
4
Opportunities for Intervention and Prevention
Moderated by planning committee member Leann Birch of the University of Georgia, Session 3 featured speakers who discussed potential opportunities for intervention and prevention based on rapidly advancing knowledge about the role of epigenetics and other factors in the early origins of obesity. This chapter summarizes the Session 3 presentations and discussion.
Reiterating what Linda Adair and Caroline Relton had voiced earlier about the importance not only of maternal over-nutrition, but also maternal under-nutrition in increasing the risk of childhood obesity (see Chapter 3 for summaries of their presentations), Karen Lillycrop of the University of Southampton summarized animal and human data suggesting that both maternal over- and under-nutrition can cause long-term metabolic changes in offspring and that many such changes are associated with altered DNA methylation patterns. She echoed Andrea Baccarelli’s and others’ hope that in the future DNA methylation differences at birth can be used as predictive biomarkers of later adiposity. Additionally, she speculated on the possibility of identifying stable epigenetic markers that could be used to monitor the success of obesity interventions over the life course. But several obstacles will need to be overcome and questions answered before the use of predictive or monitoring epigenetic biomarkers becomes a clinical reality, she said. For example, are the markers causal? What other epigenetic changes are occurring besides methylation? Which markers can be changed with nutritional or other intervention, and when and how?
Kevin Grove of the Oregon National Primate Research Center called attention to placental function and its association with a wide range of downstream metabolic complications. Based on research using a nonhuman primate model, evidence indicates that regardless of maternal body mass index (BMI), a high-fat maternal diet during pregnancy results in increased inflammatory cytokine production in the placenta and, because cytokines cross the placental barrier, in the fetus as well. Grove suggested that fetal systemic inflammation might be a potential target for therapeutic intervention. A high-fat maternal diet during pregnancy, when maintained postnatally through weaning, also has significant downstream effects on offspring neurochemistry and behavior. Grove emphasized the importance of testing any proposed intervention before implementing it, whether it involves diet and exercise, surgery, or pharmacotherapy. Results from prepregnancy bariatric surgery studies in rodents and humans have yielded conflicting risk–benefit results.
The perinatal period is sensitive not only to dietary fats, but also to leptin, a hormone produced by adipocytes. Marie-France Hivert of the Harvard Medical School discussed the physiology of leptin and summarized findings from animal and human observational studies suggesting that leptin exposure during the perinatal period is associated not only with early life weight gain, but also with long-lasting changes in the hypothalamus and other tissues. Additional human data from Hivert’s laboratory group suggest that leptin levels in offspring are regulated, at least partially, by epigenetic changes in the placenta triggered by changes in maternal glucose levels.
According to Mark Vickers of the University of Auckland, leptin was one of the first obesity interventions tested in an animal model. When results suggested that administering leptin could stop overeating in animals on high-fat diets, there were calls to add leptin to infant formula. But the effects were also shown to depend on the nutritional status of the mother and to be sex-dependent. Vickers summarized results from mostly animal obesity intervention studies testing a range of strategies from leptin to exercise. In his opinion, it is reassuring that so many different animal models, from sheep to mice, demonstrated similar reversals in metabolic dysfunction resulting from early life interventions. But researchers need to gain a better understanding of short-term versus long-term trade-offs and how to apply results from animal studies to the clinic.
There was a great deal of discussion throughout the workshop about the human microbiome and how gut microbial metabolites can impact infant metabolism and risk of childhood obesity. William Nierman of the J. Craig Venter Institute provided an overview of the human microbiome and efforts over the past decade to increase awareness of its significance in human health. He emphasized that the microbiome’s metabolic capabilities supplement the host’s metabolic capabilities—for example, by digesting
complex carbohydrates—with consequences for cancer and other disease phenotypes.
Meredith Hullar of the Fred Hutchinson Cancer Research Center expanded the discussion of microbiome to epigenetics and observed that researchers have moved beyond simply reporting associations between the microbiome and host epigenetic patterning and are beginning to study how microbial metabolism actually mediates observed epigenetic changes. For example, many host genes associated with satiety are influenced by exposure to fatty acid metabolites of the gut microbiome. Although Hullar and others have found associations between the dominant microbial makeup of the gut microbiome and adult adiposity, it is difficult at this point to conjecture how the early life microbiome affects the risk of obesity. The field is limited, in her opinion, by small sample sizes and a lack of prospective studies.
While the focus of the workshop was obesity, several speakers advocated for thinking less about size, or BMI, and more about the metabolic dysregulation that accompanies or underlies obesity. It is especially helpful to think about metabolic dysregulation when thinking about the effect of obesity on the brain, said Antonio Convit of the Nathan Kline Institute and the New York University School of Medicine. Convit spoke about how metabolic dysregulation in adolescents leads to reduced cognition and reduced hippocampal volume, with insulin resistance being the primary driver. He suggested that retinal arterial width is a potentially useful biomarker for metabolic dysfunction in the brain and identified exercise and sleep, because of their known associations with insulin resistance and metabolic dysfunction, as two “easy public health handles” for obesity intervention in adolescents.
DEVELOPMENTAL PLASTICITY: SENSITIVE PERIODS AND RISK OF OBESITY1
Traditionally it has been widely accepted that genes determine phenotype and susceptibility to chronic diseases later in life. However, rates of obesity have risen dramatically over the past 20 years in both developed and developing countries, and they are projected to rise even higher by 2030. Such a rapid rise over such a short timeframe cannot be explained solely by genetic factors, said Karen Lillycrop of the University of Southampton. It suggests that the environment also plays a role. That role may be mediated, she suggested, through altered epigenetic regulation of genes. Lillycrop went
______________
1 This section summarizes information and opinions presented by Karen Lillycrop, Ph.D., University of Southampton, Southampton, United Kingdom.
on to discuss the effects of early life nutrition on the epigenome and how altered epigenomic processes might have an impact on risk of obesity.
Animal Model Evidence That Early Environment Can Induce Epigenetic and Phenotypic Changes
Increasing evidence suggests that a number of environmental factors, including nutrition, can affect the epigenome and that the epigenome seems most susceptible to those environmental factors during the prenatal, neonatal, and pubertal periods. Most of the evidence for this comes from work with animal models showing that maternal dietary and protein restriction, as well as maternal over-nutrition, can induce long-term metabolic changes within offspring. According to Lillycrop, both maternal under- and over-nutrition often induce similar metabolic changes, with both cases being associated with dyslipidemia, insulin resistance, and obesity in offspring and with the phenotypic changes in both cases being accompanied by changes in the methylation of key metabolic genes. For example, Lillycrop and her research team demonstrated that when rats were fed a protein-restricted diet during pregnancy, their offspring exhibited alterations in methylation of a range of genes, including a gene known as PPAR-alpha (peroxisome proliferated activated receptor alpha) that encodes for a nuclear receptor known to play a key role in lipid metabolism (Lillycrop et al., 2005, 2007). Specifically, the researchers observed decreased methylation in the promoter region of PPAR-alpha in the liver of protein-restricted offspring, compared to controls, with the decreased methylation being accompanied by an increased gene expression and increased levels of beta oxidation (i.e., the metabolic process controlled by the gene). The altered methylation appeared as early as embryonic day 18 and seemed to persist through adulthood, Lillycrop said.
It is not just under-nutrition that can induce that type of epigenetic and phenotypic change, Lillycrop said. Maternal over-nutrition can do the same. When she and her team fed pregnant rats a diet high in either saturated fat or fish oil, they observed an altered fatty acid composition in the livers of the offspring, specifically decreased levels of 22:6n-3 and 20:4n-6 (Hoile et al., 2013). Additionally, upon examination of methylation and expression of Fads2, a gene that encodes the rate-limiting enzyme in polyunsaturated fatty acid synthesis, they observed decreased expression in offspring from dams who had been fed a diet high in either saturated fats or fish oil, accompanied by an increased methylation within the promoter region of Fads2. Methylation at the –394 CpG site accounted for over 56 percent of the variation in Fads2 mRNA expression among individual offspring.
Increasing evidence suggests that epigenetic plasticity extends into early postnatal life, Lillycrop stated. For example, Plagemann et al. (2009)
showed that over-nutrition in early life, which they induced by rearing the rat pups in small litters, triggered rapid fat gain and obesity in offspring and that the rapid weight gain and obesity were accompanied by alterations in the methylation of CpGs within the promoter region of the appetite control POMC gene. Specifically, they observed hyper-methylation of CpG sites in two binding sequences, Sp1 and NF-kappaB, known to be important for the induction of POMC by leptin and insulin.
Lillycrop remarked that many other studies have shown that variations in micronutrient intake, during either pregnancy or the peri-pubertal period, can similarly induce changes in gene methylation. Additionally, she said other research indicates that variations in paternal diet can induce altered methylation of a variety of genes. The affected genes often play key roles in metabolism and appetite control, suggesting to Lillycrop that the observed methylation changes underpin the long-term changes that have been observed in offspring metabolism and disease risk.
Human Evidence That Early Environment Can Induce Epigenetic and Phenotypic Changes
A number of research groups have begun to examine whether the same phenomena being observed in animals are occurring in humans. Much of the focus is on the effects of the Dutch Hunger Winter, a period of severe food shortage in the Netherlands in 1944–1945, when daily energy intakes dropped from around 1,800 kilocalories to between 400 and 800 kilocalories. Those studies, Lillycrop said, have shown that individuals born to mothers exposed to famine during pregnancy have an increased risk of heart disease, diabetes, and obesity later in life. Researchers have also reported alterations to the methylation of a number of genes in individuals exposed in utero. For example, Tobi et al. (2009) found an increase in methylation of the IL10, GNASAS, IGF2, LEP, ABCA1, and MEG3 genes and a decrease in methylation of INSIGF. Interestingly, Lillycrop noted, those measurements were made 60 years after the famine, suggesting that variations in maternal diet induce a very persistent epigenetic change in offspring.
The Mismatch Hypothesis
Revisiting some of what Linda Adair had touched on during her presentation (see Chapter 3), Lillycrop elaborated on how the ability of early life nutrition to alter the epigenome has been suggested to be part of a normal adaptive process called developmental plasticity. That is, when an organism responds to its environment in early life, it adjusts its developmental path to produce a phenotype that confers a survival or fitness advantage
(Waddington, 1942). For instance, poor maternal nutrition might signal to the fetus that nutrients are scarce, in which case the fetus will adapt its metabolism by reducing energy demands, increasing its propensity to store fat, investing less in bone and muscle mass, and preparing for an uncertain life course. If that organism then finds itself in an environment where nutrients are indeed scarce, its metabolism will be matched, and it will be at low risk of metabolic disease. If, on the other hand, it finds itself in an environment where nutrients are abundant, its metabolism will be mismatched, it will have a propensity to store fat, and it will be at an increased risk for metabolic disease. This so-called mismatch between the pre- and postnatal environment, Lillycrop explained, has been suggested to account for some of the rapid rises in rates of obesity and diabetes in the developing world as populations move from rural to urban areas.
Biomarker Detection
Given increasing evidence from both animal and human studies suggesting that early life nutrition can alter the epigenome and that reset epigenetic markers persist and contribute to alterations in metabolism and disease risk, Lillycrop concluded that it should be possible to detect such changes early in life and use them to estimate metabolic capacity and disease risk. The challenge in humans, in her opinion, is limited tissue availability. Although there are several readily available tissues that could be accessed, including the umbilical cord, cord blood, placenta, buccal cells, and blood, she said that there is a real question as to whether methylation in those cell types adequately reflects methylation in more metabolically relevant cells. She speculated that if the environmental constraint (i.e., the constraint responsible for triggering the epigenetic change) occurred early enough in development, then perhaps yes, if all three germ layers were affected.
Lillycrop and her research team wanted to see if they could find methylation differences at birth in those peripheral tissues, specifically in cord tissue, that were associated with later adiposity (Godfrey et al., 2011). Using mother and offspring cohorts based in Southampton as their study population, they started their search by sequencing the promoter regions of genes known to play roles in adipogenesis in cord tissue. They found that methylation of CpG in the promoter region of the retinoid X receptor at birth was associated with percent fat mass at the age of 9 years in one cohort and at age 6 years in another cohort. Lillycrop interpreted the findings to mean that developmentally induced epigenetic markers might indeed make a significant contribution to later phenotype.
Ideally, in Lillycrop’s opinion, methylation markers could be used not just to predict later phenotype, but also to follow the progression of an altered phenotype through the life course and to monitor the effectiveness
of interventions. Again, such a marker would have to be present in tissues that are readily accessible, such as blood or buccal cells, and it would have to be relatively stable over time. Lillycrop observed that although DNA methylation was originally thought to be a very stable marker maintained through life, a number of recent studies have suggested that epigenetic markers may be more dynamically regulated than previously thought. She mentioned that there is research indicating how just an intense bout of exercise, or even a weekend visit to the city, can change the level of methylation. If that is the case, she said, then there might be limits to the use of epigenetic markers for following the progression of a phenotype through the life course.
To address the question of stability, particularly during childhood, Lillycrop and her research team started looking at methylation of genes in blood samples collected annually from children ages 5 to 14 years. Initially they looked at PGC-1a, which plays a key role in energy sensing and in the coordination of the metabolic response. They found that methylation of the CpGs in the promoter region of PGC-1a was stable over the timeframe of the study, suggesting that, at least for these sites, methylation is set early in life—that is, before the age of 5, and stably maintained thereafter despite the onset of puberty and variations in exercise and pollution.
Having found some stable CpG methylation sites, the researchers wanted to see if the CpG sites were associated with a phenotype. They found associations between methylation of four of the CpG sites in the promoter region of PGC-1a at 5 to 7 years and percent fat mass from 9 to 14 years, again in both boys and girls. In the case of PGC-1a, the magnitude of the effect was such that, for a 10 percent difference in methylation at 5 to 7 years, percent body fat at 9 to 14 years differed from between 6.3 to 12.5 percent.
But the question remains, are the observed alterations in methylation simply markers of adiposity, or do they actually play a role in the development of obesity? To begin to address that question, Lillycrop’s team has started looking at the effect of methylation of CpG sites on the function of PGC-1a. Thus far they have found that methylation of one of the CpG sites known to be associated with adiposity leads to a decrease in promoter activity of the gene. Additionally, they have found that methylation of the CpG site located at –783 altered the binding of the HOXB9/PBX1 heterodimer, a complex known to be important for adipogenesis and an important regulator of PGC-1a. These findings suggest to Lillycrop that methylation at these sites might have a functional consequence.
Summary
In closing, Lillycrop summarized by reiterating that growing evidence indicates that early life environment is an important determinant of later obesity risk, with two pathways: under-nutrition and over-nutrition. Evidence also suggests that early life environment can alter epigenetic markers and that the altered epigenetic markers appear to be an important contributor to later phenotype. Lillycrop suggested that it may be possible to detect such markers in early life and use them to identify individuals at increased risk of obesity and its sequelae.
But there is a lot to learn, she said. For instance, little is known about how either the under-nutrition or over-nutrition pathway leads to obesity or the epigenetic changes that occur and whether the epigenetic signatures are the same for both pathways. Lillycrop remarked that the epigenetic changes at play are likely not only methylation changes, but also noncoding RNA and histone changes. Moreover, it is not clear what factors drive the changes. Nutrition is obviously very important, Lillycrop said, but what other factors are operating? Finally, are there epigenetic markers that can be changed through intervention, and, if so, when and how?
Given that researchers are now in a position to begin to address these questions, either with current cohorts or new ones, Lillycrop expressed the hope that “we should then start to be able to stem the tide of this ever-increasing rise in rates of obesity.”
MATERNAL HEALTH AND DIET’S EFFECT ON OFFSPRING’S METABOLIC FUNCTIONING2
The value added by his work, Kevin Grove of the Oregon National Primate Research Center began, is use of a nonhuman primate model to examine the impact of maternal obesity on offspring energy balance and risk of obesity. The monkey model he and his team use involves feeding adult Japanese macaques either a healthy diet or a high-fat, high-carbohydrate Western-style diet (McCurdy et al., 2009). After several years, the monkeys are bred, and sibling offspring are studied as they progress through different metabolic complications. While Grove and his group also conduct rodent studies, they are especially interested in the uncontrolled components of human studies that can be controlled using a nonhuman primate model. Initially, Grove said, he thought that the variability would make it impossible to detect anything. Then he realized, “that’s the human nature of things.” And it’s actually what he likes about the model—that is, that he
______________
2 This section summarizes information and opinions presented by Kevin Grove, Ph.D., Oregon National Primate Research Center, Beaverton, Oregon, and Novo Nordisk, Seattle, Washington.
and his research team are able to look at not only phenotype by diet, but also phenotype by a distribution of metabolic outcomes (i.e., body weight, adiposity, insulin sensitivity, triglyceride levels, and so on).
Focus on the Placenta
In Grove’s opinion, placental function is one of the most important components to understanding why many metabolic, inflammation, and brain development complications exist. There is a lot of clinical data demonstrating that clinical conditions that result in hyperinsulinemia or hyperglycemia, such as gestational diabetes, also cause complications in the placenta (e.g., Ragavendra and Tarantal, 2001). By taking a step back, Grove said, one can ask, what is it about the diet itself that might be contributing to these placental complications? Given his background in cardiovascular disease, and that he and his team study the effect of lipids on cardiovascular disease all the time, Grove views the placenta as “just a large vascular organ.” It is no surprise to him that lipids known to cause inflammation and vascular dysfunction could have a direct impact on placental function, as elegantly demonstrated by Antonio Frias, a member of his research team at the Oregon Health and Science University (OHSU). Using traditional Doppler ultrasound techniques to measure placental blood flow during the early third trimester, Frias et al. (2012) demonstrated that regardless of whether a mother was obese or lean, a high-fat diet led to reductions in placental blood flow. The resulting placental insufficiency was great enough that experimentally inducing that level would cause negative outcomes. “So just being on the diet was a problem,” Grove said.
When Frias et al. (2011) extended their study to look at some of the histological effects of a high-fat diet, they were able to characterize infarctions and calcifications in the placental tissue. A pathologist not named in the presentation was asked to analyze the blinded histological samples thought that the high-fat diet placental tissues were from preeclampsic patients, a “scary” response, Grove said, given that they were from simply either overweight or obese animals who had been fed a high-fat diet. That said, while the functional abnormalities observed by Frias et al. (2011) were caused by diet alone, the effects are exacerbated by obesity, insulin resistance, and high triglyceridemia.
Frias is currently trying to design more precise diagnostics using contrast-enhanced MRI, according to Grove. The Doppler ultrasound methodology provides a measure of average outcome across the placenta. But a dysfunctional placenta has areas that look rather normal. Rather than grinding up the entire tissue and examining the average, Grove and his team wanted to find a way to identify regional placental blood differences. So far, they have been able to identify differences between cotyledons
(subdivisions of the placenta) and have found quite a bit of variability. According to Grove, the technique can be used to image even individual spiral arteries. Preliminary results from Frias’s contrast-enhanced magnetic resonance imaging (MRI) work indicate the presence of highly localized damage. So some areas are normal, others abnormal. While these differences are useful experimentally, Grove is unsure whether they will be useful clinically, except perhaps with very-high-risk patients. But, in his opinion, at least researchers can now start correlating outcomes with biomarkers.
Summarizing Frias’s data, Grove stated that increasing dietary lipids in the maternal diet can cause placental problems, such as increased cytokine production, and that the problems can be exacerbated by adding other “hits” such as maternal obesity and hypertriglycemia. Cytokines produced in the placenta are shunted into the developing fetus, where, based on what has been learned from experimental models, they can cause all sorts of pregnancy and developmental programming complications. Other changes to nutrients in the diet—for example, a change in the n-6/n-3 fatty acid ratio—can also be transmitted through the placenta to the fetus, with important developmental consequences. N-3, for example, is important for brain development. Grove reiterated, “It may all start with the placenta.” It is not just increased dietary lipids but all sorts of maternal health conditions that have been linked to placental dysfunction, including maternal obesity, inappropriate maternal pre-pregnancy weight and pregnancy weight gain, gestational diabetes, preterm delivery, and preeclampsia. In his opinion, focusing on the placenta may be a way to improve clinical outcomes.
Turning to the downstream metabolic effects on the offspring and touching on some of what Jacob Friedman had discussed during his presentation (see Chapter 3), Grove mentioned that signs of abnormal development of metabolic systems in very young animals include increased liver triglycerides, hepatic insulin resistance, muscle insulin resistance, decreased pancreatic alpha cell mass, and cardiac hypertrophy. In Grove’s opinion, it is one thing to develop these complications at the age of 35 to 45 years, but it is another thing to “hit the ground” with that status and have to experience it throughout the entire life course. While nutritional manipulations can improve some of these metabolic outcomes, some of the underlying molecular changes still exist. Whether those changes are epigenetic in nature is unknown, Grove stated.
Turning his attention to more complex behaviors in juvenile offspring, Grove described how the offspring born either to mothers fed a healthy diet or to mothers fed a high-fat diet are kept with their mothers during the postnatal period but that upon weaning, the offspring can be maintained on either diet. So control offspring (i.e., those born to mothers who were fed a healthy diet) can be put on a high-fat diet post-weaning, and high-fat
offspring (i.e., those born to mothers fed a high-fat diet) can be put on a control diet post-weaning. Grove described some of the findings that he and his team have observed thus far as a result of switching post-weaning diets. Notably, the offspring of mothers fed a high-fat diet who were switched to a post-weaning control diet nonetheless experienced long-term persistent inflammation in the brain. That they experienced brain inflammation is not surprising, Grove said, given that cytokine production in the placenta can cause brain inflammation. But the fact that the inflammation appears to be sustained, as do some neurochemical imbalances known to be associated with hedonic feeding and appetite stress dysregulation (i.e., decreased serotonin levels, low dopamine levels, and abnormalities in the melanocortin system), suggests that the alterations persisted even after the offspring were put on healthy post-weaning diets.
Additional data from Elinor Sullivan, another Grove research team member at OHSU and the University of Portland, indicate that a high-fat diet in the same macaque model is also associated with long-term abnormalities in brain connectivity in cortical regions. Specifically, they have shown that functional connectivity in the frontal cortex decreases, while functional connectivity in the temporal cortex increases. The findings indicate long-term differences in brain function and raise questions about whether the affected regions are related to abnormal behavior.
They also raised the question for Grove, what is going on metabolically in juvenile monkeys born to mothers fed a high-fat diet? Interestingly, Grove said, animals put on a healthy diet after weaning actually normalize their body weight. So they look fairly normal, even though their liver molecular signals do not, as Jacob Friedman discussed during his presentation (see Chapter 3 for a summary of information presented by Friedman). But if the same animals are put back on a high-fat diet at the age of 1 year, they have accelerated weight gain due to increased overall food intake. Grove described the acute increase in food intake as one with almost binge-like eating episodes. If the animals are given something novel, they will lever-pull all day long, he said, and they will stuff their cheek pouches and armpits. That behavior suggests to Grove that the animals have a drive for palatable foods that they have not been getting. The control animals, on the other hand (i.e., animals born to mothers fed a healthy diet), showed no change in food preference when placed under the same conditions. Even though control infants eventually reach the same weight, Grove suspects that the acute increase in food intake in infants born to mothers fed a high-fat diet may have long-term consequences.
In terms of metabolic rate, the high-fat-diet offspring had an increased rate during their active period (i.e., during the day), and low metabolism during their inactive period (i.e., at night). While, on the one hand, the increased metabolic rate during their active period might explain why the
body weights of the high-fat-diet offspring end up being no different than those of the control offspring, Grove said that he suspects that the increased metabolic rate in the high-fat-diet offspring is a manifestation of anxiety and hyperactivity. Those offspring spend a lot of time avoiding social contacts. Not only does their metabolic rate decrease at night, but their metabolism uses fats at night, compared to carbohydrates during the day. It is not clear why they use a different fuel at night, Grove said.
Regarding the anxiety that Grove suspects may be driving the high metabolic rate during the day, Sullivan et al. (2010) showed a dramatic increase in anxiety behavior in female offspring of mothers fed high-fat diets, as measured by latency to interact with toys, and a dramatic increase in aggressive acts in males, as measured by a tendency to lash out. While the behavioral differences were sex-dependent, Grove said that he suspects that the underlying cause is probably the same, given that both sexes also had increased cortisol levels. Other social abnormalities detected in the monkeys born to mothers fed high-fat diets include more shrieks during novel peer interactions, which were not sex-dependent; less initiation of contact, but more successful contacts when they are not the initiator; more time spent alone, again regardless of sex; and less social play, in fact, almost nonexistent social play. According to Grove, evidence exists to suggest that multiple maternal health and dietary and other components are likely contributing to the abnormal behaviors.
Interventions
Grove concluded by voicing his opinion about potential interventions in humans. In his opinion, weight loss prior to pregnancy is optimal. Weight loss during pregnancy, on the other hand, typically should be avoided, although diet and exercise can be improved. The greater question for him is, can either pharmacotherapy or surgery play a role?
Regarding surgery prior to pregnancy, in a study on bariatric surgery in rats, Grayson et al. (2013) demonstrated improvements in outcomes after doing bariatric surgery prior to pregnancy, but there were some long-term complications. Several other research groups have studied bariatric surgery in women, with initial studies demonstrating dramatic improvements, including reductions in pregnancy complications and reduced macrosomia (Galazis et al., 2014; Willis et al., 2015). But more recent studies have shown a dramatic increase in preterm birth and small-for-gestational-age infants (Galazis et al., 2014; Willis et al., 2015). Grove stressed the importance of testing interventions before applying them—not just surgery, but pharmacotherapy too. He
mentioned but did not elaborate on work he and his team did with maternal resveratrol treatment during pregnancy (Roberts et al., 2014).3
The even greater question for Grove is what to do for children. He recommended against surgery and pharmacotherapy. Exercise, such as what has been tested in rodent models, may be helpful, but it needs to be tested in human, in his opinion. Some of the social issues associated with childhood obesity may be improved with enriched environments, but again, in Grove’s opinion their actual effect on obesity needs to be tested. In conclusion, he stated, “I’m just going to end this by reminding people, this is a vicious cycle.”
EARLY INFANT RAPID WEIGHT GAIN AND THE EPIGENETICS OF LEPTIN4
Marie-France Hivert of the Harvard Medical School began her presentation by asking, what is leptin? Leptin was discovered in 1994 in ob/ob mice (Zhang et al., 1994). Ob/ob mice are fully deficient in leptin, hyperphagic, and morbidly obese. Leptin was one of the first adipokine—a hormone produced by adipocytes—described in the scientific literature, Hivert said. Among the cases of leptin deficiency described in humans, Farooqi et al. (1999, 2002) demonstrated that, when provided with leptin, children fully deficient in leptin decreased their hyperphagic behavior, reduced their weight, and continued to grow in height to a point where they almost normalized their BMI.
From these early animal and human studies, researchers learned that leptin is an adipostat signal that sends messages to the hypothalamus indicating that if there is not enough adipose tissue and leptin levels fall, then one should increase his or her food intake to restore normal weight. Allard et al. (2013) demonstrated that, indeed, this negative feedback loop exists in lean individuals, and those young adults who are lean and who have relatively low leptin levels at baseline have a higher weight gain over the next 2 years. However, this does not seem to be the case in obese individuals, who seem to be resistant to leptin. Even when injected with exogenous leptin, no significant weight loss can be induced, according to Hivert (Heymsfield et al., 1999).
Leptin acts on many other systems, Hivert said, including the reproduc-
______________
3 During the panel discussion following Mark Vickers’s presentation, Jacob Friedman noted that resveratrol is a natural supplement that he and his colleagues administered to offspring of mothers fed high-fat diets. They observed reduced triglyceride levels and improved placental function among the offspring administered the supplement, but with very odd alpha- to beta-cell ratios in the developing eyelets.
4 This section summarizes information and opinions presented by Marie-France Hivert, M.D., M.M.Sc., Harvard Medical School, Boston, Massachusetts.
tive system, where it affects placental function and possibly fetal development. Leptin is actually produced by the placenta during pregnancy and not only by adipose tissue, greatly contributing to maternal circulating levels of leptin (Hauguel-de Mouzon et al., 2006). At the first trimester, long before there has been any significant pregnancy gain in adipose tissue, there is almost double the amount of maternal plasma leptin compared to the pregravid period. Within 48 hours after delivery, maternal plasma leptin levels drop significantly; the drop is due to the sudden drop in placental expulsion and the consequent lack of a placental source of leptin, Hivert said, not to a decrease in weight.
These findings raised the question for Hivert, why is the placenta producing so much leptin, given that leptin, in normal physiology, is part of a negative feedback loop on weight regulation? She and her research team have been investigating this question and trying to untangle the physiological roles of maternal versus fetal leptin. Based on data collected thus far, they think that during pregnancy, maternal circulating leptin is coming from both adipose tissue and the placenta in equal amounts, with the placenta also producing leptin that is being released on the fetal side, although Hivert sad most fetal leptin is probably being produced by fetal adipose tissue.
Using data collected from a longitudinal pregnancy cohort in Canada, Hivert and her research team measured maternal leptin levels at the first and second trimester and fetal levels at birth (circulating levels in cord blood). Hivert said that they were surprised to find that maternal leptin levels were not associated with lower subsequent gestational weight gain, but rather that higher leptin levels predicted a higher subsequent gestational weight gain, even after accounting for current maternal weight status (unpublished data). That is, the researchers observed what appeared to be a positive feedback loop, with the presence of elevated maternal leptin signaling to women that they should be eating more. The positive feedback signal was presented mainly in the second trimester and was of greater magnitude in women who were overweight. Additionally, Hivert and her team observed that maternal leptin levels were associated with both fetal adiposity, as measured by skin fold, and fetal leptin levels in cord blood at birth, once again independently from maternal weight status.
Hivert said that her interest in leptin levels in offspring in the perinatal period comes partly from animal studies published more than a decade ago that demonstrated that early leptin exposure is associated with hypothalamus development. Bouret et al. (2004) compared hypothalamic development in ob/ob mice versus normal mice and observed in the periventricular nucleus of the hypothalamus a definite lack of neuronal development in the ob/ob mice. When they injected leptin into the ob/ob mice during the perinatal period, they could rescue and reestablish density of
neuronal fiber and projection in the periventricular nucleus. When they tried to do the same in later life, again in ob/ob mice, they did not notice the same effect. Their findings suggested that leptin exposure in the perinatal period plays a critical role.
In a similar experiment, Bouyer and Simerly (2013) exposed ob/ob mice to a short course of leptin in the perinatal period and then followed the mice through life to observe their weight trajectory. Untreated ob/ob mice, who were fully leptin-deficient, became hyperphagic and morbidly obese, while ob/ob mice who were treated for only a few days in the perinatal period had a slightly lower weight trajectory. Hivert reiterated that the treated mice received only a few days of treatment and that the effects were lasting. When the researchers exposed the mice to leptin again later in life, those mice that had been exposed in the perinatal period were more sensitive to the later exposure compared to the ones who had never been exposed. The researchers also reported differences in adipocyte and adipose tissue development, with ob/ob mice exposed to leptin in the perinatal period having smaller, “healthier” adipocytes. According to Hivert, small adipocytes are more metabolically healthy and flexible than large cells. It seems to Hivert, based on these findings, that early leptin exposure is associated not only with hypothalamus development, but also adipose tissue.
In humans, as part of Project Viva, a research project led by Matthew Gillman that has been under way for more than 15 years, Parker et al. (2011) examined a series of factors in the prenatal period or at birth and their association with early infancy weight gain as measured by the change in weight-for-length from birth to 6 months. Among all of the factors that they examined, leptin emerged as a significant determinant of early-infancy weight gain, with higher leptin levels being associated with a slower weight gain between birth and 6 months of age. Mantzoros et al. (2009) reported a similar negative association at 3 years of age, with lower leptin levels in cord blood at birth being associated with a higher weight status at 3 years. However, Boeke et al. (2013) reported that higher leptin levels at 3 years of age predict a higher BMI at 7 years of age, indicating that the negative association observed earlier in life no longer exists.
So again, as with the animal data, human data suggest that the perinatal period is a critical period with respect to leptin exposure, with infants still being sensitive to leptin and with lower levels of leptin in infancy being associated with greater weight gain and adiposity at the age of 3. Hivert suggested that the association may be mediated through hypothalamic development, adipose tissue changes, or even the microbiome. Yet, the molecular mechanisms involved are still mostly unknown.
Epigenetics of Leptin in Early Life
Hivert remarked that her research group is especially interested in the impact of maternal glycemia on leptin. Bouchard et al. (2010) examined the association between maternal glucose and placental leptin gene DNA methylation in women classified as having gestational impaired glucose tolerance and found that the higher the maternal glycemia, the lower the DNA methylation in the promoter region of the fetal leptin gene but the higher the DNA methylation in the promoter region of the maternal leptin gene. Hivert suggested that if one believes that maternal leptin has a positive feedback on gestational weight gain, then the Bouchard et al. (2010) results suggest that higher maternal glycemia may lead to leptin gene DNA methylation adaptations that limit maternal leptin levels, with higher methylation in the leptin gene promoter region and therefore a lower expression of the leptin gene on the maternal side. But that possibility is only a hypothesis at this point, Hivert noted.
She and her research team used Mendelian randomization to better understand the observed association between maternal glucose and leptin epigenetic regulation. Using data from the Genetics of Glycemic Regulation in Gestation and Growth (Gen3G) cohort, the Mendelian randomization study they conducted showed that maternal glycemia is part of the causal pathway influencing offspring leptin epigenetic regulation (Allard et al., 2015) (see Chapter 3 for a summary of Caroline Relton’s discussion of Mendelian randomization). Hivert expressed hope that Mendelian randomization might help researchers to identify additional factors that have an impact on risk of excess weight at birth and in childhood, including factors that might serve as potential targets for intervention.
Hivert’s Take-Home Messages
Hivert highlighted two take-home messages. First, the perinatal period is critically sensitive to leptin, with leptin levels during that period likely affecting many tissues—not only the hypothalamus, but also adipose tissue. Based on results from both animal and human observational studies, perinatal leptin levels are associated with early life weight trajectory. Second, based on the current literature and Hivert’s laboratory observations, it seems that maternal glucose modulates the epigenetic regulation of leptin in offspring.
THERAPIES TO REVERSE METABOLIC DISTURBANCES ARISING AS A CONSEQUENCE OF DEVELOPMENTAL PROGRAMMING5
Mark Vickers of the University of Auckland in New Zealand said that it is well established that alterations early in life, particularly nutritional exposures, increase risk for a range of metabolic disorders later in life, including obesity and heart disease. There is no single cause, he said; rather, it is a complex, multifactorial process. While scientists are beginning to understand some of the underlying mechanisms, there is still a great deal to learn about how interventions early in life can diminish the incidence and severity of later disease. More importantly, Vickers said, most of what is known is derived from experimental models with limited translation in the clinical setting.
A range of interventions have been tested, including dietary interventions (e.g., lipids, pre- and probiotics, taurine, vitamins, polyphenols, methyl donors), pharmacologic strategies (e.g., leptin, growth hormone, melatonin, GLP-1 analogs, nuclear receptor agonists), and behavioral and lifestyle interventions (e.g., exercise, dietary counseling). If efficacy of behavioral and lifestyle interventions was greater than it is, Vickers remarked, then that would be the ideal option given that it is the easiest strategy to implement and does not raise the same safety issues that the other two types of interventions do.
The question for Vickers is not only how to intervene, but when to intervene. The programming window of plasticity is difficult to time correctly, especially when translating across species (e.g., from rodents to humans). Godfrey et al. (2010) described how plasticity diminishes over time and argued that the earlier the intervention, the better the effect later in life. According to Vickers, some data suggest that the best window for intervention may even be during preconception. Unfortunately, Vickers said, in some areas of New Zealand 60 to 70 percent of pregnancies, particularly among high-risk groups, are unplanned.
Regarding nutritional exposure early in life, Vickers remarked that the risk of obesity among offspring is very much a U-shaped curve, with both under- and over-nutrition resulting in increased risks (Grattan, 2008). He reminded the workshop audience to keep in mind that many cases of over-nutrition actually reflect micronutrient malnutrition and that many obese individuals are actually malnourished. He suggested that the similarity in phenotype may be due to malnutrition at both ends of the spectrum.
Before embarking on a review of intervention studies, Vickers commented on the similarity in phenotypic outcomes, in terms of efficacy in reversing metabolic disturbances, for a wide range of interventions tested
______________
5 This section summarizes information and opinions presented by Mark Vickers, M.Sc., Ph.D., University of Auckland, New Zealand.
across a range of animal models, including sheep, nonhuman primates, piglets, mice, and guinea pigs. As just one example, some of the leptin work that was originally undertaken in the rat has since been replicated in the piglet and several rodent strains.
Vickers also commented on the many different nutritional exposures, or manipulations (e.g., under-nutrition, high fat, high salt, low protein, and high sugar), that have been tested across a range of preclinical models and how they all induce a common metabolic syndrome phenotype (i.e., one featured by obesity, type 2 diabetes, heart disease, altered appetite, inflammation, and reproductive disorders). One nutritional exposure that is often overlooked, in Vickers’s opinion, is the impact of high-fructose intake during pregnancy. There is very little literature on the effect of soda intake during pregnancy. He noted that he and his research team have demonstrated that as little as two cans of soda per day during pregnancy can rewire the arcuate nucleus (an aggregation of neurons in the hypothalamus) and program leptin resistance in offspring, independent of any change in maternal body weight or body composition.
Leptin
Leptin was one of the first interventions tested as a means to reverse metabolic disturbances arising as a consequence of developmental programming. Original work by Bouret et al. (2004), which Marie-France Hivert had mentioned in her presentation (see previous section), showed that replacing leptin in leptin-deficient ob/ob mice during the first few weeks of life could restore neural network pathways from the hypothalamus and result in essentially “normal” animals, while, in contrast, post-weaning treatment had no effect. Vickers and his research team conducted a similar experiment with outbred rats and found that maternally undernourished offspring, when fed a high-fat diet, became, Vickers said, “fatter and fatter and fatter over time” (Vickers et al., 2005). But when he administered leptin for 2 weeks during their neonatal development, the maternally undernourished offspring looked like control animals. Importantly, in Vickers’s opinion, the effects were specific to animals born to undernourished mothers. Leptin had very little effect in control animals. These results have been replicated in a few other studies as well. Additionally, evidence exists to support Bouret et al.’s (2004) finding that leptin reprograms the arcuate nucleus.
Publication of the Vickers et al. (2005) report led to what Vickers described as a “big rush” of calls in the United Kingdom to add leptin to infant formula. However, subsequent studies suggested that the effects of leptin are dependent on the mother’s prior nutritional status and sex (Gluckman et al., 2007; Vickers et al., 2008). Vickers stressed the importance of examining both male and female offspring and noted that many
researchers in this field do not consider the effect of the offspring’s sex on outcome. Even Vickers et al. (2005) examined only female offspring. Leptin treatment in male neonates under the same Vickers et al. (2005) paradigm can elicit adverse metabolic phenotypes in later life, for example, with offspring becoming insulin resistant (Vickers et al., 2008).
With regards to leptin effects being dependent on the prior nutritional background of the mother, Gluckman et al. (2007) demonstrated that changes in PPAR-alpha and 11-beta-HSD2 methylation states as a result of leptin administration were directionally dependent on prior maternal nutritional status, with completely opposite effects observed depending on whether the mother was undernourished or not. Stocker et al. (2004) reported a reduction in placental 11-beta-HSD2 activity in low protein-fed mothers and a partial restoration of activity in mothers administered leptin.
GLP-1 Analogs
Vickers mentioned only one study relevant to the use of GLP-1 analogs, specifically extendin-4, as an intervention. In this study, Stoffers et al. (2003) showed that the neonate offspring of undernourished mothers had reduced beta-cell mass but that beta-cell mass and proliferation were normalized in neonates that were administered extendin-4. The normalization of beta-cell mass and proliferation was linked with a reversal of marked changes in epigenetic modifications to the pancreatic and duodenal homeobox 1 (PDX1).
Taurine
There has been quite a bit of work done on taurine, according to Vickers, including what he described as “hidden data” from two to three decades ago showing that in a rat model the administration of taurine supplement to mothers on a low-protein diet conferred completely normal pancreatic development. Taurine concentrations are known to be low in diabetic and prediabetic states, Vickers explained, with physiological plasma taurine levels playing an important role in maintaining adequate beta-cell function and insulin action. With certain liver disorders, such as maternal hepatic cholestasis, taurine is known to have a protective effect and appears to confer long-term benefits in offspring. As an example, Vickers said that members of his research group showed that 1.5 percent taurine-supplemented drinking water completely normalized the hyperinsulinemic effects of hepatic cholestasis in fructose-fed mothers (Li et al., 2015). Without taurine, the fructose diet induced a pro-inflammatory phenotype not just in the mothers but in the offspring as well, with both female and male offspring showing increased levels of inflammatory markers at birth. Importantly, in Vickers’s opinion, taurine
supplementation in the mother’s diet benefited not just the mother, but her offspring too, with normalized levels of inflammatory markers at birth.
Growth Hormone
Vickers’s group has also done some work with growth hormone. Offspring born to undernourished mothers are typically hyperleptinemic as well as hyperinsulinemic, Vickers said. They also have very low insulin-like growth factor 1 (IGF1) levels. Gray et al. (2013) observed that rats born to undernourished mothers were fatter and their adipocytes were normally larger, but that offspring administered growth hormone during the first 2 weeks of life had normal-sized adipocytes. The offspring of undernourished mothers also had markedly increased systolic blood pressure, but the offspring that had been administered growth hormone as neonates were still benefiting with normalized blood pressure and fat mass as mature adults at 150 days of age (Reynolds et al., 2013).
In Vickers’s opinion, while growth hormone administered during what, in rats, is a critical window of development is not likely to translate easily in a clinical setting, all of these findings in rats, not just with regard to growth hormone, but with leptin too, demonstrate proof of concept that a range of interventions can have lasting benefits over the life course of the offspring.
Maternal Lipid Supplementation
Vickers briefly mentioned work that his research team has done with conjugated linoleic acid, with offspring of obese mothers having significantly impaired insulin sensitivity, increased gut inflammatory markers, and altered gut taste receptors (i.e., they seem to have a preference for high-fat, high-sugar foods). But all of those effects are resolved in the offspring of mothers whose diets are supplemented with conjugated linoleic acid. The mothers benefited from the supplementation as well, with improved maternal insulin sensitivity.
Postnatal Dietary Omega-3 Supplementation
Vickers noted interesting work done by Wyroll et al. (2006) showing that offspring of pregnant rats treated with dexamethasone were hyperleptinemic by 6 months of age but that the hyperleptinemic phenotype could be completely prevented by feeding the offspring a postnatal diet rich in omega-3 (n-3) fatty acids.
Maternal Vitamin D Status
There is a high prevalence of vitamin D deficiency in New Zealand, Vickers observed, despite the amount of sun exposure, because people are always being told to cover up. The evidence for an impact of vitamin D supplementation on outcomes related to adiposity is conflicting, with the data being, in his words, “all over the place.” What is known is that pre-pregnancy obesity predicts poor vitamin D status in both mothers and neonates. Recent studies have reported that vitamin D deficiency in pregnancy can result in insulin resistance, altered inflammatory profiles, and an increased risk of early postnatal obesity in offspring (Morales et al., 2015; Zhang et al., 2014).
Dietary Methyl Donors
A range of methyl donor supplements, including folic acid, glycine, choline, and mixed supplements, have been shown to have beneficial effects on long-term cardiovascular and other outcomes in offspring (Bai et al., 2012; Carlin et al., 2013; Jackson et al., 2002; Torrens et al., 2006). As just one example, Bai et al. (2012) showed that maternal choline supplementation in rats reduces low-protein-induced elevations in systolic blood pressure and fat mass in adult offspring.
Exercise/Lifestyle Interventions
Vickers identified two critical windows where physical activity has the potential to mitigate against increased obesity in offspring who were developmentally programmed to become obese: first, maternal exercise prior to and during pregnancy and, second, exercise during childhood for those at risk (Siebel et al., 2012). He mentioned that studies in rats have shown that early exercise in offspring can reduce the adiposity associated with both maternal under-nutrition (Miles et al., 2009) and maternal obesity (Santos et al., 2015). Other studies have reported that the effects of exercise are mediated in part by improved central leptin sensitivity (Sun et al., 2013) and that they are dependent on type and duration (Hopkins et al., 2010). With respect to dietary interventions, Zambrano et al. (2010) showed that dietary intervention in obese mothers prior to pregnancy reversed metabolic programming in offspring; the effects persisted into adult life, but they were sex-specific.
Catch-Up Growth Intervention
Preventing catch-up growth is, in Vickers’s opinion, one of the easiest ways to prevent programmed postnatal obesity. Small offspring of undernourished mothers normally develop increased adiposity if they are allowed to catch up to controls. Howie et al. (2012) showed that keeping offspring born to undernourished mothers small during the pre-weaning period leads to them being completely indistinguishable from controls in adult life.
DHA Supplementation
With regard to docosahexaenoic acid (DHA) supplementation, Vickers said that studies suggest that the effects depend on dose, duration, and exposure (e.g., Carlson et al., 2013; Donahue et al., 2011), though he suggested that he and his colleague’s findings published in Albert et al. (2015) might explain some of the variation in effects being reported. They tested 32 commercially available fish oil supplements in New Zealand and found that more than 80 percent were either heavily oxidized or contained far lower levels of n-3s than marketed.
Pre- and Probiotics
There has been a lot of work done on the use of pre- and probiotics as an intervention to reverse metabolic disturbances caused by developmental programming, according to Vickers, especially in terms of reprogramming the gut flora (Luoto et al., 2010; Thum et al., 2012). Because probiotic modification of the early gut microbiota may restrain excessive weight gain during the first years of life, it has been suggested that infant formula be supplemented with oligosaccharides to compensate for the lack of some of what is naturally present in human milk.
Vickers’s Take-Home Message: One Size Does Not Fit All
The biggest take-home message from intervention studies, in Vickers’s opinion, is that interventions in “intact” systems may lead to adverse outcomes, raising the question, how can those at risk of programmed disorders be identified? Additionally, some interventions yield quite profound sex-specific differences. For example, maternal methyl-deficient diets can result in metabolic disturbances in male, but not female, rat offspring.
Predictive biomarkers are being studied as a way to identify who and when to target for intervention. For example, Vickers cited Karen Lillycrop’s work with cord blood methylation of RXR-alpha and its link to later childhood adiposity. The challenge, in his opinion, is what to do with
all the data, including how to distinguish between causative and associative links and how to weigh trade-offs. According to Vickers, epimutations with short-term trade-offs are likely associated with later negative health outcomes. For example, while maternal methyl donor supplementation can lead to a reduction in fatty liver disease, it can also lead to increased adipose tissue storage in offspring later exposed to a high-fat diet.
Vickers concluded with some comments about the generational nature of environmental exposure. A single maternal insult can affect not only the development of the fetus (the F1 generation), but also the germ cells that form the next (F2) generation. Many early studies went only to the F2 generation, which Vickers said is not truly transgenerational because F2 effects reflect the original insult. Most studies that have examined the F3 and later generations have show that in many cases the phenotype is eventually resolved, although more work in that area needs to be done in Vickers’s opinion.
In addition to what is known about maternal exposure, a large amount of data show that paternal exposure and transmission affect disease risk. For example, Ng et al. (2010) showed that a chronic high-fat diet in the father programs beta-cell dysfunction in female rat offspring. The effect can be partially reversed by exercise (McPherson et al., 2014).
In summary, Vickers said, the early life period of developmental plasticity offers the most effective avenue for intervention. Although reversal has been shown in a number of experimental models, both maternal and neonatal, direct translation to the clinic may prove difficult. Nonetheless, he said, the results will inform possible intervention strategies.
THE MICROBIOME AND OUR GENOME6
Recognition of the presence and significance of the human microbiome started to emerge around 2000, according to William Nierman of the J. Craig Venter Institute. Nierman provided a general overview of the human microbiome, beginning with what he said were “highly speculative” numbers, that is, that it contains an estimated 10 trillion bacterial cells, which amounts to about 10 times more cells than human cells and 150 times more bacterial genes than human genes. Bacteria are not the only microbial component of the human microbiome; fungi and viruses occupy our bodies as well. Altogether, the human microbiome weighs a total of approximately 3 pounds, he said.
Two striking features of the human microbiome, in Nierman’s opinion, are, first, that probably half of the microbial genes in the human body are functionally unknown and, second, that the vast majority of organisms in
______________
6 This section summarizes information and opinions presented by William Nierman, Ph.D., J. Craig Venter Institute, Rockville, Maryland.
the human microbiome have never been cultured and therefore have never been studied in the way that scientists traditionally study bacteria.
It has been clearly established by now, Nierman continued, that a healthy gut microbiome provides many metabolic capabilities that supplement its human host’s metabolic capabilities, such as digestion of complex carbohydrates, with diet playing an important role in the process. Healthy microbiomes also protect against disease. For example, a healthy stomach microbiome protects against Helicobacter pylori–mediated stomach cancer, and a healthy digestive tract microbiome protects against Clostridium difficile infection. Many disease states, particularly infectious disease states, are characterized by collapses in microbiome diversity, according to Nierman, which predisposes the human host to a number of problems, including enhanced inflammation, loss of barrier function, and increased immune dysfunction.
Of particular interest to Nierman because of his interest in the respiratory microbiome is the fact that when someone has a severe flu, the flu virus modulates the host immune system, which results in a high incidence of streptococcal pneumonia following the flu. He explained that the streptococcal bacteria are already present in the respiratory tract microbiome. In other words, people do not “catch” bacterial infections in their lungs. They might “catch” the flu virus, but streptococcal bacteria are commensal organisms that naturally live in the respiratory tract. Rather, something happens to “bring out these things.”
Because of its role in maintaining health, the microbiome can be modulated in dietary and other ways to have a therapeutic effect. For example, fecal transplants have been considered as one way to treat recurrent C. difficile infections (van Nood et al., 2013). Nierman pointed out that the idea of fecal bacteriotherapy is not new and referred workshop participants to a 1989 paper (Tvede and Rask-Madsen, 1989). Sarah Highlander, a colleage of Nierman’s at the J. Craig Venter Institute, has been exploring the use of pure cultures of lactobacilli to suppress C. difficile overgrowth and to cause quick remission of symptoms.
Humans acquire their microbiomes beginning at birth. Dominguez-Bello et al. (2010) demonstrated that the mode by which a baby is delivered (i.e., vaginal or Cesarean) affects the population structure of the infant microbiome. Koenig et al. (2011) followed infants for 2 years after birth, sampling changes in their gut microbiomes along the way, and Palmer et al. (2007) demonstrated that the gut microbiome approaches adult status by 1 to 2 years of age. The respiratory microbiome, on the other hand, shows what Nierman described as “wild variation” month to month even at 2 years of age.
Nierman emphasized the site-specific nature of the human microbiome—for example, with the respiratory, gut, and skin microbiomes each being
very different from each other. Body sites are like ecosystems, he said. You would not expect to find the same species in the tropical rainforest that you find in the desert. The same is true of the human body. Indeed, the goal of the Human Microbiome Project (HMP) was to characterize the microbiomes of healthy individuals at selected body sites in the oral cavity, skin, nasal cavity, gut, and vagina. The only observed overlap, according to Nierman, was between the external nasal site and the skin, which he said was not surprising, given that the external nares are so close to the skin. A striking finding in the oral cavity, in his opinion, was the dramatic differences between nearby sites, such as dental plaque and gums. While HMP data showed no apparent relationship between the microbiome at any site and gender, age, weight, ethnicity, or race, it did reveal tremendous variability at each site among individuals.
Regarding the usefulness of 16s rRNA profiling, one of the HMP techniques used to characterize the bacterial complexity of the microbiome, Nierman observed that such profiling provides a good view of relative abundance at the genus level but not at the species level (Keseler et al., 2013). A listing of what is present at the genus level is not very informative, in Nierman’s opinion, because of the very important functional differences that exist not just between species within a genus, but also between strains within a species. For example, the E. coli K12 strain, a lab strain, is benign and is usually found in the lower intestine, whereas the E. coli O157:H7 strain is a highly virulent pathogen that can cause severe diarrhea and kidney failure.
Obesity and the Microbiome
Although there have been several studies of an association between the microbiome and obesity, many of them in animal models, Nierman stated that no single taxonomic signature of obesity in the microbiota of the human gut has been found (Finucane et al., 2014). Generally, however, obese individuals harbor less diverse bacteria communities, and Nierman suggested that even though the mechanisms through which gut microbes influence obesity or BMI are unknown, novel therapies aimed at restoring the gut microbiota balance could potentially help to treat obesity.
In conclusion, Nierman proposed what he called the obesity–host–pathogen–microbiome–innate immune system paradigm, modeled after the host–pathogen–microbiome disease paradigm. In his “fantasy” obesity paradigm, host genetics, microbiome metabolic function, and the innate immune system would be assembled into an obesity-causing interaction triad. He suggested, based on information being presented at this workshop, that the category of host genetics should include an epigenetic component as well.
THE EPIGENETICS OF THE MICROBIOME7
“We are not alone,” Meredith Hullar of the Fred Hutchinson Cancer Research Center began. A large proportion of genomic DNA associated with humans is bacterial DNA located either within or on the human host. Hullar provided a brief overview of the epigenetic mechanisms of bacteria in the human gut, with a focus on DNA methylation; discussed how microbial metabolites of the host diet may influence host health via epigenetic mechanisms involving DNA methylation, noncoding RNAs, microRNAs (miRNA, in particular), or histone deacetylase inhibition; and concluded with a discussion of her research in obesity and the microbiome.
Epigenetics Within the Microbiome
Unlike methylated human DNA, which is mostly N5 methyl cytosine,8 bacteria have three types of methylated DNA, with the predominant one being N6 methyl adenine and the others N4 and N5 methyl cytosine. The nucleotide bases are methylated by DNA methyltransferases, with the methyl group transferred from S-adenosyl-L-methionine.
In bacteria, the predominant role of DNA methylation is to protect the host bacterium against phage attack. It does so, Hullar explained, through restriction modification systems composed of methyltransferases and restriction endonucleases. The restriction endonucleases cut DNA at the internal phosphodiester bonds and recognize unmethylated four- to six-base-pair palindrome sequences. When a phage injects its DNA into a host bacterial cell, the phage DNA, because it is not methylated, is cleaved by the restriction endonucleases so that it cannot be incorporated into the host genome. Sometimes the phages escape detection and when they do, their DNA is incorporated into the microbial genome. The phage DNA ends up with the same methylation pattern as the microbial DNA, thus, the phage escapes detection by host restriction endonucleases and the phage DNA is replicated as part of the bacterial cell cycle.
Restriction modification systems within bacteria have diversified and are broadly distributed across multiple phyla, with more than 2,000 different systems involving approximately 43,000 restriction modification enzymes (Korlach and Turner, 2012). Modification of the endonucleases creates new functions for the restriction modification systems, Hullar explained, such as the DNA adenine methylase (Dam) post-replication mismatch repair system in E. coli. The Dam system is dictated by a methylated base in the CTAG
______________
7 This section summarizes information and opinions presented by Meredith Hullar, Ph.D., Fred Hutchinson Cancer Research Center, Seattle, Washington.
8 An N5 methyl cytosine has a methyl group attached to the fifth atom in the six-atom cytosine ring.
region and affects microbial gene expression. It has been hypothesized that the changes in gene expression are associated with genes involved in phage infectivity, DNA replication, stress response, culturability, and host association (Chen et al., 2014). “These are very dynamic systems that may influence microbial function in the human gut,” Hullar said.
Methylation patterns between two bacterial strains can vary substantially and may alter bacterial fitness. In a methylome comparison of H. Pylori 26695 and J99-R3, Krebes et al. (2014) found many different methylation markers in the H. pylori genomes. They hypothesized that although methylation is a key driving force in genetic diversification, the methylation in H. pylori may reduce natural transformation. Disparate methylation patterns were also found in two strains of Bacteroides dorei, the predominant genus found in the microbial gut microbiome. One was highly methylated (20,551 methylated sites), and the other lacked a DNA adenine methyltransferase gene and lacked methylation (Leonard et al., 2014). How CTAG methylation affects these two strains is unknown. Regardless, Hullar suggested that methylation patterns may influence microbial gene expression and add another layer of complexity in understanding how microbial metabolism in the human gut affects host health.
Epigenetics Between the Host and Microbiome
Both pathogenic and commensal bacteria exploit a diverse set of epigenetic mechanisms such as DNA methylation, histone modifications, and noncoding RNAs to alter host gene expression. These epigenetic mechanisms can occur directly through infection or indirectly through microbial metabolites that interact with host genes. Altered gene expression has far-reaching implications from maintenance of gut homeostasis to changes in gene expression of human host pathways associated with inflammation and adiposity.
Commensal bacteria influence gut homeostasis through differential methylation of host genes. Although exposed to a large number of commensal gut bacteria, intestinal epithelial cells are relatively unresponsive to bacterial cell wall antigens that usually promote inflammation by interacting with host innate immunity via toll-like receptors (TLRs). Takahashi et al. (2009) found that the presence of intestinal commensal bacteria increased the methylation of host TLR genes, specifically TLR4, which in turn blunted the host inflammation response. The patterns vary between the small and large intestine in mouse models. Regulation of the host gene expression via epigenetic modifications may maintain symbiosis in the large intestine because methylation was dependent on colonization with commensal bacteria. In the small intestine, methylation was independent of bacterial colonization, and the degree of methylation varied. TLR4 genes
were more methylated in the differentiated epithelial cells at the top of the crypts than the bottom of the crypt (Takahashi et al., 2011). According to Hullar, others have found this to be the case with other TLRs (Kellermayer et al., 2011).
Some pathogenic bacteria also escape host surveillance by altering host miRNA associated with inflammation and immune response. Upon infection, miRNAs associated with some genes, Hullar explained, are up-regulated, while other miRNAs linked to inflammation response are down-regulated. This approach is employed by the well-known pathogens Listeria, Salmonella, Helicobacter, and Mycobacterium species (Maudet et al., 2014). Hullar emphasized, however, that how a host responds to a pathogen is influenced by the commensal microbiome. In a comparison of germ-free versus conventionalized mice, Archambaud et al. (2013) showed that all of the miRNAs in response to an oral Listeria infection were down-regulated in the presence of commensal bacteria, suggesting that healthy commensal microbiota can provide a barrier against some pathogens.
Host exposure to microbial fermentation products varies, in part, based on the distribution of the different microbial metabolic pathways present in the gut and may influence disease risk associated with obesity. This moves beyond the idea of caloric excess causing obesity and incorporates the idea that microbial fermentation of dietary compounds may alter host gene expression (Hullar and Fu, 2014) in host pathways associated with obesity. Compounds that escape metabolism in the small intestine enter the colon where they undergo microbial metabolism. The metabolites produced by microbial fermentation influence host pathways, and increased fermentation products from fiber have been inversely associated with obesity. Fiber is fermented into short-chain fatty acids, acetate, butyrate, and propionate. However, the fermentation process is complex, Hullar said. For example, the short-chain fatty acid butyrate can be produced via four different metabolic pathways by different groups of bacteria, although not all human guts have a microbial community with all four pathways (Vital et al., 2013). Host exposure to microbial fermentation products varies, based in part on the distribution of the different microbial metabolic pathways present in the gut, and may influence disease risk associated with obesity.
Hullar described several biological consequences of microbial fermentation in the colon and epigenetic mechanisms that influence gene regulation in host pathways in obesity. Short-chain fatty acids produced in the gut can move into the blood, enter systemic circulation, and increase lipogenesis, fatty acid oxidation, and adipogenesis. They have also been associated with increased production of the satiety peptide YY (PYY) and alter gut epithelium integrity, according to Hullar. Short-chain fatty acids modulate adiposity via a direct effect on several genes, including fasting-induced adipose factor (Fiaf) involved in fatty acid metabolism,
G-protein coupled receptor 43 (Gpr43) involved in obesity, and peroxisome proliferator-activated receptor gamma (Pparγ), which regulates fatty acid storage, and glucose metabolism histone deacetylase (Hdac) involved in epigenetic modification of the lysine residue in histones. Recently, Lukovac et al. (2014) reported that many genes associated with host fatty acid metabolism in gut tissue are influenced by exposure to short-chain fatty acids. More specifically, butyrate and propionate significantly enhanced the expression of genes associated with histone deacetylation (Hdac3, Hdac5) and fatty acid metabolism (Fiaf) and decreased expression of genes associated with fatty acid storage (Pparγ) and satiety (Gpr43).This suggests that microbiota trigger specific transcriptome responses in host epithelium, most likely depending on the metabolic products produced by different bacteria and fermentation pathways.
The studies Hullar presented pointed, she said, to a potential role of the microbiome in the epigenetics of obesity. She presented microbiome-mediated epigenetic mechanisms that alter host gene expression through three mechanisms: methylation, miRNA, and histone deacetylase (HDAC) inhibitors via microbial metabolites derived from diet. Westernized diets, which are high in fat and sugar, select for a gut microbiome composition that is associated with disease risks common to industrialized societies, such as obesity, cardiovascular disease risk, and certain cancers. Exposure to microbial metabolites from a Westernized diet may influence the epigenetic regulation of gene expression in host pathways associated with higher disease risk (Lukovac et al., 2014). In addition, several studies have shown that the consumption of a Westernized diet in primates or mice alters the microbiome composition in the offspring (Ma et al., 2014; Myles et al., 2013). Because the microbiome is transferred to the offspring from the mother, there may be a generational influence on gene expression through epigenetic modification by the microbiome, although this has not been shown in humans. Hullar said that the role of epigenetic mechanisms that influence the control of gene expression both within the microbiome and between the microbiome and host is a “burgeoning area.” She encouraged greater consideration of the interaction between microbial metabolism of the human diet and epigenetics. In her opinion, efforts are hindered by small sample sizes and the lack of prospective studies. Without prospective studies, it is difficult to know how the early life microbiome may influence obesity later in life or transgenerational effects of the microbiome on gene expression modulated by microbially mediated epigenetic mechanisms.
Diet, Obesity, and the Microbiome
Diet affects adiposity through changes in fuel availability that alters energy balance and increase lipids, adipocytes, macrophages, and inflamma-
tion. The microbiome also plays a role, and early work by Turnbaugh et al. (2009) showed an increase in microbial genes associated with carbohydrate metabolism and fatty biosynthesis in individuals who were obese. This suggested that people who are obese have different energy-gathering or energy-yielding capacity than lean people based, in part, on the composition and function of the gut microbiome. These findings led Hullar and her research team to ask whether the microbiome is associated with obesity.
To study the association between the microbiome and adiposity, Hullar and Johanna Lampe (unpublished data) collected fecal samples, measured adiposity by dual-energy X-ray absorptiometry, and obtained a 3-day food record from a cross-sectional sample of 107 women, aged 40 to 45 years. Based on an examination of 16S rRNA genes from the fecal samples, the researchers were able to group the women by the dominant microbe found in their gut. Most participants’ microbiomes were dominated by Prevotella, Bacteroides, or Pyramidobacter, and the groups were associated with dietary intake (Hullar et al., 2015). The researchers also found that the women in the Bacteroides group had increased animal protein intake and decreased fiber and vegetable protein intake and that the women whose guts were dominated by Firmicutes had increased fiber and vegetable protein intake, similar to other reports (Wu et al., 2011). Even after correcting for fiber intake (because of an observed inverse relationship between fiber intake and adiposity), Hullar and Lampe still observed a significant association between the microbiome and adiposity.
Hullar concluded by mentioning another study that she is currently collaborating on—a multiethnic cohort study on disease risk, obesity, and fat distribution. Health risk due to obesity varies among ethnic and racial groups in association with the distribution of adipose tissue (Cleary and Grossmann, 2009; Conroy et al., 2011; Cummings and Schwartz, 2003; Lim et al., 2012). The researchers will be analyzing four different potential predictors of adiposity: the exposome, genetic variation, metabolomics, and the microbiome.
TOXIC STRESS AND CHILDHOOD OBESITY9
Antonio Convit of the New York University School of Medicine remarked that he would be talking about the toxic effects of over-nutrition on the brain rather than toxic stress per se. He agreed with earlier remarks that the greater focus of discussion around childhood obesity should be on metabolic dysregulation, which he said is “certainly what has the biggest impact on the brain.” For example, data on lean versus obese children with
______________
9 This section summarizes information and opinions presented by Antonio Convit, M.D., Nathan Kline Institute and New York University School of Medicine, New York.
or without insulin resistance or type 2 diabetes reveal a step-down function of cognitive performance. Specifically, lean children performed the best in all of four school-relevant cognitive domains tested (spelling, arithmetic, general memory index, and psychomotor efficiency), obese children without marked insulin resistance performed second best, and obese children with insulin resistance had the worst marks among obese children with type 2 diabetes.
Compared to adults, Convit said, children with metabolic dysregulation are much more cognitively impaired. In a study of a group of a little more than 100 children, with the control group consisting of individuals who may meet one or two of the criteria for metabolic syndrome but not three or more criteria, Convit and his research team found that several cognitive domains were affected by metabolic syndrome: academic achievement (i.e., based on Wide Range Achievement Test), executive function, attention, and psychomotor efficiency. As the number of criteria for metabolic syndrome increased, cognitive performance decreased. With adolescents, researchers use slightly modified criteria to define metabolic syndrome, Convit explained. Instead of impaired fasting glucose, they use insulin resistance, which is a more sensitive measure obtained from the Quantitative Insulin Sensitivity Check Index (QUICKI). The other criteria are blood pressure, triglyceride levels, abdominal obesity, and high-density lipoprotein (HDL).
These findings raised the question for Convit, what is happening in the brain in children with metabolic dysregulation? He and his research team decided to address the question by using a noninvasive MRI technique called diffusor tensor imaging to examine microstructural integrity of the brain’s white matter. White matter is what connects different parts of the brain. Convit explained that a molecule in a huge vat of water can move in all directions equally. That movement is known as isotropic diffusion. But if the movement of a water drop is constrained, as it is at the top of a pipe for example, the water will move preferentially along the long axis of the pipe. Axons in the brain are basically pipes that happen to be covered with myelin. Movement of water in a pipe, or axon, is known as anisotropic diffusion, with diffusion along the x-axis (the long axis of the pipe, or axon) greater than diffusion along the y-axis (the narrow width of the pipe, or axon). Anisotropic diffusion can be measured in a magnet (i.e., a magnet in the MRI system) and a map of anisotropy produced (i.e., in the MRI image). For example, Figure 4-1 shows the difference in fractional anisotropic (FA) maps between a 16-year-old male with metabolic syndrome and a 16-year-old control male, with the red indicating where the white matter fibers are most compact (in the corpus callosum, internal capsule, etc.), that is, where water is most constrained. The yellow indicates less tightly packed white matter. The green areas are where water is least constrained, that is, in the
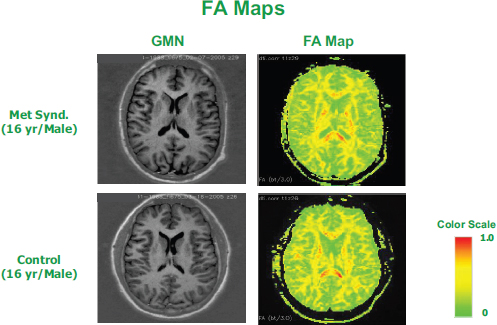
FIGURE 4-1 Gradient moment nulling (GMN) images and fractional anisotropic (FA) maps of the brains of a 16-year-old male with metabolic syndrome and a 16-year-old male without metabolic syndrome. The reduced red and yellow areas in the FA map of the child with metabolic syndrome reflect reduced white matter microstructural integrity.
SOURCE: Presented by Antonio Convit on February 26, 2015.
gray matter where the cerebrospinal fluid and neurons are located and where only membranes can prevent water from moving freely.
Using FA mapping, Convit and his team compared, voxel by voxel, the brains of children with metabolic syndrome to those without and found a significant reduction in white matter microstructural integrity in children with metabolic syndrome (Yau et al., 2012). They also measured hippocampal volume and found it was reduced in children with metabolic syndrome compared to control children. Although the same finding has been reported for both children and adults with type 2 diabetes, according to Convit, the Yau et al. (2012) results suggest that even children who are just prediabetic already have reduced hippocampal volume and more brain atrophy than children who are not.
Not all factors that contribute to metabolic syndrome are equal, in Convit’s opinion. Based on a multivariate analysis of all factors, insulin resistance appears to be the driver, not cholesterol, blood pressure, or any other measured factor. The higher the QUICKI score—so the more insulin
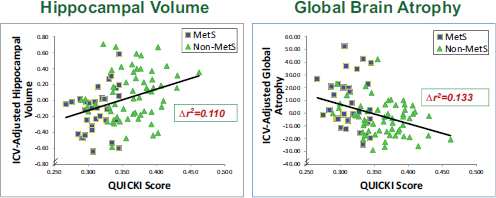
FIGURE 4-2 Associations between insulin sensitivity and hippocampal volume (left) and global brain atrophy (right).
NOTE: ICV = intracranial vault; Mets = metabolic syndrome.
SOURCE: Presented by Antonio Convit on February 26, 2015, from Yau et al., 2012.
sensitive a child is—the greater the hippocampal volume and the less global brain atrophy, with both outcomes adjusted for intracranial vault size (see Figure 4-2) (Yau et al., 2012).
In terms of a mechanism that might explain metabolic dysregulation–associated brain problems in adolescents, Convit suggested that microvascular dysfunction likely plays a role. He described what happens to your brain when you are trying to leave the house but cannot find your keys: the parts of your brain that are trying to help you find the keys get activated and start using glucose extremely avidly. However, it is known from microdialysis experiments in animals that the glucose transporter at the blood–brain barrier has a low Km value,10 meaning that it is fully saturated at normal glucose levels and, Convit said, “can’t move any faster to get you more juice to those parts of the brain that need it.” The only way to shuttle more glucose to those parts of the brain that need it is to expose more glucose transporters by relaxing the capillary bed. But with insulin resistance, endothelial dysfunction compromises the ability to relax the capillary bed. The same may be true with inflammation, he said.
One way to noninvasively examine microvascular dysfunction in children is by examining their retinas and measuring their retinal vessels. Convit explained that, developmentally, retinal vessels come from the same place that cerebral microvasculature does, and they are controlled by similar physiology. By having a child sit in a dimly lit room and dilating his or her pupils, researchers can take a picture of the eye and measure both the veins and
______________
10 The Km value is an indicator of the affinity of the transporter for glucose molecules, with a low Km value suggesting a high affinity.
arteries across the retina; the larger the artery, the healthier the individual. High blood pressure and dyslipidemia are just two of many factors that reduce the size of the retinal arteries. In a study of adolescent boys and girls, Convit and colleagues found that, when controlling for high blood pressure and other confounding factors (which together explained about 9 percent of the variance) as well as for vessel size (which explained about 33 percent of the variance), BMI and insulin resistance explained about 20 percent of the variance in arterial size (Tirsi et al., 2013).
In summary, Convit reiterated that retinal arterial size is potentially a very noninvasive way to obtain a cheap biomarker for microvascular dysfunction and, by extension, metabolic dysfunction. Using MRI technology and other methods, he and his team have found huge vascular responses in the brain under certain conditions, but with children who are insulin resistant showing blunted vascular responses.
Potential Public Health “Handles” for Intervention
Convit identified two easy public health “handles” to reduce or prevent metabolic dysfunction in adolescents: fitness and sleep. Convit and colleagues measured fitness in children in a laboratory setting and related their fitness measures to weight–height ratios and found that weight–height ratios accounted for 85 percent of the variance in fitness. The findings suggested to Convit a “very tight relationship between the weight–height ratio in the child and how fit the child is.” More importantly, in his opinion, when fitness was plotted against insulin sensitivity (QUICKI score), there was a direct correlation, with fitness increasing as insulin sensitivity increased. According to Convit, more sophisticated measures of insulin sensitivity have yielded the same results. He said, “No matter what measure of insulin sensitivity we use, we get the exact same measurements.”
Convit observed that when school budgets are cut, it is usually gym and art that are cut first, with more investment in “academic” time. The problem with that strategy, Convit said, is that according to a New York City (NYC) Department of Health and Mental Hygiene study of 550,000 students in NYC public schools, children with higher BMIs are less fit than children with lower BMIs (Egger et al., 2009). In addition to finding a correlation between BMI and fitness, when the researchers compared children with the top third fitness scores to those with the bottom third, they found statistically different grades and, more importantly in Convit’s opinion, statistically different standardized state exam results. When they compared children with the top 5 percent fitness scores to those with the bottom 5 percent, they found that the more fit children scored 36 percentile points higher on standardized tests than the less fit children. “That’s the difference between an A and a D,” Convit said. He noted that those children are at
the extremes of the distribution and that some of the unfit children may have had other illnesses, such as asthma, that could have contributed to their being unfit. Nonetheless, in his opinion, the results demonstrate that fitness is a key predictor of academic performance.
Regarding sleep, Convit said that the sleep of a healthy athletic individual can be manipulated such that within 1 week the individual will become insulin resistant. Researchers can do that in a sleep lab by disrupting sleep every time an individual enters slow wave sleep. Slow wave sleep is recuperative sleep that occurs during the first 3 to 4 hours of sleep. Based on a recent animal study, slow wave sleep appears to play an important role in cleaning toxins out of the brain. According to Convit, slow wave sleep is important not just for recuperation and cleaning toxins out of the brain, but has also been shown to be associated with insulin resistance and inflammation. The more slow wave sleep that a child has, the more insulin sensitive he or she is and the less inflammation he or she has.
Conclusion
In conclusion, Convit summarized that there are clear brain abnormalities associated with obesity and metabolic dysregulation and that the abnormalities appear to be driven by insulin resistance, with the impact of insulin resistance on cerebral vascular reactivity a plausible mechanism. In his opinion, researchers may be able to use something as simple as retinal arteriolar width as a biomarker in field trials and in other situations requiring noninvasiveness. Meanwhile, he views fitness and sleep as two interventions that could be implemented to incentivize children to stay healthier.
PANEL DISCUSSIONS WITH SPEAKERS
The Session 3 presentations were accompanied by two separate panel discussions, one following Mark Vickers’s presentation and the other following Antonio Convit’s presentation. Both panel discussions are summarized together here.
Phenotypic Outcomes Associated with Maternal Under-Nutrition Compared to Over-Nutrition
Matthew Gillman asked Karen Lillycrop and Mark Vickers why they thought similar phenotypes emerge at both “ends” of prenatal nutrition, that is, why the risk of obesity and other metabolic dysfunctions is similar for both under- and over-nutrition during pregnancy. Vickers replied that although many people associate obesity with macrosomia and fetal overgrowth, his models have associated maternal obesity with fetal growth
restriction and placental insufficiency in both structure and function. Moreover, most rat models of maternal obesity do not lead to any overgrowth or macrosomia at all. He suspected that the pathways may be similar for both under- and over-nutrition, resulting in similar phenotypic outcomes. With respect to what those outcomes are, he described offspring at both ends of the spectrum tending to be hyperleptinemic and hyperinsulinemic, with reduced birth weight and length.
Lillycrop added that it would be very interesting to compare the undernutrition and over-nutrition pathways in humans and examine epigenetic changes, or signatures, associated with each pathway. If over-nutrition is in fact a consequence of malnutrition in terms of micronutrients, she agreed that the pathways might not be so disparate.
The Mismatch Hypothesis
Gillman asked whether a high-fat diet in utero followed by a high-fat postnatal diet would be “good” based on the mismatch hypothesis, saying, “That’s a perfect match.” Kevin Grove replied that it depends on the severity. With significant placental insufficiency and a small-for-gestational-age outcome, he suggested that, yes, long-term fat may be “the answer.” The metabolic outcomes being observed suggest that high-fat-diet offspring may deal better with fat than control-diet offspring. However, the other complications that eventually emerge in association with a high-fat diet, such as inflammation and brain complications, probably end up in the long-term causing more damage.
“We don’t know,” was Vickers’s answer. He observed that many people argue that the mismatch hypothesis works for only one side of the equation, that is, under-nutrition. But, in his opinion, no one has done enough work to see if a high-fat in utero diet matched with a high-fat postnatal diet could result in healthy infants.
Lillycrop added that her research group has bred mice over four generations on a high-fat diet and has observed some adjustment in terms of metabolism. But when the mice start to age, she said, the dysfunction begins to appear.
The Placenta
The speakers were asked whether placental shape or length and width, which could be measured by ultrasound, might be useful as an indicator of risk of obesity. Grove replied that he had spoken many times with David Barker about that possibility. The challenge, Grove said, is that measurements of the size and shape—or as Barker described it, the “ovality”—of the placenta and their association with dysfunction are typically determined
postpartum. It is not clear what a dysfunctional placenta looks like in pregnancy. Moreover, placental shape and size are highly variable among women, and they depend on how many pregnancies a woman has had. He said that such an indicator would be possible, yes, but he encouraged finding a better biomarker.
Testing Dietary Interventions in Animals Versus Humans
In Jacob Friedman’s opinion, the next necessary step to studying all of the various nutritional interventions being studied in animal models is to test them in “higher” organisms and to be aware of what might be different in humans. In an effort to translate some of what has been learned in animal models to humans, his research team conducted what he said was the first randomized control diet–treated study (Hernandez et al., 2014). They randomized pregnant women diagnosed with gestational diabetes into either a “conventional” diet, that is, one that was low carbohydrate and high fat, or a high-complex-carbohydrate, low-fat diet. They found that, over time, patients on the high-complex-carbohydrate, low-fat diet had better insulin resistance scores, suppressed fat cell inflammation (based on biopsy), and fewer fat infants. Freidman emphasized that as part of the study, patients were given every single calorie of their respective diets. They did not make their own choices and then tell the researchers what those choices were. He implied that dietary interventions work, or can work, when people know what to eat. “If you’re trying to get into the real world,” he said, “this is where you have to start.”
Grove added that, in his opinion, more than being told what they need to eat, mothers who are obese need hope. He described obese mothers as feeling helpless, anxious, and depressed that their children are going to experience some of the same problems they have experienced. Part of giving them hope is telling them that something can be done and that diet is something that can be changed quickly. Then they become motivated. “I think that has to be the place we start,” he said.
In response to Friedman’s comments, Vickers remarked that the order of experiments is an interesting issue, that is, whether animal model or human studies are conducted first. He noted that a small clinical trial is under way in New Zealand testing Viagra in women at risk of very low birth weight infants, despite all animal data pointing to long-term negative consequences.
The challenge with dietary interventions in humans is not just what, but when, Marie-France Hivert reminded the workshop audience. She mentioned that screenings for gestational diabetes or gestational hyperglycemia are typically conducted in the middle of pregnancy or later. But given that the early phases of fetal development are especially sensitive to epigeneti-
cally mediated programming, perhaps these conditions should be screened and treated earlier.
Hivert’s comment prompted Lillycrop to mention a couple of diet- and exercise-based intervention trials currently under way in the United Kingdom that target obese mothers about midway through pregnancy. She noted some success with those interventions at that point in pregnancy. Given that the most plastic period of development may be earlier, she wondered if the interventions should be targeting mothers earlier during pregnancy—for example, at 6 or 7 weeks. But the challenge with that, she said, is that most high-risk women are not aware at that point that they are pregnant.
Hivert added that when conducting dietary intervention studies in humans, researchers need to start thinking about longer-term outcomes. Most outcomes being measured for dietary interventions are short-term maternal or birth outcomes. Hivert’s comment prompted Friedman to ask about the metformin trial, which he said is following children whose mothers were administered metformin and measuring outcomes. The children, he said, should be about 7 or 8 years old now. Vickers replied that while he did not know much about that particular trial, he was under the impression that the long-term outcomes were not optimal. He agreed with Hivert that, too often, people do not see past the immediate effect (of an intervention). But there are long-term trade-offs, he said. Again, he mentioned the Viagra trial under way in New Zealand and remarked that while administration of the drug allows for a fetus to be born and viable, by maturity the child will be hypertensive, obese, and insulin resistant. The long-term trade-offs are, he said, “pretty bad.” He said that the metformin trial sounds similar. He also mentioned some vitamin supplement studies in the United Kingdom, where researchers are observing long-term outcomes opposite to what was expected.
Strain Specificity in the Microbiome
Given the hundreds of thousands of microbes living in the human body, Stephen Krawetz was curious about ways to identify other functional genes besides those identified via 16S rRNA sequencing. William Nierman replied that one can analyze the microbiome at different levels depending on resources. 16S rRNA sequencing is the quickest and less expensive way to obtain information. At the other end of the scale, he said, is whole metagenomic sequencing, which provides very good coverage of most of the organisms present. Alternatively, polymerase chain reaction (PCR) assays can be used to isolate and sequence individual genes. Meredith Hullar added that in one of the studies she had described—the Vital et al. (2013) study on the production of the short-chain fatty acid butyrate by four different metabolic pathways—the researchers created sequencing primers
specific to those pathways. That seems to be the trend right now, she said. She also replied that in order to identify the functional genes identified by metagenomic sequencing, it is helpful to relate the sequences to a database of known bacterial representatives. Historically, genome sequencing was dominated by human pathogens, while the genomes of bacteria in healthy individuals were underrepresented. Despite the great progress of the HMP, Hullar replied that there is more work to be done to expand the microbial genome reference database.
Microbiome Transplants
Jacob Friedman expressed fascination with an observation made in the Netherlands that transplanting a bacterial microbiome from a healthy teenager into an individual with metabolic syndrome can lower the recipient’s insulin resistance and improve his or her health. He observed that the same thing can be done with a gnotobiotic mouse. However, eventually, with both humans and gnotobiotic mice, their health reverts to what it was before the transplant, raising the question for Friedman, how far down, taxonomically, does one need to go to understand which microbe to transplant and is it even possible to go down to the sub-species level?
Nierman replied that the work he had mentioned by Sarah Highlander involved single species management of, in many instances, near fatal C. difficile infections in antibiotic-treated humans. As far as he was aware, she identified three or four individual strains of Lactobacillus that brought about rapid remission of symptoms. He emphasized, however, that even without an intervention, after a disturbance, whether that disturbance is a severe episode of antibiotic treatment or a dramatic alteration in diet, the microbiome eventually returns on its own to something resembling its former state. “But it takes a bit of time to achieve that,” he said. He explained that the transient nature of the change observed after a microbiome transplant is a consequence of the transient proliferation of whatever it is that has been put there to compete with everything else that is there. Eventually, over time, the microbiome returns to its former state.
Effects of Metabolic Dysfunction on the Brain
In Kevin Grove’s opinion, metabolic phenotypes are “the least of our worries.” He thinks that some of the behavioral responses being observed in his animal studies are of greater concern. He said, “When you start messing with social cues and cognition, you’ve got lifelong problems in your whole community.”
Antonio Convit was asked if he had redone any of his retinal arterial or sleep analyses on adolescents after significant weight loss to see if the
weight loss affected any of the measured parameters. Convit said that he has not. He is currently in the process of trying to obtain funding for a study to longitudinally track people before and after bariatric surgery and to assess the effect of surgery on the brain. He said that existing evidence suggests that bariatric surgery and massive weight loss improve cognition, but the mechanism is unknown.
Robert Waterland asked Convit if there are any national data suggesting that the increase in adiposity over the past few decades, including among children, has affected academic performance. Convit replied that he was unaware of any nationwide longitudinal data. He noted that the National Health and Nutrition Examination Survey (NHANES) has been tracking children and contains some educational information, but he did not know what those data have shown. Also, he noted that the difference in cognition that he and his research team reported among NYC public school children was only about 10 percent. So children who have metabolic dysregulation are not necessarily performing abnormally, he said. But they are not performing as well as those who do not have metabolic dysregulation. He reiterated the importance of fitness in adolescents. Even if parents have to pay their children to play sports, he said, “that’s going to cost you a lot less than dealing with all the complications of not playing the sports.”








































