
3
Nontesting Approaches Relevant to Prediction of Acute Toxicity and Potency
The term nontesting approaches was coined during the development of the European Union Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) regulation (EU 2006; ECHA 2008) to include the search and retrieval of existing data, the identification of structural alerts1 to indicate activity, the grouping of chemicals for read-across, and the development and application of quantitative structure-activity models. In practice, nontesting approaches are used to accomplish various tasks. For example, predictions based on structure–activity relationships (SARs) and quantitative structure–activity relationships (QSARs) are used to fill specific data gaps in lieu of experimental testing, to support findings or conclusions in integrated chemical assessments, and to substantiate predictions of various properties for structurally related chemicals. A key assumption that underpins nontesting approaches is that the property of a chemical with respect to how it will interact with a defined biological system is inherent in its molecular structure; thus, similar chemicals should have similar biological activities (the similarity principle) (Raunio 2011). More detailed information about nontesting approaches can be found in Cronin and Madden (2010).
This chapter discusses nontesting approaches in the context of the conceptual framework described in Chapter 2. Box 3-1 provides definitions for some of the terms used in the chapter. The chapter discusses the use of available data to characterize chemicals of interest and in silico approaches to predict physical hazards, chemical reactivity, pharmacokinetic properties, and acute toxicity. It also provides selected examples to demonstrate how computational tools could be used in predictive toxicology.
INITIAL CHEMICAL CHARACTERIZATION
The starting point in the application of any nontesting approach for predicting acute toxicity is a preliminary search and evaluation of available data on the chemical of interest. The effort often begins with database queries, literature searches, and other approaches for finding information about the chemical’s structure, physicochemical properties, and acute toxicity (see Box 3-2). The committee notes that the forthcoming REACH regulation requires in vivo acute oral toxicity information for chemicals manufactured or imported into Europe at greater than 1 metric ton per year (ECHA 2012). It is anticipated that a large volume of in vivo oral data will be potentially disseminated publically after the REACH May 2018 deadline.
__________________
1A structural alert is a chemical structure that has been linked to toxicity or a specific toxicity end point.
BOX 3-1 Definitions of Selected Nontesting Approaches
A structure–activity relationship (SAR) is a qualitative association between a chemical (sub)structure (such as a functional group) and the potential of a chemical that contains the (sub)structure to exhibit a particular biological effect.
A quantitative structure–activity relationship (QSAR) is “a mathematical relationship between a quantifiable aspect of chemical structure and a chemical property or reactivity or a well defined biological activity, such as toxicity” (EPA 2012). QSARs can be derived to predict quantitative or qualitative end points.
A quantitative structure–property relationship (QSPR) is a special case of QSAR in which a physicochemical property is modelled as the response variable.
An expert system is a software tool that specifically encodes compilations of SARs, QSARs, or both to enable rational predictions of toxicity to be made on the basis of structure alone. Expert systems are typically categorized as statistical (for example, TOPKAT and Accelrys Inc), knowledge-based (for example, such SAR-based approaches as Derek Nexus and LHASA Ltd), or hybrid (for example, TIMES-SS).
Category approach, analogue approach, and read-across: Category and analogue approachesare techniques for grouping chemicals; read-across is a technique for filling data gaps in category and analogue approaches (ECHA 2008; OECD 2014). Read-across can be qualitative or quantitative and uses existing “information on the property of a substance (source chemical)…to make a prediction of the same property for another substance (target chemical) that is considered similar” with respect to the end point of interest (Worth 2008). Analogue approaches are used for grouping a small number of chemicals when there are no apparent trends in ti
A chemical category is “a group of chemicals whose physicochemical and human health…or environmental toxicological properties…or environmental-fate properties are likely to be similar or follow a regular pattern as a result of structural similarity” (OECD 2007a, 2014). Chemical similarity could be based on a variety of properties, including the presence of a common functional group (such as an aldehyde), common constituents or chemical classes, similar carbon range numbers, or common precursors or breakdown products.
In silico approaches include computational modeling, SAR analysis, analysis of physicochemical characteristics, and read-across techniques.
The available information can help in identifying a chemical’s potential for direct physical hazards, most relevant routes of exposure, likely bioavailability, and potential for inducing (human) toxicity (NRC 2014). It should also be considered before designing or initiating new in vitro or in vivo experimental studies, in interpreting existing empirical data, or in selecting appropriate (Q)SAR models.2
As described below, physicochemical properties of interest in predictive toxicology can be nominally categorized into three broad types: physical properties, solvation properties, and molecular attributes (NRC 2014). There are methods for empirically measuring the properties and in silico approaches for estimating their values (see Box 3-3). It can be particularly helpful to complement estimated values with experimental measurements, when that is possible.
__________________
2The committee uses the shorthand notation (Q)SAR to indicate both SAR and QSAR.
BOX 3-2 Primary Data Considered During a Preliminary Characterization of a Chemical of Interest
Chemical structure: Chemical structure is the spatial arrangement of a molecule’s constituent atoms. PubChem, DSSTox, and ChemIDplus are examples of searchable chemical-structure databases.
Physicochemical properties: Physicochemical properties contribute to the inherent hazards posed by a chemical, including its ability to interfere with normal biological processes. Physicochemical properties could also define a chemical's physical hazards of interest (such as corrosivity). Physical properties include freezing point, boiling point, melting point, infrared spectrum, electronic characteristics, viscosity, and density. Other properties of relevance include solvation properties, such as phase partitioning and solubility. One of the more important phase-partition coefficients is obtained from a system in which one solvent is water or an aqueous phase and the second is organic and hydrophobic, such as 1-octanol, that is, the octanol–water partition coefficient (Kow). An example of a database source of physicochemical-property data is the National Institute of Standards and Technology Chemistry WebBook; another is the PHYSPROP database, which is integrated into the US Environmental Protection Agency’s EPISuite software.
Acute toxicity (for example, rodent LD50 or LC50 values): These data might be available from primary sources (such as peer-reviewed literature) and secondary sources (such as the Merck Handbook). By far the most convenient sources of data are compiled databases that are readily searchable by chemical identifiers, such as chemical name, Chemical Abstracts Service registry number, or chemical structure. An example is the National Library of Medicine TOXNET® database.
See Appendix B for additional information and examples.
- Physical properties. Physical properties include such characteristics as freezing point, melting point, boiling point, vapor pressure, and viscosity. Melting point, boiling point, and vapor pressure can be used to predict a chemical’s likely physical state, which is pertinent in determining the most relevant route of exposure for any testing or indeed what practical challenges might need to be overcome in in vitro testing scenarios or even what issues to consider in interpreting in vivo results and associated testing protocols.
- Solvation properties. Solvation properties describe a chemical’s interaction with different phases and its interaction between phases (for example, logKow represents the partitioning between octanol and water). Water solubility and logKow are particularly helpful in determining the technical feasibility of performing in vitro test protocols given that they typically use aqueous media.
- Molecular attributes. Molecular attributes capture properties related to molecular shape and size. Electronic characteristics of molecules, such as frontier orbital energies and polarizability, that are related to reactivity could be considered to constitute a type of molecular attribute. They also play a role in predicting likely bioavailability and toxicity.
Physicochemical data can be used to predict a chemical’s physical hazard, reactivity, and pharmacokinetics, including absorption by different exposure routes, distribution in the body, and likely metabolites. Approaches that apply knowledge about a chemical’s physicochemical properties to predictive toxicology presume that for a chemical to exert a toxic effect, it typically must be bioavailable to such an extent that it (or its metabolite) reaches a biochemical target, where it can exert its toxic effect (Meek et al. 2013).
BOX 3-3 In Silico Approaches for Predicting Physicochemical Properties
In the absence of data on physicochemical properties, reasonable estimates based on chemical structure are feasible with the use of QSAR and QSPR models (Dearden and Worth 2007). A discussion of those methods is beyond the scope of the present activity. However, the National Research Council report A Framework to Guide Selection of Chemical Alternatives provides a succinct discussion of published models that can be used to characterize a number of physicochemical properties (NRC 2014). That report discusses methods used to estimate molecular hydrophobicity (or lipophilicity) and other physicochemical end points, such as aqueous solubility pKa and the electronic properties of molecules.
A number of software packages and algorithms are available for predicting physicochemical properties, and predictions are often in excellent agreement with experimentally derived values. For example, pKa can be estimated by using Taft and Hansch fragment coefficients, and Perrin et al. (1981) contains an extensive compilation of the fragment values and relevant equations to do so. For convenience, software tools, such as SPARC or those created by ACD Labs, contain algorithms for estimating pKa directly from chemical structure. The user of such tools, however, must have a basic understanding of the inherent advantages and limitations of the various algorithms as they are related to the accuracy of physicochemical-property prediction. In general, the QSAR models available for prediction of the key physicochemical characteristics are best suited for small organic chemicals that typically have one functional group. Other chemicals—such as pesticides, drug-like chemicals, or other pharmacological actives—typically lack published data to derive such QSARs.
Use of Physicochemical Properties to Predict Physical Hazard and Chemical Stability or Reactivity
Assessing the likely irritant or corrosive effects of a chemical would be a helpful component of a tiered evaluation strategy for predicting acute toxicity.3 In the absence of measured irritant or corrosive data, a handful of (Q)SAR approaches are useful in identifying potential irritants or corrosives. The German Federal Institute for Risk Assessment (BfR) rule base (Gerner et al. 2004; Hulzebos et al. 2005) is one example of an expert system that uses physicochemical exclusion rules and structural alert inclusion rules to determine likely skin or eye irritation hazard.4 The BfR rule base has been encoded into software tools, including the Organisation for Economic Co-operation and Development (OECD) QSAR Toolbox5 and the European Commission Joint Research Centre Toxtree (Rorije and Hulzebos 2005; Tsakovska et al. 2007). Some QSARs published for specific chemical classes have relied on such properties as pKa, dipole moment, logKow, and molecular weight or volume to estimate likely irritation potential and potency (Barratt 1996). Both pKa and dipole moment have been found to be useful measures for modeling chemical reactivity depending on whether the target substance is an acid, a base, or a neutral organic; and logKow and molecular weight have served as useful surrogates for modeling partitioning. QSARs also exist within expert
__________________
3Skin irritation or corrosion can be investigated in vitro by virtue of assays, such as Corrositex (OECD 2006) for corrosion and EpiDerm™ (OECD 2013a) for irritation. For eye irritation or corrosion, various ex vivo and in vitro assays are available, including the bovine corneal opacity permeability test (OECD 2013b), the isolated chicken-eye test (OECD 2013c), or the EpiOcular™ eye-irritation test method. A tabulation of assays for irritation and corrosion that have been validated by ECVAM or ICCVAM or that exist as test guidelines under OECD are provided on the AltTox.org Web site (AltTox 2014) and are discussed in Chapter 4.
4The BfR rule base combines two approaches: exclusion rules that use physicochemical thresholds to identify chemicals that are not skin irritants or corrosive and inclusion rules that use structural alerts to identify chemicals that are potentially irritants or corrosive (Saliner et al. 2007). The rule base assigns a regulatory classification for skin or eye irritation or corrosion.
5The committee refers here to the toolbox by its official name rather than OECD (Q)SAR Toolbox, which would be more appropriate because the Toolbox includes both SAR and QSAR approaches.
systems, such as TOPKAT and MCASE, for the prediction of irritation or corrosion. Saliner et al. (2008) reviewed the status of (Q)SAR approaches for irritation and corrosion.
Consideration should also be paid to the inherent stability or electrophilic reactivity of the chemical. A chemical might exert its effects in its parent form or be transformed abiotically or biotically to a metabolite that is a more relevant target for evaluation. Some substances are rapidly hydrolyzed; for example, acid chlorides and acid anhydrides are rapidly hydrolyzed to their corresponding carboxylic acids. Other substances are capable of being oxidized when exposed to air; for example, p-hydroquinone is rapidly oxidized to its corresponding benzoquinone, which is highly reactive. The OECD Toolbox contains simulators that help in predicting such transformations. Consideration of how a chemical might be transformed is important in interpreting experimental data, performing new testing, or using the most relevant target for (Q)SAR analyses.
Use of Physicochemical Properties to Predict Chemical Disposition and Metabolism
Pharmacokinetics describes the disposition of a chemical in an organism and considers chemical absorption, distribution, metabolism, and excretion (ADME). Pharmacokinetic properties can play an important role in the assessment of a chemical’s effects on or risks to the body. In silico approaches have been developed to predict many ADME processes; the sections below focus largely on approaches that are directly relevant for predicting acute toxicity.
Absorption: Oral
Some physicochemical properties—such as molecular weight, the number of hydrogen-bond donors and acceptors, and logKow—have been shown to be predictive of oral absorption. For example, Lipinski’s rule of 5 is considered helpful in evaluating the likely absorption, permeability, and toxicity of drug-like substances (Lipinski et al. 2001) and considers the three properties noted to make predictions about chemical behavior. Other examples of heuristic rules are provided in Table 3-1.
In addition to heuristic rules, several QSAR models have been developed to determine intestinal absorption and oral absorption. Iyer et al. (2007) used a membrane-interaction QSAR analysis to build models for human oral intestinal drug absorption. Castillo-Garit et al. (2008) developed a mathematical model that used linear indexes to predict the in vitro permeability of 157 chemicals in a Caco-2 cell model. Their mathematical model had greater than 80% accuracy in predicting how well a drug would be absorbed by Caco-2 cells. Guerra et al. (2010) developed an artificial neural network by using CODES 2D descriptions to predict oral drug absorption. Suenderhauf et al. (2011) used a broad selection of machine learning and statistical methods to derive classification and prediction models for human intestinal absorption. Several recent reviews discuss the status of such QSAR models (Xu and Mager 2011; Silva and Trossini 2014; Wang and Hou 2015).
TABLE 3-1 Examples of Heuristic Rules to Predict Oral Absorption
| Rule | Descriptiona | Reference |
| GlaxoSmithKline | Chemicals with cLogP < 4 and MW < 400 Da have superior drug-like | Gleeson 2008 |
| rule of 4/400 | properties compared with chemicals with cLogP > 4 and MW > 400 Da | |
| Pfizer rule of 3/75 | Chemicals with cLogP > 3 and total PSA < 75 Å are 2.5 times more likely | Hughes et al. 2008 |
| to have in vivo toxicity than ones with cLogP < 3 and total PSA > 75 Å | ||
| AstraZeneca | Alkalinity and increased cLogP are associated with multiple positive | Leeson and |
| responses in various toxicity assays | Springthorpe 2007 | |
There are also physiologically based packages that can predict oral absorption rates of drugs, such as GastroPlus and SimCyp (Kuentz et al. 2006; Rostami-Hodjegan and Tucker 2007; Yang et al. 2007; Simulations Plus 2010; Grbic et al. 2011). However, if the goal is to determine likely oral absorption to help to prioritize chemicals, the heuristic rules described above might be adequate for that task.
Absorption: Dermal
LogKow, water solubility, and molecular weight are also useful inputs for estimating dermal absorption characteristics of a target substance. QSAR models for predicting the dermal permeability coefficient (Kp)—a measure useful for modeling dermal penetration—typically rely on LogKow and molecular weight as input variables. Potts and Guy (1992) derived such a model that is also encoded in DERMWIN as part of EPA’s EpiSuite software. Over the years, the model derived by Potts and Guy has been modified to address limitations, and many variants now exist. Mitragotri et al. (2011) reviewed the status of models for the prediction of skin permeability in terms of their strengths, limitations, and future prospects.
Other researchers have incorporated additional information, such as degree of hydrogen bonding and melting point, to refine skin penetration estimates (Hostýnek 1997; Magnusson et al. 2004; ten Berge 2009; Dancik et al. 2013a). Models by ten Berge (2009) and Dancik et al. (2013a,b) are helpful in evaluating systemic availability as a result of dermal exposure and thus provide a means of extrapolating a dermal acute-toxicity (LD50) value from an oral acute-toxicity (LD50) value. Kasting’s model (as discussed in Dancik et al. 2013a,b) explicitly takes into account various components of the skin structure, including the stratum corneum, viable epidermis, and dermis. The model simulates one-dimensional transient passive transport into a skin slab. Some properties are also required as inputs for a simulation, including logKow, vapor pressure, melting and boiling points, molecular weight, chemical class (alcohol, hydrocarbon, or other organic), and presence of a pharmacophore6 as defined by Yamazaki and Kanaoka (2004). The model is available for use from the National Institute for Occupational Safety and Health Web site (NIOSH 2013).
Absorption and Deposition: Inhalation
For nonvolatile chemicals, particle size is an important consideration because it affects deposition in the respiratory tract and influences whether a particle poses an inhalation hazard (ECETOC 2012; Brown et al. 2013). Brown et al. (2013) predicted that about half of all 10-μm particles penetrate into the thorax and that about 20% or less of all 10-μm particles would penetrate to the extrathoracic airways and into the lower respiratory tract.
For volatile substances, physicochemical characteristics—such as vapor pressure, water solubility, and reactivity—are also important for predicting acute toxicity by the inhalation route (Veith and Wallace 2006; Veith et al. 2009).
Metabolism
Several tissues—including the lung, skin, liver, intestine, and kidney—have enzymes that can convert a parent chemical to a metabolite, for example, through oxidation and conjugation processes. Whereas parent chemical metabolism typically results in a more hydrophilic chemical that is more easily excreted, a reactive toxic metabolite is sometimes formed. Not considering that possibility and focusing solely on the parent chemical will therefore be inadequate in characterizing a chemical’s potential to elicit acute toxicity accurately.
__________________
6A pharmacophore is the collection of steric and electrostatic features of different chemicals that are necessary to ensure optimal molecular interactions with a specific biological target (Langer and Wolber 2004).
Predicting chemical metabolism requires tools that can identify the functional groups or structural components of the parent chemical that are vulnerable to metabolism (sites of metabolism) and the structure of possible metabolites. Additional information about enzyme structure and function and about the effect of metabolism on the induction or inhibition of metabolizing enzymes could also be considered, but for the purposes of predicting the potential of chemicals to elicit acute toxicity, this discussion will focus primarily on the first two factors. Determining the site of metabolism allows prediction of overall metabolic stability, such as the rate of activity (Vmax, Km) or clearance rate (Clint), that is measured either in vivo or in vitro. Predicting metabolic structures involves listing possible metabolites from reactions that the chemical could undergo (biotransformation).
Metabolism-predictive tools are based on large compilations of databases derived from metabolism information in the literature, for example, Accelrys Metabolite Database, Metabolite, MetaBase, and MetaDrug (Kirchmair et al. 2012). The metabolism information in a database can be used to identify likely metabolic sites on the basis of what is known about the target chemical structure or a similar chemical structure. The databases can also be used to predict possible metabolites by using information that describes enzyme activity, such as binding pocket sites. Most available metabolism-predictive tools consider a single aspect of metabolic reactions—such as the reaction energy barrier, geometrical properties, or pharmacokinetic properties—to predict sites of metabolism or potential metabolites. ADMET Predictor is an example of a commercial product that uses the Accelrys Metabolite Database to predict various metabolic stability values for a series of cytochromes (Simulations Plus 2010). Kirchmair et al. (2012) provide a comprehensive overview of methods for predicting sites of metabolism.
Knowledge-driven approaches, such as expert systems, allow extrapolation of structure to likely metabolites by using advanced reasoning rules and expert-system metabolite ranking, such as MetabolExpert, META, Meteor, and Metaprint2D-React. However, one problem is that they can generate a large number of metabolites from which it is difficult to determine which metabolites are the relevant and stable ones that should be considered. Other metabolism-predictive tools introduce the expert-system features with more refined computational algorithms to support the decision method and therefore limit the number of metabolites that are generated. Indeed, the software program Tissue Metabolism Simulator (TIMES) uses a comprehensive library of biotransformation information and a heuristic algorithm to generate plausible metabolic maps that are relevant to specific end points, such as skin sensitization or genotoxicity (Mekenyan et al. 2012; Patlewicz et al. 2014). Many of the TIMES metabolism simulators have been made freely available in the OECD QSAR Toolbox.
Limitations and Need for Improvement
Many tools for predicting physicochemical properties that are relevant for the evaluation of chemical disposition and distribution factors are available, but they are limited by their training sets.7 Such tools are generally most applicable for small organic chemicals—chemicals that have molecular weights of 500 Da or less (that is, not mixtures or polymers).
In vitro and in silico predictions of absorption for various routes of exposure are still crude, and current models might have little applicability to the Department of Defense (DOD).8 For oral absorption, Lipinski’s rule of 5, which is based on experience in the drug-discovery world, might
__________________
7Training sets are data that are used to develop predictive models or tools.
8For example, in vitro assays for absorption (such as Caco-2 monolayer crossing) were developed primarily to predict systemic absorption after deliberate oral dosing (Artursson and Karlsson 1991). Thus, they are expected to be much less predictive for exposure routes (dermal and inhalation) that are more relevant to acute battlefield exposure. Likewise, crossing the blood–brain barrier is especially relevant for neurotoxicity, and although there are computational approaches for predicting blood–brain barrier penetration (Gerebtzoff and Seelig 2006; Carpenter et al. 2014), no in vitro assay accurately measures this property.
provide a convenient set of heuristics for chemicals of interest to DOD but would need to be evaluated to determine its applicability. The prediction of dermal permeability at the simplest level is illustrated by QSARs that predict logKp or logJmax with such inputs as molecular weight and logKow as exemplified by Potts and Guy (1992) or Magnusson et al. (2004) (see also Fitzpatrick et al. 2004 and Mitragotri et al. 2011). Although refinements have been made to simulate penetration or systemic bioavailability (Dancik et al. 2013a,b), the underlying characteristics are still based largely on the heterogeneous dataset first compiled by Flynn (1990), which is limited in its coverage of chemicals.
Current metabolism-predictive approaches have several limitations. First, most of the existing metabolism-predictive tools were designed primarily to inform drug development. There are few examples in which such modeling tools have been used to evaluate volatile or lipophilic chemicals (Peyret and Krishnan 2012; Kirman et al. 2015). Thus, the available metabolism training sets will need to be expanded for the chemicals of interest. Second, although metabolism-predictive tools adequately predict transformations of various chemicals, they do a poor job of distinguishing differences in reactivity of closely related structural analogues. In most cases, the tools can only estimate the reactivity of the individual molecular sites. As a result, they have limited use for prioritizing a broad set of structurally related chemicals (Kirchmair et al. 2012). One interim solution that DOD might consider is to evaluate metabolites with known toxic effects and incorporate more metabolically competent test systems into its test battery. Chapter 4 describes each approach in some detail.
IN SILICO APPROACHES FOR PREDICTING TOXIC EFFECTS
In silico models incorporate a variety of physicochemical features that can be used to predict receptor binding, toxicity, and other biological outcomes. Many (Q)SAR models developed for use in toxicology have been built on a longstanding recognition that the physicochemical properties of a chemical, especially lipophilicity, are highly relevant to prediction of acute toxicity. It has been shown that the presence or absence of various physicochemical properties can be used to group chemicals into toxicity categories (Greene and Song 2011). That concept is well established and used in the pharmaceutical industry to reduce attrition in drug discovery, reduce toxicity, and improve the drug-likeness of chemicals (see Table 3-1).9 Indeed, several studies have shown how simple measures, such as logKow and total polar surface area (TPSA), provide useful indicators of potential toxicity in vivo. Hughes et al. (2008) showed for a dataset of 245 substances that substances that had low logKow and high TPSA were about 2.5 times more likely to be “clean” (nontoxic) than to be toxic. Precisely the reverse was true of chemicals that had high logKow and low TPSA, properties that increased the likelihood of chemical binding to multiple biological targets that could contribute to toxicity.
Although the chemical domain of concern for DOD goes beyond that of drug-like substances, an understanding of the type of physicochemical properties that can affect adverse outcomes and the range of property values for which the effect is likely to be substantial can offer useful insights for guiding the assessment of acute toxicity of chemicals of interest to DOD. The sections that follow describe in silico approaches that are available or could be developed for predicting acute toxicity that is relevant to DOD’s concerns.
Acute Oral Toxicity
There are a few (Q)SAR models and expert systems for prediction of in vivo acute toxicity. The predictiveness of the models, however, can be variable. For some chemicals, the models
__________________
9Drug-likeness refers to molecules that contain functional groups or have physical properties similar to those of known drugs (Walters and Murcko 2002).
provide predicted values that deviate by several orders of magnitude from the experimental data. Furthermore, efforts have focused largely on the prediction of acute rodent oral toxicity (see Appendix B for a description of various toxicity data sources). Fewer attempts have been made to derive models of acute toxicity via other routes of exposure, such as dermal or inhalation, although predictions based on extrapolation from acute oral LD50 values have been attempted.
Available (Q)SARs for acute systemic toxicity have been reviewed (Cronin and Dearden 1995; Cronin et al. 2003; Lessigiarska et al. 2005; Tsakovska et al. 2006; Devillers and Devillers 2009; Lapenna et al. 2010). Several QSAR models have identified hydrophobicity and electronic and steric effects as important model parameters. Many literature-based models were developed for a single chemical class, such as alcohols, barbiturates, pyrines and their derivatives, and benzene derivatives. Examples are provided in Table 3-2.
In contrast, models based on heterogeneous datasets have typically been incorporated into expert systems. There are, however, examples of models based on heterogeneous data that have not been incorporated into expert systems, and there are examples of models that use a hybrid approach. Table 3-3 provides several examples of various types of models and tools. There has been an evolution in the types of (Q)SAR models developed over the years to predict acute toxicity. Expert systems tended to favor large datasets (global models) that use chemistry-based descriptors to derive estimates of rodent oral toxicity. Hybrid expert systems consider biological activity, such as cytotoxicity information as described in Chapter 4, and chemistry-based descriptors as inputs. More recently, there has been a return to local (Q)SAR models; they are integrated into batteries of (Q)SARs that can predict acute toxicity of diverse chemicals.
Acute Dermal Toxicity
To the committee’s knowledge, there are no notable QSARs for the prediction of rodent dermal acute-toxicity values. Dermal LD50 values might be estimated by extrapolating from oral LD50 values in some cases by using toxicokinetic information. A case study of three cosmetic substances was performed by Gajewska et al. (2014) to evaluate such an extrapolation.
Moore et al. (2013) found that the toxicity of chemicals was usually greater by the oral route than the dermal route. They proposed that data on oral acute systemic toxicity could be used in lieu of equivalent dermal testing with little or no concern for underclassification according to the Globally Harmonized System of Classification and Labeling of Chemicals (GHS).10 For example, dermal testing of a substance that has an oral LD50 of greater than 2,000 mg/kg
TABLE 3-2 Examples of (Q)SARs for Various Chemical Classes
| Chemical Class | Description | Reference |
| Alcohols | Four molecular-structure descriptors and two indicator variables formed the basis of a categorical model that categorized 95 alcohols into ranges of LD50 values. | Guilian and Naibin 1998 |
| Barbiturates | The number of valence electrons and logKow were found to be predictive of LD50 values of a set of 11 barbiturates. | Hansch and Kurup 2003 |
| Pyrines and derivatives | The energy of the lowest unoccupied molecular orbital and logKow represented the descriptors used in a QSAR for pyrines and their derivatives. | Cronin et al. 2002 |
| Benzene derivatives | Electronegativity, dipole moment, and the presence of nitrogen-containing groups were most important in predicting the acute oral toxicity of benzene derivatives. | Toropov et al. 2008 |
__________________
10The GHS is an internationally agreed-on system created by the UN to replace the various classification and labeling standards used in different countries by using consistent criteria for classification and labeling (UNECE 2015).
TABLE 3-3 Examples of Models and Tools for Predicting Acute Oral Toxicity
| Model | Description | Outputs | Reference |
|---|---|---|---|
| Models Based on Heterogeneous Datasets That Have Been Incorporated into Expert Systems | |||
| TOxicity Prediction by KomputerAssisted Technology (TOPKAT) | 19 QSAR regression models are based on a number of structural, topological, and electropological indexes and experimental values for 4,000 chemicals in the Registry of Toxic Effects of Chemical Substances (RTECS) database. | Rat LD50 (oral) Rat LC50 (inhalation) | TOPKAT, Accelrys (reviewed in Lapenna et al. 2010) |
| Toxicity Estimation Software Tool (TEST) | Based on chemicals in the RTECS database. Uses a variety of QSAR models (hierarchical method, Food and Drug Administration [FDA] method, single-model method, group-contribution method, nearest-neighbor method, and consensus method) to yield toxicity estimates. | Rat LD50 (oral) | EPA 2014 |
| ACD/Labs Tox suite | Predictions are based on a combination of expert knowledge of various basal and extracellular effects (such as cholinesterase inhibition, ATP synthesis, and CNS disruption) and QSAR analysis of more than 10,000 chemicals. Predictions are provided with reliability estimations, and chemicals are classified into five toxicity categories. | Rat and mouse LD50 (routes include oral, intraperitoneal, subcutaneous, and intravenous) | ACD/Labs 2015 |
| ProTox | Prediction is based on the analysis of the similarity of chemicals that have known median LD50 that are taken from a dataset of 38,000 chemicals and incorporates the identification of toxic fragments. | Rodent LD50 (oral) | Drwal et al. (2014); ProTox (2015) |
| Models Based on Heterogeneous Datasets That Have Not Been Incorporated into Expert Systems | |||
| Consensus models | Use rodent in vivo acute oral data from the National Library of Medicine databases as reported in ChemIDplus. Predictions are based on different QSAR statistical techniques, including random forest, FDA MDL-QSAR program’s approach to k-nearest neighbor, and hierarchical clustering. | Rodent LD50 (oral) | Zhu et al. (2009a) |
| Multiclassification methods | Based on a dataset containing 12, 204 diverse chemicals and published acute oral rodent LD50s. Model predictions obtained with machine-learning methods, such as support vector machine, C4.5 decision tree, random forest, k-nearest neighbor, naive Bayes algorithms, and MACCS and FP4 fingerprints. | Rat LD50 (oral) | Li et al. (2014) |
| Global Hybrid Approaches |
|||
| Tiered approach | All chemicals separated into two groups: one based on the relationship between the in vitro half-maximal inhibitory concentration (IC50) and rodent LD50 and the other contained the remaining chemicals. A two-step QSAR modeling approach was then applied. The derived binary classification QSAR models predicted group membership on the basis of the in vitro–in vivo relationships, and a second QSAR model estimated the LD50s for the chemical subsets. | Rodent LD50 (oral) | Zhu et al. (2009b) |
| Chemical and biological descriptors | Dataset consisted of 67 chemicals obtained from the literature. Used structural information and in vitro basal cytotoxicity to predict human acute toxicity. | Indirect measures of human toxicity (e.g., LC50) | Lee et al. (2010) |
| Inclusion of concentration–response data derived from high-throughput screening | Used quantitative high-throughput screening concentration–response data to complement traditional chemical descriptors in the modeling of acute oral rodent LD50s. | Rodent LD50 (oral) | Sedykh et al. (2011) |
| Local Hybrid Approaches |
|||
| OASIS Pipeline | Relies on a baseline model for neutral organic substances supplemented with mechanistic SARs for different reaction-chemistry domains. The approach is underpinned by 3-D QSARs where relevant to predict acute oral-toxicity categories by using the same RTECS dataset as used by Zhu et al. (2009a). Complements the approach outlined by Koleva et al. (2011). | Mekenyan et al. (personal communication, December 2014) | |
| Local lazy method | Based on a dataset of 9,617 chemicals. Uses local structure–toxicity relationships associated with a query substance to develop acute oral LD50 models. | Rodent LD50 (oral) | Lu et al. (2014) |
aGlobal hybrid approaches use models derived on the basis of a large heterogeneous dataset that includes chemical and biological information.
bLocal hybrid approaches aim to derive models that are specific to chemical class or reaction chemistry but will still be applicable to a broad spectrum of chemicals.
would provide no added value for categorizing its hazard. Moore et al. (2013), however, reported that a majority of chemicals that they evaluated would be classified more stringently if oral classifications were applied directly to the dermal route. One approach to address the tendency for overclassification would be to consider whether a chemical is absorbed by the skin.
Acute Inhalation Toxicity
There are only a handful of QSARs for acute inhalation toxicity. One example is that for volatile substances. Veith et al. (2009) derived a baseline narcosis model that related vapor pressure (as logVP in millimeters of mercury) to the 4-hour molar logLC50 in rodents for neutral organic substances. Veith and Wallace (2006) also established relationships for reactive (electrophilic) chemicals in which reactivity, as measured in the glutathione depletion assay (Schultz et al. 2005), was related to the molar logLC50. The underlying basis of their strategy mimics the Adverse Outcome Pathway (AOP) construct as described by Ankley et al. (2010).
The expert system TOPKAT incorporates a global model for the prediction of acute inhalation toxicity. The rat inhalation LC50 module contains five models related to different chemical classes to cover a reasonable breadth of chemical coverage.
Neurotoxicity
Neurotoxicity is another debilitating effect associated with some acute exposures. A few QSARs have been derived for neurotoxicity, but they are quite limited in scope. Cronin (1996) derived a neurotoxicity QSAR for a set of 44 common solvents that depended on logKow and membrane permeability. A number of modeling approaches to derive QSAR models for organophosphorus pesticides have also been developed (Devillers 2004; Garcia-Domenech et al. 2007).
A handful of SARs exist that might identify structural alerts for neurotoxic potential. Chemical classes associated with neurotoxicity include some organic solvents, organophosphorus chemicals, and carbamates, which can induce chronic toxic encephalopathy, delayed neurotoxicity, and cholinergic effects, respectively. For example, Derek Nexus includes the following structural alerts: organophosphate (for direct and indirect anticholinesterase activity), N-methyl or N,N-dimethyl carbamate (for direct anticholinesterase activity), and gamma-diketones (for neurotoxicity) (ECHA 2014).
Neurotoxicity is clearly an effect whose mechanisms need to be better elucidated, and models developed accordingly.
Cytotoxicity
Ekwall (1983) suggested that for most chemicals, toxicity was a consequence of nonspecific alterations in cellular function; thus, evaluating the cytotoxic potential of chemicals with cytotoxicity assays could provide an indication of their potential in vivo toxicity. There have been many attempts to explore the correlation between in vivo acute toxicity and cytotoxicity data and a number of efforts to predict cytotoxicity from chemical structure. For example, as part of the Multicenter Evaluation of In Vitro Cytotoxicity program (Ekwall et al. 1998), 50 reference chemicals were tested in 61 cytotoxicity assays in the hope of predicting acute oral LD50s. The coefficients of determination (r2) were 0.61 for rat LD50s and 0.65 for mouse LD50s. And, Lessigiarska et al. (2006) demonstrated how acute toxicity in rats, mice, and humans could be predicted by using QSAR models that incorporated cytotoxicity data, other biological end points, and chemical structural descriptor data.
A host of QSAR models have been derived to predict cytotoxicity of various chemical classes. In many cases, hydrophobicity was a predominant descriptor related to cytotoxicity. For example, McKarns et al. (1997) correlated the loss of membrane integrity in rat liver epithelial cells with hydrophobicity as modeled by logKow for a series of 11 alcohols. Other QSARs, as
summarized by Tsakovska et al. (2006), have been developed for p-substituted benzyl alcohols, phenols, anilines, chlorophenols, and polybrominated diphenyl ethers. Papa et al. (2009) developed QSARs for three toxicological end points: mouse oral LD50 values, inhibition of NADH oxidase (EC50), and effect on mitochondrial membrane potential (EC50). Freidig et al. (2007) found that nonspecific cytotoxicity could help to identify irritant chemicals.
As part of the European Union framework program, ACuteTox, many investigations were performed to explore in vivo–in vitro relationships. Clothier et al. (2013) used Spearman rank-correlation analysis and hierarchical-cluster analysis to identify in vitro testing strategies for predicting acute toxicity. Classification-based and regression-based quantitative structure–toxicity relationship (QSTR) and toxicophore models were developed by Kar and Roy (2013). They used in vitro cytotoxicity data collected from the ACuteTox database.11 Their QSTR models showed that cytotoxicity was influenced by the presence of hydrophobic aliphatic groups, a ring aromatic group, and hydrogen-bond donors. The in silico models derived were considered capable of identifying the essential structural attributes and quantifying the molecular properties that drive in vitro basal cytotoxicity. Prieto et al. (2013) proposed a heuristic testing strategy for identifying potential neurotoxicants that considered octanol–water partition coefficients, the prediction results from the neutral red uptake assay performed in 3T3 cells, and in silico predictions of intestinal absorption and blood–brain barrier passage.
Limitations and Need for Improvement
The greatest focus in the literature has been on deriving QSAR models to predict oral rodent LD50s. There are some models for specific chemical classes, but there has been greater interest in exploring the feasibility of deriving global models. Many of the global models have been data-driven, although some attempts have included consideration of a chemical mechanistic approach akin to that described for acute fish toxicity (Bradbury et al. 1990; Schultz et al. 2006). More recently and in part as stimulated by work within the ACuteTox program, predicting in vivo acute toxicity has considered the use of (Q)SAR approaches in conjunction with in vitro cytotoxicity data. This shift of integrating in vitro and in silico approaches is consistent with the framework of AOPs as one means of incorporating more mechanistic information in testing and assessment approaches for different purposes.
A key issue in all approaches is their relevance and applicability to DOD chemicals. The relevance of the (Q)SAR models cited earlier would need to be probed for the types of chemicals under consideration by DOD to determine the extent to which the existing models are appropriate in light of the applicability domain and the decision context in question. If the substances of interest are entirely or mostly out of the applicability domain, new data might need to be identified or other primary sources exploited to collate and compile more relevant information for the derivation of new models or refinement of existing models. It will also be critical in such an evaluation to consider the robustness of the training set and associated data that are used to develop the (Q)SAR model. The OECD principles for the validation of (Q)SAR provide a convenient framework for assessing the validity and applicability of (Q)SAR models (OECD 2007b).
A second issue is the nonavailability of tools to assess toxicity by nonoral routes of exposure. As discussed above, most tools have been developed to evaluate the oral exposure route.
A third issue is that the existing (Q)SARs are often lacking in mechanistic basis. Future (Q)SARs could conceivably be derived to predict key events as reflected in Figure 3-1. (Q)SARs would be developed to estimate outcomes of initial or intermediate events within a pathway rather than to predict an adverse outcome directly as indicated in Figure 3-1. An example of that approach is the early work of the ACuteTox program in which QSARs were derived to estimate in vitro cytotoxicity rather than in vivo acute rat toxicity.
__________________
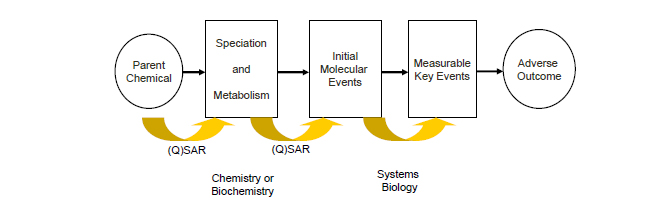
FIGURE 3-1 Conceptual framework for the future development of (Q)SARs.
Another interesting example of exploiting the framework shown in Figure 3-1 focuses on mitochondrial inhibition, a mechanism known to drive acute toxicity of some chemicals. Bhhatari et al. (2014) compared data from high-throughput screening assays of mitochondrial toxicity in HepG2 cells (see Attene-Ramos et al. 2015) with in silico data on absorption and first-pass metabolism12 and obtained promising results for predicting acute toxicity in multiple species. They found that mitochondrial inhibition predicted the minimum toxicity in fish and daphnia and that the lower the assay AC50,13 the more likely that the toxicity was driven by mitochondrial toxicity. However, mitochondrial inhibition did not often predict the toxicity of chemicals in rats because of the lack of data on oral bioavailability and first-pass metabolism. However, simulations that used in silico models for bioavailability and metabolism did improve toxicity predictions. A similar approach has been put forward by LHASA Ltd as a contribution to the OECD AOP work program (OECD 2011). A set of SARs has been derived from substances that inhibit complexes I, III, IV, and V of the electron transport chain, which characterize molecular initiating events in the pathway that leads to mitochondrial toxicity.
Another route by which models could be derived would involve integrating data from a variety of inputs (such as in vitro IC50s or AC50s and rodent LD50s) to predict acute toxicity. Several such examples have been investigated as part of the ACuteTox program as described above. Clothier et al. (2013) used Spearman rank-correlation analysis and hierarchical clustering to help to identify a combination of in vitro test systems for predicting in vivo acute toxicity. Kinsner-Ovaskainen et al. (2013) used classification and regression-tree analysis of in vitro data from the ACuteTox program to predict acute oral-toxicity categories. Kopp-Schneider et al. (2013) likewise investigated various data-mining approaches with the ACuteTox program data.
A second alternative route of developing (Q)SARs would consider the biological pathways involved in acute debilitating toxicity (such as altered oxygen transport, changes in neurotransmitter function, and disruption of cytoskeleton), which could be elucidated in a construct based on mechanistic pathways, and appropriate assays could be mapped to the key biological events. This approach would provide a different basis for development of integrated approaches, including specific (Q)SAR models that address specific key biological events of the AOP. The chemical applicability domain of the assays that characterize each key event could be extracted to inform new SARs that would facilitate profiling of untested substances. This type of approach has been attempted for skin sensitization (Patlewicz et al. 2014). Before such a strategy can be used, an approach to assessing the validity of the assays (Chapter 4), of the prediction models (data integration models) (Chapter 5), and of the pathways (Chapter 2) needs to be established (see Patlewicz et al. 2015).
__________________
12First-pass metabolism can occur at the site of chemical absorption (for example, in the gastrointestinal tract) or in the liver. In general, first-pass metabolism reduces the amount of a chemical that reaches the systemic circulation.
13AC50 is the concentration required to elicit a 50% response in an in vitro assay.
ENSURING SCIENTIFIC CONFIDENCE IN (Q)SAR MODELS
Ensuring scientific confidence in a (Q)SAR model relies on an assessment of model validity and model applicability. Both are critical for the appropriate interpretation and use of the predictions derived. The OECD validation principles for (Q)SARs provide a useful construct for evaluating and characterizing a given (Q)SAR model to determine its scientific validity. The five principles describe the need for a defined end point, an unambiguous algorithm, a defined domain of applicability, measures of performance, and a mechanistic interpretation if possible (OECD 2004, 2007b). The applicability domain,14 which involves extracting an applicability domain on the basis of the training set used to derive the QSAR model, provides a basis for judging the relevance and reliability of a prediction made for a given target substance. There are many ways to extract an applicability domain. Typical approaches include structural, mechanistic, metabolic, and descriptor considerations (Netzeva et al. 2005; Dimitrov et al. 2005). Freely available and commercial tools also exist to determine an applicability domain, namely, AMBIT Discovery and Domain Manager (Nikolova-Jeliazkova and Jaworska 2005; Patlewicz et al. 2011). Sazonovas et al. (2010) presented an alternative approach to extracting an applicability domain for an LD50 model. Each prediction was associated with a reliability index that depended on the target’s similarity to the training set and the consistency of experimental results with regard to the baseline model in the local chemical environment.
The applicability domain forms only one facet in judging the relevance and reliability of a prediction. To ensure the reliability of a prediction, one also needs to evaluate how well the models make correct predictions for similar chemicals. Such similar chemicals might be identified on the basis of the same characteristics or descriptors that were used to derive the original QSAR model or on the basis of structural similarity. The predictivity of similar analogues will form a second facet of judging the relevance of a model for the chemical of interest. The framework outlined in the REACH guidance (see Figure 3-2) might be helpful in summarizing the key considerations for the assessment of a (Q)SAR model and its associated prediction.
- Finding: Multiple databases are available for performing an initial chemical characterization of molecular structure, physicochemical properties, and available acute toxicity data. In addition, a number of in silico models are available for predicting physicochemical properties.
- Finding: Physicochemical and structural properties are critical for chemical characterization in that they can help to predict a chemical’s potential to pose a physical hazard, its reactivity, and its pharmacokinetic characteristics, such as bioavailability and likely routes of exposure.
- Finding: Although a number of tools are available to predict the site of metabolism and likely metabolic products on the basis of chemical structure and physicochemical properties, they are designed largely for pharmaceutical agents. Information about the likely metabolic products will be critical for informing experimental design, including assay selection.
- Finding: Several (Q)SAR models that use structural properties or physicochemical properties are available for predicting acute oral LD50s. Few models for predicting inhalation LC50s are available, and none for predicting dermal LD50s was identified.
- Finding: A few QSAR models for predicting neurotoxicity and cytotoxicity are available, but not for other end points that are relevant for acute, debilitating toxicity. Current research in (Q)SAR models is focusing on incorporating more biological information, such as integrating in vitro data (for example, on cytotoxicity) and information on specific AOPs.
__________________
14The applicability domain is the array of chemicals for which the (Q)SAR can confidently be applied for purposes of toxicity prediction (Aptula and Roberts 2006).
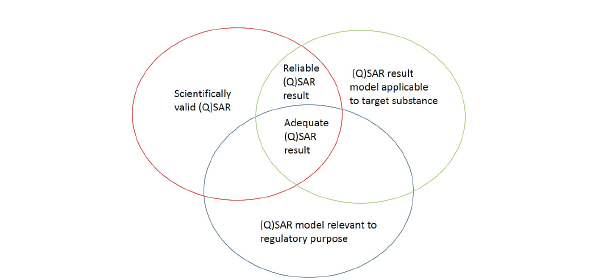
- Recommendation: DOD should evaluate the applicability, relevance, and reliability of available (Q)SAR models to meet its needs of assessing a chemical’s potential to cause acute, debilitating toxicity. OECD and REACH principles and guidelines for evaluating and characterizing (Q)SAR models might provide useful frameworks for conducting such an evaluation. Furthermore, DOD should evaluate the applicability, relevance, and reliability of models and tools for predicting physicochemical properties and metabolism.
- Recommendation: To fill remaining gaps in nontesting approaches, DOD should consider a number of options for further research and development, including extrapolation of oral LD50 to other exposure routes through pharmacokinetic models; development of new (Q)SAR models for acute lethality, focusing particularly on inhalation and dermal exposure; and development of (Q)SAR models augmented with biological information, such as in vitro data and information on targets or mechanisms of acute toxicity.
ACD/Labs. 2015. Acute Toxicity Prediction Module. Data Sheet [online]. Available: http://www.acdlabs.com/download/docs/datasheets/datasheet_acute.pdf [accessed March 24, 2015].
AltTox. 2014. Table of Validated and Accepted Alternative Methods [online]. Available: http://alttox./org/mapp/table-of-validated-and-accepted-alternative-methods/ [accessed March 18, 2015].
Ankley, G.T., R.S. Bennett, R.J. Erickson, D.J. Hoff, M.W. Hornung, R.D. Johnson, D.R. Mount, J.W. Nichols, C.L. Russom, P.K. Schmieder, J.A. Serrrano, J.E. Tietge, and D.L. Villeneuve. 2010. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29(3):730-741.
Aptula, A.O., and D.W. Roberts. 2006. Mechanistic applicability domains for nonanimal-based prediction of toxicological end points: General principles and application to reactive toxicity. Chem. Res. Toxicol. 19(8):1097-1105.
Artursson, P., and J. Karlsson. 1991. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 175(3):880-885.
Attene-Ramos, M.S., R. Huang, S. Michael, K.L. Witt, A. Richard, R.R. Tice, A. Simeonov, C.P. Austin, and M. Xia. 2015. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 123(1):49-56.
Barratt, M.D. 1996. Quantitative structure-activity relationships for skin irritation and corrosivity of neutral and electrophilic organic chemicals. Toxicol. In Vitro 10(3):247-256.
Bhhatari, B., D. Wilson, P.S. Price, M.J. Bartels, S. Chaunhuri, and E.W. Carney. 2014. Predicting Acute Toxicity Using In Vitro ToxCast HTS Mitochondrial Inhibition Assays. Presented at the Second ToxCast Data Summit, September 29-30, 2014, Durham, NC.
Bradbury, S.P., T.R. Henry, and R.W. Carlson. 1990. Fish acute toxicity syndromes in the development of mechanism specific QSARs. Pp. 295-315 in Practical Applications of Quantitative Structure Activity Relationships (QSAR) in Environmental Chemistry and Toxicology, W. Karcher, and J. Devillers, eds. Dordrecht, The Netherlands: Kluwer.
Brown, J.S., T. Gordon, O. Price, and B. Asgharian. 2013. Thoracic and respirable particle definitions for human health risk assessment. Part. Fibre Toxicol. 10:12.
Carpenter, T.S., D.A. Kirshner, E.Y. Lau, S.E. Wong, J.P. Nilmeier, and F.C. Lightstone. 2014. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 107(3):630-641.
Castillo-Garit, J.A., Y. Marrero-Ponce, F. Torrens, and R. García-Domenech. 2008. Estimation of ADME properties in drug discovery: Predicting Caco-2 cell permeability using atom-based stochastic and non-stochastic linear indices. J. Pharm. Sci. 97(5):1946-1976.
Clothier, R., M.J. Gómez-Lechón, A. Kinsner-Ovaskainen, A. Kopp-Schneider, J.E. O'Connor, P. Prieto, and S. Stanzel. 2013. Comparative analysis of eight cytotoxicity assays evaluated within the ACuteTox Project. Toxicol. In Vitro 27(4):1347-1356.
Cronin, M.T. 1996. Quantitative structure-activity relationship (QSAR) analysis of the acute sublethal neurotoxicity of solvents. Toxicol. In Vitro 10(2):103-110.
Cronin, M.T., and J.C. Dearden. 1995. QSAR in toxicology. 2. Prediction of acute mammalian toxicity and interspecies correlations. Quant. Struct. Act. Relat. 14(2):117-120.
Cronin, M.T., and J.C. Madden, eds. 2010. In Silico Toxicology: Principles and Applications. London: Royal Society of Chemistry.
Cronin, M.T., J.C. Dearden, J.C. Duff, R. Edwards, N. Manga, A.P. Worth, and A.D. Worgan. 2002. The importance of hydrophobicity and electrophilicity descriptors in mechanistically based QSARs for toxicological endpoints. SAR QSAR Environ. Res. 13(1):167-176.
Cronin, M.T., J.C. Dearden, J.D. Walker, and A.P. Worth. 2003. Quantitative structure activity relationships for human health effects: Commonalities with other endpoints. Environ. Toxicol. Chem. 22(8):1829-1843.
Dancik, Y., M.A. Miller, J. Jaworska, and G.B. Kasting. 2013a. Design and performance of a spreadsheet-based model for estimating bioavailability of chemicals from dermal exposure. Adv. Drug Deliv. Rev. 65(2):221-236.
Dancik, Y., J.A. Troutman, and J. Jaworska. 2013b. A framework incorporating the impact of exposure scenarios and application conditions on risk assessment of chemicals applied to skin. In Silico Pharmacol. 1:10; doi: 10.1186/2193-9616-1-10.
Dearden, J., and A. Worth. 2007. In Silico Prediction of Physicochemical Properties. JRC Scientific and Technical Reports EUR 23051[online]. Available: https://eurl-ecvam.jrc.ec.europa.eu/laboratoriesresearch/predictive_toxicology/doc/EUR_23051_EN.pdf [accessed March 16, 2015].
Devillers, J. 2004. Prediction of mammalian toxicity of organophosphorus pesticides from QSTR modeling. SAR QSAR Environ. Res. 15(5-6):501-510.
Devillers, J., and H. Devillers. 2009. Prediction of acute mammalian toxicity from QSARs and interspecies correlation. SAR QSAR Environ. Res. 20(5-6):467-500.
Dimitrov, S., G. Dimitrova, T. Pavlov, N. Dimitrova, G. Patlewicz, J. Niemela, and O. Mekenyan. 2005. A stepwise approach for defining the applicability domain of SAR and QSAR models. J. Chem. Inf. Model. 45(4):839-849.
Drwal, M.N., P. Banerjee, M. Dunkel, M.R. Wettig, and R. Preissner. 2014. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 42(Web Server issue):W53-W58.
ECETOC (European Centre for Ecotoxicology and Toxicology of Chemicals), 2012. Category Approaches, Read-across, (Q)SAR. Technical Report No. 116. Brussels, Belgium: ECETOC.
ECHA (European Chemicals Agency). 2008. Guidance on Information Requirements and Chemical Safety Assessment. Chapter R.6: QSARs and Grouping of Chemicals [online]. Available: http://echa.europa.eu/documents/10162/13632/information_requirements_r6_en.pdf [accessed February 12, 2015].
ECHA (European Chemicals Agency). 2012. Guidance on Registration, Version 2.0. Guidance for the Implementation of REACH. May 2012 [online]. Available:http://echa.europa.eu/documents/10162/13632/registration_en.pdf [accessed April 30, 2015].
ECHA (European Chemicals Agency) 2014. Guidance on Information Requirements and Chemical Safety Assessment, Chapter R.7a. Endpoint Specific Guidance, Version 3.0 [online]. Available: http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf.
Ekwall, B. 1983. Screening of toxic compounds in mammalian cell cultures. Ann. N.Y. Acad. Sci. 407:64-77.
Ekwall, B., F.A. Barile, A. Castano, C. Clemedson, R.H. Clothier, P. Dierickx, B. Ekwall, M. Ferro, G. Fiskesjö, L. Garza-Ocanas, M.J. Gómez-Lechón, M. Gülden, T. Hall, B. Isomaa, A. Kahru, G. Kerszman, U. Kristen, M. Kunimoto, S. Kärenlampi, L. Lewan, A. Loukianov, T. Ohno, G. Persoone, L. Romert, T.W. Sawyer, H. Segner, R. Shrivastava, A. Stammati, N. Tanaka, M. Valentino, E. Walum, and F. Zucco. 1998. MEIC evaluation of acute systemic toxicity. Part VI. Prediction of human toxicity by rodent LD50 values and results from 61 in vitro tests. ATLA 26(Suppl. 2):617-658.
EPA (US Environmental Protection Agency). 2012. Glossary of Terms: Methods of Toxicity Testing and Risk Assessment [online]. Available: http://www.epa.gov/pesticides/science/comptox-glossary.html#q [accessed May7, 2015].
EPA (US Environmental Protection Agency). 2014. Toxicity Estimation Software Tool (TEST) [online]. Available: http://www.epa.gov/nrmrl/std/qsar/qsar.html#TEST [accessed March 18, 2015].
EU (European Union). 2006. Regulation (EC) No. 1907/2006 of the European Parliament and of the Council concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No. 793/93 and Commission Regulation (EC) No. 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC.OJ E.U. 396:I1-I849 [online]. Available: http://faolex.fao.org/docs/pdf/eur68317.pdf.
Fitzpatrick, D., J. Corish, and B. Hayes. 2004. Modelling skin permeability in risk assessment--the future. Chemosphere 55(10):1309-1314.
Flynn. G.L. 1990. Physicochemical determinants of skin absorption. Pp. 93-127 in Principles of Route-to-Route Extrapolation for Risk Assessment, T.R. Gerrity, and C.J. Henry, eds. New York: Elsevier.
Freidig, A.P., S. Dekkers, M. Verwei, E. Zvinavashe, J.G. Bessems, and J.J. van de Sandt. 2007. Development of a QSAR for worst case estimates of acute toxicity of chemically reactive compounds. Toxicol. Lett. 170(3):214-222.
Gajewska, M., A. Worth, C. Urani, H. Briesen, and K.W. Schramm. 2014 Application of physiologically-based toxicokinetic modelling in oral-to-dermal extrapolation of threshold doses of cosmetic ingredients. Toxicol. Lett. 227(3):189-202.
García-Domenech, R., P. Alarcón-Elbal, G. Bolas, R. Bueno-Marí, F.A. Chordá-Olmos, S.A. Delacour, M.C. Mouriño, A. Vidal, and J. Gálvez. 2007. Prediction of acute toxicity of organophosphorus pesticides using topological indices. SAR QSAR Environ. Res. 18(7-8):745-755.
Gerebtzoff, G., and A. Seelig. 2006. In silico prediction of blood-brain barrier permeation using the calculated molecular cross-sectional area as main parameter. J. Chem. Inf. Model. 46(6):2638-2650.
Gerner, I., M.D. Barratt, S. Zinke, K. Schlegel, and E. Schlede. 2004. Development and prevalidation of a list of structure-activity relationship rules to be used in expert systems for prediction of the skin-sensitizing properties of chemicals. ATLA 32(5):487-509.
Gleeson, M.P. 2008. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 51(4):817-834.
Grbic, S., J. Parojcic, S. Ibric, and Z. Djuric. 2011. In vitro-in vivo correlation for gliclazide immediate-release tablets based on mechanistic absorption simulation. AAPS PharmSciTech. 12(1):166-171.
Green, N., and M. Song. 2011. Predicting in vivo safety characteristics using physiochemical properties and in vitro assays. Fut. Med. Chem. 3(12):1503-1511.
Guerra, A., N.E. Campillo, and J.A. Páez. 2010. Neural computational prediction of oral drug absorption based on CODES 2D descriptors. Eur. J. Med. Chem. 45(3):930-940.
Guilian, W., and B. Naibin. 1998. Structure activity relationships for rat and mouse LD50 of miscellaneous alcohols. Chemosphere 36(7):1475-1483.
Hansch, C., and A. Kurup. 2003. QSAR of chemical polarisability and nerve toxicity, 2. J. Chem. Inf. Comp. Sci. 43(5):1647-1651.
Hostynek, J.J., and P.S. Magee. 1997. Modelling in vivo human skin absorption. Quant. Struct.Act. Relat. 16(6):473-479.
Hughes, J.D., J. Blagg, D.A. Price, S. Bailey, G.A. DeCrescenzo, R.V. Devraj, E. Ellsworth, Y. Fobian, M.E. Gibbs, R.W. Gilles, N. Greene, E. Huang, T. Krieger-Burke, T. Wager, L. Whiteley, and Y. Zhang. 2008. Physiochemical drug properties associated with in vivo toxicological outcomes. Biorg. Med. Chem. Lett. 18(17):4872-4875.
Hulzebos, E., J.D. Walker, I. Gerner, and K. Schlegel. 2005. Use of structural alerts to develop rules for identifying chemical substances with skin irritation or skin corrosion potential. QSAR Comb. Sci. 24(3):332-342.
Iyer, M., Y.J. Tseng, C.L. Senese, J. Liu, and A.J. Hopfinger. 2007. Prediction and mechanistic interpretation of human oral drug absorption using MI-QSAR analysis. Mol. Pharm. 4(2):218-231.
Kar, S., and K. Roy. 2013. First report on predictive chemometric modeling, 3D-toxicophore mapping and in silico screening of in vitro basal cytotoxicity of diverse organic chemicals. Toxicol. In Vitro. 27(2):597-608.
Kinsner-Ovaskainen, A., P. Prieto, S. Stanzel, and A. Kopp-Schneider. 2013. Selection of test methods to be included in a testing strategy to predict acute oral toxicity: An approach based on statistical analysis of data collected in phase 1 of the ACuteTox project. Toxicol. In Vitro 27(4):1377-1394.
Kirchmair, J., M.J. Williamson, J.D. Tyzack, L. Tan, P.J. Bond, A. Bender, and R.C. Glen. 2012. Computational prediction of metabolism: Sites, products, SAR, P450, enzyme dynamics and mechanism. J. Chem. Inf. Model. 52(3):617-648.
Kirman, C.R., L.L. Aylward, B.A. Wetmore, R.S. Thomas, M. Sochaski, S.S. Ferguson, S.A. Csiszar, and O. Jolliet. 2015. Quantitative property-property relationship for screening-level prediction of intrinsic clearance: A tool for exposure modeling for high-throughput toxicity screening data. Appl. In Vitro Toxicol. 1(2):140-146.
Koleva, Y.K., M.T. Cronin, J.C. Madden, and J.A. Schwöbel. 2011. Modelling acute oral mammalian toxicity. 1. Definition of a quantifiable baseline effect. Toxicol. In Vitro 25(7):1281-1293.
Kopp-Schneider, A., P. Prieto, A. Kinsner-Ovaskainen, and S. Stanzel. 2013. Design of a testing strategy using non-animal based test methods: Lessons learnt from the ACuteTox project. Toxicol. In Vitro 27(4):1395-1401.
Kuentz, M., S. Nick, N. Parrott, and D. Röthlisberger. 2006. A strategy for preclinical formulation development using GastroPlus as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur. J. Pharm. Sci. 27(1):91-99.
Langer, T., and G. Wolber. 2004. Pharmacophore definition and 3D searches. Drug Discov. Today Technol. 1(3):203-207.
Lapenna, S., M. Fuart-Gatnik, and A. Worth. 2010. Review of QSAR Models and Software Tools for Predicting Acute and Chronic Systemic Toxicity. JRC Technical Report EUR 24639 EN. Luxembourg: Office of the European Union.
Lee, S., K. Park, H.S. Ahn, and D. Kim. 2010. Importance of structural information in predicting human acute toxicity from in vitro cytotoxicity data. Toxicol. Appl. Pharmacol. 246(1-2):38-48.
Leeson, P.D., and B. Springthorpe. 2007. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6(11):881-890.
Lessigiarska, I., A.P. Worth, and T.I. Netzeva. 2005. Comparative Review of QSARs for Acute Toxicity. JRC report EUR 21559 EN. European Commission, Joint Research Centre, Ispra, Italy.
Lessigiarska, I., A.P. Worth, T.I. Netzeva, J.C. Dearden, and M.T. Cronin. 2006. Quantitative structure-activity-activity and quantitative structure-activity investigations of human and rodent toxicity. Chemosphere 65(10):1878-1887.
Li, X., L. Chen, F. Cheng, Z. Wu, H. Bian, C. Xu, W. Li, G. Liu, X. Shen, and Y. Tang. 2014. In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model. 54(4):1061-1069.
Lipinski, C.A., F. Lombardo, B.W. Dominy, and P.J. Feeney. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46(1-3):3-26.
Lu, J., J. Peng, J. Wang, Q. Shen, Y. Bi, L. Gong, M. Zheng, X. Luo, W. Zhu, H. Jiang, and K. Chen. 2014. Estimation of acute oral toxicity in rat using local lazy learning. J. Cheminform. 6:26; doi:10.1186/ 1758-2946-6-26.
Magnusson, B.M., Y.G. Anissimov, S.E. Cross, and M.S. Roberts. 2004. Molecular size as the main determinant of solute maximum flux across the skin. J. Invest. Dermatol. 122(4):993-999.
McKarns, S.C., C. Hansch, W.S. Caldwell, W.T. Morgan, S.K. Moore, and D.J. Doolittle. 1997. Correlation between hydrophobicity of short chain aliphatic alcohols and their ability to alter plasma membrane integrity. Fundam. Appl. Toxicol. 36(1):62-70.
Meek, M.E., H.A. Barton, J.G. Bessems, J.C. Lipscomb, and K. Krishnan. 2013. Case study illustrating the WHO IPCS guidance on characterization and application of physiologically based pharmacokinetic models in risk assessment. Regul. Toxicol. Pharmacol. 66(1):116-129.
Mekenyan, O.G., P.I. Petkov, S.V. Kotov, S. Stoeva, V.B. Kamenska, S.D. Dimitrov, M. Honma, M. Hayashi, R. Benigni, E.M. Donner, and G. Patlewicz. 2012. Investigating the relationship between in vitro-in vivo genotoxicity: Derivation of mechanistic QSAR models for in vivo liver genotoxicity and in vivo bone marrow micronucleus formation which encompass metabolism. Chem. Res. Toxicol. 25(2):277-296.
Mitragotri, S., Y.G. Anissimov, A.L. Bunge, H.F. Frasch, R.H. Guy, J. Hadgraft, G.B. Kasting, M.E. Lane, and M.S. Roberts. 2011. Mathematical models of skin permeability: An overview. Int. J. Pharm. 418(1):115-129.
Moore, N.P., D.J. Andrew, D.L. Bjerke, S. Creton, D. Dreher, T. Homes, P. Prieto, T. Seidle, and T.G. Rowan. 2013. Can acute dermal systematic toxicity tests be replaced with oral tests? A comparison of route specific systemic toxicity and hazard classifications under the Globally Harmonised System of Classification and Labelling of Chemicals (GHS). Regul. Toxicol. Pharmcol. 66(1):30-37.
Netzeva, T., A. Worth, T. Aldenberg, R. Benigni, M. Cronin, P. Gramatica, J. Jaworska, S. Kahn, G. Klopman, C. Marchant, G. Myatt, N. Nikolova-Jeliazkova, G. Patlewicz, R. Perkins, D. Roberts, T. Schultz, D. Stanton, J. van de Sandt, W. Tong, G. Veith, and C. Yang. 2005. Current status of methods for defining the applicability domain of (quantitative) structure–activity relationships. ATLA 33(2):155-173.
Nikolova-Jeliazkova, N., and J. Jaworska. 2005. An approach to determining applicability domains for QSAR group contribution models: An analysis of SRC KOWWIN. ATLA 33(5):461-470.
NIOSH (National Institute for Occupational Safety and Health)). 2013. Skin Exposures and Effects: Finite Dose Skin Permeation Calculator [online]. Available: http://www.cdc.gov/niosh/topics/skin/finiteSkinPermCalc.html [accessed March 18, 2015].
NRC (National Research Council). 2014. Physicochemical properties and chemical fate. Pp. 47-67 in A Framework to Guide Selection of Chemical Alternatives. Washington, DC: National Academies Press.
OECD (Organisation for Economic Co-operation and Development). 2004. OECD Principles for the Validation, For Regulatory Purposes of (Quantitative) Structure-Activity Relationship Models [online]. Available: http://www.oecd.org/chemicalsafety/risk-assessment/37849783.pdf [accessed March 17, 2015].
OECD (Organisation for Economic Co-operation and Development). 2006. OECD Guideline for Testing of Chemicals No. 435: In Vitro Membrane Barrier Test Method for Skin Corrosion. Adopted July 19, 2006 [online]. Available: http://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecdtg435.pdf [accessed March 18, 2015].
OECD (Organisation for Economic Co-operation and Development). 2007a. Guidance on Grouping of Chemicals. OECD Environmental Health and Safety Publication Series on Testing and Assessment No. 80. Paris: OECD [online]. Available: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2007)28.
OECD (Organisation for Economic Co-operation and Development). 2007b. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationships [(Q)SAR] Models. OECD Environment Health and Safety Publication Series on Testing and Assessment No. 69. Paris: OECD [online]. Available: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2007)2 [accessed March 17, 2015].
OECD (Organisation for Economic Co-operation and Development). 2011. Report of the Workshop on Using Mechanistic Information in Forming Chemical Categories, December, 8-10, 2010, Crystal City, VA. OECD Environment Health and Safety Publication Series on Testing and Assessment No.138. Paris: OECD [online]. Available: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=/env/jm/mono(2011)8&doclanguage=en [accessed March 17, 2015].
OECD (Organisation for Economic Co-operation and Development). 2013a. OECD Guideline for Testing of Chemicals No. 431: In Vitro Skin Corrosion: Reconstructed Human Epidermis (RHE) Test Method. Adopted July 26, 2013 [online]. Available: https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-tg431-2013-508.pdf [accessed March 18, 2015].
OECD (Organisation for Economic Co-operation and Development). 2013b. OECD Guideline for Testing of Chemicals No. 437: Bovine Corneal Opacity and Permeability Test Method for Identifying i) Chemicals Inducing Serious Eye Damage and ii) Chemicals Not Requiring Classification for Eye Irritation or Serious Eye. Adopted July 26, 2013 [online]. Available: http://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-tg437-2013-508.pdf [accessed March 18, 2015].
OECD (Organisation for Economic Co-operation and Development). 2013c. OECD Guideline for Testing of Chemicals No. 438: Isolated Chicken Eye Test Method for Identifying i) Chemicals Inducing Serious Eye Damage and ii) Chemicals Not Requiring Classification for Eye Irritation or Serious Eye Damage.
Adopted July 26, 2013 [online]. Available: http://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd-tg438-2013-508.pdf [accessed March 18, 2015].
OECD (Organisation for Economic Co-operation and Development). 2014. Guidance on Grouping of Chemicals, Second Edition. OECD Environment Health and Safety Publication Series on Testing and Assessment No.194. Paris: OECD [online]. Available: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2014)4&doclanguage=en [accessed March 17, 2015].
Papa, E., M. Luini, and P. Gramatica. 2009. Quantitative structure-activity relationship modelling of oral acute toxicity and cytotoxic activity of fragrance materials in rodents. SAR QSAR Environ. Res. 20(7-8):767-779.
Patlewicz, G., M.W. Chen, and C.A. Bellin. 2011. Non-testing approaches under REACH--help or hindrance? Perspectives from a practitioner within industry. SAR QSAR Environ. Res. 22(1-2):67-88.
Patlewicz, G., C. Kuseva, A. Kesoba, I. Popova, T. Zhechev, T. Pavlov, D.W. Roberts, and O. Mekenyan. 2014. Towards AOP application – implementation of an integrated approach to testing and assessment (IATA) into a pipeline tool for skin sensitization. Regul. Toxicol. Pharmacol. 69(3): 529-545.
Patlewicz, G., T. Simon, R. Budinsky, J.C. Rowlands, and R.A. Becker. 2015. Proposing a scientific confidence framework to help support the application of adverse outcome pathways for regulatory purposes. Regul. Toxicol. Pharmacol. 71(3):463-477.
Perrin, D.D., B. Dempsey, and E.P. Serjeant. 1981. pKa Prediction for Organic Acids and Bases. London: Chapman and Hall.
Peyret, T., and K. Krishnan. 2012. Quantitative property-property relationship for screening-level prediction of intrinsic clearance of volatile organic chemicals in rats and its integration within PBPK models to predict inhalation pharmacokinetics in humans. J. Toxicol. 2012:286079.
Potts, R.O., and R.H. Guy. 1992. Predicting skin permeability. Pharm. Res. 9(5):663-669.
Prieto, P., A. Kinsner-Ovaskainen, S. Stanzel, B. Albella, P. Artursson, N. Campillo, R. Cecchelli, L. Cerrato, L. Díaz, E. Di Consiglio, A. Guerra, L. Gombau, G. Herrera, P. Honegger, C. Landry, J.E. O'Connor, J.A. Páez, G. Quintas, R. Svensson, L. Turco, M.G. Zurich, M.J. Zurbano, and A. Kopp-Schneider. 2013. The value of selected in vitro and in silico methods to predict acute oral toxicity in a regulatory context: Results from the European Project ACuteTox. Toxicol. In Vitro 27(4):1357-1376.
ProTox. 2015. Prediction of Rodent Oral Toxicology. Charite University of Medicine, Berlin, Germany [online]. Available: http://tox.charite.de/tox/ [accessed March 18, 2015].
Raunio, H. 2011. In silico toxicology – non-testing methods. Front. Pharmacol. 2:33.
Rorije, E., and E.M. Hulzebos. 2005. Evaluation of (Q)SARs for the Prediction of Skin Irritation/ Corrosion Potential. National Institute of Public Health and Environment (RIVM-SEC)/European Chemicals Bureau (ECB), Bilthoven, The Netherlands [online]. Available: https://eurl-ecvam.jrc.ec.europa.eu/laboratories-research/predictive_toxicology/information-sources/qsar-documentarea/Evaluation_of_Skin_Irritation_QSARs.pdf [accessed March 17, 2015].
Rostami-Hodjegan, A., and G.T. Tucker. 2007. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat. Rev. Drug Discov. 6(2):140-148.
Saliner, A.G., I. Tsakovska, M. Pavan, G. Patlewicz, and A.P. Worth. 2007. Evaluation of SARs for the prediction of skin irritation/corrosion potential: Structural inclusion rules in the BfR decision support system. SAR QSAR Environ. Res. 18(3-4):331-342.
Saliner, A.G., G. Patlewicz, and A.P. Worth. 2008. A review of (Q)SAR models for skin and eye irritation and corrosion. QSAR Comb. Sci. 27(1):49-59.
Sazonovas, A., P. Japertas, and R. Diziapetris. 2010. Estimation of reliability of predictions and model applicability domain evaluation in the analysis of acute toxicity (LD50). SAR QSAR Environ. Res. 21(1): 127-148.
Schultz, T.W., J.W. Yarbrough, and E.L. Johnson. 2005. Structure-activity relationships for reactivity of carbonyl-containing compounds with glutathione. SAR QSAR Environ. Res. 16(4):313-322.
Schultz, T.W., R.E. Carlson, M.T. Cronin, J.L. Hermens, R. Johnson, P.J. O'Brien, D.W. Roberts, A. Siraki, K.B. Wallace, and G.D. Veith. 2006. A conceptual framework for predicting the toxicity of reactive chemicals: Modeling soft electrophilicity. SAR QSAR Environ. Res. 17(4):413-428.
Sedykh, A., H. Zhu, H. Tang, L. Zhang, A. Richard, I. Rusyn, and A. Tropsha. 2011. Use of in vitro HTS derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ. Health Perspect. 119(3):364-370.
Silva, F.T., and G.H. Trossini. 2014. The survey of the use of QSAR methods to determine intestinal absorption and oral bioavailability during drug design. Med. Chem. 10(5):441-448.
Simulations Plus. 2010. Manual GastroPlus™. Simulations Plus, Inc., CA.
Suenderhauf, C., F. Hammann, A. Maunz, C. Helma, and J. Huwyler. 2011. Combinatorial QSAR modeling of human intestinal absorption. Mol. Pharm. 8(1):213-224.
ten Berge, W. 2009. A simple dermal absorption model: Derivation and application. Chemosphere 75(11): 1440-1445.
Toropov, A.A., B.F. Rasulev, and J. Leszcznski. 2008. QSAR Modeling of acute toxicity by balance of correlations. Bioorg. Med. Chem. 16(11):5999-6008.
Tsakovska, I., I. Lessigiarska, T. Netzeva, and A.P. Worth. 2006. Review of (Q)SARs for Mammalian Toxicity. EUR 22846 EN. European Commission, Directorate General, Joint Research Centre, Ispra, Italy [online]. Available: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.103.6822&rep=rep1&type=pdf [accessed March 17, 2015].
Tsakovska, I., A.G. Saliner, T. Netzeva, M. Pavan, and A.P. Worth. 2007. Evaluation of SARs for the prediction of eye irritation/corrosion potential: Structural inclusion rules in the BfR decision support system. SAR QSAR Environ. Res. 18(3-4):221-235.
UNECE (U.N. Economic Commission for Europe). 2015. Globally Harmonized System of Classification and Labelling of Chemicals (GHS) [online]. Available: http://www.unece.org/trans/danger/publi/ghs/ghs_welcome_e.html [accessed March 18, 2015].
Veith, G.D., and K.B. Wallace. 2006. Inhalation Toxicity in Fish and Rodents. Presented at the McKim Conference, June 27-29, 2006, Duluth, MN.
Veith, G.D., E.P. Petkova, and K.B. Wallace. 2009. A baseline inhalation toxicity model for narcosis in mammals. SAR QSAR Environ. Res. 20(5-6):567-578.
Walters, W.P., and M.A. Murcko. 2002. Prediction of 'drug-likeness'. Adv. Drug Deliv. Rev. 54(3):255-271.
Wang, J., and T. Hou. 2015. Advances in computationally modeling human oral bioavailability. Adv. Drug Deliv. Rev. 86:11-16.
Worth, A. 2008. QSAR and Chemical Read-Across within the Context of Non-testing Strategies. Presentation at the 1st SETAC Europe Special Science Symposium: Integrated Testing Strategies for REACH, October, 23-24, 2008, Brussels, Belgium [online]. Available: http://reach.setac.eu/embed/presentations_reach/09_WORTH_QSAR_Chem_Read_Across.pdf. [accessed March 18, 2015]
Xu, C., and D.E. Mager. 2011. Quantitative structure-pharmacokinetic relationships. Expert Opin. Drug Metab. Toxicol. 7(1):63-77.
Yamazaki, K., and M. Kanaoka. 2004. Computational prediction of the plasma protein-binding percent of diverse pharmaceutical compounds. J. Pharm. Sci. 93(6):1480-1494.
Yang, J., M. Jamei, K.R. Yeo, G.I. Tucker, and A. Rostami-Hodjegan. 2007. Prediction of intestinal first-pass drug metabolism. Curr. Drug Metab. 8(7):676-684.
Zhu, H., T.M. Martin, L. Ye, A. Sedykh, D. Young, and A. Tropsha. 2009a. Quantitative structure-activity relationship modelling of rate acute toxicity by oral exposure. Chem. Res. Toxicol. 22(12):1913-1921.
Zhu, H., L. Ye, A. Richard, A. Gobraikh, F.A. Wright, I. Rusyn, and A. Tropsha. 2009b. A novel 2-step hierarchical quantitative structure-activity relationship modelling workflow for predicting acute toxicity of chemicals in rodents. Environ. Health Perspect. 117(8):1257-1264.






















