
Evaluation of the ethical, social, and policy issues associated with mitochondrial replacement techniques (MRT) requires a comprehensive understanding of the state of the science surrounding reproductive biology and medicine, mitochondrial biology and genetics, mitochondrial DNA (mtDNA) disease, and MRT itself. In acknowledging the need for this scientific understanding, the committee also recognizes that it is the purview of the U.S. Food and Drug Administration (FDA) to thoroughly review the safety and efficacy of MRT and to determine whether the preclinical data package is sufficient for the agency to move forward with evaluation of applications for clinical investigations of MRT. Therefore, this chapter should be viewed as a nonexhaustive review of the literature surrounding MRT for the purposes of providing scientific context to inform the committee’s analysis in succeeding chapters, not a judgment of the adequacy of the state of the science. To this end, the following topics are reviewed in this chapter: (1) reproductive biology and medicine, (2) mitochondrial biology and genetics, (3) mtDNA disease, (4) MRT research to date, and (5) potential risks related to MRT. The final section of the chapter describes the policy context for this study.
INTRODUCTION TO REPRODUCTIVE BIOLOGY AND MEDICINE
A prefatory summary of concepts in reproductive biology and medicine is provided here to inform subsequent discussions of mitochondrial biology and genetics, mtDNA disease, and MRT in this chapter and the ethical
BOX 2-1
Terminology in Reproductive Biology and Medicine
Blastocyst: The stage in early embryonic development, typically 4-5 days following fertilization in vitro, when the embryo comprises approximately 200-300 cells and a hollow cavity termed the blastocoel.
Embryo: Following dissolution of the pronuclear membranes and fusion of the male and female genetic material, the zygote divides to form the two-cell embryo, each cell containing equal complements of genetic and cytoplasmic material.
Gamete: An egg (oocyte) or sperm (spermatozoa) cell. In the process of fertilization, the fusion of male and female gametes gives rise to the zygote.
Germ cells: Gametes and those cells that give rise to gametes, originating with the primordial germ cells, the common precursor of both oocytes and spermatozoa.
Germline: Collectively, germ cells make up the germline.
Pronucleus: The membrane-bound nuclear genetic material derived from the oocyte or spermatozoa following fertilization.
Somatic cells: All cells of the human body that are not germ cells.
Zygote: A single cell formed following fertilization, containing separate male and female pronuclei that replicate before fusing. The zygote is sometimes referred to as a one-cell embryo, although this report does not adopt this terminology.
analysis presented in Chapters 3 and 4. The terminology of reproductive biology and medicine used throughout the report is summarized in Box 2-1.
Formation of Embryos, Germ Cells, and Gametes
Gametes are the fundamental cells involved in human reproduction and development. The initial step in human reproduction involves the fusion of an egg (oocyte) and sperm (spermatozoa) cell (fertilization), resulting in the formation of a zygote. At this stage, the zygote contains both the male and female pronuclei and is therefore termed di-pronucleate, or 2-PN. The pronuclear genetic material first replicates before the respective nuclear membranes dissolve, followed by fusion of the male and female genetic material and equivalent division of genetic and cellular material to form the two-cell embryo. The resultant embryo will undergo rapid cell division and differentiation, acting as the fundamental precursor cells for all the cells of the human body (see Figure 2-1). Derived from the embryo—specifically, from embryonic stem cells of the inner cell mass—are two distinct cell lineages: somatic cells and germ cells. Somatic cells differentiate from embryonic stem cells to form all of the cell and tissue types of the human body; primordial germ cells differentiate from embryonic stem cells along
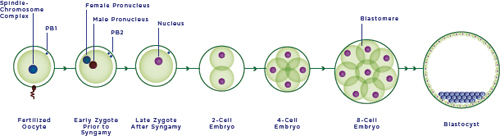
FIGURE 2-1 Embryogenesis.
NOTES: The initial step in human reproduction involves fertilization, the fusion of an oocyte and sperm cell, resulting in the formation of an early zygote. At this stage, the early zygote contains both the male and female pronuclei and is therefore termed di-pronucleate, or 2-PN. The pronuclear genetic material first replicates before the respective nuclear membranes dissolve, followed by fusion of the male and female genetic material in the late zygote, and equivalent division of genetic and cellular material to form the two-cell embryo. The resultant embryo undergoes rapid cell division, forming the 4- and 8-cell embryo and after many additional divisions, the blastocyst.
SOURCE: Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics, copyright 2005.
distinct pathways to develop into either spermatozoa in the case of males or oocytes in the case of females. The cells that make up these germ cell lineages are referred to collectively as the germline.
Changes to the genetic material of germline cells are heritable in the case of nuclear DNA (nDNA) and maternal mtDNA. Paternal mtDNA is not transmitted to offspring, and thus changes made to mtDNA in the male germline are not heritable.1 Mitochondrial and nuclear genetics and inheritance patterns are discussed later in this chapter in the section on mitochondrial biology and genetics.
In Vitro Fertilization
In vitro fertilization (IVF) is an assisted reproductive technology (ART) traditionally used to aid a woman in becoming pregnant when unassisted sexual reproduction and other ARTs, such as fertility medications and artificial insemination, fail to produce a pregnancy.
_________________
1 Although paternal transmission of mtDNA in humans was noted in a high-profile case report (Schwartz and Vissing, 2002), studies of children born following intracytoplasmic sperm injection (ICSI) have failed to detect transmission of paternal mtDNA (Houshmand et al., 1997; Marchington et al., 2002). At present, therefore, it is believed that maternal transmission is the rule in humans.
In general, IVF comprises five steps: (1) stimulation, or super ovulation, to produce a larger than normally released number of oocytes; (2) oocyte retrieval, on the order of 5-30 oocytes per stimulation cycle, requiring sedation of the woman undergoing the biopsy procedure; (3) mixing of sperm with preselected, high-quality oocytes (insemination) or, more commonly, direct injection of sperm into each oocyte, termed intracytoplasmic sperm injection (ICSI); (4) culture of the embryo to day 3 or 5 (blastocyst stage); and (5) transfer of the cultured embryo to the woman who will carry the pregnancy (i.e., the intended mother or a gestational carrier). MRT is a collective set of modified IVF techniques (see the description of MRT methodology later in this chapter).
Preimplantation Genetic Diagnosis (PGD)
PGD is a technique performed in the setting of IVF to test for a known inherited genetic disease and to allow selection of embryos for transfer to the uterus of the woman who will carry the pregnancy, with the goal of establishing a viable pregnancy and preventing transmission of that disease.2 Once a viable pregnancy has been achieved, additional prenatal diagnostic testing is essential to confirm the genetic information obtained by PGD, entailing chorionic villus sampling of fetal placental tissue, amniocentesis of discarded fetal cells, or cell-free DNA screening. The use of PGD for preventing transmission of mtDNA disease is discussed later in this chapter.
INTRODUCTION TO MITOCHONDRIAL BIOLOGY AND GENETICS
Mitochondria are microscopic organelles found in nearly all cell types of the human body,3 best known for their role in regulating cellular energy balance. They are among the most complex cellular organelles, consisting of more than 1,100 proteins that collectively support the mitochondria’s
_________________
2 Briefly, one to several single blastomeres of a post-IVF day 3 or day 5 embryo are tested in the laboratory for the known genetic condition for which the embryo is at risk. If the blastomere biopsy is performed on day 3, the embryo can remain freshly cultured in the laboratory until genetic test results are returned; the desired embryo(s) can then be transferred to the uterus on day 5. However, if the blastomere biopsy is performed on day 5—as is now more common given that embryos generally have greater viability on day 5 than on day 3—all of the embryos in that cycle are frozen until genetic testing on each is complete. At any point in the future, as soon as the following month or up to years later, the woman who will carry the pregnancy then undergoes an additional hormone preparation cycle, and the desired frozen embryo(s) are thawed and implanted into her uterus.
3 With the exception of mammalian red blood cells (erythrocytes) and mature ocular lens cells, which do not contain organelles and thus do not contain mtDNA.
myriad roles, including production of cellular energy, regulation of cellular metabolism, and assistance in control of programmed cell death (apoptosis).
According to the widely accepted endosymbiotic hypothesis, these organelles once were free-swimming bacteria, adept at harvesting energy by burning oxygen, that took up permanent residence within another cell (Vafai and Mootha, 2012). Several features of mitochondria serve as reminders of this unique ancestry. Mitochondria measure 500 nm-1 μm (approximately 1/50 the width of a human hair), have a double membrane,4 and constantly “swim” within the cells of the body—very much resembling intracellular bacteria. They have retained their own genome5 (mtDNA), another vestige of their bacterial origin. Over billions of years of evolution, virtually all of the genes once encoded by this primordial bacterial genome have been either lost or transferred to the nuclear genome. Today, human mtDNA, which is mutated in mtDNA disease, encodes 13 proteins that must operate functionally with more than 1,100 nuclear encoded proteins that are imported into mitochondria to shape the organelle’s function.
Biological Functions of Mitochondria
Cellular metabolism refers to the set of biochemical processes within a cell that generate, store, or utilize energy through the making (anabolism) or breaking (catabolism) of chemical bonds between molecules. A primary function of mitochondria is to produce the majority of energy that is needed to fuel cellular processes; thus these organelles are often referred to as “the powerhouses of the cell.” The nutrients people eat, such as carbohydrates, fats, and proteins, are broken down within the cell to form intermediate byproducts that are sent to the mitochondria, where they are processed further to produce energy in the form of adenosine triphosphate (ATP), the predominant molecule for storing and providing energy for cellular processes. This process by which ATP is produced, termed oxidative phosphorylation
_________________
4 This unique inner and outer double membrane structure allows mitochondria to compartmentalize cellular components. The space between the inner and outer membrane is termed the intermembrane space. Oxidative phosphorylation takes place by pumping protons across the inner membrane into the intermembrane space, forming the electromotive force used to drive adenosine triphosphate (ATP) synthesis. The space enclosed by the inner membrane is termed the mitochondrial matrix and is home to mtDNA, as well as the majority of mitochondrial components required for the mitochondrion to carry out its various functions. The double membrane is reflective of the ancestral bacterium from which the mitochondria derived, namely a gram negative bacterium, which also contained a double membrane.
5 The genome is the collective genetic material found within an organism. In humans, the cellular genome comprises the nuclear and mitochondrial genomes.
(OXPHOS),6 occurs at the respiratory chain7 and ATP synthase, located within the mitochondrial inner membrane. For this reason, cells with higher energy demands, such as muscle and brain cells, contain higher numbers of mitochondria so they can meet these energy requirements. In addition to this critical function, the mitochondria are principal regulators of a variety of cellular metabolic functions, play an important role in maintaining the proper intracellular environment, and are an integral component of apoptosis. The role of mitochondria in these various biological processes underscores the critical importance of proper mitochondrial function for sustaining human life.
The Respiratory Chain and Oxidative Phosphorylation
OXPHOS involves 5 protein complexes comprising a total of 90 proteins, 13 of which are encoded by mtDNA. The principal function of OXPHOS, discussed above, is to generate energy in the form of ATP. In mtDNA disease, mutations in mtDNA result in a lack or defective production of one or more mtDNA-encoded gene products, leading to varying degrees of dysfunction in respiratory chain activity and energy production.
Other Metabolic Pathways Within Mitochondria
As a result of electrons being driven through the respiratory chain in the process of OXPHOS, other metabolic processes can move forward as well, a process known as metabolic coupling. In this way, OXPHOS is coupled with other metabolic pathways within and external to the mitochondria. For example, mitochondria contain the machinery necessary to convert the fats, proteins, and carbohydrates people eat into intermediates that feed directly into the respiratory chain. Breakdown intermediates from these metabolic processes within the mitochondria can be exported back into the cytosol, where they are used as precursors for other molecules, such as sex hormones, fatty acids, DNA, and proteins. In mtDNA disease, these coupled reactions—in addition to OXPHOS—are also disrupted and can contribute to the observed disease clinical phenotypes.
_________________
6 OXPHOS is the process by which the respiratory chain generates a proton gradient across the mitochondrial inner membrane via transfer of electrons from a higher-energy donor to lower-energy cellular intermediates, terminating with formation of the terminal electron acceptor, oxygen. The electromotive force generated by this proton gradient is utilized by a protein complex, ATP synthase (complex V), to produce ATP.
7 Also known as the electron transport chain (ETC).
TABLE 2-1 Comparison of Human Nuclear and Mitochondrial Genomes
| Characteristic | Mitochondrial | Nuclear |
| Genome Structure | Circular | Linear |
| Copies of the genome per cell | 100-10,000 (more than 100,000 in mature oocytes) | 2 |
| Number of DNA base pairs | 16,569 | 3.3 billion |
| Number of coding genes | 37 | Approximately 20,000-30,000 |
| Function of gene-encoded products | OXPHOS function; mtDNA-encoded protein translation | All remaining intra-and extracellular functions required for cellular, tissue organ, and bodily functions; phenotypic traits, such as physical appearance |
| Mode of inheritance | Maternal | Biparental |
NOTE: mtDNA = mitochondrial DNA; OXPHOS = oxidative phosphorylation.
SOURCE: Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics, copyright 2005.
mtDNA Genetics and Inheritance
Mitochondria are unique in that they house mtDNA, the only ex-tranuclear source of DNA within animal cells. While mtDNA has some commonalities with nDNA, such as comprising double-stranded DNA and containing protein-encoding genes, the two differ in many ways (as summarized in Table 2-1). These differences have important implications for mtDNA disease and MRT, expanded on throughout this and subsequent sections within this chapter.
Genome Structure and Function
The mitochondrial genome contains 37 genes, 13 of which encode for proteins that are core components of the respiratory chain and OXPHOS system, with the remaining 24 assisting in the translation of OXPHOS proteins. By comparison, nDNA encodes for an estimated 20,000-30,000 protein-encoding genes. Compared with the only 2 copies of the 23 nuclear chromosomes in almost all somatic cells, mtDNA is found in high copy number,8 ranging from 2 to 10 copies per mitochondrion and 100 to
_________________
8 The copy number is the number of mtDNA molecules per cell.
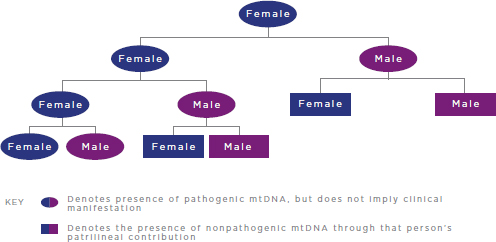
FIGURE 2-2 Inheritance of pathogenic mtDNA mutations.
NOTES: For simplicity, reproductive partners are not shown and are assumed not to carry pathogenic mtDNA mutations. mtDNA = mitochondrial DNA.
10,000 copies per cell, depending on cell type, with up to 500,000 copies in oocytes. Replication of mtDNA occurs continuously throughout the cell cycle and autonomously from nDNA, which is replicated once per cell cycle; the resulting mtDNA molecules are partitioned randomly into the daughter cells during cell division.9 While mtDNA encodes for products that are essential for the production of cellular energy, it is generally agreed that nDNA plays the predominant role in determining characteristics of anatomy, physiology, personality, and the like.
Mode of Inheritance
As noted previously, mtDNA is solely maternally inherited in humans (see Figure 2-2). Thus, only females pass their mtDNA on to offspring, both male and female; male mtDNA is not transmitted to future generations.10
_________________
9 Cell division, or mitosis, is the stage of the cell cycle that results in division of the “parent” cell into two “daughter” cells, each containing the same number of chromosomes as the parent cell.
10 Several mechanisms help ensure maternal transmission of mtDNA. First, the unfertilized oocyte has up to an estimated 500,000 copies of mtDNA, compared with approximately 100 copies of mtDNA in sperm cells, so simple dilution makes it statistically unlikely for paternal mtDNA to be transmitted. Furthermore, the mitochondria of sperm cells are “tagged” by the oocyte for degradation following fertilization (Sutovsky et al., 1999).
In contrast, nDNA is inherited both maternally and paternally, following what is known as Mendelian or biparental inheritance.11
Heteroplasmy
An additional, notable feature of mitochondrial genetics is the concept of heteroplasmy. Heteroplasmy is the state in which a cell, tissue, or person contains more than one mtDNA genotype, as opposed to the state in which all copies of the mitochondrial genome are identical, termed homoplasmy. For example, a cell whose mtDNA consists of 70 percent mutant mtDNA and 30 percent wild-type12 mtDNA is termed heteroplasmic, whereas a cell with 100 percent mutant mtDNA is termed homoplasmic. The concept of heteroplasmy and its relation to mtDNA disease and MRT is explored further in the section on complexities related to mitochondrial genetics later in this chapter.
Genetic Interactions Between nDNA and mtDNA
The mitochondrion requires extensive contributions from nDNA to perform all of its critical functions, including those encoded for by mtDNA. Eighty proteins necessary for OXPHOS function and more than 1,000 others required for mitochondrial activity and structure are encoded by the nuclear genome and imported into the mitochondria. Maintenance of this nuclear-mitochondrial cross-talk is essential for establishing and maintaining proper mitochondrial function (Lee et al., 2008). The crosstalk between the nuclear and mitochondrial genomes is an important consideration in evaluations of MRT, as its disruption could have potentially deleterious effects on overall mitochondrial and cellular health (see the section on complexities related to mitochondrial genetics later in this chapter).
mtDNA Genetic Variance in Human Populations
mtDNA molecules acquire novel mutations at a rate at least 10 times greater than that of nDNA molecules. If such mutations are acquired within oocytes, they are transmitted to any offspring conceived from those
_________________
11 This is true for the 22 autosomal, or non-sex-determining, chromosomes. The X and Y chromosomes are responsible for determining the sex of an organism—in humans, XX for females and XY for males—and can display slightly different inheritance patterns. A comprehensive overview of DNA inheritance patterns can be found at http://ghr.nlm.nih.gov/handbook/inheritance/inheritancepatterns (accessed January 15, 2016).
12 Wild-type is the most common DNA sequence found within a population, often referred to as the “normal” variant of a DNA sequence or gene.
oocytes. Lack of recombination between mtDNA molecules13 and sole matrilineal inheritance of mtDNA means that acquired mtDNA mutations can be passed down via radiating maternal lineages. From an evolutionary standpoint, the persistence of certain maternally transmitted homoplasmic mtDNA mutations has resulted in the formation of stable population subgroups, known as haplogroups, sharing the same collection of fixed mtDNA variants, or haplotypes. As the females who migrated out of Africa helped colonize the globe and novel mtDNA mutations were acquired, new haplogroups branched out from the original “macrohaplogroups” (Wallace and Chalkia, 2013). The retention of novel mtDNA mutations in evolution may have been a result of random genetic drift, in the case of neutral mtDNA mutations, or of selective pressures, in the case of mtDNA mutations that conferred advantageous traits or characteristics to individuals in novel geographic regions, wherein those haplotypes became enriched (Wallace, 1994). Continents and geographic regions are therefore associated with specific mtDNA haplogroups, which might confer certain physiological advantages to individuals who live there (Wallace and Chalkia, 2013).
A few high-profile studies have provided evidence substantiating the hypothesis that certain mtDNA haplogroups underwent positive selection as an adaptive mechanism for populations that migrated to colder climates (Mishmar et al., 2003; Ruiz-Pesini et al., 2004). These studies indicate that certain mtDNA variants result in inefficient energy production by mitochondria and concurrent generation of heat. Accordingly, theory suggests that increased heat generation conferred a selective advantage to individuals living in colder climates. Thus such variants became enriched and eventually fixed in these populations, at the expense of less efficient energy production. A complementary hypothesis is that certain mtDNA haplogroups confer an energetic advantage, such as enhanced exercise capacity, to individuals through more efficient energy production and less heat generation by mitochondria. Indeed, some studies have shown a correlation between certain mtDNA variants and relative exercise performance or aerobic capacity (see Eynon et al. [2011] for a review of the evidence).
Although intriguing, haplogroup/haplotype association studies are by nature correlative given the lack of experimental systems with sufficient sensitivity to validate the causal effect of mtDNA haplotypes on human physiology and cognition. Moreover, most of these studies to date have involved very small cohorts, have been statistically underpowered, and po-
_________________
13 During meiosis—the reductive replication and division of gametes—nDNA recombines to form new combinations of traits; however, this process does not specifically alter the sequence of nDNA through the introduction of novel mutations, but rather the combination of genetic variants. On the other hand, mtDNA does not undergo recombination, but is more prone to acquiring mutations; this allows the tracking of mtDNA variants through generations and among population subgroups.
tentially have been confounded by population stratification.14 Finally, such association studies have not found that specific mtDNA variants may confer a certain functional benefit, as a specific variation in nDNA confers a certain blood type. Rather, these studies suggest that a set of mtDNA variants are inherited together, make up a specific haplogroup, and are associated with certain functional characteristics in the context of certain populations.
Conclusion: The present state of scientific knowledge indicates that it is difficult or impossible to identify mtDNA haplogroups/haplotypes that would confer on an individual potentially advantageous traits or capacities such as enhanced exercise performance or aerobic capacity.
Mitochondrial diseases are highly heterogeneous, characterized fundamentally by a dysfunction in respiratory chain activity and corresponding reduced cellular energy production. In turn, the hallmark deleterious phenotypes of mitochondrial diseases tend to manifest in those organs with the highest energy demand, such as the brain, muscles, heart, gastrointestinal tract, and liver. At present, no FDA-approved treatment or cure exists for these diseases, and management approaches are primarily supportive and palliative. Mitochondrial disease can arise as a result of defects in nDNA or mtDNA (see the section on genetic origins of mitochondrial disease below).
Etiology, Clinical Manifestation, and Diagnosis
Genetic Origins
The respiratory chain is under dual genomic control,15 and thus mitochondrial diseases can be of nDNA or mtDNA origin. More than 275 disease-causing mtDNA mutations have been reported across every mtDNA gene since the first pathogenic mtDNA mutation was identified in 1988 (Saneto and Sedensky, 2013). Mutations in mtDNA can be categorized according to the gene-encoded products they disrupt: (1) mutations affecting OXPHOS proteins and (2) mutations affecting the translation machinery of OXPHOS proteins. Furthermore, pathogenic mtDNA mutations can either arise sporadically (de novo), originating most commonly in early development, or be inherited. Table 2-2 lists the most common maternally inherited mtDNA diseases and their associated mtDNA mutations.
_________________
14 Population stratification is differences in nuclear allele frequencies between research subjects due to systematic differences in ancestry (Price et al., 2006).
15 Control by both the nuclear and mitochondrial genomes.
TABLE 2-2 Maternally Inherited mtDNA Diseases
| mtDNA Disease | Clinical Presentation | mtDNA Gene/Genotype* |
| Leigh Syndrome | Psychomotor delay, dystonia, seizures, abnormal eye movements, recurrent vomiting, respiratory abnormalities | ATPase6: m.8993 T > G ND1, ND2, ND3, ND4, ND5, ND6, COX III, others: multiple |
| MELAS | Myopathy, encephalopathy, lactic acidosis, stroke-like episodes | TRNL1: m.3243A > G; m.3271T > C ND1 and ND5: individual mutations |
| MERRF | Myoclonic epilepsy, myopathy | TRNK: m.8344A > G; m.8356T > C |
| NARP | Neuropathy, ataxia, retinitis pigmentosa | ATP6: m. 8993T > G |
| MILS | A progressive brain-stem disorder | ATP6: m8993T > C |
| MIDD | Diabetes, deafness | TRNL1: m.3243A > G MT-RNR1: m.155A > G |
| Nonsyndromic hearing loss and deafness | Nonprogressive, moderate to profound hearing loss associated with aminoglycoside antibiotic use | MT-TS1: m.7445A > G |
| LHON | Optic neuropathy | ND1: m.3460G > A ND4: m.11778G > A ND6: m.14484T > C |
NOTES: * The most common pathological mtDNA point mutations are listed. LHON = Leber’s hereditary optic neuropathy; MELAS = mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF = myoclonic epilepsy with ragged-red fibers; MIDD = maternally inherited diabetes and deafness; MILS = maternally inherited Leigh syndrome; NARP = neuropathy, ataxia, and retinitis pigmentosa.
SOURCE: Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics, copyright 2005.
Clinical Presentation and Diagnosis
mtDNA diseases can range in severity from mild to severely debilitating or fatal, and their onset can occur in early life or adulthood. In general, mtDNA diseases tend to have later onset and to be associated with relatively milder symptoms relative to nDNA-based mitochondrial diseases, whose onset is typically earlier (often in infancy or childhood) and which
are associated with more severe phenotypes. However, at least 15 percent of pediatric-onset mitochondrial diseases are estimated to be caused by mtDNA mutations (DiMauro and Davidzon, 2005; Saneto and Sedensky, 2013), and early-onset, severe mtDNA diseases have been well documented in the clinical setting (Saneto and Sedensky, 2013). It is for this subset of mtDNA diseases that MRT would be applicable.
The principal effect of defective mtDNA is disruption of respiratory chain activity; consequent depletion of ATP levels and energy production; and eventual dysfunction and failure of cellular, tissue, and organ function. Age of onset, clinical presentation, natural history, and penetrance16 of mtDNA diseases are extremely variable, both within and across mtDNA mutations. Nonetheless, the most common disease types, such as Leigh syndrome and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), do share certain features, aiding in their clinical diagnosis. Figure 2-3 shows common clinical manifestations of adult and pediatric mtDNA diseases.
Diagnosis
Clinical diagnosis of mtDNA diseases is a complex task. However, classic diagnostic features do exist to aid physicians in making a differential diagnosis of patients with suspected mtDNA disease. These features include (1) maternal inheritance, (2) recognition of established syndromes such as MELAS, (3) recognition of characteristic clinical symptoms (e.g., biventricular cardiac hypertrophy), (4) involvement of multiple organ systems (e.g., diabetes and deafness), (5) specific combinations of symptoms (e.g., strokes, migraines, seizures, and ataxia), and (6) certain patterns of abnormal clinical and laboratory testing results (Taylor and Turnbull, 2005). Furthermore, differential diagnosis to confirm or exclude mtDNA disease may become easier with increasingly accurate and affordable sequencing technologies.
Effect of Pregnancy on Women with mtDNA Disease
The effect of pregnancy on women with mitochondrial disease in general and mtDNA disease in particular is poorly understood. As a result of the deleterious effects of mtDNA disease on cellular respiration and energy production and the concurrent increase in respiratory and energy demands
_________________
16 Penetrance is “the proportion of individuals with a mutation causing a particular disorder who exhibit clinical symptoms of that disorder; a condition is said to have complete penetrance if clinical symptoms are present in all individuals who have the disease-causing mutation, and to have reduced or incomplete penetrance if clinical symptoms are not always present in individuals who have the disease-causing mutation” (http://ghr.nlm.nih.gov [accessed January 15, 2016]).
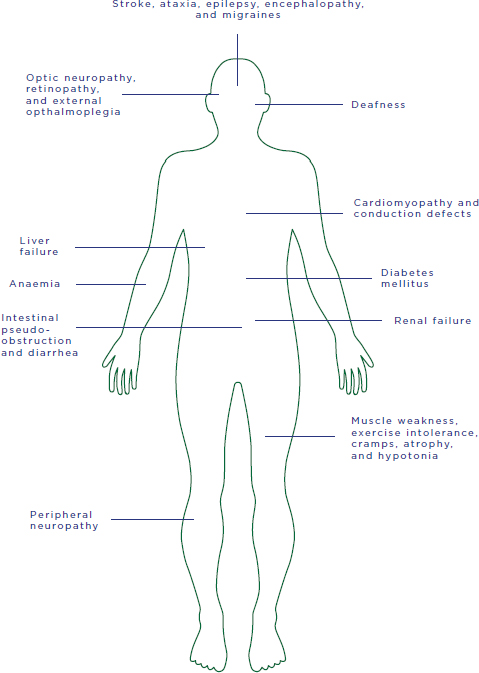
FIGURE 2-3 Potential manifestations of mtDNA diseases.
SOURCE: Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Genetics, copyright 2005.
in pregnancy, women who are at risk for or have clinically manifested mtDNA disease may develop or experience a worsening of symptoms or other obstetric complications. Given the clinical heterogeneity of mtDNA disease, the clinical course of afflicted women during pregnancy likely varies. Indeed, a review of 10 case reports of pregnancies in women with mitochondrial disease by Say et al. (2011) revealed varying levels of pregnancy complications, ranging from asymptomatic, to mild symptoms such as exercise intolerance and muscle weakness that resolved postnatally, to more serious and in some instances persistent symptoms such as kidney and nerve damage. The most commonly observed complications in this retrospective review were preterm labor and preeclampsia. To date, no cohort studies have been published on the effect of pregnancy in women with mitochondrial disease. However, an observational study currently being conducted by Robert McFarland at the University of Newcastle on Tyne is examining the incidence of pregnancy complications in patients who have mitochondrial disease or are carrying an mtDNA mutation (Feeney and McFarland, 2014). In addition, the Newcastle Mitochondrial Centre has published guidance best practices for antenatal care for women with mitochondrial disease (National Commissioning Group (NCG) for Rare Mitochondrial Diseases of Adults and Children (UK), 2013). Similarly, no studies have been published to date on the potential health effects in children gestated by women with symptomatic mtDNA disease.
Prevalence of mtDNA Disease and Pathogenic mtDNA Mutations
Determining the prevalence of mtDNA disease and prevalence of asymptomatic carriers of pathogenic mtDNA mutations has been challenging given the extensive clinical and genetic heterogeneity involved. A recent study evaluating adults (aged 16-65) referred to a mitochondrial clinic in northeast England from 1990 to 2011 estimated that at least 1 in 5,000 people harbor a pathogenic mtDNA mutation, with approximately 1 in 10,000 adults presenting with clinically manifested mtDNA disease (Gorman et al., 2015b) and 1.65 in 10,000 children and adults estimated to be at risk for development of mtDNA disease (Gorman et al., 2015b; Schaefer et al., 2008). A prospective study that evaluated the prevalence of the 10 most common pathogenic mtDNA point mutations in infants found that 0.54 percent of offspring carried at least 1 of these 10 mutations (excluding de novo mutations), suggesting that at least 1 in 200 asymptomatic people harbor a pathogenic mtDNA mutation (Elliott et al., 2008). A follow-up report to the study conducted by Gorman et al. (2015b) extrapolated from the point prevalence of pathogenic mtDNA mutations to estimate how many women may be at risk of transmitting mtDNA disease and thus could potentially benefit from MRT. Extrapolating previously ascertained
prevalence data to women of childbearing age and using fertility rates, the authors estimated that the average number of children born per year from women at risk for transmitting mtDNA disease is 152 and 778 in the United Kingdom and the United States, respectively (Gorman et al., 2015a). Such estimates are naturally tempered, however, by the fact that not all women who are at risk of transmitting mtDNA disease will decide or will be able to pursue MRT and that those who do pursue MRT may not obtain a successful pregnancy through the requisite IVF procedure.
Treatment and Prevention of Transmission of mtDNA Disease
As noted earlier, there are currently no cures or proven effective treatments for mtDNA disease (Parikh et al., 2009). Current therapeutic options for mtDNA disease focus on palliative management of an individual’s organ-specific disease symptoms as they emerge over time, rather than on targeting and correcting precise biochemical pathways (Parikh et al., 2013, 2014). This approach stems from two factors: (1) the heterogeneity of mtDNA diseases, even with respect to the same causative mtDNA mutation, which makes mutation- and patient-specific treatments highly challenging; and (2) the current lack of success in effectively delivering treatments into mitochondria with pathogenic mtDNA. Furthermore, for many women at risk of transmitting pathogenic mtDNA mutations, diagnostic techniques aimed at reliably preventing transmission of pathogenic mtDNA to future offspring (e.g., PGD or prenatal diagnosis) are not viable options, as discussed below.
Management of Symptoms
Exercise—both isotonic and aerobic, as tolerated—has been demonstrated to provide significant benefit in mtDNA disease, likely as the result of a combination of inducing the formation of new mitochondria—thereby increasing the percentage of nonpathogenic mtDNA—and preferential shifting of heteroplasmy loads toward nonpathogenic mtDNA (Tarnopolsky, 2014). A range of pharmaceuticals and nutritional supplements also are commonly prescribed to support overall mitochondrial function, despite a lack of rigorous clinical investigations validating their efficacy (Parikh et al., 2009, 2014; Pfeffer et al., 2013). Other medications have been shown to have benefit for disease-specific symptoms; examples include L-arginine to mitigate or prevent metabolic stroke (Koga et al., 2005) and folinic acid to treat changes in nervous system tissue secondary to folate deficiency (Quijada-Fraile et al., 2014). Several clinical investigations currently under way are assessing the effects of existing medications approved for other indications or of novel therapeutics developed for mtDNA disease as the
primary indication. To date, none of these therapies have been shown to have clinical efficacy or have gained FDA approval for treatment of mtDNA disease (Pfeffer et al., 2013).
Gene Editing of Somatic Cells
As with nuclear genetic diseases, gene editing of somatic cells, also sometimes known as gene transfer or gene therapy, for treatment of mtDNA disease appears to hold great promise for the clinical treatment, and potential cure, of existing mtDNA disease. In those mtDNA diseases for which the causative pathogenic mutation has been identified, gene editing would allow for precise correction of or compensation for the product of the mutated gene, thus bypassing the difficulties inherent in targeting the aberrant biochemical pathways that result from each genetic disorder. Gene-editing approaches for mtDNA disease have shown initial promise in in vitro and animal studies (Viscomi et al., 2015). However, these approaches have in general shown limited success in humans because of difficulties in delivering the therapy efficiently to the desired tissues, and in the case of mtDNA disease, in transporting the corrective/compensative material efficiently into the mitochondria containing pathogenic mtDNA.
Heteroplasmy Shift
Heteroplasmy shift is an investigational technique that selectively targets and degrades mtDNA containing pathogenic mutations, allowing for repopulation of affected cells with resident, nonpathogenic mtDNA. Cell and animal models of mtDNA disease have demonstrated its preliminary efficacy (Bayona-Bafaluy et al., 2005; Srivastava and Moraes, 2001), and more recent work has shown that it can effectively reduce heteroplasmy levels and prevent transmission of pathogenic mtDNA in mouse and mammalian oocytes and one-cell embryos. As a result, heteroplasmy shift has been proposed as an alternative to MRT for preventing maternal transmission of pathogenic mtDNA mutations that would preclude the need for the contribution of a second woman’s genetic material (Reddy et al., 2015). Unlike MRT, however, heteroplasmy shift would not be applicable for oocytes or embryos that are homoplasmic or have high heteroplasmy levels of pathogenic mtDNA, because retaining a certain baseline level of nonpathogenic mtDNA molecules in the cell is essential to enabling repopulation of the mtDNA pool and normal mitochondrial function after degradation of pathogenic mtDNA.
Preimplantation Genetic Diagnosis
PGD is a powerful technique for preventing the transmission of inherited nDNA diseases. However, only a handful of studies have evaluated PGD for selection and transfer of embryos in females at risk of transmitting known pathogenic mtDNA mutations. With at least one exception (Mitalipov et al., 2014), live-born children born following PGD generally have exhibited no adverse health outcomes, although there has been little long-term follow-up of these children beyond birth or infancy (Heindryckx et al., 2014; Monnot et al., 2011; Sallevelt et al., 2013; Steffann et al., 2006; Treff et al., 2012).
A limitation of the use of PGD to prevent transmission of mtDNA disease is that the technique involves selection of an embryo with the lowest detected heteroplasmy level; therefore, it may reduce but does not definitively eliminate the risk of transmitting mtDNA disease to offspring. Although no formal guidelines exist regarding an acceptable heteroplasmy threshold for embryo selection and transfer, Samuels et al. (2013) recently reported a model of mtDNA heteroplasmy inheritance predicting that transfer of an embryo with a heteroplasmic mutation level above 5 percent may result in a significant chance of mtDNA disease in offspring. Therefore, many families considering PGD to prevent transmission of mtDNA disease are now advised to transfer embryos with a heteroplasmic mutation level of 5 percent or less (Sallevelt et al., 2013). It is possible, however, that women at risk for transmitting mtDNA disease may not produce oocytes, and hence embryos, with low enough levels of pathogenic mtDNA molecules to be deemed acceptable for transfer. This is always the case in women who are homoplasmic for a pathogenic mtDNA mutation, all of whose oocytes will be homoplasmic for the mutation, and occurs with elevated probability in women with high heteroplasmy levels for a pathogenic mtDNA mutation, all of whose oocytes may carry the mutation to a degree that would preclude their selection and intrauterine transfer.
An additional limitation of PGD for mtDNA disease is the potential occurrence of random and rapid changes in mtDNA heteroplasmy levels following embryo implantation, a phenomenon caused by random segregation of mtDNA and the mtDNA bottleneck (see the section on complexities related to mitochondrial genetics later in this chapter), which could result in higher than expected heteroplasmy levels of pathogenic mtDNA in critical tissues of offspring born following PGD. Relatedly, while PGD may reliably reduce heteroplasmy levels of pathogenic mtDNA and prevent manifestation of mtDNA disease in offspring, females born as a result of PGD may still be at risk of transmitting mtDNA disease to offspring because of higher than expected heteroplasmy levels in their oocytes.
Given the above uncertainties, embryo selection via PGD may not
represent an effective method for reliably preventing the transmission of mtDNA disease in women who are at known risk. Recent data in human embryos suggest that refined MRT protocols would be able to produce embryos with heteroplasmy levels below recommended thresholds (see the discussion of MRT research to date below) and thus might more reliably prevent maternal transmission of pathogenic mtDNA mutations in immediate offspring and future generations.
As discussed above, PGD has limitations with respect to its efficacy for reliably preventing maternal transmission of mtDNA disease, and PGD is not a preventive option for women who are homoplasmic, and may not be an option for women who are heteroplasmic, for pathogenic mtDNA mutations. Prospective mothers who are at risk for transmitting mtDNA disease to their offspring and wish to pursue reproductive options that mitigate the risk of this transmission thus must choose among options that allow for varying degrees of nuclear genetic connection between the child and the prospective parents: using the assistance of a woman who provides an oocyte or embryo, adoption, or childlessness. Therefore, current preventive and alternative reproductive options do not fulfill the desire of prospective mothers to have an nDNA-related child at sharply reduced risk for developing mtDNA disease. MRT is being investigated as a way of providing these benefits.
Two such proposed techniques—maternal spindle transfer (MST)17 and pronuclear transfer (PNT)—involve, in principal, the formation of a reconstructed oocyte or zygote, respectively, in which the intended mother’s mutated mtDNA would effectively be replaced with an oocyte provider’s nonpathogenic mtDNA (see Figure 2-4).18 The reconstructed oocyte or zygote would contain parentally derived nDNA and would theoretically be devoid, or have very low levels, of maternally derived pathogenic mtDNA. The reconstructed embryo would then be tested by PGD to determine
_________________
17 Also known as metaphase II spindle transfer (MII-ST), spindle-chromosomal complex transfer, or spindle transfer (ST).
18 This report uses the term “nonpathogenic mtDNA” to describe mtDNA contributed from the female oocyte provider, with the understanding that following genetic testing of provided oocytes for known pathogenic mutations, any provided mtDNA would be presumed—but given the rapidly expanding and shifting knowledge of mitochondrial biology and genetics, could not be assumed—to be free of pathogenic mtDNA mutations.
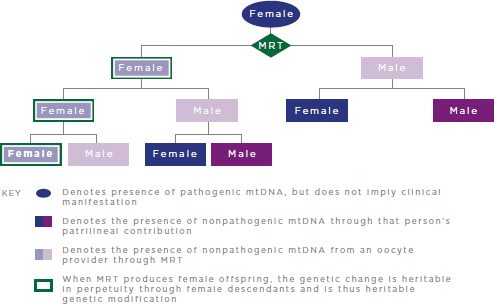
FIGURE 2-4 Heritable genetic modification via MRT.
NOTES: MRT = mitochondrial replacement techniques; mtDNA = mitochondrial DNA. MRT replaces pathogenic mtDNA from the intended mother with nonpathogenic mtDNA from an oocyte provider. For simplicity, reproductive partners are not shown and are assumed not to carry pathogenic mtDNA mutations.
heteroplasmy levels,19 as well as undergo other genetic testing for chromosomal abnormalities and sex selection (if utilized). The sections that follow describe the methodology of these techniques in more detail.
Demonstrating the safety and efficacy of MRT entails evidence of minimal pathogenic mtDNA carryover20 (and subsequent heteroplasmy), as well as normal health and growth in offspring born as a result of MRT. The high-level summary of MRT research that follows is therefore focused on those human in vitro and animal studies that were designed as proof-of-principle to demonstrate the feasibility of MRT for preventing mtDNA disease transmission and is structured to emphasize review of these outcome
_________________
19 As previously described, PGD may not be a reliable method for preventing transmission of mtDNA disease in women who are at known risk of transmitting mtDNA disease because of limitations related to complexities of mitochondrial genetics. With the advent of increasingly sensitive and accurate sequencing technologies, however, PGD is expected to be a reliable technique for determining the efficacy of MRT prior to embryo transfer.
20 As described previously, current standards of care for preventing mtDNA transmission stipulate that heteroplasmy levels in embryos should be less than 5 percent to mitigate the chance of mtDNA disease in offspring.
measures. A more detailed review of these and other studies of MRT can be found in Appendix B.
A third technique—polar body transfer (PBT)—has recently been proposed as an alternative or complement to MST and PNT. Compared with these latter two techniques, PBT has been less thoroughly investigated with respect to prevention of mtDNA disease transmission. PBT is discussed briefly in this chapter for general background purposes but is not included in the committee’s analysis of ethical, social, and policy issues associated with MRT.
Other methods involving oocyte and embryo cell modification for preventing the transmission of mtDNA disease—namely cytoplasm (ooplasm) transfer, somatic cell nuclear transfer (SCNT), embryo cell nuclear transfer, and germinal vesical transfer—have been raised in various contexts in other forums. To the committee’s knowledge, FDA currently is not considering these techniques for preventing transmission of mtDNA disease, however, so they are not discussed here.
Maternal Spindle Transfer
MST would entail removal of the nDNA (specifically, the metaphase II spindle-chromosome complex,21 or MII-SCC) from the intended mother’s oocyte and its subsequent fusion to an oocyte provided by another woman that contained nonpathogenic mtDNA and from which the nDNA had been removed.22 The reconstructed oocyte would then be fertilized with the intended father’s, or another man’s, sperm and cultured in vitro to the blastocyst stage. At this point, the blastocyst would undergo genetic testing to determine mtDNA heteroplasmy levels, chromosome abnormalities, and sex (if utilized). Embryos that met established criteria for these parameters would be transferred into the uterus of the woman intended to carry the pregnancy (see Figure 2-5). As in PNT, a small amount of cytoplasm would be carried over in the karyoplast23 removed from the intended mother’s oocyte, and thus there would be a nonzero chance for carryover of the intended mother’s pathogenic mtDNA. This and other potential risks associated with MRT are discussed later in this chapter.
_________________
21 During metaphase II, the chromosomes are attached at their centromeres to microtubules that connect to the spindle apparatus, which aids in aligning the chromosomes at the equator of the cell (the metaphase plate) in preparation for separation of the sister chromatids during anaphase II.
22 The term “enucleation” is sometimes used to describe the removal of nuclear genetic material from the metaphase II oocyte; at this meiotic stage, however, the chromosomes are not encompassed by a nuclear membrane and thus do not constitute a true nucleus.
23 Karyoplast is nuclear genetic material and cytoplasm encapsulated by a plasma membrane.
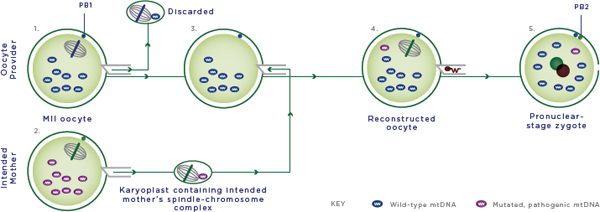
FIGURE 2-5 Maternal spindle transfer.
NOTES:
1. The spindle-chromosome complex is removed as a karyoplast from the provider oocyte and discarded.
2. The spindle-chromosome complex is removed as a karyoplast from the intended mother’s oocyte and fused to the provider oocyte from which the nuclear DNA (nDNA) material has been removed; the intended mother’s oocyte is discarded.
3. The reconstructed oocyte contains the intended mother’s nDNA and oocyte provider’s nonpathogenic mtDNA.
4. The reconstructed oocyte is fertilized by intracytoplasmic sperm injection (ICSI) with the sperm provider’s sperm.
5. The fertilized oocyte is cultured in vitro and transferred at the blastocyst stage to the woman who will carry the pregnancy. Cells and cellular contents not drawn to scale; MII oocyte = metaphase II oocyte; mtDNA = mitochondrial DNA; PB1 and PB2 = 1st and 2nd polar body.
SOURCE: Modified figure based on those appearing originally in: Richardson, J., L. Irving, L. A. Hyslop, M. Choudhary, A. Murdoch, D. M. Turnbull, and M. Herbert. 2015. Concise reviews: Assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells 33(3):639-645. License information available at: http://creativecommons.org/licenses/by/4.0.
MST in Animal Models
Wang et al. (2001) first reported MRT to be compatible with full-term mammalian development in a mouse model, wherein transfer of the MII-SCC was performed between oocytes of two genetically distinct mouse substrains. Of note is that the average body weight of the offspring at 10 days of age was within normal range for the oocyte donor substrain, which the authors suggest could indicate that factors in the oocyte donor’s cytoplasm could have an effect on the transferred nDNA. More recently, researchers at Oregon Health & Science University (OHSU), led by Shoukrat Mitalipov et al. (the OHSU Group), pioneered MST in rhesus macaque, a nonhuman primate model (Lee et al., 2012; Tachibana et al., 2009, 2013). Initial work by the OHSU group demonstrated the feasibility of MST for producing oocytes capable of fertilization and embryonic development (Tachibana et al., 2009). This study also showed that MST was capable of producing live-birth macaque offspring whose body weight was comparable to that of controls and that presented with nondetectable mtDNA carryover. A 3-year follow-up study found that these offspring were healthy, displayed no mitochondrial dysfunction, and presented with no significant change in mtDNA heteroplasmy levels in blood and skin samples over time (Tachibana et al., 2013). The OHSU group informed the United Kingdom’s Human Fertilisation and Embryology Authority (HFEA) during its most recent review of MRT that it intends to enter the macaque offspring into a breeding program to assess their fertility status, as well as to conduct more detailed investigations into the potential physiological effects of MRT (HFEA, 2014b).
Additional work by the OHSU group in macaques indicated that oocytes from females born as a result of MRT may have higher than expected levels of mtDNA carryover (Lee et al., 2012). In two female fetuses conceived by MST that were recovered preterm for analysis, mtDNA carryover was less than 0.5 percent in somatic tissues and organs. While 11 of 12 oocytes from each fetus contained less than 5.5 percent of carried-over mtDNA; 1 oocyte from each fetus contained a more substantial level of mtDNA carryover (16.2 percent and 14.1 percent). These data confirm that, while MRT would likely prevent significant mtDNA carryover and heteroplasmy in somatic tissues and organs of offspring born as a result of MRT, oocytes of females born as a result of MRT could harbor significant and clinically relevant levels of carried-over mtDNA.
MST in Human Oocytes
The OHSU group demonstrated the feasibility of MST for producing human oocytes capable of fertilization and normal embryo development in oocytes provided by healthy female volunteers (Tachibana et al., 2013).
Compared with macaque oocytes subjected to MST, whose rates of normal fertilization were comparable to those of controls, a significant proportion of human oocytes subjected to MST showed abnormal fertilization, as evidenced by an irregular number of pronuclei in the MST zygote. Of those zygotes that were normally fertilized, development to the blastocyst stage was comparable to that of controls. An average mtDNA carryover of 0.5 percent was observed in MST embryos, confirming the ability of MST to reliably limit mtDNA carryover.
A study conducted by Paull et al. (2013) at the New York Stem Cell Foundation confirmed the feasibility of MST in human oocytes, although metaphase II oocytes were parthenogenetically activated to avoid formation and destruction of potentially developmentally competent embryos. Following MST and artificial activation, an average of 0.36 percent mtDNA carryover was observed in MST zygotes. Finally, researchers at the Wellcome Trust Centre for Mitochondrial Research at Newcastle University (the Newcastle Group) have begun work on MST in human oocytes alongside PNT in zygotes to facilitate comparison of the two techniques (HFEA, 2014b). This work is still in progress.
Pronuclear Transfer
Compared with MST, wherein the transfer of genetic material would take place between metaphase II oocytes prior to fertilization, PNT would entail the transfer of nDNA between fertilized oocytes, or zygotes, prior to fusion of the pronuclei (syngamy). Specifically, the male and female pronuclei would be removed in a karyoplast from the zygote of the intended parents and fused to an enucleated zygote of the sperm provider’s sperm and the oocyte provided by a woman other than the intended mother. The reconstructed zygote would then be cultured in vitro to the blastocyst stage. At this point, the blastocyst would undergo genetic testing to determine mtDNA heteroplasmy levels, chromosome abnormalities, and sex (if utilized). Embryos that met established criteria for these parameters would be transferred into the uterus of the woman intended to carry the pregnancy (see Figure 2-6). As in MST, a small amount of cytoplasm would be transferred within the extracted karyoplast containing the pronuclei and would likely contain a variable, nonzero amount of the intended mother’s pathogenic mtDNA. This and other risks associated with PNT are discussed later in this chapter.
PNT in Animal Models
The availability of proof-of-principle studies in animal models to demonstrate the safety and efficacy of PNT is limited. Using a mouse model of mtDNA disease (“mito-mouse”) harboring a large-scale mtDNA dele-
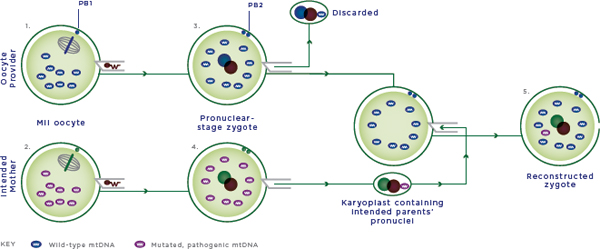
FIGURE 2-6 Pronuclear transfer.
NOTES:
1. The provider oocyte is fertilized by intracytoplasmic sperm injection (ICSI) with the sperm provider’s sperm.
2. The intended mother’s oocyte is fertilized by ICSI with the sperm provider’s sperm.
3. The male and female pronuclei are removed from the provider zygote and discarded.
4. The male and female pronuclei are removed from the intended mother’s zygote and fused to the enucleated provider zygote. The enucleated zygote of the intended mother is discarded.
5. The reconstructed zygote contains male and female nuclear DNA from the intended mother and sperm provider and nonpathogenic mtDNA from the oocyte provider. The zygote is cultured in vitro and transferred at the blastocyst stage to the woman who would carry the pregnancy.
Cells and cellular contents not drawn to scale; MII oocyte = metaphase II oocyte; mtDNA = mitochondrial DNA; PB1 and PB2 = 1st and 2nd polar body.
SOURCE: Modified figure based on those appearing originally in: Richardson, J., L. Irving, L. A. Hyslop, M. Choudhary, A. Murdoch, D. M. Turnbull, and M. Herbert. 2015. Concise reviews: Assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells 33(3):639-645. License information available at: http://creativecommons.org/licenses/by/4.0.
tion (ΔmtDNA), Sato et al. (2005) determined that PNT was effective in preventing the expected mtDNA disease phenotype in ΔmtDNA mito-mice offspring. Corresponding measurement of ΔmtDNA levels showed that the proportion of ΔmtDNA molecules increased significantly over time. As noted by the authors, however, mtDNA molecules with large-scale deletions exhibit a replicative advantage over normal mtDNA molecules, and ΔmtDNA levels therefore might be expected to increase over time. Furthermore, the authors note the limited ability to translate the findings of this study to humans given that maternal transmission of mtDNA deletions in humans is not commonly observed.
A recent study by Neupane et al. (2014) compared mtDNA carryover and developmental competence in mouse oocytes and zygotes subjected to MST and PNT, respectively. The authors found no significant difference in mtDNA carryover in MST oocytes (<2.15 percent) and PNT zygotes (<2.6 percent). In further assessment of mtDNA carryover in PNT-derived blastomeres, one blastomere contained 4.9 percent karyoplast-derived mtDNA, while the remaining seven blastomeres showed no detectable mtDNA carryover. In parthenogenetically activated MST oocytes, development to the blastocyst stage was statistically similar to that of controls. Neither cleavage rate nor blastocyst formation differed significantly between parthenogenetically activated MST and PNT embryos.
PNT in Human Zygotes
The Newcastle Group, led by Douglass Turnbull et al., pioneered PNT for the prevention of transmission of mtDNA disease. They performed initial work in fertilized zygotes,24 which are typically discarded during the course of fertility treatments (Craven et al., 2010). They found the developmental potential of reconstructed zygotes to be approximately 50 percent that of nonmanipulated abnormally fertilized control zygotes, a difference they attribute to the possibility that the reconstructed zygotes lacked the requisite complement of maternal and paternal pronuclei. Optimization of the procedure significantly minimized mtDNA carryover, which ranged from nondetectable to 11.4 percent. In response to the HFEA’s most recent scientific review, the Newcastle Group reported that they have begun to assess the efficacy of PNT in normally fertilized zygotes, and have seen reproducibly high rates of blastocyst development from PNT zygotes. The group also reported that mtDNA carryover levels were nondetectable or less than 2 percent. The researchers identified “subtle differences in embryo development” in PNT zygotes, which they are investigating (HFEA, 2014b). There
_________________
24 Zygotes that contain an abnormal number of pronuclei: one pronucleus (1N) or three pronuclei (3N), as compared with the normal complement of two pronuclei (2N).
are no published reports of PNT performed in human zygotes with the intent of preventing transmission of mtDNA disease in live-born children.25
Polar Body Transfer
A set of techniques for preventing mtDNA disease transmission related methodologically to MST and PNT—polar body 1 transfer (PB1T) and polar body 2 transfer (PB2T)—was recently documented as a potential alternative or complementary technique for preventing transmission of mtDNA disease (Wang et al., 2014). PB1T and PB2T entail the transfer of the first or second polar body to an enucleated or hemi-enucleated mature oocyte or zygote, respectively. Compared with MST and PNT, PBT has been less rigorously researched and reviewed with respect to the prevention of transmission of mtDNA disease. Furthermore, there is some reservation as to its potential future applicability given the lack of successful replication in mammals (Wolf et al., 2015). The HFEA conducted a comprehensive review of PBT for prevention of the transmission of mtDNA disease and the surrounding research landscape (HFEA, 2014a). In this review, the HFEA found that, while this research is still in its infancy as a potential MRT, PBT could potentially have advantages over MST and PNT, such as reduced mtDNA carryover, the absence of cytoskeletal inhibitors, and less invasive manipulations. More extensive preclinical research is needed in human oocytes and zygotes, however, to determine the feasibility, efficacy, and safety of PBT and whether these potential advantages would in fact be realized.
RISKS RELATED TO MRT: SCIENTIFIC COMPLEXITIES AND TECHNICAL UNKNOWNS AND UNCERTAINTIES
The clear benefit of successful implementation of MRT would be to give women who carry pathogenic mtDNA mutations the option of hav-
_________________
25 One case report documents PNT attempted in human zygotes with the intent of producing viable human offspring (Zhang et al., 2003) in a patient with a history of failed IVF treatments. Briefly, patient and provider oocytes were fertilized by ICSI, and the pronuclei from the patient’s zygotes were fused to enucleated provider zygotes via electrofusion. Five of seven successfully reconstructed zygotes were transferred to the patient’s uterus. A triplet pregnancy was achieved in the patient, but all three fetuses were lost during the pregnancy. The researchers report that all three fetuses presented with normal karyotypes, contained nDNA solely from the intended parents, and contained no detectable mtDNA from the intended mother (Zhang et al., 2003). There is some debate, however, as to whether these findings are relevant to current safety considerations for MRT. While some have suggested that the observed adverse outcome might be related to the PNT technique, others have argued that it was a result of technical error (UK Parliament House of Lords, 2015). Inclusion of this experiment in this report is not intended to convey validation or support of this case report by the committee, but to provide a more complete overview of the published literature on PNT.
ing genetically related offspring at greatly diminished risk of mtDNA disease (the potential social and ethical benefits of MRT are discussed more thoroughly in Chapters 3 and 4). This section provides a nonexhaustive overview of the risks, unknowns, and uncertainties associated with MRT.
Complexities Related to Mitochondrial Genetics
Because the mitochondrial genome is maternally inherited, exists in high copy number, and exhibits evolutionary genetics distinct from those of nDNA, several inherent complexities are associated with mitochondrial genetics that do not arise with nuclear Mendelian genetics. Three concepts of mitochondrial genetics are important considerations in MRT: heteroplasmy, mtDNA bottleneck, and mtDNA evolutionary theory (Carelli and Chan, 2014; DiMauro and Schon, 2003; DiMauro et al., 2013; Reinhardt et al., 2013). Overall, these complexities underscore the relatively unpredictable nature of mitochondrial genetics, which could complicate the ability of preclinical studies to predict with certainty the safety and efficacy of MRT in humans.
Heteroplasmy: Threshold Effect and Mitotic Segregation
As previously described, heteroplasmy is the state in which a cell, tissue, or individual contains more than one type of mtDNA genotype. In most cases, cells containing pathogenic mtDNA mutations manifest cellular dysfunction only when the levels of pathogenic mtDNA molecules accumulate to a certain threshold level at which clinical symptoms of mtDNA disease develop (threshold effect). Depending on the particular mutation, the threshold level is typically 60-90 percent mutant mtDNA. The level of heteroplasmy can also increase or decrease in different tissues of an individual at different rates as a result of shifts in the proportion of pathogenic mtDNA transmission occurring randomly during cell division, a concept known as mitotic segregation. During cell division, pathogenic mtDNA molecules can be partitioned unequally into daughter cells, shifting the level of heteroplasmy in resulting daughter cells. If this happens to a great enough extent, the level of pathogenic mtDNA molecules within a tissue can reach the threshold level for manifesting as mtDNA disease. This phenomenon underscores the difficulty of extrapolating heteroplasmy levels measured in blood to those in all potentially symptomatic tissues.
mtDNA Bottleneck
During oocyte development in the developing fetus, a phenomenon known as the prenatal mtDNA bottleneck occurs, in which only a frac-
tion of the founding pool of mtDNA molecules are partitioned to daughter oocytes (Stewart et al., 2008). It is estimated that the number of mtDNA molecules is reduced from more than approximately 100,000 in the mature oocyte to as few as 10 copies in primordial germ cells (Shoubridge and Wai, 2007). As a consequence of this mtDNA bottleneck, rapid changes in the level of mtDNA mutations from one generation to the next can occur. For example, a mother may have low-level heteroplasmy of a pathogenic mtDNA (e.g., 10 percent) but bear a child who has high levels of heteroplasmy or is homoplasmic for that pathogenic mutation. Another, less intensely studied mtDNA bottleneck is the postnatal mtDNA bottleneck, which can occur during embryonic and fetal development and results from unequal distribution or selective replication of mtDNA molecules in developing embryonic and fetal tissues.
These issues result in complexities in evaluating the risks associated with MRT. In model systems, MRT has resulted in variable levels of carryover, with the most successful experiments documented to have resulted in less than 1-2 percent carryover of mtDNA molecules from the affected female’s oocyte. This low-level carryover is expected to be compatible with clinically unaffected offspring. Because of poorly understood bottleneck effects, however, some offspring may have higher-than-expected levels of pathogenic mtDNA molecules in some tissues that could exceed the threshold level required to manifest disease. This phenomenon is exemplified by cases of cytoplasm transfer,26 a procedure used for treatment of idiopathic infertility that involved injection of cytoplasm from oocytes provided by other women into the oocytes of intended mothers (Barritt et al., 2001a,b; Brenner et al., 2000, 2001, 2004; Cohen et al., 1997, 1998; Huang et al., 1999; Lanzendorf et al., 1999). Some offspring born following cytoplasm transfer were found to have surprisingly high mtDNA levels from the provided oocytes compared with the volume of oocyte cytoplasm injected (Brenner et al., 2004). This observation may be attributable to bottleneck effects during embryonic development, but it is difficult to evaluate because these procedures were not performed quantitatively and were documented loosely. With regard to MRT, female offspring born as a result of MRT could present with low-level heteroplasmy in somatic cells but produce
_________________
26 Cytoplasm transfer was performed in the United States from 1997 to 2001 for treatment of infertility resulting from implantation failure due to poor embryo development. In a July 2001 letter to sponsors/researchers, FDA asserted jurisdiction over cytoplasm transfer on the grounds that it involved “human cells used in therapy involving the transfer of genetic material by means other than the union of gamete nuclei” (FDA, 2001a), requiring that an Investigational New Drug application be filed before clinical application of cytoplasm transfer could proceed. This effectively halted the clinical application of cytoplasm transfer, and since that time there has been no report of researchers attempting to use cytoplasm transfer for the treatment of infertility or other indications.
offspring with high levels of mtDNA mutations as a result of a potential bottleneck effect occurring in the development of their oocytes.
Evolutionary Theory: mtDNA and nDNA
Another relevant complexity is the potential for incompatibility (“haplogroup incompatibility”) between artificially combined nuclear and mitochondrial genomes from two genetically distinct individuals, as in MRT. Ample evidence in model organisms indicates that such evolutionary divergence could lead to incompatibilities between certain mtDNA and nDNA genomes. Studies of outbred strains of model organisms, for example, have identified specific mtDNA variants that are “compatible” only with certain nuclear genome backgrounds (see Reinhardt et al. [2013] and Wolff et al. [2014] for a review). Relatedly, some have suggested that co-adapted mtDNA-nDNA pairings that are advantageous to the organism are likely to be preserved, while incompatible mtDNA-nDNA pairings are likely to be selected against (Morrow et al., 2015; Reinhardt et al., 2013). Accordingly, the artificial combination of a mitochondrial genome that has not co-evolved with a provided, “foreign” nuclear genome, as in MRT, could theoretically result in disruption, and possible failure, of critical mitochondrial processes. Experts in the field of mitochondrial genetics, however, disagree as to whether these incompatibilities would manifest in humans as phenotypically relevant adverse effects. An opposing argument is the anecdotal observation that humans across vastly divergent mtDNA haplogroups have reproduced with no apparent untoward effects on human health (IOM, 2015).
Another potential impact of mtDNA-nDNA mismatch is the manifestation of male-specific deleterious phenotypes. Evolutionary theory holds that, because mtDNA is solely maternally transmitted, it could accumulate mutations that are advantageous to females but detrimental to males. In fruit flies, for example, strains containing mtDNA that is “foreign” to the nuclear genome show dramatically altered expression of genes specifically in males but not in females—particularly those genes related to male reproductive organs (Innocenti et al., 2011). Hence, evolutionary theory and model organism studies indicate that if MRT led to a mismatch between mtDNA and nDNA, male infertility would be a theoretical possibility.
A proposed solution to mitigate the uncertainty of haplogroup incompatibility is “haplogroup matching,” wherein the mtDNA of oocyte providers would be sequenced to select for those providers that were of the same haplogroup as the intended mother. The counterargument to this proposition is that haplogroup matching would not entirely mitigate the risk of mtDNA-nDNA mismatch because the genetic variants of putative
incompatibilities are poorly understood and thus may not be captured in haplogroup matching (Morrow et al., 2015).
Uncertainties and Unknowns Related to MRT Research
Certain aspects of MRT present an additional set of uncertainties and unknowns with regard to the potential safety and efficacy of first-in-human clinical investigations of the proposed techniques. These aspects include (1) limitations of current animal and in vitro models, as well as the available data, for purposes of predicting the safety and efficacy of MRT in humans; (2) the uncertainty of techniques such as PGD, amniocentesis, and chorionic villus sampling (CVS) for validating efficacy of MRT—namely for quantifying pathogenic mtDNA carryover and heteroplasmy load; and (3) the potential for yet unknown adverse effects of reagents and manipulations employed in MRT on the resulting embryo, fetus, or future child.
Limitations of the Current State of MRT Science
Research to date has provided data to support the feasibility and efficacy of MRT, although the translatability of such data is limited. The briefing document for FDA’s Cellular, Tissue and Gene Therapies (CTGT) Advisory Committee states: “These studies provide preliminary evidence that PNT and [MST] methods may be feasible. However, these data cannot be seen as traditional POC [proof-of-concept] studies. . . . Because most of these studies were not done with models of mitochondrial disease, it is not clear whether these data provide any support for the potential effectiveness of these methods in humans” (FDA Cellular Tissue and Gene Therapies Advisory Committee, 2014b). The HFEA echoes this observation in its most recent scientific review, noting that “some consulted experts recommend that as a ‘gold standard’ they would like to see experiments conducted using oocytes from women affected by mitochondrial disease to see if pathogenic mutations behave differently” (HFEA, 2014b). The HFEA also notes caveats on the implementation of this recommendation, such as the wide range of potential mtDNA mutations and the potential burden of ovarian stimulation for women with mtDNA disease.
With respect to both fundamental basic and translational science, the CTGT Advisory Committee “generally agreed that there is not sufficient animal data (particularly with regard to follow-up of offspring) to support the use of the mitochondrial manipulation technologies in first-in-human clinical trials” (FDA Cellular Tissue and Gene Therapies Advisory Committee, 2014a). This discerned lack of evidence in support of the safety and efficacy of MRT has implications for the assessment of benefits and risks inherent in the ethics of recommendations to proceed with MRT.
Efficacy: Validation of MRT
As discussed earlier in this chapter, PGD is not at present a reliable method for preventing transmission of mtDNA disease given the improbability of procuring an embryo with sufficiently low levels of heteroplasmy for transfer, as well as the potential for postnatal bottleneck amplification of pathogenic mtDNA molecules following embryo transfer. Experiments with cytoplasm transfer discussed earlier in this chapter highlighted the latter concern. Similar concerns arise regarding the ability of PGD, and correspondingly amniocentesis and CVS, to predict accurately the expected level of heteroplasmy in the tissues of offspring born as a result of MRT. As discussed earlier, current standards of care for the use of PGD to prevent transmission of mtDNA disease stipulate that heteroplasmy levels must be less than 5 percent to mitigate the chance of mtDNA disease in offspring. At present, the estimated amount of mtDNA carryover with MRT techniques is less than 1-2 percent; however, the potential for postnatal bottleneck amplification remains a concern in analyses of efficacy.
Safety: Manipulations and Reagents Used in MRT
Inadvertent physical damage or epigenetic changes to the reconstructed oocyte or zygote are a potential risk stemming from the manipulations inherent in and reagents used for MRT. Visualization of the MII-SCC in MST, for example, would require polarized light birefringence, whose safety is currently unknown. While the pronuclei in PNT would be visualized more easily than the MII-SCC, they would be larger and more difficult to manipulate, potentially resulting in greater cellular trauma (Craven et al., 2010). There could also be an increased risk for aneuploidy or chromosomal abnormalities—particularly potential loss of chromosome(s) during nuclear transfer—as a result of MRT. This risk could be augmented in MST given that the MII-SCC is not enclosed by a nuclear membrane.
Sendai virus would be used in MST and PNT for fusion of the karyoplast to the recipient oocyte or zygote. Unlike the reagents used in manufacturing processes upstream of MRT, which would be washed away or diluted in subsequent steps, Sendai virus would be injected directly into the cell, which would develop into the embryo that would subsequently be transferred into the woman who would carry the pregnancy. There could be unknown risks associated with the immunogenicity of the virus that could adversely affect the embryo or offspring. The cytoskeletal inhibitors used to aid removal of the karyoplast from the oocyte or zygote (e.g., nocodazole and cytochalasin B) could also pose an unknown risk to the oocyte or zygote. Of note, cytochalasin B would be used in both MST and PNT, and nocodazole would additionally be used in PNT.
Vitrification for Stage Matching
Matching the developmental stage of the intended mother’s oocyte or zygote and the oocyte or zygote provided by another woman is critically important, as noted by the Newcastle Group in evidence submitted to the HFEA (HFEA, 2014b). Given the potential difficulty of synchronizing oocyte retrievals for both MST and PNT, oocyte or zygote vitrification could be necessary. Work by Tachibana et al. (2013) revealed that the cytoplast may be more sensitive than the nDNA to vitrification-induced damage, at least in the macaque model, while Paull et al. (2013) provided evidence for the feasibility of using cryopreserved karyoplasts containing the MII-SCC in MST. These findings suggest an experimental design wherein the oocyte providing the nDNA of the intended mother would be cryopreserved, if necessary, to ensure that it matched the developmental stage of the provided oocyte.
Conclusion: The field of mitochondrial genetics is characterized by complexities that make predicting the behavior of mtDNA—at the cellular, tissue, and systemic levels—difficult and uncertain. Collectively, these complexities can be viewed as an unknown variable in predicting the efficacy and safety of MRT in humans. The current state of MRT science and unknown physiological impact(s) of reagents and procedures implemented in MRT present an additional set of uncertainties and unknowns. A thorough understanding of the state of the science related to the unknowns of mtDNA genetics and MRT is important for informing the benefit and risk assessment entailed in potential regulatory decisions regarding if, when, and how to proceed with MRT in first-in-human clinical investigations.
In the United States, MRT would be subject to a complex landscape of state and federal laws and regulations. The legality of the research on MRT—and perhaps even the clinical application—would vary from state to state as a result of differing laws on fetal and embryo research, including cloning. Federal funding for MRT research would likely be unavailable because of current legislative restrictions against funding research on human embryos. In the event that MRT were to move into clinical investigations, FDA has asserted regulatory jurisdiction, and a careful stepwise process, which would include FDA oversight and institutional review board (IRB) review, would be required before any form of MRT would be approved for marketing. If it were approved, there would be some potential mechanisms for oversight in the postapproval context. Potential oversight of both the
research on and clinical use of MRT would be complex, with uncertainty over the precise interpretation of how laws and regulations would apply.
Regulation of Related Technologies
Although MRT is relatively new, policies on similar technologies could apply to MRT and illustrate some of the ways in which these techniques could be regulated. Oversight of MRT would likely involve the same statutes and regulations that apply to IVF, PGD, preimplantation genetic screening (PGS), and cloning. Not only is MRT similar in some ways to IVF, PGD, and PGS, but these technologies would also be performed in conjunction with MRT.
In Vitro Fertilization
Since the 1978 birth of Louise Brown, the first baby conceived by IVF, it is estimated that more than 5 million babies have been born as a result of IVF (ESHRE, 2012). This technology, in which embryos are created outside the body and then implanted, was developed and disseminated with minimal federal oversight. In the mid-1970s, the U.S. Department of Health, Education and Welfare (DHEW) appointed an ethics advisory board (EAB) to study IVF and review proposals for federal funding for IVF research. The EAB concluded that IVF was ethically acceptable; however, the EAB no longer functioned as of 1980. Because DHEW regulations required that a federal ethics board review funding proposals, and the EAB no longer functioned, this created a de facto moratorium on federal funding for IVF research. As a result, IVF was developed with private funds and with minimal federal regulation or oversight (Knowles and Kaebnick, 2007).
FDA did not clarify its jurisdiction over IVF until 1998, when it released a proposed rule for oversight of human cellular and tissue-based products (HCT/Ps) (FDA, 1998a). The final rule, released in 2001, defines HCT/Ps as “articles containing or consisting of human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient.” The rule divides HCT/Ps into two categories with corresponding levels of regulation: minimally manipulated HCT/Ps are lightly regulated, and more-than-minimally manipulated HCT/Ps are regulated as drugs and/or biologics. Minimally manipulated HCT/Ps, which include semen, oocytes, and embryos, must be screened for communicable diseases (unless provided by an intimate partner), and manufacturers of these HCT/Ps must register with FDA (FDA, 2001a).
ART programs are subject to these HCT/Ps regulations, so they must screen gametes for communicable diseases, register with FDA, and follow guidelines for handling tissues. In addition, FDA regulates the drugs and
devices that are used in conjunction with IVF, such as drugs that stimulate production of oocytes for retrieval. According to a different law, clinics also must report their pregnancy success rates to the U.S. Centers for Disease Control and Prevention (CDC), which collects and publishes data for certain procedures performed by clinical programs conducting “treatments or procedures which include the handling of human oocytes or embryos” (42 U.S.C. 263a-1 et seq.). Clinics that do not report these data to CDC are identified as having failed to report in CDC’s publication of data and face expulsion from the Society of Assisted Reproductive Technologies (SART) for failure to report (Knowles and Kaebnick, 2007; SART, 2016).27
Preimplantation Genetic Diagnosis (PGD) and Screening (PGS)
PGD and PGS are techniques used in conjunction with IVF to test embryos for genetic disorders before intrauterine transfer. PGD involves the performance of diagnostic genetic tests to determine whether specific gene or chromosome disorders—such as a mutation that causes cystic fibrosis or an array that would determine a precise chromosomal abnormality—are present or absent in an embryo. In contrast, PGS uses biomarkers to screen for an increased risk that an embryo will harbor any chromosomal abnormality, such as trisomy 21, which causes Down syndrome; a positive biomarker screen would then need to be followed up with a definitive diagnostic test. The first successful clinical application of PGD was reported in 1990 by Handyside et al. (1990) for prevention of transmission of X-linked disorders.
The regulation of PGD and PGS is essentially identical to the regulation of IVF. Although PGD and PGS entail laboratory testing, they are not subject to the Clinical Laboratory Improvement Amendments (CLIA), which ensure the quality of laboratory testing through such requirements as specific levels of education, training, and experience for laboratory personnel (42 CFR § 493.17). Normally, laboratories that perform diagnostic tests must be compliant with CLIA to receive Medicare or Medicaid reimbursement. However, laboratories are subject to the regulations only if they perform tests on “materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings” (42 CFR § 493.2). Thus far, the Centers for Medicare & Medicaid Services (CMS) has not interpreted CLIA as applying to laboratories that perform PGD or PGS, either because an embryo is not “derived from the human
_________________
27 The 2013 Assisted Reproductive Technology Fertility Clinic Success Rates Report estimates that ART surveillance covered 98 percent of ART cycles performed in the United States in 2013 (CDC et al., 2015).
body” but is a new and unique entity (Nagy et al., 2012) or because the tests are diagnosing embryos, not “human beings” (Hudson, 2006).
Cloning
After Dolly, a sheep that was the first animal to be cloned using the nucleus from an adult somatic cell, was born in 1996, federal and state governments rushed to regulate this somatic cell nuclear transfer (SCNT) technology. Although no federal law was enacted, California passed a statute that banned reproductive cloning in 1997, and more than a dozen states followed suit with statutes banning either reproductive cloning or all SCNT, even for nonreproductive research. FDA asserted jurisdiction over cloning in 1998 with a letter to IRBs (FDA, 1998b). FDA informed IRBs that clinical research on human cloning is subject to FDA regulation, and would require the submission of an Investigational New Drug (IND) application. This letter did not analyze the specific statutory basis for FDA’s authority, but a subsequent letter in July 2001 (FDA, 2001b) pointed to the 2001 final rule on regulation of HCT/Ps (21 CFR § 1271), as well as a 1993 Federal Register notice that clarified FDA’s authority over human somatic cell therapy and gene therapy products (58 FR § 53248).
Germline Modification
Modification of the human germline—that is, modification of gametes or embryos that results in heritable genetic modification—is legal in the United States. However, several regulatory barriers have effectively prevented it from being carried out in many settings. First, the National Institutes of Health’s (NIH’s) Recombinant DNA Advisory Committee (RAC), which oversees and reviews proposals for research funded by NIH or conducted at institutions funded by NIH for similar projects that involve recombinant or synthetic DNA, has stated in guidelines since 1985 that it “will not at present entertain proposals for germ line alterations” (NIH Recombinant DNA Advisory Committee, 1985). Second, FDA, which, as discussed earlier, has regulatory authority over cell and gene therapy products, has never approved a proposal to modify the germline. Finally, the Dickey-Wicker amendment, a rider on each year’s U.S. Department of Health and Human Services (HHS) appropriations bill, prohibits the use of HHS funding for research that creates embryos for research purposes or destroys, discards, or subjects an embryo to risks with no prospect of medical benefit for the embryo. Therefore, federal funding for preclinical research on germline modification has long been unavailable. More recently, Francis Collins, director of NIH, stated that NIH “will not fund any use of gene-editing technologies in human embryos.” He noted that the
“concept of altering the human germline in embryos for clinical purposes has been debated over many years from many different perspectives, and has been viewed almost universally as a line that should not be crossed” (NIH, 2015).
This idea of “a line that should not be crossed” is reflected in the laws and regulations of many nations. Twenty-nine countries prohibit germline modification; the salient laws or regulations of 10 more countries, including the United States, are either ambiguous or would restrict but not fully prohibit it. This opposition to germline modification exists even in countries that allow other types of research involving human embryos: 13 of the countries that ban germline modification permit human embryonic stem cell research, and the United Kingdom permits MRT but prohibits all other types of germline modification (Araki and Ishii, 2014). In 2015, the International Bioethics Committee of the United Nations Educational, Scientific and Cultural Organization (UNESCO) called for a temporary ban on editing of the germline, stating that “interventions on the human genome should be admitted only for preventive, diagnostic or therapeutic reasons and without enacting modifications” that would be passed on to future generations (IBC, 2015). The U.S. National Academy of Sciences (NAS), U.S. National Academy of Medicine (NAM), Chinese Academy of Sciences, and the United Kingdom’s Royal Society convened an international summit on human gene editing in December 2015, and a committee formed by the NAS and the NAM will issue a report in 2016 on the clinical, ethical, legal, and social ramifications of both somatic and germline human gene editing.
Potential Federal Regulation of MRT
If MRT moved from preclinical to clinical research, various federal prohibitions and regulatory schemes, including those reviewed above, could apply to the techniques.
Dickey-Wicker and Federal Funding
The Dickey-Wicker Amendment states:
- None of the funds made available in this Act may be used for—
- the creation of a human embryo or embryos for research purposes; or
- research in which a human embryo or embryos are destroyed, discarded, or knowingly subjected to risk of injury or death greater than that allowed for research on fetuses in utero under 45 CFR 46.204(b) and section 498(b) of the Public Health Service Act (42 U.S.C. 289g(b)).
- For purposes of this section, the term “human embryo or embryos” includes any organism, not protected as a human subject under 45 CFR 46
as of the date of the enactment of this Act, that is derived by fertilization, parthenogenesis, cloning, or any other means from one or more human gametes or human diploid cells.
This statute prohibits the use of HHS funding for such research; however, it does not prohibit the research itself. MRT research that involved destroying embryos or manipulating embryos with no medical benefit to the embryos (i.e., if the embryos were not implanted) would be ineligible for HHS funding. Conversely, it might be the case that MRT research that involved transfer for gestation could be funded.
U.S. Food and Drug Administration Regulatory Authority
FDA does not regulate the practice of medicine itself, but instead has the authority to approve the introduction of a new drug, device, or biologic into interstate commerce28 (e.g., 21 U.S.C. 355(a)). The agency’s authority to regulate drugs and devices is found in the Federal Food, Drug, and Cosmetic (FD&C) Act, and its authority to regulate biologics is in Section 351 of the Public Health Services (PHS) Act.
In a 2001 letter to researchers, FDA asserted regulatory authority over “human cells used in therapy involving the transfer of genetic material by means other than the union of gamete nuclei,” and noted that this genetic material includes cell nuclei, oocyte nuclei, and ooplasm containing mitochondrial genetic material. The letter stated that any clinical research involving these techniques would require submission of an IND. Current MRT technologies, such as PNT, MST, and PBT, would all likely fall under this definition, thus giving FDA authority over MRT (FDA, 2001b).
As discussed above, FDA regulates standard IVF procedures as “minimal manipulation” and requires only registration of facilities and screening for communicable diseases. However, FDA has stated that the manipulation of HCT/Ps used in MRT, including “human cells used in therapy involving the transfer of genetic material (cell nuclei, oocyte nuclei, mitochondrial genetic material in ooplasm, genetic material contained in a genetic vector),” constitutes more-than-minimal manipulation and thus the manipulated HCT/Ps would be regulated as drugs and/or biologics (FDA, 2009). Whether a particular MRT technique would trigger regulation as a drug or biologic would depend on the specific technology and materials used in the technique.
_________________
28 The federal courts and FDA define “interstate commerce” broadly, and FDA asserts jurisdiction over products made from or with interstate components. See http://www.fda.gov/ICECI/ComplianceManuals/CompliancePolicyGuidanceManual/ucm073820.htm (accessed January 15, 2016).
U.S. Food and Drug Administration Regulatory Approval
Regardless of the product classification, the steps to FDA approval of MRT would be similar. Researchers wishing to conduct clinical investigations of any MRT technique would first be required to submit an IND. FDA does not regulate MRT as a technique per se, but rather the “product” that is considered a drug and/or biologic—in this case, the manipulated oocytes or zygotes (FDA, 2009). The IND includes preclinical data, information about the methods and products to be used, information about the investigators, and detailed protocols for the proposed clinical study. If the application is authorized, clinical investigations may begin. If the investigations are successful, a Biologic License Application (BLA) or a New Drug Application (NDA) can be submitted. If FDA determines, among other considerations, that the product is safe and effective and that its benefits outweigh its risks, the BLA or NDA can be approved and the product marketed in the United States.
Recent advances in the use of CRISPR-Cas9 and other tools for so-called gene editing (in which targeted changes are made in genes) have raised the question of whether this technique should ever be used in human gametes and embryos, a use that could result in intergenerational change in nDNA. To date CRISPR-Cas9 has been attempted in China with nonviable human embryos, as a demonstration of proof-of-principle, as well as demonstration of some of the technical challenges related to accuracy and precision of such changes (Liang et al., 2015). A number of countries have domestic law or have signed on to international instruments prohibiting such efforts if aimed at producing intergenerational changes in the germline (Council of Europe, 2015), and on December 18, 2015, the U.S. Congress passed an omnibus spending bill for fiscal year 201629 that would seem to forestall FDA consideration of any application to try such a technique in human clinical investigations, that is, in investigations involving transfer to a woman for gestation of the modified embryo. MRT might not result in heritable changes under all circumstances, so the applicability of the budget provision noted above to the clinical research discussed in this report is unclear, and determination of its applicability would necessarily be determined by FDA Counsel.
_________________
29 See Sec. 749, Consolidated Appropriations Act, 2016, Public Law 113, 114th Cong. (December 18, 2015). Available at: https://www.gpo.gov/fdsys/pkg/BILLS-114hr2029enr/pdf/BILLS-114hr2029enr.pdf (accessed January 11, 2016).
Recombinant DNA Advisory Committee
NIH’s RAC, which is authorized by the PHS Act,30 provides oversight and review of basic and clinical research funded by NIH or conducted at institutions funded by NIH for similar projects involving recombinant or synthetic nucleic acid molecules, which are defined as
(i) molecules that a) are constructed by joining nucleic acid molecules and b) that can replicate in a living cell, i.e., recombinant nucleic acids;
(ii) nucleic acid molecules that are chemically or by other means synthesized or amplified, including those that are chemically or otherwise modified but can base pair with naturally occurring nucleic acid molecules, i.e., synthetic nucleic acids, or
(iii) molecules that result from the replication of those described in (i) or (ii) above. (NIH, 2013)
Current MRT techniques, such as MST, PNT, and PBT, do not appear to fit this definition, as they do not involve the recombination of nucleic acid molecules or the use of synthetic nucleic acid molecules. Thus, it is unlikely that these techniques would fall under the jurisdiction of the RAC.
If the RAC were to have jurisdiction over an MRT technique, NIH’s Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules specify practices and requirements that would apply to research on the technique. NIH-funded research projects must comply with the NIH guidelines, and projects not funded by NIH must do so as well if they are conducted at or sponsored by an institution that receives NIH funds for similar projects. The guidelines require that before a clinical investigation begins, a project must (1) be approved by the Institutional Biosafety Committee (IBC); (2) be approved by the IRB; (3) obtain all applicable regulatory authorizations (e.g., IND approval); and (4) complete the RAC process, which includes initial RAC review upon submission, as well as public RAC review and discussion if deemed necessary. Once clinical investigations have begun, the RAC requires annual reports and safety reporting (NIH, 2013).
State Laws
Many states have laws regarding cloning, embryo research, stem cells, and other areas relevant to MRT. The language used in these statutes could affect whether MRT clinical research or clinical application would be legal in a state or whether certain MRT techniques would be prohibited. For example:
_________________
30 42 U.S.C. 282(b(16)).
- Arizona prohibits the creation of an embryo by any means other than the “combining of a human egg with a human sperm.”31 Under this law, both MST and PNT could potentially be illegal: MST involves fertilization of a reconstructed human oocyte with a human sperm, and PNT involves fertilization of two human oocytes, followed by transfer of the nuclear genetic material from the resulting intended parents’ zygote to the provider zygote to form a reconstructed zygote, which is cultured in vitro to a human embryo.
- California prohibits reproductive cloning and defines cloning as the transfer of a nucleus from a human cell from “whatever source” into a human oocyte for the purpose of initiating a pregnancy that could result in the birth of a human.32 Unlike other state cloning laws, the California law does not limit its prohibition on nucleus transfer to nuclei from somatic cells. Under this law, it appears that some versions of MRT would be permissible (PNT would involve transfer of a nucleus into a zygote, not an oocyte), but some would not (MST would involve transfer of nuclear genetic material into an oocyte).
- Louisiana prohibits the use of a fertilized ovum for any purpose other than “for the support and contribution of the complete development of human in utero implantation” and prohibits the creation of a fertilized ovum “solely for research purposes.” Under this law, then, MRT clinical investigations resulting in the creation of embryos for purposes other than implantation would be illegal, and clinical practice of PNT could be prohibited as well, because one fertilized ovum would not be implanted.33
- Several states have prohibitions on nontherapeutic research involving embryos, which presumably would prohibit MRT research that did not result in intrauterine transfer but permit research use of MRT if it were intended to lead to gestation and birth; these states include Michigan,34 Pennsylvania,35 and South Dakota.36
Institutional Oversight
Institutions have an important role in the oversight of research. FDA requires that any human subjects research requiring its approval be reviewed
_________________
31 Az. Rev. Stat. § 36-2311-2313.
32 Ca. Health & Safety Code § 24185.
33 La. Rev. Stat. tit. 9, §§ 122-129.
34 Mich. Comp. Laws § 333.2685 (1).
35 Pa. Cons. Stat. tit 18. § 3216 (a).
36 S.D. Codified Laws sec. 34-14-16 through 34-14-20.
by an IRB.37 IRBs are established or designated by the institutions conducting the human subjects research, but the federal government provides detailed guidance on IRB functions and duties. IRB considerations include risks, benefits, and informed consent. Any institution that receives federal funds for research involving human subjects must establish an IRB, and all such research performed at the institution must be reviewed by the IRB, regardless of its source of funding.38 As applied to MRT, an investigator would be required to obtain IRB approval for clinical research performed under an IND. IRB review would not be required for research involving purely in vitro embryo manipulation unless the research on the embryos would reveal identifiable information about the people who provided the embryos.39
For oversight of research involving recombinant or synthetic DNA, NIH requires that institutions establish an IBC. Like IRB review, IBC review is required for research at any institution that receives federal funding, regardless of the source of funding for the research (NIH, 2013). If MRT clinical research were subject to RAC oversight (see section on potential federal regulation of MRT earlier in this chapter), IBC review would be required.
Postapproval Regulation
FDA approvals are for specific indications. But even if FDA approved MRT for a specific indication, it could be used “off-label”—that is, used for an indication for which it had not been approved (see also the discussion of off-label use below). Once MRT had been approved, the FDA regulations that would apply to its clinical use would be limited to a group of postmarket measures that would be less stringent than the premarket controls. Still, there would be a few avenues for additional oversight and control of MRT, such as FDA’s Risk Evaluation and Mitigation Strategy (REMS) program or professional monitoring.
Background
As discussed above, FDA has the authority to approve MRT for a particular intended use (i.e., its labeled use), which would allow it to be marketed for that use. Marketing includes both advertising and a range of other promotional efforts. As noted, however, FDA’s regulatory authority
_________________
37 21 CFR Part 56.
38 45 CFR Part 46.
39 In practice, some institutions use the committees established to review embryonic stem cell research to review all embryo research, but this is not required by law.
wanes following approval. A clinical provider may use an approved product for an off-label purpose if, based on his or her best knowledge and clinical judgment, it is being used in the “practice of medicine” (21 CFR 312.2(d)). For example, if FDA approved MRT for the intended use of preventing the transmission of known pathogenic mtDNA mutations, a clinician could use the technique for the off-label indication of treating infertility. Although those who might receive FDA approval for mitochondrial replacement would not be permitted to market or promote a use of the product that has not been approved by FDA,40 the agency cannot prevent clinicians from using the product in any manner they deem appropriate, based on their clinical judgment. FDA does note that physicians “have the responsibility to be well informed about the product, to base its use on firm scientific rationale and on sound medical evidence, and to maintain records of the product’s use and effects” (FDA, 2014). Physicians are also subject to regulation in the form of state licensing and discipline procedures, as well as the threat of medical malpractice.
In addition to off-label clinical use, investigational use of an approved product is permitted if all of the following conditions are met:
- The new use is not intended to be submitted to FDA to support a new indication or a significant change in labeling or advertising.
- The new use does not significantly increase the risks of the product, and the investigation is conducted in compliance with IRB and informed consent protocols.
- The new use is in compliance with requirements concerning the promotion and sale of products (21 CFR 312.2(b)(1)).
As applied to MRT, these stipulations might permit a researcher to conduct clinical investigations of the use of MRT to treat such conditions as infertility without first obtaining FDA approval. Even if such use were shown to be successful, however, the product could not be marketed for that purpose without first undergoing FDA review. Notably, Shoukhrat Mitalipov, a pioneer of MRT, has announced publicly that he has submitted an IND to FDA to conduct clinical investigations of MRT for treatment of age-related infertility (Connor, 2015).
The limitations on marketing, as opposed to use, can have a significant effect on the scale of off-label use. In some areas of medicine, having marketing authority can give a sponsor, such as a pharmaceutical company, much larger market shares than would be garnered by any comparable drug
_________________
40 See, e.g., sections 505(a), 515(a), 501(f)(1), and 301(a) and (d), of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 355(a), 360e(a), 351(f)(1)) and 331(a) and (d).
without such privileges. In some areas, however, such as pediatric uses or cancer treatments, off-label use is exceedingly common.
For MRT, even off-label uses would be subject to rules concerning the safe handling of human cellular material, including oocytes, spermatozoa, and embryos. These rules are aimed at preventing communicable disease and require establishments (e.g., IVF clinics) to, for example, screen provided cells or tissues (21 CFR 1271). In addition, CDC collects and publishes pregnancy success data for ART techniques, which could include MRT because it is among the “treatments or procedures which include the handling of human oocytes or embryos” (42 U.S.C. 263a-1 et seq.).
Mechanisms for Postmarket Control
Once approved, a cell-based product, including the manipulated cells used in MRT, remains subject to controls by FDA (up to and including withdrawal of the approval), requirements for reporting to CDC, state laws governing licensing and discipline, medical malpractice suits, and the effect of insurance coverage decisions on its availability.
Risk Evaluation and Mitigation Strategy (REMS) Under the Food and Drug Administration Amendments Act of 2007, FDA has the authority to require a REMS from investigators. The REMS helps ensure that a postapproval product is used in a manner such that its benefits outweigh its risks, and applies to any use of the product, whether on- or off-label. FDA can require a REMS either as part of the approval process or after approval if new safety information emerges.
REMS programs vary significantly depending on the level of risk associated with a product. A REMS may require only that a medication guide be dispensed to patients with each prescription or it may require that the manufacturer send information on the risks of the product to health care providers and professional associations. The REMS for the acne medication isotretinoin (i.e., Accutane), for example, consists of a complex system for risk evaluation and mitigation that requires all patients, providers, and pharmacists to be registered in the iPLEDGE system in order to use, provide, or prescribe Accutane. Among other requirements, patients must demonstrate understanding of the drug’s risks and agree to use two forms of contraceptives while taking Accutane; providers must counsel patients about contraceptive use, provide scheduled pregnancy testing, and prescribe only a 30-day supply; and pharmacists must dispense the drug in a safe and systematic way (FDA, 2012).
Postmarketing requirements, commitments, and warnings FDA can require sponsors to perform postmarketing studies and postmarketing clini-
cal investigations (so-called Phase 4 investigations) for approved products (FFDCA 505(o)(3)). Such requirements can be imposed at the time of approval, or after approval if FDA becomes aware of new safety concerns. FDA can require the conduct of studies or investigations to assess a known serious risk, further examine a potential serious risk, or identify an unexpected serious risk. Each year, FDA must review the status of such studies and investigations, publish a summary in the Federal Register, and provide a report to Congress on the findings. FDA can also highlight any new concerns by communicating directly to physicians, by adding warnings to the label, or by narrowing or even completely withdrawing the approval.
State licensing Individual states have boards that license and monitor medical professionals to ensure ethical practice that meets the standard of care. Any practitioner could be disciplined for use of MRT—whether on- or off-label—that was inappropriate for the patient (e.g., overly risky or unlikely to provide benefit) or that was provided before informed and voluntary consent had been obtained. These boards vary widely in their stringency, but exist as a possible mechanism for monitoring new therapies and watching for problems.
Professional monitoring Professional societies play an important role in maintaining a standard of care in medicine. Each of the major medical societies has programs or documents that describe and periodically update the factors most salient to good practice in their field. At times these societies also have stepped in to help maintain high standards in fields that escape some of the formal mechanisms that exist for this purpose, such as surgery (which often innovates without the formal clinical investigations that trigger IRB review) and some forms of embryo research (where the absence of federal funding means far less opportunity for federal oversight). The mechanisms used by societies can range from data collection and publication of success rates, by technique and by clinic, to detailed protocols that are deemed best practices. SART is one example in the ART area, having used guidelines and recommendations for laboratory personnel, procedures, and safety, as well as membership for clinics that follow these voluntary measures, to steer practice in the appropriate direction (SART, 2007, 2008). More than 90 percent of ART clinics in the country are SART members (SART, 2015).
Insurance coverage Insurance availability can have a strong influence on how often a product or procedure is used, whether on- or off-label. In general, insurance companies will cover a product or procedure only if it is “medically necessary” (Bergthold, 1995; IOM, 2012) and considered a therapy rather than an experimental procedure. This affects uptake two
ways. First, it means coverage often is not available for uses that are off-label (thus lacking FDA approval) and not yet employed widely enough to have generated the data necessary to persuade the insurer that the off-label use is a proven therapy. Second, insurers distinguish among indications, so that, for example, they might cover a procedure if used to circumvent a disease such as MELAS but not cover the same procedure if used to circumvent a natural condition such as menopause. In this way, what some have called “enhancement” applications may well lack coverage. Lack of coverage, of course, will limit the number of patients who can afford a procedure.
Araki, M., and T. Ishii. 2014. International regulatory landscape and integration of corrective genome editing into in vitro fertilization. Reproductive Biology and Endocrinology 12:108.
Barritt, J. A., S. Willadsen, C. Brenner, and J. Cohen. 2001a. Cytoplasmic transfer in assisted reproduction. Human Reproduction Update 7(4):428-435.
Barritt, J. A., C. A. Brenner, H. E. Malter, and J. Cohen. 2001b. Mitochondria in human offspring derived from ooplasmic transplantation: Brief communication. Human Reproduction 16(3):513-516.
Bayona-Bafaluy, M. P., B. Blits, B. J. Battersby, E. A. Shoubridge, and C. T. Moraes. 2005. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proceedings of the National Academy of Sciences of the United States of America 102(40):14392-14397.
Bergthold, L. A. 1995. Medical necessity: Do we need it? Health Affairs 14(4):180-190.
Brenner, C. A., J. A. Barritt, S. Willadsen, and J. Cohen. 2000. Mitochondrial DNA heteroplasmy after human ooplasmic transplantation. Fertility and Sterility 74(3):573-578.
Brenner, C. A., J. A. Barritt, J. Cohen, and K. E. Pierce. 2001. Quantification of mitochondrial DNA heteroplasmy following ooplasmic transplantation. Human Reproduction 16(Suppl. 1):66-68.
Brenner, C. A., H. M. Kubisch, and K. E. Pierce. 2004. Role of the mitochondrial genome in assisted reproductive technologies and embryonic stem cell-based therapeutic cloning. Reproduction, Fertility, and Development 16(7):743-751.
Carelli, V., and D. C. Chan. 2014. Mitochondrial DNA: Impacting central and peripheral nervous systems. Neuron 84(6):1126-1142.
CDC (U.S. Centers for Disease Control and Prevention), ASRM (American Society for Reproductive Medicine), and SART (Society for Assisted Reproductive Technology). 2015. 2013 Assisted reproductive technology fertility clinic success rates report. Atlanta, GA: CDC.
Cohen, J., R. Scott, T. Schimmel, J. Levron, and S. Willadsen. 1997. Birth of infant after transfer of anucleate donor oocyte cytoplasm into recipient eggs. Lancet 350(9072):186-187.
Cohen, J., R. Scott, M. Alikani, T. Schimmel, S. Munné, J. Levron, L. Wu, C. Brenner, C. Warner, and S. Willadsen. 1998. Ooplasmic transfer in mature human oocytes. Molecular Human Reproduction 4(3):269-280.
Connor, S. 2015. Scientist who pioneered “three-parent” IVF embryo technique now wants to offer it to older women trying for a baby. The Independent, February 8.
Council of Europe. 2015. Statement on Genome Editing Technologies. http://www.coe.int/en/web/bioethics/-/gene-editing?redirect= (accessed December 18, 2015).
Craven, L., H. A. Tuppen, G. D. Greggains, S. J. Harbottle, J. L. Murphy, L. M. Cree, A. P. Murdoch, P. F. Chinnery, R. W. Taylor, R. N. Lightowlers, M. Herbert, and D. M. Turnbull. 2010. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465(7294):82-85.
DiMauro, S., and G. Davidzon. 2005. Mitochondrial DNA and disease. Annals of Medicine 37(3):222-232.
DiMauro, S., and E. A. Schon. 2003. Mitochondrial respiratory-chain diseases. New England Journal of Medicine 348(26):2656-2668.
DiMauro, S., E. A. Schon, V. Carelli, and M. Hirano. 2013. The clinical maze of mitochondrial neurology. Nature Reviews Neurology 9(8):429-444.
Elliott, H. R., D. C. Samuels, J. A. Eden, C. L. Relton, and P. F. Chinnery. 2008. Pathogenic mitochondrial DNA mutations are common in the general population. The American Journal of Human Genetics 83(2):254-260.
ESHRE (European Society of Human Reproduction and Embryology). 2012. The world’s number of IVF and ICSI babies has now reached a calculated total of 5 millions. ScienceDaily, July 2. http://www.sciencedaily.com/releases/2012/07/120702134746.htm (accessed December 22, 2015).
Eynon, N., M. Moran, R. Birk, and A. Lucia. 2011. The champions’ mitochondria: Is it genetically determined? A review on mitochondrial DNA and elite athletic performance. Physiological Genomics 43(13):789-798.
FDA (U.S. Food and Drug Administration). 1998a. Establishment registration and listing for manufacturers of human cellular and tissue-based products (21 CFR Parts 207, 807, 1271). Federal Register 63(93):26744-26755.
FDA. 1998b. Letter about human cloning, October 26, 1998. http://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/ucm150508.htm (accessed November 9, 2015).
FDA. 2001a. Human cells, tissues, and cellular and tissue-based products: Establishment registration and listing. Federal Register 66(13):5447-5469.
FDA. 2001b. Letter to sponsors/researchers: Human cells used in therapy involving the transfer of genetic material by means other than the union of gamete nuclei. http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ucm105852.htm (accessed January 9, 2015).
FDA. 2009. FDA regulation of human cells, tissues, and cellular and tissue-based products (HCT/Ps) product list. http://www.fda.gov/BiologicsBloodVaccines/TissueTissueProducts/RegulationofTissues/ucm150485.htm (accessed November 10, 2015).
FDA. 2012. Risk Evaluation and Mitigation Strategy (REMS): The iPLEDGE Program. Single Shared System for Isotretinoin. http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM234639.pdf (accessed January 9, 2015).
FDA. 2014. “Off-label” and investigational use of marketed drugs, biologics, and medical devices—information sheet. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126486.htm (accessed July 9, 2015).
FDA Cellular Tissue and Gene Therapies Advisory Committee. 2014a. Cellular, Tissue and Gene Therapies Advisory Committee Meeting Summary Minutes, February 25, Gaithersburg, MD.
FDA Cellular Tissue and Gene Therapies Advisory Committee. 2014b. Oocyte modification in assisted reproduction for the prevention of transmission of mitochondrial disease or treatment of infertility. FDA briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/CellularTissueandGeneTherapiesAdvisoryCommittee/UCM385461.pdf (accessed December 23, 2015).
Feeney, C., and R. McFarland. 2014. Complications of pregnancy in patients with mitochondrial disease (observational study). In UK Clinical Research Network Study Portfolio. London, UK: National Institute for Health Research Clinical Research Network Coordinating Centre. http://public.ukcrn.org.uk/search/StudyDetail.aspx?StudyID=12853 (accessed December 23, 2015).
Gorman, G. S., J. P. Grady, Y. Ng, A. M. Schaefer, R. J. McNally, P. F. Chinnery, P. Yu-Wai-Man, M. Herbert, R. W. Taylor, R. McFarland, and D. M. Turnbull. 2015a. Mitochondrial donation—how many women could benefit? New England Journal of Medicine 372(9):885-887.
Gorman, G. S., A. M. Schaefer, Y. Ng, N. Gomez, E. L. Blakely, C. L. Alston, C. Feeney, R. Horvath, P. Yu-Wai-Man, P. F. Chinnery, R. W. Taylor, D. M. Turnbull, and R. McFarland. 2015b. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of Neurology 77(5):753-759.
Handyside, A. H., E. H. Kontogianni, K. Hardy, and R. M. L. Winston. 1990. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 344(6268):768-770.
Heindryckx, B., J. Neupane, M. Vandewoestyne, C. Christodoulou, Y. Jackers, J. Gerris, E. Van den Abbeel, R. Van Coster, D. Deforce, and P. De Sutter. 2014. Mutation-free baby born from a mitochondrial encephalopathy, lactic acidosis and stroke-like syndrome carrier after blastocyst trophectoderm preimplantation genetic diagnosis. Mitochondrion 18:12-17.
Houshmand, M., E. Holme, C. Hanson, U.-B. Wennerholm, and L. Hamberger. 1997. Is paternal mitochondrial DNA transferred to the offspring following intracytoplasmic sperm injection? Journal of Assisted Reproduction and Genetics 14(4):223-227.
Huang, C. C., T. C. Cheng, H. H. Chang, C. C. Chang, C. I. Chen, J. Liu, and M. S. Lee. 1999. Birth after the injection of sperm and the cytoplasm of tripronucleate zygotes into metaphase II oocytes in patients with repeated implantation failure after assisted fertilization procedures. Fertility and Sterility 72(4):702-706.
Hudson, K. L. 2006. Preimplantation genetic diagnosis: Public policy and public attitudes. Fertility and Sterility 85(6):1638-1645.
HFEA (Human Fertilisation and Embryology Authority). 2014a. Review of the safety and efficacy of polar body transfer to avoid mitochondrial disease: Addendum to “Third scientific review of the safety and efficacy of methods to avoid mitochondrial disease through assisted conception: 2014 update.” London, UK: HFEA.
HFEA. 2014b. Third scientific review of the safety and efficacy of methods to avoid mitochondrial disease through assisted conception: 2014 update. London, UK: HFEA.
IBC (International Bioethics Committee). 2015. Report of the IBC on updating its reflection on the human genome and human rights (SHS/YES/IBC-22/15/2 REV.2). Paris, France: United Nations Educational, Scientific and Cultural Organization.
Innocenti, P., E. H. Morrow, and D. K. Dowling. 2011. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 332(6031):845-848.
IOM (Institute of Medicine). 2012. Essential health benefits: Balancing coverage and cost. Washington, DC: The National Academies Press.
IOM. 2015. Science and Medicine Panel Discussion. Public Workshop of the Committee on Ethical and Social Policy Considerations of Novel Techniques for Prevention of Maternal Transmission of Mitochondrial DNA Diseases, March 31, Washington, DC.
Knowles, L. P., and G. E. Kaebnick (Eds.). 2007. Reprogenetics: Law, policy, and ethical issues. Baltimore, MD: The Johns Hopkins University Press.
Koga, Y., Y. Akita, J. Nishioka, S. Yatsuga, N. Povalko, Y. Tanabe, S. Fujimoto, and T. Matsuishi. 2005. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 64(4):710-712.
Lanzendorf, S. E., J. F. Mayer, J. Toner, S. Oehninger, D. S. Saffan, and S. Muasher. 1999. Pregnancy following transfer of ooplasm from cryopreserved-thawed donor oocytes into recipient oocytes. Fertility and Sterility 71(3):575-577.
Lee, H. S., H. Ma, Rita C. Juanes, M. Tachibana, M. Sparman, J. Woodward, C. Ramsey, J. Xu, E.-J. Kang, P. Amato, G. Mair, R. Steinborn, and S. Mitalipov. 2012. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell Reports 1(5):506-515.
Lee, H. Y., J. Y. Chou, L. Cheong, N. H. Chang, S. Y. Yang, and J. Y. Leu. 2008. Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 135(6):1065-1073.
Liang, P., Y. Xu, X. Zhang, C. Ding, R. Huang, Z. Zhang, J. Lv, X. Xie, Y. Chen, Y. Li, Y. Sun, Y. Bai, Z. Songyang, W. Ma, C. Zhou, and J. Huang. 2015. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & Cell 6(5):363-372.
Marchington, D. R., M. S. Scott Brown, V. K. Lamb, R. J. van Golde, J. A. Kremer, J. H. Tuerlings, E. C. Mariman, A. H. Balen, and J. Poulton. 2002. No evidence for paternal mtDNA transmission to offspring or extra-embryonic tissues after ICSI. Molecular Human Reproduction 8(11):1046-1049.
Mishmar, D., E. Ruiz-Pesini, P. Golik, V. Macaulay, A. G. Clark, S. Hosseini, M. Brandon, K. Easley, E. Chen, M. D. Brown, R. I. Sukernik, A. Olckers, and D. C. Wallace. 2003. Natural selection shaped regional mtDNA variation in humans. Proceedings of the National Academy of Sciences of the United States of America 100(1):171-176.
Mitalipov, S., P. Amato, S. Parry, and M. J. Falk. 2014. Limitations of preimplantation genetic diagnosis for mitochondrial DNA diseases. Cell Reports 7(4):935-937.
Monnot, S., N. Gigarel, D. C. Samuels, P. Burlet, L. Hesters, N. Frydman, R. Frydman, V. Kerbrat, B. Funalot, J. Martinovic, A. Benachi, J. Feingold, A. Munnich, J.-P. Bonnefont, and J. Steffann. 2011. Segregation of mtDNA throughout human embryofetal development: m.3243A >G as a model system. Human Mutation 32(1):116-125.
Morrow, E. H., K. Reinhardt, J. N. Wolff, and D. K. Dowling. 2015. Risks inherent to mitochondrial replacement. EMBO Reports 16(5):541-544.
Nagy, Z. P., A. C. Varghese, and A. Agarwal (Eds.). 2012. Practical manual of in vitro fertilization: Advanced methods and novel devices. New York: Springer Science+Business Media, LLC.
National Commissioning Group (NCG) for Rare Mitochondrial Diseases of Adults and Children (UK). 2013. Newcastle mitochondrial disease guidelines: Pregnancy in mitochondrial disease. http://www.mitochondrialncg.nhs.uk/documents/Pregnancy_Guidelines_%202011.pdf (accessed December 22, 2015).
Neupane, J., M. Vandewoestyne, S. Ghimire, Y. Lu, C. Qian, R. Van Coster, J. Gerris, T. Deroo, D. Deforce, P. De Sutter, and B. Heindryckx. 2014. Assessment of nuclear transfer techniques to prevent the transmission of heritable mitochondrial disorders without compromising embryonic development competence in mice. Mitochondrion 18:27-33.
NIH (National Institutes of Health). 2013. NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules. Bethesda, MD: HHS, NIH, Office of Biotechnology Activities.
NIH. 2015. Statement on NIH funding of research using gene-editing technologies in human embryos, edited by F. Collins. http://www.nih.gov/about-nih/who-we-are/nih-director/statements/statement-nih-funding-research-using-gene-editing-technologies-humanembryos (accessed December 22, 2015).
NIH Recombinant DNA Advisory Committee. 1985. Points to consider in the design and submission of human somatic cell gene therapy protocols. Recombinant DNA Technical Bulletin 8(4):181-186.
Parikh, S., R. Saneto, M. J. Falk, I. Anselm, B. H. Cohen, and R. Haas. 2009. A modern approach to the treatment of mitochondrial disease. Current Treatment Options in Neurology 11(6):414-430.
Parikh, S., A. Goldstein, M. K. Koenig, F. Scaglia, G. M. Enns, and R. Saneto. 2013. Practice patterns of mitochondrial disease physicians in North America. Part 2: Treatment, care and management. Mitochondrion 13(6):681-687.
Parikh, S., A. Goldstein, M. K. Koenig, F. Scaglia, G. M. Enns, R. Saneto, I. Anselm, B. H. Cohen, M. J. Falk, C. Greene, A. L. Gropman, R. Haas, M. Hirano, P. Morgan, K. Sims, M. Tarnopolsky, J. L. Van Hove, L. Wolfe, and S. DiMauro. 2014. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genetics in Medicine 17(9):689-701.
Paull, D., V. Emmanuele, K. A. Weiss, N. Treff, L. Stewart, H. Hua, M. Zimmer, D. J. Kahler, R. S. Goland, S. A. Noggle, R. Prosser, M. Hirano, M. V. Sauer, and D. Egli. 2013. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493(7434):632-637.
Pfeffer, G., R. Horvath, T. Klopstock, V. K. Mootha, A. Suomalainen, S. Koene, M. Hirano, M. Zeviani, L. A. Bindoff, P. Yu-Wai-Man, M. Hanna, V. Carelli, R. McFarland, K. Majamaa, D. M. Turnbull, J. Smeitink, and P. F. Chinnery. 2013. New treatments for mitochondrial disease—no time to drop our standards. Nature Reviews: Neurology 9(8):474-481.
Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick, and D. Reich. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38(8):904-909.
Quijada-Fraile, P., M. O’Callaghan, E. Martin-Hernandez, R. Montero, A. Garcia-Cazorla, A. M. de Aragon, J. Muchart, I. Malaga, R. Pardo, P. Garcia-Gonzalez, C. Jou, J. Montoya, S. Emperador, E. Ruiz-Pesini, J. Arenas, M. A. Martin, A. Ormazabal, M. Pineda, M. T. Garcia-Silva, and R. Artuch. 2014. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome. Orphanet Journal of Rare Diseases 9:217.
Reddy, P., A. Ocampo, K. Suzuki, J. Luo, S. R. Bacman, S. L. Williams, A. Sugawara, D. Okamura, Y. Tsunekawa, J. Wu, D. Lam, X. Xiong, N. Montserrat, Concepcion R. Esteban, G.-H. Liu, I. Sancho-Martinez, D. Manau, S. Civico, F. Cardellach, M. del Mar O’Callaghan, J. Campistol, H. Zhao, J. M. Campistol, C. T. Moraes, and J. C. Izpisua Belmonte. 2015. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161(3):459-469.
Reinhardt, K., D. K. Dowling, and E. H. Morrow. 2013. Mitochondrial replacement, evolution, and the clinic. Science 341(6152):1345-1346.
Richardson, J., L. Irving, L. A. Hyslop, M. Choudhary, A. Murdoch, D. M. Turnbull, and M. Herbert. 2015. Concise reviews: Assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells 33(3):639-645.
Ruiz-Pesini, E., D. Mishmar, M. Brandon, V. Procaccio, and D. C. Wallace. 2004. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303(5655):223-226.
Sallevelt, S. C. E. H., J. C. F. M. Dreesen, M. Drüsedau, S. Spierts, E. Coonen, F. H. J. van Tienen, R. J. T. van Golde, I. F. M. de Coo, J. P. M. Geraedts, C. E. M. de DieSmulders, and H. J. M. Smeets. 2013. Preimplantation genetic diagnosis in mitochondrial DNA disorders: Challenge and success. Journal of Medical Genetics 50(2):125-132.
Samuels, D. C., P. Wonnapinij, and P. F. Chinnery. 2013. Preventing the transmission of pathogenic mitochondrial DNA mutations: Can we achieve long-term benefits from germ-line gene transfer? Human Reproduction (Oxford, England) 28(3):554-559.
Saneto, R. P., and M. M. Sedensky. 2013. Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics 10(2):199-211.
SART (Society for Assisted Reproductive Technology). 2007. Requirements for SART membership. http://www.sart.org/uploadedFiles/Affiliates/SART/Members/Membership%20Requirements%2007_08.pdf (accessed July 9, 2015).
SART. 2008. Revised guidelines for human embryology and andrology laboratories. http://www.sart.org/uploadedFiles/ASRM_Content/News_and_Publications/Practice_Guidelines/Guidelines_and_Minimum_Standards/Revised_guidelines_for_human(1).pdf (accessed July 9, 2015)
SART. 2015. What is SART? http://www.sart.org/What_is_SART (accessed July 9, 2015).
SART. 2016. Join SART: Requirements for SART Membership. http://www.sart.org/membership (accessed January 11, 2016).
Sato, A., T. Kono, K. Nakada, K. Ishikawa, S.-I. Inoue, H. Yonekawa, and J.-I. Hayashi. 2005. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proceedings of the National Academy of Sciences of the United States of America 102(46):16765-16770.
Say, R. E., R. G. Whittaker, H. E. Turnbull, R. McFarland, R. W. Taylor, and D. M. Turnbull. 2011. Mitochondrial disease in pregnancy: A systematic review. Obstetric Medicine: The Medicine of Pregnancy 4(3):90-94.
Schaefer, A. M., R. McFarland, E. L. Blakely, L. He, R. G. Whittaker, R. W. Taylor, P. F. Chinnery, and D. M. Turnbull. 2008. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 63(1):35-39.
Schwartz, M., and J. Vissing. 2002. Paternal inheritance of mitochondrial DNA. New England Journal of Medicine 347(8):576-580.
Shoubridge, E. A., and T. Wai. 2007. Mitochondrial DNA and the mammalian oocyte. In Current Topics in Developmental Biology, Vol. 77, edited by G. Schatten and J. St. John. Waltham, MA: Academic Press. Pp. 87-111.
Srivastava, S., and C. T. Moraes. 2001. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Human Molecular Genetics 10(26):3093-3099.
Steffann, J., N. Frydman, N. Gigarel, P. Burlet, P. F. Ray, R. Fanchin, E. Feyereisen, V. Kerbrat, G. Tachdjian, J. P. Bonnefont, R. Frydman, and A. Munnich. 2006. Analysis of mtDNA variant segregation during early human embryonic development: A tool for successful NARP preimplantation diagnosis. Journal of Medical Genetics 43(3):244-247.
Stewart, J. B., C. Freyer, J. L. Elson, and N. G. Larsson. 2008. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nature Reviews Genetics 9(9):657-662.
Sutovsky, P., R. D. Moreno, J. Ramalho-Santos, T. Dominko, C. Simerly, and G. Schatten. 1999. Ubiquitin tag for sperm mitochondria. Nature 402(6760):371-372.
Tachibana, M., M. Sparman, H. Sritanaudomchai, H. Ma, L. Clepper, J. Woodward, Y. Li, C. Ramsey, O. Kolotushkina, and S. Mitalipov. 2009. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 461(7262):367-372.
Tachibana, M., P. Amato, M. Sparman, J. Woodward, D. M. Sanchis, H. Ma, N. M. Gutierrez, R. Tippner-Hedges, E. Kang, H.-S. Lee, C. Ramsey, K. Masterson, D. Battaglia, D. Lee, D. Wu, J. Jensen, P. Patton, S. Gokhale, R. Stouffer, and S. Mitalipov. 2013. Towards germline gene therapy of inherited mitochondrial diseases. Nature 493(7434):627-631.
Tarnopolsky, M. A. 2014. Exercise as a therapeutic strategy for primary mitochondrial cytopathies. Journal of Child Neurology 29(9):1225-1234.
Taylor, R. W., and D. M. Turnbull. 2005. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics 6(5):389-402.
Treff, N. R., J. Campos, X. Tao, B. Levy, K. M. Ferry, and R. T. Scott, Jr. 2012. Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertility and Sterility 98(5):1236-1240.
UK (United Kingdom) Parliament House of Lords. 2015. Human fertilisation and embryology (mitochondrial donation) regulations 2015—motion to approve amendment to the motion. http://www.publications.parliament.uk/pa/ld201415/ldhansrd/text/150224-0002.htm (accessed October 13, 2015).
Vafai, S. B., and V. K. Mootha. 2012. Mitochondrial disorders as windows into an ancient organelle. Nature 491(7424):374-383.
Viscomi, C., E. Bottani, and M. Zeviani. 2015. Emerging concepts in the therapy of mitochondrial disease. Biochimica et Biophysica Acta (BBA)—Bioenergetics 1847(6-7):544-557.
Wallace, D. C. 1994. Mitochondrial DNA sequence variation in human evolution and disease. Proceedings of the National Academy of Sciences of the United States of America 91(19):8739-8746.
Wallace, D. C., and D. Chalkia. 2013. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harbor Perspectives in Biology 5(11): a021220.
Wang, M. K., D. Y. Chen, J. L. Liu, G. P. Li, and Q. Y. Sun. 2001. In vitro fertilisation of mouse oocytes reconstructed by transfer of metaphase II chromosomes results in live births. Zygote 9(1):9-14.
Wang, T., H. Sha, D. Ji, Helen L. Zhang, D. Chen, Y. Cao, and J. Zhu. 2014. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 157(7):1591-1604.
Wolf, D. P., N. Mitalipov, and S. Mitalipov. 2015. Mitochondrial replacement therapy in reproductive medicine. Trends in Molecular Medicine 21(2):68-76.
Wolff, J. N., E. D. Ladoukakis, J. A. Enríquez, and D. K. Dowling. 2014. Mitonuclear interactions: Evolutionary consequences over multiple biological scales. Philosophical Transactions of the Royal Society B: Biological Sciences 369(1646).
Zhang, J., G. Zhuang, Y. Zeng, C. Acosta, Y. Shu, and J. Grifo. 2003. Pregnancy derived from human nuclear transfer. Fertility and Sterility 80:56.



















































