absorption was slightly lower, approximately 70 percent of the total dosage administered. Similarly, in hamsters, 75 percent of a single oral dose of [3H]-TCDD administered in olive oil (650 µg/kg) was absorbed, and in the guinea pig, approximately 50 percent of a single oral dose of TCDD in acetone/corn oil was absorbed. Thoracic duct-cannulated rats showed that intestinal absorption of [14C]-TCDD led to the transfer of the radioactive label to chylomicrons, which presumably transported the absorbed TCDD via the lymphatics into the circulation (Lakshman et al., 1986).
In Fischer 344 rats, dermal application of 60 µl of a TCDD solution (0.00015-1.0 µmol/kg in acetone) to a shaved 1.8-cm2 area of the animal's back, which was subsequently covered with a perforated stainless steel cap (to prevent confounding effects due to animal grooming), revealed several trends (Brewster et al., 1989). First, the percentage of the total TCDD dose absorbed decreased as the dosage increased. Second, the absolute absorbed amount of TCDD increased nonlinearly with dose. Lastly, the majority of the applied dose remained at the site of application, associated primarily with the stratum corneum, the uppermost layer of the epidermis, and did not penetrate through to the dermis. Absorption kinetic studies over 120 hours in the Fischer 344 rat model following application of 200 pmol of TCDD (64 µg/l acetone) indicated that the rate of dermal absorption was very slow,
with an absorption rate constant of approximately 0.005 pmol/hour (Banks and Birnbaum, 1991). After dermal application of 26 ng of TCDD in 50 µl methanol, approximately 14 percent of the total dose was found associated with the liver at 24 hours, compared to 40 percent (in 50 percent ethanol vehicle) after administration of an equal dose of TCDD orally (Poiger and Schlatter, 1980). Studies using soil-bound TCDD showed that a marked decrease occurred in the percentage of TCDD absorbed (approximately 1 percent of total applied dose), compared to when methanol was used as the vehicle, as determined by the total amount of hepatic TCDD (Shu et al., 1988a,b). In an attempt to simulate dermal exposure from contaminated soil, TCDD was applied in a soil-water paste to rats for 24 hours. Only 2 percent of the applied dosage was detected in hepatic tissues, suggesting very poor absorption (Poiger and Schlatter, 1980). Studies using a number of other vehicles suggest that the percentage of TCDD absorbed dermally is dependent on formulation. Taken together, these findings indicate poor dermal absorption of TCDD.
Little information is available pertaining to pulmonary absorption of TCDD; however, it is believed to be very high. Intratracheal instillation of 1 nmol/kg TCDD in Emulphor into male Fischer 344 rats resulted in approximately 92 percent absorption (Diliberto et al., 1991).
Distribution
Once absorbed, the distribution of xenobiotics occurs through body fluids, primarily the lymphatics and blood, where agents either can be transported in the aqueous phase or are free to associate with various lipids and proteins that can serve as endogenous carriers. Following gastrointestinal uptake, TCDD enters the lymphatics where approximately 96 percent is found to be associated with the chylomicron fraction in thoracic duct-cannulated rats (Lakshman et al., 1986). TCDD is transported in this manner into the circulation. Disappearance of TCDD from plasma followed first-order kinetics, with the first 67 percent of absorbed TCDD leaving the blood compartment rapidly (half-life = 0.81 min). The majority of absorbed TCDD was found to be distributed to the liver and adipose tissue.
The amount of an agent distributed to any given tissue is dependent on a number of factors, including the amount of blood flow to that tissue and overall tissue size. The primary sites of initial TCDD distribution from the blood, in terms of percentage of total administered dose, are the liver, adipose tissue, skin, and muscle during the first hour following administration. However, within days the majority of TCDD redistributes to the liver and adipose tissue, the primary sites of TCDD deposition. This general profile of distribution for TCDD has been observed in a variety of animal species including mice, rats, nonhuman primates, guinea pigs, and hamsters (Rose
et al., 1976; Kociba et al., 1978; Gasiewicz and Neal, 1979; Olson et al., 1980a; Gasiewicz et al., 1983; Birnbaum, 1986; Pohjanvirta et al., 1990). Whole-body autoradiography has also revealed that in both mice and rats, in addition to the liver and adipose tissue, there was a distinct localization of 14C-TCDD in the nasal olfactory mucosa (Appelgren et al., 1983; Gillner et al., 1987). The nasal olfactory mucosa was probably not identified in previous biodistribution studies because it is such an unlikely site for TCDD deposition, and therefore was most likely not previously examined.
There is evidence to suggest that the profile of TCDD tissue deposition may also be governed by the temporal kinetics of TCDD administration and the magnitude of the administered dose. Some studies suggest that TCDD tissue distribution is dose-dependent. Biodistribution studies following a single intraperitoneal administration of TCDD in rats revealed a marked increase in the concentration of TCDD in liver at doses greater than 10 ng/kg, with a concomitant decrease in adipose tissue-associated TCDD (Abraham et al., 1988). Similarly, following administration of single doses of TCDD, a dose-related increase was observed in the proportion of TCDD distributed to the liver as compared to adipose tissue (Poiger et al., 1989). Although the mechanism for this phenomenon is unclear, it may be partially related to the fact that rats also exhibit a concomitant and dose-dependent loss of adipose tissue. Other evidence suggests that an increase in hepatic TCDD retention is mediated by a liver-associated binding species. Several laboratories have demonstrated that this binding species is TCDD inducible (Poland et al., 1989a; Curtis et al., 1990; Leung et al., 1990). Pretreatment of rats with 5 or 15 µg/kg of TCDD increased the accumulation of TCDD in hepatic tissue in a dose-dependent manner, when followed by subsequent oral administration of TCDD. Similarly, increased hepatic uptake of TCDD-related compounds was observed after pretreatment with TCDD (Poland et al., 1989b; Leung et al., 1990). Findings by several independent laboratories suggest that the hepatic binding species is cytochrome P4501A2 (Voorman and Aust, 1987, 1989; Poland et al., 1989a,b). As would be expected for cytochrome P4501A2 involvement, Poland and coworkers (1989a) found that the TCDD-binding species was associated primarily with the microsomal fraction of the liver and was heat and trypsin sensitive, inactivated by mercurials, and liver specific. The prospect that cytochrome P4501A2 can act as a TCDD-binding protein is also consistent with the fact that the only other site at which this P450 isozyme is TCDD inducible other than the liver is the nasal olfactory mucosa, a tissue that exhibits high TCDD bioaccumulation (Tuteja et al., 1985; Gillner et al., 1987). Contrary to the premise that cytochrome P4501A2 represents the TCDD hepatic binding species was the observation by Poland and colleagues (1989b) that dietary administration of the cytochrome P4501A2 inducer, isosafrole, did not increase hepatic uptake of TCDD.
In contrast to studies describing dose-dependent tissue distribution of TCDD, findings from several other studies do not support this trend (Rose et al., 1976; Clark et al., 1991c; Tritscher et al., 1992).
Species and tissue-related differences exist for TCDD retention time. In rats, TCDD is more persistent in adipose tissue than in liver (Abraham et al., 1988), whereas in the mouse, TCDD has a similar half-life in adipose and hepatic tissue (Birnbaum, 1986). In nonhuman primates such as the rhesus monkey, TCDD is exceptionally persistent in adipose tissue (Bowman et al., 1989). Adding to this complexity of TCDD retention are biodisposition studies suggesting that the rate of TCDD decay from liver, adipose tissue, and other tissue may not remain constant with time (Birnbaum et al., 1980; Olson et al., 1980a; Birnbaum, 1986; Pohjanvirta et al., 1990; Neubert et al., 1990a).
Likewise, there is also evidence to suggest that TCDD retention in the rat liver may be cell-type specific. Four days after TCDD exposure, approximately 60 percent of TCDD associated with the liver was retained in parenchymal cells (half-life ~ 13 days) and 12 percent with nonparenchymal cells.
Metabolism
TCDD is biotransformed to water-soluble metabolites in a wide range of mammalian species (Poiger and Schlatter, 1979; Ramsey et al., 1979, 1982; Olson et al., 1980a; Poiger et al., 1982; Gasiewicz et al., 1983; Kleeman et al., 1988; Sijm et al., 1990). In a number of rodent species including the rat, mouse, hamster, and guinea pig, more than 90 percent of the TCDD that undergoes urinary and biliary excretion is in a polar biotransformed form. In fact, excretion of absorbed TCDD is metabolism dependent, with the exception of nonabsorbed compound that undergoes direct intestinal excretion. In dogs, the effects of pretreatment with mixed-function oxidase (MFO) inducers, either phenobarbital or TCDD, on the biliary excretion of subsequently administered 3H-TCDD were investigated (Poiger and Schlatter, 1985). Without pretreatment, 24.5 percent of the absorbed TCDD was excreted in bile within 110 hours. Phenobarbital pretreatment produced no effect on the rate of TCDD biliary excretion, whereas TCDD pretreatment (a single 10-µg/kg dose 9 days earlier) resulted in a doubling in the biliary TCDD elimination rate. These results illustrate the important role the MFOs play in the rate of TCDD elimination.
Although the metabolism of TCDD has been somewhat enigmatic, a number of metabolites have been identified. Six TCDD metabolites were detected in the bile of dogs that had received a lethal dose (Poiger et al., 1982). The major metabolite was 1,3,7,8-tetrachloro-2-hydroxydibenzo-p-dioxin. Additionally, 3,7,8-trichloro-3-hydroxydibenzo-p-dioxin and 1,2-dichloro-4,5-hydroxybenzene
were identified as minor metabolites. The structure of the three remaining metabolites was not confirmed; however, it was believed that two of the metabolites were trichlorohydroxydibenzo-p-dioxins, with the third possibly being chlorinated 2-hydroxydiphenyl ether. In the rat, trichlorodihydroxydibenzo-p-dioxin and tetrachlorodihydroxydiphenyl ether were the major metabolites identified in bile (Poiger and Buser, 1984). Additionally, what were believed to be glucuronide conjugates were identified in rat but not dog bile. In vitro studies utilizing isolated rat hepatocytes in culture identified two glucuronide conjugates as the major metabolites of 2,3,7,8-TCDD (Sawahata et al., 1982). Deconjugation of the metabolites with β-glucuronidase yielded 1-hydroxy-2,3,7,8-TCDD and 8-hydroxy-2,3,7-trichloro-3-hydroxydibenzo-p-dioxin. It is generally believed that the major route of metabolism in the rat involves oxygenation of the unsubstituted carbon nearest the bridging oxygen in 2,3,7,8-TCDD.
Metabolic biotransformation of TCDD is generally accepted as being a detoxification reaction. This premise is supported by a number of different studies using a variety of approaches. For example, bile extracts from TCDD-treated dogs administered to guinea pigs were found to be 100 times less toxic than orally administered TCDD itself (Weber et al., 1982). Structure-activity relationship studies using synthesized congeners of known TCDD metabolites found those compounds to be toxicologically inactive even at very high concentrations (i.e., up to 5,000 µg/kg), suggesting that TCDD itself is the active species (Mason and Safe, 1986). Poland and Glover (1979), investigating the potential for in vivo bioactivation of TCDD to reactive intermediates, demonstrated that very low amounts of TCDD actually formed DNA adducts (i.e., 8 1 DNA adduct/35 cells). These findings suggest that the covalent binding of TCDD to DNA is not likely to be responsible for its oncogenic effects, and further support the premise that TCDD metabolism is primarily a detoxification mechanism.
Excretion
The rate and primary route of TCDD excretion has been found to differ among animal species. After a single dose, TCDD undergoes a first-order elimination process exhibiting very slow excretion kinetics. In the hamster, the half-life for elimination has been estimated at approximately 11 days. The mean half-life of TCDD in the guinea pig is approximately 94 days (Olson, 1986). In the rat, following repeated oral dosing (0.1-1.0 µg/kg), 5 days per week for 7 weeks, the half-life for elimination ranged from 16 to 37 days (Rose et al., 1976). From drug disposition studies in the rat, Rose and colleagues (1976) concluded that based on calculated steady-state values, it was unlikely that TCDD would continue to accumulate indefinitely in the tissues of animals exposed chronically to low levels of the compound.
However, in nonhuman primates, TCDD was found to be highly persistent (Bowman et al., 1989; Neubert et al., 1990a).
Species differences also exist with respect to the route of elimination of TCDD. In the hamster and mouse, excretion of TCDD occurs via both feces and urine (Olson et al., 1980a; Gasiewicz et al., 1983; Birnbaum, 1986). Conversely, in all other species, excretion occurs primarily through feces (Piper et al., 1973a; Allen et al., 1975; Rose et al., 1976; Gasiewicz and Neal, 1979). In virtually all rodent studies, results indicate that all of the TCDD excreted in urine and bile is in the form of TCDD metabolites. In the rat, hamster, and mouse, approximately 15-35 percent of TCDD in feces is unmetabolized, whereas in the guinea pig, approximately 81 percent was unmetabolized in feces (Olson et al., 1980a; Neal et al., 1982; Gasiewicz et al., 1983; Olson, 1986). Unmetabolized TCDD in feces is believed to be primarily a result of direct intestinal elimination since no parent form of the compound is normally observed in bile. Minimal excretion of 14C-TCDD has also been reported in expired air (Piper et al., 1973a). The relationship between administered dose and excretion rate is limited; however, little change in excretion of 2,3,7,8-TBDD (2,3,7,8-tetrabromodibenzo-p-dioxin) was observed between 1- and 100-nmol/kg doses (Kedderis et al., 1991). Based on present data, no significant correlations have been made between metabolism and disposition of TCDD and strain- or species-specific toxicity.
Mechanism of Action
Introduction
A great deal of research has gone into determining the mechanism of TCDD toxicity in order to determine the plausible biologic activity of the molecule. Most of this research has focused on identification and characterization of the interaction of TCDD with an intracellular protein called the Ah receptor. An Ah receptor protein can interact with a TCDD molecule when it enters a cell, and then translocate to the nucleus where the TCDD-receptor complex can interact with specific sites on DNA. Interaction with specific sites on DNA may have an effect on the regulation of DNA expression, affecting a wide range of mechanisms that regulate normal cellular activity.
Receptor-mediated events are generally characterized by the following: (1) they are restricted to cells that express the receptor; (2) there is a structure-activity relationship (i.e., molecules that bind have a specific geometric configuration, and their potency varies with deviations in this geometric configuration); (3) binding occurs at relatively low concentrations of the ligand (the molecule that binds the receptor); (4) binding is reversible; and (5) the magnitude of the response is proportional to the number of receptors
occupied by the ligand. For Ah receptor-mediated events, TCDD meets many of these criteria.
Not all effects of TCDD are mediated through the binding of TCDD to the Ah receptor, including neurotoxicity and in vitro immunotoxicity to B cells. The mechanism by which these effects are elicited by TCDD is currently unknown; therefore, the emphasis in this section is on effects mediated by the Ah receptor.
Ah Receptor
Early studies by Poland demonstrated that TCDD saturably binds an intracellular protein with a high affinity. Further characterization of the binding properties indicated that the ligand binding exhibited stereospecificity (i.e., planar molecules with at least three halogen atoms were bound) (Poland and Glover, 1973; Poland and Knutson, 1982). Additional studies showed that the binding affinity of various congeners for the soluble receptor correlated well with the ability of the molecules to elicit a biological response. In addition, genetic strains of mice were identified whose Ah receptor had a lower affinity for TCDD. These mouse strains had a decreased sensitivity to the toxic effects of TCDD. Crossbreeding studies indicate that the sensitive phenotype segregates as an autosomal dominant phenotype. Further genetic studies identified the "Ah locus" as the area of the genome that encodes for the Ah receptor (Poland and Knutson, 1982; Nebert, 1989). Therefore, biochemical and genetic evidence indicates that the cytosolic protein Ah is the receptor for TCDD. Although this protein has a high affinity for TCDD, recent studies have identified possible naturally occurring high affinity ligands for the receptor (Gillner et al., 1985, 1987; Rannug et al., 1987; Bjeldanes et al., 1991).
Human cells from a variety of tissue types contain an intracellular protein that resembles the Ah receptor in animals (Manchester et al., 1987; Cook and Greenlee, 1989; Harris et al., 1989; Roberts et al., 1990; Lorenzen and Okey, 1991; Waithe et al., 1991). The isolated receptor was shown to have approximately the same sedimentation rate, molecular weight, and binding specificity as the murine Ah receptor (Harper et al., 1988). The human Ah receptor has a binding affinity 5-10 times higher than mouse (5-10 nM versus 0.8-3 nM in the murine hepa 1 cell line) (Manchester et al., 1987; Roberts et al., 1991; Waithe et al., 1991). In addition, human cells have a lower sensitivity to enzyme induction than murine cells (Harper et al., 1991; Roberts et al., 1991). The properties of this receptor have not been extensively characterized, but it is likely that, as in mice, the human population will be polymorphic with respect to the structure, function, and ligand affinity of the Ah receptor (Nebert et al., 1991).
Complementation studies conducted using variant cells that are defective
in either TCDD binding or translocation of the receptor-ligand complex to the nucleus, which occurs as part of the signal transduction process, indicate that more than one gene contributes to receptor function (Hankinson, 1983; Miller et al., 1983; Whitlock, 1990). Prior to ligand binding, the receptor is cryptic and contains the 90-Kd heat shock protein, whose release is necessary to unmask the functional activity of the receptor (Poellinger et al., 1991).
Studies were conducted to determine the ligand characteristics important for binding and eliciting a biologic response. There is good correlation between the binding affinities of various TCDD congeners for the Ah receptor and the induction of enzyme (aryl hydrocarbon hydroxylase, AHH) activity (Poland et al., 1979). Analogous structure-activity studies implicate the Ah receptor in a broad number of biochemical, morphological, immunologic, neoplastic, and reproductive effects (Poland and Knutsen, 1982; Safe, 1986). However, some responses do not have a clear relationship to Ah receptor binding and therefore may not be mediated by the Ah receptor (Rozman et al., 1993).
The Ah receptor is a soluble intracellular protein that, upon binding to TCDD, acquires a high affinity for DNA and accumulates in the nucleus (Denison et al., 1989; Hapgood et al., 1989; Nemoto et al., 1990; Saatcioglu et al., 1990a,b; Cuthill et al., 1991; Denison and Yao, 1991). The transformation of the Ah receptor into a DNA-binding form involves multiple events and interactions, including a conformational change measured by several parameters (Denison et al., 1987; Gasiewicz and Bauman, 1987; Kester and Gasiewicz, 1987; Henry et al., 1989). Evidence from a variety of sources indicates that the DNA-binding form of the receptor is composed of at least two different proteins (Elferink et al., 1990; Gasiewicz et al., 1991). One protein, termed ''Arnt," that does not bind TCDD is associated with the liganded Ah receptor and may be either the DNA-binding component of the receptor or associated with translocation of the receptor from the cytoplasm to the nucleus (Hoffman et al., 1991). In addition, the ligand-binding portion of the Ah receptor appears to have been identified (Bradfield et al., 1991, Burbach et al., 1992; Ema et al., 1992).
Function of the Ah Receptor Binding of TCDD to its receptor, subsequent translocation to the nucleus, and DNA-binding result in a number of biologic effects. Many genes have elements associated with them that are responsive to TCDD (dioxin-responsive enhancers, DRE; J.M. Fisher et al., 1989; Whitlock, 1990). For example, TCDD induces AHH activity (a drug-metabolizing enzyme) by stimulating the transcription of the CYP1A1 gene, which encodes for the hydroxylase protein, through a means that does not require protein synthesis and is receptor dependent. The liganded receptor binds to a transcriptional enhancer regulatory element, DRE, upstream from
the CYP1A1 gene (Jones et al., 1986; Neuhold et al., 1986; Fujiisawa-Sehara et al., 1987; Fisher et al., 1990). This liganded receptor recognizes a specific nucleotide sequence (5'-TGCGTG-3'), which occurs in multiple copies within the enhancer region (Denison et al., 1989; Hapgood et al., 1989; Saatcioglu et al., 1990a,b). The activity of a transcriptional promotor is also required to enhance the appropriate transcription of the gene that is being affected by TCDD (Jones and Whitlock, 1990).
Studies have characterized the interaction between the liganded-Ah receptor and the DRE element. As stated above, a specific nucleotide sequence was identified that occurs multiple times within the DRE. Further studies, using either DNA fragments or intact cells, show that the binding of the TCDD-Ah receptor complex to DNA occurs within the major DNA groove and contacts four guanines of the recognition sequence (Shen and Whitlock, 1989; Neuhold et al., 1989). Binding of the TCDD-Ah receptor to DRE and the function of DRE are diminished by methylation of the cytosine nucleotide within the recognition sequence. This may be one mechanism by which differences in responsivity to TCDD occur between tissues (Shen and Whitlock, 1989). Finally, binding of the liganded receptor to the DRE may alter the configuration of the DNA and chromosome structure (Durrin and Whitlock, 1989; Elferink and Whitlock, 1990; Wu and Whitlock, 1992).
As indicated above, the induction of the transcription of a variety of genes is mediated by TCDD binding to the Ah receptor and subsequent binding of the liganded receptor to DNA at an element similar to that found upstream of the CYP1A1 gene. These genes include a cytochrome P4501A2 gene, a glutathione S-transferase Ya subunit gene, an aldehyde dehydrogenase gene, and a quinone reductase gene (Dunn et al., 1988; Jaiswal et al., 1988; Telakowski-Hopkins et al., 1988; Quattrochi and Tukey, 1989; Favreau and Pickett, 1991). Recent studies indicate that TCDD also induces the transcription of plasminogen activator inhibitor-2 and interleukin-1β as well as other unidentified genes (Sutter et al., 1991). However, the mechanism by which this induction occurs is not as well defined as that described above.
In response to the induction of gene expression and primary effects on the cell, compensatory effects may occur. For example, TCDD affects the levels of steroid hormones and growth factors in rodents (Umbreit and Gallo, 1988; Ryan et al., 1989; Sunahara et al., 1989; Harris et al., 1990; Choi et al., 1991). These direct alterations in gene transcription lead to a variety of effects that are not mediated directly by the Ah receptor. TCDD may therefore induce a cascade of biochemical changes and thereby produce a biological response, such as cancer, by several different mechanisms that can affect different tissues. TCDD may either induce a gene for a growth factor and directly affect tissue proliferation, or induce the gene for the growth factor receptor and increase the sensitivity of the cell to the growth factor
signal. Alternatively, TCDD may lead to tissue destruction, and the compensatory cellular proliferation may make permanent a genetic defect that allows cellular transformation and consequently neoplasia. Additional mechanisms have also been proposed for the induction of neoplastic tissue in the intact animal. Which of these mechanisms are actually involved is not clear, and they may vary among tissues and animal species.
Linearity of Response or Threshold Scientists hold vastly different opinions about the existence of a threshold effect, that is, whether there is a point below which no effect of the chemical exists, for the activity of TCDD. There are those who argue that all events that occur up to and including the induction of gene transcription have a linear dose-response curve; others argue that more complex events that require the concordance of two or more events, such as cell proliferation, may have a threshold. This seems to be the most favored view, and it is supported by the available data. This does not mean, however, that there is a threshold for the biological effects of TCDD, simply that the response is receptor-mediated (Portier et al., 1993).
From this latter view, however, two further divergent views are defined. One view is that (1) all events that occur in response to the binding of TCDD to its Ah receptor have a linear dose-response curve; (2) the observance of a threshold is due to background levels of the response, which obscure the detection of any TCDD-induced response at low doses; and (3) a linear dose-response curve would be observed if the method of measurement could be more refined (Silbergeld, 1991). Scientists who hold this view note that (1) there is a linear dose-response curve for the effects of TCDD; (2) it is not clear what causes the dose-response curve to become nonlinear as the complexity of the event mediated by TCDD increases; (3) TCDD is acting in addition to a natural ligand for the Ah receptor that has yet to be defined; and (4) the concentration of the natural ligand within the cell may influence the response to TCDD.
The other view suggests that a finite amount of TCDD is necessary to elicit detectable effects due to the possibility that (1) events subsequent to receptor binding may not have a linear response to TCDD; (2) activation of transcription of a gene does not correlate with the binding affinity for DNA; and (3) the concentration of TCDD required to produce a biological effect varies among species and among individuals within a species (Whitlock, 1991; Shen and Whitlock, 1992). Scientists who hold this view believe that a threshold amount of TCDD is required to produce any of its biological effects, but they do not believe that currently available data are sufficient to set the dose for this threshold.
Some scientists feel that there may be a threshold for the biological effects of TCDD, but are unwilling to set a level for this threshold; they feel
that the current body burden for TCDD (when added to the toxic equivalency factors for other compounds that bind to the Ah receptor) in industrialized nations may exceed this threshold, and therefore the point is of only theoretical and not practical significance.
Non-Ah-Mediated Toxicity
In order to establish that an observed toxicity is mediated through an Ah receptor, certain criteria must be met. Currently, these are (1) a structure-activity relationship when using ligands of lower affinity for the Ah receptor, and (2) differential sensitivities when using mice congenic at the Ah locus. However, some toxicologic effects do not meet these criteria and therefore cannot be considered to be mediated through the Ah locus. Examples of this are immune suppression after in vitro exposure to TCDD and neurotoxicity. The mechanism by which these events occur is unknown at this time, but this does indicate that some actions of TCDD are not mediated through the mechanism discussed above.
Health Outcomes in Animal Studies
Carcinogenicity
Carcinogenicity Bioassays Long-term carcinogenicity bioassays of TCDD have been conducted in rats, mice, and hamsters. A total of eight studies have been reported; these are summarized in Table 4-2. Routes of exposure have included oral, intraperitoneal, dermal, and subcutaneous. Increased tumor rates have been reported at several sites; the only consistent site among species and studies has been the liver. The results of each of these bioassays are described below.
Kociba et al., (1978) The Kociba and colleagues (1978) bioassay of TCDD is the most cited and that on which the Environmental Protection Agency cancer potency estimate is based. Groups of male and female Sprague-Dawley rats received 0, 0.001, 0.01, or 0.1 µg TCDD/kg body weight/day in the diet for two years. The results of this study were based on histopathologic evaluations of tissue samples performed by Dr. R.J. Kociba. These tissues were subsequently evaluated independently by Dr. R.A. Squire (Squire, 1980). Both investigators used the criteria for evaluating hepatocellular lesions described by Squire and Levitt (1975) and ILAR (1979). These criteria have been revised since their evaluations, based upon continuing studies of hepatocellular proliferative lesions (Maronpot et al., 1986; NTP, 1984). Because of these revisions, an independent panel review of the liver histopathology slides from the Kociba study has recently been conducted by PATHCO, Inc. (1990a; Goodman and Sauer, 1992). The results
of this and the Squire review are at variance with each other and with the original results, all of which are summarized for female rats in Table 4-6.
Table 4-6 shows the incidences of foci of hepatocellular proliferation, hepatocellular adenoma, and carcinoma for each dose group. It is apparent from the table that few of the lesions identified by the original investigators as malignant were confirmed as such in the more current evaluation. Increased liver tumor rates were not observed in male rats.
Hepatic toxicity was observed frequently among treated female rats (Table 4-6). These lesions included necrosis, vacuolization, cellular enlargement, multinucleated cells, infiltration of inflammatory cells, fatty changes, oval cell proliferation, and regenerative hyperplasia. Grading of the toxic lesions was performed by PATHCO, Inc. (1990b; Goodman and Sauer, 1992), based on a scale of 1 (minimal), 2 (mild), 3 (moderate), or 4 (marked), reflecting increased incidence and severity of the lesions noted above. Nonspecific (minimal) changes were noted primarily in the control and low dose groups, although several animals in the low dose group exhibited mild hepatotoxicity as well. In the mid- and high dose groups, there was a clear increase in the incidence and severity of hepatotoxicity. Interestingly, tumors in these groups were observed only in animals exhibiting toxicity.
In addition to liver tumors in female rats, a significantly increased incidence of squamous cell carcinoma of the nasal turbinates/hard palate in both sexes, as well as of the tongue in males, was identified. These tumors are rare in Sprague-Dawley rats. A statistically significant increase in the incidence of keratinizing squamous cell carcinoma of the lung was also detected in female rats at the high dose. In addition, significantly reduced incidences in tumors of the uterus, pancreas, and the pituitary, mammary, and adrenal glands were reported.
National Toxicology Program (NTP, 1982a) In the NTP (1982a) standard carcinogenicity bioassay, groups of male and female Osborne-Mendel rats and male B6C3F1 mice received doses of 0, 5, 25, or 250 ng TCDD/kg body weight by gavage twice weekly for two years; female mice received 0, 20, 100, or 1,000 ng TCDD/kg. These doses corresponded to average daily doses of 0, 1.4, 7.1, or 71 ng/kg for rats and male mice, and 0, 5.7, 28.6, or 286 ng/kg for female mice. Survival was not affected, and there was an increased incidence of both adenomas and carcinomas of the liver at the high dose in male and female mice. In female rats, the increased incidence of hepatocellular adenomas and carcinomas was significant when combined. Other positive results included increased rates of thyroid follicular cell adenoma in male rats at all dose levels, of combined adenomas and carcinomas of the adrenal gland in high dose female rats, of subcutaneous fibrosarcomas in high dose rats and female mice, and of lymphoma in high dose female mice.
National Toxicology Program (NTP, 1982b) Male Swiss-Webster mice received doses of 0.001 µg TCDD applied to the skin 3 days per week for
TABLE 4-6 Results of Kociba et al. (1978) TCDD Bioassay in Female Rats: Liver Lesions
|
|
|
Dose (ng/kg/day) |
|
|
|
|
Pathologist |
Lesion |
0 |
1 |
10 |
100 |
|
Kociba |
Hyperplastic nodule |
8/86 (9.3) |
3/50 (6) |
18/50 (36) |
23/50 (46) |
|
|
Hepatocellular carcinoma |
1/86 (1.2) |
0/50 (0) |
2/50 (4) |
11/50 (22) |
|
Squire |
Hyperplastic nodule and hepatocellular carcinoma |
16/86 (18.6) |
8/50 (16) |
27/50 (54) |
33/47 (70.2) |
|
PATHCO (Goodman and Sauer) |
0.6 (57) |
1.2 (88) |
2.3 (96) |
3.6 (100) |
|
|
|
Foci of cellular alteration (eosinophilic foci)c |
31/86 (36) |
23/50 (46) |
37/50 (74) |
40/45 (80) |
|
|
Hepatocellular adenomad |
2/86 (2.3) |
1/50 (2) |
9/50 (18) |
14/45 (28) |
|
|
Hepatocellular carcinoma |
0/86 (0) |
0/50 (0) |
0/50 (0) |
4/45 (8.9) |
|
NOTE: Results are given as number of animals with lesion/number of animal evaluated (percent). a Severity was graded on a scale of 1 (minimal), 2 (mild), 3 (moderate), or 4 (marked). b In some cases the number of animals examined for hepatotoxicity differed slightly from the number examined for other lesions due to autolysis and leukemia. c May include animals with hepatocellular adenoma or carcinoma. d Only the most malignant tumor in each animal was counted. |
|||||
104 weeks. Female mice received 0.005 µg TCDD per application. One control group remained untreated and one received acetone instead of TCDD. Treatment with TCDD significantly increased the incidence of skin fibrosarcomas in female mice. This effect was not significant in male mice.
Rao et al. (1988) Groups of male Syrian golden hamsters received two to six doses of 50 or 100 µg TCDD/kg body weight by either intraperitoneal or subcutaneous injection over a 4-week period. Animals were sacrificed after 12-13 months. Those that received the highest dose (total dose 600 µg/kg) developed squamous cell carcinomas of the facial skin at a rate of 4/18 (22 percent) by intraperitoneal injection and 3/14 (21 percent) by subcutaneous injection. The tumors were large, with extensive necrosis and some metastasis to the lung. This tumor is so rare that none could be identified in a study of control hamsters of this strain. No other treatment groups developed this or other exposure-related tumors.
Della Porta et al. (1987) Groups of male and female B6C3F1 and B6CF 1 mice received intraperitoneal doses of 0, 1, 30, or 60 µg TCDD/kg body weight in corn oil once weekly for 5 weeks, starting at 10 days of age. The mice were observed until they reached 78 weeks of age. Only the liver, kidney, and organs with gross pathologic changes were examined histologically. Thymic lymphomas occurred at a statistically elevated rate in both sexes of both strains receiving the highest dose. In addition, an increased rate of hepatocellular adenomas and carcinomas occurred in B6C3F1 males. In another study, groups of male and female B6C3F1 mice received 0, 2.5, or 5.0 µg TCDD/kg body weight by gavage once weekly, starting at 6 weeks of age, for 52 weeks. Mice were observed until 110 weeks of age, and complete histopathology was performed. An increased incidence of hepatocellular carcinomas was observed for both sexes at both doses.
Van Miller et al. (1977) The Van Miller study was intended to be a range-finding study and, as such, used few rats and was poorly reported (U.S. EPA, 1985). Male Sprague-Dawley rats were fed a diet containing 0, 0.001, 0.005, 0.05, 0.5, 1.0, 5.0, 50, 500, or 1,000 parts per billion (ppb) of TCDD for 78 weeks. This regimen corresponded to approximate dose levels of 0, 0.0003, 0.001, 0.01, 0.1, 0.4, 2.0, 24, 240, and 500 µg TCDD/kg body weight/week. Surviving animals were observed for an additional 17 weeks. All animals receiving 1 ppb TCDD or more were dead by week 90. Survival at lower doses was unaffected, although degenerative changes in the kidneys were observed. A statistically elevated total tumor incidence was reported in rats receiving 0.005 ppb or more, along with an elevated incidence of squamous cell tumors of the lungs, hepatic neoplastic nodules, and cholangiocarcinomas in the 5-ppb dose group. However, no tumors were reported to have occurred in control animals, which is very unusual for Sprague-Dawley rats. The validity of these results is thus questionable.
Toth et al. (1979) Groups of male Swiss/H/Riop mice received doses of
0, 0.007, 0.7, or 7.0 µg TCDD/kg body weight weekly by gavage for one year. There were two vehicle and two untreated control groups. Animals were observed for their life spans. Liver tumors (combined adenomas and carcinomas) occurred at a high spontaneous rate in control groups (18-33 percent) and were positively correlated with survival. A statistically elevated liver tumor incidence was observed in the middle-dose group, although the ratio of benign to malignant tumors was unaffected. A nonstatistically elevated liver tumor incidence was observed in the highest dose group. The latter group also had reduced survival, however, and time-to-tumor data, if they had been available, were likely to have shown a decrease in time-to-tumor compared to controls. The results of this study are thus suggestive of an effect of TCDD on liver tumor incidence.
Initiation/Promotion Bioassays Several bioassays have been performed to assess the ability of TCDD to promote the carcinogenicity of an initiating chemical or treatment. Initiation/promotion bioassays recognize that there are discrete stages in carcinogenesis, and understanding the stage or stages that are affected by an agent gives clues to its mechanism of action. The initiation/promotion paradigm for carcinogenesis postulates that a cell undergoes genetic alteration at a critical site on DNA (initiation) and that the altered cell undergoes cell proliferation and clonal expansion (promotion), which can be followed by additional rounds of DNA damage and cell proliferation (progression) until a tumor is formed. Skin tumor initiation/promotion bioassays usually start with a single dose of a genotoxic chemical, followed by repeated applications of the test chemical to determine if it is a promoter. Liver tumor initiation/promotion protocols generally involve treatment with a genotoxic chemical and partial hepatectomy to make the genetic damage permanent, followed by treatment with the test agent. An agent is considered to promote carcinogenesis if it increases tumor incidence compared to that expected to be induced by the initiator alone. Chemicals that test positive may be considered tumor promoters within the operational definition of these protocols.
Skin The skin tumor-promoting ability of TCDD was tested in the presence and absence of initiation by dimethylbenzanthracene (NTP, 1982b). Male Swiss-Webster mice received doses of 0.001 µg TCDD applied to the skin 3 days per week for 104 weeks. Female mice received 0.005 µg TCDD per application. Half the animals received a single application of 50 µg dimethylbenzanthracene one week prior to beginning TCDD treatment. One control group remained untreated, and one received acetone instead of TCDD. Treatment with TCDD significantly increased the incidence of skin fibrosarcomas in female mice, in both the presence and the absence of dimethylbenzanthracene (8/29 and 8/27, respectively, versus 2/41 in controls). This effect was not significant in male mice.
In another study, Poland et al. (1982) administered a single initiating dose of N-methyl-N-nitrosoguanidine to the skin of hairless mice, followed by twice weekly doses of 0, 3.75, 7.5, 15, or 30 ng TCDD for 20 weeks. Papilloma formation occurred at all doses in a dose-dependent manner. Tumors were infrequent in mice that received either compound alone.
The tumor-initiating ability of TCDD has also been tested, by using an initiation/promotion protocol with phorbol ester (TPA) as the promoter. TCDD had weak or no initiating activity in this system (DiGiovanni et al., 1977).
The skin tumor-promoting activity of TCDD is dependent on the presence of the Ah receptor and segregates with the hr locus (Poland and Knutson, 1982; Poland et al., 1982).
Liver In a bioassay reported by Pitot and colleagues (1980), female Sprague-Dawley rats received an initiating dose of diethylnitrosamine followed by 0.14 or 1.4 µg TCDD/kg body weight subcutaneously once every two weeks for seven months (equivalent to 10 and 100 ng/kg/day, the same as the medium and high doses in the Kociba et al. 1978 bioassay). No liver tumors occurred in groups of rats that received either diethylnitrosamine or TCDD alone, or in the group that received diethylnitrosamine and the low dose of TCDD. Five of seven rats that received diethylnitrosamine and the high dose of TCDD developed hepatocellular carcinomas, however. Altered hepatic foci (eosinophilic) also occurred much more frequently in this treatment group than in the others. These results have been confirmed by other studies, which have also demonstrated that TCDD's promoting effect in rats is dependent on the presence of ovaries (Graham et al., 1988; Clark et al., 1991b; Flodstrom and Ahlborg, 1991; Lucier et al., 1991, 1992; Dragan et al., 1992). This finding is consistent with the results of long-term studies showing that TCDD is a hepatocarcinogen in female but not male rats. Interestingly, although the absence of ovaries appeared to protect against TCDD-induced liver tumors, lung tumors appeared only in ovariectomized rats treated with diethylnitrosamine and TCDD. Complex hormonal interactions clearly are involved in the site specificity of TCDD-induced carcinogenesis.
Mechanistic Studies TCDD has a wide range of effects on growth regulation, hormone systems, and other factors associated with the control of cell proliferation and differentiation. Several of those effects, and the roles they may play in TCDD-mediated carcinogenesis, are described below.
Genotoxicity The preponderance of data indicates that TCDD is not genotoxic: it does not form detectable adducts with DNA (U.S. EPA, 1985; Turteltaub et al., 1990); it has produced negative results in batteries of tests for genotoxicity (NTP, 1984); and although it is an effective tumor promoter (see previous discussion), it is only a weak initiator. There is no
consistent evidence for increased frequencies of chromosomal aberrations among humans exposed to TCDD either occupationally or accidentally (Shu et al., 1987). The results of specific genotoxicity tests of TCDD have been thoroughly reviewed by Shu and colleagues (1987).
Enzyme Induction The induction of cytochrome P450 isoenzymes CYP1A1 and CYP1A2 by TCDD has been studied extensively (Whitlock, 1990). Information is available on isoenzyme specificity; the time-course and dose-response aspects of induction; molecular mechanisms of transcriptional activation; and species, tissue, and cell specificity. Several studies have shown that TCDD also induces the activity of at least one isoenzyme of uridine 5'-diphosphate glucuronosyltransferase (UDPGT) (Lucier et al., 1986). This effect, like that on P450, is Ah receptor dependent (Bock, 1991). Both P450 and UDPGT are responsible for conjugating numerous substrates, both endogenous and exogenous, rendering them water-soluble and excretable in urine. A mechanistic relationship among cytochrome P450, UDPGT induction, and carcinogenesis, or any other end point of toxicity, has not been established, however.
The controversy regarding the relevance of P450 induction to carcinogenesis is based on the notion that inducing P450 would enhance the rate at which it activates other carcinogens to form DNA-reactive metabolites. However, P450 also deactivates reactive metabolites, and most evidence indicates that the carcinogenic potency of many chemicals is diminished by P450 induction. For example, both the carcinogenic potencies of benzo[a]pyrene and dimethylbenzanthracene are decreased by TCDD exposure, as are their rates of DNA adduct formation, presumably because deactivation of their reactive metabolites is enhanced (Cohen et al., 1979). While it is possible that P450 induction could increase the formation of reactive metabolites and lead to increased rates of DNA adduct formation, in general, P450 induction diminishes the carcinogenicity of a wide variety of chemicals (Miller et al., 1958; Wattenburg, 1978, 1985). Generalizations are not possible, however, and predictions cannot be made for specific chemicals in the absence of experimental data.
Another possible role of P450 in TCDD-mediated carcinogenesis is the ability of CYP1A2 to convert estrogens to catechol estrogens in the liver (Graham et al., 1988). Catechol estrogens are thought to possess DNA-damaging capability as a consequence of free-radical generation (Metzler, 1984; Li and Li, 1990). Inducing CYP1A2 may thus increase DNA damage by estrogens, which may play a part in TCDD-induced carcinogenesis in the female rat liver. This possibility is consistent with the observations that CYP1A2 is found only in the liver, that ovariectomy protects against TCDD-induced hepatocarcinogenesis in female rats, and that male rats are not susceptible.
Evaluating the possible role of P450 induction in TCDD-mediated carcinogenesis
is complicated by the heterogeneity of hepatocyte responses to TCDD: P450 activity in some hepatocytes is maximally induced by low doses of TCDD, whereas others do not respond. Increasing the dose of TCDD increases the number of cells responding, not the amount of induction in responding cells (Bars and Elcombe, 1991; Tritscher et al., 1992). In addition, cells that exhibit TCDD-mediated increases in P450 induction are different from those that exhibit TCDD-induced increases in DNA replication (Lucier et al., 1992).
Estrogen Receptor-Mediated Responses The results of the Kociba and colleagues (1978) bioassay indicated that TCDD can increase liver tumor incidence in rats while it decreases tumor incidence in organs such as the mammary gland, uterus, and pituitary gland. These results may be affected by the interaction of TCDD and the estrogen receptor. The estrogen receptor-ligand complex is capable of reversibly binding with DNA; this interaction is responsible for the transcriptional activation of estrogen-responsive genes. TCDD decreases the binding capacity of estrogen receptors in the liver and uterus, leading to decreased estrogen concentrations and activity (Romkes et al., 1987). This effect is also Ah receptor dependent (Lin et al., 1991a). The role of this response in TCDD-mediated carcinogenesis is unknown, however.
Cell Proliferation TCDD-mediated hepatocarcinogenesis may be related to its ability to increase rates of hepatocellular proliferation in that organ. Both of these effects are dependent on the presence of ovaries (Clark et al., 1991b). Increased rates of cell proliferation may be related to TCDD's effects on the epidermal growth factor (EGF) receptor. EGF is a potent mitogen that affects both normal and neoplastic cells (Stoschek and King, 1986). The EGF receptor and its ligands also play a role in cell transformation and tumorigenesis (Marti et al., 1989; Velu, 1990). Several studies have shown that TCDD mimics EGF, decreasing the binding capacity of the plasma membrane EGF receptor for its ligand without changing its binding affinity (Hudson et al., 1985; Abbot and Birnbaum, 1990; Astroff et al., 1990), although TCDD does not bind with the receptor itself. TCDD produces a functional change by decreasing EGF-stimulated autophosphorylation of the receptor (Clark et al., 1991b). In addition, TCDD induces the production of transforming growth factor-α (TGF-α), which does bind to the EGF receptor, leading to increased mitogenic signals (Choi et al., 1991). TCDD also has effects on a number of other EGF receptor pathways (Cochet et al., 1984; Beguinot et al., 1985; Astroff et al., 1990). Increasing cell proliferation rates occur at higher doses than loss of the EGF receptor (Lucier et al., 1992), however, so TCDD's effects on the EGF receptor probably have a number of consequences in addition to cell proliferation. Nonetheless, these responses are consistent with the observation that TCDD has mitogenic effects, which may play a role in its tumor-promoting ability.
Immune Suppression As discussed further below, there is ample evidence that administration of TCDD to animals results in suppression of the immune response. Some studies in animals and humans have shown that the exposure to immunosuppressive agents increases the incidence of tumors. As discussed further below, administration of TCDD did increase the incidence of tumors in animals injected with tumor cells. This may add another mechanism by which exposure to TCDD could increase the incidence of tumors in treated animals.
Conclusions Chronic rodent bioassays have demonstrated that exposure to TCDD can enhance tumor rates in a variety of target organs. TCDD is not a genotoxic carcinogen, although it may enhance the DNA-damaging properties of other chemicals. In multistage models of carcinogenesis, TCDD acts as a tumor promoter and has little, if any, tumor-initiating activity. TCDD mediates carcinogenesis through a variety of biochemical effects that are dependent on the presence of the Ah receptor. These effects involve multiple pathways that play roles in regulating cell proliferation and differentiation. The multiple site specificity of TCDD-mediated carcinogenesis is likely to reflect its multiple mechanisms of action.
Immunotoxicity
Introduction The immune system is pivotal in the maintenance of health, resistance to infection, and surveillance for some types of altered cells. Suppression of the immune system can result in an increase in the incidence and severity of infectious disease, as well as an increase in the incidence of some types of neoplasia. Enhancement of the immune response can result in the development or exacerbation of allergy and autoimmune disease. Therefore, an alteration in the level of immune responsiveness, either increased or decreased, could result in an increase in susceptibility to disease and should be considered toxic.
The immune system is a complex network that involves the interaction of different types of cells and soluble mediators. Because of the consequences of alterations in immune function, this system is highly regulated in the extent and duration of the response to a given antigen. Since a chemical may act at a given step in the generation of an immune response, the timing of exposure to the chemical or antigen to which a response is generated is critical, and must be considered in determining the applicability of animal studies on chemical exposure to epidemiologic evaluation of the effects of environmental toxicants on the human immune system.
Numerous studies in several species have indicated that the immune system is highly sensitive to the effects of exposure to TCDD and related compounds. However, because of the wide variation in the experimental
design, exposure protocols, and immunologic assays used, these studies are only summarized.
In Vivo Immunotoxicity Following exposure to relatively high levels of TCDD, the amount of lymphoid tissue is depleted, with the thymus being most sensitive to its effects (Vos et al., 1973; Vos, 1977; McConnell and Moore, 1979; Exon et al., 1987; Van Loveren et al., 1991). Alterations in immune effector function and host resistance occur at doses well below those that cause lymphoid tissue depletion. Multiple cellular targets, which are described below, have been implicated in the immunotoxic effects of TCDD. In addition, there is evidence that some of the effects of TCDD on the immune system are due to indirect actions on nonlymphoid tissues.
Cellular Immunity Results of studies of immune function in mice, rats, guinea pigs, and nonhuman primates indicate that TCDD suppresses cell-mediated and humoral immune functions in a dose-related manner (reviewed by Kerkvliet, 1984; Exon et al., 1987; Vos and Luster, 1989; Holsapple et al., 1991a,b). Because there are extensive scientific reviews available of the effects of TCDD on specific immune functions, this information is only summarized here.
A common observation in rodents following adult and pre- or postnatal exposure to TCDD has been the suppression of cell-mediated immunity. Immune functions suppressed following exposure to TCDD include delayed-type hypersensitivity, proliferative responses to mitogen and allogeneic lymphocytes, graft-versus-host disease, allograft rejection, and generation of cytotoxic T lymphocytes to allogeneic tumor (Vos et al., 1973; Faith and Moore, 1977; Sharma and Gehring, 1979; Thomas and Hinsdill, 1979; Luster et al., 1980a,b; Mantovani et al., 1980; Vecchi et al., 1980; Clark et al., 1981; Kerkvliet et al., 1990a). Of interest, especially in light of the effects observed in human studies, proliferative responses of rodent splenocytes to mitogen were shown to be elevated following exposure to low doses of TCDD (Luster et al., 1980a,b).
Humoral Immunity Multiple studies have shown that TCDD suppresses the generation of a humoral immune response, which results in the generation of monospecific antibodies from terminally differentiated B cells, in mice and guinea pigs (Vos et al., 1973; Thomas and Hinsdill, 1979; Hinsdill et al., 1980; Vecchi et al., 1980, 1983; Davis and Safe, 1988; Kerkvliet and Brauner, 1990; Kerkvliet et al., 1985, 1990b). Either the generation of specific serum antibody titer or the number of plaque-forming cells produced after immunization with sheep red blood cells (SRBCs), a T cell-dependent antigen, is markedly suppressed following TCDD exposure. In adult mice, the generation of a humoral immune response appears to be more sensitive to TCDD than a cell-mediated immune response. There is a differential sensitivity in the generation of humoral immune responses to
antigens (SRBCs, dinitrophenyl-Ficoll, and trinitrophenyl-lipopolysaccharide) that correlates with the degree of T cell involvement in the generation of the immune response (House et al., 1990; Kerkvliet et al., 1990b). Although a direct effect on B cell function has been shown after in vitro exposure or in vivo exposure and ex vivo immunization, these data suggest that TCDD may affect regulatory T cell function when the humoral immune response is generated in vivo (Holsapple et al., 1986; Dooley and Holsapple, 1988; Luster et al., 1988; Morris et al., 1991).
Macrophage and Polymorphonuclear Neutrophil Function and Inflammation Macrophage functions have also been examined following TCDD exposure and generally found to be resistant to suppression by TCDD when assessed ex vivo (Vos et al., 1978; Mantovani et al., 1980). In contrast, there is a selective inhibition of the anti-tumor cytolytic and cytostatic activity of polymorphonuclear neutrophils (PMNs) (Ackermann et al., 1989).
On the other hand, the pathology associated with TCDD toxicity includes neutrophilia and an inflammatory response characterized by activated macrophage and PMN accumulation (Vos et al., 1973, 1974; Weissberg and Zinkl, 1973; Puhvel and Sakamoto, 1988). This effect may be due to a normal inflammatory response to tissue injury or to a specific effect on inflammatory cells. Recent studies indicate that TCDD increased the respiratory burst of rat peritoneal macrophages (Alsharif et al., 1990). In addition, the inflammatory response that occurs following intraperitoneal injection of SRBCs, as measured by infiltration of PMNs and macrophages and alterations in macrophage cell surface proteins, was increased (Kerkvliet and Brauner, 1990; Kerkvliet and Oughton, 1993). One mechanism by which TCDD and related halogenated aromatic hydrocarbons may augment the inflammatory response is through enhanced production of inflammatory mediators such as interleukin-1 and tumor necrosis factor (TNF). On the other hand, serum complement has been reported to be suppressed in TCDD-treated mice (White et al., 1986). Recent studies indicate that the hypersensitivity of TCDD-treated mice to endotoxin may be due to an increase in the production of these inflammatory cytokines (Thomas and Hinsdill, 1978, 1979; Vos et al., 1978; Loose et al., 1979; Taylor et al., 1990; Clark et al., 1991a; Hoglen et al., 1992). In addition, the wasting syndrome and the mortality observed following exposure to TCDD were reduced after treatment of mice with pharmacologic levels of anti-TNF antibodies, suggesting a role for this inflammatory mediator in the process (Taylor et al., 1992). Recent studies show that TCDD may affect the transcription of the interleukin-1 gene directly through interaction with a DRE (Sutter et al., 1991).
Interspecies Sensitivity Studies have indicated that there is a difference in the sensitivity of various species to the immunotoxic effects of TCDD; however, most studies use different species and antigens, and are therefore not completely comparable. In one report, the effects of TCDD on the
immune system of rats, mice, and guinea pigs were examined, and even then, different immunologic parameters were assessed and different antigens were used (Vos et al., 1973). For example, the delayed-type hypersensitivity (DTH) response was the cell-mediated immune response parameter measured in rats and guinea pigs, and graft-versus-host response was measured in mice. From this study, it was shown that guinea pigs were more sensitive to the effects of TCDD than rats, which is consistent with other toxic effects of TCDD (McConnell et al., 1978a; Poland and Knutson, 1982). The mouse cell-mediated immune system also seems to be more sensitive to TCDD than the rat's; however, different antigens and end points were tested, and therefore the results cannot be compared. Studies by Clark and colleagues (1981) indicated that the sensitivities of immune responses of different antigens to TCDD in the same species and the same study are not equivalent.
In Utero Exposure Several studies have examined immune function in rodents following exposure to TCDD during fetal development (Vos et al., 1973; Faith and Moore, 1977; Thomas and Hinsdill, 1979; Luster et al., 1980a; Clark et al., 1983). These studies indicate that the fetus is much more sensitive to the effects of TCDD than the adult animal. The most sensitive indicator of TCDD immunotoxicity in these studies was a decrease in the host resistance to tumor. The ability of the offspring of TCDD-exposed dams to generate a cell-mediated and humoral immune response to antigen was also suppressed. When the mice were exposed only in utero and not during lactation, significant thymic atrophy and suppressed cell-mediated immunity were observed (Holladay et al., 1991).
Host Resistance Models The ability of an animal to eliminate viral, bacterial, and parasitic infections, as well as neoplastic diseases, is determined by both nonspecific and specific immunologic functions. A decrease in the functional activity of any immunologic compartment may result in increased susceptibility to disease. Host resistance models allow investigators to determine whether alterations in specific immunologic parameters result in increased susceptibility to disease.
TCDD exposure increases the susceptibility to challenge with gramnegative bacteria (Thigpen et al., 1975; Hinsdill et al., 1980). In addition, increased mortality was observed in animals infected with Streptococcus pneumonia after subchronic administration of TCDD (White et al., 1986). Enhanced susceptibility to viral and parasitic disease has also been reported after TCDD administration (Clark et al., 1983; Tucker et al., 1986; House et al., 1990). An increase in the growth of transplanted tumors has been demonstrated in mice treated with TCDD in utero (Luster et al., 1980a,b). This exposure protocol resulted in an increased incidence of PYB6 tumors in pups from dams receiving repeated doses of TCDD.
Role of Ah Locus in Immunotoxicity Many studies have been conducted to define the involvement of the Ah receptor, the protein that is thought to mediate the translocation of TCDD from the cytosol to the nucleus and its binding to DNA, in the mediation of TCDD immunotoxicity. Two lines of investigation have been undertaken to assess this contribution: comparison of the potency of the immunotoxicity of various congeners; and studies using mice of different genetic background known to differ at the Ah locus. Though the use of TCDD-sensitive (B6) and resistant (D2) mice, which differ in the affinity of their Ah receptor for TCDD, and mice that are congenic at the Ah locus, the generation of cell-mediated and humoral immune responses in vivo seems to be Ah receptor-mediated (Silkworth and Grabstein, 1982; Vecchi et al., 1983; Kerkvliet et al., 1990a,b). The involvement of the Ah receptor in the immunotoxic effect of TCDD was further confirmed by structure-activity relationships (Kerkvliet et al., 1985; Davis and Safe, 1988-1990). These studies all involved acute or subacute exposure to TCDD in vivo.
The contribution of the Ah receptor to the immunotoxicity observed after chronic exposure to TCDD is less clear. Recent studies showed that the sensitivity of D2 mice to TCDD-induced immunosuppression increased when TCDD was administered daily over 14 days rather than as a single dose (Morris et al., 1992). The effects of this treatment regime on the sensitivity of B6 mice are unclear in this study. In addition, the early studies of Vecchi, which indicated that the Ah receptor was involved in the immunotoxicity of TCDD, were conducted following multiple exposures to TCDD (Vecchi et al., 1983). Therefore, further studies are required to define the contribution of the Ah receptor to immunotoxicity after chronic exposure.
Some, but not all, in vitro studies of the effects of TCDD on the generation of a humoral immune response indicate that the Ah receptor may not be involved in the observed immunosuppression (Holsapple et al., 1986; Tucker et al., 1986; Davis and Safe, 1991). The basis for these variable effects following in vitro exposure to TCDD is unknown at this time, and the relevance of these negative data in light of the in vivo results is uncertain.
Contribution of the Endocrine System Many studies of the effects of TCDD and congeners that bind the Ah receptor following in vivo administration and immunization indicate that there may be an indirect component to the immunosuppression observed. Studies by Kerkvliet and colleagues (1990a) indicated a measurable elevation in serum corticosterone levels that correlated with the suppression of the generation of a cytotoxic T lymphocyte (CTL) response after exposure to hexachlorobiphenyl. However, further studies using adrenalectomized mice or rats and a glucocorticoid receptor antagonist suggest that this increase in serum corticosterone
is probably not involved (Van Logten et al., 1980; DeKrey et al., 1990). Alternatively, comparison of the sensitivity of male and female mice to hexachlorobiphenyl immunotoxicity, and the partial protection of male mice to the immunotoxicity of hexachlorobiphenyl by castration, suggest a role for testosterone in the suppression of the CTL response (Kerkvliet and Baecher-Steppan, 1988; DeKrey et al., 1992). These data suggest that the hormonal status of the animal may contribute to the immunotoxic effects observed after exposure.
Immunotoxicity of TCDD in Nonhuman Primates Some studies have been conducted on the effects of TCDD on the immune system of nonhuman primates. Immunologic effects were described in rhesus monkeys and their offspring chronically exposed to TCDD (Hong et al., 1989). In the mothers, the total number of T cells and CD8+ cells was increased and the number of CD4+ cells was decreased. No effect was observed on immune function. The ability of the offspring to generate a humoral immune response to tetanus toxoid was significantly increased.
Acute administration of TCDD to marmosets resulted in a decrease in CD4+ T cells (especially CD4+CDw29+) and CD20+ B cells, and an increase in the percentage of CD8+ cells and CD4+45RA+ (Neubert et al., 1990b). Chronic exposure of marmosets to a low dose of TCDD resulted in the opposite effects on the subpopulation of CD4+ cells (i.e., CD4+CDw29+ cells increased, and CD4+45RA+ cells decreased; Neubert et al., 1992). On the other hand, when the dose of TCDD was increased in this study, the results were consistent with those observed after acute administration of TCDD. These data are extremely important with regard to observations in the human population after chronic low dose exposure to TCDD. As the authors state, ''Extrapolations of the results obtained at higher doses to very low exposures is not justified with respect to the effects induced by TCDD on the immune system of marmosets" (Neubert et al., 1992).
Hepatotoxicity
Introduction In animal species that exhibit sensitivity to TCDD and related halogenated aromatic hydrocarbons (HAHs), the liver represents one of the primary target organs. From animal studies, a characteristic profile of liver-associated changes at the cellular, biochemical, and molecular levels has been identified. It is important to emphasize that almost without exception, structure-activity studies indicate that liver alterations produced by HAH congeners are associated with the Ah locus. The most extensive characterization of liver-associated alterations in response to TCDD and related compounds has been in rodents, primarily mice and rats; however, some hepatotoxicity data are available in other mammalian species. This
section describes the profile of hepatic changes induced by TCDD as identified in laboratory animals.
Morphologic Changes Significant inter- and intraspecies variation has been observed in the severity of hepatic lesions produced by TCDD. Without exception, all mammalian species investigated experimentally exhibit some degree of hepatomegaly even at TCDD doses that are sublethal (Kociba et al., 1976; McConnell, 1985). Hepatomegaly appears to be a direct result of hyperplasia (an increase in number) and hypertrophy (increase in size) of parenchymal cells (hepatocytes) (Fowler et al., 1973; Hinton et al., 1978). Additionally, administration of lethal doses of TCDD has been shown to produce degenerative and necrotic changes in a variety of rat strains, which can be accompanied by mononuclear cell infiltration, multinucleated giant hepatocytes, an increase in hepatic smooth endoplasmic reticulum, and increased numbers of mitotic figures (Gupta et al., 1973; Jones and Butler, 1973; Kociba et al., 1976; Hinton et al., 1978). Other lesions varying among species include focal, centrilobular lesions in the mouse (Vos et al., 1974) and widespread necrosis in the rabbit (Vos and Beems, 1971). Neither the guinea pig nor the hamster displays severe morphological alterations in the liver following TCDD administration. In light of this, although liver lesions may contribute to TCDD lethality in laboratory animals, the cause of death cannot be explained on the basis of liver lesions alone.
Changes in Hepatic Function Alterations in liver morphology following TCDD treatment are accompanied by impaired hepatobiliary function including increased microsomal monooxygenase activity, liver enzyme leakage, impaired plasma membrane function, porphyria, hyperlipidemia, hyperbilirubinemia, hyperproteinemia, and increased regenerative DNA synthesis. As stated above, in most cases, the magnitude of these changes is Ah related and has been demonstrated in numerous studies using Ah-responsive and nonresponsive mouse strains (C57/BL/6J and DBA/2J, respectively).
Hyperlipidemia Recently, studies comparing the hepatotoxic effects of TCDD in Ah-responsive and nonresponsive mice demonstrated that C57/BL/6J mice, administered a single dose of 3 µg/kg TCDD, developed mild to moderate hepatic lipid accumulation but no inflammation. Severe fatty change, mild inflammation, and necrosis were observed in C57/BL/6J mice following a single dose of 150 µg/kg (Shen et al., 1991). Conversely, DBA/2J mice that received a single dose of 30 µg/kg developed hepatocellular necrosis and inflammation, and exhibited no hepatic lipid accumulation; only slight lipid accumulation was observed at high doses of TCDD (600 µg/kg). Induction of fatty liver following exposure to TCDD and related compounds has also been demonstrated in a number of other animal species including rat, chicken, and human. Sublethal doses of TCDD in rats have been reported to produce an increase in total hepatic lipid content.
Specifically, triglycerides and free fatty acids increased while sterol esters decreased. Lethal doses of TCDD increased cholesterol esters and free fatty acids (Albro et al., 1978). Dose-dependent increases in total and high-density serum lipoproteins in rats administered sublethal concentrations of TCDD have also been reported (Poli et al., 1980). No difference in lipid synthesis between control and TCDD-treated rats has been found (Cunningham and Williams, 1972). Studies with polychlorobiphenyl-treated rats, which were also found not to have increased lipid synthesis, provided evidence that hepatic lipids had an increased half-life (Hinton et al., 1978). Further evidence supporting a decrease in lipid utilization following TCDD treatment was obtained in studies by Albro and coworkers (1978) in which ultrastructural changes in liposomes and reduced levels of ester hydrolases were reported. Taken together, these studies suggest that fatty liver following exposure to TCDD is due to decreased lipid utilization rather than increased lipid biosynthesis.
Porphyria Accumulation of porphyrins (a family of compounds derived from tetrapyrrole, which is found in hemoglobin, myoglobin, and most cytochromes) in liver as well as spleen and kidney is stimulated by subchronic or chronic treatment with TCDD and related compounds, and is also accompanied by an increase in urinary porphyrin excretion (Goldstein et al., 1982). Cantoni and colleagues (1981) demonstrated that oral exposure of rats to 0.01, 0.1, or 1 µg TCDD/kg body weight/week for 45 weeks produced an increase in coproporphyrin concentrations at all dose levels.
Porphyria cannot be induced by a single dose of TCDD or related compounds. The mechanism responsible for HAH-mediated hepatic porphyria, although not elucidated, may occur partially through a decrease in uroporphyrinogen decarboxylase activity as suggested from work by Elder (1978). Jones and Sweeney (1977) also reported a decrease in this enzyme activity as well as elevated levels of carboxylated porphyrins following several weeks of TCDD treatment in mice. Additionally, TCDD-mediated induction of hepatic porphyria and inhibition of uroporphyrinogen decarboxylase activity in C57/BL6 mice were markedly reduced by iron deficiency (Sweeney et al., 1979). In contrast to these studies, Yao and Safe (1989) recently demonstrated in C57/BL6 mice that 6-methyl-1,3,8-trichlorodibenzofuran (MCDF), an inactive HAH congener, was capable of antagonizing the induction of porphyria by TCDD; however, MCDF was incapable of antagonizing the inhibition of uroporphyrinogen decarboxylase or the induction of both AHH and ethoxyresorufin O-deethylase (EROD) activity. These studies suggest that inhibition of uroporphyrinogen decarboxylase is not the primary event responsible for porphyria. Another enzyme also implicated in the mechanism associated with TCDD-induced hepatic porphyria is δ-aminolevulinic acid (ALA) synthetase, the enzyme responsible for the initial and rate-limiting step in heme biosynthesis. ALA
synthetase is markedly induced by TCDD exposure. In rodents, ALA synthetase is not increased following acute TCDD treatment but is elevated following subchronic or chronic exposure. Goldstein and colleagues (1982) reported that after 16 weeks of chronic exposure, ALA synthetase was increased only in animals that were porphyric. However, the role of ALA synthetase in the mechanism of porphyria is questionable, according to several reports. Goldstein and colleagues (1974) reported that elevation in ALA synthetase occurs only after the onset of porphyria. Additionally, studies by Jones and Sweeney (1980) demonstrated that porphyria could be produced by TCDD in mice without increased ALA synthetase activity. In light of these findings, the induction of ALA synthetase does not appear to be a necessary event in the induction of porphyria.
Impaired Cell Membrane Function The effects of TCDD on hepatocyte plasma membrane function have been investigated in a number of experimental systems. In rodents, TCDD markedly inhibited the binding and/or expression of a number of receptors for regulatory factors in hepatocytes including epidermal growth factor receptor (Madhukar et al., 1988; Lin et al., 1991a), glucocorticoid receptor (Lin et al., 1991b), estrogen receptor (Lin et al., 1991b), and low-density lipoprotein (LDL) receptor (Bombick et al., 1984). TCDD produced an 80-90 percent decrease in the maximum binding capacity (both high- and low-affinity sites) of the hepatic EGF receptor in female Ah-responsive and Ah-nonresponsive mice. However, the ED50 (dose at which 50 percent of animals experience the effect) for the effects of TCDD on EGF binding was tenfold higher in the Ah-nonresponsive mice (Lin et al., 1991a). Additionally, TCDD did not affect the hepatic content of EGF receptor MRNA, indicating that the effects on the EGF receptor are not pretranslational. TCDD administered to mice also was found to produce approximately a 30 percent decrease in the maximal binding capacity of both the hepatic glucocorticoid and the estrogen receptors. A concomitant 30 percent decrease in tyrosine aminotransferase activity, which is regulated by the glucocorticoid receptor was also observed (Lin et al., 1991b). A greater sensitivity to inhibition by TCDD of the binding of estrogen to the hepatic estrogen receptor was demonstrated in Ah-responsive than in Ah-nonresponsive mice. Conversely, TCDD produced a comparable inhibition of glucocorticoid receptor binding and tyrosine aminotransferase activity in Ah-responsive and nonresponsive mice. These results suggest that the Ah receptor regulates the effects of TCDD on the binding of estrogen to the hepatic estrogen receptor, but the decreased binding of the hepatic glucocorticoid receptor does not appear to be mediated directly by the Ah locus. Acute TCDD exposure of guinea pigs and rats also produces a significant reduction in binding of LDL to its receptor on the hepatic plasma membrane (Bombick et al., 1984). It is important to emphasize that this reduction in LDL binding was not caused
by a decrease in food intake by treated animals as demonstrated by comparison to pair-fed control animals.
Biliary Excretion A number of studies have investigated the effect of TCDD and related compounds on bile flow. Structure-activity studies indicated that treatment of rats with HAH congeners decreased bile flow as measured by the clearance of ouabain, a model compound for neutral nonmetabolized substrates (Yang et al., 1983). The magnitude of depression by HAH congeners of ouabain excretion was closely associated with their potency in inducing AHH activity. Bile duct cannulation of TCDD-treated rats followed by segmented retrograde intrabiliary injection of radiolabeled morphine, imipramine, or ouabain demonstrated that TCDD inhibited the canalicular transport of glucuronide metabolites of morphine and imipramine into the bile (Berman et al., 1986). Likewise, TCDD inhibited the canalicular transport of ouabain into bile. Acute TCDD exposure in rats was also found to produce a greater than twofold increase in hepatic copper (compared to pair-fed controls; i.e., animals possessing the same dietary intake), a trace metal whose homeostasis depends on its biliary excretion (Elsenhans et al., 1991). These data suggest that TCDD and related compounds decrease bile flow.
Enzyme Induction The most extensively studied biochemical effects on hepatic function associated with exposure to TCDD and related compounds are the marked changes this class of chemicals produces on enzyme activity. The most profound effect is an increase in microsomal mixed-function oxidase activity in parenchymal cells. HAHs markedly induce the cytochrome P450 isozyme CYP1A1, which is most often monitored experimentally through the measurement of AHH and EROD activity. Numerous in vivo and in vitro structure-activity studies have investigated the relative potency of HAH compounds, including the chlorinated dioxins, biphenyls, and dibenzofurans, in their ability to induce AHH and EROD activities (reviewed: Safe, 1990). A structural relationship was identified between the arrangement of halogen atoms on the dibenzo-p-dioxin molecule and the ability of these congeners to induce AHH and EROD. The sites identified as confirming the greatest biological potency were the four lateral ring positions. Additionally, congeners that were halogenated at all four lateral ring positions, the 2,3,7,8-sites, demonstrated more biological activity than those that were halogenated at only three of the four positions. Halogenation at only two of the four lateral positions results in loss of biological activity. On a molecular basis, 2,3,7,8-TCDD is the most potent inducer of AHH activity among the HAH congeners. A strong correlation for HAHs has also been established between AHH induction and their Ah-binding affinity. A similar correlation has been established between AHH induction by chlorinated dibenzo-p-dioxins, chlorinated dibenzofurans, and polychlorinated biphenyls and their respective acute toxicities (Poland and Glover, 1973; Safe, 1990).
There are two obvious concerns pertaining to the relevance of MFO induction. First, MFO induction can result in a greater rate of metabolism of endogenous substrates. Secondly, and perhaps of even greater significance is the potential for increased metabolism of exogenous compounds that undergo metabolic bioactivation to a more toxic form. However, detoxification of exogenous substrates is also an important role played by MFO, so inducing MFO can also reduce the toxicity of a variety of compounds.
The most widely studied experimental models of MFO induction have been murine (i.e., rats and mice). In rats, TCDD has been shown to increase a variety of oxidative and conjugative enzymes involved in drug metabolism and elimination including CYP1A1 and CYP1A2, aniline hydroxylase, AHH, biphenyl hydroxylase, ethoxycoumarin O-deethylase (ECOD), EROD, and UDPGT. Additionally, a number of non-drug metabolizing enzymes have also been identified as being inducible by TCDD (ornithine decarboxylase, prostaglandin synthetase, porphyrinogen carboxylase, transglutaminase, aldehyde dehydrogenase, δ-aminolevulinic acid synthetase).
In the mouse, AHH induction by TCDD and related compounds is under the control of the Ah gene locus, which is believed to encode a soluble receptor that binds to inducers and mediates an increase in cytochrome P450 gene transcription (Taylor, 1984—see mechanism of action section above). Identification of the Ah locus was achieved primarily from enzyme induction studies using various mouse strains differing at this locus. TCDD as well as other inducers of AHH (i.e., 3-methylcholanthrene, benzo[a]pyrene, etc.) are 10 times as potent, or more, in inducing hepatic CYP1A1 in C57BL/6J mice than in DBA/2J mice (Poland and Knutson, 1982; Nebert, 1989).
Data on enzyme induction in other species are less well-characterized. The guinea pig, which is the most sensitive species to TCDD, at least in terms of lethality, shows only a very slight induction of MFO (Beatty and Neal, 1977). Similarly, enzyme induction in the rabbit is not well-characterized. In the Syrian golden hamster, TCDD was found to induce MFO and to reduce NAD(P):menadione oxidoreductase [NAD(P) = nicotinamideadenine dinucleotide (phosphate)] and ECOD (Gasiewicz et al., 1986).
Reproductive and Developmental Toxicity
Introduction TCDD has been found to have a number of effects on reproductive and developmental function in laboratory animals. Although a number of studies have addressed the effects of TCDD on the fertility of male laboratory animals, there are no data relating male animal exposure to TCDD and developmental end points such as congenital anomalies, cancer, and growth retardation, which are the end points of interest in humans. Extrapolating observations from laboratory animals to humans is not straightforward, however, due to important species determinants for both reproductive
and developmental end points. In addition, little information is available on the cellular and molecular mechanisms of action of TCDD that play a role in these effects. This section summarizes the information that is available in laboratory animals for each end point.
Reproductive Toxicity
Male A number of studies have demonstrated that overtly toxic doses of TCDD can affect several parameters that play a role in the reproductive function of male laboratory animals. TCDD administration to adult animals can decrease the weight of testes and accessory sex organs, affect testicular morphology, decrease spermatogenesis, and reduce fertility (Allen and Lalich, 1962; Allen and Carstens, 1967; Khera and Ruddick, 1973; Kociba et al., 1976; Van Miller et al., 1977; McConnell et al., 1978a; Moore et al., 1985; Chahoud et al., 1989, 1992; Morrisey and Schwetz, 1989; Rune et al., 1991a,b). These effects have been reported in rats, mice, guinea pigs, marmosets, monkeys, and chickens, but have generally been observed only at doses that also decrease body weight and feed intake.
TCDD's effects on spermatogenesis include loss of germ cells, appearance of degenerating spermatocytes and spermatozoa, and reduced numbers of seminiferous tubules containing mature spermatozoa. The lowest total dose at which effects on spermatogenesis have been observed was 3 µg TCDD/kg, which was administered as a single subcutaneous injection to rats and reduced the number of spermatids per testis to 60 percent that of controls (Chahoud et al., 1992). No other toxic effects were reported at this dose. In another study in rats, daily gavage doses totaling 65 µg TCDD/kg, administered over 13 weeks, also reduced spermatogenesis (Kociba et al., 1976); at this dose, however, body weights and feed consumption were reduced as well.
The effects of TCDD on fertility were evaluated in male rats exposed to either a single subcutaneous dose of 25 µg/kg followed by weekly maintenance doses of 5 µg/kg, or a single dose of 75 µg/kg followed by maintenance doses of 15 µg/kg. Rats were treated for 10 weeks prior to mating, as well as throughout the 12-week mating period. Female rats were untreated. Mortality was 93 percent in the high dose group, and body weights were significantly decreased in both groups. At the low dose, 15 percent of animals were found to be sterile, morphological changes of the testes were apparent, and the mating and fertility indices were somewhat reduced (84 percent and 95 percent of controls, respectively). The pregnancy index was not affected, however (Chahoud et al., 1991).
At lower doses of TCDD (15 µg/kg), androgenic deficiencies in rats have been detected, including decreased plasma testosterone levels, decreased testicular responsiveness to luteinizing hormone, and increased pituitary responsiveness to feedback inhibition by androgens and estrogens (Moore et
al., 1989, 1991, 1992; Bookstaff et al., 1990a,b; Kleeman et al., 1990). Decreased testosterone levels apparently result from inhibition of testosterone biosynthesis due to inhibition of the mobilization of free cholesterol, a testosterone precursor (Moore et al., 1991). Normally, an increase in plasma levels of luteinizing hormone would be expected to facilitate testicular compensation for decreased plasma testosterone; in TCDD-treated rats, however, this increase does not occur, due to an enhanced ability of testosterone and its metabolites to inhibit luteinizing hormone secretion (Moore et al., 1989; Bookstaff et al., 1990a,b). In addition, TCDD treatment prevents the increase in the number of pituitary gland receptors for gonadotropin-releasing hormone that normally occurs in response to decreased plasma androgen concentrations (Bookstaff et al., 1990b).
In general, both the morphologic and the biochemical effects of TCDD on fertility-related parameters occur in laboratory animals at doses that also induce overt toxicity. Only one study demonstrated an effect on spermatogenesis in rats at a dose that was not associated with other toxicity (Chahoud et al., 1992); the consequence of this effect on fertility was not evaluated. Much higher doses of TCDD did not affect male rat fertility (Chahoud et al., 1991). The reproductive systems of adult male laboratory animals thus appear to be relatively insensitive to TCDD.
Female Reduced female fertility has been demonstrated in a number of studies of rodents exposed to TCDD. Murray and colleagues (1979) exposed both male and female rats to 0, 0.001, 0.01, or 0.1 µg TCDD/kg body weight/day in the diet over three generations. The female fertility index, defined as the ratio of the number of females confirmed pregnant to the number of females for which mating was confirmed, varied among both control and exposed animals, with a reduced number of impregnated animals reported in the exposed groups. Reduced fertility and litter size were reported in the F0 generation at the 0.1- µg/kg daily exposure level and in the F1 and F2 generations at the 0.01- µg/kg daily exposure level. Kociba and colleagues (1976, 1978) reported suppression of the estrous cycle, absence of ovulation, and signs of ovarian dysfunction in female rats exposed to 1-2 µg TCDD/kg/day for 13 weeks, but not in rats exposed to 0.001-0.01 µg/kg/day for two years.
Reduced female fertility, detected as a reduced ability to conceive and give birth, has also been demonstrated in a series of studies in rhesus monkeys, although many of those exposed also experienced maternal toxicity (Allen et al., 1977, 1979; Barsotti et al., 1979; Schantz et al., 1979; Bowman et al., 1989; Schantz and Bowman, 1989). In these experiments, female monkeys received TCDD in their diets at levels ranging from 5 to 500 ppt, and were mated to unexposed males after seven months of exposure. No effects on reproductive function were detected at the lowest dose. A reduced ability to give birth was also seen when monkeys were fed a total
dose of 1 µg TCDD/kg during the first trimester of pregnancy; no effect was observed at lower doses (McNulty, 1984).
These limited experiments on the effects of TCDD on female reproductive function indicate that in laboratory animals, TCDD can decrease fertility, decrease the ability to remain pregnant throughout gestation, and in the case of rats, decrease litter size, although some of these effects may have been secondary to maternal toxicity. Mechanistic information is confined to the effects of TCDD on ovarian function suggested by Kociba and colleagues (1976), and to studies of serum hormone levels in monkeys reported by Allen and colleagues (1979) and Barsotti and colleagues (1979), in which reductions in estradiol and progesterone concentrations were correlated with dietary levels of TCDD as well as with reduced fertility and increased spontaneous abortion. No such alterations in hormone levels were detected in rats, however, which suggests species differences (Shiverick and Muther, 1983). Whether TCDD's effects on hormone levels result from their increased metabolism (due to induced hepatic P450 levels) or to an effect on the responsiveness of gonadal tissue itself is unknown. Antiestrogenic effects of TCDD in rats and mice include decreased uterine weight and a decreased concentration of tissue progesterone receptor levels; when TCDD and 17β-estradiol were coadministered, TCDD prevented the usual 17β-estradiol-induced increases in uterine weight and progesterone receptor levels (Gallo et al., 1986; Astroff et al., 1990; Safe et al., 1991). This effect is age dependent and is not observed in immature animals (Safe et al., 1991).
Developmental Toxicity Prenatal exposure to TCDD has been associated with both increased fetal mortality and increased developmental abnormalities in laboratory animals. Increased fetal mortality is dose related and depends on the timing of exposure: embryos are more susceptible to TCDD-induced mortality at certain stages of development than others (Couture et al., 1990a). The embryolethal dose of TCDD is generally one to two orders of magnitude lower than the dosage that is lethal to adults; however, increased embryolethality generally occurs at doses that also induce nonlethal maternal toxicity (Sparschu et al., 1971; Khera and Ruddick, 1973; Courtney, 1976; Giavini et al., 1982). Other common fetotoxic effects of TCDD are thymic hypoplasia, subcutaneous edema, decreased growth, and hematologic alterations. Species-specific effects include cleft palate formation and hydronephrosis in the mouse, extra ribs in the rabbit, and intestinal hemorrhage in the rat (Courtney and Moore, 1971; Courtney, 1976; Giavini et al., 1982; Couture et al., 1990b). Evidence in the mouse indicates that the developmental effects of TCDD may be mediated by the Ah receptor (Poland and Knutson, 1982). Mouse strains with Ah receptors that have a relatively high affinity for TCDD respond to lower doses than strains with a relatively low-affinity (Poland and Glover, 1980; Hassoun et al., 1984).
The tissue specificity of the response to TCDD cannot be attributed solely to the presence of Ah receptors, however; other as yet unidentified factors also play a role.
Neurotoxicity
Introduction The physiological and structural function of the nervous system is preserved across species in terms of the mechanisms by which neurons generate and propagate action potentials, the mechanisms that transmit chemically encoded information across synaptic clefts, and the manner in which organized functional groups of neurons or CNS nuclei communicate and influence each other. Behavioral actions and interactions at their most basic levels are also conserved phylogenetically. Thus there are common themes that are apparent in the ways in which laboratory animals and humans sense, integrate, and respond to external stimuli. These similarities include similarities in cognitive function, learning, and memory. Because of the structural and functional similarities of the nervous system across species, extrapolation of experimentally produced neurotoxicity from laboratory animals to humans is easier than for many other end points of toxicity.
Exposure Levels Although TCDD and related chemicals have an affinity for accumulating in lipid-rich tissues, the concentrations of these materials in the brain after systemic exposure are low (Piper et al., 1973a; Van Miller et al., 1976; Gasiewicz and Neal, 1979; Olson et al., 1980a; Gasiewicz et al., 1983; Abraham et al., 1990). Although different methods have been used to investigate the TCDD concentrations in the brain, the actual levels reported are quite close among rodent species. For example, within 1-3 days after receiving a dose of TCDD, the concentrations of TCDD (percentage of dose per gram of tissue) in the brain were rat—0.06; hamster—0.05; guinea pig—8 0.25; and mouse—0.1-0.4. TCDD levels in adult primates have been reported to be lower than those found in the rat (0.006 versus 0.13 percent of dose/g tissue), but infant monkeys have higher levels than adults (0.018 versus 0.006 percent of dose/g tissue) (Van Miller et al., 1976). Brain TCDD concentrations do not appear to differ significantly in TCDD-sensitive mice when compared to TCDD-insensitive mice (Gasiewicz et al., 1983). Initial brain levels of TCDD do not appear to be affected by redistribution of TCDD from adipose tissue during the weeks following a single systemic exposure (Piper et al., 1973a; Olson et al., 1980a; Gasiewicz et al., 1983).
Direct injection (8 µg/kg) of TCDD into the lateral ventricle of Sprague-Dawley rats results in total brain concentrations that are 100 times greater than those achieved after intravenous injection (72 µg/kg) (Stahl and Rozman, 1990). Total brain concentrations of 356 ppb TCDD following intraventricular
injections of 8 µg/kg were not associated with systemic toxicity or neurotoxicity (Stahl and Rozman, 1990).
Ah Receptors in the Nervous System Relatively little work has been done to quantify the concentration of Ah receptors in the nervous system (Gasiewicz and Rucci, 1984). In the CNS of the guinea pig, a species that is sensitive to TCDD-induced acute toxicity, Ah receptors have been identified in the cerebrum and cerebellum at similar concentrations (11-12 fmol/mg cytosolic protein). The hamster, which is resistant to TCDD-induced acute toxicity, showed a similar concentration of receptors in the cerebrum, but none were detected in the cerebellum. No Ah receptors were detected in the midbrain, medulla, or hypothalamus of either species. The CNS Ah receptor concentrations (where detectable) were approximately 20 percent of those seen in the livers of the guinea pig and hamster. The presence of Ah receptors in the peripheral nervous system has not been investigated, and Ah receptors have not been detected in skeletal muscle tissue (Gasiewicz and Rucci, 1984). Because high concentrations of Ah receptors have been associated primarily with epithelial structures (Gasiewicz and Rucci, 1984), the most likely region of the brain to have higher concentrations of Ah receptors would be the epithelium of the choroid plexus.
Neurotoxicity Studies Following Acute and Subchronic Exposure A large number of acute toxicity studies have been conducted with TCDD, but most of these studies were not designed specifically to investigate neurotoxicity. None of these studies specifically noted frank signs of neurotoxicity associated with TCDD exposure.
Sirkka and colleagues (1992) studied a series of neurological end points in male Han/Wistar rats given 1,000 µg/kg of TCDD intraperitoneally (i.p.). This dose level is equivalent to one-third of the i.p. LD50 for this strain of rat. Controls included both ad libitum and pair-fed controls for the majority of end points examined. The tests were conducted at varying times after exposure and were designed to detect changes in motor control, nociception, anxiety level, and learning. TCDD exposure had no effect on the ability of rats to complete these tests, with the possible exception of a transient decrement in coordination on the narrow-bridge portion of the horizontal-bridge test 16 hours after exposure. At 8 days, there was no performance decrement, and at 16 hours, performance on the broad-bridge portion of the test and on the more difficult rotating-rod test were normal. Overall, these results indicated no significant neurological impairment in animals that developed significant decreases in body weight following TCDD exposure.
Allen and colleagues (1977) studied the clinical and morphological effects associated with feeding diets containing TCDD [500 parts per trillion (ppt)] to female rhesus monkeys for nine months. The only morphological effect reported in the CNS was hemorrhage into the Virchow-Robin space.
This effect was associated with significant bone marrow toxicity and hemorrhage in many other organ systems. In this study, hemorrhage was probably an agonal lesion since no other effects were reported in the adjacent neuropil.
Role of Neurotoxicity in the Wasting Syndrome Because appetite is significantly regulated by CNS processes (Morley and Levine, 1985), there has been interest in trying to determine if CNS dysfunction is involved in causing TCDD-induced hypophagia. The TCDD-induced wasting syndrome has been observed in several different species (Kelling et al., 1985), but the relationship between the syndrome and central nervous system function has been studied primarily in rats. Seefeld and colleagues (1984) showed that hypophagia induced by TCDD in Sprague-Dawley rats was associated with a change in feeding behavior (increased spillage) that occurred in a dose-dependent fashion 7 days after TCDD administration and continued to increase for 15 days after dose administration. By 15 days, TCDD-treated rats were spilling 2 to 3 times more feed than the controls. The TCDD-induced depression of feed intake also began immediately after dosing. Hypophagia in these animals was not associated with an inability to feed, since rats given TCDD and placed on a feed-restricted diet prior to TCDD administration exhibited a significant degree of hyperphagia and a greater rate of weight gain than unrestricted animals (Seefeld et al., 1984). Thus TCDD did not appear to impair the rats' ability to feed but may have affected the set point at which body weight was maintained, which is under CNS control.
In light of pharmacokinetic data suggesting that little administered TCDD reaches the brain (Piper et al., 1973a; Lakshman et al., 1986), Pohjanvirta and colleagues (1989) compared the differences in toxicity of TCDD when administered intracerebrally (left ventricle) or subcutaneously via implanted minipumps in TCDD-susceptible Long-Evans rats and TCDD-resistant Han/Wistar rats. The pumps were set to deliver 20-21 µg/kg/day of TCDD. In both rat strains, intracerebral TCDD was more effective at reducing feed intake and body weight gain than subcutaneous TCDD. While the authors interpreted these data to imply that the CNS has a specific role to play in induction of TCDD toxicity, this interpretation is highly speculative in the absence of mechanistic or pharmacokinetic data linking the route of administration to a particular tissue target site or showing that the administered TCDD was not transported out of the brain.
Pohjanvirta and Tuomisto (1990) reported that TCDD-resistant Han/Wistar rats given a sublethal dose of TCDD showed residual alterations in feeding behavior following a variety of feeding regulatory challenges. After groups of 11 male rats were given a single high dose of TCDD (1,000 µg/kg body weight) or corn oil (vehicle), the TCDD-dosed rats began losing
body weight by about day 6 and continued to lose body weight for approximately 5 weeks, at which time the body weight reached a plateau (12.5 percent maximum loss) that was maintained until the end of the study (week 15). Both the TCDD-treated and the control groups were apparently fed ad libitum. Because the TCDD-dosed animals would be expected to reduce their feed consumption in response to TCDD and the controls were not pair-fed, a confounding variable may have been introduced into this study. In response to a sodium chloride challenge (1 percent of body weight of 1 M NaCl), water consumption was stimulated equivalently in both groups. In response to glucose deprivation induced by 2-deoxyglucose (400 mg/kg body weight), TCDD-treated rats ate little, while controls increased their feed consumption 10 times. The response to insulin-induced (10 µg/kg body weight) hypoglycemia was reduced in TCDD-treated rats, two of which died because they failed to eat following insulin dosing. Deprivation of rat chow induced feeding in both control and TCDD-treated rats, but naloxone (an antagonist of the endogenous opioid peptidergic feeding system) suppressed the feeding response to fasting in controls and to a lesser extent in the TCDD-treated rats. TCDD-dosed rats, deprived of rat chow for 24 hours and then fed, showed a suppression of feeding when given glucose (1.36 g/kg body weight) or fructose (1.36 g/kg body weight) i.p. Feed intake was not similarly suppressed in control rats. A challenge with sodium 2-mercaptoacetate (MA), an inhibitor of mitochondrial β-oxidation of fatty acids, following feeding with a high-fat diet resulted in a decrease in intake of a high-fat diet rather than an expected increase. When TCDD-dosed rats were challenged with both MA and 2-deoxyglucose, feed consumption was increased, although the response was delayed and attenuated. The abnormal responses imply that TCDD is capable of producing long lasting impairments to metabolic challenges, which are at least in part mediated by the CNS. The data are not sufficient to determine whether there was a CNS component involved in causing the effects observed, however.
The impact of pharmacologic agents on TCDD-induced wasting was investigated by Pohjanvirta and colleagues (1988b). The TCDD model that was used in this study included TCDD-sensitive Long-Evans rats given a dose of TCDD (20 µg/kg body weight i.p.) that induced hypophagia and death. TCDD-dosed rats were exposed to a series of pharmacologic agents for periods of 3-14 days. The agents were given at dose levels and for time periods that were expected to increase feed consumption in TCDD-dosed animals. Because aminergic and serotonergic neurotransmission are involved in control of feed consumption, agents affecting one or the other of these systems were included in the treatment program.
The treatments included dopamine antagonists (haloperidol and amperozide), an α-adrenoceptor-blocking agent (phenoxybenzamine), a 5-hydroxy tryptophan (HT) synthesis inhibitor [p-chlorophenylalanine (PCPA)], a β-blocker
of sympathetic stimulation of brown adipose tissue (sotalol), a steroidal inducer of gluconeogenesis (dexamethasone), a hypoglycemia-inducing agent (insulin), doses of other modifiers of aminergic neurotransmission that have been reported to increase feeding behavior (amphetamine, morphine, reserpine, chlordiazepoxide, and clonidine), and indomethacin at a dose level that has been reported to increase food consumption. None of the drug treatments lessened or prevented the induction of hypophagia or weight loss leading to death. None of the treatments affected the nocturnal feeding reduction induced by TCDD, although some of the treatments altered the daytime TCDD-induced hypophagia. For example, reserpine and PCPA further reduced feeding in TCDD-treated rats, and insulin, which increased daytime feeding in TCDD-treated rats, was associated with aphagia on discontinuation of treatment. None of the treatments were regarded as suggesting a crucial role for aminergic or serotonergic regulatory systems in the mechanism of action of TCDD-induced hypophagia.
To investigate further the role of aminergic neurotransmission in TCDD-induced hypophagia and lethality, Tuomisto and colleagues (1990) determined the levels of noradrenalin, dopamine, dihydroxyphenylacetic acid, homovanillic acid, 5-hydroxytryptamine, 5-hydroxyindoleacetic acid, tryptophan, and histamine in several areas of the brain at various times (1-76 hours) after i.p. administration of a lethal dose (50 µg/kg body weight) of TCDD to TCDD-sensitive Long-Evans rats. Control rats were treated with the vehicle used to deliver the TCDD (corn oil) and were apparently fed ad libitum. Except for a slight increase in tryptophan concentrations in several areas of the brain, there were no consistent or readily interpretable changes in other parameters.
Bestervelt and colleagues (1991) investigated the role of hypothalamic endorphin and mu opioid receptor levels in Sprague-Dawley rats given a high but sublethal oral dose of TCDD (50 µg/kg body weight). Two control groups received the vehicle (acetone and corn oil 1:2); one group was fed ad libitum and one was pair-fed with the TCDD group. Pair feeding did not alter receptor levels. TCDD-dosed rats initially showed a relative increase in hypothalamic β-endorphin (βE) concentrations followed by depressed concentrations 2 and 3 days after dosing. The mean absolute βE concentrations were 6.0, 12.4, and 11.6 pg in the controls on postdosing days 1, 2, and 3, respectively, and 10.0, 4.8, and 5.6 pg in the TCDD-dosed rats. Thus the actual concentrations showed a significant amount of overlap and were not unequivocally different. Three days after TCDD administration, the brain mu receptor number was 60 percent higher in the TCDD-dosed rats than in controls, but the binding affinity of the mu opioid receptor was not changed. The changes in receptor number coincided with the beginning of hypophagia in the rats and may indicate that TCDD can act as an opioid antagonist.
The effects of TCDD on serum levels of melatonin and the morphology of the pineal gland by light and electron microscopy were investigated by Linden and colleagues (1991) in TCDD-resistant Han/Wistar rats. Serum melatonin levels decreased within 24 hours after an i.p. dose of 50 µg/kg TCDD and remained reduced for 28 days. The change in serum melatonin level was not associated with morphological changes in the pineal glands even when rats were given a higher dose of TCDD (1,000 µg/kg i.p.). The reduction in serum melatonin levels (˜50 percent of control values) could not be correlated with hypophagia or changes in body weight.
Russell and colleagues (1988) investigated hypothalamic factors that could account for the reduction of serum concentrations of prolactin in Sprague-Dawley rats given a lethal dose of TCDD (50 µg/kg body weight) i.p. This dose level of TCDD results in a marked inhibition of prolactin release within 4 hours of dosing. Exposure to pimozide, a dopamine receptor antagonist, 30 minutes prior to TCDD administration prevented TCDD-induced suppression of hormone release. The pimozide effect was presumed to have been the result of antagonism of the inhibitory effect of dopamine on prolactin release from the anterior pituitary (Russell et al., 1988). Measurement of dopamine concentrations in the median eminence of TCDD-dosed rats indicated that TCDD increased dopamine levels by 15 percent. The rate of dopamine synthesis and the rate constant for decline of dopamine in the median eminence were increased. Similar changes in norepinephrine levels were not observed. Exposure of pituitary tissue to TCDD in vitro did not alter the secretion rate of prolactin. Taken together, these data imply that exposure to TCDD at dose levels that can produce the wasting syndrome can increase dopamine synthesis and turnover in the hypothalamus and that, through increased release of dopamine, TCDD can inhibit the release of prolactin.
In a series of papers Stahl and Rozman (1990), Weber et al. (1991), Rozman et al. (1991), and Stahl et al. (1991) have shown that the hypophagia induced by TCDD is most likely not due to a direct effect of TCDD on the brain, but is related to changes that occur in the liver affecting the peripheral feedback mechanisms involved in appetite control. Stahl and Rozman (1990) attempted to induce hypophagia in Sprague-Dawley rats by direct intraventricular injection of 3H-labeled TCDD (8 µg/kg body weight) into the brain, leading to TCDD levels in various regions of the brain that were increased 3-400 times. For a comparison, a second group of rats was given 72 µg/kg TCDD intravenously. Levels of TCDD in the brain after intraventricular injection ranged from 30 to 1,220 ppb and after intravenous injection from 2.4 to 5.2 ppb. Levels in the liver were much higher in rats given TCDD intravenously. In spite of the much higher brain levels of TCDD after intraventricular injection, hypophagia and lethality were only seen in the group given TCDD intravenously. These data demonstrated that TCDD
accumulation in the brain is not associated with appetite suppression or the wasting syndrome.
Rozman and colleagues (1991) found that administration of 125 µg/kg body weight TCDD i.p. to Sprague-Dawley rats increased plasma tryptophan (a serotonin precursor) and brain levels of tryptophan, serotonin, and 5-hydroxyindoleacetic acid (a serotonin metabolite). No changes were observed in brain levels of norepinephrine and dopamine in the hypothalamus. The increase in the plasma levels of tryptophan and the brain concentrations of serotonin, its precursor, and its metabolite was later shown to be associated with impairment of several enzymes involved in gluconeogenesis. The most important of these enzymes appears to be phosphoenolpyruvate carboxykinase (PEPCK), although pyruvate carboxylase and glucose-6-phosphatase were also altered. Inhibition of PEPCK results in increased tryptophan levels in the blood and brain. Increases in tryptophan levels can result in increased levels of serotonin, which can act as an appetite suppressant. These data indicate that peripheral mechanisms may play an important role in the wasting syndrome.
Stahl and colleagues (1991) showed that central serotonergic pathways may not be involved in the wasting syndrome. Prior to dosing with TCDD, rats were given a dose of 5,7-dihydroxytryptamine to cause central 5-HT (a serotonin metabolite) depletion (up to 90 percent). The 5-HT depletion did not affect the outcome of TCDD exposure, suggesting that although TCDD increases 5-HT levels in the brain, this may not be the cause of the wasting syndrome.
While lethal or near lethal dose levels of TCDD may be associated with neurochemical alterations or changes in the responsiveness of neurochemical processes in the central nervous system, the changes observed may also be regulatory responses occurring secondary to changes induced in other organ systems. The available data imply that CNS alterations play at most a secondary role in the pathogenesis of the wasting syndrome induced by TCDD.
Metabolic Toxicity
Toxicity Associated with Intermediary Metabolism Animal studies, primarily those performed in the rat, have identified a number of changes in intermediary metabolism following treatment with TCDD. These include effects on body temperature and serum and tissue concentrations of glucose, insulin, glycogen, somatostatin, and thyroid hormones. There is some evidence suggesting that TCDD-mediated alteration in intermediary metabolism may be linked to the wasting syndrome described in a number of rodent species following treatment with TCDD and related compounds.
Blood Glucose Lethal doses of TCDD produce a rapid onset of severe
hypoglycemia in the rat (Zinkl et al., 1973; Gasiewicz et al., 1980; Potter et al., 1983; Gorski and Rozman, 1987; Gorski et al., 1988a,b). The direct cause of hypoglycemia, although initially suspected to be the progressive starvation stress associated with TCDD treatment in rodents, cannot be attributed to a decrease in food consumption. Gasiewicz and coworkers (1980) demonstrated that TCDD-treated rats that received continuous intravenous feeding exhibited hypoglycemia as well as other signs of TCDD toxicity, including lethality, while maintaining normal weight gain. These findings suggest that rats treated with TCDD are capable of efficiently utilizing glucose and amino acids. Similarly, pair-feeding studies clearly showed that although TCDD-treated rats developed hypoglycemia (Christian et al., 1986a), control rats in a comparable state of nutrition maintained normal blood glucose levels.
In pair-fed control studies, concomitant to hypoglycemia, TCDD-treated rats also exhibited a decrease in serum and pancreatic insulin (Potter et al., 1983; Gorski and Rozman, 1987). This decrease is unusual because hyperglycemia normally would be expected in the presence of hypoinsulinemia. Comparable onset kinetics were observed with respect to both decreased insulin and decreased serum glucose following TCDD treatment (Gorski and Rozman, 1987). TCDD treatment of rats was also found to induce insulin hypersensitivity. Injection of nontoxic doses of insulin 3 days after administration of 125 µg/kg TCDD resulted in 80 percent mortality of rats within 24 hours. Insulin hypersensitivity preceded both hypoglycemia and hypoinsulinemia.
Blood glucose is derived from three sources: dietary intake, glycogen, and gluconeogenesis. Prior to TCDD-induced death, rats cease to eat, which results in an exhaustion of glycogen stores. However, neither the reduction in glycogen stores nor the decrease in dietary intake appears to account for the decrease in blood glucose. As described above, although TCDD-treated rats exhibited severe hypoglycemia, pair-fed control rats remained normoglycemic (Christian et al., 1986a). There is evidence to suggest that decreased blood glucose following TCDD treatment in rodents may be mediated through the disruption of gluconeogenesis. TCDD treatment of rats was found to produce an elevation in serum alanine, suggesting a decrease in the conversion of alanine to glucose (Christian et al., 1986a). At a lethal dose of TCDD (125 µg/kg), conversion of 14C-alanine to 14C-glucose was significantly reduced compared to pair-fed controls (Gorski et al., 1990). Following a lethal dose of TCDD, rats also maintained a low respiratory quotient (the ratio of CO2 output to oxygen usage), whereas pair-fed control rats showed an enhanced respiratory quotient (Muzi et al., 1989), suggesting protein utilization for maintenance of normal blood glucose concentration in untreated rats. Additionally, a congener of TCDD, 3,3',4,4'-tetrachloroazobenzene, was found to inhibit markedly several key enzymes required
for hepatic gluconeogenesis (Hsia and Kreamer, 1985), further supporting a role for altered gluconeogenesis as a mechanism for TCDD-induced hypoglycemia.
Somewhat paradoxically, one laboratory using two independent methods has reported corticosterone, a stimulator of gluconeogenesis, to be greatly elevated in TCDD-treated hypoglycemic rats (Gorski et al., 1988d). This observation is contrary to results reported by Balk and Piper (1984), who found approximately a 70 percent decrease in corticosterone 14 and 21 days after a single oral dose of TCDD (25 µg/kg) in rats. A second major effect of corticosterone on intermediary metabolism, in addition to stimulating gluconeogenesis, is to decrease peripheral utilization of glucose. In light of this, corticosterone reduction in rats by adrenalectomy significantly increased the rate of TCDD lethality, as compared to rats without this surgical alteration (Neal et al., 1979; Gorski et al., 1988a). Likewise, corticosterone replacement offered partial protection to TCDD-treated adrenalectomized rats, as compared to TCDD-treated adrenalectomized rats not receiving corticosterone (Gorski et al., 1988a). The protective effects of supplemental corticosterone may be mediated through an increase in peripheral glucose utilization and therefore a reduction in hypoglycemia.
Somatostatin is known to decrease serum glucose, as well as various hormones including insulin and glucagon. A single nonlethal dose of TCDD (45 µg/kg) in rats had no effect on somatostatin concentrations in serum, liver, or pancreas (Potter et al., 1983), thus ruling out a role for somatostatin of TCDD-induced hypoglycemia.
Fatty Acid Biosynthesis Thyroid-derived hormones are known to regulate de novo fatty acid synthesis. Within 7 days after TCDD treatment (45 µg/kg), rats exhibited hypothyroidism (Potter et al., 1983) followed by changes in serum concentrations of certain thyroid-derived hormones. TCDD treatment has no known direct effect on thyroid-stimulating hormone. Conversely, total thyroxine (TT4) and free thyroxine (FT4 ) were decreased somewhat dose dependently beginning around 2-4 days after a single dose of TCDD and returned to control levels by day 32. Similar findings were observed for reverse triiodothyronine (Rt 3) following TCDD administration. One study has reported no effect of TCDD on total triiodothyronine (TT3) (Potter et al., 1983), whereas a second study found that TCDD increased TT3 (Bastomsky, 1977). One role of thyroid-derived hormones is the regulation of de novo fatty acid synthesis, especially by triiodothyronine (T3), which is stimulatory. Following TCDD treatment, rats exhibited an increase in plasma fatty acids and de novo fatty acid synthesis in liver. Conversely, there was a decrease in de novo fatty acid synthesis in interscapular brown adipose tissue. The mechanisms for differences in de novo fatty acid synthesis between liver and interscapular brown adipose tissue are unknown; however, these findings may be related to findings by Bianco and Silva (1987),
which indicated that T4 (thyroxine), but not T3, penetrates interscapular brown adipose tissue. Once T4 has penetrated into interscapular brown adipose tissue, it is converted to T3, which in turn stimulates de novo fatty acid synthesis. In light of this, Gorski et al. (1988c) have speculated that a decrease in T4 by TCDD may account for the decrease in de novo fatty acid synthesis in interscapular brown adipose tissue.
Body Temperature Potter and colleagues (1983) reported that within 2 weeks of a single 90-µg/kg i.p. dose of TCDD, rats exhibited a body temperature of less than 35°C, with the lowest mean value of 34.5°C recorded on day 16. Mean body temperatures for control rats ranged from 36.8 to 37.5°C.
Gastrointestinal Toxicity
Although research in this area has been limited, gastrointestinal (GI) associated changes have been reported following TCDD administration in a number of animal models (i.e., rat, rabbit, monkey, chicken, hairless mouse).
Stomach-related effects of TCDD have been among the most widely studied. A greater than threefold decrease in gastric antrum somatostatin, a hormone possessing moderate inhibitory effects on gastric secretion, was observed in rats following a single nonlethal dose of TCDD (45 µg/kg) and was accompanied by a 29 percent increase in stomach dry weight (Potter et al., 1983). The mechanism for the increase in stomach dry weight is unknown. Theobald and colleagues (1991) also reported a decrease in both antral gastrin and somatostatin in rats following TCDD treatment (100 µg/kg) and a seven- to tenfold elevation in serum gastrin on day 14 posttreatment. Histologic examination revealed increased antral mucosal height on day 14 in TCDD-treated rats and increased antral wet weight. Because the TCDD ED50 values that produced decreased antral somatostatin concentrations were less than those that produced increased gastrin—and, secondly, the decrease in antral somatostatin in TCDD-treated rats occurred one week before the hypergastrinemia—it is unlikely that decreased antral somatostatin levels initiate the increase in gastrin. It has been suggested that the main cause of TCDD-induced hypergastrinemia is decreased gastric acid secretion (Mably et al., 1990).
Chronic exposure to TCDD and related compounds has been associated with hyperplasia and metaplasia within the oxyntic gland of the stomach of monkeys and rats (Norback and Allen, 1973; Becker and McNulty, 1984). The height of the oxyntic gland mucosa in TCDD-treated monkeys is greatly enhanced, with extensions of gastric glands into the submucosa and formation of submucosal cysts (Norback and Allen, 1973; Becker et al., 1979; Becker and McNulty, 1984). Likewise, TCDD has been shown to cause hyperplasia of the ileal mucosa in the hamster (Olson et al., 1980b).
TCDD has also been reported to interfere with intestinal absorption. In pair-fed control studies, TCDD-treated rats (125 µg/kg) exhibited decreased glucose absorption as measured in isolated perfused jejunal segments. The greatest magnitude of inhibition was measured 2 days following TCDD treatment (30 percent), with partial resolution occurring by day 14 (14 percent inhibition) (Richter et al., 1992), compared to vehicle controls (ad libitum fed or pair-fed). No effect was observed on sodium or calcium transfer; however, potassium transfer was markedly increased, which paralleled the inhibition in glucose absorption (Richter et al., 1992). Measurements of 3-O-methylglucose uptake in mucosal tissue suggested that TCDD inhibition of glucose uptake occurred at the basolateral membrane.
Respiratory Tract Associated Toxicity
The effects of TCDD on the respiratory tract have not been widely studied and have focused primarily on enzyme induction, most notably cytochrome P450 in animal lung tissue. As discussed earlier, one of the primary toxicologic issues associated with the phenomenon of enzyme induction pertains to the fact that a wide variety of carcinogenic agents, although relatively inert in their parent form, can be metabolically activated to a carcinogenic form by cytochrome P450 isozymes. Hence, an increase in cytochrome P450 enzyme activity can allow a tissue (e.g., lung) to activate carcinogens more efficiently. Conversely, increasing P450 activity can also increase the rate at which carcinogens are detoxified. This is especially a concern in terms of the respiratory tract because it is a major site of deposition of inhaled xenobiotics.
TCDD treatment in both the rabbit and the rat resulted in significant induction of cytochrome P450 isozymes in the lung (Hooker et al., 1975; Domain et al., 1986; Overby et al., 1992). Examination of rat lung cytosol revealed the presence of a high affinity, low-capacity TCDD-binding complex (Kurl et al., 1986). In the rabbit, immunochemical examination and in situ hybridization have identified a number of different cell types containing the TCDD-inducible isoform of P450, P4501A1, including endothelial cells of the entire vascular bed of rabbit lung, Clara cells at all levels of airway, type 2 cells, alveolar macrophages, and, to a lesser degree, ciliated cells (Overby et al., 1992). Interestingly, cytochrome P450 reductase, which is required for P450 activity, although present in Clara cells, alveolar macrophages, and type 2 cells (Domain et al., 1986), was not present in endothelium of rabbit lung, which raises the question of what role this isozyme may play in this tissue (Overby et al., 1992). Intraperitoneal treatment of rats with TCDD also produced a twofold induction of AHH activity in rat nasal tissue. These studies indicate that exposure to TCDD causes a modest induction of AHH enzyme activity in the rat and rabbit respiratory tract.
Cardiovascular Toxicity
Currently, there is a very limited amount of information pertaining to whether administration of TCDD to laboratory animals can produce cardiovascular toxicity. To date only two studies have been published in this area, whose results are described below.
Mechanical responses of isolated atria to (–)-isoproterenol were assessed in rats 7 days following TCDD treatment (6.25, 25, or 100 µg/kg) compared to vehicle-treated rats with unlimited access to feed. In TCDD-treated rats (100 µg/kg), the basal rate of spontaneously beating right atria was significantly decreased, whereas for the left atria, maximal inotropic responses to (–)-isoproterenol and 1-methyl-3-isobutylxanthine were enhanced to the same degree (Kelling et al., 1987). The changes in a trial function were not secondary to loss in body weight as determined by pair-fed controls. There was no effect of TCDD exposure on the ratio of heart ventricular mass to body weight or on the activities of pyruvate kinase and citrate synthase in homogenates prepared from heart ventricular muscle. The authors concluded that overtly toxic doses of TCDD in the rat did not depress mechanical function of the heart.
As discussed earlier, TCDD treatment in many animal species induces an increase in serum triglyceride concentrations. In rabbits, a single i.p. injection of TCDD (1 or 50 µg/kg) produced elevated triglycerides by day 10 (Brewster et al., 1988). By day 20 in the 50-µg/kg treated rabbits, electron microscopic examination of aortic arches revealed alterations resembling preatherosclerotic lesions, which included ruffling, denudation, and sloughing off of the cell surface. Additionally, there was an appearance of macrophage-like structures in the intima and the media of endothelial cells.
Corticosteroids
Many of the toxic effects observed after acute exposure to near lethal levels of TCDD (e.g., alterations in metabolic homeostasis and hypercholesterolemia) may be mediated by changes in endocrine function. Studies have included examination of the effects of high doses of TCDD on the levels of pituitary, thyroid, and adrenal hormones. The data presented in the literature are conflicting. Since the dosing regimens (i.e., acute administration of lethal doses of TCDD) used in such studies are not relevant to the questions being examined in this report and there is no obvious explanation for the conflicting data (although variations in protocol such as the age of the animal, feeding regimen, length of time between dosing and sacrifice, and time of sacrifice, may contribute), a short summary of the data is presented with no attempt to resolve this controversy.
The effect of TCDD on serum levels of corticosteroids is variable. Both an increase and a decrease in the level of corticosteroids have been observed (Neal et al., 1979; van Logten et al., 1980; Balk and Piper, 1984; DiBartolomeis et al., 1984, 1987; Gorski et al., 1988d). This variation may be due to the time at which the serum was harvested (diurnal variations) (DiBartolomeis et al., 1987). DiBartolomeis and colleagues observed that administration of TCDD depresses the evening peak in corticosteroid secretion and elevates the peak observed early during the light phase. These authors also felt that the early rise in corticosteroid levels was due to nutritional deprivation. However, this same diurnal variation was not observed by other laboratories (Gorski et al., 1988d; Kerkvliet et al., 1990a).
There is also controversy as to the mechanism by which the level of serum corticosterone is altered. Again, this controversy may be due to the length of time between the dosing and the test. At least two laboratories indicate that the mechanism by which corticosterone levels are altered is an indirect response to other physiologic alterations. Jones and colleagues (1987) observed a delay in the evening peak and an increase in the early peak on day 1 after dosing. These alterations were preceded by a decrease in prolactin, which may regulate the response of the adrenals to adrenocorticotropin. On the other hand, as discussed in more detail above, Rozman and colleagues (1992) and Stahl and colleagues (1992) showed a decrease in the MRNA, protein, and enzyme activity of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase, within 4-8 hours after oral dosing with TCDD. The alterations in insulin and the increase in corticosterone 4-8 days after TCDD administration occurred as a response to this primary defect in PEPCK activity. Although the alterations observed in insulin and corticosterone (key regulators of PEPCK activity) should act to increase PEPCK activity, this does not occur. The authors suggest that TCDD alters the responsivity of the PEPCK gene to these endogenous regulatory agents.
Other investigators (DiBartolomeis et al., 1986a-c) suggest that the alterations in corticosterone levels (from the laboratory that observes a decrease in levels) are due to alterations in cholesterol accumulation and enzyme levels involved in adrenal hormone synthesis. Synthesis of adrenal steroids employs a series of distinct cytochrome P450-dependent mixed-function oxidases located in the mitochondria and endoplasmic reticulum of cells. In the rat, TCDD may alter adrenal steroidogenesis by modulating the level of perturbation of adrenal cellular cholesterol homeostasis and the inhibition of cholesterol side chain cleavage (DiBartolomeis et al., 1986a-c).
Dermal Toxicity
Chloracne is considered the most consistent response to—and the hallmark of—TCDD toxicity in humans. Although chloracne is considered a
symptom of exposure to a certain class of chemicals, including TCDD, other polychlorinated dibenzodioxins, and polychlorinated biphenyls, the absence of chloracne does not indicate that no exposure occurred. Early on, it was established that these compounds are capable of eliciting a chloracnegenic response in animals (rabbit ear bioassay) as well as humans (Jones and Krizek, 1962). However, the skin of laboratory animals generally does not exhibit a similar sensitivity to chlorinated compounds. In fact, cutaneous changes are often seen as the last manifestation of systemic toxic effects following exposure to a compound that generates chloracne (Kimbrough, 1974).
The biological response of the cutaneous tissue to TCDD observed in certain animals (but not all) is the elicitation of hyperkeratosis and comedone formation. The formation of an acnegenic response after exposure to TCDD was shown to occur in the rabbit, monkey, and hairless mouse (Jones and Krizek, 1962; Knutson and Poland, 1982; Puhvel et al., 1982). The skin areas affected all lacked major hair growth, which suggested to some that longer hair shafts may reduce the formation of chloracne lesions (Kociba and Schwetz, 1982).
Structure-activity relationships and genetic studies by Knutson and Poland (1982) indicate that in hairless mice (hr/hr, hairless and nu/nu, nude), the ability of TCDD to produce epidermal hyperplasia, hyperkeratosis, and sebaceous gland metaplasia is mediated through the Ah receptor described above. However, in this study, an additional genetic locus (hr) seems to regulate the expression of the chloracne lesion. In these mice, the formation of chloracne was observed on a gross level if the animal was homozygous for the hr gene. However, if the animals were heterozygous at this gene (i.e., only a single genetic difference between the two mouse strains and therefore the same at the Ah locus), no visible scaliness or hyperkeratosis was observed. Although the dermal reaction was not observed, the binding and persistence of TCDD on the skin and the induction of liver enzymes by TCDD were the same in both strains. These studies indicate that TCDD is biologically active in the hr/+ mouse; however, additional events must occur (which, in this case, may be mediated by the hr gene locus) for the chloracne lesion to be evident.
Further studies show that some proliferation of the epidermal cells (hyperplasia) following histopathologic examination of the hr/+ mice was observed after TCDD exposure, but this proliferation alone did not result in the formation of a chloracne lesion. TCDD generated sebaceous gland metaplasia, inflammation, and hyperkeratosis only in mice homozygous at the hr locus (Molloy et al., 1987). Biochemical analysis of the keratins in the skin of TCDD-treated mice from the hr/+ and hr/hr strains indicated a difference between these two strains in the changes in keratins that occur after TCDD exposure. The alterations in keratin observed in the hr/+ mice
were consistent with an increase in the proliferative response to TCDD. On the other hand, the alterations in keratin observed in the hr/hr mice were consistent with an increase in proliferation and differentiation in these mice.
Studies of the generation of hyperkeratosis after TCDD administration in neonatal mice (which have hair in both hr/+ and hr/hr strains) show that this genetic control at the hr locus is evident even prior to hair loss (Puhvel and Sakamoto, 1988). This observation suggests that the differential response to TCDD is under genetic control and not a secondary effect due to phenotypic differences. However, other studies (Puhvel and Sakamoto, 1987) showed that the response to TCDD of keratinocytes from hr/+ and hr/hr mice after in vitro exposure could not be distinguished. The authors suggest that physiologic factors beyond the epidermal cells themselves may be involved in the expression of the different responses observed in the skin of mice exposed to TCDD in vivo. However, as described above with regard to the immunotoxicity of TCDD and the Ah receptor, the concentration of TCDD to which the cells are exposed in vitro may be so high compared to the amount in vivo that any difference due to strain sensitivity and gene loci may be overwhelmed by the amount of compound available.
Lastly, one study examined the contribution of diet to the ability of TCDD to generate chloracne lesions in hairless mice (Puhvel et al., 1991). In this study, mice of both the +/+ and the hr/hr strains were placed on a diet deficient in vitamin A. Depletion of vitamin A from the diet did not lead to an observable response in the +/+ mice, but did increase the hyperkeratinization observed in hr/hr mice. These studies show that the toxicity of TCDD can be modulated by the diet of the animal.
In summary, the formation of chloracne lesions after administration of TCDD is observed in some laboratory animals. The lesions are formed in areas of reduced hair growth or in strains of animals in which hair growth is reduced. Studies indicate that there may be an additional gene locus, besides the Ah locus, that regulates the differentiation (but not proliferation) of keratinocytes and the formation of a chloracne lesion in response to TCDD exposure. In addition, deficiency of vitamin A in the diet may increase the expression of chloracne lesions in animals genetically predisposed to the formation of such lesions.
TOXICITY PROFILE OF 2,4-DICHLOROPHENOXYACETIC ACID
Introduction
2,4-D (2,4-dichlorophenoxyacetic acid; Chemical Abstracts Service (CAS) No. 94-75-7; Figure 4-1) has been used commercially in the United States since World War II to control the growth of broadleaf plants and weeds on
range lands, lawns, golf courses, forests, roadways, parks, and agricultural land (CCT, 1987). Formulations used include 2,4-D amine and alkali salts and esters, which are mobile in soil and easily absorbed through both the leaves and the roots of many plants.
2,4-Dichlorophenoxyacetic acid is an odorless (when pure), white to yellow (the yellow color is due to phenolic impurities) crystalline powder. The melting point of 2,4-D is 138°C, and the free acid is corrosive to metals. It is soluble in water and a variety of organic solvents (e.g., acetone, alcohols, ketones, ether, and toluene).
Pharmacokinetics
The pharmacokinetics of 2,4-D resemble those of other phenoxy acid herbicides. Absorption of oral doses is rapid and complete, whereas absorption of dermal doses is much slower (Arnold and Beasley, 1989). Only 4.5-6.4 percent of a dermal dose of 2,4-D is thought to be absorbed by humans (Feldmann and Maibach, 1984; H.L. Fisher et al., 1989; Harris and Solomon, 1992). Absorption following inhalation is less well-studied, but experiments in rats indicate that inhaled 2,4-D is absorbed rapidly (WHO, 1984). Studies with human volunteers have also demonstrated that single doses of ingested 2,4-D are absorbed rapidly (Sauerhoff et al., 1977). Experiments in rats confirm that dermal absorption of 2,4-D is much slower than gastrointestinal absorption (Knopp and Schiller, 1992). Accumulation of 2,4-D in the brain has been reported in rats, mice, and rabbits (Kim et al., 1988; Schulze and Dougherty, 1988).
2,4-D undergoes no notable transformations in animals. Its amines and salts are hydrolyzed rapidly, whereas its esters are hydrolyzed more slowly (Arnold and Beasley, 1989). Some conjugation with amino acids can occur, and 2,4-D in serum is protein bound (Arnold and Beasley, 1989). 2,4-D is not metabolized to reactive intermediates capable of interacting with DNA.
2,4-D is excreted rapidly in urine, predominantly in its unmetabolized form (Sauerhoff et al., 1977), although both dose and formulation can affect elimination rate (Bjorklund and Erne, 1966). Human urinary excretion of 2,4-D is diurnal and continues for many days after the initial exposure, so one sample is unlikely to be representative (WHO, 1984). In addition, its distribution and elimination are dose-dependent, characterized by a sigmoidal relationship; extrapolation from high to low exposure doses is thus problematic (Gehring and Betso, 1978). A half-life in humans following single doses of 2,4-D has been estimated to be approximately 18 to 20 hours (Sauerhoff et al., 1977; WHO, 1984); a half-life in humans based on multiple doses has not been estimated with certainty (Ibrahim et al., 1991).
Carcinogenicity
Several studies of the carcinogenicity of 2,4-D have been performed in laboratory animals. In contrast to more recent studies, those conducted before 1986 are considered inadequate because they do not meet current guidelines for bioassay quality (Sontag et al., 1976; Huff, 1982). The earlier studies were universally negative, whereas the later studies provide limited but not conclusive evidence of carcinogenicity. These studies are described below.
Bionetics (1968a) and Innes and colleagues (1969) reported a study in which 18 male and 18 female mice of strain (C57BL/6×C3H/Anf)F1 or strain (C57BL/6×AKR)F1 received commercial (90 percent pure) 2,4-D according to the following dosing regimen: a dose of 46.4 mg/kg body weight by gavage at 7 days of age, the same amount unadjusted for increasing body weight daily up to 28 days of age, then 149 mg/kg of diet until about 78 weeks of age. Another group of 18 males and 18 females of strain (C57BL/6×AKR)F1 received 100 mg/kg by gavage from days 7 to 28 of age, followed by 323 mg 2,4-D/kg diet until about 78 weeks of age. These doses were considered to be the maximum tolerated doses. No differences in tumor rates were detected compared to untreated or vehicle controls.
The same investigators also administered single doses of 215 mg/kg body weight of commercial (90 percent) 2,4-D in dimethyl sulfoxide (DMSO) subcutaneously or by gavage on day 28 of age to groups of 18 male and 18 female mice of strain (C57BL/6×C3H/Anf)F1 or of strain (C57BL/6×AKR)F1 (Bionetics, 1968a). Animals were observed up to 78 weeks of age. Again, no differences in tumor rates were detected compared to untreated or vehicle controls.
In another study, groups of 25 male and 25 female Osborne-Mendel rats received 0, 5, 25, 125, 625, or 1,250 parts per million (ppm) 96.7 percent pure 2,4-D/kg diet for two years (Hansen et al., 1971). No effect on growth rate, survival, organ weights, or hematologic parameters was observed. There were no statistically significant elevations in tumor rates at any site, although complete pathologic examinations were not performed on every animal and the maximum tolerated dose was not achieved.
In the first study that conformed to guidelines for the conduct of bioassays (Sontag et al., 1976; Huff, 1982), groups of 60 male and 60 female Fischer 344 rats received doses of 0, 1, 5, 15, or 45 mg 2,4-D/kg body weight in the diet for two years (Hazleton, 1986). Six male rats receiving the highest dose developed brain tumors (astrocytomas), compared to one control rat. None of the other treated rats had an excess incidence of this tumor, and no other excess tumor incidences were observed. In a second study, groups of 60 male and 60 female B6C3F1 mice received 0, 1, 15, or 45 mg 2,4-D/kg body weight in the diet for two years (Hazleton, 1987). No
excess tumor incidences of any kind were observed. These studies have been criticized for possibly failing to identify and use the maximum tolerated dose, although body weights of the high dose females were significantly reduced. An expert panel that reviewed the results of these studies concluded that they provided weak evidence that exposure to 2,4-D causes astrocytomas in male rats, noting that the background incidence of this tumor is variable and that there was no apparent explanation for the absence of such tumors in female rats because there are no known sex differences in its absorption, distribution, metabolic fate, or elimination (Ibrahim et al., 1991). It is possible that high doses of 2,4-D overwhelm the male rat's excretion capability and no tumors would be expected at lower doses that did not affect normal excretion rates. Pharmacokinetic studies of this possibility are needed. Other factors that precluded a strong association between 2,4-D exposure and astrocytoma formation were the absence of preneoplastic lesions or target organ toxicity, decreased tumor latency, and certain histologic characteristics that usually distinguish agent-induced brain tumors from those that occur spontaneously (CCT, 1987; Munro et al., 1992).
A recent study evaluated the ability of 2,4-D to enhance lung tumor initiation by urethan in mice (Blakley et al., 1992). Groups of 25 male CD-1 mice received Weed-no-More in their drinking water for 15 weeks at concentrations of 0, 0.0325, 0.08125, or 0.163 percent (equivalent to doses of 0-50 mg 2,4-D/kg body weight/day). After 3 weeks of treatment, 20 of the mice in each group received a single intraperitoneal dose of 1.5 mg urethan/kg body weight. 2,4-D had no effect on the metabolism of urethan or on the size of the lung adenomas induced. The number of adenomas was somewhat enhanced, although not in a dose-related manner, suggesting that 2,4-D may have a weak co-carcinogenic effect on urethan-induced adenomas in mice. In a simultaneous study, the effect of 2,4-D on spontaneous leukemia incidence in CD-1 mice was evaluated (this strain has a spontaneous leukemia incidence of 50 percent). After one year of treatment with the same dose levels used in the urethan study, no effect on mortality or survival was seen, and more untreated mice died of leukemia than did treated mice.
Hayes and colleagues (1991) reported the results of a case-control study in which the incidence of malignant lymphoma among dogs kept as pets was found to have a positive association with owners' use of 2,4-D on lawns. Information from a self-administered questionnaire and telephone interviews was used to show that households with dogs that developed malignant lymphoma used 2,4-D or a commercial lawn service somewhat more frequently than control households (odds ratio = 1.3). The risk rose to a twofold excess in households with four or more annual applications of 2,4-D. These results are of interest because of the histologic and epidemiologic similarity between canine and human malignant lymphoma, and because of
suggestive epidemiologic evidence linking human lymphoma with 2,4-D exposure. The results do not establish a clear association between 2,4-D exposure and lymphoma in dogs, however, because of the likelihood that both owners and lawn services used a number of chemicals in addition to 2,4-D, and because no actual exposure data were available. In addition, the etiology of malignant lymphoma is unknown. Also, given the nature of the data collection methodology, the possibility of recall bias cannot be excluded. The results are in contrast to those of Hansen and colleagues (1971), who administered 2,4-D to 3 male and 3 female beagle dogs in their diet for two years (0, 10, 50, 100, or 500 ppm) and reported no malignant tumors. Two years is generally not long enough to obtain tumors in dogs, however.
In conclusion, studies in animals indicate that there is suggestive, but not compelling, evidence that 2,4-D exposure may be associated with the development of astrocytoma in male rats and malignant lymphoma in dogs. 2,4-D has equivocal genotoxic effects (see below), is not metabolized to an active intermediate, is rapidly eliminated, and does not accumulate in body tissues (Munro et al., 1992). There are no significant mechanisms of action that indicate a likely risk of tumorigenesis from 2,4-D. 2,4-D thus presents a possible, but not probable, risk of cancer to humans.
Genotoxicity
A variety of in vitro tests of the mutagenicity and clastogenicity of 2,4-D have been conducted, with both positive and negative results reported. 2,4-D is not a classic mutagen. The majority of test results for both 2,4-D and its compounds are negative, including those in Salmonella typhimurium and Escherichia coli (IARC, 1977; Mortelmans et al., 1984; Ibrahim et al., 1991). Some positive results for mutagenicity have been reported at high doses (> 10 µg/ml) in plant cells and in V79 Chinese hamster fibroblast cells (Khalatkar and Bhargara, 1982; Pavlica et al., 1991), but the relevance of positive results in plants or in immortalized cell lines in vitro to normal human cells in vivo is not known. An in vivo study in mice found that topically applied 2,4-D was weakly positive in the hair follicle nuclear aberration assay and negative in the bone marrow micronucleus test (Schop et al., 1990). In contrast, 2,4-D was weakly clastogenic to the bone marrow cells of rats treated twice by i.p. injection with doses of 35 or 70 mg/kg body weight (Adhikari and Grover, 1988). Weak positive results in tests for sister chromatid exchanges in cultured human lymphocytes have also been reported (Linnainmaa, 1984; Turkula and Jalal, 1985), but the majority of such tests, conducted in rat, mouse, hamster, and human cells, have been negative (IARC, 1977; Ibrahim et al., 1991). In vivo tests for sister chromatid exchange and chromosomal aberrations have also been negative, including in exposed humans (Linnainmaa, 1983; Mustonen et al., 1986, 1989;
Galloway et al., 1987, Ibrahim et al., 1991). This positive result is of some interest in view of the suggested target sites observed in epidemiologic studies, although the negative results in the overwhelming majority of both in vitro and in vivo studies of lymphocytes and bone marrow cells make conclusions difficult to support. In addition, the phenomenon of sister chromatid exchange has not been correlated to carcinogenicity and its clinical significance is unknown.
Acute Toxicity
The acute toxicity of 2,4-D varies a great deal among species, with the variation apparently related to its plasma half-life (Nielsen et al., 1965). Its LD50 ranges from 100 mg/kg in dogs to 540 mg/kg in chickens and 375-2,000 mg/kg in rats (Bjorklund and Erne, 1966; Nielsen et al., 1965; IARC, 1977). The target organ for acute toxicity in humans is the central nervous system (Flanagan et al., 1990). In a case report of attempted suicide, Friesen and colleagues (1990) noted prominent CNS depression and muscle damage, but no liver or kidney damage or electrocardiogram (EKG) abnormalities, at a dose sufficient to produce a serum level of 392 mg/liter (approximately 50 g). Studies in cats and dogs also indicate that the CNS is the principal target organ for acute 2,4-D toxicity in mammals, and suggest that the primary site of action is the cerebral cortex or the reticular formation (Dési et al., 1962a,b; Arnold et al., 1991).
Chronic Systemic Toxicity
Studies in laboratory animals have demonstrated that chronic exposure to 2,4-D can elicit effects in a number of organs. For example, chronic exposure to 2,4-D has produced a wide variety of hepatotoxic effects in rodents, including subacute toxic hepatitis, local necrosis, centrilobular atrophy, elevated peroxisomes, elevated mixed-function oxidases, and other enzyme and glycogen changes (IARC, 1977; WHO, 1984). Other studies have shown that 2,4-D is only a weak inducer of mixed-function oxidases, and that the pattern of induction differs from that of TCDD (Mustonen et al., 1989; Chaturvedi et al., 1991; Knopp and Schiller, 1992). Some renal and hematologic effects of 2,4-D have also been shown in rodents: increased kidney weights and cortical and subcortical pathology were seen at doses of 15 mg/kg/day or higher, and reductions in mean hemoglobin, mean hematocrit and red blood cell levels, and mean reticulocyte levels were observed at doses of 5 mg/kg/day or higher in rats and at 15 mg/kg/day in mice (Hazleton, 1986; Gorzinski et al., 1987a). Renal toxicity in rats was also reported in the Hazleton (1986) study described in the carcinogenicity section; a no-observed-effect level of 1 mg 2,4-D/kg body weight was identified.
Lukowicz-Ratajczak and Krechniak (1988) concluded that the renal toxicity of 2,4-D in rats is due to damage to the loop of Henle and not to the proximal tubule.
Neurobehavioral toxicity was reported in rats that received daily subcutaneous injections of 150-250 mg 2,4-D/kg body weight over four successive 14 day periods (Schulze and Dougherty, 1988). The extent of toxicity was affected by the formulation used and by food deprivation (Schulze, 1987). This toxicity was reversible, however, due to an apparent tolerance that developed with repeated exposure. A functional/cellular mechanism was thought to be responsible for the developed tolerance.
Reproductive and Developmental Toxicity
Tests of the developmental toxicity of 2,4-D have produced both positive and negative results. Administration of 90 percent pure 2,4-D, its isopropyl or butyl ester (99 percent pure), or its isooctyl ester (97 percent pure) to pregnant BL6, AKR, or C3H mice during days 6-14 of gestation increased the incidence of fetal anomalies. No effects were seen in B6AK or A/Ha mice (Bionetics, 1968a,b). No malformations or effects on litter size were observed among the offspring of Sprague-Dawley rats that received doses of 1,000 mg/liter drinking water 2,4-D (purity unspecified) during pregnancy and for an additional 10 months. The mothers also remained normal. The offspring then received 2,4-D for up to two years, and exhibited retarded growth and increased mortality; no unequivocal clinical or morphologic changes were observed, however (Bjorklund and Erne, 1966). Similar results were obtained by Hansen and colleagues (1971), who fed diets containing 0, 100, 500, or 1,500 ppm 2,4-D (96.7 percent pure) to male and female Osborne-Mendel rats over three successive generations (further detail not provided). No effects on fertility were reported, and average litter sizes were normal, although offspring mortality was increased and growth rates were decreased at the highest dose.
In another study, female rats received 0, 1,000, or 2,000 ppm 2,4-D (purity unspecified) in the diet for 95 days and were then mated with untreated males, continuing on the diet throughout pregnancy and lactation. The offspring of the rats fed the highest dose were small, and most died before weaning (IARC, 1977). In contrast, Schwetz and colleagues (1971) reported no effects on fertility, gestation, lactation, or mortality when Sprague-Dawley rats received the maximum tolerated dose (87.5 mg/kg/day) of 98.7 percent pure 2,4-D or its esters on days 6-15 of gestation. This dose was not teratogenic but was lethal to embryos, produced some delayed ossification, and had growth retarding effects on the offspring, however. No effect on fertility was noted in a multigenerational study in rats that received 0, 5, or 20 mg 2,4-D/kg/day (Mullison, 1986).
When 2,4-D (purity unspecified) was fed at a dose of 500 mg/kg diet to a pig throughout its pregnancy, anorexia was observed and the piglets were underdeveloped and apathetic; most died within 24 hours of birth (Bjorklund and Erne, 1966). No consistent embryotoxic effects were observed when 2,4-D was administered orally to hamsters on days 6-10 of gestation at doses up to 100 mg/kg (Collins and Williams, 1971).
Taken together, these studies suggest that 2,4-D does not affect fertility and is not a teratogen in laboratory animals, but it can reduce growth rates and increase mortality among exposed offspring. Very high doses are required to elicit these effects, however.
Immunotoxicity
Little work has been performed to evaluate the potential immunotoxicity of 2,4-D. There have been a number of case reports of allergic skin reactions involving 2,4-D exposure (Cushman and Street, 1982). One clinical study suggested that 2,4-D could produce contact sensitization among exposed farmers: 3 of 30 farmers patch-tested with a 1 percent solution of 2,4-D (purity unspecified) tested positive, whereas no positive reactions were observed among dermatitis-free controls (Sharma and Kaur, 1990).
Blakley (1986) administered single doses of 2,4-D butyl ester at 50-200 mg/kg body weight to female BDF1 mice by gavage and reported enhanced antibody production against SRBCs, as well as a stimulated lymphoproliferative response to lipopolysaccharide. These doses also produced clinical signs of toxicity and CNS pathology, however. In contrast, after dermal application of similarly toxic doses, a suppressed antibody response to SRBCs was noted (Blakley and Schiefer, 1986). Repeated administration of lower doses by either route for 3 weeks failed to produce either toxicity or immunologic effects. In these cases, the immunologic effects appear to have occurred as an indirect consequence of the clinical toxicity of 2,4-D.
TOXICITY PROFILE OF 2,4,5-TRICHLOROPHENOXYACETIC ACID
Introduction
Like 2,4-D, 2,4,5-T (2,4,5-trichlorophenoxyacetic acid; CAS No. 93-76-5; Figure 4-1) was developed during World War II as an herbicide to control the growth of broadleaf plants and weeds on range lands, lawns, golf courses, forests, roadways, parks, and agricultural land (CCT, 1987). Most of these uses were banned by the U.S. Department of Health, Education and Welfare, the Department of Agriculture, or the Environmental Protection Agency between 1969 and 1972 because of the suspicion that 2,4,5-T
might be a human health hazard. The subsequent discovery that 2,4,5-T is generally contaminated with TCDD has led many to conclude that the reported adverse effects were due to this contaminant (Lilienfeld and Gallo, 1989). The registration for 2,4,5-T was canceled by the EPA in 1978.
When purified, 2,4,5-T is an odorless, white to light-tan solid with a melting point of 158°C. This compound is noncorrosive, and is soluble in alcohol and water. 2,4,5-T reacts with organic and inorganic bases to form salts, and with alcohol to form esters. As stated, commercial formulations of 2,4,5-T used during the Vietnam war contained TCDD as a contaminant from the manufacturing process.
Pharmacokinetics
The pharmacokinetics of 2,4,5-T resemble those of other phenoxy acid herbicides. Experiments in rats, dogs, and humans show that absorption of oral doses is rapid and complete, whereas absorption of dermal doses is much slower (Leng et al., 1984; Arnold and Beasley, 1989). Pharmacokinetic modeling has indicated that 97 percent of the 2,4,5-T absorbed through the skin is cleared, primarily through the urine, within one week (Leng et al., 1984). The pharmacokinetics of inhalation exposure have not been studied.
Distribution of 2,4,5-T occurs quickly, and the parent compound is eliminated via the urine, undegraded, as the free acid (Gehring et al., 1973; Piper et al., 1973b). Salts of 2,4,5-T are hydrolyzed prior to excretion, and a small amount may be conjugated (Gehring et al., 1973). Rates of clearance from plasma and urinary excretion depend on dose and are species-specific. Doses greater than 50 mg/kg body weight saturate the renal clearance mechanism for 2,4,5-T in rats (Piper et al., 1973b). Following a single oral dose in humans, 2,4,5-T was found to have a plasma half-life of about 19-23 hours, although interindividual variation was substantial; its urinary excretion was relatively rapid and fluctuated diurnally (Gehring et al., 1973; Kohli et al., 1974). Gastrointestinal absorption kinetics, plasma clearance, and urinary elimination are all first-order processes (Gehring et al., 1973; Piper et al., 1973b). Because the distribution and elimination of 2,4,5-T are dose-dependent, characterized by a sigmoidal relationship, extrapolation from high to low exposure doses is problematic (Gehring and Betso, 1978).
Carcinogenicity
Several studies of the carcinogenicity of 2,4,5-T have been performed in laboratory animals; these are described below. Only one study conforms to current standards for the conduct of carcinogenicity bioassays (Sontag et al., 1976; Huff, 1982). All produced negative results. There appears to be no evidence of or mechanistic basis for the carcinogenicity of 2,4,5-T in laboratory animals, although thorough testing has not been performed.
Bionetics (1968a) and Innes and colleagues (1969) reported a study in which 18 male and 18 female mice of strain (C57BL/6×C3H/Anf)F1 or strain (C57BL/6×AKR)F1 received commercial (98 percent pure) 2,4,5-T according to the following dosing regimen: a dose of 21.5 mg/kg body weight by gavage at 7 days of age, the same amount unadjusted for increasing body weight daily up to 28 days of age, then 60 mg/kg of diet until about 78 weeks of age. No differences in tumor rates were detected when compared to untreated or vehicle controls.
The same investigators also administered single doses of 21.5 mg/kg body weight of commercial (98 percent pure) 2,4,5-T in DMSO subcutaneously on day 28 of age to groups of 18 male and 18 female mice of strain (C57BL/6×C3H/Anf)F1 or of strain (C57BL/6×AKR)F1 (Bionetics, 1968a). Animals were observed up to 78 weeks of age. Again, no differences in tumor rates were detected compared to untreated or vehicle controls.
In another experiment, 20 male and 19 female 6-week-old inbred XVII/G mice received a concentration of 100 mg 2,4,5-T (containing 8 0.05 mg chlorinated dibenzodioxins)/liter of drinking water for two months, followed by 80 mg/kg diet for their life spans (Muranyi-Kovacs et al., 1976). Treated animals survived longer than did controls, and an increase in the incidence of spontaneous lung tumors was observed, although it was attributable to the longer life span of the treated animals. A group of 22 male and 25 female C3Hf mice received the same treatment, but treated males experienced reduced survival compared to controls. In addition, the total number of tumors in treated females was greater than that in female controls, although poor reporting prevented an analysis of tumor sites.
Kociba and colleagues (1979) provided groups of 60 male and 60 female Sprague-Dawley rats with diets containing 3, 10, or 30 mg 2,4,5-T/kg body weight/day for up to two years. A group of 96 males and 96 females served as untreated controls. An interim sacrifice was performed on 10 animals of each sex from each group after 118-119 days of treatment. Some toxicity was observed at the highest dose, indicating that the maximum tolerated dosage was achieved. No effect on tumor incidence was observed.
Two studies of the ability of 2,4,5-T to modulate carcinogenesis have been performed. In one, Abdellatif and colleagues (1990) used an initiation/selection/promotion protocol for the induction of liver tumors in Wistar rats to test the promoting ability of 2,4,5-T. Rats received an initiating dose of diethylnitrosamine, followed by a diet containing 2-acetylaminofluorine, and in the middle of the latter treatment, they received a necrogenic dose of carbon tetrachloride. Finally, diets containing 0.05 percent 2,4,5-T were provided for the remaining 23 weeks of the experiment. Rats receiving 2,4,5-T had an incidence of hepatocellular carcinoma of 16 percent, compared to the control incidence of 0 percent. The relevance of this result to human carcinogenesis is not known. In the second study, Mirvish and
colleagues (1991) sought to test the suspected relationship between 2,4,5-T exposure and non-Hodgkin's lymphoma in humans by providing 600 mg/kg diet to MRC-Wistar rats simultaneously with 75 mg of 2-hydroxyethylnitrosourea/liter of drinking water. The 2,4,5-T was 98 percent pure and contained 1-4 µg/kg each of TCDD and 2,3,7,8-tetrachlorodibenzofuran. 2-Hydroxyethylnitrosourea is a known inducer of B cell lymphoma. Coadministration of 2,4,5-T did not affect the incidence of lymphoma induced by the nitrosamine, nor did it produce tumors at any other site.
Genotoxicity
2,4,5-T is not genotoxic. Tests for mutagenicity in Salmonella typhimurium, Escherichia coli WP2, Serratia marcescens a21, and Saccharomyces cerevisiae D4 have been negative for both 2,4,5-T and its compounds (IARC, 1977; Mortelmans et al., 1984; Rashid et al., 1984). 2,4,5-T produced an increase in the frequency of chromosomal aberrations in Chinese hamster ovary cells and of clastogenicity in Mongolian gerbil bone marrow cells only at very high concentrations (1.75 mg/ml and 250 mg/kg for 5 days, respectively) (Majumdar and Hall, 1973; Galloway et al., 1987). It did not produce sex-linked recessive mutations in Drosophila (Zimmering et al., 1984) or micronuclei in bone marrow erythrocytes of mice (Jenssen and Renberg, 1976).
Acute Toxicity
The acute toxicity of 2,4,5-T varies among species, with the variation thought to be related to plasma half-life (Lilienfeld and Gallo, 1989). The LD50 of 2,4,5-T ranges from 100 mg/kg body weight in dogs to between 389 and 940 mg/kg in mice (Rowe and Hymas, 1954; IARC, 1977). Bjorklund and Erne (1966) gave single oral doses of 100 mg 2,4,5-T to pigs, and reported anorexia, vomiting, diarrhea, ataxia, hemorrhagic enteritis, and congestion of the liver and kidneys. Myotonia and anorexia have also been observed in dogs that received 100 mg 2,4,5-T/kg body weight (Drill and Hiratzka, 1953).
Chronic Systemic Toxicity
A number of effects have been reported in laboratory animals chronically exposed to 2,4,5-T. For example, some minor liver congestion was reported in dogs that received doses of 20 mg/kg for 90 days (Drill and Hiratzka, 1953). Hepatic inflammation, biliary hyperplasia, and renal disease were also reported in Sprague-Dawley rats that received diets containing 30 mg 2,4,5-T/kg/day for two years; only minimal changes occurred at lower doses (Kociba et al., 1979).
Reproductive and Developmental Toxicity
Several studies have indicated that 2,4,5-T can be fetotoxic in rodents. In general, doses of 2,4,5-T greater than 20 mg/kg body weight administered on days 6-15 of pregnancy can retard growth, and increase embryolethality and the frequency of cleft palates among mice, both in the presence and in the absence of maternal toxicity (Bionetics, 1968b; Roll, 1971; Neubert and Dillmann, 1972; Seidenberg et al., 1986; Seidenberg and Becker, 1987; Holson et al., 1992). Bazare and colleagues (1990) found that the half-life of 2,4,5-T is increased in pregnant mice, which may contribute to its toxicity. Malformations have also been produced in hamsters treated with 100 mg 2,4,5-T/kg body weight orally on days 6-10 of pregnancy (Collins and Williams, 1971). Doses of at least 400 mg 2,4,5-T/kg body weight were required to induce significant embryotoxicity in rats in the study of Wilson and colleagues (1971), although other studies have reported embryotoxicity and reduced fetal mortality in rats at lower dosages (Sparschu et al., 1971; Smith et al., 1981). Some behavioral toxicity has also been associated with intrauterine exposure of rats to 2,4,5-T or to a 2,4-D/2,4,5-T mixture (Rogers, 1983; Mohammad and St. Omer, 1986). No developmental or fetotoxic effects of 2,4,5-T have been reported in rabbits, sheep, or monkeys, however (IARC, 1977).
The purity of 2,4,5-T appears to affect its fetotoxicity and teratogenicity in hamsters but not in mice. In general, 2,4,5-T contaminated with TCDD was fetocidal and teratogenic to hamsters at doses of 20 mg/kg or higher, whereas that containing no detectable TCDD elicited effects only at a dose of 100 mg/kg (Collins and Williams, 1971). Both technical grade and analytical grade 2,4,5-T (differing tenfold in TCDD content) produced similar frequencies of embryolethality, cleft palate, and kidney malformations in mice at the same dose levels, however (Courtney and Moore, 1971; Nelson et al., 1992). In addition, the embryotoxicity of doses of 100 mg 2,4,5-T/kg body weight was not increased by the simultaneous administration of 1 µg TCDD/kg in mice (Courtney and Moore, 1971).
No studies of the reproductive toxicity of 2,4,5-T could be found.
Immunotoxicity
No studies of the immunotoxicity of 2,4,5-T could be found.
TOXICITY PROFILE OF CACODYLIC ACID
Introduction
Cacodylic acid (hydroxydimethylarsine oxide; dimethylarsinic acid; CAS No. 75-60-5; Figure 4-1) is a nonselective, postemergence contact herbicide.
It is currently a registered herbicide in the United States and is a List B chemical under the Federal Insecticide, Fungicide, and Rodenticide Act registration, with data development and review ongoing. The herbicide formulation used in Vietnam in defoliation and crop destruction missions (Agent Blue or Phytar 560-G) contained 26.4 percent sodium cacodylate and 4.7 percent cacodylic acid as the active ingredients (Hood, 1985). Sodium cacodylate and cacodylic acid are likely to have similar toxicologic characteristics.
The solid form is an odorless, colorless crystal with a melting point of 195-196°C. In aqueous solution, the chemical is mildly corrosive. It is soluble in alcohol, acetic acid, and solutions that are 50 percent aqueous.
Pharmacokinetics
Cacodylic acid is one of the primary metabolites of inorganic arsenic: pentavalent inorganic arsenic is reduced to the trivalent form [As(III)], then methylated to methanearsonic acid and subsequently to dimethylarsinic acid (cacodylic acid) (Yamanaka et al., 1991). There is no evidence in the recent literature that inorganic arsenic is released into the body after exposure to cacodylic acid. Cacodylic acid has been used in the past as a pharmacologic agent, and it was believed that both the efficacy of this agent and its toxicity were due to inorganic arsenic liberated by hydrolysis in the stomach. Cacodylic acid is, however, very resistant to hydrolysis, and studies with improved analytical methodology have not demonstrated the metabolism of cacodylic acid to arsenic (Hood, 1985). For example, Marafante and colleagues (1987) studied the metabolism of orally administered cacodylic acid in mice, hamsters, and humans, and found that in mice and hamsters, 80-85 percent of the dose was eliminated as unmetabolized cacodylic acid, and 13-15 percent as a cacodylic acid complex; in mice and hamsters respectively, 3.5 percent and 6.4 percent of the doses were excreted as trimethylarsine oxide (TMAO), whereas in humans, about 80 percent of the dose was excreted as cacodylic acid and 4 percent as TMAO. No demethylation of cacodylic acid to inorganic arsenic was observed in any species.
Studies in rats indicate that cacodylic acid is absorbed more slowly following oral administration than intratracheal administration, becomes bound to red blood cells, and is excreted readily in urine. Clearance from the rat red blood cell is very slow, with the half-life of cacodylic acid in rat erythrocytes being approximately the same as that of the erythrocytes (95 days), indicating irreversible binding of cacodylic acid to rat hemoglobin. Rat erythrocytes were found to bind cacodylic acid much more readily than rabbit or guinea pig erythrocytes, however, and human red blood cells had the least binding, demonstrating substantial species differences (Stevens et al., 1977).
In another study of metabolism, cacodylic acid was administered orally to hamsters (50 mg/kg). Peak blood levels were reached 6 hours after administration and declined rapidly thereafter. The cacodylic acid was excreted relatively rapidly by the hamster (80 percent after 24 hours following oral administration), primarily in urine and feces. Some of the cacodylic acid (approximately 26 percent of the total arsenic in the whole blood) was further methylated to a trimethylarsinic compound, which was excreted primarily in the urine (Yamahuchi and Yamamura, 1984). Humans also excrete cacodylic acid predominantly in the urine following oral dosing (Buchet et al., 1981).
Carcinogenicity
Very sparse animal oncogenicity data are available for cacodylic acid. The one limited study conducted in mice showed no evidence of oncogenic potential; however, conclusions should be drawn with caution because of its inadequacies. In this study, male and female pathogen-free mice (strain unspecified) were administered cacodylic acid (purity unspecified) in distilled water at a dose of 46.4 mg/kg from days 7 to 28 of age, and then received 121 ppm (approximately 18 mg/kg/day) in the diet for 18 months. EPA concluded that this dose was close to a maximum tolerated dose; however, EPA did not state the basis for this conclusion. No evidence of oncognicity was observed in this study, based on statistical analyses for the cacodylic acid treated mice versus pooled negative controls for four tumor categories, which included hepatoma, pulmonary tumors, lymphoma, and total tumors. Although this study was negative for oncogenicity, it should be noted that it was a screening test with relatively small numbers of animals (not specified) and only a single dose level. This study does not meet NTP guidelines for oncogenicity testing (Innes et al., 1969; Sontag et al., 1976; Huff, 1982).
There is speculation that pulmonary carcinogenesis could result from high dose exposure to cacodylic acid because both inorganic arsenic and cacodylic acid share dimethyl- and trimethylarsine as metabolites. Dimethylarsine has been associated with DNA damage in both rat and mouse lung tissue following high dose oral acute exposures. Excess lung cancer has been reported in epidemiologic studies of smelter workers occupationally exposed primarily to pentavalent arsenic (U.S. EPA, 1981).
Genotoxicity
In general, the genotoxicity profile for cacodylic acid is mixed. It is negative in bacterial tests for mutagenicity, positive for mutagenicity and clastogenicity in yeast, negative in the dominant lethal test in mice, negative
for sister chromatid exchange in Chinese hamster ovary cells, positive for the mouse erythrocyte micronucleus test, and negative for unscheduled DNA syntheses in human diploid fibroblasts (U.S. EPA, 1981). Positive studies suggest that cacodylic acid poses a genotoxic risk only at high doses. For example, the mouse lymphoma assay showed mutagenic effects only at cytotoxic doses, and clastogenic activity was observed in the erythrocytes of mice only at acutely toxic doses (Jotz and Mitchell, 1980; Kirkhart, 1980).
Recent data implicate the cacodylic acid metabolite dimethylarsine as a mutagen in bacterial and mammalian systems (Yamanaka et al., 1989a,b). In particular, DNA single-strand breaks were induced in rat and mouse lung after oral administration of high doses of cacodylic acid (1,500 mg/kg). In vitro follow-up experiments showed that the breaks were caused by active oxygen species (superoxide anion radical produced by the one-electron reduction of molecular oxygen) and/or dimethylarsinic peroxyl radicals, both resulting from the presence of dimethylarsine. Dimethylarsine was stated to be a volatile metabolite of cacodylic acid excreted in the expired air in the in vivo phase of the study (Yamanaka et al., 1989a,b, 1991).
Acute Toxicity
Human case studies have included reports of nausea and gastrointestinal distress following exposures to cacodylic acid (Peoples et al., 1979). Data from farm animal studies have shown diarrhea and anorexia resulting from exposure to organic arsenicals. Hemorrhaging in the intestinal tract was noted at necropsy. Cacodylic acid was the least toxic of the arsenicals evaluated, with a 10 day exposure to 25 mg/kg considered ''marginally toxic" to cattle (Hood, 1985).
Sedation was noted among surviving rats in an acute LD50 study with cacodylic acid; however, at doses of 0.6-1.35 g/kg/day, this appears to be a nonspecific effect of the test material rather than a sign of neurotoxicity (Ansul, 1967).
An acute dermal irritation study in rabbits showed that cacodylic acid was essentially nonirritating when applied dermally in a single acute exposure (Ansul, 1967). Female rates exposed to 6.94 mg/liter air of cacodylic acid in acute inhalation toxicity experiments developed erythematous lesions on the feet and ears (Stevens et al., 1979).
Chronic Systemic Toxicity
Studies of the chronic systemic toxicity of cacodylic acid are extremely limited. Reports of early clinical experience with cacodylic acid showed renal damage, including nephritis, in some cases (Hood, 1985). Cattle given
cacodylic acid at 10-20 mg/kg/day for 4 weeks showed renal tubular degeneration in four of the five animals treated (Dickinson, 1975).
Reproductive and Developmental Toxicity
Limited data are available for assessing the potential effects of cacodylic acid on male-mediated reproductive toxicity. No studies of sperm morphology, viability, or sperm counts have been done, and reproductive toxicity studies are absent. In addition, no studies have been reported that assess potential reproductive toxicity in females or offspring.
The studies of cacodylic acid in male animals are limited to a subacute dermal study in a single male rabbit, which showed decreased spermatogenesis at a high dose level (1 g/kg), and to a 21 day feeding study in Sprague-Dawley rats, which showed seminiferous tubule atrophy and decreased spermatogenesis at 226 mg/kg/day, with a no-observed-adverse-effect level for this effect of 118 mg/kg/day (Ansul, 1967). The high dose levels at which these effects were observed suggest that there is likely to be a relatively low-risk of reproductive toxicity to humans in actual exposure situations.
Exposure to cacodylic acid has been associated with fetotoxicity in laboratory animals, although these effects occurred only at doses that also produced maternal toxicity. Fetotoxic effects of cacodylic acid in rats, mice, and hamsters include reduced fetal weight, decreased ossification, and increased incidences of cleft palate, irregular palatine rugae, micrognathia, hypoplastic lungs, and other major malformations (Chernoff and Rogers, 1975; WARF, 1976; Rogers et al., 1981; Hood et al., 1982, Kavlock et al., 1985). In each of these studies, overt maternal toxicity was also apparent.
Immunotoxicity
The potential immunotoxicity of cacodylic acid has not been evaluated.
TOXICITY PROFILE OF PICLORAM
Introduction
Picloram (4-amino-3,5,6-trichloropicolinic acid; CAS No. 1918-02-1; trade name Tordon; Figure 4-1) is a systemic herbicide used to control broadleaf and woody plants. Picloram was combined with 2,4-D to generate the formulation termed Agent White, an herbicide used during the war in Vietnam. The toxicological data base for picloram is limited. Picloram is a colorless (off-white to brown, if contaminants are present) powder or crystal with a chlorine-like odor. Its melting point is 218°C. It is soluble in water and a variety of organic solvents (e.g., acetone, alcohols, and benzene).
Pharmacokinetics
The only information that could be located on the pharmacokinetics of picloram was a study in which 15 rats received either an oral or an intravenous dose, 82 percent of which was detected in urine and 15.5 percent in feces within 48 hours (U.S. EPA, 1988a,c).
Carcinogenicity
Three somewhat limited studies of the carcinogenicity of picloram have been performed, with one study producing equivocal positive results and the rest producing negative results. The positive results were attributed to hexachlorobenzene contamination, however. These studies are described below.
Fischer 344 rats (50 of each sex per group) were administered 0, 20, 60, or 200 mg/kg/day of technical picloram in the diet for two years. No clinical signs of toxicity were observed in any dose group, indicating that the maximum tolerated dose was not reached, although a number of hepatocellular alterations were detected. No oncogenic effects were observed (Stott et al., 1990).
In another study, male and female Osborne-Mendel rats were administered 0, 10,000 (500 mg/kg), or 20,000 ppm (1,000 mg/kg) of picloram in the diet for 39 weeks; due to signs of overt toxicity, the dosages were lowered to 5,000 (250 mg/kg) and 10,000 ppm (500 mg/kg) for an additional 41 weeks. Animals were administered control diets for a recovery period of 33 weeks. An increased incidence of follicular hyperplasia, C cell hyperplasia, and C cell adenoma of the thyroid was observed in both sexes of treated rats compared to controls; however, the authors concluded that these effects were not associated with picloram treatment. An increased incidence of pituitary chromophobe adenoma was observed for females in the treated groups, compared to the matched controls but not compared to pooled controls; this lesion is common in aging rats. Focal cellular changes were noted in treated male rat livers. A greater number of liver changes and an increased incidence of neoplastic nodules were observed for females at the high dose. One hepatocellular carcinoma was observed in a low dose male and one in a high dose female. The authors concluded that picloram was carcinogenic to Osborne-Mendel female rats based on the increased incidence of liver neoplastic nodules in the high dose group (NCI, 1978). An EPA peer review committee reassessed the tumor incidence and agreed that there was a statistically significant (p<0.05) increase in liver adenomas and in combined liver adenomas and carcinomas for high dose females; however, the hexachlorobenzene (HCB, 130 ppm) contaminating the test material was believed to be responsible for the liver tumors observed. (It
should be noted that this study was done in 1978 with technical picloram; therefore, the picloram used in Vietnam probably contained a similar level of HCB.) EPA also concluded that the biologic significance of the thyroid lesions could not be determined because of the small control group.
Finally, male and female B6C3F1 mice were administered 0, 2,500 (357 mg/kg/day), or 5,000 ppm (714 mg/kg/day) of picloram in the diet for 79 weeks and then allowed to recover for 10 weeks prior to sacrifice. Mean body weights of the treated mice were unaffected by treatment, and no oncogenic response was observed (NCI, 1978).
Genotoxicity
Information on the potential genotoxic effects of picloram is meager: it did not produce cytogenetic effects in rats exposed to single doses up to 2,000 mg/kg (U.S. EPA, 1988c), and it did not produce mutagenic effects when tested in Salmonella typhimurium in the absence of metabolic activation (U.S. EPA, 1988c).
Acute Toxicity
The available information on the acute toxicity of picloram is also paltry. The dermal LD50 for technical grade picloram and a formulation of picloram referred to as Tordon K+ salt liquor in rabbits is greater than 2,000 mg/kg (U.S. EPA, 1988c). Some erythema, but no signs of toxicity, were observed at this dose. Some neurologic effects, including hyperactivity, ataxia, and tremors, were reported in pregnant rats exposed to 750 or 1,000 mg picloram/kg (Thompson et al., 1972).
Chronic Systemic Toxicity
Several studies have reported various effects of technical grade picloram on the livers of rats. In the carcinogenicity bioassay conducted by Stott and colleagues (1990) described above, treatment-related hepatomegaly was noted in the 60 and 200 mg/kg/day dose groups, along with hepatocellular swelling and altered tinctorial properties in the central regions of the liver lobules. In addition, increased liver weights were observed for males and females at the high dose compared to the controls. The NOEL was 20 mg/kg/day, and the lowest effect level (LEL) was 60 mg/kg/day for histological changes in centrilobular hepatocellular tissues. According to EPA, the levels of hexachlorobenzene (197 ppm), a contaminant in the technical picloram tested, were probably not responsible for the liver effects (U.S. EPA, 1988c). Gorzinski and colleagues (1987b) also reported a dose-related increase in liver weights, hepatocellular hypertrophy, and changes in centrilobular
tinctorial properties in male and female F344 rats at doses of 150 mg/kg/day and higher in the diet for 13 weeks. In a 90 day study, cloudy swelling in the liver cells and bile duct epithelium occurred in male and female F344 rats administered 0.3 or 1 percent technical picloram in the diet (U.S. EPA, 1988c).
Liver effects have also been reported in dogs exposed to picloram: increased liver weights were reported in beagles that received 35 mg/kg/day or more in the diet for six months (U.S. EPA, 1988c).
No other effects of chronic exposure to picloram have been reported.
Reproductive and Developmental Toxicity
The reproductive toxicity of picloram was evaluated in a two-generation study; too few animals were evaluated, and no toxicity was detected at the highest dose tested (150 mg/kg/day) (U.S. EPA, 1988c).
Some developmental toxicity was produced in rabbits exposed to 400 mg picloram/kg/day by gavage on days 6 through 18 of gestation. Fetal abnormalities included single litter incidences of forelimb flexure, fused ribs, hypoplastic tail, and omphalocele (John-Greene et al., 1985). Some maternal toxicity was observed at this dose, however, and EPA concluded that these malformations were not treatment related, based on the low litter incidence of these findings (U.S. EPA, 1988c).
No teratogenic effects were produced in the offspring of rats administered doses of picloram by gavage up to 1,000 mg/kg/day on days 6 to 15 of gestation, although the occurrence of bilateral accessory ribs was significantly increased at this dose (Thompson et al., 1972).
Immunotoxicity
Studies of the potential immunotoxicity of picloram are limited to dermal sensitization. In one study, 53 human volunteers were administered nine 24-hour applications of 0.5 ml of a 2 percent potassium picloram solution on the skin of both upper arms. Each volunteer received challenge doses from 17 to 24 days later. This formulation of picloram (its K+ salt) was not a skin sensitizer or an irritant (U.S. EPA, 1988c). In a similar study, a 5 percent solution of picloram (M-2439, Tordon 101 formulation) produced slight dermal irritation and caused a sensitization response in 6 of the 69 volunteers exposed. When the individual components of M-2439 were tested separately [picloram, triisopropanolamine (TIPA) salt, and 2,4-D TIPA salt], no sensitization reaction occurred, however (U.S. EPA, 1988c). Tordon K+, but not technical grade picloram, was also found to be a skin sensitizer in guinea pigs (U.S. EPA, 1988c).
REFERENCES
Abbott BD, Birnbaum LS. 1990. TCDD-induced altered expression of growth factors may have a role in producing cleft palate and enhancing the incidence of clefts after coadministration of retinoic acid and TCDD. Toxicology and Applied Pharmacology 106:418-432.
Abdellatif AG, Preat V, Vamecq J, Nilsson R, Roberfroid M. 1990. Peroxisome proliferation and modulation of rat liver carcinogenesis by 2,4-dichlorophenoxyacetic acid, 2,4,5-trichlorophenoxyacetic acid, perfluorooctanoic acid and nafenopin. Carcinogenesis 11:1899-1902.
Abraham K, Krowke R, Neubert D. 1988. Pharmacokinetics and biological activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin. I. Dose-dependent tissue distribution and induction of hepatic ethoxyresorufin O-deethylase in rats following a single injection. Archives of Toxicology 62:359-368.
Abraham K, Weismüller T, Hagenmaier H, Neubert, D. 1990. Distribution of PCDDs and PCDFs in various tissues of marmoset monkeys. Chemosphere 20:1071-1078.
Ackermann MF, Gasiewicz TA, Lamm KR, Germolec DR, Luster MI. 1989. Selective inhibition of polymorphonuclear neutrophil activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 101:470-480.
Adhikari N, Grover IS. 1988. Genotoxic effects of some systemic pesticides: in vivo chromosomal aberrations in bone marrow cells in rats. Environmental and Molecular Mutagenesis 12:235-242.
Albro PW, Corbett JT, Harris M, Lawson LD. 1978. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on lipid profiles in tissue of the Fischer rat. Chemico-Biological Interactions 23:315-330.
Allen JR, Carstens LA. 1967. Light and electron microscopic observations in Macaca mulatta monkeys fed toxic fat . American Journal of Veterinary Research 28:1513-1526.
Allen JR, Lalich JJ. 1962. Response of chickens to prolonged feeding of crude "toxic fat." Proceedings of the Society for Experimental Biology and Medicine 109:48-51.
Allen JR, Van Miller JP, Norback DH. 1975. Tissue distribution, excretion, and biological effects of [14C]tetrachlorodibenzo-p-dioxin in rats. Food and Cosmetics Toxicology 13:501-505.
Allen JR, Carstens LA, Abrahamson LJ. 1976. Responses of rats exposed to polychlorinated biphenyls for fifty-two weeks. I. Comparison of tissue levels of PCB and biological changes. Archives of Environmental Contamination and Toxicology 4:409-419.
Allen JR, Barsotti DA, Van Miller JP, Abrahamson LJ, Lalich JJ. 1977. Morphological changes in monkeys consuming a diet containing low levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Food and Cosmetics Toxicology 15:401-410.
Allen JR, Barsotti DA, Lambrecht LK, Van Miller JP. 1979. Reproductive effects of halogenated aromatic hydrocarbons on nonhuman primates. Annals of the New York Academy of Sciences 320:419-425.
Alsharif NZ, Lawson T, Stohs SJ. 1990. TCDD-induced production of superoxide anion and DNA single-strand breaks in peritoneal macrophages of rats. Toxicologist 10:278.
Ansul Company. 1967. Toxicological Data: Methanearsonic Acid and Dimethylarsinic Acid. Marinette, WI: Ansul.
Appelgren LE, Brandt I, Brittelos EB, Gillner M, Gustafsson SA. 1983. Autoradiography of 2,3,7,8-tetrachloro-14C-dibenzo-p-dioxin (TCDD): accumulation in the nasal mucosa. Chemosphere 4/5:545-548.
Arnold EK, Beasley VR. 1989. The pharmacokinetics of chlorinated phenoxy acid herbicides: a literature review. Veterinary and Human Toxicology 31:121-125.
Arnold EK, Beasley VR, Parker AJ, Stedelin JR. 1991. 2,4-D toxicosis II: a pilot study of clinical pathologic and electroencephalographic effects and residues of 2,4-D in orally dosed dogs. Veterinary and Human Toxicology 33:446-449.
Astroff B, Rowlands C, Dickerson R, Safe S. 1990. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibition of 17-beta-estradiol-induced increases in rat uterine epidermal growth factor receptor binding activity and gene expression. Molecular and Cellular Endocrinology 72:247-252.
Balk JL, Piper WN. 1984. Altered blood levels of corticosteroids in the rat after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochemical Pharmacology 33:2531-2534.
Banks YB, Birnbaum LS. 1991. Absorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) after low dose dermal exposure. Toxicology and Applied Pharmacology 107:302-310.
Bars RG, Elcombe CR. 1991. Dose-dependent acinar induction of cytochromes P450 in rat liver. Evidence for a differential mechanism of induction of P450IA1 by beta-naphthoflavone and dioxin. Biochemical Journal 277:577-580.
Barsotti DA, Abrahamson LJ, Allen JR. 1979. Hormonal alterations in female rhesus monkeys fed a diet containing 2,3,7,8-tetrachlorodibenzo-p-dioxin. Bulletin of Environmental Contamination and Toxicology 21:463-469.
Bastomsky CH. 1977. Enhanced thyroxine metabolism and high uptake goiters in rats after a single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Endocrinology 101:292-296.
Bazare JJ Jr, Nelson CJ, Young JF. 1990. Pharmacokinetics of 2,4,5-T in mice as a function of dose and gestational status . Reproductive Toxicology 4:137-144.
Beatty P, Neal RA. 1977. Factors affecting the inductions of DT-diaphorase by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochemical Pharmacology 27:505-510.
Beatty PW, Vaughn WK, Neal RA. 1978. Effect of alteration of rat hepatic mixed-function oxidase (MFO) activity on the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicology and Applied Pharmacology 45:513-519.
Becker GM, McNulty WP. 1984. Gastric epithelial cell proliferation in monkeys fed 3,4,3,4'-tetrachlorobiphenyl. Journal of Pathology 143:267-274.
Becker GM, McNulty WP, Bell M. 1979. Polychlorinated biphenyl-induced morphologic changes in the gastric mucosa of the rhesus monkey. Laboratory Investigations 40:373-383.
Beguinot L, Hanover JA, Ito S, Richert MD, Willingham MC, Pastan I. 1985. Phorbol esters induce internalization without degradation of unoccupied epidermal growth factor receptors. Proceedings of the National Academy of Sciences (USA) 82:2774-2778.
Berman EF, Schaus P, Fujimoto JM. 1986. Comparison of the inhibition of biliary excretion produced by certain inducing agents including 2,3,7,8-tetrachlorodibenzo-p-dioxin. Journal of Toxicology and Environmental Health 17:395-403.
Bestervelt LL, Nolan CJ, Cai Y, Maimansomsuk P, Mousigan CA, Piper WN. 1991. Tetrachlorodibenzo-p-dioxin alters rat hypothalamic endorphin and mu opioid receptors. Neurotoxicology and Teratology 13:495-497.
Bianco AC, Silva JE. 1987. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. Journal of Clinical Investigation 79:295-300.
Bionetics Research Laboratories. 1968a. Evaluation of Carcinogenic, Teratogenic and Mutagenic Activities of Selected Pesticides and Industrial Chemicals. Vol. 1, Carcinogenic Study. Bethesda, MD: Bionetics Research Laboratories. NTIS PB 223 159.
Bionetics Research Laboratories. 1968b. Evaluation of Carcinogenic, Teratogenic and Mutagenic Activities of Selected Pesticides and Industrial Chemicals. Vol. 2, Teratogenic Study of Mice and Rats. Bethesda, MD: Bionetics Research Laboratories. NTIS PB 223 160.
Birnbaum LS. 1985. The role of structure in the disposition of halogenated aromatic xenobiotics. Environmental Health Perspectives 61:11-20.
Birnbaum LS. 1986. Distribution and excretion of 2,3,7,8-tetrachlorodibenzo-p-dioxin in congenic strains of mice which differ at the Ah locus. Drug Metabolism and Disposition 14:34-40.
Birnbaum LS, Decad GM, Matthews HB. 1980. Distribution and excretion of 2,3,7,8-tetrachlorodibenzo-p-dioxin
in the rat. Toxicology and Applied Pharmacology 55:342-352.
Bjeldanes LF, Kim J-Y, Grose KR, Bartholomew JC, Bradfield CA. 1991. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proceedings of the National Academy of Sciences (USA) 88:9543-9547.
Bjorklund N, Erne K. 1966. Toxicological studies of phenoxyacetic herbicides in animals. Acta Veterinaria Scandinavica 7:364-390.
Blakley BR. 1986. The effect of oral exposure to the p-butyl ester of 2,4-dichlorophenoxyacetic acid on the immune response in mice. International Journal of Immunopharmacology 8:93-99.
Blakley BR, Schiefer BH. 1986. The effect of topically applied n-butyl ester of 2,4-dichlorophenoxyacetic acid on the immune response in mice. Journal of Applied Toxicology 6:291-295.
Blakley BR, Gagnon JM, Rousseaux CG. 1992. The effect of a commercial 2,4-D formulation on chemical- and viral-induced tumor production in mice. Journal of Applied Toxicology 12:245-249.
Bock KW. 1991. Roles of UDP-glucuronosyltransferases in chemical carcinogenesis. Critical Reviews in Biochemistry and Molecular Biology 26:129-150.
Bombick DW, Matsumura F, Madhukar BV. 1984. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) causes reduction in the low-density lipoprotein (LDL) receptor activities in the hepatic plasma membrane of the guinea pig and rat. Biochemical and Biophysical Research Communications 118:548-554.
Bookstaff RC, Moore RW, Peterson RE. 1990a. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases the potency of androgens and estrogens as feedback inhibitors of luteinizing hormone secretion in male rats. Toxicology and Applied Pharmacology 104:212-224.
Bookstaff RC, Kamel F, Moore RW, Bjerke DL, Peterson RE. 1990b. Altered regulation of pituitary gonadotropin-releasing hormone (GNRH) receptor number and pituitary responsiveness to GNRH in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated male rats. Toxicology and Applied Pharmacology 105:78-92.
Bowman RE, Schantz SL, Weerasinghe NCA, Gross ML, Barsotti DA. 1989. Chronic dietary intake of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) at 5 and 25 parts per trillion in the monkey: TCDD kinetics and dose-effect estimate of reproductive toxicity. Chemosphere 18:243-252.
Bowman RE, Schantz SL, Ferguson SA, Tong HY, Gross ML. 1990. Controlled exposure of female rhesus monkeys to 2,3,7,8-TCDD; cognitive behavioral effects in their offspring. Chemosphere 20:7-9.
Bradfield CA, Glover E, Poland A. 1991. Purification and N-terminal amino acid sequence of the Ah receptor from the C57B1/6J mouse. Molecular Pharmacology 39:13-19.
Brewster DW, Bombick DW, Matsumura F. 1988. Rabbit serum hypertriglyceridemia after administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Journal of Toxicology and Environmental Health 25:495-507.
Brewster DW, Banks YB, Clark AM, Birnbaum LS. 1989. Comparative dermal absorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin and three polychlorinated dibenzofurans. Toxicology and Applied Pharmacology 97:156-166.
Bruckner JV, Khanna KL, Cornish HH. 1973. Biological responses of the rat to polychlorinated biphenyls . Toxicology and Applied Pharmacology 24:434-448.
Buchet JP, Lauwerys R, Roels H. 1981. Comparison of the urinary excretion of arsenic metabolites after a single dose of sodium arsenite, monomethylarsonate, or dimethylarsinate in man. International Archives of Occupational and Environmental Health 48:71-79. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental
Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
Burbach KM, Poland A, Bradfield CA. 1992. Cloning of the Ah receptor cDNA reveals a distinctive ligand-activated transcription factor. Proceedings of the National Academy of Sciences (USA) 89:8185-8189.
Canadian Centre for Toxicology (CCT). 1987. Expert Panel Report on Carcinogenicity of 2,4-D. Guelph, Ontario: CCT.
Cantoni L, Salmona M, Rizzardini M. 1981. Porphyrogenic effect of chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin in female rats. Dose-effect relationship following urinary excretion of porphyrins. Toxicology and Applied Pharmacology 57:156-163.
Chahoud I, Krowke R, Schimmel A, Merker HJ, Neubert D. 1989. Reproductive toxicity and pharmacokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin. 1. Effects of high doses on the fertility of male rats. Archives of Toxicology 63:432-439.
Chahoud I, Krowke R, Bochert G, Burkle B, Neubert D. 1991. Reproductive toxicity and toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin. 2. Problem of paternally-mediated abnormalities in the progeny of rat. Archives of Toxicology 65:27-31.
Chahoud I, Hartmann J, Rune GM, Neubert D. 1992. Reproductive toxicity and toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin. 3. Effects of single doses on the testis of male rats. Archives of Toxicology 66:567-572.
Chapman DE, Schiller CM. 1985. Dose-related effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57BL/6J and DBA/2J mice. Toxicology and Applied Pharmacology 78:147-157.
Chaturvedi AD, Kuntz D, Rao N. 1991. Metabolic aspects of the toxicology of mixtures of parathion, toxaphene and/or 2,4-D in mice. Journal of Applied Toxicology 11:245-251.
Chernoff N, Rogers EH. 1975. Effects of cacodylic acid on prenatal development of rats and mice. Substitute Chemical Program, the First Year of Progress: Proceedings of a Symposium. Vol. II. Toxicological Methods and Genetic Effects Workshop. U.S. EPA. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental Fate . Washington, DC: Veterans Administration, Agent Orange Projects Office.
Choi EJ, Toscano DG, Ryan JA, Riedel N, Toscano WA Jr. 1991. Dioxin induces transforming growth factor-alpha in human keratinocytes. Journal of Biological Chemistry 266:9591-9597.
Christian BJ, Menahan LA, Peterson RE. 1986a. Intermediary metabolism of the mature rat following 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Toxicology and Applied Pharmacology 83:360-378.
Christian BJ, Inhorn SL, Peterson RE. 1986b. Relationship of the wasting syndrome to lethality in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 82:239-255.
Clark DA, Gauldie J, Szewczuk MR, Sweeney G. 1981. Enhanced suppressor cell activity as a mechanism of immunosuppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proceedings of the Society for Experimental Biology and Medicine 168:290-299.
Clark DA, Sweeney G, Safe S, Hancock E, Kilburn DG, Gauldie J. 1983. Cellular and genetic basis for suppression of cytotoxic T cell generation by haloaromatic hydrocarbons . Immunopharmacology 6:143-153.
Clark GC, Taylor MJ, Tritscher AM, Lucier GW. 1991a. Tumor necrosis factor involvement in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated endotoxin hypersensitivity in C57B1/6J mice congenic at the Ah locus. Toxicology and Applied Pharmacology 111:422-431.
Clark GC, Tritscher AM, Maronpot R, Foley J, Lucier GW. 1991b. Tumor promotion by TCDD in female rats. In: Gallo MA, Scheuplein RJ, van der Heijden KA, eds. Biological Basis for Risk Assessment of Dioxin and Related Compounds. Plainview, NY: Cold Spring Harbor Laboratory. Banbury Report 35.
Clark GC, Tritscher AM, McCoy Z. 1991c. Dose-response relationships for chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in a rat liver tumor promotion model: 1. Relationships of TCDD tissue concentrations to serum clinical chemistry cell proliferation and preneoplastic foci. In: Proceedings of the 11th International Symposium on Chlorinated Dioxins and Related Compounds, Dioxin. Research Triangle Park, NC.
Cochet C, Gill GN, Meisenhelder J, Cooper JA, Hunter T. 1984. C-kinase phosphorylates the epidermal growth factor receptor and reduces its epidermal growth factor-stimulated tyrosine protein kinase activity. Journal of Biological Chemistry 259:2553-2558.
Cohen SM, Ellwein LB. 1990. Cell proliferation in carcinogenesis. Science 249:1007-1011.
Cohen GM, Bracken WM, Iyer RP, Berry DL, Selkirk JK, Slaga TJ. 1979. Anticarcinogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on benzo{a}pyrene and 7,12-demethylbenz{a}anthracene tumor initiation and its relationship to DNA-binding. Cancer Research 39:4027-4033.
Collins TFX, Williams CH. 1971. Teratogenic studies with 2,4,5-T and 2,4-D in the hamster. Bulletin of Environmental Contamination and Toxicology 6:559-567.
Cook JC, Greenlee WF. 1989. Characterization of a specific binding protein for 2,3,7,8-tetrachlorodibenzo-p-dioxin in human thymic epithelial cells. Molecular Pharmacology 35:713-719.
Courtney KD. 1976. Mouse teratology studies with chlorodibenzo-p-dioxins. Bulletin of Environmental Contamination and Toxicology 16:674.
Courtney KD, Moore JA. 1971. Teratology studies with 2,4,5-trichlorophenoxyacetic acid and 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 20:396-403.
Couture LA, Harris MW, Birnbaum LS. 1990a. Characterization of the peak period of sensitivity for the induction of hydronephrosis in C57B1/6N mice following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundamental and Applied Toxicology 15:142-150.
Couture LA, Abbott BD, Birnbaum LS. 1990b. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology 42:619-627.
Crampton MA, Rogers LJ. 1983. Low doses of 2,4,5-trichlorophenoxyacetic acid are behaviorally teratogenic in rats. Experientia 39:891-892.
Cunningham HM, Williams DT. 1972. Effects of tetrachlorodibenzo-p-dioxin on growth rate and the synthesis of lipids and proteins in rats. Bulletin of Environmental Contamination and Toxicology 7:45-51.
Curtis LR, Kerkvliet NI, Baecher-Steppan L, Carpenter HM. 1990. 2,3,7,8-Tetrachlorodibenzo-p-dioxin pretreatment of female mice altered tissue distribution but not hepatic metabolism of a subsequent dose. Fundamental and Applied Toxicology 14:523-531.
Cushman JR, Street JC. 1982. Allergic hypersensitivity to the herbicide 2,4-D in BALB/c mice. Journal of Toxicology and Environmental Health 10:729-741.
Cuthill S, Wilhelmsson A, Poellinger L. 1991. Role of the ligand in intracellular receptor function: receptor affinity determines activation in vitro of the latent dioxin receptor to a DNA-binding form. Molecular and Cellular Biology 11:401-411.
Davis D, Safe S. 1988. Immunosuppressive activities of polychlorinated dibenzofuran congeners: quantitative structure-activity relationships and interactive effects. Toxicology and Applied Pharmacology 94:141-149.
Davis D, Safe S. 1989. Dose-response immunotoxicities of commercial polychlorinated biphenyls (PCBs) and their interaction with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology Letters 48:35-43.
Davis D, Safe S. 1990. Immunosuppressive activities of polychlorinated biphenyls in C57B1/6N mice: structure-activity relationships as Ah receptor agonists and partial antagonists. Toxicology 63:97-111.
Davis D, Safe S. 1991. Halogenated aryl hydrocarbon-induced suppression of the in vitro
plaque-forming cell response to sheep red blood cells is not dependent on the Ah receptor. Immunopharmacology 21:183-190.
DeKrey GK, Steppan LB, Deyo JA, Kerkvliet NI. 1990. Adrenalectomy (ADX) and 3,4,5,3',4',5'-hexachlorobiphenyl (HXCB) suppression of cytotoxic T lymphocyte (TL) response to P815 allogeneic tumor in C57B1/6 mice. Toxicologist 10:290.
DeKrey GK, Deyo JA, Kerkvliet NI. 1992. Castration (ODX) partially alleviates the suppression of cytotoxic T lymphocyte (CTL) activity by 3,3',4,4',5,5'-hexachlorobiphenyl (HxCB). Toxicologist 12:132.
Della Porta G, Dragani TA, Sozzi G. 1987. Carcinogenic effects of infantile and long-term 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment in the mouse. Tumori 73:99-107.
Denison MS, Yao EF. 1991. Characterization of the interaction of transformed rat hepatic cytosolic Ah receptor with a dioxin-responsive transcriptional enhancer. Archives of Biochemistry and Biophysics 284:158-166.
Denison MS, Vella LM, Okey AB. 1987. Structure and function of the Ah receptor: sulfhydryl groups required for binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to cytosolic receptor from rodent livers. Archives of Biochemistry and Biophysics 252:388-395.
Denison MS, Fisher JM, Whitlock JP Jr. 1989. Protein-DNA interactions at recognition sites for the dioxin-Ah receptor complex. Journal of Biological Chemistry 264:16478-16482.
Dési I, Sos J, Nikolits I. 1962a. New evidence concerning the nervous site of action of a chemical herbicide causing professional intoxication. Acta Physiologica Academiae Scientarum Hungaricae 22:73-80.
Dési I, Sos J, Olasz J, Sule F, Markus V. 1962b. Nervous system effects of a chemical herbicide. Archives of Environmental Health 4:95-102.
DiBartolomeis MJ, Jefcoate CR, Christian BJ, Moore RW, Peterson RE. 1984. The effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on adrenal steroidogenesis, in vivo. Toxicologist 4:187.
DiBartolomeis MJ, Moore RW, Peterson RE, Jefcoate CR. 1986a. Hypercholesterolemia and the regulation of adrenal steroidogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Toxicology and Applied Pharmacology 85:313-323.
DiBartolomeis MJ, Williams C, Jefcoate CR. 1986b. Inhibition of ACTH action on cultured bovine adrenal cortical cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin through a redistribution of cholesterol. Journal of Biological Chemistry 261:4432-4437.
DiBartolomeis MJ, Christou M, Jefcoate CR. 1986c. Regulation of rat and bovine adrenal metabolism of polycyclic aromatic hydrocarbons by adrenocorticotropin and 2,3,7,8-tetrachlorodibenzo-p-dioxin . Archives of Biochemistry and Biophysics 246:428-438.
DiBartolomeis MJ, Moore RW, Peterson RE, Christian BJ, Jefcoate CR. 1987. Altered regulation of adrenal steroidogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Biochemical Pharmacology 36:59-67.
Dickinson JO. 1975. Toxicity of cacodylic acid silvicide in cattle. West Vet 3:1-4. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
DiGiovanni J, Viaje A, Berry DL, Slaga T, Juchau MR. 1977. Tumor-initiating ability of TCDD and Aroclor 1254 in the two stage system of mouse skin carcinogenesis. Bulletin of Environmental Contamination and Toxicology 18:552-557.
Diliberto JJ, Jackson JA, Birnbaum LS. 1991. 2,3,7,8-Tetrabromodibenzo-p-dioxin in rats. Toxicologist 11:272.
Domain BA, Devereux TR, Philpot RM. 1986. The cytochrome P-450 monooxygenase system of rabbit lung: enzyme components, activities and induction in the nonciliated bronchiolar epithelial (Clara) cell, alveolar type II cell, and alveolar macrophage. Molecular Pharmacology 30:296-303.
Dooley RK, Holsapple MP. 1988. Elucidation of cellular targets responsible for tetrachlorodibenzo-p-dioxin
(TCDD)-induced suppression of antibody responses: the role of the B lymphocyte. Immunopharmacology 16:167-180.
Dougherty J, Schulze G, Taylor R. 1984. Effects of 2,4-D on scheduled-controlled behavior. Neurobehavioral Toxicology and Teratology 6:177.
Dragan YP, Xu XH, Goldsworthy TL, Campbell HA, Maronpot RR, Pitot HC. 1992. Characterization of the promotion of altered hepatic foci by 2,3,7,8-tetrachlorodibenzo-p-dioxin in the female rat. Carcinogenesis 13:1389-1395.
Drill VA, Hiratzka T. 1953. Toxicity of 2,4-dichlorophenoxyacetic acid and 2,4,5-trichlorophenoxyacetic acid. A report on their acute and chronic toxicity in dogs. Archives of Industrial Hygiene and Occupational Medicine 7:61-67.
Dunn TJ, Lindahl R, Pitot HC. 1988. Differential gene expression in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Noncoordinate regulation of a TCDD-induced aldehyde dehydrogenase and cytochrome P-450c in the rat. Journal of Biological Chemistry 263:10878-10886.
Durrin LK, Whitlock JP Jr. 1989. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible aryl hydrocarbon receptor-mediated change in CYP1A1 chromatin structure occurs independently of transcription. Molecular and Cellular Biology 9:5733-5737.
Elder GH. 1978. Porphyria caused by hexachlorobenzene and other polyhalogenated aromatic hydrocarbons. In: DeMatteis F, Aldridge WN, eds. Heme and Hemoproteins. New York: Springer-Verlag. 157-200.
Elferink CJ, Whitlock JP Jr. 1990. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible, Ah receptor-mediated binding of enhancer DNA. Journal of Biological Chemistry 265:5718-5721.
Elferink CJ, Gasiewicz TA, Whitlock JP Jr. 1990. Protein-DNA interactions at a dioxin-responsive enhancer. Evidence that the transformed Ah receptor is heteromeric. Journal of Biological Chemistry 265:20708-20712.
Elsenhans B, Forth W, Richter E. 1991. Increased copper concentrations in rat tissues after acute intoxication with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Archives of Toxicology 65:429-432.
Ema M, Sogawa K, Wanatabe N, Chujoh Y, Matsushita N, Gotch O, Funae Y, Fujii-Kuriyama Y. 1992. cDNA cloning and structure of mouse putative Ah receptor. Biochemical and Biophysical Research Communications 184:246-253.
Exon JH, Kerkvliet NI, Talcott PA. 1987. Immunotoxicity of carcinogenic pesticides and related chemicals. Environmental Carcinogenesis Reviews C5.
Faith RE, Moore JA. 1977. Impairment of thymus-dependent immune functions by exposure of the developing immune system to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Journal of Toxicology and Environmental Health 3:451-464.
Favreau LV, Pickett CB. 1991. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene: identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. Journal of Biological Chemistry 266:4556-4561.
Feldmann RJ, Maibach HI. 1984. Percutaneous penetration of some pesticides and herbicides in man. Toxicology and Applied Pharmacology 28:126-132.
Fishbein L. 1987. Health risk estimates for 2,3,7,8-tetrachlorodibenzo-p-dioxin: an overview. Toxicology and Industrial Health 3:91-134.
Fisher HL, Most B, Hall LL. 1989. Dermal absorption of pesticides calculated by deconvolution. Journal of Applied Toxicology 5:163-177.
Fisher JM, Jones KW, Whitlock JP Jr. 1989. Activation of transcription as a general mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin action. Molecular Carcinogenesis 1:216-221.
Fisher JM, Wu L, Denison MS, Whitlock JP Jr. 1990. Organization and function of a dioxin-responsive enhancer. Journal of Biological Chemistry 265:9676-9681.
Flanagan RJ, Meredith TJ, Ruprah M, Onyon LJ, Liddle A. 1990. Alkaline dieresis for acute poisoning with chlorophenoxy herbicides and ioxynil. Lancet 335:454-458.
Flodstrom S, Ahlborg UG. 1991. Promotion of hepatocarcinogenesis in rats by PCDDs and PCDFs. In: Gallo MA, Scheuplein RJ, van der Heijden KA, eds. Biological Basis for Risk Assessment of Dioxins and Related Compounds. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 35. 405-414.
Fowler BA, Lucies GW, Brown HW, McDaniel OS. 1973. Ultrastructural changes in rat liver cells following a single oral dose of TCDD. Environmental Health Perspectives 5:141-148.
Friesen EG, Jones GR, Vaughan D. 1990. Clinical presentation and management of acute 2,4-D oral ingestion. Drug Safety 5:155-159.
Fujiisawa-Sehara A, Sogawa K, Yamane N, Fujii-Kuriyama Y. 1987. Characterization of xenobiotic responsive elements upstream from the drug-metabolizing cytochrome P450c gene: a similarity to glucocorticoid regulatory elements. Nucleic Acids Research 15:4179-4191.
Gallo MA, Hesse EJ, Macdonald GJ, Umbreit TH. 1986. Interactive effects of estradiol and 2,3,7,8-tetrachlorodibenzo-p-dioxin on hepatic cytochrome P-450 and mouse uterus. Toxicology Letters 32:123-132.
Galloway SM, Armstrong MJ, Reben C, Colman S, Brown B, Cannon D, Bloom AD, Nakamura F, Ahmed M, Duk S, Rimpo J, Margolin BH, Resnick MA, Anderson KB, Zeiger E. 1987. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: evaluations of 108 chemicals. Environmental and Molecular Mutagenesis 10:1-175.
Gasiewicz TA, Bauman PA. 1987. Heterogeneity of the rat hepatic Ah receptor and evidence for transformation in vitro and in vivo. Journal of Biological Chemistry 262:2116-2120.
Gasiewicz TA, Neal RA. 1979. 2,3,7,8-Tetrachlorodibenzo-p-dioxin tissue distribution, excretion, and effects on clinical chemical parameters in guinea pigs. Toxicology and Applied Pharmacology 51:329-339.
Gasiewicz TA, Rucci G. 1984. Cytosolic receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Evidence for a homologous nature among various mammalian species. Molecular Pharmacology 26:90-98.
Gasiewicz TA, Holscher MA, Neal RA. 1980. The effect of total parenteral nutrition on the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Toxicology and Applied Pharmacology 54:469-488.
Gasiewicz TA, Geiger LE, Rucci G, Neal RA. 1983. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metabolism and Disposition 11:397-403.
Gasiewicz TA, Rucci G, Henry EC, Baggs RB. 1986. Changes in hamster hepatic cytochrome P-450, ethoxycoumarin O-deethylase, and reduced NAD(P): menadione oxidoreductase following treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Partial dissociation of temporal and dose-response relationships from elicited toxicity. Biochemical Pharmacology 35:2737-2742.
Gasiewicz TA, Elferink CJ, Henry EC. 1991. Characterization of multiple forms of the Ah receptor: recognition of a dioxin-responsive enhancer involves heteromer formation. Biochemistry 30:2909-2916.
Gehring PJ, Betso JE. 1978. Phenoxy acids: effects and fate in mammals. Ecological Bulletins 27:122-133.
Gehring PJ, Kramer CG, Schwetz BA, Rose JQ, Rowe VK. 1973. The fate of 2,4,5-trichlorophenoxyacetic acid (2,4,5-T) following oral administration to man. Toxicology and Applied Pharmacology 26:352-361.
Giavini E, Prati M, Vismara C. 1982. Rabbit teratology study with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environmental Research 27:74-78.
Gillner M, Bergman J, Cambillau C, Fernstrom B, Gustafsson JA. 1985. Interactions of indoles
with specific binding sites for 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat liver. Molecular Pharmacology 28:357-363.
Gillner M, Brittebo EB, Brandt I, Soderkvist P, Appelgren LE, Gustafsson JA. 1987. Uptake and specific binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the olfactory mucosa of mice and rats. Cancer Research 47:4150-4159.
Goldstein JA, Hickman P, Jue DL. 1974. Experimental hepatic porphyria induced by polychlorinated biphenyls. Toxicology and Applied Pharmacology 27:437-448.
Goldstein JA, Linko P, Bergman H. 1982. Induction of porphyria in the rat by chronic versus acute exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochemical Pharmacology 31:1607-1613.
Goldstein JA, Lin FH, Stohs SJ, Graham M, Clarke G, Birnbaum L, Lucier G. 1990. The effects of TCDD on receptors for epidermal growth factor, glucocorticoid, and estrogen in Ah-responsive and -nonresponsive congenic mice and the effects of TCDD on estradiol metabolism in a liver tumor promotion model in female rats. Progress in Clinical and Biological Research 331:187-202.
Goodman DG, Sauer RM. 1992. Hepatotoxicity and carcinogenicity in female Sprague-Dawley rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): a pathology working group reevaluation. Regulatory Toxicology and Pharmacology 15:245-252.
Gorski JR, Rozman K. 1987. Dose-response and time-course of hypothyroxinemia and hypoinsulinemia and characterization of insulin hypersensitivity in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicology 44:297-307.
Gorski JR, Rozman T, Greim H, Rozman K. 1988a. Corticosterone modulates acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in male Sprague-Dawley rats. Fundamental and Applied Toxicology 11:494-502.
Gorski JR, Lebofsky M, Rozman K. 1988b. Corticosterone decreases toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in hypophysectomized rats. Journal of Toxicology and Environmental Health 25:349-360.
Gorski JR, Weber LW, Rozman K. 1988c. Tissue-specific alterations of de novo fatty acid synthesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Archives of Toxicology 62:146-151.
Gorski JR, Muzi G, Weber LW, Pereira DW, Iatropoulos MJ, Rozman K. 1988d. Elevated plasma corticosterone levels and histopathology of the adrenals and thymuses in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Toxicology 53:19-32.
Gorski JR, Weber LW, Rozman K. 1990. Reduced gluconeogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Archives of Toxicology 64:66-71.
Gorzinski SJ, Kociba RJ, Campbell RA, Smith FA, Nolan RJ, Eisenbrandt DL. 1987a. Acute, pharmacokinetic, and subchronic toxicological studies of 2, 4-dichlorophenoxyacetic acid. Fundamental and Applied Toxicology 9:423-435.
Gorzinski SJ, Johnson KA, Campbell RA, Landry TD. 1987b. Dietary toxicity of picloram herbicide in rats. Journal of Toxicology and Environmental Health 20:367-377.
Graham MJ, Lucier GW, Linko P, Maronpot RR, Goldstein JA. 1988. Increases in cytochrome P-450 mediated 17- beta-estradiol 2-hydroxylase activity in rat liver microsomes after both acute administration and subchronic administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin in a two-stage hepatocarcinogenesis model. Carcinogenesis 9:1935-1941.
Greig JB, Jones G, Butler WH, Barnes JM. 1973. Toxic Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Food and Cosmetics Toxicology 11:585-595.
Gupta BN, Vos JG, Moore JA, Zinkl JG, Bullock BC. 1973. Pathologic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals. Environmental Health Perspectives 5:125-140.
Haag D, Goerttler K, Preiss D. 1975. The influence of non-cytotoxic concentrations of the herbicide 2,4-dichlorophenoxyacetic acid on the DNA synthesis in cultured vertebrate cells. Archives of Toxicology 33:91-102.
Hankinson O. 1983. Dominant and recessive aryl hydrocarbon hydroxylase-deficient mutants of the mouse hepatoma line, Hepa 1, and assignment of the recessive mutants to three complementary groups. Somatic Cell Genetics 9:497-514.
Hansen WH, Quaife ML, Habermann RT, Fitzhugh OG. 1971. Chronic toxicity of 2,4-dichlorophenoxyacetic acid in rats and dogs. Toxicology and Applied Pharmacology 20:122-129.
Hapgood J, Cuthill S, Denis M, Poellinger L, Gustafsson JA. 1989. Specific protein-DNA interactions at a xenobiotic-responsive element: copurification of dioxin receptor and DNA-binding activity. Proceedings of the National Academy of Sciences (USA) 86:60-64.
Harper PA, Golas CL, Okey AB. 1988. Characterization of the Ah receptor and aryl hydrocarbon hydroxylase induction by 2,3,7,8-tetrachlorodibenzo-p-dioxin and benz(a)anthracene in the human A431 squamous cell carcinoma line. Cancer Research 48:2388-2395.
Harper PA, Prokipcak RD, Bush LE, Golas CL, Okey AB. 1991. Detection and characterization of the Ah receptor for 2,3,7-8-tetrachlorodibenzo-p-dioxin in the human colon adenocarcinoma cell line LS180. Archives of Biochemistry and Biophysics 290:27-36.
Harris M, Piskorska-Pliszczynska J, Zacharewski T, Romkes M, Safe S. 1989. Structure-dependent induction of aryl hydrocarbon hydroxylase in human breast cancer cell lines and characterization of the Ah receptor. Cancer Research 49:4531-4535.
Harris M, Zacharewski T, Safe S. 1990. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds on the occupied nuclear estrogen receptor in MCF-7 human breast cancer cells. Cancer Research 50:3579-3584.
Harris SA, Soloman KR. 1992. Percutaneous penetration of 2,4-dichlorophenoxyacetic acid and 2,4-D dimethylamine salt in human volunteers. Journal of Toxicology and Environmental Health 36:233-240.
Hassoun E, d'Argy R, Dencker L, Lundin LG, Borwell P. 1984. Teratogenicity of 2,3,7,8-tetrachlorodibenzofuran in BXD recombinant inbred strains. Toxicology Letters 23:37-42.
Hayes HM, Tarone RE, Cantor KP, Jessen CR, McCurnin DM, Richardson RC. 1991. Case-control study of canine malignant lymphoma: positive association with dog owner's use of 2,4-D herbicides. Journal of the National Cancer Institute 83:1226-1231.
Hazleton Laboratories America. 1986. Combined Toxicity and Oncogenicity Study in Rats with 2,4-Dichlorophenoxyacetic Acid (2,4-D). Final Report. Prepared for the Industry Task Force on 2, 4-D Research Data.
Hazleton Laboratories America. 1987. Oncogenicity Study in Mice with 2,4-Dichlorophenoxyacetic Acid (2,4-D) Final Report. Prepared for the Industry Task Force on 2,4-D Research Data.
Henck JM, New MA, Kociba RJ, Rao KS. 1981. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: acute oral toxicity in hamsters. Toxicology and Applied Pharmacology 59:405-407.
Henry EC, Rucci G, Gasiewicz TA. 1989. Characterization of multiple forms of the Ah receptor: comparison of species and tissues. Biochemistry 28:6430-6440.
Hinsdill RD, Couch DL, Speirs RS. 1980. Immunosuppression in mice induced by dioxin (TCDD) in feed. Journal of Environmental Pathology and Toxicology 4:401-425.
Hinton DE, Glaumann H, Trump BF. 1978. Studies on the cellular toxicity of polychlorinated biphenyls (PCBs). I. Effects of PCBs on microsomal enzymes and on synthesis and turnover of microsomal and cytoplasmic lipids of rat liver: a morphological and biochemical study. Virchows Archiv B. Cell Pathology 27:279-306.
Hochstein JR, Aulerich RJ, Bursian SJ. 1988. Acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin to mink. Archives of Environmental Contamination and Toxicology 17:33-37.
Hoffman EC, Reyes H, Chu F-F, Sander F, Conley LH, Brooks BA, Hankinson O. 1991. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 252:954-958.
Hoglen N, Swim A, Roberston L, Shedlofsky S. 1992. Effects of xenobiotics on serum tumor necrosis factor (TNF) and interleukin 6 (IL-6) release after LPS in rats. Toxicologist 12:290.
Holladay SD, Lindstrom P, Blaylock BL, Comment CE, Germolec DR, Heindell JJ, Luster MI. 1991. Perinatal thymocyte antigen expression and postnatal immune development altered by gestational exposure to tetrachlorodibenzo-p-dioxin (TCDD). Teratology 44:385-393.
Holsapple MP, Dooley RK, McNerney PJ, McCay JA. 1986. Direct suppression of antibody responses by chlorinated dibenzodioxins in cultured spleen cells from (C57BL/6 × C3H)F1 and DBA/2 mice. Immunopharmacology 12:175-186.
Holsapple MP, Morris DL, Wood SC, Snyder NK. 1991a. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: possible mechanisms. Annual Review of Pharmacology and Toxicology 31:73-100.
Holsapple MP, Snyder NK, Wood SC, Morris DL. 1991b. A review of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: 1991 update. Toxicology 69:219-255.
Holson JF, Gaines TB, Nelson CJ, LaBorde JB, Gaylor DW, Sheehan DM, Young JF. 1992. Developmental toxicity of 2,4,5-trichlorophenoxyacetic acid (2,4,5-T): I. Multireplicated dose-response studies in four inbred strains and one outbred stock of mice. Fundamental and Applied Toxicology 19:286-297.
Hong R, Taylor K, Abonour R. 1989. Immune abnormalities associated with chronic TCDD exposure in rhesus. Chemosphere 18:313-320.
Hood RD. 1985. Cacodylic Acid: Agricultural Uses, Biological Effects, and Environmental Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
Hood RD, Harrison WP, Vedel GC. 1982. Evaluation of arsenic metabolites for prenatal effects in the hamster. Bulletin of Environmental Contamination and Toxicology 29:679-687.
Hooker GER, Haseman JK, Lucier GW. 1975. Induction and suppression of hepatic and extrahepatic microsomal foreign compounds metabolizing enzyme systems by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemico-Biological Interactions 10:199-214.
Hori S, Obana H, Kashimoto T, Otake T, Nishimura H, Ikegami N, Kunits N, Uda H. 1982. Effect of polychlorinated biphenyls and polychlorinated quaterphenyls in cynomolgus monkey (Macaca fasicularis). Toxicology 24:123-139.
House RV, Lauer LD, Murray MJ, Thomas PT, Ehrlich JP, Burleson GR, Dean JH. 1990. Examination of immune parameters and host resistance mechanisms in B6C3F1 mice following adult exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Journal of Toxicology and Environmental Health 31:203-215.
Hsia MT, Kreamer BL. 1985. Delayed wasting syndrome and alterations of liver gluconeogenic enzymes in rats exposed to the TCDD congener 3,3',4,4'-tetrachloroazoxybenzene. Toxicology Letters 25:247-258.
Hudson LG, Toscano WA Jr, Greenlee WF. 1985. Regulation of epidermal growth factor binding in a human keratinocyte cell line by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 77:251-259.
Huff J. 1982. Carcinogenesis bioassay results from the National Toxicology Program. Environmental Health Perspectives 45:185-198.
Huff J. 1992. 2,3,7,8-TCDD: a potent and complete carcinogen in experimental animals. Chemosphere 25:173-176.
Ibrahim MA, Bond GG, Burke TA, Cole P, Dost FN, Enterline PE, Gough M, Greenberg RS, Halperin WE, McConnell E. 1991. Weight of the evidence on the human carcinogenicity of 2,4-D. Environmental Health Perspectives 96:213-222.
Institute of Laboratory Animal Resources (ILAR). 1979. Histologic typing of liver tumors in rats. Journal of the National Cancer Institute 64:179-206.
Innes JR, Ulland BM, Valerio MG, Petrucelli L, Fishbein L, Hart ER, Pallotta AJ, Bates RR,
Falk HL, Gart JJ, Klein M, Mitchell I, Peters J. 1969. Bioassay of pesticides and industrial chemicals for tumorigenicity in mice: a preliminary note. Journal of the National Cancer Institute 42:1101-1114.
International Agency for Research on Cancer (IARC). 1977. Some Fumigants, the Herbicides 2,4-D and 2,4,5-T, Chlorinated Dibenzodioxins and Miscellaneous Industrial Chemicals. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man, Vol. 15. Lyon: IARC. 111-138, 273-299.
International Agency for Research on Cancer. 1986. Occupational Exposures to Chlorophenols. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Vol. 41. Lyon: IARC.
Jaiswal AK, McBride OW, Adesnik M, Nebert DW. 1988. Human dioxin-inducible cytosolic NAD(P)H:menadione oxidoreductase. cDNA sequence and localization of gene to chromosome 16. Journal of Biological Chemistry 263:13572-13578.
Jenssen D, Renberg L. 1976. Distribution and cytogenetic test of 2,4-D and 2,4,5-T phenoxyacetic acids in mouse blood tissues. Chemico-Biological Interactions 14:291-299.
John-Greene JA, Ouellette JH, Jeffries TK, Johnson KA, Rao KS. 1985. Teratological evaluation of picloram potassium salt in rabbits. Journal of Food and Chemical Toxicology 23:753-756.
Jones EL, Krizek HA. 1962. A technic for testing acnegenic potency in rabbits applied to the potent acnegen 2,3,7,8-tetrachlorodibenzo-p-dioxin. Journal of Investigative Dermatology 39:511-517.
Jones G, Butler WH. 1973. A morphological study of rat liver lesion induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Journal of Pathology 112:93-97.
Jones KG, Sweeney GD. 1977. Association between induction of aryl hydrocarbon hydroxylase and depression of uroporphyrinogen decarboxylase activity. Research Communications in Chemical Pathology and Pharmacology 17:631-637.
Jones KG, Sweeney GD. 1980. Dependence of the porphyrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin upon inheritance of aryl hydrocarbon hydroxylase responsiveness. Toxicology and Applied Pharmacology 53:42-49.
Jones KW, Whitlock JP Jr. 1990. Functional analysis of the transcriptional promoter for the CYP1A1 gene. Molecular and Cellular Biology 10:5098-5105.
Jones MK, Weisenburger WP, Sipes IG, Russell DH. 1987. Circadian alterations in prolactin, corticosterone, and thyroid hormone levels and down-regulation of prolactin receptor activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 87:337-350.
Jones PB, Durrin LK, Galeazzi DR, Whitlock JP Jr. 1986. Control of cytochrome P1-450 gene expression: analysis of a dioxin-responsive enhancer system . Proceedings of the National Academy of Sciences (USA) 83:2802-2806.
Jotz M, Mitchell AD. 1980. An Evaluation of Mutagenic Potential of Cacodylic Acid Employing the L5178y TK Mouse Lymphoma Assay. SRI Project No. LSU 7558. Report to the U.S. EPA. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
Kavlock RJ, Chernoff N, Rogers EH. 1985. The effect of acute maternal toxicity on fetal development in the mouse. Teratogenesis, Carcinogenesis, and Mutagenesis 5:3-13.
Kedderis LB, Diliberto JJ, Birnbaum LS. 1991. Disposition and excretion of intravenous 2,3,7,8-tetrabromodibenzo-p-dioxin (TBDD) in rats. Toxicology and Applied Pharmacology 108:397-406.
Kelling CK, Christian BJ, Inhorn SL, Peterson RE. 1985. Hypophagia induced weight loss in mice, rats, and guinea pigs treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundamental and Applied Toxicology 5:700-712.
Kelling CK, Menahan LA, Peterson RE. 1987. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment on mechanical function of the rat heart. Toxicology and Applied Pharmacology 91:497-501.
Kerkvliet NI. 1984. Halogenated aromatic hydrocarbons (HAH) as immunotoxicants. In: Kende M, Gainer J, Chirigos ME, eds. Chemical Regulation of Immunity in Veterinary Medicine. New York: Alan R. Liss. 369-387.
Kerkvliet NI, Baecher-Steppan L. 1988. Suppression of allograft immunity by 3,4,5,3',4',5'-hexachlorobiphenyl. I. Effects of exposure on tumor rejection and cytotoxi T cell activity. Immunopharmacology 16:1-12.
Kerkvliet NI, Brauner JA. 1990. Functional analysis of antigen presenting cells following antigen challenge: influence of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicologist 10:289.
Kerkvliet NI, Oughton JA. 1993. Acute inflammatory response to sheep red blood cell challenge in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): phenotypic and functional analysis of peritoneal exudate cells. Toxicology and Applied Pharmacology 119:248-257.
Kerkvliet NI, Brauner JA, Matlock JP. 1985. Humoral immunotoxicity of polychlorinated diphenyl ethers, phenoxyphenols, dioxins and furans present as contaminants of technical grade pentachlorophenol. Toxicology 36:307-324.
Kerkvliet NI, Baecher-Steppan L, Smith BB, Youngberg JA, Henderson MC, Buhler DR. 1990a. Role of the Ah locus in suppression of cytotoxic T lymphocyte activity by halogenated aromatic hydrocarbons (PCBs and TCDD): structure-activity relationships and effects in C57B1/6 mice congenic at the Ah locus. Fundamental and Applied Toxicology 14:532-541.
Kerkvliet NI, Steppan LB, Brauner JA, Deyo JA, Henderson MC, Tomar RS, Buhler DR. 1990b. Influence of the Ah locus on the humoral immunotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: evidence for Ah receptor-dependent and Ah receptor-independent mechanisms of immunosuppression. Toxicology and Applied Pharmacology 105:26-36.
Kester JE, Gasiewicz TA. 1987. Characterization of the in vitro stability of the rat hepatic receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Archives of Biochemistry and Biophysics 252:606-625.
Khalatkar AS, Bhargara YR. 1982. 2,4-Dichlorophenoxyacetic acid: a new environmental mutagen. Mutation Research 103:111-114.
Khera KS, Ruddick JA. 1973. Polychlorodibenzo-p-dioxins: Perinatal effects and the dominant lethal test in Wistar rats. In: Blair EH, ed. Chlorodioxins: Origin and Fate. Washington, DC: American Chemical Society. 70-84.
Kim CS, Keizer RF, Pritchard JB. 1988. 2,4-Dichlorophenoxyacetic acid intoxication increases its accumulation within the brain. Brain Research 440:216-226.
Kimbrough RD. 1974. The toxicity of polychlorinated polycyclic compounds and related chemicals. CRC Critical Reviews in Toxicology 2:445-498.
Kirkhart B. 1980. Micronucleus Test on Cacodylic Acid. SRI Project No. LSU 7558-19. Report to the U.S. EPA. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
Kleeman JM, Olson JR, Peterson RE. 1988. Species differences in 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and biotransformation in fish. Fundamental and Applied Toxicology 10:206-213.
Kleeman JM, Moore RW, Peterson RE. 1990. Inhibition of testicular steroidogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats: evidence that the key lesion occurs prior to or during pregnenolone formation. Toxicology and Applied Pharmacology 106:112-125.
Knopp D, Schiller F. 1992. Oral and dermal application of 2,4-dichlorophenoxyacetic acid
sodium and dimethylamine salts to male rats: investigations on absorption and excretion as well as induction of hepatic mixed-function oxidase activities. Archives of Toxicology 66:170-174.
Knutson JC, Poland A. 1982. Response of murine epidermis to 2,3,7,8-tetrachlorodibenzo-p-dioxin: interaction of the Ah and hr loci. Cell 30:225-234.
Kociba RJ, Schwetz BA. 1982. Toxicity of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD). Drug Metabolism and Disposition 13:387-406.
Kociba RJ, Keeler PA, Park, Gehring PJ. 1976. 2,3,7,8-tetrachlorodibenzo-p-dioxin: results from a 13-week oral toxicity study in rats. Toxicology and Applied Pharmacology 35:553-574.
Kociba RJ, Keys DG, Beyer JE, Careon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnar SD, Hummel RA, Humiston CG. 1978. Results of a two year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicology and Applied Pharmacology 46:279-303.
Kociba RJ, Keyes DG, Lisowe RW, Kalnins RP, Dittenber DD, Wade CE, Gorzinski SJ, Mahle NH, Schwetz BA. 1979. Results of a two year chronic toxicity and oncogenic study of rats ingesting diets containing 2,4,5-trichlorophenoxyacetic acid (2,4,5-T). Food and Cosmetics Toxicology 17:205-221.
Kohli JD, Khanna RN, Gupta BN, Dhar MM, Tandon JS, Sircar KP. 1974. Absorption and excretion of 2,4,5-trichlorophenoxy acetic acid in man. Archives Internationales de Pharmacodynamie et de Therapie 210:250-255.
Kozuka H, Yamada J, Horie S, Watanabe T, Suga T, Ikeda T. 1991. Characteristics of induction of peroxisomal fatty acid oxidation-related enzymes in rat liver by drugs. Relationships between structure and inducting activity. Biochemical Pharmacology 41:617-623.
Kurl RN, Chaudhary KC, Villee CA. 1986. Characterization and control of cytosolic binding proteins for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat lung. Pharmacology 33:181-189.
Lakshman MR, Campbell BS, Chirtel SJ, Ekarohita N, Ezekiel M. 1986. Studies on the mechanism of absorption and distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Journal of Pharmacology and Experimental Therapeutics 239:673-677.
Leng ML, Ramsey JC, Braun WH, Lavy TL. 1982. Review of studies with 2,4,5-trichlorophenoxyacetic acid in humans including applicators under field conditions. In: Plimmer JR, ed. Pesticide Residues and Exposure. ACS Symposia Series 182:133-156.
Leung HW, Poland A, Paustenbach DJ, Murray FJ, Andersen ME. 1990. Pharmacokinetics of (125I)-2-iodo-3,7,8-trichlorodibenzo-p-dioxin in mice: analysis with a physiological modeling approach. Toxicology and Applied Pharmacology 103:411-419.
Li JJ, Li SA. 1990. Estrogen carcinogenesis in hamster tissues: a critical review. Endocrine Reviews 11:524-531.
Lilienfeld DE, Gallo MA. 1989. 2,4-D, 2,4,5-T, and 2,3,7,8-TCDD: an overview. Epidemiologic Review 11:28-58.
Lin FH, Clark G, Birnbaum LS, Lucier GW, Goldstein JA. 1991a. Influence of the Ah locus on the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the hepatic epidermal growth factor receptor. Molecular Pharmacology 39:307-313.
Lin FH, Stohs SJ, Birnbaum LS, Clark G, Lucier GW, Goldstein JA. 1991b. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the hepatic estrogen and glucocorticoid receptors in congenic strains of Ah-responsive and Ah-nonresponsive C57BL/6J mice. Toxicology and Applied Pharmacology 108:129-139.
Linden J, Pohjanvirta R, Rahko T, Tuomisto J. 1991. TCDD decreases rapidly and persistently serum melatonin concentration without morphologically affecting the pineal gland in TCDD-resistant Han/Wistar rats. Pharmacology and Toxicology 69:427-432.
Linnainmaa K. 1983. Sister chromatid exchanges among workers occupationally exposed to
phenoxy acid herbicides 2,4-D and MCPA. Teratogenesis, Carcinogenesis, and Mutagenesis 3:269-279.
Linnainmaa K. 1984. Induction of sister chromatid exchanges by the peroxisome proliferators 2,4-D, MCPA, and clofibrate in vivo and in vitro. Carcinogenesis 5:703-707.
Loose LD, Silkworth JB, Mudzinski SP, Pittman KA, Benitz KF, Mueller W. 1979. Environmental chemical-induced immune dysfunction. Ecotoxicology and Environmental Safety 2:173-198.
Lorenzen A, Okey AB. 1991. Detection and characterization of Ah receptor in tissue and cells from human tonsils. Toxicology and Applied Pharmacology 107:203-214.
Lucier GW, Rumbaugh RC, McCoy Z, Hass R, Harvan D, Albro P. 1986. Ingestion of soil contaminated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters hepatic enzyme activities in rats. Fundamental and Applied Toxicology 6:364-371.
Lucier GW, Tritscher A, Goldsworthy T, Foley J, Clark G, Goldstein J, Maronpot R. 1991. Ovarian hormones enhance 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated increases in cell proliferation and preneoplastic foci in a two-stage model for rat hepatocarcinogenesis. Cancer Research 51:1391-1397.
Lucier GW, Clark G, Tritscher A, Foley J, Maronpot R. 1992. Mechanisms of dioxin tumor promotion: implications for risk assessment. Chemosphere 25:177-180.
Lukowicz-Ratajczak J, Krechniak J. 1988. Effects of sodium 2,4-dichlorophenoxyacetate on renal function in the rat. Bulletin of Environmental Contamination and Toxicology 41:815-821.
Luster MI, Boorman GA, Harris MW, Moore JA. 1980a. Laboratory studies on polybrominated biphenyl-induced immune alterations following low-level chronic and pre/postnatal exposure. International Journal of Immunopharmacology 2:69-80.
Luster MI, Boorman GA, Dean JH, Harris MW, Luebke RW, Padarathsingh ML, Moore JA. 1980b. Examination of bone marrow, immunologic parameters and host susceptibility following pre- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). International Journal of Immunopharmacology 2:301-310.
Luster MI, Germolec DR, Clark G, Wiegand G, Rosenthal GJ. 1988. Selective effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and corticosteroid on in vitro lymphocyte maturation. Journal of Immunology 140:928-935.
Mably TA, Theobald HM, Ingall GB, Peterson RE. 1990. Hypergastrinemia is associated with decreased gastric acid secretion in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Toxicology and Applied Pharmacology 106:518-528.
Mably TA, Moore RW, Peterson RE. 1992a. In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. 1. Effects on androgenic status. Toxicology and Applied Pharmacology 114:97-107.
Mably TA, Moore RW, Goy RW, Peterson RE. 1992b. In utero and lactational exposure of male rats to 2,3,7,8-tetrachlorodibenzo-p-dioxin. 2. Effects on sexual behavior and the regulation of luteinizing hormone secretion in adulthood. Toxicology and Applied Pharmacology 114:108-117.
Madhukar BV, Ebner K, Matsumura F, Bombick DW, Brewster DW, Kawamoto T. 1988. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes an increase in protein kinases associated with epidermal growth factor receptor in the hepatic plasma membrane. Journal of Biochemical Toxicology 3:261-277.
Majumdar SK, Hall RC. 1973. Cytogenetic effects of 2,4,5-T on in vivo bone marrow cells of Mongolian gerbils. Journal of Heredity 64:213-216.
Manchester DK, Gordon SK, Golas CL, Roberts EA, Okey AB. 1987. Ah receptor in human placenta: stabilization by molybdate and characterization of binding of 2,3,7,8-TCDD, 3-methyl-cholanthrene, and benzo[a]pyrene. Cancer Research 47:486-488.
Mantovani A, Vecchi A, Luini W, Sironi M, Candiani GP, Spreafico F, Garattini S. 1980.
Effect of 2,3,7,8-tetrachlorodibienzo-p-dioxin on macrophage and natural killer cell-mediated cytotoxicity in mice. Biomedicine 32:200-204.
Marafante E, Vahter M, Norin H, Envall J, Sandstrom M, Christakopoulos A, Ryhange R. 1987. Biotransformation of dimethylarsinic acid in mouse, hamster and man. Journal of Applied Toxicology 7:111-117.
Maronpot RR, Montgomery CA, Boorman GA, McConnell EE. 1986. National Toxicology Program nomenclature for hepatoproliferative lesions of rats. Toxicologic Pathology 14:263-273.
Marti M, Burwen S, Jones A. 1989. Biological effects of epidermal growth factor, with emphasis on the gastrointestinal tract and liver: an update. Hepatology 9:126-139.
Mason G, Safe S. 1986. Synthesis, biologic and toxic effects of the major 2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites in the rat. Toxicology 41:153-159.
Mattsson JL, Johnson KA, Albee RR. 1986. Lack of neuropathologic consequences of repeated dermal exposure to 2,4-dichlorophenoxyacetic acid in rats. Fundamental and Applied Toxicology 6:175-181.
McConnell EE. 1985. Comparative toxicity of PCBs and related compounds in various species of animals. Environmental Health Perspectives 60:29-33.
McConnell EE, Moore JA. 1979. Toxico-pathology characteristics of the halogenated aromatics. Annals of the New York Academy of Sciences 320:138-150.
McConnell EE, Moore JA, Haseman JK, Harris MW. 1978a. The comparative toxicity of chlorinated dibenzo-p-dioxins in mice and guinea pigs. Toxicology and Applied Pharmacology 44:335-356.
McConnell EE, Moore JA, Dalgard DW. 1978b. Toxicity of 2,3,7,8-tetrachlorodibenzop-dioxin in rhesus monkeys (Macaca mulatta) following a single oral dose. Toxicology and Applied Pharmacology 43:175-187.
McNulty WP. 1984. Fetotoxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) for rhesus macaques (Macaca mulatta). American Journal of Primatology 6:41-47.
Metzler M. 1984. Metabolism of stilbene estrogens and steroidal estrogens in relation to carcinogenicity. Archives of Toxicology 55:104-109.
Miller AG, Israel D, Whitlock JP Jr. 1983. Biochemical and genetic analysis of variant mouse hepatoma cells defective in the induction of benzo(a)pyrene-metabolizing enzyme activity. Journal of Biological Chemistry 258:3523-3527.
Miller EC, Miller JA, Brown RR, MacDonald JC. 1958. On the protective action of certain polycyclic aromatic hydrocarbons against carcinogenesis by aminoazo dyes and 2-acetylaminofluorene. Cancer Research 18:469.
Mirvish SS, Nickols J, Weisenburger D, Joshi SS, Kaplan P, Gross M, Tong HY. 1991. Effects of 2,4,5-trichlorophenoxyacetic acid, pentachlorophenol, methylprednisolone, and Freund adjuvant on 2-hydroxyethylnitrosourea carcinogenesis in MRC-Wistar rats. Journal of Toxicology and Environmental Health 32:59-74.
Mohammad FK, St Omer VEV. 1986. Behavioral and developmental effects in rats following in utero exposure to 2,4-D/2,4,5-T mixture. Neurobehavioral Toxicology and Teratology 8:551-560.
Molloy CJ, Gallo MA, Laskin JD. 1987. Alterations in the expression of specific epidermal keratin markers in the hairless mouse by the topical application of the tumor promoters 2,3,7,8-tetrachlorodibenzop-dioxin and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate. Carcinogenesis 8:1193-1199.
Moore RW, Potter CL, Theobald HM, Robinson JA, Peterson RE. 1985. Androgenic deficiency in male rats treated with 2,3,7,8-tetrachlorodibenzop-dioxin. Toxicology and Applied Pharmacology 79:99-111.
Moore RW, Parsons JA, Bookstaff RC, Peterson RE. 1989. Plasma concentrations of pituitary
hormones in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated male rats. Journal of Biochemical Toxicology 4:165-172.
Moore RW, Jefcoate CR, Peterson RE. 1991. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits steroidogenesis in the rat testis by inhibiting the mobilization of cholesterol to cytochrome P450(SCC). Toxicology and Applied Pharmacology 109:85-97.
Moore RW, Bookstaff RC, Mably TA, Peterson RE. 1992. Differential effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on responsiveness of male rats to androgens, estradiol, luteinizing hormone, gonadotropin-releasing hormone, and progesterone. Chemosphere 25:91-94.
Morley JE, Levine AS. 1985. The pharmacology of eating behavior. Annual Review of Pharmacology and Toxicology 25:127-146.
Morris DL, Jordan SD, Holsapple MP. 1991. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on humoral immunity. I. Similarities to Staphylococcus aureus cowan strain i (sac) in the in vitro T-dependent antibody response. Immunopharmacology 21:159-169.
Morris DL, Snyder NK, Gokani V, Blair RE, Holsapple MP. 1992. Enhanced suppression of humoral immunity in DBA/2 mice following subchronic exposure to 2,3,7,8-tetracholorodibenzo-p-dioxin (TCDD). Toxicology and Applied Pharmacology 112:128-132.
Morrisey RE, Schwetz BA. 1989. Reproductive and developmental toxicity in animals. In: Kimbrough RD, Jensen AA, eds. Halogenated Biphenyls, Terphenyls, Naphthalenes, Dibenzodioxins and Related Products. 2nd ed. Amsterdam: Elsevier. 195-225.
Mortelmans K, Haworth S, Speck W, Zeiger E. 1984. Mutagenicity testing of Agent Orange components and related chemicals. Toxicology and Applied Pharmacology 75:137-146.
Mullison WR. 1986. An Interim Report Summarizing 2,4-D Toxicological Research Sponsored by the Industry Task Force on 2,4-D Research Data and A Brief Review of 2,4-D Environmental Effects.
Munro IC, Carlo GL, Orr JC, Sund KG, Wilson RM, Kennepohl E, Lynch BS, Jablinske M, Lee NL. 1992. A comprehensive, integrated review and evaluation of the scientific evidence relating to the safety of the herbicide 2,4-D. Journal of the American College of Toxicology 11:559-664.
Muranyi-Kovacs I, Rudali G, Imbert J. 1976. Bioassay of 2,4,5-trichlorophenoxyacetic acid for carcinogenicity in mice. British Journal of Cancer 33:626-633.
Murray FJ, Smith FA, Nitschke KD, Humiston CG, Kociba RJ, Schwetz BA. 1979. Three-generation reproduction study of rats given 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the diet. Toxicology and Applied Pharmacology 50:241-252.
Mustonen R, Kangas J, Vuojolahti P, Linnainmaa K. 1986. Effects of phenoxyacetic acids on the induction of chromosome aberrations in vitro and in vivo. Mutagenesis 1:241-245.
Mustonen R, Elovaara E, Zitting A, Linnainmaa K, Vainio H. 1989. Effects of commercial chlorophenolate, 2,3,7,8-TCDD, and pure phenoxyacetic acids on hepatic peroxisome proliferation, xenobiotic metabolism and sister chromatid exchange in the rat. Archives of Toxicology 63:203-208.
Muzi G, Gorski JR, Rozman K. 1987. Composition of diet modifies toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in cold adapted rats. Archives of Toxicology 61:34-39.
Muzi G, Gorski JR, Rozman K. 1989. Mode of metabolism is altered in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicology Letters 47:77-86.
National Cancer Institute (NCI). 1978. Bioassay of Picloram for Possible Carcinogenicity. Report Series No. 23. Bethesda, MD: NCI.
National Toxicology Program (NTP). 1982a. Carcinogenesis Bioassay of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (CAS No. 1746-01-6) in Osborne-Mendel Rats and B6C3F1 Mice (Gavage Study). Research Triangle Park, NC. NTP-80-31; NIH/PUB-82-1765 1746-01-6. 197 pp.
National Toxicology Program. 1982b. Carcinogenesis Bioassay of 2,3,7,8-Tetrachlorodibenzo-p-dioxin
(CAS No. 1746-01-6) in Swiss-Webster Mice (Dermal Study). Research Triangle Park, NC. NTP-80-32; NIH/PUB-82-1757. 115 pp.
National Toxicology Program. 1984. Report of the NTP Ad Hoc Panel on Chemical Carcinogenesis Testing and Evaluation. Research Triangle Park, NC: U.S. Department of Health and Human Services.
Neal RA, Beatty PW, Gasiewicz TA. 1979. Studies of the mechanisms of toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Annals of the New York Academy of Sciences 320:204-213.
Neal RA, Olson JR, Gasiewicz TA, Geiger LE. 1982. The toxicokinetics of 2,3,7,8-tetrachlorodibenzo-p-dioxin in mammalian systems. Drug Metabolism Review 13:355-385.
Nebert DW. 1989. The Ah locus: genetic differences in toxicity, cancer, mutation, and birth defects. Critical Reviews in Toxicology 20:153-174.
Nebert DW, Petersen DD, Puga A. 1991. Human Ah locus polymorphism and cancer: inducibility of CYP1A1 and other genes by combustion products and dioxin. Pharmacogenetics 1:68-78.
Nelson CJ, Holson JF, Gaines TB, LaBorde JB, McCallum WF, Wolff GL, Sheehan DM, Young JF. 1992. Developmental toxicity of 2,4,5-trichlorophenoxyacetic acid: II. Multireplicated dose-response studies with technical and analytical grades of 2,4,5-T in four-way outcross males. Fundamental and Applied Toxicology 19:298-306.
Nemoto T, Mason GG, Wilhelmsson A, Cuthill S, Hapgood J, Gustafsson JA, Poellinger L. 1990. Activation of the dioxin and glucocorticoid receptors to a DNA-binding state under cell-free conditions. Journal of Biological Chemistry 265:2269-2277.
Neubert D, Dillmann I. 1972. Embryotoxic effects in mice treated with 2,4,5-trichlorophenoxyacetic acid and 2,3,7,8-tetrachlorodibenzo-p-dioxin. Naunyn-Schmiedeberg's Archiv fur Experimentelle Pathologie und Pharmakologie 272:243-264.
Neubert D, Wiesmuller T, Abraham K, Krowke R, Hagenmaier H. 1990a. Persistence of various polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDDs and PCDFs) in hepatic and adipose tissue of marmoset monkeys. Archives of Toxicology 64:431-442.
Neubert R, Jacob-Muller U, Stahlmann R, Helge H, Neubert D. 1990b. Polyhalogenated dibenzo-p-dioxins and dibenzofurans and the immune system. 1. Effects on peripheral lymphocyte subpopulations of a nonhuman primate (Callithrix jacchus) after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Archives of Toxicology 64:345-359.
Neubert R, Golor G, Stahlmann R, Helge H, Neubert D. 1992. Polyhalogenated dibenzo-p-dioxins and dibenzofurans and the immune system. 4. Effects of multiple-dose treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on peripheral lymphocyte subpopulations of a nonhuman primate (Callithrix jacchus). Archives of Toxicology 66:250-259.
Neuhold LA, Gonzalez FJ, Jaiswal AK, Nebert DW. 1986. Dioxin-inducible enhancer region upstream from the mouse P(1)450 gene and interaction with a heterologous SV40 promoter. DNA 5:403-411.
Neuhold LA, Shirayoshi Y, Ozato K, Jones JE, Nebert DW. 1989. Regulation of mouse CYP1A1 gene expression by dioxin: requirement of two cis-acting elements during induction. Molecular and Cellular Biology 9:2378-2386.
Nielsen K, Kaempe B, Jensen-Holm J. 1965. Fatal poisoning in man by 2,4-D. Acta Pharmacologica et Toxicologica 22:224-234.
Norback DH, Allen JR. 1973. Biological responses of the nonhuman primate, chicken, and rat to chlorinated dibenzo-p-dioxin ingestion. Environmental Health Perspectives 5:233-240.
Olson JR. 1986. Metabolism and disposition of 2,3,7,8-tetrachlorodibenzo-p-dioxin in guinea pigs. Toxicology and Applied Pharmacology 85:263-273.
Olson JR, Gasiewicz TA, Neal RA. 1980a. Tissue distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the golden Syrian hamster. Toxicology and Applied Pharmacology 56:78-85.
Olson JR, Holscher MA, Neal RA. 1980b. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the golden Syrian hamster. Toxicology and Applied Pharmacology 55:67-78.
Olson JR, Gasiewicz TA, Geiger LE, Neal RA. 1983. The metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in mammalian systems. In: Coulston P, Pocchiari F, eds. Accidental Exposure to Dioxins: Human Health Aspects. New York: Academic Press. 81-100.
Overby LH, Nishio S, Weir A, Carver GT, Plopper CS, Philpot RM. 1992. Distribution of cytochrome P-450 1A1 and NADPH-cytochrome P-450 reductase in lungs of rabbits treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin: ultrastructural immunolocalization in in situ hybridization. Molecular Pharmacology 41:1039-1046.
PATHCO. 1990a. Pathology Working Group Report. 2,3,7,8-Tetrachlorodibenzo-p-dioxin in Sprague-Dawley Rats. Ijamsville, MD. Submitted to the Maine Scientific Advisory Panel.
PATHCO. 1990b. Hepatotoxicity in Female Sprague-Dawley Rats Treated with 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Ijamsville, MD: PATHCO.
Pavlica M, Papes D, Nagy B. 1991. 2,4-Dichlorophenoxyacetic acid causes chromatin and chromosome abnormalities in plant cells and mutation in cultured mammalian cells. Mutation Research 263:77-81.
Peoples SA, Maddy KT, Peifer WR, Edmiston S. 1979. Occupational exposures to pesticides containing organoarsenicals in California. Veterinary and Human Toxicology 21:417-421.
Piper WN, Rose JQ, Gehring PJ. 1973a. Excretion and tissue distribution of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the rat. Environmental Health Perspectives 5:241-244.
Piper WN, Rose JQ, Leng ML, Gehring PJ. 1973b. The fate of 2,4,5-trichlorophenoxyacetic acid (2,4,5-T) following oral administration to rats and dogs. Toxicology and Applied Pharmacology 26:339-351.
Pitot HC, Goldsworthy T, Campbell HA, Poland A. 1980. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethylnitrosamine. Cancer Research 40:3616-3620.
Poellinger L, Wilhelmsson A, Cuthill S, Pongratz I, Mason GGF, Gustafsson J-A. 1991. Modulation of dioxin receptor function by protein-protein interaction. In: Gallo MA, Scheuplein RJ, van der Heijden KA, eds. Biological Basis for Risk Assessment of Dioxins and Related Compounds. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 35. 311-320.
Pohjanvirta R, Tuomisto J. 1987. Han/Wistar rats are exceptionally resistant to TCDD. II. Archives of Toxicology Supplement 11:344-347.
Pohjanvirta R, Tuomisto J. 1990. Remarkable residual alterations in responses to feeding regulatory challenges in Han/Wistar rats after recovery from the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Food and Chemical Toxicology 28:677-686.
Pohjanvirta R, Juvonen R, Karenlampi S, Raunio H, Tuomisto J. 1988a. Hepatic Ah receptor levels and the effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on hepatic microsomal monooxygenase activities in a TCDD-susceptible and -resistant rat strain. Toxicology and Applied Pharmacology 92:131-140.
Pohjanvirta R, Tuomisto J, Vikkula K. 1988b. Screening of pharmacological agents given peripherally with respect to TCDD-induced wasting syndrome in Long-Evans rats. Pharmacology and Toxicology 63:240-247.
Pohjanvirta R, Tuomisto L, Tuomisto J. 1989. The central nervous system may be involved in TCDD toxicity. Toxicology 58:167-174.
Pohjanvirta R, Vartiainen T, Uusi-Rauva A, Monkkonen J, Tuomisto J. 1990. Tissue distribution, metabolism, and excretion of 14C-TCDD in a TCDD-susceptible and a TCDD-resistant rat strain alpha. Pharmacology and Toxicology 66:93-100.
Poiger H, Buser HR. 1984. The metabolism of TCDD in the dog and rat. In: Poland A,
Kimbrough RD, eds. Biological Mechanisms of Dioxin Action. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 18. 39-48.
Poiger H, Schlatter CH. 1979. Biological degradation of TCDD in rats. Nature 281:706-707.
Poiger H, Schlatter CH. 1980. Influence of solvents and adsorbents on dermal and intestinal absorption of TCDD . Food and Cosmetics Toxicology 18:477-481.
Poiger H, Schlatter C. 1985. Influence of phenobarbital and TCDD on the hepatic metabolism of TCDD in the dog. Experientia 41:376-378.
Poiger H, Buser HR, Weber H, Zweifel U, Schlatter C. 1982. Structure elucidation of mammalian TCDD-metabolites. Experientia 38:484-486.
Poiger H, Pluess N, Buser HR. 1989. The metabolism of select PCDFs in the rat. Chemosphere 18:259-264.
Poland A, Glover E. 1973. Chlorinated dibenzo-p-dioxins: potent inducers of 6-aminolevulinic acid synthetase and aryl hydrocarbon hydroxylase. II. A study of the structure-activity relationship. Molecular Pharmacology 9:736-747.
Poland A, Glover E. 1979. An estimate of the maximum in vivo covalent binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to rat liver protein, ribosomal RNA, and DNA. Cancer Research 39:3341-3344.
Poland A, Glover E. 1980. 2,3,7,8,-Tetrachlorodibenzo-p-dioxin: segregation of toxicity with the Ah locus . Molecular Pharmacology 17:86-94.
Poland A, Knutson JC. 1982. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annual Review of Pharmacology and Toxicology 22:517-554.
Poland A, Greenlee WF, Kende AS. 1979. Studies on the mechanism of action of the chlorinated dibenzo-p-dioxins and related compounds. Annals of the New York Academy of Sciences 320:214-230.
Poland A, Palen D, Glover E. 1982. Tumor promotion by TCDD in skin of HRS/J hairless mice. Nature 300:271-273.
Poland A, Teitelbaum P, Glover E. 1989a. [125I]2-Iodo-3,7,8-trichlorodibenzo-p-dioxin-binding species in mouse liver induced by agonists for the Ah receptor: characterization and identification. Molecular Pharmacology 36:113-120.
Poland A, Teitelbaum P, Glover E, Kende A. 1989b. Stimulation of in vivo hepatic uptake and in vitro hepatic binding of [125I]2-iodo-3,7,8-trichlorodibenzo-p-dioxin by the administration of agonist for the Ah receptor. Molecular Pharmacology 36:121-127.
Poli A, Franceschini G, Puglisi L, Sirtori CR. 1980. Increased total and high-density lipoprotein cholesterol with apoprotein changes resembling streptozotocin diabetes in tetrachlorodibenzodioxin (TCDD) treated rats. Biochemical Pharmacology 29:835-838.
Portier C, Tritscher A, Kohn M, Sewall C, Clark G, Edler L, Hoel D, Lucier G. 1993. Ligand/receptor binding for 2,3,7,8-TCDD: implications for risk assessment. Fundamental and Applied Toxicology 20:48-56.
Potter CL, Sipes IG, Russell DH. 1983. Hypothyroxinemia and hypothermia in rats in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin administration. Toxicology and Applied Pharmacology 69:89-95.
Puhvel SM, Sakamoto M. 1987. Response of murine epidermal keratinocyte cultures to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): comparison of haired and hairless genotypes. Toxicology and Applied Pharmacology 89:29-36.
Puhvel SM, Sakamoto M. 1988. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on murine skin. Journal of Investigative Dermatology 90:354-358.
Puhvel SM, Sakamoto M, Ertl D, Reisner RM. 1982. Hairless mice as models for chloracne: a study of cutaneous changes induced by topical application of established chloracnegens. Toxicology and Applied Pharmacology 64:492-503.
Puhvel SM, Connor MJ, Sakamoto M. 1991. Vitamin A deficiency and the induction of cutaneous
toxicity in murine skin by TCDD. Toxicology and Applied Pharmacology 107:106-116.
Quattrochi LC, Tukey RH. 1989. The human cytochrome Cyp1A2 gene contains regulatory elements responsive to 3-methylcholanthrene. Molecular Pharmacology 36:66-71.
Ramsey JC, Hefner JG, Karbowski RJ, Braun WH, Gehring PJ. 1979. The in vivo biotransformation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat. Toxicology and Applied Pharmacology 48:A162.
Ramsey JC, Hefner JG, Karbowski RJ, Braun WH, Gehring PJ. 1982. The in vivo biotransformation of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat. Toxicology and Applied Pharmacology 65:180-184.
Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, Grafstrom AK. 1987. Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. Journal of Biological Chemistry 262:15422-15427.
Rao MS, Subbarao V, Prasad JD, Scarpelli DG. 1988. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the Syrian golden hamster. Carcinogenesis 9:1677-1679.
Rashid KA, Babish JG, Mumma RO. 1984. Testing of 2,4,5-T-amino acid conjugates for mutagenic activity in Salmonella typhimurium strains. Mutation Research 136:217-221.
Richter E, Hunder G, Forth W. 1992. Inhibition of intestinal glucose absorption in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Veterinary and Human Toxicology 34:123-126.
Roberts EA, Johnson KC, Harper PA, Okey AB. 1990. Characterization of the Ah receptor mediating aryl hydrocarbon hydroxylase induction in the human liver cell line hep g2. Archives of Biochemistry and Biophysics 276:442-450.
Roberts EA, Johnson KC, Dippold WG. 1991. Ah receptor mediating induction of cytochrome P450IA1 in a novel continuous human liver cell line (mz-Hep-1). Detection by binding with (3H)2,3,7,8-tetrachlorodibenzo-p-dioxin and relationship to the activity of aryl hydrocarbon hydroxylase. Biochemical Pharmacology 42:521-528.
Rogers EH, Chernoff N, Kavlock RJ. 1981. The teratogenic potential of cacodylic acid in the rat and mouse. Drug and Chemical Toxicology 4:49-61.
Rogers L. 1983. Herbicides and the development of brain and behavior: a study in behavioral toxicology. In: Kidman AD, Morris TJ, Cooper CA, eds. Molecular Pathology of Nerve and Muscle: Noxious Agents and Genetic Lesions. Clifton, NJ: Humana Press.
Roll R. 1971. The teratogenic effects of 2,4,5-T in mice. Food and Cosmetics Toxicology 9:671-676. [In German]
Romkes M, Piskorska-Pliszczynska J, Safe S. 1987. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on hepatic and uterine estrogen receptor levels in rats. Toxicology and Applied Pharmacology 87:306-314.
Rose JQ, Ransy JC, Wentzler TH, Hummel RA, Gehring PJ. 1976. The fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin following single and repeated oral doses to the rat. Toxicology and Applied Pharmacology 36:209-226.
Rosin DL, Martin BR. 1981. Neurochemical and behavioral effects of polychlorinated biphenyls in mice. Neurotoxicology 2:749-764.
Rowe VK, Hymas TA. 1954. Summary of toxicological information on 2,4-D and 2,4,5-T type herbicides and an evaluation of the hazards to livestock associated with their use. American Journal of Veterinary Research 15:622-629.
Rozman K, Greim H. 1986. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in cold-adapted rats. Archives of Toxicology 59:211-215.
Rozman K, Rozman T, Greim H. 1984. Effect of thyroidectomy and thyroxine on 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induced toxicity. Toxicology and Applied Pharmacology 72:372-376.
Rozman K, Pfeifer B, Kerecsen L, Alper RH. 1991. Is a serotonergic mechanism involved in
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced appetite suppression in the rat? Archives of Toxicology 65:124-128.
Rozman K, Lebofsky M, Stahl BU, Weber LWD. 1992. The role of insulin and corticosterone in the toxicity of dioxins. Chemosphere 25:79-82.
Rozman K, Roth WL, Greim H, Stahl BU, Doull J. 1993. Relative potency of chlorinated dibenzo-p-dioxins in acute, subchronic and chronic (carcinogenicity) toxicity studies: implications for risk assessment of chemical mixtures. Toxicology 77:39-50.
Rune GM, Desouza P, Krowke R, Merker HJ, Neubert D. 1991a. Morphological and histochemical effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on marmoset (Callithrix jacchus) testes. Archives of Andrology 26:143-154.
Rune GM, Desouza P, Krowke R, Merker HJ, Neubert D. 1991b. Morphological and histochemical pattern of response in rat testes after administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Histology and Histopathology 6:459-467.
Russell DH, Buckley AR, Shah GN, Sipes IG, Blask DE, Benson B. 1988. Hypothalamic site of action of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 94:496-502.
Ryan RP, Sunahara GI, Lucier GW, Birnbaum LS, Nelson KG. 1989. Decreased ligand binding to the hepatic glucocorticoid and epidermal growth factor receptors after 2,3,4,7,8-pentachlorodibenzofuran and 1,2,3,4,7,8-hexachlorodibenzofuran treatment of pregnant mice. Toxicology and Applied Pharmacology 98:454-464.
Saatcioglu F, Perry DJ, Pasco DS, Fagan JB. 1990a. Multiple DNA-binding factors interact with overlapping specificities at the aryl hydrocarbon response element of the cytochrome P450IA1 gene. Molecular and Cellular Biology 10:6408-6416.
Saatcioglu F, Perry DJ, Pasco DS, Fagan JB. 1990b. Aryl hydrocarbon (Ah) receptor DNA-binding activity. Sequence specificity and Zn2+ requirement. Journal of Biological Chemistry 265:9251-9258.
Safe SH. 1986. Comparative toxicology and mechanism of action of polychlorinated dibenzo-p-dioxins and dibenzofurans. Annual Reviews of Pharmacology and Toxicology 26:371-399.
Safe S. 1990. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs). Critical Reviews in Toxicology 21:51-88.
Safe S, Astroff B, Harris M, Zacharewski T, Dickerson R, Romkes M, Biegel L. 1991. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and related compounds as antioestrogens: characterization and mechanism of action. Pharmacology and Toxicology 69:400-409.
Sanderson CA, Rogers LJ. 1981. 2,4,5-Trichlorophenoxyacetic acid causes behavioral effects in chickens at environmentally relevant doses. Science 211:593-595.
Sauerhoff MW, Braun WH, Blau GE, Gehring PJ. 1977. The fate of 2,4-dichlorophenoxyacetic acid (2,4-D) following oral administration to man. Toxicology 8:3-11.
Sawahata T, Olson JR, Neal RA. 1982. Identification of metabolites of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) formed on incubation with isolated rat hepatocytes. Biochemical and Biophysical Research Communications 105:341-346.
Schantz SL, Bowman RE. 1989. Learning in monkeys exposed perinatally to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Neurotoxicology and Teratology 11:13-19.
Schantz SL, Barsotti DA, Allen JR. 1979. Toxicological effects produced in nonhuman primates chronically exposed to fifty parts per trillion 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicology and Applied Pharmacology 48:A180.
Schantz SL, Laughlin NK, Van Valkenberg HC, Bowman RE. 1986. Maternal care by rhesus monkeys of infant monkeys exposed to either lead or 2,3,7,8-tetrachlorodibenzo-p-dioxin. Neurotoxicology 7:637-650.
Schop RN, Hardy MH, Goldberg MT. 1990. Comparison of the activity of topically applied pesticides and the herbicide 2,4-D in two short-term in vivo assays of genotoxicity in the mouse. Fundamental and Applied Toxicology 15:666-675.
Schulze GE. 1987. Formulation and food deprivation affects 2,4-D neurobehavioral toxicity in rats. Neurotoxicology and Teratology 9:363-367.
Schulze GE, Dougherty JA. 1988. Neurobehavioral toxicity and tolerance to the herbicide 2,4-dichlorophenoxyacetic acid-n-butyl ester (2,4-D ester). Fundamental and Applied Toxicology 10:413-424.
Schwetz BA, Sparschu GL, Gehring PJ. 1971. The effect of 2,4-dichlorophenoxyacetic acid (2,4-D) and esters of 2,4-D on rat embryonal, fetal and neonatal growth and development. Food and Cosmetics Toxicology 9:801-817.
Schwetz BA, Norris JM, Sparschu GL, Rowe UK, Gehring PJ, Emerson JL, Gerbig GC. 1973. Toxicology of chlorinated dibenzo-p-dioxins. Environmental Health Perspectives 5:87-99.
Seefeld MD, Corbett SW, Keesey RE, Peterson RE. 1984. Characterization of the wasting syndrome in rats treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 73:311-322.
Seegal RF, Bush B, Brosch KO. 1985. Polychlorinated biphenyls induce changes in brain norepinephrine concentrations in adult rats. Neurotoxicology 6:13-24.
Seegal RF, Brosch KO, Bush B. 1986. Polychlorinated biphenyls produce regional alterations of dopamine metabolism in rat brain. Toxicology Letters 30:197-202.
Seegal RF, Bush B, Shain W. 1990. Lightly chlorinated ortho-substituted PCB congeners decrease dopamine in nonhuman primate brain and in tissue culture. Toxicology and Applied Pharmacology 106:136-144.
Seidenberg JM, Becker RA. 1987. A summary of the results of 55 chemicals screened for developmental toxicity in mice. Teratogenesis, Carcinogenesis, and Mutagenesis 7:17-28.
Seidenberg JM, Anderson DG, Becker RA. 1986. Validation of an in vivo developmental toxicity screen in the mouse. Teratogenesis, Carcinogenesis, and Mutagenesis 6:361-374.
Sharma RP, Gehring PJ. 1979. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on splenic and lymphocyte transformation in mice after single and repeated exposures. Annals of the New York Academy of Sciences 320:487-497.
Sharma VK, Kaur S. 1990. Contact sensitization by pesticides in farmers. Contact Dermatitis 23:77-80.
Shen ES, Whitlock JP. 1989. The potential role of DNA methylation in the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Journal of Biological Chemistry 264:17754-17758.
Shen ES, Whitlock JP. 1992. Protein-DNA interactions at a dioxin-responsive enhancer: mutational analysis of the DNA-binding site for the liganded Ah receptor. Journal of Biological Chemistry 267:6815-6819.
Shen ES, Gutman SI, Olson JR. 1991. Comparison of 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated hepatotoxicity in C57B1/6J and DBA/2J mice. Journal of Toxicology and Environmental Health 32:367-381.
Shiverick KT, Muther TF. 1983. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) effects on hepatic microsomal steroid metabolism and serum estradiol of pregnant rats. Biochemical Pharmacology 32:991-995.
Shu H, Paustenbach DJ, Murray FJ. 1987. A critical evaluation of the use of mutagenesis, carcinogenesis, and tumor promotion data in a cancer risk assessment of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Regulatory Toxicology and Pharmacology 7:57-88.
Shu H, Teitelbaum AS, Webb AS, Marple L, Brunck B, Dei Rossi D, Murray FJ, Paustenbach D. 1988a. Bioavailability of soil-bound TCDD: dermal bioavailability in the rat. Fundamental and Applied Toxicology 10:335-343.
Shu H. Paustenbach D, Murray FJ, Marple L, Brunck B, Dei Rossi D, Teitelbaum P. 1988b.
Bioavailability of soil-bound TCDD: oral bioavailability in the rat. Fundamental and Applied Toxicology 10:648-654.
Sijm DTHM, Yarechewski AL, Muir DCG, Webster GRB, Seinen W, Opperhuizen A. 1990. Biotransformation and tissue distribution of 1,2,3,7-tetrachlorodibenzo-p-dioxin, 1,2,3,4,7-pentachlorodibenzo-p-dioxin and 2,3,4,7,8-pentachlorodibenzofuran in rainbow trout. Chemosphere 21:845-866.
Silbergeld EK. 1991. Dioxin: receptor-based approaches to risk assessment. In: Gallo MA, Scheuplein RJ, van der Heijden KA, eds. Biological Basis for Risk Assessment of Dioxins and Related Compounds. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 35. 441-458.
Silkworth JB, Grabstein EM. 1982. Polychlorinated biphenyl immunotoxicity: dependence on isomer planarity and the Ah gene complex. Toxicology and Applied Pharmacology 65:109-115.
Sirkka U, Pohjanvirta R, Nieminen SA, Tuomisto J, Ylitalo P. 1992. Acute neurobehavioral effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in Han/Wistar rats. Pharmacology and Toxicology 71:284-288.
Smith FA, Murray FJ, John JA, Nitschke KD, Kociba RJ, Schwetz BA. 1981. Three-generation reproduction study of rats ingesting 2,4,5-trichlorophenoxyacetic acid in the diet. Food and Cosmetics Toxicology 19:41-45.
Sontag JM, Page NP, Saffiotti U. 1976. Guidelines for Carcinogen Bioassay in Small Rodents. Carcinog. Technical Report Series 1. DHEW Publ. (NIH 76-801). Bethesda, MD: National Cancer Institute.
Sparschu Gl, Dunn FL, Lisowe RW, Rowe VK. 1971. Study of the effects of high levels of 2,4,5-trichlorophenoxyacetic acid on fetal development in the rat. Food and Cosmetics Toxicology 9:527-530.
Squibb RE, Tilson HA, Mitchell CL. 1983. Neurobehavioral assessment of 2,4-dichlorophenoxyacetic acid (2,4-D) in rats. Neurobehavioral Toxicology and Teratology 5:331-335.
Squire RA. 1980. Pathological Evaluations of Selected Tissues from the Dow Chemical TCDD and 2,4,5-T Rat Studies. Submitted to the U.S. EPA, Carcinogen Assessment Group, Washington, DC. Under contract no. 68-01-5092.
Squire RA, Levitt MH. 1975. Report of a workshop on classification of specific hepatocellular lesions in rats. Cancer Research 35:3214-3223.
Stahl BU, Rozman K. 1990. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-induced appetite suppression in the Sprague-Dawley rat is not a direct effect on feed intake regulation in the brain. Toxicology and Applied Pharmacology 106:158-162.
Stahl BU, Alper RH, Rozman K. 1991. Depletion of brain serotonin does not alter 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced starvation syndrome in the rat. Toxicology Letters 59:65-72.
Stahl BU, Beer DG, Weber LW, Lebofsky M, Rozman K. 1992. Decreased hepatic phosphoenolpyruvate carboxykinase gene expression after 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment: implications for the acute toxicity of chlorinated dibenzo-p-dioxins in the rat. Archives of Toxicology Supplement 15:151-155.
Stevens JT, Hall LL, Farmer JD, DiPasquale LC, Chernoff N, Durham WF. 1977. Disposition of 14C or 74As-cacodylic acid in rats after intravenous, intratracheal, or peroral administration. Environmental Health Perspectives 19:151-157.
Stevens JT, DiPasquale LC, Farmer JD. 1979. The acute inhalation toxicology of the technical grade organoarsenical herbicides, cacodylic acid and disodium methanearsonic acid: a route comparison. Bulletin of Environmental Contamination and Toxicology 21:304-311.
St. Omer VEV, Mohammad FF. 1987. Ontogeny of swimming behavior and brain catecholamine turnover in rats prenatally exposed to a mixture of 2,4-dichlorophenoxyacetic and 2,4,5-trichlorophenoxyacetic acids. Neuropharmacology 9:1351-1358.
Stoschek C, King L. 1986. Role of epidermal growth factor in carcinogenesis. Cancer Research 46:1030-1037.
Stott WT, Johnson KA, Landry TD, Gorzinski SJ, Cieszlak FS. 1990. Chronic toxicity and oncogenicity of picloram in Fischer 344 rats. Journal of Toxicology and Environmental Health 30:91-104.
Sunahara GI, Lucier GW, McCoy Z, Bresnick EH, Sanchez ER, Nelson KG. 1989. Characterization of 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated decreases in dexamethasone binding to rat hepatic cytosolic glucocorticoid receptor. Molecular Pharmacology 36:239-247.
Sutter TR, Guzman K, Dold KM, Greenlee WF. 1991. Targets for dioxin: genes for plasminogen activator inhibitor-2 and interleukin-1 beta. Science 254:415-418.
Sweeney GD, Jones KG, Cole FM, Basford D, Krestynski F. 1979. Iron deficiency prevents liver toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Science 204:332-335.
Taylor BA. 1984. The aryl hydrocarbon hydroxylase inducibility locus (Ah) of the mouse, a genetic perspective . In: Poland A, Kimbrough RD, eds. Biological Mechanisms of Dioxin Action. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 18. 97-108.
Taylor MJ, Clark GC, Atkins ZZ, Lucier GW, Luster MI. 1990. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases the release of tumor necrosis factor-alpha (TNF-a) and induces ethoxyresorufin-O-deethylase (EROD) activity in rat Kupffer's cells (KCs). Toxicologist 10:276.
Taylor MJ, Lucier GW, Mahler JF, Thompson M, Lockhart AC, Clark GC. 1992. Inhibition of acute TCDD toxicity by treatment with anti-tumor necrosis factor antibody or dexamethasone. Toxicology and Applied Pharmacology 117:126-132.
Telakowski-Hopkins CA, King RG, Pickett CB. 1988. Glutathione S-transferase Ya subunit gene: identification of regulatory elements required for basal level and inducible expression. Proceedings of the National Academy of Sciences (USA) 85:1000-1004.
Theobald HM, Ingall GB, Mably TA, Peterson RE. 1991. Response of the antral mucosa of the rat stomach to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology and Applied Pharmacology 108:167-179.
Thigpen JE, Faith RE, McConnell EE, Moore JA. 1975. Increased susceptibility to bacterial infection as a sequela of exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Infection and Immunity 12:1319-1324.
Thomas PT, Hinsdill RD. 1978. Effect of polychlorinated biphenyls on the immune responses of rhesus monkeys and mice. Toxicology and Applied Pharmacology 44:41-51.
Thomas PT, Hinsdill RD. 1979. The effect of perinatal exposure to tetrachlorodibenzo-p-dioxin on the immune response of young mice. Drug and Chemical Toxicology 2:77-98.
Thompson DJ, Emerson JL, Strebing RJ, Gerbig CG, Robinson VB. 1972. Teratology and postnatal studies on 4-amino-3,5,6-trichloropicolinic acid (picloram) in the rat. Food and Cosmetics Toxicology 10:797-803.
Toth K, Somfai-Relle S, Sugar J, Bence J. 1979. Carcinogenicity testing of herbicide 2,4,5-trichlorophenoxyethanol containing dioxin and of pure dioxin in Swiss mice. Nature 278:548-549.
Travis CC, Hattemer-Frey HA, Silbergeld E. 1989. Dioxin, dioxin everywhere. Environmental Science and Technology 23:1061-1063.
Tritscher AM, Goldstein JA, Portier CJ, McCoy Z, Clark GC, Lucier GW. 1992. Dose-response relationships for chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin in a rat tumor promotion model: quantification and immunolocalization of CYP1A1 and CYP1A2 in the liver. Cancer Research 52:3436-3442.
Truelove J, Grant D, Mes J, Tryphonas H, Tryphonas L, Zawidzka Z. 1982. Polychlorinated biphenyl toxicity in the pregnant cynomolgus monkey: a pilot study. Archives of Environmental Contamination and Toxicology 11:583-588.
Tryphonas H, Hayward S, O'Grady, Loo JC, Arnold DL, Bryce F, Zawidzka ZZ. 1989. Immunotoxicity
studies of PCB (Aroclor 1254) in the adult rhesus (Macaca mulatta) monkey: preliminary report. International Journal of Immunopharmacology 11:199-206.
Tryphonas H, Luster MI, White KL, Naylor PH, Erdos MR, Burleson GR, Germolec D, Hodgen M, Hayward S, Arnold DL. 1991. Effects of PCB (Aroclor 1254) on non-specific immune parameters in rhesus (Macaca mulatta) monkeys. International Journal of Immunopharmacology 13:639-648.
Tucker AN, Vore SJ, Luster MI. 1986. Suppression of B cell differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Molecular Pharmacology 29:372-377.
Tuomisto J, Pohjanvirta R. 1987. The Long-Evans rat: a prototype of an extremely TCDD-susceptible strain variant. Pharmacology and Toxicology 60(Suppl. I):72.
Tuomisto J, Pohjanvirta R, MacDonald E, Tuomisto L. 1990. Changes in rat brain monoamines, monoamine metabolites and histamine after a single administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Pharmacology and Toxicology 67:260-265.
Turkula TE, Jalal SM. 1985. Increased rates of sister chromatid exchanges induced by the herbicide 2,4-D. Journal of Heredity 76:213-214.
Turteltaub KW, Felton JS, Gledhill BL, Vogel JS, Southon JR, Caffee MW, Finkel RC, Nelson DE, Proctor ID, Davis JC. 1990. Accelerator mass spectrometry in biomedical dosimetry: relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proceedings of the National Academy of Sciences (USA) 87:5288-5292.
Tuteja N, Gonzalez FJ, Nebert DW. 1985. Developmental and tissue-specific differential regulation of the mouse dioxin-inducible P1-450 and P3-450 genes. Developmental Biology 112:177-184.
Umbreit TH, Gallo MA. 1988. Physiological implications of estrogen receptor modulation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology Letters 42:5-14.
U.S. Environmental Protection Agency (EPA). 1980. Dioxins. Cincinnati: EPA, ORD. EPA publication 600/2-80-197.
U.S. Environmental Protection Agency. 1981. Cacodylic Acid: Decision Document. Washington, DC: EPA, Office of Pesticide Programs.
U.S. Environmental Protection Agency. 1985. Health Assessment Document for Polychlorinated Dibenzo-p-dioxins.
U.S. Environmental Protection Agency. 1988a. Guidance for the Reregistration of Pesticide Products Containing Picloram as the Active Ingredient. Washington, DC: EPA, Office of Pesticide Programs.
U.S. Environmental Protection Agency. 1988b. Health Advisory for 2,4,5-Trichlorophenoxyacetic Acid. EPA, Office of Drinking Water.
U.S. Environmental Protection Agency. 1988c. Toxicology Chapter of the Registration Standard for Picloram. Washington, DC: EPA, Office of Pesticide Programs.
U.S. Environmental Protection Agency. 1992. Workshop Review Draft of Health Assessment for 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and Related Compounds. Chapter 3, Acute, Subchronic, and Chronic Toxicity. Washington, DC: EPA, Office of Research and Development.
Vainio H, Nichels J, Linnainmaa K. 1982. Phenoxy acid herbicides cause peroxisome proliferation in Chinese hamsters. Scandinavian Journal of Work, Environment, and Health 8:70-73.
van Logten MJ, Gupta BN, McConnell EE, Moore JA. 1980. Role of the endocrine system in the action of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the thymus. Toxicology 15:135-144.
Van Loveren H, Schuurman H-J, Kampinga J, Vos JG. 1991. Reversibility of thymic atrophy induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and bis(tri-n-butyltin) oxide (TBTO). International Journal of Immunopharmacology 13:369-377.
Van Miller JP, Marlar RJ, Allen JR. 1976. Tissue distribution and excretion of tritiated tetrachlorodibenzo-p-dioxin in nonhuman primates and rats. Food and Cosmetics Toxicology 14:31-34.
Van Miller JP, Lalich JJ, Allen JR. 1977. Increased incidence of neoplasms in rats exposed to low levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chemosphere 9:537-544.
Vecchi A, Mantovani A, Sironi M, Luini W, Spreafico F, Garattini S. 1980. The effect of acute administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on humoral antibody production and cell-mediated activities in mice. Archives of Toxicology Supplement 4:163-165.
Vecchi A, Sironi M, Canegrati MA, Recchia M, Garattini S. 1983. Immunosuppressive effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in strains of mice with different susceptibility to induction of aryl hydrocarbon hydroxylase. Toxicology and Applied Pharmacology 68:434-441.
Velu T. 1990. Structure, function and transforming potential of the epidermal growth factor receptor. Molecular and Cellular Endocrinology 70:205-216.
Voorman R, Aust SD. 1987. Specific binding of polyhalogenated aromatic hydrocarbon inducers of cytochrome P-450D to the cytochrome and inhibition of its estradiol 2-hydroxylase activity. Toxicology and Applied Pharmacology 90:69-78.
Voorman R, Aust SD. 1989. TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) is a tight binding inhibitor of cytochrome P-450D. Journal of Biochemical Toxicology 4:105-109.
Vos JG. 1977. Immune suppression as related to toxicology. CRC Critical Reviews in Toxicology 5:67-101.
Vos JG, Beems RB. 1971. Dermal toxicity studies of technical polychlorinated biphenyls and fractions thereof in rabbits. Toxicology and Applied Pharmacology 19:617-633.
Vos JG, Luster MI. 1989. Immune alterations. In: Kimbrough RD, Jensen AA. Halogenated Biphenyls, Terphenyls, Naphthalenes, Dibenzodioxins and Related Products. New York: Elsevier. 295.
Vos JG, Moore JA, Zinkl JG. 1973. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the immune system of laboratory animals. Environmental Health Perspectives 5:149-162.
Vos JG, Moore JA, Zinkl JG. 1974. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in C57B1/6 mice. Toxicology and Applied Pharmacology 29:229-241.
Vos JG, Kreeftenberg JG, Engel HW, Minderhoud A, van Noorle Jansen LM. 1978. Studies on 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced immune suppression and decreased resistance to infection: endotoxin hypersensitivity, serum zinc concentrations and effect of the thymosin treatment. Toxicology 9:78-86.
Waithe WI, Michaud M, Harper PA, Okey AB, Anderson A. 1991. The Ah receptor, cytochrome P450IA1 mRNA induction, and aryl hydrocarbon hydroxylase in a human lymphoblastoid cell line. Biochemical Pharmacology 41:85-92.
Walden R, Schiller CM. 1985. Short communications. Comparative toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in four (sub)strains of adult male rats. Toxicology and Applied Pharmacology 77:490-495.
WARF Institute. 1976. Reproduction and Teratology Study of Cacodylic Acid. Report to the Ansul Company, Marinette, WI. As cited in Hood, RD. 1985. Cacodylic Acid: Agricultural Uses, Biologic Effects, and Environmental Fate. Washington, DC: Veterans Administration, Agent Orange Projects Office.
Wattenberg LW. 1978. Inhibition of chemical carcinogenesis. Journal of the National Cancer Institute 60:11-18.
Wattenberg LW. 1985. Chemoprevention of cancer. Cancer Research 45:108.
Weber H, Poiger H, Schlatter C. 1982. Acute oral toxicity of TCDD-metabolites in male guinea pigs. Toxicology Letters 14:117-122.
Weber LWD, Stahl BU, Lebofsky M, Alper RH, Kerecsen L, Rozman, K. 1991. Inhibition of phosphoenolpyruvate carboxykinase activity appears to be the key biochemical lesion in the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Chemosphere 23:1957-1962.
Weissberg JB, Zinkl JG. 1973. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin upon hemostasis and hematologic function in the rat. Environmental Health Perspectives 5:119-123.
White KL Jr, Lysy HH, McCay JA, Anderson AC. 1986. Modulation of serum complement levels following exposure to polychlorinated dibenzo-p-dioxins. Toxicology and Applied Pharmacology 84:209-219.
Whitlock JP Jr. 1990. Genetic and molecular aspects of 2,3,7,8-tetrachlorodibenzo-p-dioxin action. Annual Review of Pharmacologic Toxicology 30:251-277.
Whitlock JP. 1991. Mechanism of dioxin action: relevance to risk assessment. In: Gallo MA, Scheuplein RJ, van der Heijden KA, eds. Biological Basis for Risk Assessment of Dioxins and Related Compounds. Plainview, NY: Cold Spring Harbor Laboratory Press. Banbury Report 35. 351-366.
Wilson JG, Boutwell RK, Davis DE, Dost FN, Hayes WJ, Kalter H, Loomis TA, Schubert A, Sterling TD, Bowen DL. 1971. Report of the Advisory Committee on 2,4,5-T to the Administrator of the Environmental Protection Agency. Washington, DC: U.S. GPO.
World Health Organization (WHO). 1984. 2,4-Dichlorophenoxyacetic Acid (2,4-D). Geneva: WHO. Environmental Health Criteria 29.
Wu L, Whitlock JP. 1992. Mechanism of dioxin action: Ah receptor-mediated increase in promoter accessibility in vivo. Proceedings of the National Academy of Sciences (USA) 89:4811-4815.
Yamahuchi H, Yamamura Y. 1984. Metabolism and excretion of orally administered dimethylarsinic acid in the hamster. Toxicology and Applied Pharmacology 74:134-140.
Yamanaka K, Hasegawa A, Sawamura R, Okada S. 1989a. Dimethylated arsenics induce DNA strand breaks in lung via the production of active oxygen in mice. Biochemical and Biophysical Research Communications 165:43-50.
Yamanaka K, Hasegawa A, Sawamura R, Okada S. 1989b. DNA strand breaks in mammalian tissues induced by methylarsenics. Biological Trace Element Research 21:413-417.
Yamanaka K, Hoshino M, Okamoto M, Sawamura R, Hasegawa A, Okada S. 1991. Induction of DNA damage by dimethylarsine, a metabolite of inorganic arsenics, is for the major part due to its peroxyl radical. Biochemical and Biophysical Research Communications 168:58-64.
Yang KH, Yoo BS, Choe SY. 1983. Effects of halogenated dibenzo-p-dioxins on plasma disappearance and biliary excretion of ouabain in rats. Toxicology Letters 15:259-264.
Yao C, Safe S. 1989. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced porphyria in genetically inbred mice: partial antagonism and mechanistic studies. Toxicology and Applied Pharmacology 100:208-216.
Young AL, Calcagni JA, Thalken CE, Tremblay JW. 1978. The Toxicology, Environmental Fate, and Human Risk of Herbicide Orange and Its Associated Dioxin. Brooks AFB, TX: Air Force Occupational and Environmental Health Lab. USAF OEHL-TR-78-92. 262 pp.
Zimmering S, Mason JM, Valencia R, Woodruff RC. 1984. Chemical mutagenesis testing in Drosophila. II. Results of 20 coded compounds tested for the National Toxicology Program. Environmental Mutagenesis 6:189-202.
Zinkl JG, Vos JG, Moore, JA, Gupta BN. 1973. Hematologic and clinical chemistry effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals. Environmental Health Perspectives 111-118.














































































































