
4
Assays for Predicting Acute Toxicity
The future path of toxicity testing was foreshadowed early in the 2000s with publication of frameworks or roadmaps that called for an increased emphasis on the use of in vitro assays that evaluate key biological pathways and molecular mechanisms linked to human disease (EPA 2003; NTP 2004). High-throughput testing would allow less expensive, rapid screening of large numbers of chemicals to set testing priorities on the basis of predicted adverse health effects. The National Research Council (NRC) report Toxicity Testing in the 21st Century: A Vision and a Strategy (NRC 2007) built on the early publications and gave rise to a variety of large-scale initiatives to see how in vitro testing methods can be used to predict human toxicity. To implement the strategy outlined in the NRC report, a collaboration was formed between the National Toxicology Program (NTP), the US Environmental Protection Agency (EPA) National Center for Computational Toxicology, and the National Institutes of Health National Chemical Genomics Center (NCGC)1 to identify mechanisms of chemically induced biological activity, to set priorities among chemicals for more extensive toxicological evaluation, and to develop more predictive models of human biological response (MOU 20082; Austin et al. 2008; Kavlock et al. 2009; Krewski et al. 2009). The collaboration is now formally referred to as the Tox21 program.
The EPA-sponsored ToxCast program (Dix et al. 2007; Kavlock et al. 2012), the Tox21 program, and the European ACuteTox program (Clemedson et al. 2007; Clemedson 2008) are specific examples of large-scale initiatives to evaluate in vitro testing methods for their ability to predict human toxicity. Phase I of the ToxCast program evaluated about 300 conventional pesticide active ingredients in a battery of cell-free and cell-based assays (Judson et al. 2010). In phase II, the chemical space was broadened to include chemicals used in consumer products and industrial processes and unmarketed drugs donated by pharmaceutical companies (Kavlock et al. 2012; Sipes et al. 2013). The completed first phase of the Tox21 screening program tested about 2,800 chemicals—including solvents, fire retardants, dyes, preservatives, plasticizers, therapeutic agents, inorganic and organic pollutants, drinking water-disinfection byproducts, and natural products—in 50 assays (Shukla et al. 2010; Attene-Ramos et al. 2013). The studies laid the groundwork for efforts to characterize the ability of cell-free and cell-based assays and data-modeling approaches to predict activity and potency in selected biochemical targets. Most of the assays developed and validated for high-throughput screening (HTS) applications like the ToxCast program provide information about activation of molecular-receptor families or biochemical activities that are of interest to the pharmaceutical industry. In fact, for most assays, there is not a direct linkage between specific cells or tissue and chemical or mechanistic targets associated with acute lethal or debilitating effects outlined in Table 2-1. Thus, the assays might be of less use for identifying chemicals that potentially can cause acute, debilitating injuries in deployed personnel.
__________________
1NCGC is now part of the National Center for Advancing Translational Sciences.
2High Throughput Screening, Toxicity Pathway Profiling, and Biological Interpretation of Findings, Memorandum of Understanding Between NTP, NCGC and EPA, January 2008. Available: http://www.niehs.nih.gov/about/highlights/assets/docs/memorandum_of_understanding_508.pdf.
This chapter reviews currently available tools and their limitations for immediate implementation (in the next 3-10 years) by the US Department of Defense (DOD) to screen chemicals of interest to DOD for acute toxicity.
Numerous in vitro screening assays have been developed for measuring specific biological activities of chemicals in specific organs or cell types with an eye to elucidating mechanisms of action. For the purposes of hazard assessment, biological activity in an in vitro system can identify a mechanism of action or response that could be extrapolated to an in vivo end point. This section reviews relevant in vitro biochemical assays that could potentially be used to assess acute toxicity.
Specific-Protein Assays
Testing whether a chemical inhibits a particular enzyme or binds to a particular receptor or other biomolecule is the most direct way to test a chemical for a specific mechanism of action at the molecular level. Enzyme and receptor-binding assays tend to be reliable, to exhibit relatively good agreement between different laboratories, and to be well suited for high-throughput formats. The specific protein, protein complex, receptor, or other biomolecule of interest can be provided in the assay as a pure molecule, as a partially purified complex, or as a component of living cells.3
Given their high value for predicting specific molecular effects, protein assays are the most frequent type of assay in the ToxCast program (Figure 4-1). Some of the ToxCast assays have direct relevance to predicting acute toxicity of chemicals that could be used as warfare agents. For example, assays of acetylcholinesterase activity are applicable to cholinesterase-inhibiting nerve agents, and mitochondrial electron-transport assays are applicable to cyanide. Specific-protein assays in the ToxCast and ACuteTox studies that were designed to detect nervous system effects, such as modulation of ion-channel activity, are also relevant for predicting acute toxicity. However, it is important to recognize that, like many of the other assays in ToxCast, most of the specific-protein assays included in ToxCast were designed for needs other than predicting acute toxicity—for example, to predict endocrine-disrupting activity—and might have little value for predicting acute toxicity.
Limitations and Needs for Improvement of Protein Assays
A major limitation is the biological space that is covered by specific-protein assays that are designed to assess the actions of a chemical on specific enzymes or receptors. More complicated or nonspecific mechanisms might also be involved in acute toxicity. For example, vesicant action on the skin is not mediated by the action of a chemical on a specific enzyme or receptor. Toxic chemicals that act through nonspecific mechanisms might not be identified in specific-protein assays or might be identified in multiple assays but with poor correlation between dose and response, which potentially confuses analyses. Another limitation is that assays for particular acute toxic mechanisms (such as activity on particular ion channels) might be missing from the suite of assays or might be unreliable. A final limitation is that some specific-protein assays involve general mechanisms that are common to many cell types and thus might not predict particular organ toxicities themselves.
__________________
3Assays that use living cells to evaluate the effects of chemicals on specific proteins could have been considered in the section that follows, but for simplicity, this section reviews all assays whose purpose is to measure effects of a chemical on the biochemical activity of a specific protein or other biomolecule.
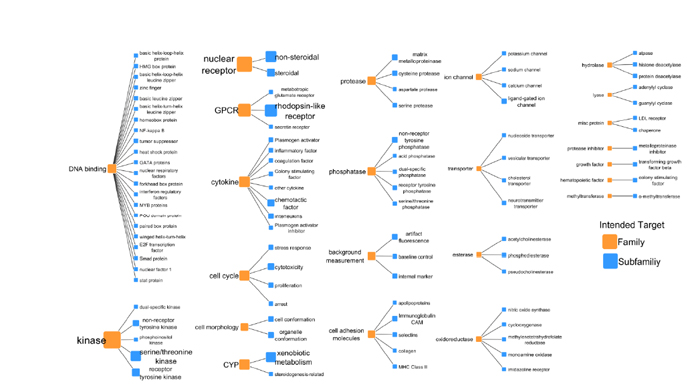
FIGURE 4-1 Intended target families and subfamilies for the ToxCast program. The number of assays for each intended target is represented by the sizes and font sizes of the orange nodes. For each intended target family, the assays are subdivided into assay end points (see EPA 2014a, p.13).
To make specific-protein assays more useful for DOD’s needs than they are now, there is a need to identify the subset of existing specific-protein assays that are directly mechanistically and quantitatively relevant to debilitating injuries, to fill gaps in assay types (for example, to develop assays for perturbation of particular ion channels that are not included in current platforms), to develop methods for identifying and classifying chemicals that have potent but nonspecific toxic actions, and to evaluate performance and predictive value of specific-protein assays for predicting acute toxicity by using a panel of reference and test chemicals relevant to DOD’s interests.
Cell-Based Phenotypic Assays
This section describes high-throughput assays that use cultured cells and measure some overall phenotypic output that is relevant to predicting acute toxicity, such as cell proliferation, plasma membrane permeability, or adenosine triphosphate (ATP) content. There is a large scientific literature on the application of cell-based assays to drug development and a growing literature on their application to toxicology. The ToxCast program includes more than 100 cell-based assays whose purpose is to measure cytotoxicity and other aspects of cellular phenotype. Simple cytotoxicity assays have long been used, with partial success, to predict animal and human toxicity and to estimate starting concentrations for animal toxicity studies.
A key consideration in all in vitro cell-based phenotypic assays is the choice of cell type. The general approach is to attempt to mimic specific human cell types; however, few data suggest, for example, that hepatocytes best predict liver injury or that renal tubule cells best detect renal injury (Lin and Will 2012). Assays are conducted with cells that are grown in a single layer (cell monolayer culture), and these conditions inevitably provide only a crude approximation of in vivo tissue environments, cell types, and cell–cell interactions. Furthermore, immortalized (often cancer-derived) and other cell lines have provided the mainstay of cell-based assays for decades because they are convenient for obtaining large numbers of cells in a standard state and for enabling cell lines to be readily shared between laboratories. However, the cells are often cultured using different media conditions, and this can affect cell response to chemicals. For example, high glucose concentrations in the growth media might increase the resistance of neural cells to the mitochondrial toxicant 1-methyl-4-phenylpyridinium (Mazzio et al. 2010). Similarly, cytotoxicity results from assays that evaluate mitochondrial disruption can be influenced by the Crabtree effect, in which cancer cells preferentially use glycolysis instead of oxidative phosphorylation for ATP production (Marroquin et al. 2007). Another limitation related to cell type is that many in vitro studies use cell lines that do not reflect the variation that is found in the human population. For some chemicals, sensitivity to cytotoxicity varied by as much as a factor of 100 in a single cell type taken from a broad population sample (Abdo et al. 2015).
There is an increasing shift away from cell lines toward cell-based assays that use cell types that are more physiologically relevant, such as animal or human primary cells, and human induced pluripotent stem (iPS) cells that have differentiated into specific cell types (Kraushaar et al. 2012; Godoy et al. 2013). However, unless great care is taken to mimic tissue-relevant physical and chemical environments, those cell types might provide little improvement over cell lines in physiological relevance. That consideration has led to an increasing push toward the use of organotypic model systems (discussed below).
Another key technical consideration is the measurement of assay results (readout). Readouts can be broadly divided into ones that average the response of a number of cells in a tissue culture well and ones that assess individual cell behavior, the latter sometimes called high-content assays (discussed below in the section “Emerging Technologies”). Whole-well readouts are most commonly used and have the advantage of being fast and simple, and it is straightforward to generate statistical metrics of assay performance and chemical activity. High-content readouts have the advantage, in principle, of providing much more information—for example, on
cell-to-cell heterogeneity in response and on specific cytotoxic mechanisms—but scoring and interpreting the additional information is computationally challenging (Wink et al. 2014).
Few studies have evaluated a large number (hundreds) of chemicals for their ability to predict acute toxicity (O’Brien et al. 2006; Xu et al. 2008; Lin and Will 2012; Porceddu et al. 2012). Most studies examine only a handful of chemicals, and studies that evaluate only specific classes of chemicals, such as endocrine disruptors or hepatotoxicants, substantially bias estimates of model performance (Thomas et al. 2012).
Cytotoxicity Assays
Measurements of cell life or death in culture have a long history in toxicology research (Ekwall 1983). Methods for measuring cytotoxicity in cell culture usually involve direct measurement of the fraction of cells that have intact membranes, for example, with neutral red uptake or fluorescent DNA dye uptake; measurement of the metabolism of surviving cells, for example, with reduction of 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), reduction of Alomar blue, or uridine uptake; or measurement of ATP content, cell number, total DNA content, total protein content, or cell proliferation. Cytotoxicity assays are normally run for a day or more (3 days is common), so viable cells will proliferate and increase the live-cell signal in control wells. Thus, the assays usually combine measurements of acute cell lethality, cell proliferation, or cell metabolism and might represent the simultaneous occurrence of several mechanisms (Huang et al. 2008). The longer an assay is run, the more it tends to factor in effects on cell proliferation unless it is run on nonproliferating cell types. Short-term (1-hour) assays are also used but need to include sensitive measures of cell injury, such as mitochondrial membrane potential or ATP content. Some assays are based on ultraviolet (UV) or fluorescence readouts and warrant caution because some chemicals can artificially interfere with UV- or fluorescence-based assays. In the pharmaceutical industry, luminescent readouts have largely replaced UV- and fluorescence-based assays.
In vitro cytotoxicity tests have been recommended as an adjunct to animal tests to improve initial dose selection and modestly reduce the number of animals used. The registry of cytotoxicity (RC) prediction model has been recommended (NIEHS 2001a,b) for evaluating the predictive accuracy of candidate cytotoxicity assays.4 Many RC prediction models evaluate the correlation between in vivo LD50s and in vitro cytotoxicity IC50s.5 The BALB/c 3T3 and normal human keratinocyte (NHK) neutral red uptake (NRU) cytotoxicity assays have been used to predict acute toxicity (NIEHS 2001a,b), but a major drawback of those cell-based assays is that they are difficult to automate. Recent advances have led to the development of commercially available “ready-to-go” cell plates that simply need to be defrosted and fed with media. The plates can then be used in the conduct of high-throughput assays.
Recent systematic studies that evaluated the predictiveness of in vitro assays for acute toxicity included several cell-based cytotoxicity assays partly because of their ease of use, reproducibility, and proven (albeit limited) predictive value. For example, of the 53 assays in the European Union (EU) ACuteTox project, seven constitute either a cell lethality assay (for example, NRU) or a metabolism assay (for example, MTT reduction or 2-deoxyglucose and uridine uptake) that effectively measures the number of living cells remaining after treatment. Notably, several cytotoxicity assays—an MTT assay in primary rat hepatocytes; a cytotoxicity panel that measures intracellular Ca2+ levels, mitochondrial membrane potential, and plasma membrane potential in HepG2, SHSY5Y and A.704 cells; and a basal cytotoxicity NRU assay in BALB/3T3 cells—generated data of sufficient quality to be considered in future acute-toxicity testing strategies (Kinsner-Ovaskainen et al. 2013).
__________________
4Halle (Spielmann et al. 1999; Halle 2003) describes inclusion criteria for data in the RC database.
5IC50 is the chemical concentration at which 50% inhibition is achieved.
Gene-Expression and Protein-Secretion Assays
Gene-expression and protein-secretion assays aim to identify a specific toxic mechanism by measuring expression of a specific gene or secretion of a specific protein that has been implicated as a biomarker of a particular toxic mechanism in humans. Gene-expression readouts can use engineered reporter genes, whereby an artificial promoter drives expression of an easily assayed reporter gene (such as luciferase) in response to activation of a toxic pathway or endogenous gene circuits. Engineered reporter genes provide easily standardized, reliable readouts, and a panel of such assays can be used to cover multiple biological pathways.
In recent years, several vendors have offered reporter assays for a variety of biological pathways involved in toxicity, such as inflammation, apoptosis, and endoplasmic reticulum stress. Their main limitation is that an artificial promoter in a generic cell line (such as HEK cells) might not indicate pathway activity in the same way as the natural pathway in a specific human cell type; that is, the assays could fail to capture biological context dependence. Reporter gene assays are of particular value for predicting endocrine disrupters because they work by reporting the expression of genes that are naturally regulated by sex hormones. However, endocrine disruption does not qualify as a likely mechanism of systemic toxicity that would result in an acute, debilitating injury in deployed personnel.
Expression of many endogenous genes can be simultaneously analyzed at the mRNA or protein level to create a “toxicogenomic signature” (discussed below under “Emerging Technologies”). Assays for induction of endogenous genes can be applied to primary cells that synthesize specific proteins in response to chemicals. A major advantage of this approach over artificial promoter constructs is that it measures endogenous gene-expression pathways and is thus more likely to indicate physiologically relevant pathway perturbation. A major disadvantage is that separate readouts must be developed for each protein, and this makes the approach more complex and labor-intensive than measuring the expression of a reporter like luciferase from engineered gene-expression constructs.
Protein-secretion assays are of particular value for measuring the activity of immunotoxins that induce expression and secretion of specific inflammatory cytokines, such as TNF-α, IL1, and IL6 from white blood cells. Cytokine measurements are usually made with an immune assay, typically a capture ELISA assay that uses two antibodies to provide high specificity and sensitivity. The relevance of the cytokine assays for evaluating acute toxicity associated with chemicals of relevance to DOD is limited.
Several recent systematic studies have combined multiple high-throughput assays, including assays for the expression of single endogenous genes in primary cells with mRNA or protein readouts. For example, the ACuteTox project included four cytokine secretion assays performed on primary white blood cells isolated from human blood and four assays that measured synthesis of neuronal and glia proteins, at the mRNA level, from aggregates of primary cells derived from rat brain (Kinsner-Ovaskainen et al. 2013). A meta-analysis found a variety of problems with all the assays, including failure to measure in the correct range for immunoassays and lack of reproducibility of primary cell aggregates. The ToxCast program evaluated the performance of the Biologically Multiplexed Activity Profiling (BioMAP) human primary cell disease models with the ToxCast phase I library (Houck et al. 2009). The chemicals that were tested generated relatively weak signatures compared with the reference pharmacological probes and drugs, and this raised the question of whether the concentrations that were used (up to 40 μM) were adequate. Furthermore, a follow-up study demonstrated that some chemicals with known biological activity, such as pharmaceuticals, gave false-negative results in the assays (Kleinstreuer et al. 2014). All the problems highlighted here indicate the difficulty of working with complex primary cells in a high-throughput assay format.
Limitations and Needs for Improvement of Cell-Based Phenotypic Assays
Cell-based assays are efficient in assessing chemical mechanisms of action. They have also been useful for predicting some mechanisms of chronic toxicity, such as endocrine disruption, but might have less applicability to acute-toxicity prediction. Furthermore, conventional cell-based cytotoxicity assays typically lack metabolic competence and often miss chemicals that require bioactivation. They also fail to provide data on some of the most important toxic mechanisms, notably ones that involve organ- or cell-type specific physiology. For example, they miss chemicals that perturb synaptic transmission and neurotransmitter metabolism.
Responses seen in cytotoxicity assays can also be influenced by the choice of cell used. In the Tox21 cytotoxicity profiling screen, cytotoxic response patterns varied by cell type and indicated differences in sensitivity and kinetics of the response (Xia et al. 2008). Overall, the human blood-derived cells (Jurkat), neuron-derived cells (SH-SY5Y), and rodent cells (N2a, H-4-II-E and NIH 3T3) were most sensitive to chemical-induced cytotoxicity, and human fibroblastic, endothelial, and skin cells (HUV-EC-C, BJ, and MRC-5) were least sensitive. Another finding from the study was the lack of similarity in the patterns of chemical activity in cells that were derived from the same tissue but from different species, for example, human HepG2 and rat H-4II-E hepatoma cells.
To improve the utility of cell-based assays for predicting acute toxicity, several aspects of experimental design need to be considered. First, cell-based assays that express the relevant biological processes and toxicologic mechanisms under study need to be selected. In some cases, that might require using primary cell cultures rather than cultured immortalized cells. Second, relevant chemical concentrations and exposure durations need to be used, and appropriate test and reference chemicals need to be identified for evaluating the performance and predictive value of the assays. Ultimately, the readout of a cell-based assay must be mechanistically and quantitatively linked to a chemical’s toxicity phenotype in humans if it is to be truly predictive.
Organotypic Models
Many of the limitations of cell-based assays, as noted above, arise from the fact that single cells growing on a dish are inevitably poor models for human tissues. That is the case even if the cells themselves were derived from human or animal tissue because the artificial in vitro environment changes their phenotype in hours. The culture-induced changes affect cells’ responses to chemicals and the cells’ (especially liver cells’) ability to biotransform chemicals into more or less toxic species. To address the limitation, researchers have been developing approaches for co-culture of multiple cell types or for cultures of whole organs as slices or cell aggregates. Those approaches are collectively referred to as organotypic models.6 The discussion below highlights a few organ systems that are relevant to acute toxicity of chemicals.
Skin Organotypic Models
Skin, as the largest human organ, is important for the absorption of many classical chemical-warfare agents and constitutes the principal barrier to and defense against absorption of toxic lipophilic chemicals. In addition, the skin is the primary site of action of acute blistering agents. The military’s historical reliance on chemical-protective boots, suits, and gloves emphasizes the importance placed on dermal protection against chemical exposure. Furthermore, one of the first
__________________
6EPA defines organotypic culture models as “tissue culture models that mimic in vivo tissue architecture through interactions of heterotypic cell types (e.g., epithelium-stroma) and extracellular matrices (ECM). They can be established from isolated cells or from tissue fragments harvested in vivo, and will bridge the gap between conventional monolayer cell cultures and whole-animal systems” (EPA 2013).
questions that the “warfighter” asks when told that a potent, toxic chemical can be absorbed through intact skin is, How much and how quickly? Although definitive dermal absorption studies can be conducted on animals by using small numbers of chemicals that are synthesized with radiolabels, various in vitro models have been developed by using instrumented flow cells with human or porcine skin explants. Those in vitro models are far more amenable to high-throughput screening than are dermal absorption studies with radiolabeled chemicals (Basketter et al. 2012).
Organotypic cultures of skin in formats that are applicable to screening thousands of chemicals are relatively advanced. The field has benefited from the relatively simple anatomical organization of skin, from the fact that proliferating human keratinocytes are relatively easy to grow, from the abundant availability of human tissue from minor surgical procedures, from the need for artificial skin for treating burn patients, and, not least, from huge investment by the cosmetics industry. Especially in the European Union, cosmetics manufacturers have been under pressure to increase safety testing while reducing animal use. In an example that is generally encouraging for toxicity prediction, scientists at L’Oreal, Inc. in France have used the EpiSkinSM model to predict irritant activity (Cotovio et al. 2005, 2008). The model consists of primary human keratinocytes that have been induced to self-organize into a multilayered structure similar to skin by use of bioengineered substrates. MTT reduction and release of the inflammatory cytokine IL-1α were measured. The model accurately (> 80%) predicted irritant activity of 184 cosmetic ingredients (Cotovio et al. 2008). The study suggests that good—although not perfect—predictions can be made for acute skin irritants by using a sophisticated organotypic culture model. In contrast, numerous in vitro and in vivo models have been examined in attempts to emulate the blistering (vesication) seen in human skin on exposure to sulfur mustard or lewisite, most with little or no success. Chemical-induced skin blistering might be limited to humans, or it requires epithelial and fibroblast immune cell functions that are not accounted for in current organotypic skin models.
Eye Organotypic Models
Loss of vision would be an incapacitating effect of concern to DOD. Testing for eye irritation has benefited from investment by the cosmetics industry, although current organotypic cornea models are generally less advanced than skin models. Commercial models are available and include the EpiOcular™ OCL-200 tissues from MatTek Corporation (Ashland, MA) and the SkinEthic™ Reconstituted Human Corneal Epithelium developed by a consortium of European cosmetics companies in response to banning of rabbit testing. Systematic evaluation of the systems continues but mostly in the chemical space relevant to cosmetics. Both systems achieved benchmarks for between-laboratory reproducibility and are undergoing tests of predictive value (Alépée et al. 2013; Pfannenbecker et al. 2013). On the basis of the results with skin, a reasonable degree of predictive power is expected.
Lung Epithelium Organotypic Models
The lung is a relevant organ for both chemical absorption and acute toxicity. Multiple 3-D organotypic models have been described, including the commercial EpiAirway™ (MatTek Corporation, Ashland, MA) and MucilAir™ (Epithelix Sàrl, Geneva, Switzerland) systems. The systems use primary cells cultured at an air–liquid interface, and they model airway function better than traditional submerged tissue culture. A recent paper evaluated the models relative to each other and to two conventional submerged tissue-culture systems for their ability to predict rodent lung toxicity of 19 chemicals (Sauer et al. 2015). None of the systems performed well in
predicting lung toxicity in rats, and the 3-D organotypic systems performed no better than conventional tissue-culture models. The findings suggest that organotypic lung epithelium models lag behind skin models in predictive value. The poor predictivity probably reflects the greater complexity of airway epithelium or perhaps differences in investment.
There does not appear to be a particularly good in vitro model for lung damage associated with chemicals (for example, phosgene) that are known to increase pulmonary permeability that results in noncardiogenic pulmonary edema. The development of in vitro assays to evaluate similar compounds identified as chemicals of interest, for example, on the basis of chemical structure or quantitative structure–activity relationships will require further DOD investment to replace animal inhalation-toxicity studies.
Liver Organotypic Models
The liver is especially relevant to biotransformation of chemicals into more or less toxic metabolites and is an important site of acute toxic action of some chemicals. Hepatocytes are the major biochemical engine of the liver and are responsible for metabolism and excretion of many xenobiotics. Hepatocytes can be cultured in standard 2-D formats, but their phenotype with respect to xenobiotic metabolism and responses changes rapidly under culture conditions. Immortalized cell lines derived from hepatocellular carcinoma (such as HepG2) have often been used as a surrogate for hepatocytes, but their biology is even more distant from hepatocytes in situ. Because of the relevance of liver toxicity to drug development, the pharmaceutical industry has made major investments in modeling liver biology in 2-D cell cultures, 2-D co-cultures, 3-D cell cultures, and engineered organotypic systems (Godoy et al. 2013).
Multiple approaches have been taken to build organotypic models of human and rodent liver, and substantial improvements over 2-D hepatocyte culture systems in recapitulating normal liver biology, drug metabolism, and drug responses have been noted (Khetani and Bhatia 2008; Godoy et al. 2013; Messner et al. 2013). Despite improvements, predictive-toxicology studies still tend to focus on 2-D cultures of primary hepatocytes of hepatocellular carcinoma-derived cell lines. For example, the EU-funded LIINTOP project is evaluating multiple 2-D culture models for liver and intestine (Gómez-Lechón et al. 2010), and the EU ACuteTox project included five assays with HepG2 cells (Kinsner-Ovaskainen et al. 2013).
The continued reliance on 2-D cultures and hepatoma-derived cell lines is problematic because testing in more robust in vitro models would probably generate more-predictive data. For example, Khetani and Bhatia (2008) showed that a microengineered 2-D culture system in which hepatocytes are co-cultured with stromal cells provided better modeling of gene expression, metabolism, and drug action than conventional 2-D cultures. Another promising system involves co-culture of hepatocytes with Kupffer cells in small spheroids (Messner et al. 2013). The microengineered systems often have lower throughput and are more expensive than conventional 2-D cultures, but given the importance of the liver in toxicology the expense might be worthwhile. Although it remains to be seen how their overall predictivity differs from that of simpler models, the use of complex liver models should improve the recognition of inflammation and other mechanisms of toxicity that are not easily detected in simpler hepatocyte cultures. For example, Khetani et al. (2013) demonstrated that using co-cultured hepatocytes better predicted drug-induced liver injury in a small test set of 45 chemicals.
Godoy et al. (2013) provides an exceptionally comprehensive overview of hepatic models. It is interesting to note, given that the liver is perhaps the most thoroughly characterized of the organotypic model systems, that the authors concluded that “one key message is that despite our enthusiasm for in vitro systems, we must never lose sight of the in vivo situation. Although hepatocytes have been isolated for decades, the hunt for relevant alternative systems has only just begun.”
Neural Organotypic Models
The brain has multiple neuronal and glial subtypes, complicated neuronal networks that have different types of chemical synapses, important cell–cell interactions, and myelinated axons. That cellular complexity helps to make it an important site of action for many acute toxicants and makes it difficult to identify neurotoxic effects with conventional cytotoxicity assays or other in vitro systems. Brain aggregate cultures replicate some organotypic structural and functional features and have been used as a model system for neurotoxicity testing. For example, the EU ACuteTox project included seven assays of cell aggregates derived from rat brain as part of a battery of 50 assays that included many involving 2-D cultures of neurons (Forsby et al. 2009). The authors concluded that “using aggregate cell cultures prepared from embryonic rat brain and a multiparametric endpoint scheme, all chemicals known to be highly toxic in humans also showed high toxicity (significant effects in the lower micromolar range) to extreme toxicity (significant effects at nanomolar concentrations) in aggregate cultures” (Honneger et al. 2009). They also noted that inclusion of data on metabolism and pharmacokinetics of the blood–brain barrier would likely improve predictive value.
Limitations and Needs for Improvement of Organotypic Models
Additional organotypic models have been developed for the heart, kidney, and skeletal muscle. Organotypic models have high potential for predicting acute toxicity and potentially can recapitulate the metabolism and biological activity of a chemical. That said, the science of accurately modeling human organs in a culture dish, especially in formats suitable for high-throughput testing, is still in its infancy. Progress is being made, but much caution is warranted, particularly for acute-toxicity prediction, which has not been the goal of most studies. Organotypic assays are much more complex and expensive than pure protein-based and cell-based assays and less robust than rodent models because they do not integrate multiple physiological systems. Their reliability has often been called into question in systematic studies. Their predictivity is also far from guaranteed; for example, the lack of success in predicting vesicant activity by using organotypic skin cultures is troubling.
For organotypic cultures to be used in DOD screening for acutely toxic chemicals, there is a need to evaluate the potential of organotypic assays for acute-toxicity prediction and to invest further in the basic science of organotypic cultures. The potential usefulness is high, even if current systems are far from ideal.
NONMAMMALIAN IN VIVO ANIMAL MODELS
The use of an in vivo approach facilitates the crucial understanding of how chemicals affect complex metabolic targets and pathology at the cell and organ level. Mice, rats, rabbits, and other laboratory mammals have been used extensively to study chemical toxicity. However, in vivo assays with those species are often expensive, use large amounts of toxic test chemicals, and are difficult to use in a moderate-throughput to high-throughput manner. Those drawbacks have led toxicologists to develop alternative animal models for chemical testing. Many alternative test organisms share biological processes with rodents and other mammals, including humans. Three test platforms of note that can be adapted to high-throughput screening rely on insect, nematode, and zebrafish models (Giacomotto and Ségalat 2010). They can complement other cell-based in vitro test systems, and data from assays that use the alternative animals could be used to set priorities among chemical candidates for future traditional animal testing. Development and application of the nonmammalian models could help DOD to screen for pathway-specific effects and could demonstrate how chemical toxicity varies among species.
This review of alternative animal models is not intended to be exhaustive; rather, the committee has focused on end points that are relevant to acute toxicity and on selected examples that illustrate how these systems could be adapted for high-throughput testing of acute effects.
Fruit Fly Models
The fruit fly (Drosophila melanogaster) has been used as a model organism in studies of genetics and developmental biology for over 100 years (Rubin and Lewis 2000). Fruit flies have a fully mapped genome, and many protocols for biochemical and genetic analysis are well established. More than 60% of human genes have functional orthologs in D. melanogaster (Bier 2005). A variety of molecular tools, including mutagenesis and RNAi, are available for modifying fruit fly genetics. A dedicated Web-based database (FlyBase) contains information relative to fruit fly genetics and its molecular biology (Drysdale 2008).
Fruit flies have the potential to be used for chemical-toxicity screens (Nichols 2006; Whitworth et al. 2006; Segalat 2007). In particular, fruit flies and other insect models have improved our understanding of the molecular action of pyrethroids, which act on both mammalian and insect sodium channels, and other insecticides (Peterson et al. 2008). Despite their use in neurotoxicology research, there are important limitations (Rand 2010). For example, γ-aminobutyric acid, acetylcholine, and other neurotransmitters often have roles in the insect nervous system that are different from their roles in vertebrate nervous systems (Peterson et al. 2008).
Specialized video-based equipment has been developed to assess flying, chemotaxis, geotactic climbing,7 and other behaviors (Sawin-McCormack et al. 1995; Rand 2010; Sokolowski 2001; Podratz et al. 2011; Gregory et al. 2012; Podratz et al. 2013). In addition, eclosion8 and adult lethality are simple end points that can be assessed without the need of a microscope. However, the transition from larva to adult fly is complex and occurs by mechanisms distinct from those seen in mammals; this draws into question the utility of LD50s obtained for this developmental stage (Rand 2010). There are other limitations of the use of fruit flies. For example, chemical administration to the fly embryo must overcome the barriers presented by the hydrophobic vitelline membrane (Limbourg and Zalokar 1973; Rand 2010). In addition, fly toxicokinetics of xenobiotics can differ substantially from that in mammals, including humans.
Nematode Models
The most widely used nematode model for biomedical research is likely Caenorhabditis elegans. C. elegans have a fully mapped genome (C. elegans Sequencing Consortium 1998), and more than 50% of human genes have functional orthologs in C. elegans (Harris et al. 2004). Genetic or genomic manipulation—such as knockouts, knockdown via RNAi, and transgenic strains—is routinely available. A variety of bioinformatics tools have been developed to support high-throughput genomic studies with this organism (Cho et al. 2014). In addition, a dedicated Web site (Wormbase) allows investigators access to microarray data and comprehensive data on gene structures, mutants, RNAi phenotypes, and protein–protein interactions (Chen et al. 2005).
Numerous studies have shown that C. elegans and humans share many essential biological characteristics. C. elegans has a rudimentary nervous system, exhibits behavior, and is capable of rudimentary learning and memory functions. The anatomy of C. elegansis well understood. It contains 959 somatic cells, including about 300 neurons that are microscopically visible. The developmental cell lineage and the neural wiring diagram of C. elegans have been completely mapped. Many vertebrate neurotransmitters are well conserved in this nematode (Villatte et al.
__________________
7Drosophila instinctively climb against gravity (geotaxis).
8Eclosion is the hatching of adults from the pupal stage.
1998; McVey et al. 2012). The presence of a functional nervous system has been exploited by neurotoxicologists to study the acute neurotoxic effects of pesticides and other chemicals on this nematode (Williams and Dusenbery 1990; Ruan et al. 2009; Avila et al. 2012; McVey et al. 2012; Meyer and Williams 2014). End points evaluated include changes in survival, behavior, locomotion, life span, cell death, neurotransmitter concentration, and function. Image-tracking systems have also been developed for assessing C. elegans locomotion (Feng et al. 2004). Melstrom and Williams (2007) reported a strong correlation between LC50s determined for C. elegans and LD50s identified in rodents after exposure to cholinesterase-inhibiting pesticides. End points examined by Melstrom and Williams included depression of acetylcholinesterase activity and decreased movement.
Zebrafish Models
As a vertebrate, zebrafish (Danio rerio) have substantial physiological, anatomic, and genetic homology with humans (Barbazuk et al. 2000; Howe et al. 2013; Chakravarthy et al. 2014). Zebrafish are amenable to gene manipulation, have a short generation time, and have well-characterized rapid developmental stages; these characteristics have led to their growing use in developmental-toxicity studies (de Esch et al. 2012; Raldúa and Piña 2014). Its application to the study of developmental toxicity has garnered the interest of regulatory bodies. For example, the Organisation for Economic Co-operation and Development (OECD) has recently formulated guidelines for using zebrafish embryos for testing acute toxicity of 119 chemicals and for developmental toxicology (OECD 2013a,b). Zebrafish can be bred in large numbers with minimal maintenance cost (Raldúa and Piña 2014). They are a cost-effective in vivo model for screening drugs and other chemicals and meet many objectives of a high-throughput screening assay (Taylor et al. 2010; Tsang 2010; Lessman 2011). High-throughput zebrafish assays have also been used for microarray and proteomic studies (Love et al. 2004). Because zebrafish larvae are transparent, they are ideal for in vivo imaging without the use of invasive techniques (Knudsen et al. 2011; Raldúa and Piña 2014). Another advantage of the zebrafish larva model is that up to 4 days after fertilization they are not treated as vertebrates by US Institutional Animal Care and Use Committees because they retain a yolk and higher-order neuronal functions are generally absent.
Zebrafish are used in safety pharmacology studies to screen for arrhythmogenicity (Langheinrich et al. 2003; Milan et al. 2003; Burns et al. 2005), nephrotoxicity (Hentschel et al. 2005; Wu et al. 2012), hepatotoxicity (Vliegenthart et al. 2014), and neurotoxicity (de Esch et al. 2012; Legradi et al. in press). Some drugs that affect human cardiac function and structure are known to have similar effects in zebrafish (Milan et al. 2003). Heart-specific expression of the green fluorescent protein in zebrafish has been accomplished by using the cardiac myosin light chain 2 promoter (Huang et al. 2003). Specialized equipment exists to monitor changes in zebrafish heart rate after chemical exposure (Burns et al. 2005; Simoneschi et al. 2014). Behavioral assays of swimming behaviors have been developed for zebrafish (Ali et al. 2012; Bichara et al. 2014). Driessen and co-workers (2013) have shown good concordance in histopathological responses and gene expression profiles between zebrafish embryos and mice exposed to known hepatotoxic chemicals. Yen et al. (2011) evaluated changes in zebrafish larva survival, acetylcholinesterase activity, and behavior after exposure to three organophosphorus pesticides. That type of study might be useful for nerve agents and other chemicals that have a similar mechanism of action. In vivo zebrafish assays with reverse dosimetry have also been used to develop human oral-dose hazard values (Perkins et al. 2013).
Limitations and Needs for Improvement of Nonmammalian In Vivo Animal Models
Nonrodent animal models have the potential to assist in characterizing the acute toxicity of chemicals. One advantage of such systems is their ability to identify whole-animal and organ-
level responses. The exploration of nontraditional in vivo models for assessing acute-toxicity potential, however, will need to consider species differences in metabolism and cellular targets and other issues related to interspecies and in vitro–to–in vivo extrapolations. Other factors to consider include differences in organ composition (multiple cell types), cell organization or structure, and gradual enzyme expression in different tissue regions. Another challenge is related to the extrapolation of aqueous (as in the case of zebrafish) or medium (as in the case of C. elegans) concentrations to exposure concentrations that are relevant to humans. Alternative animal model in vivo assays have considerably lower throughput than other assay systems considered by the committee and are likely to be used in the later stages of the assessment process. Little work has been performed regarding the applicability of such models to assess the acute toxicity of chemicals relevant to DOD, and they have been incompletely analyzed for their predictive validity with respect to acute toxic effects or identification of affected organ systems.
The drive to develop nonanimal methods for toxicity testing is still in its infancy, and new technologies are continually being developed. Most are aimed at commercial applications, particularly for safety assessment in the pharmaceutical and cosmetics industries, but a subset of the new technologies will also be useful for predicting acute toxicity. In this section, emerging technologies are broadly divided into ones that generate large amounts of information per sample by multiplexed or image-based measurements and ones, such as organ-on-a-chip and induced pluripotent stem (iPS) cell technologies, that aim to model human tissues more accurately.
Multiplexed Assays
Multiplexed assays allow the measurement of dozens, hundreds, or even thousands of end points simultaneously on a single sample. In general, they seek to provide more biological data per sample than traditional assays that measure a single end point. Conceptually, obtaining data on many end points is expected to help in deciphering chemical mechanisms and identifying new biological targets for development of more specific assays (Larson et al. 2011). The most used, and best understood, multiplexed readout is a gene-expression profile, in which the amounts of hundreds or thousands of mRNAs in a sample are measured in parallel (Fabian et al. 2011; Klaper et al. 2014). Microarrays have been popular for measuring gene expression, but the decreasing cost of DNA sequencing is leading to a gradual replacement of microarray and related technologies with RNAseq approaches. There is increasing interest in multiplexed measurement of micro-RNAs, whose expression also reflects the state of a cell or tissue.
Protein and metabolite measurements can also be multiplexed. For example, multiplexed immunoassays can be used to measure the concentration of tens or hundreds of cytokines or other proteins in a single sample. The main limitation of such assays is in developing high-quality antibody pairs to capture and quantify a given protein with high sensitivity and specificity. Given recent developments in proteomics technology, it is possible that mass-spectrometry–based measurements will gradually replace immunoassays for multiplexed protein measurements (Fu et al. 2010; Potts et al. 2011). Modern multiplexed proteomics methods allow quantification of thousands of proteins in tens of samples in a single spectrometry run (McAlister et al. 2014). However, protein measurements are inevitably more difficult and less sensitive than nucleic acid measurements because proteins cannot be amplified by replication and have different physical properties and abundances. Metabolites can also be profiled by using a coupled chromatography–mass spectrometry technique.
Questions remain regarding how useful multiplexed measurements will be for predicting acute toxicity, whether they are at the level of RNA, proteins, or metabolites. Most relevant information is currently available for mRNA profiling because it has the longest history, but de-
pending on the toxic mechanism, profiling at the protein or metabolite level might be equally or more informative. Published examples show that comparison of mRNA profiles allows identification of chemicals that have common actions on cells in culture, including mechanisms that could cause acute toxicity (Lamb et al. 2006; Ravindranath et al. 2015). Those examples are encouraging, but it is important to recognize that they highlight specific success stories, not systematic toxicity prediction.
Another relevant literature is on toxicogenomic approaches to predicting toxicity of chemicals in liver and other organs. Those studies typically involve treating rodents with chemicals, harvesting organs, and using microarrays combined with pathology reports to classify effects on the liver and other organs. Pharmaceutical companies and governments have invested a great deal of resources in this approach, and major databases have recently been released to the public. The results have been mixed: considerable improvement in understanding toxic actions and identifying biomarkers but far from a complete solution to the problem of predicting liver toxicity (Chen et al. 2012). A recently created database includes expression signatures for 1,000 genes, using the L1000 assays, for treatment of tens of cell lines with thousands of chemicals (Duan et al. 2014). Analysis of that dataset should help to clarify the potential value of highly multiplexed gene-expression signatures in predictive toxicology.
In principle, multiplexed measurements at the protein or metabolite level might be expected to reveal a chemical’s mechanisms of action more effectively, and with more predictive value, than gene-level measurements. More ambitiously, a combination of multiplexed measurements of mRNA, protein, and metabolite levels in parallel would in principle cover the most ground with respect to producing mechanistic information relevant to prediction of acute toxicity. That kind of integrated –omics approach has been shown to improve understanding of specific toxic mechanisms (Wilmes et al. 2013, in press) but is expensive and unproven in its usefulness for systematic testing of acute-toxicity potential. However, the data can be used to design new high-throughput assays that are mechanistically relevant to critical processes or pathways targeted by chemical-warfare agents.
High-Content Screening Assays
High-content screening (HCS) assays make multiple measurements of cell biology at the level of single cells by using microscopy or other imaging technologies. Typically, cells grown on multiwell plates are treated with a chemical, stained with several fluorescent markers that report on various aspects of metabolism and organelle health, imaged, and detected with some type of automated algorithm. HCS technology can provide much information that is relevant to specific pathways or organelles in a single assay, and it is faster and less expensive than multiplexed assays of gene or protein expression. It has been used in drug development for some time but only recently applied to toxicology. Recent studies show high potential (O’Brien 2014; Persson et al. 2014). In particular, some mechanisms of acute toxicity that might be poorly detected in gene-expression assays, such as damage to cellular membranes or organelles, can be directly assessed with HCS assays. So far, too few studies have been published to evaluate this promising technology, and key questions, such as reproducibility between laboratories, need to be addressed.
Imaging is a useful way to screen for numerous mechanistic end points at the same time, such as cell death, apoptosis, oxidative stress, mitochondrial membrane potential, DNA damage, and cell-cycle inhibition. Algorithms have been developed, for example, to predict human liver injury. Predictivity is achieved by testing enough positive and negative chemicals in the system to know the specificity and sensitivity of the platform or any particular assay. In general, these assays have high specificity but low sensitivity. In fact, in the absence of consideration of exposure, predictivity of drug-induced liver injury rarely gets above 50% (Xu et al. 2008). In addition, because these mechanistic end points are common end points of cell injury and death, it is difficult to predict particular organ toxicities. For example, the liver toxicant troglitazone and the cardiac toxicant doxorubicin both cause oxidative stress and mitochondrial dysfunction in vitro.
Other factors that contribute to toxicity in humans—such as inflammation, use of multiple drugs, and genetics—are difficult to model in simple cell systems.
Organ-on-a-Chip, Microphysiological Systems, and Advanced Organotypic Assays
The field of tissue engineering has advanced rapidly in recent years, having been stimulated especially by advances in microfabrication technology, such as micropatterning and microfluidics. The traditional goal of the tissue-engineering field is to develop replacement organs, but a shorter-term goal of generating tissue models for drug and toxicity testing has emerged (Alepee et al. 2014; Jennings 2015). Increasingly, proponents of this kind of technology are promoting “organ-on-a-chip” models as the ultimate systems for determining toxicity mechanisms in organ systems (Huh et al. 2010, 2012; Esch et al. 2011; Godoy et al. 2013; NAS 2014; Pamies et al. 2014; Sung et al. 2014). Recently, Maschmeyer et al. (in press) reported creation of long-term microphysiological systems that more closely mimic the human liver, intestinal barrier, and skin in vivo. Those systems might have broader applications in toxicology.
The considerations in evaluating the potential of these technologies are similar to those already discussed for organotypic models given that they are a modern extension of the organotypic models. Typically, organ-on-a-chip models are much more complex and expensive than simple cell-culture models. In theory, and in some studies, their predictive value is higher than that of simple cell culture, but their reliability needs to be evaluated, and their cost per data point might exceed that of animal models, especially if human primary cells are needed.
Induced Pluripotent Stem Cell–Derived Primary Cells
An iPS cell is a type of human pluripotent stem cell that can be differentiated into multiple types of tissue cells in cell culture. iPS cells are derived from adult tissues by forced expression of stem-cell transcription factors—an approach pioneered by Shinya Yamanaka (Takahashi and Yamanaka 2006) that avoids use of cells derived from human embryos. iPS cells can in principle provide a renewable source of almost any human primary cell type without requiring an embryo donor. iPS cells should work well with organotypic cultures, organ-on-a-chip technologies, and other biomimetic approaches that provide more realistic cell-culture models (Mathur et al. 2013). Another advantage to using iPS cells, in principle, is that they can be derived from donors who have different genotypes and might respond differently to toxicants, for example, different cytochrome p450 alleles that cause differences in drug metabolism. Thus, the long-term potential for application of iPS cells to toxicity testing is high. Nevertheless, many hurdles must be overcome, including development of methods for reliable differentiation of iPS cells into cell types that are relevant to acute toxicity. The field is promising but unlikely to be useful for chemical testing in the next 5 years. One cell type that is relevant to acute toxicity and is relatively easy to generate from iPS cells is human cardiomyocytes (Kraushaar et al. 2012); cardiotoxicity testing will therefore be a field to monitor for application of iPS technology to toxicology.
Chemical metabolism is an important in vivo biological process that should be considered during the interpretation of in vitro testing data. There is a large capacity for metabolism in the body; metabolizing enzymes are present in the liver, lung, nasal region, and other tissues. Metabolism can convert parent chemicals into toxic metabolites (metabolic activation), into nontoxic metabolites (metabolic inactivation), or into metabolites that are rapidly removed from circulation (detoxification). Some in vitro systems are metabolically competent and thus provide at least some metabolism in an assay. Others are less metabolically competent and should use parent chemicals and metabolites as test agents to ensure that the appropriate chemicals are assessed. The use of structural identification tools described in Chapter 3 can help to predict toxic metabo-
lites. However, it can be difficult to identify a biologically active molecule without knowing the metabolic pathway in advance. This section summarizes metabolic assays and considerations that can complement in vitro testing strategies that focus on activity of a parent chemical. Although the most effective classical chemical-warfare agents do not require metabolic activation, a clear understanding of the role of chemical metabolism in toxicity has the potential to refine acute-toxicity assessments when incorporated with other testing strategies.
Assessing Reactive-Metabolite Formation
Many structural alerts9 that indicate the formation of reactive metabolites have been identified. Reactive metabolite formation, however, is not necessarily sufficient for toxicity to occur; in fact, many marketed drugs, which are considered safe at therapeutic dosages, contain such alerts (Kalgutkar and Dalvie 2014). Thus, it is difficult to assign toxicity to metabolites in the absence of in vitro experimentation. An initial strategy that could be used in the assessment of metabolite toxicity could be to identify potential metabolites by nontesting approaches (such as the use of quantitative structure–activity relationships) and then to confirm the presence of predicted chemical moieties experimentally. Formation of human metabolites can be assessed with in vitro systems (such as incubations with liver microsomes or hepatocytes) coupled with mass-spectrometric detection and measurement of metabolite formation. Thus, screening for toxicity can be based directly on a known metabolite or determined analytically by measuring metabolites formed during an assay.
Some chemical moieties (such as quinones and epoxides) are known to elicit toxicity. Highly electrophilic metabolites are known to be reactive with reduced glutathione (GSH), an endogenous nucleophile that plays a key role in xenobiotic metabolism and detoxification. Detection of GSH conjugates in in vitro assays that incorporate hepatocytes or liver microsomes provide indirect evidence that a reactive metabolite was formed.
In Vitro Systems to Study the Role of Metabolism in Toxicity Potential
A variety of in vitro model systems have been developed to study metabolism and include “precision-cut tissue slices, subcellular fractions such as the microsomal fraction, primary cells in suspension, primary monolayers of cells in culture, continuous cell lines, immortalized primary cells, liver-derived cell lines re-expressing biotransformation enzymes and genetically-engineered cell lines expressing biotransformation enzymes” (Combes et al. 2006). Tissue fractions that contain metabolic enzymes, such as microsomes or S9 fractions, can also be introduced into an assay system to increase its metabolic competence (Glatt et al. 1989). Encapsulation of S9 in hydrogel microbeads has been recently introduced into cytotoxicity assays as one method of reducing leakage of potentially toxic microsomal lipid peroxides (Yamamoto et al. 2011).
Despite the availability of in vivo rodent acute-toxicity data on some chemicals, the studies will not identify toxic metabolites and resulting toxicity that are elicited via human-specific metabolic pathways not present in laboratory animal species. One approach to evaluating species differences in metabolism is to use primary human hepatocytes or other human-origin in vitro systems. Another approach to elucidating metabolic pathways relies on evaluating cell responses in the presence and absence of a P450 inhibitor, such as 1-aminobenzotriazole (ABT). If the formation of metabolites is inhibited by ABT, isoform-specific inhibitors can be used to identify specific isoforms involved. An individual cDNA-expressed human P450 isoforms system can be used to confirm the results of P450 isoform-specific inhibitors.
__________________
9As noted in Chapter 3, a structural alert is a chemical structure that has been linked to toxicity or a specific toxicity end point.
Current State of Metabolic Competence in in Vitro Testing Approaches
Because of their robust proliferative potential and sensitivity to toxic effects, immortalized cell lines have been identified as the system of choice for high-throughput assays. However, most of the cell lines have little or no metabolic enzyme content (that is, they are metabolically incompetent) and respond differently from tissue slices, primary cells, or tissue that is exposed to the same stimuli in vivo. For example, breast-cancer cell lines, such as MCF-7, are more prone to toxic effects than normal breast cells. Furthermore, although the hepatoma-derived HepG2 cells have many liver-specific functions and express conjugating enzymes, they lack functional expression of almost all the relevant human xenobiotic metabolizing enzymes in the cytochrome P450 family (Donato et al. 2008). Primary cells that are derived directly from animal or human tissues are more metabolically competent than immortalized cell lines but have much shorter half-lives and require much more care to be sustained for toxicity screening. More recent advances in cell-culture technology have improved metabolic capability in in vitro systems. HepaRG cells cultured in 3-D spinner-bioreactors are an attractive tool for toxicological studies and show an expression of CYP450 enzymes and phase II metabolism that more closely mimics in vivo conditions (Leite et al. 2012). Multicellular 3-D human primary liver cell cultures that contain hepatocytes, fibroblasts, stellate cells, and Kupffer cells have also demonstrated increased metabolic activity in the presence of fluid flow (Esch et al. 2015). In summary, cell systems have their own advantages and drawbacks, and understanding their enzymatic content will be important for estimating the effect of metabolism on in vitro screening data.
In the absence of metabolic activity, in vitro assays might still be used effectively in screening for biological activity of the parent chemical. However, in vitro assays designed to assess chemical metabolism or enzyme involvement might not translate directly to in vivo effects in that they might not recapitulate the endogenous concentrations or physiological distribution of enzymes in vivo. Although high-throughput screening focuses on targeted assay systems, pharmacokinetic information can be integrated independently, as further discussed in Chapter 5.
ASSAY CONSIDERATIONS FOR IMPROVING PREDICTION OF ACUTE TOXICITY
The limitations of specific in vitro assays have been discussed above, and here the focus is on broad steps that could be taken to improve the ability of screening assays to predict acute toxicity. The largest improvement needed is the demonstration of a linkage of assay measurements to relevant mechanisms of toxicity that quantitatively reflect an in vivo toxicity phenotype in target cell types. Prediction of acute toxicity would also be improved if the route of exposure were considered in assay design. The committee assumes that the relevant exposure routes are dermal and inhalation, and most assays have been designed to model oral and intravenous exposures. The issues surrounding exposure route increase as assays become higher throughput and less metabolically competent. As mentioned earlier, future assays should also take chemical metabolism into account. Those and other considerations are discussed in more detail below.
Quantitative Linkage of Assay Measurements to in Vivo Phenotype
Validated alternative test methods are needed for evaluating the safety of chemicals, cosmetics, and drugs. To address that need, the EU ACuteTox program assessed the ability of in vitro and in silico tools and assays to predict specific organ and system toxicity (such as hematotoxicity, neurotoxicity, nephrotoxicity, and hepatotoxicity) and intestinal absorption, distribution, and metabolism. The first “prevalidation” phase of the ACuteTox project tested a set of 57 chemicals in 50 in vitro and in silico assays or approaches and used published toxicity data as the phenotypic anchor for statistical analyses. That phase identified eight assays that showed some value in predicting acute toxicity (Clemedson et al. 2006); Box 4-1 provides more detailed information. The ACuteTox project identified several broad kinds of improvement that would need to be con-
sidered in the design of future in vitro screening systems, namely, improved consideration of mechanistic data and increased use of pharmacokinetic data to enhance toxicity estimates (ACuteTox 2010).
One example of mechanistically informed assay design comes from the field of testing of mitochondrial toxicity. Mitochondrial toxicity is a major contributor to organ toxicity, such as toxicity in liver, kidney, heart, muscle, and the central nervous system (Dykins and Will 2008). Acute effects can be measured rapidly in 96-well formats by using isolated mitochondrial and soluble-oxygen sensor technology (Luxcel Biosciences, Cork, Ireland) or cell-based assays that evaluate mitochondrial respiration and glycolysis (Seahorse Biosciences, Bellerica, MA) (Will and Dykens 2014). Those assays potentially can be multiplexed with readouts of mitochondrial membrane potential dissipation or cytotoxicity (ATP content), and incubation times can be tailored to be from 1–24 hours after drug exposure (Porceddu et al. 2012). In the absence of pharmacokinetic data, most biochemical and cell-based assays remain primarily ranking tools for hazard identification, not predictors of true risk.
Some in vitro assays have been sufficiently validated that they can largely replace animal testing. For example, the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee endorsed the EPISKIN test and the EpiDerm method as scientifically valid replacements in a tiered testing strategy for the rabbit skin irritation method and for identifying skin irritants, respectively (Spielmann et al. 2007). In addition, the use of the Cultex® Radial Flow System to assess acute pulmonary toxicity of fine dusts and nanoparticles could possibly be adapted to test for chemicals that might be relevant to DOD (Steinritz et al. 2013). In vivo–in vitro comparison of acute respiratory tract toxicity using human 3-D airway epithelial models and human A549 and murine 3T3 monolayer cell systems has also been reported (Sauer et al. 2013).
BOX 4-1 ACuteTox Testing Strategy
The goal of the EU ACuteTox project was to evaluate whether regulatory animal tests for acute systemic toxicity could be replaced with a combination of in vitro assays. The ACuteTox program assessed the correlation of concentrations for in vitro activity with effective doses derived from whole-animal studies and evaluated a series of assays and physicochemical properties to determine how well they predicted in vivo acute systemic toxicity. On the basis of statistical analysis, eight test methods were found to be promising for inclusion in the testing strategy and, therefore, selected for participation in the pre-validation study:
- The neutral red uptake assay that uses the 3T3 fibroblast cell line (3T3/NRU).
- The cytokine release assay that uses human whole blood (IL-1, IL-6, and TNF-α).
- Inhibition of colony-forming-unit efficiency in human cord blood–derived cells stimulated ith CFU-GM CBC/CFU-GM
- Gene expression (GFAP, HSP-32, MBP, and NF-H) and uridine incorporation measuring total mRNA synthesis in primary rat brain aggregate cultures.
- A panel that measures oxidative stress (intracellular peroxidative activity, intracellular concentrations of superoxide anion, and oxidized DNA base 8-oxoguanine) and cytotoxicity screening (intracellular Ca2+ concentrations, mitochondrial membrane potential, and plasma membrane potential) in HepG2, SH-SY5Y, and A.704 cells.
- The MTT assay that uses primary rat hepatocytes.
- Kinetic parameters (volume of distribution, protein binding, clearance, and oral absorption using Caco-2 cells and neuronal networks) for estimating the oral dose on the basis of the effective concentration observed in vitro.
- Estimation of chemical passage through the blood–brain barrier using neuronal networks (for neurotoxicity assays).
Sources: ACuteTox (2010); Combes et al. (2006).
In summary, evaluations of in vitro assays for predicting acute toxicity have focused on nonmechanistic indicators of toxicity, such as cytotoxicity assays, or low-throughput measurements. Few in vitro assays have been developed with quantitative linkage to any phenotype (acute or chronic). Screening for acute toxicity by using mechanisms that are likely to cause debilitating injury (Table 2-1) will require assays that are purposefully selected for their biochemical targets and characterized for their potential value in predicting human toxicity.
Assay Considerations: Lessons Learned from High-Throughput Screening Programs
The recent investment in EPA’s ToxCast program and the broader Tox21 Initiative has allowed important progress in the development and use of HTS platforms to assess biological activity and potential mechanisms of action for industrial and environmental chemicals. Such programs provide rapid screening of hundreds of chemicals for dozens of cellular targets and relevant pathways. However, some chemical, cellular, and assay conditions need to be considered if one is to use and interpret the data appropriately.
First, for various reasons, the nominal chemical concentration added to the assay well is not necessarily representative of the concentration at which chemical bioactivity is observed (Groothuis et al. 2015). Chemical purity must be confirmed and solubility in the assay medium checked to determine that the initial applied concentration is accurately known. Chemical stability also needs to be monitored over the course of the assay so that chemical stability and availability can be tracked. Labile chemicals can degrade rapidly on exposure to light, aqueous conditions, or other constituents of the media or in vitro system. In fact, MacArthur et al. (2009) conducted chemical stability studies with cytochrome P450 assays at NCGC and found decreased chemical potency over time and lower efficacy of older samples stored in dimethyl sulfoxide (DMSO). For that reason, test chemicals at NCGC are used for no longer than 4–6 months. If degradation does occur, assay bioactivity (or lack thereof) might be inaccurately attributed to the parent chemical. Alternatively, the chemical might bind to plastics, cellular constituents, or proteins in the in vitro system and render it unavailable to elicit any effect in the test system (Blaauboer 2010).
Second, there are limitations of current cellular systems. The cells used in HTS assays typically are selected for their proliferative capacity, adherent properties, and ease of growth in high-throughput plates and systems (Shukla et al. 2010). Immortalized cancer cell lines—such as MCF-7 (breast cancer), A549 (lung cancer), and HepG2 (liver cancer)—are commonly used. It is possible—or perhaps likely—that assessments in the limited cellular space might fail to detect chemical activities or effects that might occur in normal (nontumor) differentiated cells. In addition, proliferative cell lines have a reduced ability to metabolize parent chemicals. To address those issues, new hepatic cell lines are being developed to be more metabolically competent and are discussed further in the next paragraph.
Third, assay reproducibility can be an issue. Chemical autofluorescence and cytotoxicity are common causes of assay interference that can lead to false positives or false negatives (Huang et al. 2011). Furthermore, cells can have different levels of activity or responsiveness, depending on whether they are primary cells, differentiated cells, or immortalized cells and on how many times they have been passaged.10 Variability in metabolic capabilities among various sources of isolated primary hepatocytes is well documented and is due to numerous factors, including isolation issues and donor variability. Recently, the HepaRG cell line has been introduced as a hepatic cell line that has a degree of metabolic competence and is amenable to use in a 96-well high-throughput system (Guillouzo et al. 2007). However, maintenance of the HepaRG cells in a differentiated state requires the use of high concentrations of DMSO, which has substantial cellular effects, including inhibition of metabolizing enzymes of the cytochrome P450 family and alteration of membrane
__________________
10Cultured cells are routinely transferred and replated (subcultured) to avoid the senescence associated with high cell density. Passage number refers to the number of times that the cells have been subcultured.
permeability and antioxidant status. Whereas the HepaRG cells are recognized as metabolically competent, the competence is on a much lower scale than that of fresh or cryopreserved primary hepatocytes (Kanebratt and Andersson 2008; Lubberstedt et al. 2011).
Fourth, interpretation of activity or effective concentrations from HTS assays should be carefully considered. The concentration at which bioactivity is observed should be considered in the context of the complete activity profile among all assays tested and the range of the dose-response relationships. Review of the ToxCast data has revealed that a burst of activity in many assays might result at concentrations close to or approaching cytotoxicity (EPA 2014b). Activity measurements at high concentrations probably represent nonspecific effects and offer little information about specific bioactivity. Likewise, a lack of response can be due to tested concentrations below bioactivity, lack of representation of the biological target, or assay unreliability. The finding that cytotoxicity of some chemicals varies with the cell type emphasizes the need to characterize biological activity over a broad concentration range (Xia et al. 2008).
Fifth, assays should always include positive-control reference chemicals, whose activity can be used to determine assay variability, sensitivity, and specificity. Some of those considerations were discussed by Judson et al. (2013) and Patlewicz et al. (2013). Indeed, efforts to characterize and document nonguideline in vitro assays, including high-throughput and high-content assays, have been made under the auspices of OECD, which has recently published a guidance document (OECD 2014).
- Finding: On the basis of the results of HTS programs, in vitro assays have demonstrated some predictive value for acute toxicity; therefore, an in vitro screening approach for predicting the potential for chemicals to cause acute, debilitating injuries is theoretically feasible.
- Finding: Current assays were not developed for predictions of acute toxicity (particularly lethality) and have generally not dealt with chemicals that are acutely toxic or debilitating. The few evaluations of in vitro assays to predict acute toxicity have focused primarily on nonmechanistic indicators of toxicity, such as cytotoxicity assays, or on low-throughput measurements.
- Finding: There is little evidence that results of in vitro assays are predictive of in vivo outcomes of concern to DOD. A screening program for acute toxicity will require the development of new in vitro assays that are mechanistically relevant to critical processes or pathways that are related to acute, debilitating toxicity.
- Finding: Evaluations of in vitro assays have focused on oral and intravenous exposure. The evaluation and development of in vitro assays that address dermal and inhalation exposures and contact toxicity will require additional research to understand absorption and permeability in the skin and lung.
- Finding: Most in vitro assays do not account for important pharmacokinetic characteristics, such as metabolism, that can influence in vivo toxicity. Although in vitro assays lacking metabolic capacity can effectively screen for biological activity of the parent chemical, the pharmacokinetic relationship between exposure and concentration at a target site needs to be addressed.
- Finding: In vitro testing and screening programs (Tox21, ToxCast, and ACuteTox) offer a number of useful lessons regarding assay reliability, chemical solubility or purity, standardized reference chemicals, dose–response experimental designs, and standardized data processing.
- Recommendation: The experience of HTS programs should be considered in the design of an in vitro screening program to predict acute, debilitating toxicity. Because of the potential need to include highly toxic agents, if only as reference chemicals, such a screening program will need to consider health, safety, and environmental issues associated with handling highly toxic and threat agents.
Abdo, N., M. Xia, C.C. Brown, O. Kosyk, R. Huang, S. Sakamuru, Y.H. Zhou, J. Jack, P. Gallins, K. Xia, Y. Li, W.A. Chiu, A. Motsinger-Reif, C.P. Austin, R.R. Tice, I. Rusyn, and F.A. Wright. 2015. Population-based in vitro hazard and concentration-response assessment of chemicals: The 1000 Genomes High-Throughput Screening Study. Environ. Health Perspect. 125(5):458-466.
ACuteTox. 2010. ACuteTox: Prevalidation, Highlights and Main Achievements of the Project. News Letter [online]. Available: http://www.acutetox.eu/Final_newsletter_Oct_2010.pdf [accessed May 7, 2015].
Alépée, N., S. Bessou-Touya, J. Cotovio, A. de Smedt, B. de Wever, C. Faller, P. Jones, B. Le Varlet, M. Marrec-Fairley, U. Pfannenbecker, M. Tailhardat, F. van Goethem, and P. McNamee. 2013. Cosmetics Europe multi-laboratory pre-validation of the SkinEthic reconstituted human corneal epithelium test method for the prediction of eye irritation. Toxicol. In Vitro 27(5):1476-1488.
Alépée, N., A. Bahinski, M. Daneshian, B. De Wever, E. Fritsche, A. Goldberg, J. Hansmann, T. Hartung, J. Haycock, H. Hogberg, L. Hoelting, J.M. Kelm, S. Kadereit, E. McVey, R. Landsiedel, M. Leist, M. Lubberstedt, F. Noor, C. Pellevoisin, D. Petersohn, U. Pfannenbecker, K. Reisinger, T. Ramirez, B. Rothen-Rutishauser, M. Schafer-Korting, K. Zeilinger, and M.G. Zurich. 2014. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. ALTEX 31(4):441-477.
Ali, S., D.L. Champagne, and M.K. Richardson. 2012. Behavioral profiling of zebrafish embryos exposed to a panel of 60 water-soluble compounds. Behav. Brain Res. 228(2):272-283.
Attene-Ramos, M.S., N. Miller, R. Huang, S. Michael, M. Itkin, R.J. Kavlock, C.P. Austin, P. Shinn, A. Simeonov, R.R. Tice, and M. Xia. 2013. The Tox21 robotic platform for the assessment of environmental chemicals - from vision to reality. Drug Discov. Today 18(15-16):716-723.
Austin, C., R. Kavlock., and R. Tice. 2008. Tox21: Putting a Lens on the Vision of Toxicity Testing in the 21st Century [online]. Available: http://alttox.org/tox21-putting-a-lens-on-the-vision-of-toxicitytesting-in-the-21st-century/ [accessed March 19, 2014].
Avila, D., K. Helmcke, and M. Aschner. 2012. The Caenorhabiditis elegans model as a reliable tool in neurotoxicology. Hum. Exp. Toxicol. 1(3):236-243.
Barbazuk, W.B., I. Korf, C. Kadavi, J. Heyen, S. Tate, E. Wun, J.A. Bedell, J.D. McPherson, and S.L. Johnson. 2000. The syntenic relationship of the zebrafish and human genomes. Genome Res. 10(9):1351-1358.
Basketter, D., D. Jírova, and H. Kandárová. 2012. Review of skin irritation/corrosion hazards on the basis of human data: A regulatory perspective. Interdiscip. Toxicol. 5(2):98-104.
Bichara, D., N.B. Calcaterra, S. Arranz, P. Armas, and S.H. Simonetta. 2014. Set-up of an infrared fast behavioral assay using zebrafish (Danio rerio) larvae, and its application in compound biotoxicity screening. J. Appl. Toxicol. 4(2):214-219.
Bier, E. 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 6(1):9-23.
Blaauboer, B.J. 2010. Biokinetic modeling and in vitro-in vivo extrapolations. Toxicol. Environ. Health B Crit. Rev. 13(2-4):242-252.
Burns, C.G., D.J. Milan, E.J. Grande, W. Rottbauer, C.A. MacRae, and M.C. Fishman. 2005. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat. Chem. Biol. 1(5):263-264.
C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 282(5396):2012-2018.
Chakravarthy, S., S. Sadagopan, A. Nair, and S.K. Sukumaran. 2014. Zebrafish as an in vivo high-throughput model for genotoxicity. Zebrafish 11(2):154-166.
Chen, M., M. Zhang, J. Borlak, and W. Tong. 2012. A decade of toxicogenomic research and its contribution to toxicological science. Toxicol. Sci. 130(2):217-228.
Chen, N., T.W. Harris, I. Antoshechkin, C. Bastiani, T. Bieri, D. Blasiar, K. Bradnam, P. Canaran, J. Chan, C.K. Chen, W.J. Chen, F. Cunningham, P. Davis, E. Kenny, R. Kishore, D. Lawson, R. Lee, H.M. Muller, C. Nakamura, S. Pai, P. Ozersky, A. Petcherski, A. Rogers, A. Sabo, E.M. Schwarz, K. Van Auken, Q. Wang, R. Durbin, J. Spieth, P.W. Sternberg, and L.D. Stein. 2005. WormBase: A comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 33(Database Issue):D383-D389.
Cho, A., J. Shin, S. Hwang, C. Kim, H. Shim, H. Kim, H. Kim, and I. Lee. 2014. WormNet v3: A network-assisted hypothesis-generating server for Caenorhabditis elegans. Nucleic Acids Res. 42(Web Server issue):W76-W82.
Clemedson, C. 2008. The European ACuteTox project: A modern integrative in vitro approach to better prediction of acute toxicity. Clin. Pharmacol. Ther. 84(2):200-202.
Clemedson, C., B. Blaauboer, J.V. Castell, P. Prieto, L. Risteli, J.A, Vericat, and A. Wendel. 2006. ACuteTox – Optimation and pre-validation of an in-vitro test strategy for predicting human acute toxicity. ALTEX 23(Suppl.):254-258.
Clemedson, C., A. Kolman, and A. Forsby. 2007. The integrated acute systemic toxicity project (ACuteTox) for the optimisation and validation of alternative in vitro tests. ATLA 35(1):33-38.
Combes, R., S. Coecke, M. Jacobs, E. Brandon, U. Bernauer, and W. Janssens. 2006. The Use of Metabolizing Systems for in Vitro Testing of Endocrine Disruptors [online]. Available: http://www.oecd.org/chemicalsafety/testing/36280446.pdf [accessed May 7, 2015].
Cotovio, J., M.H. Grandidier, P. Portes, R. Roguet, and G. Rubinstenn. 2005. The in vitro skin irritation of chemicals: Optimisation of the EPISKIN prediction model within the framework of the ECVAM validation process. ATLA 33(4):329-349.
Cotovio, J., M. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, and J. Leclaire. 2008. In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy: Results and performances with 184 cosmetic ingredients. AATEX 14(Special Issue):351-358 [online]. Available: http://altweb.jhsph.edu/wc6/paper351.pdf [accessed March 19, 2015].
de Esch, C., R. Slieker, A. Wolterbeek, R. Woutersen, and D. de Groot. 2012. Zebrafish as potential model for developmental neurotoxicity testing: A mini review. Neurotoxicol. Teratol. 34(6):545-553.
Dix, D.J., K.A. Houck, M.T. Martin, A.M. Richard, R.W. Setzer, and R.J. Kavlock. 2007. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 95(1):5-12.
Donato, M.T, A. Lahoz, J.V. Castell, and M.J. Gomez-Lechon. 2008. Cell lines: A tool for in vitro drug metabolism studies. Curr. Drug Metab. 9(1):1-11.
Driessen, M., A.S. Kienhuis, J.L. Pennings, T.E. Pronk, E.J. van de Brandhof, M. Roodbergen, H.P. Spaink, B. van de Water, and L.T. van der Ven. 2013. Exploring the zebrafish embryo as an alternative model for the evaluation of liver toxicity by histopathology and expression profiling. Arch. Toxicol. 87(5):807-823.
Drysdale, R. 2008. FlyBase: A database for the Drosophila research community. Methods Mol. Biol. 420:45-59.
Duan, Q., C. Flynn, M. Niepel, M. Hafner, J.L. Muhlich, N.F. Fernandez, A.D. Rouillard, C.M. Tan, E.Y. Chen, T.R. Golub, P.K. Sorger, A. Subramanian, and A. Ma’ayan. 2014. LINCS Canvas Browser: Interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 42(Web Server Issue):W449-W460.
Dykens, J.A., and Y. Will. 2008. Drug-Induced Mitochondrial Dysfunction. Hoboken, NJ: Wiley.
Ekwall, B. 1983. Screening of toxic compounds in mammalian cell cultures. Ann. N.Y. Acad. Sci. 407:64-77.
EPA (US Environmental Protection Agency). 2003. Framework for Computational Toxicology Research Program in ORD. EPA/600/R-03/065. Office of Research and Development, US Environmental Protection Agency, Washington, DC [online]. Available: http://epa.gov/comptox/download_files/basic_information/comptoxframework06_02_04.pdf [accessed May 6, 2015].
EPA (US Environmental Protection Agency). 2013. Organotypic Culture Models for Predictive Toxicology Center: Synopsis of Program [online]. Available: http://epa.gov/ncer/rfa/2013/2013_star_ocm.html [accessed March 19, 2015].
EPA (US Environmental Protection Agency). 2014a. ToxCast Assay Annotation Version 1.0 Data User Guide. National Center for Computational Toxicology, US Environmental Protection Agency, Research Triangle Park, NC. October 2014 [online]. Available: http://epa.gov/ncct/toxcast/files/ToxCast%20Assays/ToxCast_Assay_Annotation_Data_Users_Guide_20141021.pdf [accessed March 19, 2015].
EPA (US Environmental Protection Agency). 2014b. Meeting on Scientific Issues Associated with Integrated Endocrine Bioactivity and Exposure based Prioritization and Screening, December 2-5, 2014. FIFRA Scientific Advisory Panel, US Environmental Protection Agency [online]. Available: http://www.epa.gov/scipoly/sap/meetings/2014/december/120214transcript.pdf [accessed March 26, 2015].
Esch, M.B., T.L. King, and M.L. Shuler. 2011. The role of body-on-a-chip devices in drug and toxicity studies. Annu. Rev. Biomed. Eng. 13:55-72.
Esch, M.B., J.M. Prot, Y.I. Wang, P. Miller, J.R. Llamas-Vidales, B.A. Naughton, D.R. Applegate, and M.L. Shuler. 2015. Multi-cellular 3D human primary liver cell culture elevates metabolic activity under fluidic flow. Lab Chip. 15(10):2269-2277.
Fabian, G., N. Farago, L.Z. Feher, L.I. Nagy, S. Kulin, K. Kitajka, T. Bito, V. Tubak, R.L. Katona, L. Tiszlavicz, and L.G. Puskas. 2011. High-density real-time PCR-based in vivo toxicogenomic screen to predict organ-specific toxicity. Int. J. Mol. Sci. 12(9):6116-6134.
Feng, Z., C.J. Cronin, J.H. Wittig, Jr., P.W. Sternberg, and W.R. Schafer. 2004. An imaging system for standardized quantitative analysis of C. elegans behavior. BMC Bioinformatics 5:115.
Forsby, A., A.K. Bal-Price, A. Camins, S. Coecke, N. Fabre, H. Gustafsson, P. Honegger, A. Kinsner-Ovaskainen, M. Pallas, V. Rimbau, E. Rodríguez-Farré, C. Suñol, J.A. Vericat, and M.G. Zurich. 2009. Neuronal in vitro models for the estimation of acute systemic toxicity. Toxicol. In Vitro 23(8):1564-1569.
Fu, Q., F.S. Schoenhoff, W.J. Savage, P. Zhang, and J.E. Van Eyk. 2010. Multiplex assays for biomarker research and clinical application: Translational science coming of age. Proteomics Clin. Appl. 4(3):271-284.
Giacomotto., J, and L. Ségalat. 2010. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 160(2):204-216.
Glatt, H., R. Padykula, G.A. Berchtold, G. Ludewig, K.L. Platt, J. Klein, and Oesch. 1989. Multiple activation pathways of benzene leading to products with varying genotoxic characteristics. Environ. Health Perspect. 82:81-89.
Godoy, P., N.J. Hewitt, U. Albrecht, M.E. Andersen, N. Ansari, S. Bhattacharya, J.G. Bode, J. Bolleyn, C. Borner, J. Böttger, A. Braeuning, R.A. Budinsky, B. Burkhardt, N.R. Cameron, G. Camussi, C.S. Cho, Y.J. Choi, J.C. Rowlands, U. Dahmen, G. Damm, O. Dirsch, M.T. Donato, J. Dong, S. Dooley, D. Drasdo, R. Eakins, K.S. Ferreira, V. Fonsato, J. Fraczek, R. Gebhardt, A. Gibson, M. Glanemann, C.E.P. Goldring, M.J. Gómez-Lechón, G.M.M. Groothuis, L. Gustavsson, C. Guyot, D. Hallifax, S. Hammad, A. Hayward, D. Häussinger, C. Hellerbrand, P. Hewitt, S. Hoehme, H.G. Holzhütter, J.B. Houston, J. Hrach, K. Ito, H. Jaeschke, V. Keitel, J.M. Kelm, B.K. Park, C. Kordes, G.A. Kullak-Ublick, E.L. LeCluyse, P. Lu, J. Luebke-Wheeler, A. Lutz, D.J. Maltman, M. Matz-Soja, P. McMullen, I. Merfort, S. Messner, C. Meyer, J. Mwinyi, D.J. Naisbitt, A.K. Nussler, P. Olinga, F. Pampaloni, J. Pi, L. Pluta, S.A. Przyborski, A. Ramachandran, V. Rogiers, C. Rowe, C. Schelcher, K. Schmich, M. Schwarz, B. Singh, E.H.K. Stelzer, B. Stieger, R. Stöber, Y. Sugiyama, C. Tetta, W.E. Thasler, T. Vanhaecke, M. Vinken, T.S. Weiss, A. Widera, C.G. Woods, J. J. Xu, K.M. Yarborough, and J.G. Hengstler. 2013. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 87(8):1315-1530.
Gómez-Lechón, M.J., L. Tolosa, J.V. Castell, and M.T. Donato. 2010. Mechanism-based selection of compounds for the development of innovative in vitro approaches to hepatotoxicity studies in the IIINTOP project. Toxicol. In Vitro 24(7):1879-1889.
Gregory, J.M., T.P. Barros, S. Meehan, C.M. Dobson, and L.M. Luheshi. 2012. The aggregation and neurotoxicity of TDP-43 and its ALS-associated 25 kDa fragment are differentially affected by molecular chaperones in Drosophila. PLoS One 7(2):e31899.
Groothuis, F.A., M.B. Heringa, B. Nicol, J.L. Hermens, B.J. Blaauboer, and N.I. Kramer. 2015. Dose metric considerations in in vitro assays to improve quantitative in vitro-in vivo dose extrapolations. Toxicology 332(5):30-40.
Guillouzo, A., A. Corlu, C. Aninat, D. Glaise, F. Morel, and C. Guguen-Guillouzo. 2007. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 168(1):66-73.
Halle, W. 2003. The Registry of Cytotoxicity: Toxicity testing in cell cultures to predict acute toxicity (LD50) and to reduce testing in animals. ATLA 31(2):89-198.
Harris, T.W., N. Chen, F. Cunningham, M. Tello-Ruiz, I. Antoshechkin, C. Bastiani, T. Bieri, D. Blasiar, K. Bradnam, J. Chan, C.K. Chan, W.J. Chen, P. Davis, E. Kenny, R. Kishore, D. Lawson, R. Lee, H.M. Muller, C. Nakamura, P. Ozersky, A. Petcherski, A. Rogers, A. Sabo, E.M. Schwarz, K. Van Auken, Q. Wang, R. Durbin, J. Spieth, P.W. Sternberg, and L.D. Stein. 2004. WormBase: A multi-species resource for nematode biology and genomics. Nucleic Acids Res. 32(Database Issue):D411-D417.
Hentschel, D.M., K.M. Park, L. Cilenti, A.S. Zervos, I. Drummond, and J.V. Bonventre. 2005. Acute renal failure in zebrafish: A novel system to study a complex disease. Am. J. Physiol. Renal. Physiol. 288(5):F923-F929.
Honegger, P., F. Monnet-Tschudi, and M.G. Zurich. 2009. Brain Aggregate Cell Cultures as an In Vitro Model for Neurotoxicity Testing [online]. Available: http://alttox.org/brain-aggregate-cell-culturesas-an-in-vitro-model-for-neurotoxicity-testing/ [accessed March 19, 2015].
Houck, K.A., D.J. Dix, R.S. Judson, R.J. Kavlock, J. Yang, and E.L. Berg. 2009. Profiling bioactivity of the ToxCast chemical library using BioMAP primary human cell systems. J. Biomol. Screen. 14(9): 1054-1066.
Howe, K, M.D. Clark, C.F. Torroja, J. Torrance, C. Berthelot, M. Muffato, J.E. Collins, S. Humphray, K. McLaren, L. Matthews, S. McLaren, L. Sealy, M. Caccamo, C. Churcher, C. Scott, J.C. Barrett, R. Koch, G.J. Rauch, S. White, W. Chow, B. Kilian, L.T. Quintais, J.A. Guerra-Assunção, Y. Zhou, Y. Gu, J. Yen, J.H. Vogel, T. Eyre, S. Redmond, R. Banerjee, J. Chi, B. Fu, E. Langley, S.F. Maguire, G.K. Laird, D. Lloyd, E. Kenyon, S. Donaldson, H. Sehra, J. Almeida-King, J. Loveland, S. Trevanion, M. Jones, M. Quail, D. Willey, A. Hunt, J. Burton, S. Sims, K. McLay, B. Plumb, J. Davis, C. Clee, K. Oliver, R. Clark, C. Riddle, D. Elliot, G. Threadgold, G. Harden, D. Ware, S. Begum, B. Mortimore, G. Kerry, P. Heath, B. Phillimore, A. Tracey, N. Corby, M. Dunn, C. Johnson, J. Wood, S. Clark, S. Pelan, G. Griffiths, M. Smith, R. Glithero, P. Howden, N. Barker, C. Lloyd, C. Stevens, J. Harley, K. Holt, G. Panagiotidis, J. Lovell, H. Beasley, C. Henderson, D. Gordon, K. Auger, D. Wrigh, J. Collins, C. Raisen, L. Dyer, K. Leung, I. Robertson, K. Ambridge, D. Leongamornlert, S. McGuire, R. Gilderthorp, C. Griffiths, D. Manthravadi, S. Nichol, G. Barker, S. Whitehead, M. Kay, J. Brown, C. Murnane, E. Gray, M. Humphries, N. Sycamore, D. Barker, D. Saunders, J. Wallis, A. Babbage, S. Hammond, M. Mashreghi-Mohammadi, L. Barr, S. Martin, P. Wray, A. Ellington, N. Matthews, M. Ellwood, R. Woodmansey, G. Clark, J. Cooper, A. Tromans, D. Grafham, C. Skuce, R. Pandian, R. Andrews, E. Harrison, A. Kimberley, J. Garnett, N. Fosker, R. Hall, P. Garner, D. Kelly, C. Bird, S. Palmer, I. Gehring, A. Berger, C.M. Dooley, Z. Ersan-Ürün, C. Eser, H. Geiger, M. Geisler, L. Karotki, A. Kirn, J. Konantz, M. Konantz, M. Oberländer, S. Rudolph-Geiger, M. Teucke, C. Lanz, G. Raddatz, K. Osoegawa, B. Zhu, A. Rapp, S. Widaa, C. Langford, F. Yang, S.C. Schuster, N.P. Carter, J. Harrow, Z. Ning, J. Herrero, S.M. Searle, A. Enright, R. Geisler, R.H. Plasterk, C. Lee, M. Westerfield, P.J. de Jong, L.I. Zon, J.H. Postlethwait, C. Nüsslein-Volhard, T.J. Hubbard, H. Roest Crollius, J. Rogers, and D.L. Stemple. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496(7446):498-503.
Huang, C.J., C.T. Tu, C.D. Hsiao, F.J. Hsieh, and H.J. Tsai. 2003. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev. Dyn. 228(1):30-40.
Huang, R., N. Southall, M.H. Cho, M. Xia, J. Inglese, and C.P. Austin. 2008. Characterization of diversity in toxicity mechanism using in vitro cytotoxicity assays in quantitative high throughput screening. Chem. Res. Toxicol. 21(3):659-667.
Huang, R., M. Xia, M.H. Cho, S. Sakamuru, P. Shinn, K.A. Houck, D.J. Dix, R.S. Judson, K.l. Witt, R.J. Kavlock, R.R. Tice, and C.P. Austin. 2011. Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ. Health Perspect. 119(8):1142-1148.
Huh, D., B.D. Matthews, A. Mammoto, M. Montoya-Zavala, H.Y. Hsin, and D.E. Ingber. 2010. Reconstituting organ-level lung functions on a chip. Science 328(5986):1662-1668.
Huh, D., D.C. Leslie, B.D. Matthews, J.P. Fraser, S. Jurek, G.A. Hamilton, K.S. Thorneloe, M.A. McAlexander, and D.E. Ingber. 2012. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 4(159):159ra147.
Jennings, P. 2015. The future of in vitro toxicology. Toxicol. In Vitro 29(6):1217-1221.
Judson, R.S., K.A. Houck, R.J. Kavlock, T.B. Knudsen, M.T. Martin, H.M. Mortensen, D.M. Reif, D.M. Rotroff, I. Shah, A.M. Richard, and D.J. Dix. 2010. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ. Health Perspect. 118(4):485-492.
Judson, R., R. Kavlock, M. Martin, D. Reif, K. Houck, T. Knudsen, A. Richard, R.R. Tice, M. Whelan, M. Xia, R. Huang, C. Austin, G. Daston, T. Hartung, J.R. Fowle III, W. Wooge, W. Tong, and D. Dix. 2013. Perspectives on validation of high-throughput assays supporting 21st century toxicity testing. ALTEX 30(1):51-56.
Kalgutkar, A.S., and D. Dalvie. 2014. Predicting toxicities of reactive metabolite-positive drug candidates. Annu. Rev. Pharmacol. Toxicol. 55:35-54.
Kanebratt, K.P., and T.B. Andersson. 2008. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 36(7):1444-1452.
Kavlock, R.J., C.P. Austin, and R.R. Tice. 2009. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal. 29(4):485-487.
Kavlock, R.J., K. Chandler, K. Houck, S. Hunter, R. Judson, N. Kleinstreuer, T. Knudsen, M. Martin, S. Padilla, D. Reif, A. Richard, D. Rotroff, N. Sipes, and D. Dix. 2012. Update on EPA’s ToxCast program: Providing high throughput decision support tools for chemical risk management. Chem. Res. Toxicol. 25(7):1287-1302.
Khetani, S.R., and S.N. Bhatia. 2008. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 26(1):120-126.
Khetani, S.R., C. Kanchagar, O. Ukairo, S. Krzyzewski, A. Moore, J. Shi, S. Aoyama, M. Aleo, and Y. Will. 2013. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol. Sci. 132(1):107-117.
Kinsner-Ovaskainen, A., P. Prieto, S. Stanzel, and A. Kopp-Schneider. 2013. Selection of test methods to be included in a testing strategy to predict acute oral toxicity: An approach based on statistical analysis of data collected in phase 1 of the ACuteTox project. Toxicol. In Vitro 27(4):1377-1394.
Klaper, R., D. Arndt, J. Bozich, and G. Dominguez. 2014. Molecular interactions of nanomaterials and organisms: Defining biomarkers for toxicity and high-throughput screening using traditional and next-generation sequencing approaches. Analyst. 139(5):882-895.
Kleinstreuer, N.C., J. Yang, E.L. Berg, T.B. Knudsen, A.M. Richard, M.T. Martin, D.M. Reif, R.S. Judson, M. Polokoff, D.J. Dix, R.J. Kavlock, and K.A. Houck. 2014. Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol. 32(6):583-591.
Knudsen, T.B., R.J. Kavlock, G.P. Daston, D. Stedman, M. Hixon, and J.H. Kim. 2011. Developmental toxicity testing for safety assessment: New approaches and technologies. Birth Defects Res. B Dev. Reprod. Toxicol. 92(5):413-420.
Kraushaar, U., T. Meyer, D. Hess, L. Gepstein, C.L. Mummery, S.R. Braam, and E. Guenther. 2012. Cardiac safety pharmacology: From human ether-a-gogo related gene channel block towards induced pluripotent stem cell based disease models. Expert Opin. Drug Saf. 11(2):285-298.
Krewski, D., M.E., Andersen, E. Mantus, and L. Zeise. 2009. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal. 29(4):474-479.
Lamb, J., E.D. Crawford, D. Peck, J.W. Modell, I.C. Blat, M.J. Wrobel, J. Lerner, J.P. Brunet, A. Subramanian, K.N. Ross, M. Reich, H. Hieronymus, G. Wei, S.A. Armstrong, S.J. Haggarty, P.A. Clemons, R. Wei, S.A. Carr, E.S. Lander, and T.R. Golub. 2006. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 313(5795):1929-1935.
Langheinrich, U., G. Vacun, and T. Wagner. 2003. Zebrafish embryos express an orthologue of HERG and are sensitive toward a range of QT-prolonging drugs inducing severe arrhythmia. Toxicol. Appl. Pharmacol. 193(3):370-382.
Larson, B., P. Banks, S. Shultz, M. Sobol, and J.J. Cali. 2011. The utility of semi-automating multiplexed assays for ADME/Tox applications. Comb. Chem. High Throughput Screen. 14(8):658-668.
Legradi, J., N. El Abdellaoui, M. van Pomeren, and J. Legler. In press. Comparability of behavioural assays using zebrafish larvae to assess neurotoxicity. Environ. Sci. Pollut. Res.
Leite, S.B., I. Wilk-Zasadna, J.M. Zaldivar, E. Airola, M.A. Reis-Fernandes, M. Mennecozzi, C. Guguen-Guillouzo, C. Chesne, C. Guillou, P.A. Alves, and S. Coecke. 2012. Three-dimensional HepaRG model as an attractive tool for toxicity testing. Toxicol. Sci. 130(1):106-116.
Lessman, C.A. 2011. The developing zebrafish (Danio rerio): A vertebrate model for high-throughput screening of chemical libraries. Birth Defects Res. Part C Embryo Today 93(3):268-280.
Limbourg, B., and M. Zalokar. 1973. Permeabilization of Drosophila eggs. Dev. Biol. 35(2):382-387.
Lin, Z., and Y. Will. 2012. Evaluation of drugs with specific organ toxicities in organ-specific cell lines. Toxicol. Sci. 126(1):114-127.
Love, D.R., F.B. Pichler, A. Dodd, B.R. Copp, and D.R. Greenwood. 2004. Technology for high-throughput screens: The present and future using zebrafish. Curr. Opin. Biotechnol. 15(6):564-571.
Lubberstedt, M., U. Muller-Vieira, M. Mayer, K.M. Biemel, F. Knospel, D. Knobeloch, A.K. Nussler, J.C. Gerlach, and K. Zeilinger. 2011. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J. Pharmacol. Toxicol. Methods 63(1):59-68.
MacArthur, R., W. Leister, H. Veith, P. Shinn, N. Southall, C.P. Austin, J. Inglese, and D.S. Auld. 2009. Monitoring compound integrity with cytochrome P450 assays and qHTS. J. Biomol. Screen 14(5):538-546.
Marroquin, L.D., J. Hynes, J.A. Dykens, J.D. Jamieson, and Y. Will. 2007. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 97(2):539-547.
Maschmeyer, I., T. Hasenberg, A. Jaenicke, M. Lindner, A.K. Lorenz, J. Zech, L.A. Garbe, F. Sonntag, P. Hayden, S. Ayehunie, R. Lauster, U. Marx, E.M. Materne. In press. Chip-based human liver-intestine and liver-skin co-cultures - A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm.
Mathur, A., P. Loskill, S. Hong, J. Lee, S.G. Marcus, L. Dumont, B.R. Conklin, H. Willenbring, L.P. Lee, and K.E. Healy. 2013. Human induced pluripotent stem cell-based microphysiological tissue models of myocardium and liver for drug development. Stem Cell Res. Ther. 4(Suppl. 1):S14.
Mazzio, E.A., Y.I. Soliman, and K.F. Soliman. 2010. Variable toxicological response to the loss of OXPHOS through 1-methyl-4-phenylpyridinium-induced mitochondrial damage and anoxia in diverse neural immortal cell lines. Cell Biol. Toxicol. 26(6):527-539.
McAlister, G.C., D.P. Nusinow, M.P. Jedrychowski, M. Wühr, E.L. Huttlin, B.K. Erickson, R. Rad, W. Haas, and S.P. Gygi. 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86(14):7150-7158.
McVey, K.A., J.A. Mink, I.B. Snapp, W.S. Timberlake, C.E. Todt, R. Negga, and V.A. Fitsanakis. 2012. Caenorhabditis elegans: An emerging model system for pesticide neurotoxicity. J. Environ. Anal. Toxicol. 2:125, doi: 10.4172/2161-0525.S4-003.
Melstrom, P.C., and P.L. Williams. 2007. Reversible AChE inhibitors in C. elegans vs. rats, mice. Biochem. Biophys. Res. Commun. 357(1):200-205.
Messner, S., I. Agarkova, W. Moritz, and J.M. Kelm. 2013. Multi-cell type human liver in microtissues for hepatotoxicity testing. Arch. Toxicol. 87(1):209-213.
Meyer, D., and P.L. Williams. 2014. Toxicity testing of neurotoxic pesticides in Caenorhabditis elegans. J. Toxicol. Environ. Health B Crit. Rev. 17(5):284-306.
Milan, D.J., T.A. Peterson, J.N. Ruskin, R.T. Peterson, and C.A. MacRae. 2003. Drugs that induce repolarization abnormalities cause bradycardia in zebrafish. Circulation.107(10):1355-1358.
NAS (National Academy of Sciences). 2014. The Potential of the Tissue Chip for Environmental Health Studies. Emerging Science For Environmental Health Decisions Newsletter Issue II [online]. Available: http://nas-sites.org/emergingscience/files/2013/05/tissue-chips-final.pdf [accessed March 20, 2015].
Nichols, C.D. 2006. Drosophila melanogaster neurobiology, neuropharmacology, and how the fly can inform central nervous system drug discovery. Pharmacol. Ther. 112(3):677-700.
NIEHS (National Institute of Environmental Health Sciences). 2001a. Report of the International Workshop on In Vitro Methods for Assessing Acute Systemic Toxicity. NIH Publication 01-4499. National Toxicology Program [online]. Available: http://ntp.niehs.nih.gov/pubhealth/evalatm/test-method-evaluations/acute-systemic-tox/in-vitro/wksp-rpt-recs/index.html#Complete-Report [accessed March 20, 2015].
NIEHS (National Institute of Environmental Health Sciences). 2001b. Guidance Document on Using In Vitro Data to Estimate In Vivo Starting Doses for Acute Toxicity Based on Recommendations from an International Workshop Organized by ICCVAM and NICEA TM. NIH Publication 01-4500. National Toxicology Program [online]. Available: http://ntp.niehs.nih.gov/pubhealth/evalatm/test-method-evaluations/acute-systemic-tox/in-vitro/guidance-document/index.html [accessed March 20, 2015].
NRC (National Research Council). 2007. Toxicity Testing in the 21st Century. Washington, DC: The National Academies Press.
NTP (National Toxicology Program). 2004. A National Toxicology Program for the 21stCentury: A Roadmap for the Future [online]. Available: https://ntp.niehs.nih.gov/ntp/about_ntp/ntpvision/ntproadmap_508.pdf [accessed March 23, 2015].
O’Brien, P. 2014. High-content analysis in toxicology: Screening substances for human toxicity potential, elucidating subcellular mechanisms and in vivo use as translational safety biomarkers. Basic Clin. Pharmacol. Toxicol. 115(1):4-17.
O’Brien, P.J., W. Irwin, D. Diaz, E. Howard-Cofield, C.M. Krejsa, M.R. Slaughter, B. Gao, N. Kaludercic, A. Angeline, P. Bernardi, P. Brain, and C. Hougham. 2006. High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening. Arch. Toxicol. 80(9):580-604.
OECD (Organization for Economic Co-operation and Development). 2013a. Test No. 210: Fish, Early-life Stage Toxicity Test. OECD Guidelines for the Testing of Chemicals, Section 2. Paris, France: OECD Publishing.
OECD (Organization for Economic Co-operation and Development). 2013b. Test No. 236: Fish Embryo Acute Toxicity (FET) Test. OECD Guidelines for the Testing of Chemicals, Section 2. Paris, France: OECD Publishing.
OECD (Organization for Economic Co-operation and Development). 2014. Guidance Document for Describing Non-guideline In Vitro Test Methods. Series on Testing and Assessment No. 211 [online]. Available: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2014)35&doclanguage=en [accessed March 24, 2015].
Pamies, D., T. Hartung, and H.T. Hogberg. 2014. Biological and medical applications of a brain-on-a-chip. Exp. Biol. Med. 239(9):1096-1107.
Patlewicz, G., T. Simon, K. Goyak, R.D. Phillips, J.C. Rowlands, S.D. Seidel, and R.A. Becker. 2013. Use and validation of HT/HC assays to support 21st century toxicity evaluations. Regul. Toxicol. Pharmacol. 65(2):259-268.
Perkins, E.J., G.T. Ankley, K.M. Crofton, N. Garcia-Reyero, C.A. LaLone, M.S. Johnson, J.E. Tietge, and D.L. Villeneuve. 2013. Current perspectives on the use of alternative species in human health and ecological hazard assessments. Environ. Health Perspect. 121(9):1002-1010.
Persson, M., A. Løye, M. Jacquet, N. Mow, A. Thougaard, T. Mow, and J. Hornberg. 2014. High-content analysis/screening for predictive toxicology: Application to hepatotoxicity and genotoxicity. Basic Clin. Pharmacol. Toxicol. 115(1):18-23.
Peterson, R.T., R. Nass, W.A. Boyd, J.H. Freedman, K. Dong, and T. Narahashi. 2008. Use of nonmammalian alternative models for neurotoxicological study. Neurotoxicology 29(3):546-555.
Pfannenbecker, U., S. Bessou-Touya, C. Faller, J. Harbell, T. Jacob, H. Raabe, M. Tailhardat, N. Alepee, A. De Smedt, B. De Wever, P. Jones, Y. Kaluzhny, B. Le Varlet, P. McNamee, M. Marrec-Fairley, and F. Van Goethem. 2013. Cosmetics Europe multi-laboratory pre-validation of the epiocular reconstituted human tissue test method for the prediction of eye irritation. Toxicol. In Vitro 27(2):619-626.
Podratz, J.L., N.P. Staff, D. Froemel, A. Wallner, F. Wabnig, A.J. Bieber, A. Tang, and A.J. Windebank. 2011. Drosophila melanogaster: A new model to study cisplatin-induced neurotoxicity. Neurobiol. Dis. 43(2):330-337.
Podratz, J.L., N.P. Staff, J.B. Boesche, N.J. Giorno, M.E. Hainy, S.A. Herring, M.T. Klennert, C. Milaster, S.E. Nowakowski, R.G. Krug II, Y. Peng, and A.J. Windebank. 2013. An automated climbing apparatus to measure chemotherapy-induced neurotoxicity in Drosophila melanogaster. Fly (Austin) 7(3):187-192.
Porceddu, M., N. Buron, C. Roussel, G. Labbe, B. Fromenty, and A. Borgne-Sanchez. 2012. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol. Sci. 129(2):332-345.
Potts, S.J., T.D. Johnson, F.A. Voelker, H. Lange, and G.D. Young. 2011. Multiplexed measurement of proteins in tissue in a clinical environment. Appl. Immunohistochem. Mol. Morphol. 19(6):494-498.
Raldúa, D., and B. Piña. 2014. In vivo zebrafish assays for analyzing drug toxicity. Expert Opin. Drug Metab. Toxicol. 10(5):685-697.
Rand, M.D. 2010. Drosophotoxicology: The growing potential for Drosophila in neurotoxicology. Neurotoxicol. Teratol. 32(1):74-83.
Ravindranath, A.C., N. Perualila-Tan, A. Kasim, G. Drakakis, S. Liggi, S.C. Brewerton, D. Mason, M.J. Bodkin, D.A. Evans, A. Bhagwat, W. Talloen, H.W. Göhlmann, Z. Shkedy, and A. Bender. 2015. Connecting gene expression data from connectivity map and in silico target predictions for small molecule mechanism-of-action analysis. Mol. BioSyst. 11(1):86-96.
Ruan, Q.L., J.J. Ju, Y.H. Li, R. Liu, Y.P. Pu, L.H. Yin, and D.Y. Wang. 2009. Evaluation of pesticide toxicities with differing mechanisms using Caenorhabditis elegans. J. Toxicol. Environ. Health A. 72(11-12):746-751.
Rubin, G.M., and E.B. Lewis. 2000. A brief history of Drosophila's contributions to genome research. Science 24(5461):2216-2218.
Sauer. U.G., S. Vogel, A. Hess, S.N. Kolle, L. Ma-Hock, B. van Ravenzwaay, and R. Landsiedel. 2013. In vivo-in vitro comparison of acute respiratory tract toxicity using human 3D airway epithelial models and human A549 and murine 3T3 monolayer cell systems. Toxicol. In Vitro 27(1):174-190.
Sauer, U.G., A. Aumann, L. Ma-Hock, R. Landsiedel, and W. Wohlleben. 2015. Influence of dispersive agent on nanomaterial agglomeration and implications for biological effects in vivo or in vitro. Toxicol. In Vitro 29(1):182-186.
Sawin-McCormack, E.P., M.B. Sokolowski, and A.R. Campos. 1995. Characterization and genetic analysis of Drosophila melanogaster photobehavior during larval development. J. Neurogenet. 10(2):119-135.
Segalat, L. 2007. Invertebrate animal models of diseases as screening tools in drug discovery. ACS Chem. Biol. 2(4):231-236.
Shukla, S.J., R. Huang, C.P. Austin, and M. Xia. 2010. The future of toxicity testing: A focus on in vitro methods using a quantitative high throughput screening platform. Drug Discov. Today 15(23-24):997-1007.
Simoneschi, D., F. Simoneschi, and N.E. Todd. 2014. Assessment of cardiotoxicity and effects of malathion on the early development of zebrafish (Danio rerio) using computer vision for heart rate quantification. Zebrafish 11(3):275-280.
Sipes, N.S., M.T. Martin, P. Kothiya, D.M. Reif, R.S. Judson, A.M. Richard, K.A. Houck, D.J. Dix, R.J. Kavlock, and T.B. Knudsen. 2013. Profiling 976 ToxCast chemicals across 331 enzymatic and receptor signaling assays. Chem. Res. Toxicol. 26(6):878-895.
Sokolowski, M.B. 2001. Drosophila: Genetics meets behaviour. Nat. Rev. Genet. 2(11):879-890.
Spielmann, H., E. Genschow, M. Leibsch, and W. Halle. 1999. Determination of the starting dose for acute oral toxicity (LD50) testing in the up and down procedure (UDP) from cytotoxicity data. ATLA 27(6):957-966.
Spielmann, H., S. Hoffmann, M. Liebsch, P. Botham, J.H. Fentem, C. Eskes, R. Roguet, J. Cotovio, T. Cole, A. Worth, J. Heylings, P. Jones, C. Robles, H. Kandarova, A. Gamer, M. Remmele, R. Curren, H. Raabe, A. Cockshott, I. Gerner, and V. Zuang. 2007. The ECVAM international validation study on in vitro tests for acute skin irritation: Report on the validity of the EPISKIN and EpiDerm assays and on the Skin Integrity Function Test. ATLA 35(6):559-601.
Steinritz, D., N. Möhle, C. Pohl, M. Papritz, B. Stenger, A. Schmidt, C.J. Kirkpatrick, H. Thiermann, R. Vogel, S. Hoffmann, and M. Aufderheide. 2013. Use of the Cultex® Radial Flow System as an in vitro exposure method to assess acute pulmonary toxicity of fine dusts and nanoparticles with special focus on the intra- and inter-laboratory reproducibility. Chem. Biol. Interact. 206(3):479-490.
Sung, J.H., B. Srinivasan, M.B. Esch, W.T. McLamb, C. Bernabini, M.L. Shuler, and J.J. Hickman. 2014. Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med. (Maywood) 239(9):1225-1239.
Takahashi, K., and S. Yamanaka. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4):663-676.
Taylor, K.L., N.J. Grant, N.D. Temperley, and E.E. Patton. 2010. Small molecule screening in zebrafish: An in vivo approach to identifying new chemical tools and drug leads. Cell Commun. Signal 8:11.
Thomas, R.S., M.B. Black, L. Li, E. Healy, T.M. Chu, W. Bao, M.E. Andersen, and R.D. Wolfinger. 2012. A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol. Sci. 128(2):398-417.
Tsang, M. 2010. Zebrafish: A tool for chemical screens. Birth Defects Res. Part C Embryo Today 90(3):185-192.
Villatte, F., V. Marcel, S. Estrada-Mondaca, and D. Fournier. 1998. Engineering sensitive acetylcholinesterase for detection of organophosphate and carbamate insecticides. Biosens. Bioelectron. 13(2):157-164.
Vliegenthart, A.D., C.S. Tucker, J. Del Pozo, and J.W. Dear. 2014. Zebrafish as model organisms for studying drug induced liver injury. Br. J. Clin. Pharmacol. 78(6):1217-1227.
Whitworth, A.J., P.D. Wes, and L.J. Pallanck. 2006. Drosophila models pioneer a new approach to drug discovery for Parkinson's disease. Drug Discov. Today 11(3-4):119-126.
Will, Y., and J. Dykens. 2014. Mitochondrial toxicity assessment in industry–a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 10(8):1061-1067.
Williams, P.L., and D.B. Dusenbery. 1990. A promising indicator of neurobehavioral toxicity using the nematode Caenorhabditis elegans and computer tracking. Toxicol. Ind. Health 6(3-4):425-440.
Wilmes, A., A. Limonciel, L. Aschauer, K. Moenks, C. Bielow, M.O. Leonard, J. Hamon, D. Carpi, S. Ruzek, A. Handler, O. Schmal, K. Herrgen, P. Bellwon, C. Burek, G.L. Truisi, P. Hewitt, E. Di Consiglio, E. Testai, B.J. Blaauboer, C. Guillou, C.G. Huber, A. Lukas, W. Pfaller, S.O. Mueller, F.Y. Bois, W. Dekant, and P. Jennings. 2013. Application of integrated transcriptomic, proteomic and metabolomic profiling for the delineation of mechanisms of drug induced cell stress. J. Proteomics. 79:180-194.
Wilmes, A., C. Bielow, C. Ranninger, P. Bellwon, L. Aschauer, A. Limonciel, H. Chassaigne, T. Kristl, S. Aiche, C.G. Huber, C. Guillou, P. Hewitt, M.O. Leonard, W. Dekant, F. Bois, and P. Jennings. In press. Mechanism of cisplatin proximal tubule toxicity revealed by integrating transcriptomics, proteomics, metabolomics and biokinetics. Toxicology In Vitro.
Wink, S., S. Hiemstra, S. Huppelschoten, E. Danen, M. Niemeijer, G. Hendriks, H. Vrieling, B. Herpers, and B. van de Water. 2014. Quantitative high content imaging of cellular adaptive stress response pathways in toxicity for chemical safety assessment. Chem. Res. Toxicol. 27(3):338-355.
Wu, T.S., J.J. Yang, F.Y. Yu, and B.H. Liu. 2012. Evaluation of nephrotoxic effects of mycotoxins, citrinin and patulin, on zebrafish (Danio rerio) embryos. Food Chem. Toxicol. 50(12):4398-4404.
Xia, M., R. Huang, K.L. Witt, N. Southall, J. Fostel, M.H. Cho, A. Jadhav, C.S. Smith, J. Inglese, C.J. Portier, R.R. Tice, and C.P. Austin. 2008. Compound cytotoxicity profiling using quantitative high-throughput screening. Environ. Health Perspect. 116(3):284-291.
Xu, J.J., P.V. Henstock, M.C. Dunn, A.R. Smith, J.R. Chabot, and D. de Graaf. 2008. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci. 105(1):97-105.
Yamamoto, N., K. Komori, K. Montagne, H. Matsui, H. Nakayama, S. Takeuchi, and Y. Sakai. 2011. Cytotoxicity evaluation of reactive metabolites using rat liver homogenate microsome-encapsulated alginate gel microbeads. J. Biosci. Bioenerg. 111(4):454-458.
Yen, J., S. Donerly, E.D. Levin, and E.A. Linney. 2011. Differential acetylcholinesterase inhibition of chlorpyrifos, diazinon and parathion in larval zebrafish. Neurotoxicol. Teratol. 33(6):735-741.





























