
WORKSHOP SUMMARY
A long-held goal in oncology has been to develop therapies that target the specific abnormalities in each patient’s cancer rather than simply treating cancers based on the tissue of origin. Early pioneering efforts with cancer drugs such as Gleevec and Herceptin have shown how effective it can be to treat tumors based on the genetic anomalies they harbor. In the past decade, advances in technology have enabled researchers to relatively quickly and inexpensively determine, in minute detail, the genetic makeup of tumors. Studies using this new technology have garnered greater knowledge about the molecular underpinnings of cancer, uncovering specific genetic alterations that drive the growth of individual tumors. Consequently, the rationale for and feasibility of developing molecularly targeted cancer therapies has never been stronger. Although relatively few targeted cancer therapies are currently available in the clinic and it is not yet clear whether all cancers are driven by genetic changes that can be targeted, there is widespread optimism in the cancer community that this new ability to assess the genetic abnormalities in tumors will ultimately lead to better cancer treatments and improved patient outcomes. There are hundreds of candidate targeted drugs in the development pipeline and several new cancer drugs targeting specific genetic alterations have entered the market in the past 2 years.
However, many challenges remain in effectively and efficiently developing new targeted cancer therapies and the biomarker tests that indicate
which patients will be responsive to them, and in implementing them appropriately in clinical practice. These challenges include many policy issues, such as the level of oversight needed for test development and use, levels of evidence necessary for reimbursement decisions, and ways to meet informational needs of patients and care providers. New paradigms may be needed for assessing the efficacy of targeted therapies as well as the clinical validity and usefulness of biomarker tests. The standard approach of defining treatment based on the anatomic origin of cancer is becoming less tenable now that genomic tests are stratifying cancers into rare subsets defined instead by the molecular drivers of the tumors. As use of these tumor profiles has become more common and extensive, clinicians may need more clarity on how to interpret and act on them in the clinic. In addition, the marked complexity and rapidly evolving nature of the latest genomic tests have raised questions about whether new standards and methods are needed for assessing their validity and clinical utility, as well as for making regulatory and reimbursement decisions.
Review and oversight of test development is currently quite variable. Most tests used in clinical practice have never been reviewed by the Food and Drug Administration (FDA), but rather are offered as laboratory-developed tests (LDTs). Laboratories that perform these tests are subject to quality assurance requirements under the Clinical Laboratory Improvement Amendments (CLIA),1 but the tests are not subject to FDA review. Even when a drug and a biomarker test are co-developed and co-approved by FDA, with the companion diagnostic listed in the drug label, clinical laboratories can quickly develop similar tests as LDTs and offer them to patients without FDA review. The LDT pathway can facilitate rapid innovation in test development, but concerns have been raised about whether greater oversight is necessary for more complex tests. FDA has recently announced the intent to develop a risk-based approach to the oversight of LDTs (FDA, 2014).
Neither development pathway, as an LDT or as an FDA-approved diagnostic test, requires evidence of clinical usefulness (clinical utility), which is often expected for reimbursement. Furthermore, there is concern that prevailing reimbursement rates for diagnostic tests often do not reflect the value of clinically useful biomarker tests. Thus, developers may be
_______________
1 See http://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html (accessed March 18, 2015).
reluctant to invest the time and resources necessary to demonstrate clinical utility and support reimbursement decisions.
The Institute of Medicine’s (IOM’s) National Cancer Policy Forum has been organizing a series of workshops focused on these policy issues in the development of new cancer therapies. The first, held in 2009, examined a broad range of issues in developing “personalized” or “precision” therapy for cancer (IOM, 2010a). The most recent, held in Washington, DC, on November 10 and 11, 2014, entailed a 2-day workshop on “Policy Issues in the Development and Adoption of Biomarkers for Molecularly Targeted Cancer Therapies.”2 The next workshop in the series will focus on policy issues in the development of immunotherapies for cancer, a rapidly developing therapeutic area in oncology.
At the November 2014 workshop, subject-matter experts and members of the public discussed recent trends in the development and implementation of molecularly targeted cancer therapies and explored potential policy actions to address specific challenges. Topics included
- Recent advances in tumor biomarker tests and the developmental, regulatory, clinical, and reimbursement challenges they pose;
- FDA regulation of tumor biomarker tests and how it is evolving;
- Innovative trial designs, databases, and other potential ways to generate evidence to support reimbursement decisions;
- Practice guidelines and treatment pathways that can influence clinical implementation of molecularly targeted therapies; and
- Education and research needs to support the ongoing molecular biology revolution in oncology.
This report is a summary of the presentations and discussions at the workshop. A broad range of views and ideas were presented and a summary of suggestions from individual participants is provided in Box 1. Additional details and context for these suggestions can be found throughout the workshop summary. The workshop Statement of Task and agenda can be found
_______________
2 This workshop was organized by an independent planning committee whose role was limited to the identification of topics and speakers. This workshop summary was prepared by the rapporteurs as a factual summary of the presentations and discussions that took place at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants, and are not necessarily endorsed or verified by the IOM or the National Cancer Policy Forum; and should not be construed as reflecting any group consensus.
BOX 1
Suggestions Made by Individual Workshop Participants
Develop New Standards for Biomarker Tests
- Standardize specimen sampling, processing, and storage. Matthias Holdhoff
- Raise the bar for proficiency testing for genomic profiling and make the results public. Mickey Williams
- Establish reporting standards both for test methods and results, including minimum reporting requirements for publishing next-generation sequencing data. Mickey Williams
- Develop standards and processes for annotation of genetic variants in tumors and for reporting a genetic variant as clinically actionable. Patricia Ganz, Mia Levy, Federico Monzon, Richard Schilsky, Deborah Schrag, Mickey Williams
- Create standards for matching treatments with genomic test results. Richard Schilsky, Mickey Williams
- Harmonize global regulation for biomarker tests. Karen Long, Anne-Marie Martin
Generate New Evidence to Support Clinical Use of Tests
- Establish a single public curated databse for annotated data on cancer mutations identified in clinical studies. Matthias Holdhoff, Richard Schilsky, Mickey Williams
- Develop policies that support data sharing among laboratories, pharma and diagnostic companies, and health care providers to help advance the clinical knowledge base. Bruce Johnson, Mia Levy, Federico Monzon, Richard Schilsky, Mickey Williams
- Develop an app to help clinicians and patients identify clinical trials relevant to the results of tumor profiling tests. Lillian Siu
- Conduct more dynamic trials in which tumors are extensively profiled both at baseline and when the cancer progresses, or that entail frequent sampling of circulating tumor DNA. Matthias Holdhoff, Lillian Siu
- Include more data on patient characteristics, such as ethnicity, smoking history, weight, etc., as well as all relevant outcomes in databases and in the annotation of stored tumor specimens. Garnet Anderson, Patricia Ganz
- Use subgroup analysis in trials to identify specific mutations associated with response to targeted therapies. David Solit
- Support postmarket research and Coverage with Evidence Development to better assess clinical utility of biomarker tests. Donna Messner, Federico Monzon, Sean Tunis
- Include adverse-event reports in a transparent public registry under the Clinical Laboratory Improvement Amendments (CLIA). Federico Monzon
- Conduct prospective studies to gather data on who is using genomic tests, patient and provider perspectives on test results, as well as the outcomes, benefits, and costs. Kathryn Phillips
- Use “root-cause analysis” to assess whether a test addresses a clinical problem, provides results that are useful for patient management, and improves existing outcomes. David Eberhard
- Establish reimbursement science. Sean Tunis
Facilitate Innovation in Test Development
- Consult with the Food and Drug Administration (FDA) early in the test development process. David Litwack
- Make CLIA regulations more stringent rather than shifting test oversight to FDA. Federico Monzon
- Provide greater clarity on how laboratories are reimbursed for the services and innovation they provide. Dane Dickson, Federico Monzon
- Evaluate the impact of the changing health care policy environment, such as new current procedure terminology codes for diagnostic tests and the rise in accountable care organizations. Kathryn Phillips
Increase Patient and Provider Knowledge About Tumor Profiling Tests
- Create publicly available databases of test availability, cost, and value. Kathryn Phillips
- Develop and assess patient education strategies and tools for different levels of health literacy. Mia Levy, Patricia LoRusso
- Develop guidance on how to structure and frame information about genomic test results to facilitate communication and decision making with patients. Kathryn Phillips
- Develop educational materials for health care providers with different learning styles. Mia Levy
in the Appendix. The speakers’ biographies and presentations (as PDF and audio files) have been archived at http://www.iom.edu/Activities/Disease/NCPF/2014-NOV-10.aspx (accessed March 18, 2015).
MOLECULAR BIOLOGY REVOLUTION IN CANCER DIAGNOSIS AND TREATMENT
A molecular biology revolution that has changed the way in which cancer is diagnosed and treated began in earnest in the 1990s and early 2000s when several molecularly targeted therapies became available. Biomarker tests were used to assess the likelihood of responding to specific treatments targeted to the genetic alterations in the tumors that drive their growth. Each of these tests detect only one specific biomarker of tumor response and thus are considered “single analyte” tests. Such tests have been followed by the development of more comprehensive genomic profiling enabled by “next-generation” sequencing technology. Other novel techniques such as RNA sequencing tests and “liquid biopsies” that sample the DNA of tumor cells circulating in the blood are also being developed as methods for molecularly profiling cancers. The rapidly changing nature of the technologies used to develop tests adds to the complexity of assessing new tests as they arise.
Lessons Learned from Single Analyte Tests
Single analyte tests and the targeted treatments associated with them have led to remarkable improvements in treatment response, noted Adrian Senderowicz, president of Oncology Drug Development, LLC. He pointed out as an example that in 2000, the response rate of advanced refractory lung cancer to standard chemotherapy was usually in the single digits and median survival was less than 6 months. But 10 years later, following the introduction of therapies targeting the epidermal growth factor receptor (EGFR), the response rate increased to about 60 percent for patients with certain EGFR mutations, and the median duration of those responses was 48 weeks (Camidge et al., 2012).
Similarly, a therapy for melanoma targeting the BRAF gene led to dramatic improvements in patients whose tumors had the variant form of the BRAF targeted by the drug, with nearly all of those patients experiencing a regression of their tumors (Sosman et al., 2012). Researchers then discovered that 1 percent of lung cancer patients have BRAF driver mutations
in their tumors and these patients also responded to the BRAF-targeted therapy. In addition, 2 percent of lung cancer patients have mutations in the HER2 gene, which had previously been identified as an effective target for certain patients with breast cancer (Gandhi et al., 2013). By 2010 researchers had identified six genetic variants in the tumors of lung cancer patients that indicated likelihood of responding to specific treatments as well as a KRAS variant that indicated a lack of response to a group of targeted treatments known as tyrosine kinase inhibitors (see Figure 1).
As noted by Mia Levy, director of cancer clinical informatics at Vanderbilt-Ingram Cancer Center, the dramatic responses achieved with targeted treatments changed the approach to treating lung cancer. Prior to the development of targeted therapies, lung cancer patients were divided up into two main groups based on the appearance of their tumor cells, with the majority being classified as non-small-cell lung cancers. All patients with this type of lung cancer were given the same treatments before 2000. But now “we have predictive biomarkers to segment out this population so instead of treating everybody the exact same way, we treat them differently with targeted therapy. So instead of having response rates of 30 percent or
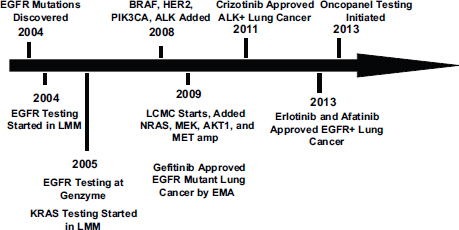
FIGURE 1 Genotyping time line for non-small-cell lung cancer.
NOTE: BRAF, EGFR, HER2, KRAS, MEK, NRAS, and PIK3CA are genes detected by biomarker tests; EMA = European Medicines Agency; LCMC = Lung Cancer Mutation Consortium; LMM = Laboratory of Molecular Medicine at the Harvard Medical School.
SOURCE: Johnson presentation, November 10, 2014.
worse [to first line therapy], we’re getting response rates of close to 80 percent in this population,” Levy said. Roy Herbst, Ensign Professor of Medicine and Professor of Pharmacology, chief of medical oncology, Yale Cancer Center and Smilow Cancer Hospital, and associate director for translational research, Yale Cancer Center, added, “We really have taken lung cancer and made it a disease where we are focusing on more and more pieces of the pie.”
Similar scenarios have evolved in the treatment of many other cancers, including breast, melanoma, lung, and colon cancers. Using tests to identify molecular drivers of tumor growth in these cancers is often key to selecting a treatment. Senderowicz stressed that “the segmentation of these patient [populations] is very important because based on the segmentation, you can treat different patients with different agents.” Therefore, it is crucial that the tests that indicate the segmentation be accurate, he added, because false negatives will prevent patients who need these more effective targeted therapies from receiving them. “It’s a big responsibility for the manufacturers of these tests, for the physicians who order them, for the pathologists who perform them, and for the regulatory agencies who regulate them,” he said. “This creates a lot of challenges for different stakeholders.”
Even when single analyte tests are accurate, patients can still be resistant to the targeted therapies or acquire such resistance after a favorable initial response to treatment, Levy pointed out. Understanding the cause of the primary or acquired resistance of these patients is now possible due to technological advances that have made it feasible and economical to decipher much or all of the entire genome of tumor cells, she added. Such “next-generation sequencing” has uncovered co-occurring genetic mutations in tumors, including molecular backup pathways that can emerge when a major tumor driver is blocked from acting by a specific treatment.
Bruce Johnson, chief clinical research officer and professor of medicine, Harvard Medical School, noted that the more detailed sequencing of tumor DNA by next-generation sequencing can preclude the need to acquire additional tumor tissue for testing to determine why patients are not responding to targeted treatments, by revealing before such treatment even begins the co-mutations that can prevent a durable treatment response. “Look at how many times you can spare yourself from having to go back and do another test as you give second and third line treatments to these patients,” he said. Lilian Siu, senior staff physician, division of medical oncology and hematol-
ogy, Princess Margaret Hospital in Toronto, agreed, adding “These genetic panels are important so that we have almost everything that we want done in one shot with one specimen.”
Levy stressed, “Genomic profiling in cancer is here to stay. Instead of just testing for a single biomarker that’s going to drive your decision for therapy, we can test for multiple types of alterations.” Anne-Marie Martin, head, molecular medicine and precision medicine & diagnostics, GlaxoSmithKline (GSK), noted, “The technology is allowing us to generate comprehensive data in much smaller patient samples.” However, Mickey Williams, director, molecular characterization laboratory, Frederick National Laboratory for Cancer Research, added the caveat that next-generation sequencing is fostering the development of tests that do not just detect a handful of genetic defects but screen the more than three billion bases in DNA for alterations. “That’s a lot of analytes and to be able to demonstrate that you can do this accurately is a daunting task,” he said.
Next-generation sequencing can determine the sequence of a portion or all of the DNA in a tumor sample, but not all that DNA will be transcribed into RNA and then into proteins that play an active role in tumor cells. To focus sequencing efforts on the genes that are being transcribed and are thus more likely to have an effect on tumors, some researchers do another type of genomic tumor testing, known as RNA sequencing. Neil Hayes, associate professor, clinical research, hematology/oncology, University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, stressed that although RNA is more difficult to work with than DNA, “RNA is actually where the action is, whereas most of the genome is not transcribed and not of interest.”
According to Hayes, another advantage to working with RNA as opposed to DNA is that most mutations in oncogenes are easier to detect in RNA. One study he conducted showed that RNA sequencing integrated with DNA sequencing improved the mutation detection rate in samples with low-purity tumor cells (Wilkerson et al., 2014). Hayes said it is also less expensive and easier to find repeated or deleted sequences and other structural alterations to the genome with RNA versus DNA sequencing. This technology could be helpful in detecting genetic alterations in the many patients for which DNA analysis has not revealed mutations that are driving their cancers, he said. For example, he said that for about half of all
lung cancer patients who have had their tumor DNA sequenced, no known driver mutations were detected. In addition, RNA sequencing might prove useful in detecting altered expression of immune system components that are the targets of several new immunotherapies in development for cancer.
Tests for Circulating Tumor DNA
Other innovative biomarker tests on the horizon are those that measure tumor DNA circulating in the blood. Called liquid biopsies, these tests analyze a small blood sample to detect and screen the naked DNA released by tumor cells during cell turnover. Although these DNA fragments are small, they can contain genetic mutations, according to Matthias Holdhoff, assistant professor of oncology, Johns Hopkins Medicine. For these tests, circulating tumor DNA must be separated from the DNA of normal cells in the bloodstream, which can be like finding the proverbial needle in a haystack. But Holdhoff said one of his studies showed that using a polymerase chain reaction (PCR) to duplicate DNA sequences so they are easier to find, combined with flow cytometry to sort and quantify them, has a detection rate of 1 in 10,000 or better, which is akin to that of standard PCR-based assays (Holdhoff et al., 2009).
According to Holdhoff, such liquid biopsies are advantageous because they are non-invasive and they enable the collection of multiple specimens with minimal burden to patients. These tests can also be done on fresh samples, whereas many biomarker tests are done on paraffin-embedded samples, in which the DNA may be degraded. In addition, the DNA from multiple genetically diverse metastatic tumors can be collected in a single blood sample, unlike surgical biopsies that only sample the DNA of the specific tumor site they biopsy.
Liquid biopsy tests have many potential uses in oncology, Holdoff said, including using them to determine the mutation status of the tumor, to monitor tumor burden, and to track the development of resistance to targeted therapies. He added that if the tests are sensitive enough, oncologists could also potentially use liquid biopsies to detect residual disease after treatment, as well as early recurrence. Circulating tumor DNA could also reveal how the genetics of the tumor changes over time, and to track the emergence of new mutations that might influence response to treatment.
But Holdhoff noted that circulating tumor DNA tests might not be the best tests for every type of cancer. Initial studies of these tests in solid tumors found that although they appear to work well for bladder, colorec-
tal, breast, lung, and other cancers, they do not work as well for brain and some other types of cancer (Bettegowda et al., 2014). Tumor stage also seems to be important, with greater detection rates for higher stage tumors (Bettegowda et al., 2014).
Business Climate for Developing Diagnostic Tests
Due to the reduced costs and increased efficiency of genomic sequencing tests, and the advent of other new technologies, said Federico Monzon from Invitae, the current business climate for developing diagnostic tests is encouraging. However, he also noted increased competition in the field due to smaller laboratories having access to better technology, which has “leveled the playing field” and enabled a lot of laboratories to do genomic testing. Prior to recent technological developments fostered by the Human Genome Project, genetic testing was mainly the purview of large academic medical centers or specialized laboratories, he said. Now, smaller academic hospitals are expressing interest in doing tumor profiling, which increasingly is being required for clinical trials with targeted agents. In addition, he said the recent Supreme Court ruling that invalidated patents of isolated genes that occur in nature also triggered greater interest in developing genetic tests (U.S. Supreme Court, 2013).
Consequently, Monzon said, there is a healthy trend in investments in diagnostics, and there are projections that the field will go from a $15 billion market to a $25 billion market by the end of the decade (Personalized medicine, 2012). Currently the United States represents about half of the market. Molecular diagnostics has been the fastest growing segment within clinical diagnostics in the past decade, Monzon reported (Budel, 2013; DeciBio, 2013; Shields and Deshmukh, 2013).
But there are also reasons to be concerned about business opportunities in diagnostics, Monzon noted, including the pricing for new molecular codes by the Centers for Medicare & Medicaid Services (CMS) in 2014. These new codes caused the median price to drop by 15 percent compared to how CMS previously reimbursed molecular tests, with many prices dropping by more than 50 percent, he said (Malone, 2014).
Another area of uncertainty is how CMS will price reimbursements for molecular tests in the future, which will also impact pricing from the private payer sector. With the new “Doc Fix” law, starting in 2017, Medicare will rely on an average of private payer rates to set its fee schedule, and give special treatment to single-source proprietary tests. The Doc Fix law limits how
deep market-based rates can cut the current fee schedule for the first 6 years. From 2017 to 2019, CMS cannot reduce the payment for an individual test more than 10 percent per year, and from 2020 to 2022, not more than 15 percent per year (Malone, 2014). “There needs to be clarity on reimbursement and a path forward to actually get laboratories reimbursed for the services we provide and the innovations,” Monzon said.
He also noted concerns about FDA’s recent announcement that it will develop a risk-based approach to oversight of laboratory-developed tests, and how that could reshape the diagnostics industry. “Everyone is bracing for these increased regulations,” Monzon said.
CHALLENGES IN BIOMARKER TEST DEVELOPMENT
Workshop participants described numerous technical challenges in biomarker test development, including a lack of standards and reference materials, difficulty in gathering the evidence to assess a test’s validity and utility in the clinic, and the need for greater cooperation and sharing of data to gather that evidence.
Several presenters noted the lack of standards for developing biomarker tests. Williams pointed out the need for test reference materials for quality assurance purposes and to enable comparison of tests across different laboratories. “Every cancer center is doing next-generation sequencing, but we really don’t know if we’re getting the same results because we have an urgent need for reference materials,” he said. Williams noted that the National Institute of Standards and Technology can provide a certified reference human genome that laboratories can sequence to assess if their assays are accurate. But he added that current proficiency tests have a low bar that should be raised for genomic profiling, and that results should be made public. Holdhoff said there was a need for better standardization of specimen sampling, processing, and storage. David Solit, Geoffrey Beene Chair in Cancer Research, and director, Marie-Josée and Henry R. Kravis Center for Molecular Oncology at Memorial Sloan Kettering Cancer Center, said that the Actionable Genome Consortium aims to establish various standards for cancer genomics (see Box 2).
Williams also pointed out that there are no standard operating procedures followed by all laboratories for the same test. For example, some labs
may only sequence samples containing 20 percent tumor material, while others may sequence samples with 50 percent. With such variation, “there’s no guarantee that every lab is going to see the same genetic mutations reproducibly,” he said. Hayes agreed, noting that there is little published data on the tissue requirements and quality needed for accurate RNA sequencing tests, although various thresholds are used by different researchers. “The bottom line is more tissue is better, higher percentage tumor is better, but setting that threshold is very challenging,” he said. However, Solit cautioned that setting those thresholds might “allow the perfect to be the enemy of the good. Are we going to throw away samples and not analyze them if tumor content is too low?” he asked. Williams noted that for the National Cancer Institute’s Molecular Analysis for Therapy Choice (MATCH) Program trial (see the section on basket trials), tumor samples that do not meet threshold minimums are still analyzed, but the results are reported separately from the others.
BOX 2
Actionable Genome Consortium
The Actionable Gene Consortium was formed in 2014 to create and publicize standards for cancer genetics. Composed of representatives from the National Cancer Institute, the Memorial Sloan Kettering Cancer Center, the MD Anderson Cancer Center, the Broad Institute of the Massachusetts Institute of Technology, Cancer Research UK, Fred Hutchinson Cancer Research Center, Princess Margaret Hospital Cancer Center, the Dana-Farber Cancer Research Center, and the gene sequencing company Illumina, the consortium aims to demonstrate clinical utility, democratize genomic testing so it is more widely available to patients, contain costs, and define and standardize what actionable genes are across institutions. The Consortium aims to develop standards for sample processing, tumor content, sequencing, data analysis and reporting. All of the Consortium’s standards, standard operating procedures, analytic tools, results, and conclusions will be published and made available to the public.
SOURCES: Solit presentation, November 10, 2014; Actionable Genome Consortium to guide NGS in cancer, 2014.
Williams suggested establishing reporting standards both for methods and results, including minimum reporting requirements for publishing next-generation sequencing data, so others can reproduce the test and determine whether they get the same results. “There are just way too many parameters and if they aren’t reported we’ll never be able to know how these tests are done,” he said. He also suggested standards for matching treatments with sequencing test results.
Herbst pointed out that when different tests for the same genetic alteration first emerge from several different laboratories, the way in which the test is done and the cut-off standards used for reporting results can be quite variable. Samir Khleif, director of the Georgia Health Sciences University Cancer Center, Georgia Regents University Cancer Center, agreed this is a major problem and that when different tests are used in the same clinical studies, they can have discordant results. But Khleif also noted that better standardization in the early development of tests would require competing companies to cooperate and share their data prior to their tests entering the market, which they are unlikely to do without regulation and/or incentives to do so.
Assessing Analytical Validity, Clinical Validity, and Clinical Utility
Developers must validate their tests before they can be used for clinical care. The validation process begins with analytical validation. This reveals how accurately the test detects the specific analytes it was designed to detect, and includes assessment of the test’s range, accuracy, precision, bias, and reproducibility when used by different operators or instruments, or in different settings (Febbo et al., 2011; IOM, 2010a; Woodcock, 2010).
Clinical validation is also essential in the test development process. Clinical validity is a measure of the accuracy of a test for a specific clinical purpose, such as selection of targeted therapy in a specific patient population (IOM, 2010a, 2012). Such validation involves assessment of the sensitivity, specificity, cut-offs, and other parameters of a test (Febbo et al., 2011; Woodcock, 2010). Williams outlined the steps for validating a next-generation sequencing assay system for use in a clinical trial, as shown in Box 3.
Generally, establishing clinical validity for a test involves showing that it is “fit for purpose,” a process that relies on data collected from clinical trials or from archived samples that are well annotated with outcomes and other clinical information. More recently, sponsors have been submitting
BOX 3
Steps to Validating a Next-Generation Sequencing Assay System for Use in a Clinical Trial
Mickey Williams outlined the steps needed to clinically validate a test for use in clinical research trials, as follows:
Define Intended Use
The intended use determines what must be demonstrated for validation. Researchers may use tests in clinical studies for pure discovery purposes, such as to discover a new treatment response biomarker or to determine patient enrollment or treatment selection, the latter of which would have more stringent performance requirements.
Define the Test System
Tests are systems that include all the steps involved, from biopsy through test result reporting. Because next-generation sequencing tests are complex, any deviation from standard operating procedures can confound the data. An important step is to specify and not deviate from any aspects of the defined system during the validation process, even if improvements are later identified that could potentially make the test better. Locking down the test system in this way is challenging for genomic tests because the technology for these tests is changing rapidly, Williams noted.
Conduct Initial Feasibility Tests
These tests should reveal the strengths and weaknesses of the test in a clinical setting.
Consult with FDA
This is important if the specimens will be collected as part of a clinical trial specifically for assessing the test.
Conduct Analytical Validation of Assay Performance
This assesses how well the test measures the molecular event of interest, including its range, accuracy, and precision under conditions that replicate the clinical setting in which the test is intended to be used.
SOURCE: Williams presentation, November 10, 2014.
case studies, results from database analyses, and medical literature reviews to FDA to establish the clinical validity of their tests, noted David Litwack, Personalized Medicine Staff at FDA. “As long as the evidence is good, there’s no reason why it has to be a clinical study, and the use of databases is going to be a very important part of FDA regulation in the future that will hopefully ease the pathway for everybody,” he said. Dane Dickson, director of clinical science, MolDx, Palmetto GBA, suggested that genetic panels be disease-specific and recognize that mutations in a gene such as BRAF, although relevant to determining treatment in a melanoma patient, may not indicate proper treatment in a colon cancer patient. But Levy noted that it would be difficult for diagnostic companies providing genomic panels to clinically validate them for each individual cancer type. Rather, what they tend to do is run the comprehensive genetic test, but only report and charge for the genetic results relevant to the particular tumor sample being tested. This also helps prevent clinicians from being overwhelmed by an excessive amount of genomic information, she added.
Several speakers noted other challenges in assessing the clinical validity of biomarker tests that are frequently used by oncologists. Hayes pointed out that most archived samples are not available due to proprietary claims of the institutions that house them, and it is time consuming and expensive for a diagnostic company to collect their own samples. Johnson added that tests to assess the clinical validity of biomarker tests are often done on cancer cell lines, rather than clinical tumor specimens, which are more relevant for assessing clinical validity. He also noted that when assessing clinical validity, laboratories tend to use clinical specimens enriched with the mutations the test is designed to detect, and he questioned the relevance of those results to clinical settings.
Both clinicians and insurers need to know a test’s clinical utility in order to assess its value for certain cancers. Clinical utility is a measure of whether clinical use of the test improves patient outcomes for a specific indication. Several speakers noted that many diagnostic companies and laboratories do not assess the clinical utility of their tests before they are used in the clinic. Next-generation sequencing tests especially tend to provide extensive information on genetic variants in a sample, but little to no supporting information on which of those alterations are “actionable,” that is, indicate specific clinical interventions. Williams called for guidelines for identifying and reporting a genetic variant as clinically actionable. These guidelines could specify what level of evidence is needed to take clinical action in response to a test result.
Dickson added that because “no two next-generation sequencing methods are the same, if one lab is doing it one way and has a greater sensitivity and lower specificity than another lab, how can I aggregate that information to determine clinical utility?” He also stressed that an increased sensitivity will not necessarily translate into better clinical outcomes. David Eberhard, director, Pre-Clinical Genomic Pathology, Lineberger Comprehensive Cancer Center, and associate professor, Departments of Pathology and Pharmacology, UNC at Chapel Hill, added that the sensitivity needed in a test can vary depending on how large tumor samples are likely to be. Lung cancer biopsies tend to be tiny, for example, so would require greater sensitivity to be clinically useful, he said, as would samples with a low percentage of tumor cells.
Assessing clinical utility of tests usually requires applying the test to a large number of patients or patient samples, which can be challenging to do given the rarity of some of the mutations that the tests are designed to detect. Some relevant mutations, such as those in BRAF, occur in only 1 percent or less of lung cancer patients. Johnson noted that for one study, it took 3 years at major medical centers to accrue 50 lung cancer patients with such rare mutations. Martin added that in one of GSK’s studies, researchers had to screen more than 11,000 lung cancer patients to enroll 23 patients with a specific BRAF mutation (V600E) (Marchetti et al., 2011; Paik et al., 2011).
“As we get into more complicated mutation sequencing, we are going to get into smaller and smaller datasets,” said Dickson. “It is unlikely that we are going to be able to really get some of these good datasets we have traditionally used for determining if a test is appropriate. That is a problem because even though we agree it is hard to get those levels of information in a molecular test, we also need to recognize that when we are taking people away from well-established interventions based on limited datasets, we could potentially really harm a patient.”
Eberhard reported that nearly 20 years ago the Tumor Marker Utility Grading System was developed to define levels of evidence for tumor markers (Hayes et al., 1996). The grades given in this system were determined to a large degree by the types of studies used to assess the markers. Subsequently, some consideration has also been given to how the samples were obtained and how the tests were performed to create the evidence. More recently the National Comprehensive Cancer Network (NCCN) created categories of evidence for tumor markers to aid decision making of practicing oncologists, ranging from high-level evidence that leads to
uniform NCCN consensus to weaker evidence that results in major NCCN disagreement regarding an intervention (Febbo et al., 2011).
But neither the Tumor Marker Utility Grading System nor the NCCN guidelines adequately address whether a biomarker test is medically necessary, which is a key component of clinical utility, Eberhard pointed out. To assess this aspect of molecular diagnostics, he suggested using a problem-solving approach called “root-cause analysis,” which is outlined in Box 4.
Clinical utility also entails feasibility of clinical implementation. That aspect of fitness for purpose depends on the platform the test is performed on and how robust, complex, and suitable both the platform and the test are to the clinical purpose at hand, Eberhard noted. Sample characteristics also influence clinical utility as well as how results are interpreted. The final results of a test must indicate specific actions to have clinical utility, Eberhard pointed out. But the results from molecular diagnostic tests often fall into large, uninterpretable gray zones due to a lack of evidence on the clinical significance of the alterations detected. For example, Oncotype Dx, a biomarker test for breast cancer that measures the expression of 21 genes, has a large gray zone of results called “intermediate” for which there is no one clear treatment recommended. For patients given this result, the test currently has no clinical utility (a clinical trial called TAILORx is ongoing to assess the utility of the test for this patient population). There are no standards for what size of gray zone is acceptable for a test to enter the market, Eberhard noted. Next-generation sequencing also often identifies genetic variants of unknown significance. “So if we have a variant of unknown significance, what should we tell the oncologist?” Eberhard asked.
Determining clinical validity and utility of biomarker tests would be greatly aided if companies and institutions amassing tumor profiling data and samples collaborated more and shared information, several participants suggested. “We have a great opportunity to work across different pharmaceutical and diagnostic companies’ interest and in the patients’ interest to work collaboratively to be certain that we’re bringing these tests into the clinic, and at the end of the day doing no harm, but actually really pushing the field forward,” said Williams. Monzon added, “We need to develop policies that support data sharing among laboratories and health care providers to help advance the clinical knowledge base.”
Root-cause analysis aims to a find a cause for a problem, which when removed, prevents an undesirable event from occurring. This analysis is often used in quality assurance programs.
Root-cause analysis is performed systematically with conclusions and causes backed up by documented evidence. There may be more than one root cause for a problem. The goal is to identify solutions to prevent recurrence at lowest cost in the simplest way. If there are alternatives that are equally effective, then the simplest or lowest cost approach is preferred. Root causes identified depend on the way in which the problem or event is defined. Root-cause analysis should establish a sequence of events to understand relationships among contributory (causal) factors, root cause(s), and the defined problem, and can potentially address problems before they occur or escalate rather than reacting to problems as they occur.
Eberhard gave an example of a root-cause analysis undertaken to address the problem that diagnosis of non-small-cell lung cancer based on how it appears under a microscope is imprecise and does not recognize subtypes. The root-cause analysis of this problem identified that accurate and reproducible subtyping can be compromised by samples that are too small, by inexperienced interpretation, or by being unable to distinguish poorly differentiated adenocarcinomas from squamous cell carcinomas (Grilley-Olson et al., 2013; Thunnissen et al., 2014). The solution to this problem could be new diagnostics that can distinguish adenocarcinomas from squamous cell carcinomas, which may be useful if they can provide the same result on small biopsies as what would have been obtained from larger definitive samples of the same tumor, Eberhard noted.
SOURCE: Eberhard presentation, November 10, 2014.
Schilsky noted that various databases for genomic information are being acquired through next-generation sequencing in clinical studies, including some that are publicly available, but there are no standards for how that information is reported and annotated. He suggested that a public agency develop a genetic variant annotation process and database for the community that it curated and updated. Levy responded that the National
Institutes of Health (NIH) and the National Human Genome Research Institute are already providing such public annotated databases for inherited germ-line mutations. But she added that more extensive clinical outcome data are needed for tumor mutations.
Williams suggested the American Society of Clinical Oncology (ASCO) and NCCN could be involved in creating such a clinically annotated public database for cancer mutations. “I think the feeling is shared that the time is now, everybody is acting on information, and if we could have some common data that everybody could point to so that we knew we were acting identically as we get this information is extremely important,” he said. Holdhoff also advocated for having one major database that is housed and curated by a government agency, which could have the advantage of being unbiased and long lasting. “Everyone wants to have their own database, but it’s the public trust we really have to respond to so there should be one major database that will last for the next hundred years or so and outlast everybody’s individual careers,” Holdhoff said.
Levy pointed out that as an oncologist receiving genomic profiling data, she has found a lot of detailed information missing from the reports provided by the molecular diagnostics lab that conducted the testing. She added that there needs to be a new paradigm for making data public, and pointed out that some companies have the largest collection of data on specific tumors or mutations, but those data are not accessible. She also noted that some patients have been uploading their own data onto websites that researchers can access, so more progress can be made in treating their disease.
Johnson pointed out that the Lung Cancer Mutation Consortium, a group of 16 centers across the United States, is assembling detailed sequencing information (BAM files3) and ensuring they are reproducible across institutions so they can be shared. Hayes added that BAM files can be entered into a public database known as DbGap, but because of formatting and consent issues it is difficult to do so. “We need an easier way to get these BAM files out,” he said.
_______________
3 A BAM file is the binary version of a SAM file. A SAM file is a tab-delimited text file that contains sequence alignment data.
REGULATORY OVERSIGHT CHALLENGES
Clinical tests are usually done in laboratories accredited by the College of American Pathologists (CAP). A goal of accreditation is to ensure the quality of testing systems and the reproducibility of results. Another goal is to ensure that the results from one lab are comparable to that of other laboratories, through proficiency testing of reference samples provided by CAP or other large collegiate organizations. All clinical labs must have proper CLIA certification to receive Medicare or Medicaid payments.
There are two types of laboratories: (1) laboratories affiliated with a health care institution that provide testing directly to patients in a clinical setting, and (2) those that are known as reference laboratories, which are laboratories to whom samples are sent for testing by clinician providers. But there are also hybrids, such as large institutional reference laboratories that have outreach programs to acquire samples from the community. Monzon said that reference labs can be more efficient and reduce cost compared to institution-affiliated laboratories, and can be more proficient as well because of the high volume of tests they conduct. Reference labs tend to perform tests for esoteric conditions, that is, to diagnose rare disorders that only a thousand patients may have. Because these patients are scattered across the country, there is an advantage to having only one central reference lab that offers the test for a rare disease, Monzon pointed out. But the disadvantage of reference lab tests is that because they are not offered onsite, there can be delays due to shipping samples and reporting results.
How a test is regulated is determined by how it comes to market, Hayes reported. A test may be marketed as a commercial test “kit,” a group of reagents used in the processing of samples that are packaged together and sold to multiple labs. More commonly, a test comes to market as an LDT, where the test is developed and performed by a single laboratory, and where specimen samples are sent to that laboratory to be tested. FDA regulates only tests sold as kits and, to date, has practiced “enforcement discretion” for LDTs, which it defines as in-vitro diagnostics manufactured, developed, validated, and offered by a single laboratory. There are tens of thousands of LDTs in clinical use, and most cancer diagnostics are LDTs, Monzon said.
According to FDA, LDTs are supposed to be simple, well-understood pathology tests, tests used to diagnose rare diseases, or those for which testing outside the institution would be prohibitive to patient care due to delays between test ordering and delivery of test results. FDA does not consider a diagnostic test an LDT if it was designed or manufactured completely or
partly outside of the laboratory that offers and uses them. But Hayes said that next-generation sequencing tests are neither simple nor well understood. He added that sequencing tests are often conducted by large reference laboratories, to which institutions throughout the entire country send their samples, rather than by a single institution as part of the patient care services they offer. This can cause clinically significant delays, Hayes noted. “Our pathologists who are reading cases coming out of the operating room get a sample on Monday and they need to report a result on Thursday because that patient wants to get treated for their cancer within a few days. But for the LDTs that have to be sent out, there can be very extensive delays that can be prohibitive,” he said, noting that part of those delays are due to a lack of coordination in information management. He stressed that “the [LDT] regulatory issues need legislation because it’s going to be hard for us to solve this as physicians and scientists.”
FDA approval of diagnostics tests involves more rigorous oversight along two main regulatory pathways. One, called the premarket notification (510k) process, requires showing that the test (which is considered a device) is substantially equivalent to a device that is already in the market or was on the market before 1976, and that the test meets quality standards set by FDA. The 510k pathway can only be employed for tests with moderate levels of risk linked to their use. For more complex tests that pose more risk to patients, manufacturers must submit an application for Premarket Approval (PMA) to FDA that details the safety and effectiveness of their test. The test cannot enter the market until FDA reviews and approves this application. In its review of tests, FDA considers analytical and clinical validity, but not clinical utility (IOM, 2010a, 2012).
Conducting the studies required for either the 510K or PMA regulatory pathways can be quite expensive. Hayes noted although the 510K route is the less expensive route, it can still cost millions of dollars to carry out, and no NIH grants or other public funds are allocated for this purpose, so it requires private-sector involvement. Part of the expense of acquiring FDA approval for a biomarker test can be due to having to submit to FDA review not just the test, but the platform on which the test was done. For RNA sequencing tests, for example, FDA has only reviewed one machine used for the tests, but there are several other platforms on which the tests can be run, Hayes noted. Patricia LoRusso, professor of medicine and associate director of innovative medicine at Yale Cancer Center, added, “Not all platforms are created equal, even if you are going after the same targets.” Hayes noted that “the FDA hasn’t looked at Illumina sequencers for RNA or multiplex
PCR machines, so if you want to take a test forward in the full regulatory path, you’re going to spend a lot of money getting that machine approved as well, and that’s one of our challenges.”
Monzon was also critical of having to specify and acquire FDA approval for the instrument on which the test is performed. “Response to therapy is linked to the presence of a mutation or biomarker and not to the actual result of the instrument,” he said. Monzon suggested setting standards for the minimum performance needed to achieve a positive or negative result, but not requiring specific instrumentation because “innovation allows us to move forward and do better testing with better devices.” Martin noted that her group is working with FDA to “establish a novel regulatory framework that considers both the PMA predictive or clinical claims as well as the analytical claims so as to move from one test-one drug to one test-multiple drugs.”
Evolving Regulation of Laboratory-Developed Tests
Senderowicz noted that when FDA regulations for devices were implemented in 1976, a decision was made to exercise enforcement discretion with regard to premarket review of LDTs because most LDTs were relatively simple and considered low risk. Recognizing how tests have evolved since then, becoming both more complex and higher risk, in July 2014 FDA submitted a letter to Congress with proposals for regulating some LDTs. This letter noted the problems that FDA has identified with several high-risk LDTs, including claims not adequately supported with evidence, lack of appropriate controls in studies to evaluate the test, erroneous results, and falsification of data. This has resulted in faulty LDTs that could have led to patients being over- or undertreated for heart disease, cancer patients being exposed to inappropriate therapy or not receiving effective therapy, and incorrect diagnoses of serious conditions, such as autism, FDA stated. “So it’s a serious issue and I foresee there’s going to be significant changes in the regulation of LDTs,” Senderowicz said.
Litwack reported that FDA’s current proposal for regulating LDTs is to collect basic information on all LDTs through a new notification process and to use a public process (i.e., advisory committees) to obtain input on risk and priority for regulation. FDA would then phase in a new regulatory framework based on risk over a period of about 9 years, with regulatory guidances for LDTs considered highest risk issued first, followed by regulations governing those of more moderate and then those of lowest risk.
FDA would continue some enforcement discretion for specific categories it determines to be in the best interest of public health. It would also consider tests for unmet needs as well as other factors that might require special regulation.
In contrast, Monzon proposed maintaining LDT regulatory oversight within CMS rather than shifting it to FDA, and instead making CLIA regulations more stringent, with adverse-event reports part of the CLIA registry. He also said the registry should be made public and transparent. He stressed the importance of enabling continued innovation, such as allowing academic labs to use new discoveries of genetic drivers of tumor growth by quickly translating them into LDTs performed in a CLIA environment. “Reimbursement and regulatory pressures could constrain our ability to remain in a leadership position in diagnostics development,” he warned.
Senderowicz stressed that as the science evolves, development pathways and regulation also evolve. He cited the development of the breast cancer drug Herceptin and the associated biomarker tests as an example. The first diagnostic test for this drug was introduced in 1998 as a test for HER2 protein overexpression, but now there are 10 diagnostic tests to guide decisions about treatment with Herceptin. Some of these use different methods for detecting HER2 protein overexpression, while others use various techniques to detect amplification of the HER2 gene in tumors. In addition, next-generation sequencing is revealing new mutations in HER2 that previous tests could not detect, Monzon and Solit pointed out.
Litwack noted that the use of different FDA-approved tests can affect the quality of clinical trials, which often use local test results for patient accrual and subgroup analyses. Test results obtained with different technologies may not be comparable, and can affect clinical trial results. “We need to be aware of these issues and when we see a response rate in a clinical trial, it would be good to ask to what degree is that variability in response underlain by the test,” he said.
Given the evolving nature of test technology and FDA regulations, Litwack suggested that test developers consult with FDA early in the development process. “There’s no rule about when you have to meet with FDA—you can meet with them fairly early, even during the conceptual phase,” he said. He also stressed there is a lot of back-and-forth discussion between FDA and test developers during the review process, and that developers also can use FDA resources posted online, including relevant guidances on devices, and information about FDA’s Center for Devices and Radiological Health (CDRH).
In response to a question from Monzon asking how FDA would regulate a multigene panel, Litwack noted that although FDA traditionally has reviewed the data for each analyte separately in a multianalyte test, such data could not be expected for large gene panel tests. Instead FDA may request that a representative group of analytes be validated. He stressed that it would depend on a number of factors and recommended consulting with FDA to determine what data will be required for the test to be approved.
He also said, “Just because you’re using really new technology for a serious illness doesn’t actually mean you would necessarily need an investigational device exemption (IDE). If you or your institutional review board (IRB) determined it was a significant risk study, then we would require an IDE submission,” he said. But he noted that the level of acceptable risk can vary depending on the disease and the patient, and that risk is considered along with the potential benefit during the review. Just using a test to select therapy for a patient does not necessarily indicate a high-risk situation, he pointed out, and for patients who have exhausted all other treatment options, the risk is much less than it would be for patients in which the test would be used to determine first line treatment of breast or other cancers for which several effective therapies exist. The toxicity of the drug treatment that the test would indicate would be another factor considered, he added.
Litwack reported on how FDA cleared an innovative test that detects 139 variants of the cystic fibrosis (CF) gene. The test was run on a DNA sequencing instrument called MiSeqDx. FDA separated its review of the test from its review of the instrument on which the test was run, requiring only analytical validation for the latter. That validation was done using cell-line samples for normal controls and a representative set of samples with characteristic genetic variants to assess the performance capabilities of the instrument under different scenarios, such as sequencing regions rich in certain bases, sequences from different chromosomes with different proximities to the centromere, etc. After this testing was accomplished successfully, FDA cleared the sequencing device to be used for detecting hereditary disorders from genetic sequences in blood samples. But as Litwack noted, the device was not cleared for a particular indication for a specific disease, so it cannot be used without an FDA-cleared test. “You can’t just buy the MiSeqDx and start running tests on it and assume you’re compliant with the FDA—you need to develop a specific test for hereditary disease to be used on it,” he said.
Both analytical and clinical validation were assessed for the 139-variant-CF test run on MiSeqDx. The sponsor conducted the analytical validation
using samples with the 139 variants and normal sequences. Clinical validation was done using a CF database housed at Johns Hopkins University (see Table 1). According to Litwack, the database had several features that made it useful for regulatory purposes, including being expertly curated with preclinical and clinical data, having cooperation of the patient community, and being sustainably supported through various public and private grants. In addition, because there are good CF preclinical models, researchers could take a variant of interest discovered from the database, create the same variant in a cell line, and test whether the variant affected function.
TABLE 1 Clinical and Functional Translation of CFTR (CFTR2) Database
| Data Type | Information Gathered |
| Mutation Name/Associated Nomenclature | Provides a standardized mutation name and mutation by amino acid and nucleotide number (relative to the CFTR gene). |
| Associated Clinical Characteristics/Validation |
Provides the following relevant clinical characteristics:
|
| Functional Testing/Validation of Mutation | Notes the results of in vitro laboratory tests performed for applicable mutations. Specifically, assesses protein processing and maturation, CFTR dependent chloride current, and gene splicing. |
| Literature Review | Notes research previously completed on this particular mutation. |
| Annotation History | Provides a history of changes and timestamps of any revisions to the annotation. |
NOTE: CFTR = cystic fibrosis transmembrane conductance regulator.
SOURCES: Litwack presentation, November 10, 2014; http://www.cftr2.org (accessed March 18, 2015).
Based on this experience, FDA is currently considering the essential characteristics of a “regulatory-grade” database that sponsors could use to support the clinical validity claims of their tests. Such a database would probably have to be sustainable, well annotated, follow good practices, and ensure the quality of the testing data entered into it, according to Litwack.
Often biomarker tests in oncology are designed to be used in conjunction with specific targeted treatments, and both the test and the experimental treatment can be co-developed and tested simultaneously in clinical trials. Safety and efficacy of the new drug and of the new diagnostic are typically demonstrated in the same clinical trial, with the goal of simultaneous FDA registration for both the drug and diagnostic. Biomarker tests that are co-developed and co-approved by FDA with a drug in this way are known as companion in vitro diagnostics (IVDs). To date, this approach has been used to gain FDA approval for less than 20 companion diagnostics in oncology (see Table 2), including tests for BRAF, HER2-neu, and EGFR,4 Monzon said. Companion diagnostics provide information that FDA considers essential for the safe and effective use of a corresponding therapeutic product, and are intended for use in the collection, preparation, and examination of specimens taken from the human body. Approved drugs and their companion diagnostics refer to each other in their labels, as indicated in FDA guidance (FDA, 2011). An example of a co-development strategy was described by Karen Long, divisional vice president, medical, regulatory, and clinical affairs at Abbott Molecular (see Figure 2).
With the companion diagnostic pathway, manufacturers have to submit to FDA the analytic and clinical validity of their tests, their intended uses, and the settings in which the devices will be used, that is, in a clinical laboratory or point-of-care setting. If a diagnostic guides patient care, that is, has substantial importance in “diagnosing, curing, mitigating, or treating disease,” then manufacturers of the diagnostic must apply for an IDE so their diagnostic can be tested in clinical trials as part of the companion diagnostic co-development process.
FDA will grant this exemption if it determines the benefits of the test, such as indicating effective treatment, likely outweigh the risks, which could
_______________
4 See http://www.fda.gov; http://www.captodayonline.com (accessed March 18, 2015); http://www.ncbi.nlm.nih.gov/gtr (accessed March 18, 2015).
TABLE 2 List of FDA Approved Companion Diagnostic Devices for Oncology
| Companion Diagnostic | Device Manufacturer | Drug(s) |
| BRACAnalysis CDx™ | Myriad Genetic Laboratories, Inc. | Olaparib |
| Therascreen KRAS RGQ PCR Kit | Qiagen Manchester, Ltd. | Cetuximab, Panitumumab |
| DAKO EGFR PharmDx Kit | Dako North America, Inc. | Cetuximab, Panitumumab |
| Therascreen EGFR RGQ PCR Kit | Qiagen Manchester, Ltd. | Afatinib |
| DAKO C-KIT PharmDx | Dako North America, Inc. | Imatinib mesylate |
| INFORM HER-2/NEU | Ventana Medical Systems, Inc. | Trastuzumab |
| PATHVYSION HER-2 DNA Probe Kit | Abbott Molecular Inc. | Trastuzumab |
| PATHWAY ANTI-HER-2/NEU | Ventana Medical Systems, Inc. | Trastuzumab |
| INSITE HER-2/NEU KIT | Biogenex Laboratories, Inc. | Trastuzumab |
| SPOT-LIGHT HER2 CISH Kit | Life Technologies, Inc. | Trastuzumab |
| Bond Oracle Her2 IHC System | Leica Biosystems | Trastuzumab |
| HER2 CISH PharmDx Kit | Dako Denmark A/S | Trastuzumab |
| INFORM HER2 DUAL ISH DNA Probe Cocktail | Ventana Medical Systems, Inc. | Trastuzumab |
| HERCEPTEST | Dako Denmark A/S | Trastuzumab, Pertuzumab, Adotrastuzumab emtansine |
| HER2 FISH PharmDx Kit | Dako Denmark A/S | Trastuzumab, Pertuzumab, Adotrastuzumab emtansine |
| THxID™ BRAF Kit | bioMérieux Inc. | Tramatenib, Dabrafenib |
| cobas EGFR Mutation Test | Roche Molecular Systems, Inc. | Erlotinib |
| VYSIS ALK Break Apart FISH Probe Kit | Abbott Molecular Inc. | Crizotinib |
| COBAS 4800 BRAF V600 Mutation Test | Roche Molecular Systems, Inc. | Vemurafenib |
SOURCE: Adapted from http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.html (accessed March 18, 2015).
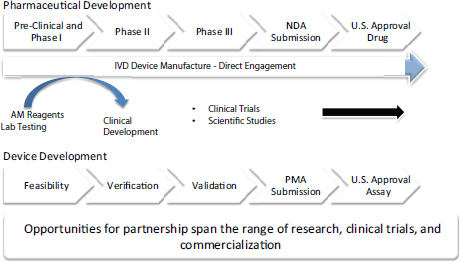
FIGURE 2 High-level strategy for drug and test co-development.
NOTE: AM = Abbott Molecular; IVD = in vitro diagnostic; NDA = new drug application; PMA = premarket approval.
SOURCE: Long presentation, November 11, 2014.
stem from false positives indicating unneeded treatments or false negatives that would prevent patients from having an appropriate treatment. Such risks and benefits vary according to the disease involved and its standard of care, Senderowicz noted. He added that researchers working with a drug that requires an IVD to predict response are advised to consider the companion diagnostic pathway and begin consulting with not only FDA’s Center for Drug Evaluation and Research or Center for Biologics Evaluation and Research, but also with CDRH, to facilitate submission of an IDE as soon as possible, enabling simultaneous review and approval of both the drug and diagnostic.
Litwack noted that historically, FDA regulation of companion diagnostics was designed for single analyte tests that indicated treatment with a specific companion drug, and not for next-generation sequencing-based tests with multiple analytes that could indicate use of multiple drugs. “We’re talking about over three billion bases in the human genome with millions of variants that any individual can have, so how are we going to analytically and clinically validate those?” Litwack asked. Monzon added, “The model for companion diagnostics is not sustainable in the era of multianalyte tests.”
Even using the existing co-development pathway for single analyte companion diagnostics can take a long time, Johnson pointed out, noting that FDA did not approve EGFR-targeting drugs for lung cancer until nearly 10 years after researchers first discovered EGFR mutations that could drive the growth of some of these lung cancers. However, Long said that such co-development can slash 18 to 24 months off the development time line for both the diagnostic and the drug. She pointed out that the relatively quicker companion diagnostic regulatory pathway requires close coordination and communication between those developing the diagnostic and those developing the therapeutic. “We talk to our partners on a weekly basis. If it’s a fast-paced study, we want to make sure all the resources are in the right place to make sure the project is successful,” she said, adding, “We fast-track everything we need to make sure that we can file our final application with the U.S. FDA at the same time the New Drug Application is filed.” There also is close cooperation with FDA early on in the development process to “keep everybody in the loop” and ensure the correct approach is being taken, Long said.
Monzon pointed out that after a test for a specific genetic variant is approved as a companion diagnostic for one type of cancer, evidence often surfaces suggesting it is also likely to be effective as a companion diagnostic for the same treatment used in a different type of cancer. But manufacturers have to clinically validate the tests for this new use in order for it to be listed on the drug’s label. Monzon said this requires finding a pharmaceutical partner willing to share patient specimens. Such sharing is often hindered by the rareness of the specimens and informed consent limitations, he said. “This is a huge challenge,” he said.
Litwack stressed that often DNA sequencing tests in oncology are used for discovery purposes, “and you don’t know exactly what it is you’re going to end up diagnosing.” The intended use of a test often determines its regulatory path, but sometimes the intended use will change during the course of development and review, he stressed. “When you run next-generation sequencing on somebody, you may have incidental findings and diagnose diseases or conditions other than the ones you originally started testing for, and how do we apply our regulatory framework when you don’t even want to define a very precise population?” he asked.
But it is possible to make changes during a test’s development process, Senderowicz noted, giving the example of the drug Crizotinib. This drug was at first thought to specifically target the enzymes MET and ALK, which are part of a tumor growth-promoting molecular pathway. A Phase I clinical
study was started with eligibility dependent on testing positive for genetic alterations of MET and ALK to select an enriched patient population likely to respond, but only a minimal response was seen in these patients. Then researchers discovered that a specific genetic rearrangement involving the EML4 and ALK gene was a driver of tumor growth. Consequently, researchers developed an LDT that detects this ALK gene rearrangement and enriched their next clinical trial of the drug with patients who tested positive for this biomarker. This trial showed more favorable results that led to the drug’s accelerated approval by FDA. But he stressed that the latter ALK-based test system still had to be “locked down” and could no longer be modified before being used in the registration clinical trial for FDA approval of the test.
Litwack also noted that next-generation sequencing tests are frequently modified due to rapidly evolving technology, and those modifications can affect performance. But he added that it is critical to preserve the ability to make modifications to allow for innovation and to accommodate specific testing needs (e.g., detection of single-base mutations versus detection of multiple copies or deletions of longer genetic sequences). In addition, there are no FDA-cleared next-generation sequencing testing systems for oncology, although there are a few cleared instruments. So laboratories are essentially cobbling together different components and customizing software to create their tests in what he termed a “mix and match” fashion. This can lead to a lot of variability in a test. Finally, he noted that some genetic variants are so rare that it is not possible to gather sufficient evidence of their clinical validity.
Another major challenge is the development of harmonized global regulation for genomic tests, as most pharmaceutical companies and diagnostic developers operate on a global scale. Martin noted that testing platforms need to be used and accessed worldwide, and thus the regulatory path for these diagnostics needs to be consistent globally. “We want to be able to work across not only the FDA, but to engage other health authorities, especially in Europe and Japan, who are seeking to gain more regulation for companion diagnostic tests,” he said. Long agreed, stressing, “We develop one product that is sold worldwide so we are dealing with many regulatory bodies around the world.”
However, the multiple agencies worldwide that regulate biomarker tests use different approaches to regulating products, Long said. For example, some countries approve a companion diagnostic without any clinical utility data, but once those data accrue from studies of clinical use, the label for
the diagnostic is adjusted accordingly. Japan and China both have complex regulatory approval processes and require country-specific clinical studies, Long noted, while other countries will accept certification of U.S. approval along with a technical file submitted to a regulatory authority.
CLINICAL IMPLEMENTATION CHALLENGES
Speakers at the workshop noted several challenges to effectively implementing molecularly targeted cancer diagnostics and therapies in the clinic, including
- Insufficient or inadequate tissue specimens;
- An overwhelming amount of data that are difficult to interpret and relay to patients;
- A lack of standards for and comparative effectiveness data on diagnostics;
- Time delays in acquiring test results;
- Lack of financial resources and a testing infrastructure; and
- Uncertainty over how to address incidental findings and report them to patients.
Insufficient or Inadequate Specimens
Hayes noted that many medical oncologists want to test their patients’ tumor samples to discover genetic alterations that may suggest treatment avenues. However, surgeons acquire those samples, and their primary goal is to minimize harm to the patient while doing a biopsy of the tissue. Consequently, the samples obtained for testing may be of poor quality and are often insufficient for conducting the multiple tests frequently needed to select treatment. Martin emphasized that this is why it is increasingly important to use a comprehensive test that can simultaneously detect a multitude of biomarkers to guide patient treatment or to direct patients to different clinical studies. Johnson agreed, noting, “The thing to do is to try to test for every gene you think you might need to know . . . in one fell swoop.”
Doing so, Levy pointed out, can result in “a tsunami of genetic data entering the clinic at a pace that we’ve never seen before as providers. Now with next-generation sequencing, we’re testing hundreds of genes all at the same time and we do not know what to do with all of that information that’s coming into the clinic. Clinicians are clearly overwhelmed and some are staying away from genomic tests for that reason. We’re stuck with having this massive knowledge gap of what we’re supposed to do with all this information coming at us today.”
Levy added that each of the markers associated with treatment response has variable levels of evidence, ranging from only preclinical data to data demonstrating clinical validity or utility, so it is not clear what the quality of the test is and what can be done with the information it provides. There is an urgent need for bioinformatics experts to analyze the data so they can be more useful in a clinical setting, several presenters suggested.
Siu agreed that clinical implementation of targeted cancer treatments is currently hampered by inadequate matching of those treatments to the genetic alterations detected in genomic tests. She cited one study of breast cancer patients that found genomic profiling of tumors only led to targeted treatment selection in 48 out of 404 patients. Such targeted treatments only proved beneficial in 13 (3 percent) of the patients (André et al., 2014).
Many patients’ tumors are not matched to treatments because of a lack of awareness of what treatments might work for their particular genomic profile, according to Siu. To increase that awareness, her institution, Princess Margaret Hospital, created a spreadsheet for each cancer patient that delineates all the individual genetic alterations detected in the tumor and the currently available and relevant clinical trials the patient could be enrolled in based on that genotyping. “We send this to our clinicians on a regular basis so they are constantly reminded that if they have a patient with this profile, they need to think about the clinical trial,” Siu said.
She suggested developing an app that can generalize this information and make it available across the entire country “so clinicians don’t forget to match their patients or try to find treatments for their patients.” She added that “a ‘genetic variant’ to a clinician means relatively little and to a patient, it almost means zero. It is important for us to use that information and make it work by finding an action that comes after the variant is discovered.”
Choosing the best test can also be challenging now that multiple tests often provide the same type of information, yet the comparative effectiveness of these diagnostics has yet to be determined. Furthermore, it is generally unclear how extensive a review process the test has undergone, several speakers noted. “How do we know, as the oncologist consumer who is ordering the test, that [the test] is really up to snuff?” asked Patricia Ganz, Distinguished Professor, University of California, Los Angeles (UCLA), Fielding School of Public Health, and director, Cancer Prevention & Control Research, Jonsson Comprehensive Cancer Center, UCLA. “Has the test actually undergone FDA review or is it just done in a CLIA-certified lab? How can the buyer beware?”
Monzon responded that CLIA regulations mandate that tests must be validated before they are put into clinical use, but the standards and criteria for test validation are determined by the medical director of each laboratory. To make that validation more transparent, NIH developed its publicly available Genetic Test Registry in which all genetic laboratories now provide their test validation data, he added.5 He suggested oncologists try to understand what type of test a lab is performing and request validation data. “It may be onerous, but it is very important so you can have the assurance that the laboratory that you are using has that appropriate validation,” he said.
But Ganz responded that genetic counselors and oncologists do not have the expertise nor the time to evaluate the validity of tests. “We are getting hounded with marketing by all of the companies that are doing the tests and we don’t have the time to investigate the quality, so there needs to be some quality control, some standards. There may have to be evaluations of the testing platforms, etc. The average oncology consumer is not going to be able to make those discriminations.”
Monzon agreed with the need to better define standards for tests and how their performance information is shared. Litwack added that how good a test is also depends on the disease it will be used for and other factors. He noted that FDA publicly posts its decision summaries regarding tests. Eberhard pointed out that some tests, though more accurate, are more difficult to conduct or require frozen tissue rather than the more common paraffin-embedded tissue, so they are a less favorable option for clinicians.
As for assessing the comparative effectiveness of tests, Holdhoff said
_______________
5 See http://www.ncbi.nlm.nih.gov/gtr (accessed March 18, 2015).
that would require a prospective study that compared different commercially available tests on the same patient samples, and companies are not likely to conduct such studies unless a regulatory body specifies that they must. But Herbst noted that companies were unexpectedly willing to collaborate in his Lung Cancer Master Protocol (Lung-MAP) study, which compares various targeted therapies for lung cancer.
As previously noted, because many tumor profiling tests must be performed in reference laboratories that are not located at the point of care, delays may occur that can limit the usefulness of some tests. Herbst said that is especially problematic when tumors from patients with advanced cancer are tested to assess their eligibility for clinical trials of targeted treatments. They may wait 2 weeks for a test result that ends up being negative, and then have to wait another 2 weeks for another test result. This is vexing for patients who have diseases in which the predicted survival is just a matter of months, Herbst pointed out. “There needs to be a better way to find the right drug for the right patient at the right time,” he said.
Solit noted that next-generation sequencing of tumors is currently done primarily at large academic medical centers. “It’s not that the general community does not feel that the testing is useful. It is more an issue that we can’t provide this testing to all of the patients because we don’t have the financial resources or even the infrastructure in place,” he said. “There are not enough sequencing centers set up or informatics experts, so we need to ration who is going to be tested. The way it is being rationed right now is whether you can afford to pay for it, or if you happen to be lucky enough to be at a medical center that has the technical capability and the institutional support to do it,” he added.
Several speakers pointed out challenges in reporting genomic test results to both physicians and their patients. “Digesting this information in a way that people, particularly people with very little time or background in this, can understand is a real challenge,” said Litwack. Deborah Schrag,
chief, Division of Population Sciences, professor of medicine, Department of Medical Oncology, Harvard Medical School, Dana-Farber Cancer Institute, called for reporting standards for genomic test results that would make it easier for clinicians to process them. She pointed out that there is consistent reporting of the results of a Complete Blood Count test of blood components. For example, the white blood cell count is always listed at the top of such a report. But reporting of the results of biomarker panels and their significance is quite variable. “Who is going to take ownership of this taxonomy?” she asked. “It can take a half hour sometimes to process these reports. I know where to find a platelet count, but I don’t always know where to find a BRAF mutation,” she said.
Monzon responded that both the American College of Medical Genetics and Genomics (ACMG) and CAP have standards for what information is reported, but there are no standards for the organization of that information. He said there are clear standards on how to categorize and report germ-line genetic variants, but there are no such standards for the genetic variations seen in tumor cells that are not congenital. He suggested that ASCO, the American Association for Cancer Research (AACR), CAP, and other relevant organizations develop those standards.
Levy noted that when Oncotype DX was developed, the usability of 80 different report formats were tested prior to entering the market. “This is an active area of research and not just in molecular labs. How do we best deliver this type of information? We are so early in the game that we are going to see a lot of variability in how we report these results and hopefully there will be organizations coming together to try to come up with these types of reporting standards,” Levy said. Monzon added that consumers have many different preferences on how they want to see information reported.
Another challenge is to decide whether and how to report incidental findings to patients. Incidental findings can result from a deliberate search for pathogenic or likely pathogenic alterations in genes that are not apparently relevant to the diagnostic indication for which sequencing test was ordered. LoRusso noted that incidental findings may be of medical value or utility to the ordering physician and patient. One survey LoRusso cited found that most breast cancer patients would want genomic profiling if it
were offered to them, even if it might discover incidental findings or findings relevant to their tumor for which there is no targeted treatment.6
Incidental findings are likely to be relatively rare, but not insignificant, LoRusso pointed out. One study entailing germline whole exome sequencing of 1,000 patients found that the rate of incidental findings was 2.3 percent, she noted (Dorschner et al., 2013). “It’s going to lend somewhat of a paradigm shift to how we can practice oncology and what knowledge base we will need to know as medical oncologists or clinicians who have to deliver this data. We have to be proactive and think about this now rather than later,” she said.
In 2013, ACMG published recommendations for the reporting of incidental findings in genomic sequencing tests, but there were no oncologists or members from cancer professional societies in the working group that developed these recommendations, LoRusso pointed out. The guidelines identified a list of 56 mutations not related to a cancer diagnosis that should be reported by the lab to the ordering clinician regardless of the indication for which the sequencing was ordered. ACMG guidelines also stated that the ordering clinician or research team should be responsible for providing comprehensive pre- and posttest counseling to the patient.
The recommendations reflected the limitations of current technology and focused on disorders caused by single-base mutations or the insertion or deletion of a small number of bases often seen in inherited disorders, and not the structural variations and copy-number alterations that are commonly found in tumors. The fifth major recommendation was that the working group refine and update the list of actionable mutations at least annually, and in 2014 that working group added the recommendation that patients should have the opportunity to opt out of a search for medically actionable germ-line variants unrelated to the condition that led the patient to undergo a genomic sequencing test.
But the ACMG guidelines left many clinical questions unanswered, LoRusso noted, especially how to report incidental findings to patients. “As a medical oncologist, I don’t feel comfortable discussing incidental findings with my patients given that they all have advanced metastatic disease,” she said.
Many in the oncology community view the ACMG recommendations as unwarranted or excessively burdensome, LoRusso pointed out, and are critical of ACMG viewing tumor profiling tests to be equivalent
_______________
6 Personal communication, E. Hofstatter, Yale University, 2014.
to germ-line tests. Such cancer tests already have significant technical and interpretative challenges due to tumor heterogeneity and genetic changes over time, and huge datasets that require appropriate bioinformaticians. “When you are dealing with that massive amount of data, adding incidental findings is another major hurdle,” she said. Test reports on somatic (nonhereditary) genetic variants in tumors typically involve no specific analysis or annotation of germ-line variants, and most labs doing such testing do not have germ-line reporting capabilities because tumor and germ-line sequencing require analogous but different setups, including distinct bioinformatics methods and specialized personnel for interpretation and clinical reporting, one study noted (Parsons et al., 2014). Consequently, detecting and reporting germ-line variants likely will be more time consuming and twice as costly to do, with part of that cost possibly not being reimbursed, compared with detecting just tumor variants, LoRusso pointed out. “The patient could die before we get all these results back,” she said.
Particularly troublesome are incidental germ-line mutations that have treatment implications not just for the patient being tested, but for their families as well. Siu noted that in her clinic, patients indicate prior to being tested if they wish to be informed of incidental germ-line findings and if they do, genetic counselors convey those results to them. Martin added that at GSK, when researchers discover germ-line mutations in genetic testing, they notify the IRB that is overseeing the research and then the investigators so there is a consistent process to follow if they need to notify the patient. “But what is the physician’s obligation to report data to the family of a patient who has died?” LoRusso asked. She noted that for many patients with advanced cancer, non-cancer related diagnoses are probably not that meaningful, but they might be meaningful for their families.
LoRusso stressed that reporting incidental findings to patients will require genetic counselors and additional clinic time. Clinics will need to have the expertise and resources to provide follow-up care for those patients found to have incidental findings outside the realm of cancer. “You either have to refer that patient out to another institution or to another investigator within your own institution, or you have to start developing a team approach to treating these cancer patients,” LoRusso said.
No studies have been done to show that the benefits of reporting incidental findings to patients outweigh their costs, she said, although a few preliminary studies have found that patients or their families would like to know or appreciate knowing about these findings. Few data are available on how to educate patients and communicate genetic findings, nor
are there many communication or education tools to aid this endeavor. What remains unclear is whether it is useful to give patients limited datasets or whether it is better to provide more complete data that they could use to seek second opinions. However, as LoRusso noted, even clinicians are not given complete datasets and the relevance of a lot of those data are unknown. “These genetic differences may not be relevant ever or we may find their relevance somewhere down the line,” she noted. In addition, genetic findings from tumors can change over time as the cancer evolves with new mutations, yet “when you give the patients the results, many of them stick to them as if they are the Bible for their survival, so we really have to watch how we end up reporting these data to the patients and what we promise them as a result of getting these data,” LoRusso said.
Another challenge is when a test shows an unexpected genetic variant suggesting that a patient’s tumor is likely to respond to a treatment that is not yet approved for that indication, such as Herceptin for lung cancer, and thus may not be reimbursed by an insurer. “Once you have the data, what is your obligation?” asked LoRusso. “We do try to get these drugs on a compassionate-use basis, but a lot of times there are standard-of-care drugs and it’s difficult to get reimbursement for others,” she added.
Katharine Phillips, professor of health economics and health services research at the University of California, San Francisco (UCSF), and director of the UCSF Center for Translational and Policy Research on Personalized Medicine, noted that the media and the diagnostic companies tend to portray the concept that more genetic information is better than less and that it can be inexpensively acquired, giving patients unrealistic expectations of genomic profiling. LoRusso agreed, saying, “They talk about the $1,000 genome, but they don’t tell you that it takes $10 million to buy the equipment to give you the $1,000 genome. They advertise personalized medicine like it’s the cure for cancer, but they don’t tell you all the challenges that we are still facing trying to get the answers to those questions.”
Bill Gradishar, Lurie Comprehensive Cancer Center of Northwestern University Division of Hematology/Oncology, agreed, saying, “The key missing link is that often there is not a lot that is actionable yet unless you are willing to send your patients to the other side of the country, even though the companies are promoting these tests as the be-all and end-all.” LoRusso added that many community oncologists or even sometimes internists “are buying into profiling, even though they cannot act on most of the results. A study from the Cancer Treatment Centers of America, which profiles all their patients, found they could only act on 2 or 3 percent of the
profiles they acquired. But they advertise on all the billboards ‘we profile your tumor.’ It’s a lot of money spent for nothing.”
Molecular profiling tests for cancer offer major reimbursement challenges, including
- A lack of evidence regarding clinical validity and utility;
- Off-label and investigational uses;
- Downstream costs, including the costs of dealing with incidental findings; and
- Greater expense without evidence of offering greater value.
Donna Messner, vice president and senior research director, Center for Medical Technology Policy, said that payers expect data to show that a test is medically necessary and that the results are actionable and improve clinical outcomes, as well as data on how the test compares to others already used in standard care, or to use of no test at all. For their reimbursement determinations, payers rely on evidence from a number of sources:
- Peer-reviewed studies published in medical journals
- A review of available studies on a particular topic, such as reviews done by the Agency for Healthcare Research and Quality, Blue Cross/Blue Shield Technology Evaluation Center, or the Duke Evidence-based Practice Center
- Evidence-based consensus statements or guidelines from professional societies or other nationally recognized health care organizations, such as ASCO or NCCN
According to Messner, payers prefer regulatory mechanisms to ensure adequate clinical validation. “FDA-cleared tests with drug label information are typically persuasive for payers, but not always,” she said. When the strength of the evidence is not ideal, expert clinical opinion and physician practice patterns can be persuasive, especially when the medical need is great, she added.
But many biomarker tests are LDTs that do not undergo regulatory
review, and these LDTs are increasing in number, complexity, and cost, Messner pointed out. Many validation studies done on tests are inadequate or flawed, she said, and there is a lack of shared understanding of what kind of evidence is needed to show the value of such a test in the clinic. “We are swimming in new genetic variants that are being discovered all the time, with the potential downstream consequences of wrong decisions uppermost in the mind of payers. Closer scrutiny is needed even as these tests proliferate,” she said.
Messner reported on a 2012 study of payer policies which concluded that “the low number of disease-related genomic tests considered for coverage by insurers is likely due to the few studies published demonstrating clinical utility, the often small role of genetics in complex diseases, and availability of alternative effective screening methods.” Sean Tunis, President and Chief Executive Officer, Center for Medical Technology Policy, added, “It’s not that payers are looking for a way to not pay for these tests as much as they are looking for some kind of consensus that there is an evidentiary framework that distinguishes tests that are useful from tests that are not. If there were more regulation of tests by the FDA, there probably would be a lot more willingness for payers to reimburse them. I don’t think most payers feel like they have the technical abilities to fill in for the FDA.”
Phillips said her study found that although payers covered tests for BRCA 1 and 2 mutations to assess genetic susceptibility for developing breast and ovarian cancer, they often did not cover genetic panels that included BRCA gene testing, and 64 percent of payers with reimbursement policies on gene panel tests classified these types of tests as investigational, and thus non-reimbursable (Clain et al., 2015). “But everyone is moving to panels, even Myriad, which developed the BRCA tests, so clearly this needs to change,” she said.
Phillips asserted that one of the major barriers to reimbursement for innovative tests has been that payers cannot tell what tests are being done when, how, on whom, and what the outcomes are because there are no established codes for the tests that enable tracking of the tests in databases. The advent of the new CPT codes might help payers better assess the clinical utility of the test, she suggested. Dickson noted that the new codes categorize tests so that payers can distinguish tests that sequence 5 genes from those that sequence 50, for example, but how much sequencing is needed for each type of cancer is not known. He also stressed that the codes do not indicate performance variability between laboratories testing for the same analytes. He suggested that it may be warranted to develop a specific code
for each individual test, to better assess clinical utility. “The new CPT codes are a step forward, but they probably don’t address the payers’ needs yet,” he said. Messner added that coding helps payers to know more accurately what they are paying for, but ultimately it is still evidence reviews that tell payers whether or not a test or treatment has clinical utility.
Investigational and Off-Label Uses
Several presenters stressed that Medicare cannot reimburse for experimental treatments, and will not reimburse for associated biomarker tests that are not standard of care. Messner also noted that other payers are reluctant to reimburse off-label, test-directed treatments for cancer because such practices have yet to be clinically validated. Solit said even if the treatments are effective, they might not be covered. For example, he said a study had indicated that about half of patients with Erdheim-Chester disease7 have BRAF mutations. But he was unable to acquire reimbursement for an Erdheim-Chester patient who tested positive for the mutation and had a complete response to subsequent treatment that has lasted more than 2 years. The insurer said that BRAF testing and treatment was not standard of care for the disease so therefore it could not pay for it.
Next-generation sequencing is generally seen as investigational by payers, but Messner noted that one payer, Priority Health in Michigan, is in the process of developing reimbursement policies that would cover such sequencing tests for specific clinical situations as well as test-directed, off-label use of targeted therapies in the context of clinical trials, akin to coverage with evidence development. Dickson noted that the Affordable Care Act requires coverage of routine care costs associated with enrollment in clinical trials, but not for experimental treatments or tests. Thus, screening tests to determine eligibility for a trial would likely not be covered. “It depends on the protocol,” he said. This is a dilemma because academic institutions and cancer centers often require screening biomarker tests to determine eligibility for clinical trials of targeted cancer treatments, but payers will often not reimburse such testing, noted Jennifer Malin, medical director of oncology for WellPoint, Inc. When she asked who is paying for such testing then, Herbst responded that his institution “eats a lot of the cost.”
_______________
7 A rare disease characterized by the abnormal multiplication of a specific type of white blood cells called histiocytes.
Phillips reported that when she met with a payer board that included representatives from all eight of the largest U.S. private health plans, she found that they see the potential benefits of next-generation tumor sequencing and recognize it as a revolutionary trend in health care. “But they are not quite sure how to pay for it at this point,” she said. Eighty percent said such genetic tests do not fit their definition of medical necessity, and 70 percent viewed gene panels as bundles of individual gene tests for which every gene marker in the bundle needed to be evaluated separately, versus viewing the overall genetic pattern as more important, with the sum being greater than the parts. Seventy percent were skeptical about the new evidentiary methods being used to evaluate biomarker panels. She quoted one payer as saying, “Personalized medicine has a lot of promise, but nothing frightens managed-care folks more than hearing the word ‘promising’ from an oncologist. That means that they want us to pay for something expensive that they don’t have any supporting data for yet” (Trosman et al., 2015).
Another dilemma payers have is determining the minimum number of genes that need that to be analyzed to select the best treatment, and how to prioritize genetic findings, that is, which genetic alterations are drivers of cancer growth and should be targeted with treatment, and which are inconsequential passengers, several participants noted. As Dickson said, “At some point it will be the same cost to test 5 genes as it is to test 20 or 500 genes. Does that mean that we need to be looking at 500 genes and actually be paid more than for 5 genes?”
Schilsky said that payers often will not pay for follow-up of incidental findings on genomic tests, and he questioned why this is so, giving the example of computerized tomography (CT) abdominal scans for cancer that often reveal incidental findings in organs other than the original organ of interest. “Payers don’t say, ‘Why should I be paying for these images of the kidneys and the lymph nodes, when all the doctor really wants to know is what is happening in the liver?’ Why is genomic profiling different? We are getting more information than perhaps the doctor is immediately going to use. We are finding incidental findings. It’s exactly the same as when CT scanning was introduced in the 1970s,” he said.
Phillips responded that “we’re really talking about two different things. We’re talking about payers covering panels where you are deliberately getting more information than you are looking for, versus when we happen to be
looking for something and we discover an incidental finding. It’s important to keep those two separate, but this issue is not really brand new and there should be lessons we can learn from previous experience that we can take forward to resolve these issues.” Malin gave another example, saying it was once routine to order a test called a Chem-20 that often indicated abnormal protein albumin ratios. “We routinely did bone marrow biopsies on a whole bunch of people who didn’t need it just because we scattershot-ordered the test. Costs aside, I think there are reasons to be clinically thoughtful about when it makes sense to order a scattershot test versus when we should really be hypothesis-driven,” she said.
Phillips noted that NIH is currently funding the development of a publicly available registry of reimbursement coverage policies that will include what and where tests are available, how much they cost, and what insurers are paying for them.
Greater Expense, But Not Necessarily Greater Value
Although Solit suggested that if diagnostic tests were less expensive, insurers would be more likely to reimburse them, Messner stressed that cost effectiveness is not part of the initial evidence review payers conduct. However, Malin said that for WellPoint-affiliated health plans, the costs of drugs for cancer patients comprise about one-quarter of the total cost of cancer care, while physician visits only comprise about 3 percent, so there is a great need to figure out if the resources being spent on biomarker tests and the associated targeted treatments are contributing to greater value and outcomes for patients.
Part of the reason more genetic tests have not been reimbursed may be because most of the “actionable” genetic alterations these tests uncover do not yet translate into better clinical outcomes, Gradishar noted. Malin pointed out that most new targeted treatments for cancer are expensive without dramatically improving clinical outcomes. In 2012, for example, FDA approved 13 new targeted treatments, but only 1 extended survival by more than 6 months, and only 2 extended survival by more than 4 to 6 weeks, at an average cost of $6,000 per month (Emanuel, 2014). Older studies found that although outcomes in lung cancer patients did not substantially vary by treatment, the cost did (Patel et al., 2012; Reck et al., 2010; Sandler et al., 2006; Scagliotti et al., 2008; Socinski et al., 2012).
Phillips added that she has found “almost no evidence on whether these genes provide value, not only in the clinically actionable sense, but
in the economic trade-off of benefits and cost. There is a huge dearth of knowledge.” She stressed that “we need more evidence,” including evidence of a test’s operational utility, meaning can providers and clinics use the test and do they have the infrastructure to support it? There is still a lack of agreement about what the standard of evidence should be for genetic tests, she said.
Phillips added that another study she conducted found that genetic tests in general, not just in oncology, provide benefits, but at a higher cost. This is also true for most pharmaceuticals, she noted (Phillips et al., 2014). “Very few interventions in health care save money. They provide better health at higher cost,” she said. Payers are generally willing to pay for a higher cost of treatment if that treatment has greater value than standard of care, but Phillips noted it is challenging to figure out the economic value of multiplex and genetic panel tests because each marker has its own pathway of benefits and costs, and one must consider not only the test itself, but every treatment that happens downstream from test use. Other variables to consider are how the benefits and costs are going to vary based on how the test is used, what sequencing and analytic methods are used in the test, and how results are reported. In addition, there is a “personal utility” value of a test that should be factored in if patients see a value to knowing a test’s results even if they are not actionable. Benefits and costs are also accrued to family members for germ-line mutations.
Phillips also noted that payers are perhaps most concerned about the downstream cost of genetic testing, including additional tests, off-label treatments, etc. “In talking with payers it really doesn’t seem like they are concerned about the cost of the test, but about what occurs once you have given the test,” Phillips said, because many new targeted cancer therapies are very costly (IOM, 2014). On the other hand, tests can potentially spare patients the cost and side-effects of using therapies that are not likely to benefit them.
GATHERING THE EVIDENCE: INNOVATIVE CLINICAL TRIALS
The ability to detect numerous genetic anomalies in cancers is not equally matched by the ability to understand what those molecular flaws mean clinically, several presenters pointed out. Clinical studies are needed to assess this, but few studies are being conducted.
Siu pointed out that many cancer patients whose tumors have been profiled opt not to participate in a clinical trial of a targeted therapy for
various reasons, such as a lack of relevant experimental drugs, a decline in their performance status, or unwillingness to travel to a trial site. But many patients do not participate in clinical studies because they and their providers are not aware of what studies are available and for which they might be eligible, Siu said. One study found that among 283 patients with advanced cancers whose tumors were genetically profiled, only 30 percent were matched to a targeted treatment being tested in a clinical trial (Ferté et al., 2013). At her own institution, Princess Margaret Hospital in Ontario, only about 6 percent of the 2,000 cancer patients enrolled in the genetic profiling programs known as IMPACT or COMPACT ultimately are entered into early-phase clinical trials of a targeted therapy. “Many institutions across the world already have genetic profiling programs, but taking the next step to translate that profiling into benefit is much harder—showing what we are doing is important to the care of the patients is a much higher bar to reach and is where we should be focusing our time and energy,” Siu said.
She showed how researchers are trying to better match their genetically profiled cancer patients to experimental therapies by using two types of innovative clinical trial designs, umbrella trials and basket trials (see Table 3). An umbrella trial enrolls patients with one specific tumor type, such as refractory lung cancer, profiles the tumors, and treats them with different therapies, each targeting a different biomarker profile, in the same trial. By contrast, a basket trial usually groups together patients with several different types of cancers, but with a similar biomarker profile in their tumors so they can receive a treatment that targets those molecular drivers.
Siu noted that people with rare tumors are likely to be studied in basket trials, whereas people with more common cancers might do better in umbrella trials. Solit added that basket studies offer an opportunity for patients to receive targeted therapies for less common but not necessarily rare tumors, such as bladder cancers, because pharmaceutical companies have focused on testing targeted treatments on more prevalent cancers. Both basket and umbrella trials enable the testing of multiple treatments using the same protocol in the clinic. “Basket and umbrella trials allow us to physically and financially be sustainable because they are more economical,” Siu said.
Solit provided a general overview of basket trials along with some examples of specific trials under way. Solit said that basket trials evolved in part
TABLE 3 Selected International Trials to Match Patients Based on Molecular Profile
| Program Name | Led By | Tumor Types | Trial Type |
| I-SPY 2 | NIH | Breast | Umbrella |
| Lung-MAP | NCI | Squamous lung | Umbrella |
| ALCHEMIST | NCI | Adenocarcinoma lung | Umbrella |
| FOCUS 4 | Cancer Research UK | Colorectal | Umbrella |
| ASSIGN | NCTN | Colorectal | Umbrella |
| SAFIR-01 | Gustave Roussy | Breast | Umbrella |
| NCI-MATCH | NCI | Advanced solid tumors | Basket |
| NCI-M-PACT | NCI | Advanced solid tumors | Basket |
| Signature | Novartis | Advanced solid tumors | Basket |
| My Pathway | Genentech | Advanced solid tumors | Basket |
| Princess Margaret Mobility Series | 002-Bedard (GSK) 003-Razak (BI) | Pancreas/GI Advanced solid tumors | Basket |
NOTE: BI = Boehringer Ingelheim; GI = gastrointestinal; GSK = GlaxoSmithKline; NCI = National Cancer Institute; NCTN = National Clinical Trial Network; NIH = National Institutes of Health.
SOURCE: Siu presentation, November 10, 2014.
from the discovery of what he called “extraordinary responders.” These are patients who respond remarkably well to a targeted treatment even though most other patients with their type of cancer do not. Genetic profiling done on such outliers often reveals a mutation that is rare for their particular type of tumor, yet from a molecular pathology standpoint is well matched to the targeted treatment to which they responded. The study of extraordinary responders led to the awareness that the same driver mutation can occur in different tumor types, but may be quite rare in some tumor types.
For example, Solit described a patient with recurrent ovarian cancer who was enrolled in a clinical trial of a drug that targeted the tumor driver MEK1. She was the only patient who had a complete response, which so far has lasted 5 years, and there were few other partial responses, with most of the trial results considered disappointing. When Solit used next-generation sequencing to analyze the tumor of the patient who responded so well, he discovered that she had a deleted sequence of bases in a gene that encodes MEK1. Laboratory studies determined that this mutation locks the MEK1
protein into a constitutively active conformation that can be inhibited by the MEK1-targeted treatment. Subsequent sequencing studies discovered melanoma and lung cancer patients who had the same MEK1 mutation and were likely to respond to the same MEK1 inhibitor to which the patient with ovarian cancer responded.
To find and test other patients with rare mutations, Solit has since developed basket trials for patients with all types of cancers that harbor specific mutations. For example, in one study, patients found to have BRAF mutations in their tumor were treated with an experimental therapy to inhibit the BRAF protein. Similarly, he designed another basket trial of neratinib, which dually inhibits the tumor driver genes HER2 and EGFR (see Figure 3). He described one breast cancer patient in this study who, despite her tumor testing negative for HER2 amplification, had a specific and rare type of mutation that caused excessive HER2 activation. This patient had a complete response 2 months after treatment was begun, he said.
Williams also described the NCI-MATCH basket trial. This complex study has 22 different treatment arms in which the mutations present in
FIGURE 3 Neratinib basket study schema.
NOTE: EGFR = epidermal growth factor receptor; HER = human epidermal growth factor receptor; MSKCC = Memorial Sloan Kettering Cancer Center; OS = overall survival; PFS = progression-free survival.
SOURCE: Solit presentation, November 10, 2014.
FIGURE 4 NCI-MATCH study schema.
NOTE: CR = complete response; PD = progressive disease; PR = partial response; RECIST = Response Evaluation Criteria In Solid Tumors; SD = stable disease.
1 CR, PR, SD, and PD as defined by RECIST.
2 Stable disease is assessed relative to tumor status at re-initiation of study agent.
3 Rebiopsy; if additional mutations, offer new targeted therapy.
SOURCE: Williams presentation, November 10, 2014.
patients’ tumor samples are matched to a targeted treatment, Williams reported (see Figure 4). Genetic alterations identified by next-generation sequencing in tumor samples include mutations, gene amplifications, and fusions, and will determine eligibility in the trial and which treatment patients receive. Patients whose cancers progress while on experimental therapy have the option of being rebiopsied and being placed in a different treatment arm if the mutations detected suggest that another treatment arm would be appropriate.
Williams said all four participating clinical laboratories at different institutions will be running the same next-generation sequencing tests on the tumor samples. A component of test validation entails demonstrating that each lab produces the same sequencing results on a given sample. The test detects a minimum of 4,048 known and annotated genetic variants, but at FDA’s suggestion in presubmission consultations, the researchers will only be demonstrating analytical performance with a representative subset of these variants, including those likely to occur most frequently, as well as those most difficult to sequence. Each laboratory will determine the
sensitivity of the test on five gene variant classes and the specificity on all reportable variants using five cell lines. Repeatability and reproducibility on different instruments will also be assessed, and researchers at the MD Anderson Cancer Center will conduct a “fit-for-purpose” analysis by running specimens through the entire research pipeline prior to launching the study.
Anticipating that germ-line mutations will be detected in the trial, there is an established working group on incidental findings that has engaged with genetic counselors, reported Barbara Conley, associate director, Cancer Diagnosis Program (CDP), Division of Cancer Treatment and Diagnosis (DCTD), NCI.
Herbst described the Lung-MAP trial as a Phase II/III trial that entails a public-private collaboration among institutions participating in NCI’s National Clinical Trials Network, nonprofit organizations such as the Foundation for the National Institutes of Health and the Friends of Cancer Research, and several drug companies, all of which will provide funding for the $150 million trial. This trial’s ultimate objective is to identify efficacious targeted therapies matched to response-predictive biomarkers in patients’ lung tumors and quickly gain FDA approval. The study was designed as a model umbrella study whose protocol could be adapted for use in similar trials, Herbst noted.
The Lung-MAP protocol compares new targeted therapies to the standard of care for lung cancers. All patients’ tumors will be screened for trial entry and placement in subgroups based on the molecular alterations detected. By providing a “one-stop” platform, the trial aims to improve the time lines for clinical trials by maximizing the number of eligible patients in a single protocol, Herbst said. The protocol schematic can be seen in Figure 5.
Herbst described several advantages of the protocol, including
- Enrollment efficiency—Grouping multiple drugs and biomarkers under a single trial increases the probability that patients will be eligible for the trial based on their biomarker profile.
- Operational efficiency—The single master protocol can be amended as needed as drugs enter and exit the study.
- Consistency—Every drug entered into the trial will be tested in an identical manner.
- Predictability—If prespecified efficacy and safety criteria are met, the drug and accompanying companion diagnostic will be approved by FDA.
- Patient benefit—the goal is to bring safe and effective drugs to patients sooner than they might otherwise be available.
Following discussions with more than 20 drug companies, a drug selection committee (composed of lung cancer experts and stakeholders not employed by the drug companies developing the experimental therapies being considered) chose 5 experimental therapies from 5 companies to be tested in Lung-MAP, as shown in Table 4. The experimental drugs were chosen based on Phase I and II data showing some evidence that they were safe and active in lung cancer. The companies whose drugs will be tested are providing three-quarters of the funding for the trial, including funding for the sequencing and new biopsies when patients progress on therapy. This will enable Lung-MAP to accrue patients at community sites without the resources to do such screening and repeat biopsies.
Lung-MAP began accruing patients in June 2014. As of November 2014, the trial had IRB approvals at 353 sites. Thirty-five sites have
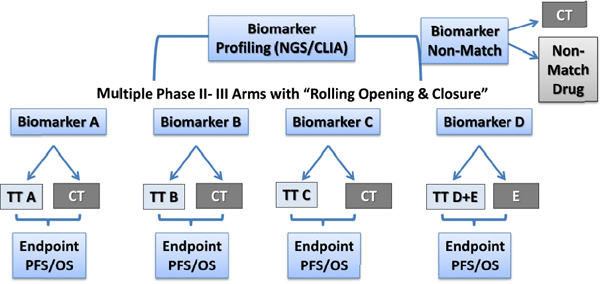
FIGURE 5 Lung-MAP study schema.
NOTE: CLIA = Clinical Laboratory Improvement Amendments–certified laboratory; CT = chemotherapy (docetaxel); E = erlotinib; NGS = next-generation sequencing; OS = overall survival; PFS = progression-free survival; TT = targeted therapy.
SOURCE: Herbst presentation, November 10, 2014.
TABLE 4 Lung-MAP Drug Selection Committee Nominations
| Drug | Company | Target |
| AZD4547 | AstraZeneca | Fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor |
| GDC-0032 | Genentech | PI3K pathway inhibitor |
| MEDI4736 | MedImmune | Anti-PD-L1 monoclonal antibody |
| Palbociclib | Pfizer | CDK 4/6 inhibitor |
| Rilotumumab | Amgen | Hepatocyte growth factor receptor/c-met inhibitor |
SOURCE: Herbst presentation, November 10, 2014.
accrued at least 1 patient, with nearly 100 patients enrolled within the first 3 months. Lung-MAP eventually will run at more than 500 sites in the United States and Canada.
Challenges with Basket and Umbrella Trials
Basket and umbrella trials present several challenges that researchers are trying to overcome, including
- The difficulty of finding enough patients who have the rare molecular subtypes of cancers likely to respond to experimental targeted treatments;
- The need for competing drug sponsors and research institutions to collaborate;
- The difficulty in treating a moving target, given the heterogeneous and dynamic nature of tumors; and
- The uncertainty about how to prioritize genetic targets for treatment.
Sufficient Patients for Trials
Most cancer patients are not aware their tumors have rare mutations that might respond to certain targeted therapies. Solit said that nearly all breast cancer patients with HER2 mutations he identified in his study had
no prior knowledge of that fact because standard breast cancer tests only detect HER2 amplification or overexpression, but not specific mutations within the gene. Consequently, a key challenge for basket trials is doing the widespread genomic screening that enables clinicians to detect such rare mutations that may respond well to approved or experimental targeted therapies.
Solit estimates that to complete the neratinib study outlined in Figure 3, 30,000 to 40,000 patients will have to be screened, an enormous undertaking that cannot be accomplished at a single institution. Consequently, in addition to screening cancer patients at his own institution, Memorial Sloan Kettering Cancer Center, he is collaborating with the MD Anderson Cancer Center, Foundation Medicine, and other institutions or companies that do genomic profiling of large numbers of patients regularly. This screening can identify patients that have any mutations in HER2 who can then enter the neratinib trial. “Once you start screening every single patient for mutations, finding things that are rare becomes relatively easy,” he said. But Siu raised the issue that there may not be enough resources and patients to do an umbrella or basket study for every question clinical trialists would like to answer.
In the neratinib study, Solit is also assessing outcomes for subgroups of enrolled patients according to the type of HER2 mutation they have. He suggested that a subgroup response rate that is high enough and durable enough might be sufficient to warrant an immediate change in clinical practice given that there never will be enough patients with these rare mutations to support a randomized clinical trial of the treatments. He estimates only 100 patients per year in the entire country may have such rare mutations, so the only feasible way to run such a trial and other similar basket studies is to separate the screening protocol from the treatment protocol. This is a polarizing concept, he noted, because it raises the issue of who is going to pay for the screening. Drug companies generally will not pay for it if the screening is not part of the therapeutic protocol, he noted, and often the institutions where the trials are run or the screening is done have to financially support the genomic profiling.
To deal with the challenge of finding patients with rare mutations to enroll in trials, GSK, Pfizer, and other companies recently collaborated to develop a global master screening network, Martin reported. The pharmaceutical companies are gathering input from multiple precision medicine stakeholders, including NCI, academia, patient advocacy groups, as well as regulatory agencies and payers, with the goal of enabling standardized and
shared genomic screening of patients among pharmaceutical companies to create more efficient and cost-effective clinical testing, especially in regard to identifying rare patient populations. Another goal of the network is to advance a regulatory framework to support the dissemination of next-generation sequencing in the marketplace, Martin reported.
The master screening network Martin described, which is still being developed, will use laboratories throughout the globe to collect and profile a pool of patient samples using the same standard operating procedures at central reference labs (see Figure 6). It will also provide a way for clinical centers that lack the ability to run genomic profiling tests to send in their samples for testing at another facility that is part of the network. Commercialization of tests in the future could then be expediently accomplished using the same labs in the network once the associated targeted therapies achieve regulatory approval.
Need for Collaboration
Basket and umbrella trials require collaboration across institutions, companies, and areas of expertise. Siu noted that although companies traditionally have not shared their compounds and data with other companies, that non-sharing culture is changing. “We already see companies coming together to be part of the same umbrella or within the same basket to test their drugs at the same time,” she said. But getting multiple disease teams to work together within a basket trial can also be challenging, Solit noted. “The breast group wants their own study, the lung group wants their own study, etc.,” he said. Regular communication among collaborating institutions is essential, Siu pointed out. Herbst said that for the Lung-MAP Trial, “It wasn’t easy working with both the public and private groups, bringing all the academics into one trial, and bringing all the Cooperative Groups together, but it is working and I hope that this sort of cooperation and solutions to some of the financial issues we have worked out will help other groups.”
Tumor Heterogeneity
A particularly difficult challenge is the heterogeneity of tumors and their evolving genetic makeup. Meeting this challenge might require simultaneously sampling multiple tumor sites in the same patient, as well as resampling over time, to ensure a representative sample of all the molecular alterations that therapies need to target, Siu pointed out. “Eventually can-
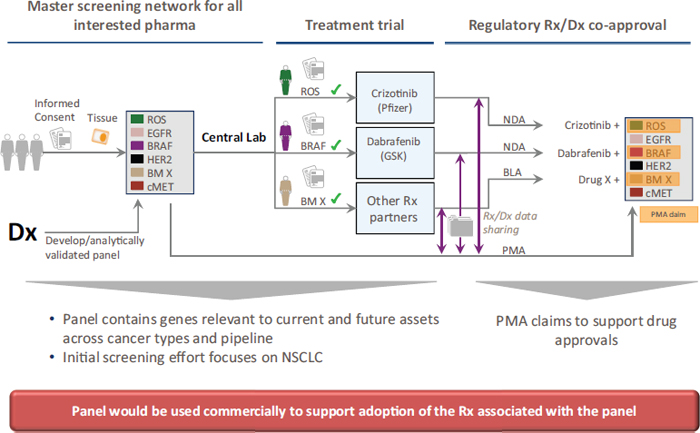
FIGURE 6 Proposed collaborative multipharma network to advance multibiomarker diagnostics.
NOTE: BLA = biologic license application; Dx = diagnostic; NDA = new drug application; NSCLC = non-small-cell lung cancer; PMA = premarket approval; Rx = drug.
SOURCE: Martin presentation, November 10, 2014.
cers are going to evolve and change to avoid the treatment’s effectiveness,” she said, and pointed out a study that found an approximately 80 to 90 percent concordance rate for some major cancer driver genes, such as KRAS and BRAF, in primary and metastatic tumors taken from the same patient, but only about a 50 to 60 percent concordance rate for some other genes, such as PIK3CA (Kopetz et al., 2014).
“Obviously if you profiled only one sample, it is possible that you would not be catching what is actually active and relevant at the present time,” Siu said, and noted that the emerging drug resistance that commonly occurs after treatment with targeted therapies is probably due to new mutations, which perhaps could be detected early on in circulating tumor DNA. She suggested having more dynamic trials in which patients’ tumors would be extensively profiled both at baseline and when they become resistant to an experimental therapy, along with frequent sampling of circulating tumor DNA at regular intervals. “We need to understand the dynamics of the cancer and not just one specific static point in time,” she said.
Prioritizing Molecular Targets
Another challenge Siu discussed is understanding how to prioritize molecular targets, not just in terms of their frequency, but more importantly their functionality and how much they drive growth of the tumor. Solit agreed, noting that it can be difficult to assess the ultimate influence of co-mutations. For example, a co-mutation of KRAS is likely to confer resistance to HER2 inhibition, but it is not known for certain, so “the question is would you put a patient with both mutations into a clinical trial testing neratinib?” Solit asked. For some patients, only a subset of their cancer cells is likely to have a mutation that will respond to a treatment that targets that mutation, making it questionable whether these patients will have a significant response to the treatment, he added. In the Lung-MAP trial, patients with two or more response biomarkers will be randomized to relevant treatment arms, skewing toward the arm with the less prevalent marker so as to increase the chances of accruing enough patients with that marker for treatment, Herbst noted.
GATHERING THE EVIDENCE: COVERAGE DECISIONS
Tunis noted that patient access to new products is not ensured by FDA approval, but rather by payer coverage determinations, and “there are many
different payers with many different standards.” Consequently, he said there is a great need for “reimbursement science,” which he defined as the science of developing new tools, standards, and approaches to assess the comparative effectiveness and value of products covered by public and private health plans. Such a science would have to consider all the social objectives impacted by reimbursement, such as access to therapy, innovation, safety and effectiveness, and value for money and affordability. “The job of the payers is not to pay for everything, but to allocate resources where they are most efficiently used. Reimbursement affects things like innovation, affordability, and cost of care, but there is no platform where these issues of reimbursement science get discussed,” Tunis noted. “Who will advance reimbursement science to speed innovation, improve reimbursement decision making, and get products to people in need to improve population health outcomes and efficient [use of] resources?” he asked.
Tunis pointed out that in 2008, the Secretary’s Advisory Committee on Genomics in Health and Society also identified this problem and recommended that the U.S. Department of Health and Human Services (HHS) create a public–private entity of stakeholders to establish evidentiary standards and levels of certainty required for different situations, but no such entity was ever formed. Given that HHS has not taken the reins in this regard, he suggested that one or more multistakeholder partnerships undertake this effort to establish reimbursement science, analogous to FDA’s concept of “regulatory science” (IOM, 2011). “I think it’s doable, but the only way we are going to get progress with this is a mechanism for sustaining dialogue with multiple stakeholders that gets into technical details, and is highly iterative. That is the only way we are going to make sure that it is not just the payers dictating what the standard should be, but the payers in dialogue with all the other key stakeholders who have equally legitimate social objectives,” Tunis concluded.
Coverage with Evidence Development
Several speakers suggested that one useful option for gathering the evidence needed to assess the clinical utility of molecular diagnostics would be Coverage with Evidence Development (CED). This approach to reimbursement is linked to patient participation in a clinical study, Tunis reported. The payer determines which technologies CED is applied to, what the research questions will be, and what the study design will be.
CED was developed to give payers some leverage in making sure stud-
ies are designed to answer the sorts of questions they are interested in, such as clinical utility, he said. The CED that Medicare has implemented also enables earlier than normal patient access to promising technologies while promoting studies that determine whether they benefit patients, Tunis added. Medicare has used CED for a number of indications, including gene testing for warfarin sensitivity and molecular diagnostics for prostate cancer.
To be considered for CED, a medical diagnostic or treatment must address an important health need and/or specific payer priority, and the existing evidence on the intervention must be adequate to conclude that the technology is promising. In addition, the proposed study must generate valid and relevant evidence to inform future clinical and policy decisions, and the study must be reasonably likely to be feasible. There also must be a credible process for assessing all these necessary elements, Tunis reported.
The trade-off for CED is that it usually takes longer to acquire the evidence than it does to gather the data needed for standard coverage decisions, he added. CED can also be difficult to carry out. “Although CED is intuitively attractive, it is a bear to actually make it work. The highways are littered with the carcasses of CED efforts, both in the United States and internationally,” Tunis said. He noted that it is a lot of work for payers and companies to make decisions about what constitutes medical necessity and to establish the criteria for what is considered “promising.” It is also difficult to ascertain the kind of study that will provide sufficient evidence for permanent coverage of the intervention.
Tunis noted that past justifications CMS has used to authorize CED for cancer diagnostics make it difficult to predict future determinations from the agency. These reasons sometimes included perceived level of physician enthusiasm for a test based on surveys, or inclusion of the test in an NCCN or an ASCO guideline, even though these guidelines do not always agree with each other. Consequently, “It is pretty difficult for someone in the venture capital community or in the diagnostics industry to come up with a clear clinical development plan,” Tunis said. “They have no idea what is necessary to do [to increase the] likelihood that payers will reimburse their tests. It is sort of undefined.”
The MolDx Approach to Test Reimbursement
MolDx was started in November 2011 as a CMS pilot project by Elaine Jeter, M.D. The goal of this program is to develop and use an evidence framework to evaluate molecular diagnostics for CMS, with an emphasis on
clinical utility evaluations. More specifically, MolDx performs the following basic functions8:
- Facilitate claims processing and track use of molecular diagnostic tests;
- Establish clinical utility expectations;
- Technically assess published test data to assess clinical utility and coverage; and
- Establish reimbursement.
As Dickson reported, MolDx recognizes the new paradigms posed by genomic tests, including the likelihood that large clinical trials of these tests are not likely to be conducted or duplicated due to the difficulty of enrolling enough patients now that many targeted treatments are directed at rare patient subsets, as well as the often low or negative return on investment on research in advanced molecular testing. This has resulted in “less than perfect science” for molecular diagnostics in areas where there are substantial unmet needs, Dickson said.
Consequently, MolDx created a new paradigm for reimbursement. Instead of dividing up interventions into the traditional two categories, “standard” and “experimental,” MolDx recognized a new category, called “transitional” for treatments and tests that are in between, and established a pathway for provisional coverage for tests that fall into this transitional space. To determine which genetic tests are considered transitional, MolDx has high-level discussions with multiple stakeholders with the aid of its “Partner Specialty Societies.” A MolDx Executive Committee selected from this group makes the coverage decisions in an expedited pathway, as outlined in Figure 7. The committee is composed mostly of molecular pathologists from academic institutions.
As Dickson reported, the MolDx Executive Committee reviews every dossier to identify tests that are considered reasonable and necessary, meaning they are tests that address a significant unmet clinical need, could potentially dramatically improve patient care, and have widespread acceptance by the medical community, but do not yet have robust evidence of clinical utility. These experts then decide if the tests should be covered, have “limited coverage,” or have “coverage with data development,” the latter of
_______________
8 See http://www.palmettogba.com/palmetto/MolDX.nsf/DocsCatHome/MolDx (accessed March 19, 2015).
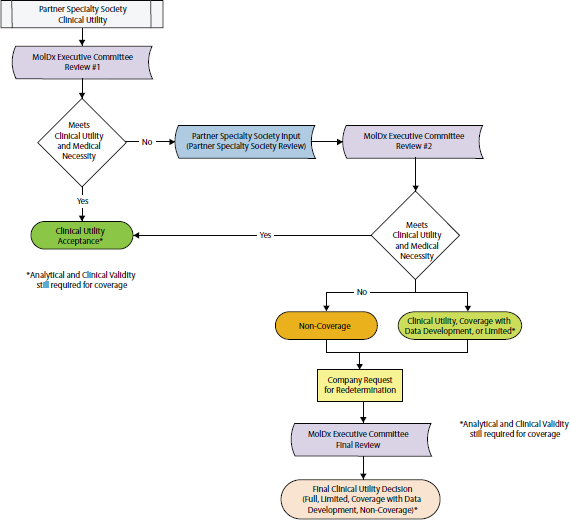
FIGURE 7 MolDx: Algorithm for expedited review.
SOURCE: Dickson presentation, November 10, 2014.
which is akin to the CMS approach to coverage with evidence development. Once those data are collected, a decision is made to continue or suspend coverage for the diagnostic, depending on what the data reveal. Analytical and clinical validity are still required for coverage with data development. Limited coverage is reserved for diagnostics for which the clinical utility data are too difficult to acquire. Instead, strict limits are put on its use; that is, it is limited to a very specific group of patients in a very specific way to make sure it is reasonably safe, Dickson said.
MolDx plans to evaluate each genetic panel and laboratory in which it is performed separately until there are accepted standards for these tests. Each genetic panel test or groups of tests must show analytic and clinical validity as well as clinical utility, and be disease specific. MolDx also rec-
ognizes the need for hierarchical reporting of what is considered standard, transitional, or experimental, Dickson said.
Monzon said he was enthusiastic about this concept of transitional standards because often researchers uncover more uses for a genetic test beyond the original intended use when they have the opportunity to study clinical correlations. But Dickson stressed that data have to be collected in a controlled manner when there is coverage with data development. He also cautioned that insurers have to be consistent, and if they pay for experimental genetic retesting of tumors for patients who do not respond to current treatments, for example, then they should also pay for it for patients with any chronic disease, such as rheumatoid arthritis, who are in a similar situation.
Green Park Collaborative Recommendations
The Green Park Collaborative is a group of stakeholders, including payers, life sciences companies, patients, clinicians, researchers, and regulators, who were brought together by the Center for Medical Technology Policy to craft recommendations on generating the evidence needed to inform both clinical and payment decisions for use of molecular diagnostics in adult oncology. The recommendations are aimed at researchers and are detailed in a document published online9 in 2013. They recognize the time and expense involved in doing randomized controlled trials and, in addition to the use of such trials, also offer alternative methods where appropriate, including prospective-retrospective studies, in which biomarkers can be validated using patient samples archived from previous trials of treatment efficacy. If no stored samples are available, the Collaborative recommends conducting single-arm studies with non-contemporaneous controls. Another alternative it proposed was conducting high-quality prospective observational studies, in which patient outcome measures are determined prior to the start of the study and there are sufficient numbers of patients to power the study so that conclusions drawn from the data are reliable. In situations in which there are multiple data sources, the Collaborative recommended modeling techniques in which it might be possible to “connect the dots to infer patient benefit,” even if there is no direct evidence of clinical utility, Messner said.
_______________
9 See http://www.cmtpnet.org/docs/resources/MDX_EGD.pdf (accessed March 19, 2015).
But the white paper did not address issues specific to next-generation sequencing, and after a series of meetings with stakeholders, there are plans to draft new recommendations to address that gap by the summer of 2015, Messner reported. This new paper will address the types of evidence standards and assessments needed for genomic panels, and whether “interim standards” could be considered when information is lacking but patient need is great, such as case reports for rare or newly discovered biomarkers. In addition, the Collaborative will consider standards for interpretation and integration with other data, and whether standards should be established for reporting to payers the breadth of sequencing in genetic panels and the types of variants detected.
GATHERING THE EVIDENCE: DATABASES AND REGISTRIES
High-quality genetic databases that are well annotated with clinical outcomes data and patient characteristics are a valuable resource for gathering the evidence needed to assess the clinical utility of genetic tests and targeted treatments, several speakers noted. The observational studies done using these databases can often capture a larger and more representative group of patients being treated in community settings than clinical trials run in academic or large cancer care institutions, noted Garnet Anderson, senior vice president and director, Public Health Sciences, Fred Hutchinson Cancer Research Center. They can also capture a wider range of therapies in their analyses, and can enable rapid evaluation of new biomarkers for lower cost than randomized controlled trials, she added. In addition, databases can aid physician decision making and enrollment in clinical trials by indicating approved or experimental targeted treatments relevant to the genetic alterations detected. Databases can also collect information on tests and treatments used in clinical care, which payers can use to determine their reimbursement policies. Speakers at the workshop reported on several genetic databases relevant to cancer that have recently been established, including My Cancer Genome, ClinGen, and the ASCO Targeted Agent and Molecular Profiling Utilization Registry (TAPUR). They also discussed the challenges and limitations of using databases and registries to assess clinical utility.
The mission of My Cancer Genome is to curate and disseminate knowledge regarding the clinical significance of genomic alterations in cancer, including those that predict response to therapy as well as prognostic and diagnostic biomarkers, Levy reported. It is a publicly available website that covers more than 400 different disease-gene variant relationships for 56 cancer-related genes found in more than 20 different cancers. For every genetic alteration, the database provides information on its location, frequency in particular cancers, and how it affects the sensitivity or resistance to particular targeted therapeutics. The database is a worldwide collaborative effort with 65 contributors from 21 institutions in 10 countries.
Levy said that My Cancer Genome includes information derived from various sources that provide different levels of evidence. “We are not trying to be an FDA approval process,” Levy added. “As long as a drug is being used in a human clinical study, we include it in our database so people can understand the potential actionability of some of these alterations they are finding.” Some preclinical findings are also included, she said.
My Cancer Genome has a cancer drug targets list with more than 500 cancer-related drugs and their respective genetic targets. In addition, it has a rare mutation database that includes case reports of drug response. My Cancer Genome also can indicate relevant clinical trials as it is tied to the Physician Data Query (PDQ) clinical trial database for physicians and patients who want to find experimental treatments that target specific genetic alterations in tumors. This NCI database lists more than 40,000 clinical trials for more than 135 different cancer diagnoses and 500 cancer genes.
The types of genetic alterations in the database include base point mutations, insertions and deletions, as well as rearranged bases, and gene fusions and duplications (amplification). Altered protein expression is also documented, for which immunohistochemistry is a standard form of testing. In addition, the database documents co-mutations that affect response to treatment. The importance of those co-mutations was underscored by Levy, who gave the example of the L858R mutation on the EGFR gene, which confers sensitivity to treatment with erlotinib, but is attenuated by an additional mutation called T790M. “You can’t just say ‘I’m EGFR-positive.’ You really have to know the co-occurring alterations and how that might impact the sensitivity to the drug,” she said.
My Cancer Genome makes its resources publicly available on its website and on a mobile app. The database can also suggest clinical actions
when it is integrated with clinical institutions’ electronic health records, Levy said. At Vanderbilt University, for example, electronic health records indicate what genetic variants patients test positive for, and when clinicians click on those variants they are taken to the record of their clinical significance as documented in the My Cancer Genome database. A commercial partner also can integrate the database into laboratory reporting.
Levy said that clinicians can also use My Cancer Genome as a knowledge base of clinically relevant variants for interpretation of next-generation sequencing cancer panels. For such test results, it lists the genetic variants with therapeutic significance for the patient’s tumor, the types of drugs that target those mutations, and potentially relevant clinical trials.
In closing, Levy stressed, “We need to be able to aggregate all of our data together on the clinical outcomes of patients and their genomic alterations because the number of patients with these very rare alterations is just staggeringly small, and no one institution is going to have enough patients with each of these variants to be able to do any real discovery with it.” To address that challenge, she advocated for learning health care systems in which the care of a patient seen today is informed by data collected on all similar patients treated before, and the data collected on today’s care is then used to inform future care (IOM, 2010b).
ClinGen, funded by NIH, is a resource dedicated to harnessing both research data and the data from the hundreds of thousands of clinical genetics tests being performed each year. The database has expert curation to determine which variants are most relevant to patient care. In 2013, the National Human Genome Research Institute and the Eunice Kennedy Shriver National Institute of Child Health and Human Development awarded three grants totaling more than $25 million to support a consortium of research groups to design and implement a framework for evaluating which variants play a role in disease and identifying those that are relevant to patient care. The groups will work closely with the National Center for Biotechnology Information of the National Library of Medicine, which will distribute this information through its ClinVar database.10
As Monzon reported, ClinGen is an effort to bring all the information from many laboratory providers together in a single database that can be
_______________
10 See http://www.iccg.org/about-the-iccg/clingen (accessed March 19, 2015).
used to determine how relevant genetic mutations are to disease. It has 353 supporters for its large, worldwide network submitting genetic information. “We need to encourage reporting and sharing of test performance and response-to-therapy data in tumor tests,” he stressed.
Targeted Agent and Molecular Profiling Utilization Registry
TAPUR is hosted by ASCO and researchers plan to use it to conduct a prospective, observational, non-randomized clinical study that aims to describe the performance (both safety and efficacy) of commercially available, targeted anticancer drugs prescribed for patients with advanced cancer showing a potentially actionable genomic variant.11 Schilsky said targeted therapies are often prescribed off-label when tumor profiling suggests that a drug targeting a specific genetic alteration in a patient’s tumor might be beneficial for the patient, but the drug is not FDA approved for that indication. The outcomes for these patients are not being captured fully now in a way that physicians and payers can learn from them.
Off-label prescribing is legal, but it may not be reimbursed. The goal of TAPUR is to collect evidence to inform reimbursement decisions and to simplify patient access to off-label targeted therapies; pharmaceutical companies will provide their drugs at no cost to patients who consent to participation in an IRB-approved protocol for data collection in the TAPUR registry. Physicians will have to submit the required follow-up data specified in the protocol. ASCO is hopeful that payers will reimburse for the routine clinical care costs, Schilsky said. Another participant noted that the Affordable Care Act requires reimbursement of routine clinical care costs of patients enrolled in clinical trials.
Schilsky said the study protocol does not specify a particular test for genomic profiling; the treating physician can choose a test offered by any laboratory that is accredited by CAP, is CLIA-certified, and has a McKesson Bioscience Z-code indicating it is a unique and vetted test. Only patients with advanced cancer for whom no standard treatment options exist are eligible to enroll in the study, and only if their tumors have a genetic variant that could be targeted by one of the drugs available through the registry protocol.
Physicians will submit the genomic profiling test results for their eligible patients to TAPUR, along with a treatment plan using one of the drugs
_______________
11 See http://www.asco.org/practice-research/targeted-agent-and-profiling-utilization-registry-study (accessed March 19, 2015).
in the protocol. A group of experts known as the Molecular Tumor Board will review this information and will either confirm the treatment plan or recommend an alternative. This review step is taken to protect patients from undergoing inappropriate treatments based on physician misunderstanding of drug action or misinterpretation of molecular test results, Schilsky noted.
In the study, eight patients will be enrolled for each combination of tumor type, genetic variant, and drug used to treat it (e.g., eight bladder cancer patients with a BRAF V600E mutation treated with a BRAF inhibitor). If there is at least 1 response in each group of 8 patients, an additional 16 will be enrolled in the group. Treatment responses will be considered significant and worth pursuing further only if at least 4 out of 24 patients respond. If no responses are seen in the first eight patients, treatment will end.
As Schilsky noted, “This is very much a hypothesis-generating study. We are not going to prove anything, but are looking for signals that are based on real-world assessments.” In addition to generating hypotheses to inform new studies, TAPUR could potentially be used for a variety of purposes, including modifying drug labels for safety issues or new indications, modifying compendiums or treatment guidelines, and aiding reimbursement policy determinations and doctor–patient decision making. It could potentially benefit all stakeholders:
- Patients would receive a targeted agent matched to their specific molecular profile.
- Physicians would receive interpretation of molecular test results, guidance in treatment recommendations, and access to drugs.
- Pharmaceutical companies would receive data on their drugs’ use and outcomes to inform their research and development plans.
- Payers would receive data on test and drug use and outcomes to inform coverage decisions.
- Regulators would receive data on the extent and outcomes of off-label drug and test use and additional safety information.
TAPUR has defined clinically actionable variants and criteria for its drug selection as can be seen in Box 5.
Speakers noted several challenges in using databases or registries to support clinical utility assessments. For example, databases may not capture all relevant outcomes or may not be well annotated with high-quality data, and
Definition of Clinically Actionable Variants
- Gene variant is the target of an approved drug for any cancer indication
- Activating mutations in genes upstream of the molecular target of an approved drug
- Inactivating mutations in genes that result in unique susceptibility to a specific molecular intervention (for example, BRCA1 mutation and poly ADP ribose polymerase inhibitors)
- Other genes of interest that have appropriate justification for selection based on published scientific evidence regarding susceptibility to a specific molecularly targeted therapy
Criteria for Drug Selection
- Level 1: Agent met a clinical end point (objective response, progression-free survival or overall survival) in a clinical trial testing the agent in the patient’s tumor type harboring the mutation of interest
- Level 2: Agent is commercially available for use in any tumor type with the specific genomic variant identified in the patient’s tumor
- Level 2: Agent has demonstrated evidence of clinical activity against the patient’s tumor type based on published literature
- Level 3: Agent has demonstrated preclinical evidence of antitumor activity and evidence of target inhibition in model systems of patient’s tumor type
SOURCE: Schilsky presentation, November 10, 2014.
there is a lack of high-quality repositories with broad patient consents that allow patient samples collected for one study to be used in other studies. The data can also have confounders that lead to errors in interpretation.
Ganz suggested that annotation of tumor specimens should include
not only information regarding genetic variants, but also information about patients that might be relevant to their tumor pathology, such as whether they smoked, their ethnicity, gender, etc. “We would like to prevent those advanced cancers and know something about the context in which the mutations occur,” Ganz said. “Not collecting additional data about the host in which the cancer occurs seems to be a missed opportunity.” Martin responded, “The key element in the future will be the ability to tie the genomic data back to electronic health record data where you have much more information along the lines of what you are suggesting.” Siu noted that in a recent clinical trial with genetic profiling, she included a companion epidemiological survey that was completed by more than 95 percent of the patients because “this is information they realize is very important to collect.”
A few participants suggested that registries are best developed in academic settings. “Academic settings are the better place to develop these resources when we are so early on in this process, as they have few conflicts of interest, clear oversight mechanisms, and more open access,” Anderson said.
But even high-quality databases and registries can generate faulty findings if researchers do not make a special effort to consider confounders and other issues that can bias the results of observational studies. Such confounders include co-occurring illnesses and the type of supportive care and surveillance received that might differ between the two groups being compared in a study, Anderson noted. For example, she pointed out that the disagreement between the observational and randomized controlled trial results for post-menopausal hormonal therapy were largely due to time-dependent effects. The randomized controlled trials showed that adverse cardiovascular effects from this therapy tended to occur within the first 2 years of taking hormones. But this was followed by a gradual reduction in the increased risk over time, with the potential for overall reduced risk of cardiovascular disease after 5 years (Prentice et al., 2005).
In most cases it is possible to align randomized and non-randomized results, but that alignment depends on capturing all noteworthy potential confounders in the data and creating a natural experiment within the observational study. This experiment could involve using a similar study population that met the same eligibility criteria as that of the randomized study, and having parallel, high-quality follow-up as well as a pseudo-intention-to-treat analysis, Anderson said. She suggested that when researchers use databases to evaluate therapies, they emulate a randomized trial as much
as possible, and gave an example of this approach taken by Miguel Hernán from Harvard. Hernán simulated multiple clinical trials within the Nurse’s Health Study (Hernán et al., 2008). Anderson also suggested that when databases are developed de novo, they should have randomized trials at their core, or at least as their intent.
The effort needed to control potential bias in observational studies using databases depends on how strong the study results are, Anderson also pointed out. “If you really have a home run, maybe you don’t need to go to all this effort. The more modest effects—say 20 or 30 percent changes in event rates—can easily be masked or lost through the confounding or other issues that can exist in an observational study,” she said.
Frequent sources of errors in interpretation of genetic profiling studies are multiple subgroup analyses, which can be challenging to deal with statistically even when they are conducted on randomized trial databases, such as patient sample repositories, Anderson pointed out. Subgroup analyses tend to increase the probability of interpreting a chance finding as a significant difference. They also tend to miss true differences that are there because they were not considered in the original study design (Wang et al., 2007). But subgroup analyses can lead to “hints or ideas” that might guide future research, Anderson said.
EDUCATION AND CLINICAL DECISION SUPPORT NEEDS
Studies suggest that a lack of awareness or understanding of tumor biomarker tests has impeded their use by patients and their physicians. “Patients want tests when they understand them,” Phillips said. A survey of the general population she conducted found that three quarters of respondents did not know what the term “personalized medicine” meant. But when they were told what it meant, then 95 percent responded that they wanted it. The survey also found that if a genetic test indicated they would not benefit from a cancer treatment, 84 percent of respondents indicated they would either want the treatment anyway, or they would seek a second opinion to assess if they should have the treatment. “Patients don’t trust test results and they want it all, especially if they don’t have to pay for it,” she said, adding that other studies find that patients’ willingness to pay for genetic tests varies by how much the test will cost and how much income they have (Garfield et al., 2015). “They are happy to know everything, as long as somebody else is paying for it,” Phillips said.
She suggested putting more emphasis on engaging patients than on
educating them about genetic tests. “A lot of patients want the doctor to make the decision, but they want to be engaged in the decision and understand it. They may not care about understanding the genetics. The average American has about a seventh-grade reading level. They don’t understand statistics and probabilities so you can’t really educate in that way, but you can engage them. I absolutely think we have to engage patients,” she said. LoRusso added that she relies on genetic counselors in her clinical trials to educate patients on the meaning of their genetic test results. Jane Perlmutter, patient advocate, who is involved in the I-SPY trial for targeted breast cancer treatments, said that when they return incidental results from that trial, investigators will also rely on genetic counselors, who provide patient educational materials and are developing a webinar to train clinicians on how to convey that information.
Physicians also need better education on cancer genetic tests, Phillips reported. Even at a cutting-edge institution such as Dana-Farber Cancer Center, a survey of their oncologists found wide variation in multiplex tumor testing use, genomic knowledge confidence, and disclosure of results (Gray et al., 2014). Another study found that oncologists were enthusiastic about germ-line and tumor testing, but they often did not understand these genetic tests and were not comfortable with them (Dressler et al., 2014). “Most of us aren’t equipped to have that conversation with patients about what the results mean. We also don’t have the infrastructure and driven environment to, in an efficient and sympathetic way, deal with the information,” Gradishar said.
This is problematic given that patients may come to their physicians’ office with company marketing materials about genomic tests, and physicians often assume that the information presented in these materials has been subject to FDA review, even though that often is not the case, Dickson pointed out. He added that some medical oncologists distrust pathology results and that might make them more willing to rely on genomic test results. Herbst added that sometimes even internists are ordering genomic tests, although they are not trained to interpret or act on those results. Malin responded that some tests, such as Oncotype Dx, specify that it is to be ordered by the physician who is going to be making the treatment decision as opposed to the surgeon or internist, who is not necessarily going to have a conversation with the patient about how the information will be used.
One study found that community oncologists were more likely to use genomic tests than academic medical centers (Dressler et al., 2014). Monzon said academic oncologists can often offer oncology patients who
have exhausted standard treatment options the opportunity to participate in clinical trials available at their own institutions. But such trials may not be available in the community where most oncology patients are treated, so tumor profiling is seen as the most accessible way to help select the next plan for care. “Your patient is demanding what the next step is. They want to know what else you can do for them,” Monzon pointed out.
But Ganz noted that palliative care is a worthwhile option that can be discussed with end-stage cancer patients instead of “promising them a genetic test is going to be the key to their survival. We need to have an open discussion about what our care can and cannot do. We are always hoping for a miracle, but most people [with advanced cancer] are going to die from their cancer and sequencing everyone is not going to be the solution,” she said.
Solit responded by saying that sequencing data are useful not only for determining treatment, but also for clarifying diagnoses and preventing unnecessary treatment, and that many of his end-stage patients who do not have an actionable mutation do select hospice care.
Workshop participants discussed various approaches for educating care providers about genomic profiling tests. Senderowicz said, “Practicing oncologists don’t know much about this . . . and who is going to educate these clinical oncologists? Should we include this in the fellowship program and make it mandatory? Should it be part of the certification for the boards?” Gradishar responded, “We’re all getting retrofitted in our generation to have some superficial understanding of this. But if it’s really going to be ingrained; it has to come from early training and this will be incumbent upon the medical boards, etc.” LoRusso noted that there is a new curriculum at Yale for medical students and it includes modules on genetic tests.
But Phillips noted that “you can’t educate people about everything,” and suggested providing guidance and structure and framing of information so that patients and their providers can make decisions without requiring a high-level education in genetic testing. She noted that studies in behavioral economics show that the way in which information is framed to patients and providers, meaning whether information is framed as a gain or a loss, greatly impacts their behavior. She suggested creating guidelines to structure decision making, and creating publicly available databases of test availability, test price, insurer policies, and economic evidence of their value.
Johnson added, “There’s a lot of different decisions providers need to make when they’re trying to navigate this sea of biomarkers—what test to order and when, how to interpret and report those results, and most
importantly how to apply those results to patients. There’s a lot of different types of decision support you can provide, but when and how to provide that decision support and to whom are all open challenges.” Two forms of decision support for providers discussed at the workshop were practice guidelines and treatment pathways.
Gradishar said NCCN guidelines set the standard for clinical care and policy in oncology in the United States. He said the development of these guidelines is transparent and objectively based on the current evidence base, with reliance on experts to consider the gaps and provide expert consensus where evidence is not strong. To develop the guidelines, NCCN has established 48 multidisciplinary panels with 25 to 30 experts per panel. These experts have developed more than 160 practice algorithms that are updated continuously, published on the Internet, and serve as the basis for insurance coverage policy and quality evaluation. The guidelines cover the full spectrum of cancer care, from detection to end-of-life care, and rate the strength of the supporting evidence based on the quality, extent, and consistency of clinical studies for each intervention.
In 2012, NCCN also launched a compendium for biomarker tests, including genetic tests, that are used to make clinical decisions related to screening, diagnosis, monitoring, or providing predictive or prognostic information in oncology. The same rating system was used for the supporting evidence, and the compendium is regularly updated as more information becomes available, usually several times per year, Gradishar reported.
Tests used for clinical decision making are included in the compendium, while those used for research purposes only usually are not. NCCN criteria for clinical usefulness include data demonstrating that the biomarker affects clinical decisions and/or divides patients into clinically relevant subgroups, and documentation that the test is widely available and reliable. However, the platform or the methods used to conduct the biomarker tests listed in the compendium are often not defined, according to Gradishar, and when multiple tests serve the same purpose, it usually does not indicate which test might be the best. Eventually NCCN hopes to create electronic versions of its software so that its algorithms can be incorporated into electronic health records, he said.
Levy noted there can be inconsistency between FDA-approved labels and NCCN guidelines for biomarker tests because of different levels of
evidence accepted for each. Dickson pointed out there are even inconsistencies among NCCN guidelines with regard to when tests can be used for different types of cancer because of disagreements among guideline committees. Gradishar responded that although consistency is the ideal to strive for, when options are more limited for certain disease sites, experts tend to be more liberal in their interpretation of “what we should be doing for patients when there is a paucity of options. In these situations, people are more willing to adopt strategies that have less evidence to support them.”
Guidelines are often broad and do not provide enough guidance for physicians to select the most effective treatment, Malin pointed out. New guidelines suggest more than 50 treatment options for lung cancer, for example. Treatment pathways tend to be more specific and are another option for guiding physicians to use cancer biomarker tests and targeted treatments appropriately. Such pathways are usually developed by insurers or provider organizations and are specific to those entities. Malin also noted that treatment pathways are widely discussed as potential solutions to the escalating cost of cancer care. A study undertaken by US Oncology found that pathway use was associated with a 30 percent decrease in care costs at 1 year, with no difference in survival between patients treated on and off pathway. To use a treatment regimen other than the one indicated by the pathway, a treating physician had to have one of his or her partners co-sign the orders and agree that the alternative treatment was reasonable (Neubauer et al., 2010).
When US Oncology was acquired by McKesson, its treatment pathway was incorporated into a new version developed by McKesson in collaboration with NCCN, Malin reported. Some treatment pathways developed by care providers are designed to be integrated within their own specific health care programs and are implemented through their electronic health records. By contrast, treatment pathways developed by WellPoint (now known as Anthem) are publicly available. Anthem manages 14 of the state Blue Cross/Blue Shield plans and has 36 million members across the United States, Malin reported.
A team of internal oncologists and pharmacists at Anthem reviewed nationally based guidelines, such as those of NCCN and ASCO, as well as peer-reviewed evidence from clinical trials to summarize the clinical benefits, side effects and toxicity, and costs of treatments, as well as the
strength of national guideline recommendations. Then an external group of academic and community-based oncology experts added their input to the pathway development. The end results are treatment pathways specific to tumor type, biomarkers, and patient characteristics. These pathways are updated quarterly based on new information in the medical literature, changes to NCCN and ASCO guidelines, and new FDA-approved indications.
Anthem’s pathways are optional, but physicians who select a treatment listed in a pathway are eligible for an additional payment for treatment planning and care coordination. That additional $350 per month is about equivalent to the profit margin oncologists in private practice typically make on the drug and is provided “to level the playing field so that choices can be made primarily on the clinical basis and practices are not penalized for practicing cost-effectively,” Malin said. She said treatments included in the pathway have been shown to be clinically effective, have a favorable side effect profile, and are cost-effective. When physicians enter patient results from biomarker tests into the electronic health records, they are informed of the most appropriate treatment choices. But the pathways do not affect coverage decisions, which continue to be based on medical policy, Malin said.
Anthem reviews adherence to its pathways and will be assessing regimens used off pathway to determine if they suggest a trend, such as a subset of the population for which a new treatment pathway should be created. Anthem also considers comments from the public on its website and reviews them at its advisory meetings. “We have had some good dialogues with some of the patient advocacy groups and have made changes to our treatment pathways based on their input,” Malin said.
In addition to gathering the evidence needed to assess the clinical validity and utility of biomarker tests, speakers suggested a few other research opportunities. Phillips suggested that prospective studies should also gather data on the entire pathway of events, including who is using the tests, how patients and providers feel about the tests, and the information the tests provide, in addition to the outcomes, benefits, and costs. “It could be very easy to make these studies more useful by gathering a full range of data from patients and providers,” she said. She also suggested studies on the changing policy environment, such as the new CPT codes for molecular diagnostic
tests, and the rise in accountable care organizations. Herbst noted that as part of the Lung-MAP study, physicians and patients are being surveyed to assess what they know and understand about tumor profiling tests, to get a better sense of the education needed in this regard.
LoRusso pointed to the need to do research on how best to educate the patient, including what types of tools work best for what types of patients. “Patients are not created equal in terms of knowledge base and background,” she said. Levy agreed, noting that cancer patients have different degrees of health literacy and different learning styles, and those should be considered when helping them understand their test results. She suggested developing and evaluating patient-focused and provider-focused educational content. She also suggested that new approaches be used, such as online educational videos.
Monzon suggested supporting postmarket research to better assess clinical utility. Holdhoff noted that circulating tumor DNA tests are in an early stage of development and there is a need for large-scale clinical prospective studies to assess their value in clinical practice. He suggested providing funding and resources to enable the inclusion and evaluation of circulating tumor DNA tests within large-scale prospective clinical trials, and involving experts in that technology early in clinical trial design.
In closing, Planning Committee Chair Adrian Senderowicz thanked the speakers and participants for their contributions to the many fruitful discussions during the meeting. Noting that many views had been heard and many useful suggestions had been made (see Box 1), he said that “we heard a very significant update of what is new in the field and all the growing pains that this field is experiencing. . . . There are many different challenges in this new field that we need to understand. I think when we go back to our jobs tonight, tomorrow, we should discuss these with our stakeholders and try to see how we can speed progress in the field to improve patient outcomes.”
Actionable Genome Consortium to guide NGS in cancer. 2014. Nature Biotechnology 32(10):965.
André, F., T. Bachelot, F. Commo, M. Campone, M. Arnedos, V. Dieras, M. Lacroix-Triki, L. Lacroix, P. Cohen, D. Gentien, J. Adélaide, F. Dalenc, A. Goncalves, C. Levy, J.-M. Ferrero, J. Bonneterre, C. Lefeuvre, M. Jimenez, T. Filleron, and H. Bonnefoi. 2014. Comparative genomic hybridization array and DNA sequencing to direct treatment of metastatic breast cancer: A multicenter, prospective trial (SAFIR01/UNICANVER). Lancet Oncology 15(3):267-274.
Bettegowda, C., M. Sausen, R. J. Leary, I. Kinde, Y. Wang, N. Agrawal, B. R. Bartlett, H. Wang, B. Luber, R. M. Alani, E. S. Antonarakis, N. S. Azad, A. Bardelli, H. Brem, J. L. Cameron, C. C. Lee, L. A. Fecher, G. L. Gallia, P. Gibbs, D. Le, R. L. Giuntoli, M. Goggins, M. D. Hogarty, M. Holdhoff, S.-M. Hong, Y. Jiao, H. H. Juhl, J. J. Kim, G. Siravegna, D. A. Laheru, C. Lauricella, M. Lim, E. J. Lipson, S. K. Nagahashi Marie, G. J. Netto, K. S. Oliner, A. Olivi, L. Olsson, G. J. Riggins, A. Sartore-Bianchi, K. Schmidt, I.-M. Shih, S. M. Oba-Shinjo, S. Siena, D. Theodorescu, J. Tie, T. T. Harkins, S. Veronese, T.-L. Wang, J. D. Weingart, C. L. Wolfgang, L. D. Wood, D. Xing, R. H. Hruban, J. Wu, P. J. Allen, C. M. Schmidt, M. A. Choti, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, N. Papadopoulos, and L. A. Diaz, Jr. 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Science Translational Medicine 6(224):224ra24.
Budel, S. 2013. NGS in the clinic at JPM—JP Morgan Conference Highlights. DeciBio (press release). January 2013. http://decibio.com/blog/ngs-in-the-clinic-at-jpm-jp-morgan-conference-highlights (accessed March 19, 2015).
Camidge, D. R., Y. J. Bang, E. L. Kwak, A. J. Iafrate, M. Varella-Garcia, S. B. Fox, G. J. Riely, B. Solomon, S. H. Ou, D. W. Kim, R. Salgia, P. Fidias, J. A. Engelman, L. Gandhi, P. A. Jänne, D. B. Costa, G. I. Shapiro, P. LoRusso, K. Ruffner, P. Stephenson, Y. Tang, K. Wilner, J. W. Clark, and A. T. Shaw. 2012. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a Phase 1 study. Lancet Oncology 13(10):1011-1019.
Clain, E., J. R. Trosman, M. P. Douglas, C. B. Weldon, and K. A. Phillips. 2015 (unpublished). Emergence of new BRCA1/2 tests and gene panels: Availability & payer coverage policies.
DeciBio. 2013. Clinical diagnostics. http://decibio.com/clinical-diagnostics.php (accessed February 18, 2015).
Dorschner, M. O., L. M. Amendola, E. H. Turner, P. D. Robertson, B. H. Shirts, C. J. Bennett, K. L. Jones, M. J. Tokita, J. T. Bennett, J. H. Kim, E. A. Rosenthal, D. S. Kim, H. K. Tabor, M. J. Bamshad, A. G. Motulsky, C. R. Scott, C. C. Pritchard, T. Walsh, W. Burke, W. H. Raskind, P. Byers, F. M. Hisama, D. A. Nickerson, and G. P. Jarvik. 2013. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. American Journal of Human Genetics 9(4):631-640.
Dressler, L. G., A. M. Deal, J. Patel, J. Markey, M. van Riper, and H. L. McLeod. 2014. Cancer pharmacogenomics, adoption by oncologists and patient benefit. Personalized Medicine 11(2):143-153.
Emanuel, E. J. 2014. A plan to fix cancer care. The New York Times, March 23, SR14.
FDA (Food and Drug Administration). 2011. Draft guidance on in vitro companion diagnostic devices. http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM262327.pdf (accessed February 18, 2015).
FDA. 2014. Framework for regulatory oversight of laboratory developed tests (LDTs). http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM416685.pdf (accessed January 30, 2015).
Febbo, P. G., M. Ladanyi, K. D. Aldape, A. M. De Marzo, M. E. Hammond, D. F. Hayes, A. J. Iafrate, R. K. Kelley, G. Marcucci, S. Ogino, W. Pao, D. C. Sgroi, and M. L. Birkeland. 2011. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. Journal of the National Comprehensive Cancer Network (Suppl 5):S1-S3.
Ferté, C., J. Guinney, J. Dry, R. McEwen, G. Manceau, K. J. Kao, K.-M. Chang, C. Bendtsen, K. Hudson, E. Huang, B. Dougherty, M. Ducreux, J.-C. Soria, S. Friend, J. Derry, and P. Laurent-Puig. 2013. Modeling RAS phenotype in colorectal cancer uncovers novel molecular traits of RAS dependency and improves prediction of response to targeted agents in patients. Clinical Cancer Research 20:265.
Gandhi, L., R. Bahleda, S. M. Tolaney, E. L. Kwak, J. M. Cleary, S. S. Pandya, A. Hollebecque, R. Abbas, R. Ananthakrishnan, A. Berkenblit, M. Krygowski, Y. Liang, K. W. Turnbull, G. I. Shapiro, and J.-C. Soria. 2013. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2—dependent and other solid tumors. Journal of Clinical Oncology 32(2):68-75.
Garfield, S., M. P. Douglas, K. V. MacDonald, D. A. Marshall, and K. A. Phillips. 2015. Consumer familiarity, perspectives and expected value of personalized medicine with a focus on applications in oncology. Personalized Medicine 12(1):13-22.
Gray, S. W., K. Hicks-Courant, A. Cronin, B. J. Rollins, and J. C. Weeks. 2014. Physicians’ attitudes about multiplex tumor genomic testing. Journal of Clinical Oncology 32:1317.
Grilley-Olson, J. E., D. N. Hayes, D. T. Moore, K. O. Leslie, M. D. Wilkerson, B. F. Qaqish, M. C. Hayward, C. R. Cabanski, X. Yin, M. A. Socinski, T. E. Stinchcombe, L. B. Thorne, T. C. Allen, P. M. Banks, M. B. Beasley, A. C. Borczuk, P. T. Cagle, T. Christensen, T. V. Colby, G. G. Deblois, G. Elberger, P. Graziano, C. F. Hart, K. D. Jones, D. M. Maia, K. V. Nance, W. D. Travis, and W. K. Funkhouser. 2013. Validation of interobserver agreement in lung cancer assessment: Hematoxylin-eosin diagnostic reproducibility for non-small-cell lung cancer: The 2004 World Health Organization classification and therapeutically relevant subsets. Archives of Pathology & Laboratory Medicine 137(1):32-40.
Hayes, D. F., R. C. Bast, C. E. Desch, H. Fritsche, Jr., N. E. Kemeny, J. M. Jessup, G. Y. Locker, J. S. Macdonald, R. G. Mennel, L. Norton, P. Ravdin, S. Taube, and R. J. Winn. 1996. Tumor marker utility grading system: A framework to evaluate clinical utility of tumor markers. Journal of the National Cancer Institute 88(20):1456-1466.
Hernán, M. A., A. Alonso, R. Logan, F. Grodstein, K. B. Michels, W. C. Willett, J. E. Manson, and J. M. Robins. 2008. Observational studies analyzed like randomized experiments: An application to postmenopausal hormone therapy and coronary heart disease. Epidemiology 19(6):766-779.
Holdhoff, M., K. Schmidt, R. Donehower, and L. A. Diaz, Jr. 2009. Analysis of circulating tumor DNA to confirm somatic KRAS mutations. Journal of the National Cancer Institute 101(18):1284-1285.
IOM (Institute of Medicine). 2010a. Policy issues in the development of personalized medicine in oncology: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2010b. A foundation for evidence-driven practice: A rapid learning system for cancer care: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2011. Building a national framework for the establishment of regulatory science for drug development: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2012. Evolution of translational omics: Lessons learned and the path forward. Washington, DC: The National Academies Press.
IOM. 2014. Ensuring patient access to affordable cancer drugs: Workshop summary. Washington, DC: The National Academies Press.
Kopetz, S., M. J. Overman, K. Chen, A. K. Lucio-Eterovic, B. K. Lee, D. R. Fogelman, A. Dasari, K. P. S. Raghav, E. V. Sanchez, J. Phillips, I. Shureiqi, C. R. Garrett, R. A. Wolff, K. Patel, K. D. Aldape, R. Luthra, M. Routbort, D. M. Maru, F. Meric-Bernstam, and C. Eng. 2014. Mutation and copy number discordance in primary versus metastatic colorectal cancer (mCRC). Paper presented at the annual meeting of the American Society of Clinical Oncology, Chicago, IL, May 30-June 3.
Malone, B. 2014. A new era for lab reimbursement. Clinical Laboratory News, June 1. https://www.aacc.org/publications/cln/articles/2014/june/lab-reimbursement (accessed March 19, 2015).
Marchetti, A., L. Felicioni, S. Malatest, M. D. Sciarrotta, L. Guetti, A. Chella, P. Biola, C. Pullara, F. Mucilli, and F. Buttitta. 2011. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. Journal of Clinical Oncology 29(26):3574-3579.
Neubauer, M. A., J. R. Hoverman, M. Kolodziej, L. Reisman, S. K. Gruschkus, S. Hoang, A. A. Alva, M. McArthur, M. Forsyth, T. Rothermel, and R. A. Beveridge. 2010. Cost effectiveness of evidence-based treatment guidelines for the treatment of non-small-cell lung cancer in the community setting. Journal of Oncology Practice 6(1):12-18.
Paik, R. K., M. E. Arcila, M. Fara, C. S. Sima, V. A. Miller, M. G. Kris, M. Ladanyi, and G. J. Riely. 2011. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. Journal of Clinical Oncology 29(15):2046-2051.
Parsons, D. Williams, A. Roy, and S. E. Plon. 2014. Clinical tumor sequencing: An incidental casualty of the American College of Medical Genetics and Genomics recommendations for reporting of incidental findings. Journal of Clinical Oncology 32(21):2203-2205.
Patel, J. P., M. Gönen, M. E. Figueroa, H. Fernandez, Z. Sun, J. Racevskis, P. van Vlierberghe, I. Dogalev, S. Thomas, O. Aminova, K. Huberman, J. Cheng, A. Viale, N. D. Socci, A. Heguy, A. Cherry, G. Vance, R. R. Higgins, R. P. Ketterling, R. E. Gallagher, M. Litzow, M. R. van den Brink, H. M. Lazarus, J. M. Rose, S. Luger, A. Ferrando, E. Paiette, M. S. Tallman, A. Melnick, O. Abdel-Wahab, and R. L. Levine. 2012. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New England Journal of Medicine 366(12):1079-1089.
Personalized medicine: Trends and prospects for the new science of genetic testing and molecular diagnostics. 2012. Minnetonka, MN: UnitedHealth Group.
Phillips, K. A., J. A. Sakowski, J. Trosman, M. P. Douglas, S.-Y. Liang, and P. Neumann. 2014. The economic value of personalized medicine tests: What we know and what we need to know. Genetics in Medicine 16(3):251-257.
Prentice, R. L., R. Langer, M. L. Stefanick, B. V. Howard, M. Pettinger, G. Anderson, D. Barad, J. D. Curb, J. Kotchen, L. Kuller, M. Limacher, and J. Wactawski-Wende. 2005. Combine postmenopausal hormone therapy and cardiovascular disease: Toward resolving the discrepancy between observational studies and the women’s health initiative clinical trial. American Journal of Epidemiology 162(5):404-414.
Reck, M., J. von Pawel, P. Zatloukal, R. Ramlau, V. Gorbounova, V. Hirsch, N. Leighl, J. Mezger, V. Archer, N. Moore, and C. Manegold. 2010. Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: Results from a randomised Phase III trial (AVAiL). Annals of Oncology 21(9):1804-1809.
Sandler, A. R. Gray, M. C. Perry, J. Brahmer, J. H. Schiller, A. Dowlati, R. Lilenbaum, and D. H. Johnson. 2006. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. New England Journal of Medicine 355:2542-2550.
Scagliotti, G. V., P. Parikh, J. von Pawel, B. Biesma, J. Vansteenkiste, C. Manegold, P. Serwatowski, U. Gatzemeier, R. Digumarti, M. Zukin, J. S. Lee, A. Mellemgaard, K. Park, S. Patil, J. Rolski, T. Goksel, F. de Marinis, L. Simms, K. P. Sugarman, and D. Gandara. 2008. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naïve patients with advanced-stage non-small-cell lung cancer. Journal of Clinical Oncology 26(21):3543-3551.
Shields, D., and R. Deshmukh. 2013. Biomarkers market (Diagnostic applications in risk assessment, molecular diagnostics, disease diagnosis, drug discovery & development, drug formulation, forensic applications and others)—global industry analysis, emerging technologies, competitive intelligence, growth trends, size, share, opportunities and forecast, 2013-2020. Portland, OR: Allied Market Research.
Socinski, M. A., I. Bondarenko, N. A. Karaseva, A. M. Makhson, I. Vynnychenko, I. Okamoto, J. K. Hon, V. Hirsch, P. Bhar, H. Zhang, J. L. Iglesias, and M. F. Renschler. 2012. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: Final results of a Phase III trial. Journal of Clinical Oncology 30(17):2055-2062.
Sosman, J. A., K. B. Kim, L. Schuchter, R. Gonzalez, A. C. Pavlick, J. S. Weber, G. A. McArthur, T. E. Hutson, S. J. Moschos, K. T. Flaherty, P. Hersey, R. Kefford, D. Lawrence, I. Puzanov, K. D. Lewis, R. K. Amaravadi, B. Chmielowski, H. J. Lawrence, Y. Shyr, F. Ye, J. Li, K. B. Nolop, R. K. Lee, A. K. Joe, and A. Ribas. 2012. Survival in BRAF V600—mutant advanced melanoma treated with vemurafenib. New England Journal of Medicine 366:707-714.
Thunnissen, E., M. Noguchi, S. Aisner, M. B. Beasley, E. Brambilla, L. R. Chirieac, J. H. Chung, S. Dacic, K. R. Geisinger, F. R. Hirsch, Y. Ishikawa, K. M. Kerr, S. Lantegoul, Y. Matsuno, Y. Minami, A. L. Moreira, G. Pelosi, I. Peterson, V. Roggli, W. D. Travis, I. Wistuba, Y. Yatabe, R. Dziadziuszko, B. Witte, M. S. Tsao, and A. G. Nicholson. 2014. Reproducibility of histopathological diagnosis in poorly differentiated NSCLC: An international multiobserver study. Journal of Thoracic Oncology 9(9):1354-1362.
Trosman, J. R., C. B. Weldon, R. K. Kelley, and K. A. Phillips. 2015. Challenges of coverage policy development for next-generation tumor sequencing panels: Experts and payers weigh in. Journal of the National Comprehensive Cancer Network.
U.S. Supreme Court. 2013. Association for Molecular Pathology et al. v. Myriad Genetics, Inc. et al. http://www.supremecourt.gov/opinions/12pdf/12-398_1b7d.pdf (accessed January 30, 2015).
Wang, R., S. W. Lagakos, J. H. Ware, D. J. Hunter, and J. M. Drazen. 2007. Statistics in medicine—reporting of subgroup analyses in clinical trials. New England Journal of Medicine 357:2189-2194.
Wilkerson, M. D., C. R. Cabanski, W. Sun, K. A. Hoadley, V. Walter, L. E. Mose, M. A. Troester, P. S. Hammerman, J. S. Parker, C. M. Perou, and D. N. Hayes. 2014. Integrated RNA and DNA sequencing improves mutation detection in low purity tumors. Nucleic Acids Research 42(13):e107.
Woodcock, J. 2010. Assessing the clinical utility of diagnostics used in drug therapy. Clinical Pharmacology & Therapeutics 88:765-773.















































































