
8
Governing Gene Drive Research and Applications
The governance of science ensures that research, whether in a laboratory or in the field, is conducted with appropriate oversight and in accordance with societal values. Governance of technology has a similar role in regard to how the products of research and innovation enter society and the environment. Thus, the governance of science and technology concerns questions about who conducts and oversees research activities, who benefits from scientific advances, mechanisms to ensure that members of the public are protected, and mechanisms to include communities, stakeholders, and publics in making decisions about research and its applications. The accelerated pace of gene drive research, combined with the ease of use of molecular technologies to create gene drives, has prompted discussion of the capacity of existing professional and regulatory mechanisms to govern these activities. The novelty of this technology also provides an opportunity to reflect more generally on the principles governing scientific research and suggest areas for improvement.
The previous chapters of this report identify values and ethical questions reflected in and challenged by gene drive research and its related applications. Through a set of case studies we also explored ways to assess risk and principles for how and why to engage affected communities, other stakeholders, and broader publics, in discussions about gene drive research. This chapter builds upon those themes to answer two primary questions:
- What general principles could guide the evaluation and improvement of governance systems as gene drive research matures?
- Do existing governance systems in the United States and abroad adequately promote and protect public health, the environment, and other societal interests?
These questions are critical for the future of gene drive research and the potential release of gene-drive modified organisms into the environment.
WHAT IS GOVERNANCE?
The definition of governance varies by scholarly discipline, politics, and culture. Governance includes standards—voluntary norms and policies that arise from tradition or consensus processes that are often widely accepted, but not enforceable by law. It also includes regulation—mandatory policies agreed upon by legislative authorities that are enforceable by law. For the purposes of this report, the committee adopts a broad definition that is derived from the World Bank’s World Wide Governance Indicators1 (World Bank, 2015):
The process of exercising oversight through regulations, standards, or customs through which individuals and communities are held accountable. This includes:
- the process by which authorities are selected, monitored, and replaced;
- the capacity of governing authorities to formulate and implement sound policies; and
- the respect of governed communities for the authorities and processes that govern their activities.
___________________
This definition encompasses a wide spectrum of policy tools, including norms and guidelines that stretch from traditional customs to regulation.
Governance of Science and Technology
The importance of governing science has been broadly accepted since the development of the Nuremberg Code after World War II (Annas and Grodin, 1992). The governance of science in the post-WWII United States has included federal and state legislation and other governmental regulations, professional and institutional codes of conduct for scientists, systems of professional certification and accreditation of the education of scientists and manufacturers, public engagement in discourse over science, and other mechanisms to align scientific activities with societal interests in health, environmental integrity, or other social goods (NRC, 2015).
The governance of science consists of both a set of policy tools for self-governance developed by the scientific community, and mandatory policy tools developed by entities outside the scientific community. In self-governance, the scientific community itself defines, establishes, and enforces professional codes of conduct and guidelines that define and govern best practices and unacceptable behavior. These differ from systems of public regulation, wherein national or state authorities have legal powers to oversee the processes and products of research and technology. There is a middle ground in which governments create guidelines that shape the behavior of scientists and research institutions by creating norms and expectations of good practice. Table 8-1 provides some examples of policy tools that govern scientists, research institutions, and applications of science and technology.
TABLE 8-1 Examples of Policy Tools Used to Govern Science and Technology
| Policy Tool | Description | Examples |
| Professional Scientific Standards or Norms | Self- governing mechanisms within the scientific community | Hippocratic Oath, the Nuremberg Code; American Society of Microbiology’s Code of Ethicsa |
| Guidelines on the Practice of Scientific Research | Developed by recognized scientific authority | World Health Organization 2014 Guidance Framework for Testing Genetically Modified Mosquitoes |
| Requirements of Research Funders and Sponsors | Enacted in funding agreements rather than through formal law, and often implemented at the institutional level | US National Institutes of Health Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules; Institutional Biosafety Committees; Institutional Animal Care and Use Committees |
| Regional-Level Regulation of Science and Technology | State or national regulation with binding legal force | California Department of Fish and Game |
| National-Level Regulation of Science and Technology | Governmental regulation with binding legal force | Human subjects research protections in all federally funded research (i.e., the Common Rule and related regulations) |
| International Agreements | Regulatory and non-regulatory agreements between countries. | International Plant Protection Convention to protect cultivated and wild plants by preventing the introduction and spread of pests |
aASM, 2005.
The Spectrum of Governance for Biotechnology: From Prevention to Promotion
The regulation of biotechnology is seldom straightforward. Certain biotechnologies have been controversial precisely because there are disagreements about the levels of risk and uncertainty that they involved, as well as what uncertainty should mean for decision makers (Tait, 2014). Uncertainty attends all governance decisions about safety and hazards because the probabilities produced in risk assessment are never zero or 100% (Charo, 2015). Existing governance for biotechnology products is context dependent, and there does not have to be only one approach to the governance of all biotechnology. Governance tools often take different policy directions across national systems (Tait, 2008). Different societies will tolerate different levels of uncertainty under different circumstances, which results in diverse stances on how to manage innovation, the process through which knowledge is converted into potentially useful applications. However, common stances can be organized into four general categories (Charo, 2015; see Box 8-1).
Innovation and precaution can be complementary with public understanding and effective oversight creating the public confidence needed to support risk-taking and novel technologies (Baltimore et al., 2015; Carroll and Charo, 2015). Nonetheless, some have challenged whether the precautionary principle can truly be implemented (Sunstein, 2009; El-Zahabi-Bekdash and Lavery, 2010). Oversight, however, must be balanced with the potential benefits of innovation. Regulatory regimes that are designed to be adaptive to the lessons of future experience and unexpected harms or benefits could enable the continued development of the science and technology with increased capacity to deliver benefits to society in the future.
KEY CONSIDERATIONS FOR GOVERNING GENE DRIVES
Gene drives have two major features that distinguish them from other types of biotechnology: they intentionally spread a genetic trait through a population, and their effects on ecosystems are potentially irreversible. These two features carry important implications for the governance of gene drive research and related applications.
First, it is the goal of using a gene drive to spread a genetic trait through a population. Intentional spread challenges current governing systems for biotechnology predicated on managing risk by containing genetically modified organisms through physical, biological, or environmental methods. A mechanism designed to spread genetic information has consequences associated with accidental release that differ from other genetically modified organisms. Unexpected gene flow is a concern that regulators of current genetically modified organisms seek to mitigate, whereas such flow is expected and even intended for organisms bearing gene drive constructs. In addition gene-drive modified organisms are expected, at least under some conditions, to cross legal boundaries and territories. Actions with trans-border effects complicate already difficult questions of governance, e.g., who should make decisions, who should be consulted, who is accountable to whom, and how liability should be handled as a legal matter. Thus, the anticipated transboundary effects of gene-drive modified organisms give rise to the need for international policies or regulation that build agreements between countries.
Second, gene drives heighten concerns about irreversibility. Once a gene-drive modified organism is released into the environment, any unintended effects on other species or ecosystems could be potentially irreversible. Thus, it will be important for governance to take into account the potential need to (1) stop the spread of a gene-drive modified organism that has been released; (2) mitigate harm and restore the environment; and (3) provide compensation for harms that cannot be addressed by mitigation or ecological restoration measures. The characteristic of biological irreversibility has important implications not only for physical-material risk, but also for the perception and communication of harms and benefits. Public perception of technological risk tends to respond to known factors that raise special concern. Technologies that are novel and less well-known, whose use is not directly perceptible, or which have delayed outside effects, also tend to be of higher public concern (Slovic, 1987).
General Principles for Governance of Gene Drives
Developing effective governance of science and technology, in general, is challenging because these frameworks must reflect the values of multiple publics, stakeholders, and communities. Some sets of values may align readily, for example, that we should combat human disease and promote and protect human well-being (see Chapter 4). Other sets of values may be in tension or conflict with one another, for example, that ecosystems should be protected and that humans should not tamper with nature (see Chapter 4). An ideal governance framework seeks to ensure that science and technology are safe for people and the environment, deliver the expected benefits, and are developed and used responsibly following high ethical standards. For instance, in some fields, a technical risk assessment of an experiment’s potential harms and benefits is a foundation for decision making (Emanuel et al., 2000). Furthermore, it is clear that governance is a joint responsibility involving the collaboration of a broad range of publics—including public, private, governmental, lay, and professional individuals and organizations.
Based on the distinctive features of gene drives and the discussion of values, risk assessment, and public engagement in previous chapters, several desirable features can be identified for their governance, prior to examining whether existing mechanisms include these qualities (see Box 8-2).
First, risk assessment is thorough and includes a variety of experts. As indicated in Chapter 6, robust models of risk assessment can inform decision makers at each level of governance. Risk assessment will need to be generally informed by the diverse forms of expertise that gene drive technology requires, including knowledge on best practices in laboratory and field research. Furthermore, it is important that risk assessments identify, and when possible, account for sources of uncertainty, confounders, and other limitations. The release of gene-drive modified organisms requires predicting the consequences of genetic modifications in complex environments. This is and will likely remain an imperfect task; sources of uncertainty and ignorance will need to be clear to decision makers.
Second, a process to engage affected communities and broader publics feeds into the governance process. The anticipation of those affected by these decisions is a central tenet of democracy, and, public engagement processes can be useful for bridging gaps between researchers, communities, other stakeholders, and broader publics (see Chapter 7). Communication among scientists, risk assessors, and policy makers with communities has long been seen as an important component of the governance of risk—not just of decision making, but also in the characterization of risk (NRC, 1996). Effective governance creates and sustains effective mechanisms for ongoing conversations with communities, especially those proximate in time and place to proposed activities, before and after decisions are made about research and technology. Applying this principle here—if and when gene drive research moves outside the laboratory, iterative communication with affected communities will be a key part of the risk assessment process. Engagement with broader publics is also essential when important new questions about science and technological governance arise, especially because gene drive technologies are often envisioned to spread beyond the boundaries of discrete human communities.
Third, clear lines of authority and responsibility and methods for accountability are essential to good governance. Due to the distinctive forms of harms and benefits entailed in using gene drives, and the growing public interest in the technology, clear lines of authority and responsibility will be even more important, both in terms of the effects of gene drives and decision making about them. Accountability as a norm aims at generating desired performance through control and oversight, facilitating ethical behavior, and promoting democratic governance through institutional reforms (Dubnick and Frederickson, 2010).
Fourth, proportionality is another central characteristic of the effective governance of technology, with the level of oversight proportionate to the risks involved in the technology as well as sensitive to the ways that regulation can restrict innovation. It is possible, in other words, to “over regulate.” Governance has an important relationship to innovation. Certain forms of governance and regulatory approaches, based on different responses to uncertainty, may adversely impact the development of important new technologies and their potential benefits to society. That being said, the protection of society’s interests and values, as well as public perceptions, may require rigorous oversight in some cases. Proportionality may be especially important to seeing that a single level of oversight should not necessarily be applied across functions and across levels of integration with the environment.
Fifth, good systems of governance are adaptive in the face of scientific and social developments. In arenas of biotechnology like gene-drive modified organisms, the technological frontier will shift constantly. A rigid approach that cannot adapt to changing technological and institutional conditions will quickly become outdated and potentially harmful to the interests it was designed to protect.
Finally, the ability to anticipate trans-boundary movements of gene-drive modified organisms will be critical. Trans-boundary effects, especially harms, can give rise to complex legal and political controversies. Therefore, as a principle, the governance system will need to be conducive to multilateral approaches to governance—including mechanisms, agreements, or norms—in order to encourage cooperation across borders.
RELEVANT GOVERNANCE FRAMEWORKS FOR GENE DRIVE RESEARCH AND APPLICATIONS IN THE UNITED STATES
Biotechnology emerged in the early 1970s with the development of recombinant DNA (rDNA) technology. This new technology allowed the movement of genes from one organism to another to create “engineered” organisms containing genetic combinations that did not exist in nature. From the beginning, rDNA research raised concerns about the potential harms posed by such organisms. After a 1973 conference on rDNA research helped spur a National Academy of Sciences inquiry into its potential hazards (Krimsky, 1982), biologist Paul Berg assembled a
team of distinguished scientists to plan what would become the 1975 Asilomar Conference on Recombinant DNA. The 1980s and early 1990s saw intense debate on the appropriate form of regulation for genetically modified organisms, leading to divergent regulatory approaches in the United States and the European Union characterized as product based and process based, respectively (Tait, 2008). Regulation based on the potential function of a gene drive has now been proposed, where risk is defined as “the ability to influence any key biological component the loss of which would be sufficient to cause harm to humans or other species of interest” (Oye et al., 2014). This is essentially a product-based approach that embraces a case-by-case risk assessment of gene drive technologies. However, the concept of function usefully underscores how important it is that regulatory assessments capture the potential harms to human and environmental health posed by the intended uses of gene drives in their social and ecological contexts.
Coordinated Framework for the Regulation of Biotechnology
In the United States, regulation of gene-drive modified organisms will most likely fall under the Coordinated Framework for the Regulation of Biotechnology. Crafted in 1986 and updated in the 1990s, the Coordinated Framework outlines a comprehensive regulatory policy for ensuring the safety of biotechnology products based on their intended use. Regulatory authority for genetically modified organisms under the Coordinated Framework is shared across the US Food and Drug Administration (FDA), the US Department of Agriculture (USDA), and the US Environmental Protection Agency (EPA). FDA has regulatory oversight over genetically modified foods, or any modified organisms interpreted to contain an “animal drug.” USDA oversees regulation of any organisms that are potential plant pests. EPA has oversight over products perceived to be pesticides. If biotechnology products have potential environmental consequences, all three agencies must adhere to National Environmental Policy Act (NEPA). A fourth agency, the Centers for Disease Control and Prevention, has regulatory authority if/when public health is threatened; for example, if a gene drive intended to prevent the spread of dengue (Case Study 1, Chapter 3), caused the Asian tiger mosquito to be a more effective transmitter of another disease, such as chikungunya.
The regulatory landscape pertinent to gene drive technologies is itself evolving, as the US system of regulating biotechnology is currently being reassessed. Pending changes stem from awareness within government, industry, and civil society that there are potential inconsistencies and gaps that require clarification and adjustment. In July 2015, the Obama administration issued a memorandum directing the “primary agencies that regulate the products of biotechnology—EPA, FDA, and USDA—to update the Coordinated Framework, develop a long-term strategy to ensure that the Federal biotechnology regulatory system is prepared for the future products of biotechnology, and commission an expert external analysis of the future landscape of biotechnology products to support this effort” (Holdren et al., 2015).
An Examination of Governance Mechanisms Through a Phased Testing Pathway
This section canvasses the national and international oversight mechanisms that are most relevant for research on gene-drive modified organisms and potential applications of the technology. The committee uses this landscape to consider the adequacy of US and global capacity to protect public health and the environment from the potential harms of gene-drive modified organisms, and to identify major concerns or gaps. The governance landscape in this section is described through the lens phased testing pathway from laboratory-based research to field trials to environmental release described in Chapter 5 (see Figure 5-1). To aid the committee’s analysis, Case Studies (see Chapter 3, Box 3-1) of likely gene drive applications are used along with more hypothetical examples to discuss considerations for and gaps in governance.
Governance Mechanisms for Phase 1 (Laboratory-Based Research)
In academic settings, laboratory experiments on gene drive technologies are overseen at the institutional level through Institutional Biosafety Committees (IBCs). These committees are the cornerstone of institutional oversight of recombinant DNA research, and are the primary oversight mechanism for research involving genetic modification at National Institutes of Health (NIH)-funded institutions. IBCs work with researchers to develop appropriate protections of health and environmental safety for experiments involving biotechnology. These committees assess the risk of proposed experiments and recommend containment mechanisms based on categories of risk.
For research funded by NIH, the NIH Office of Biotechnology Activities ultimately oversees practices for the safe containment of basic research involving the creation and use of organisms and viruses containing recombinant or synthetic nucleic acid molecules. IBCs are accountable to the NIH Office of Biotechnology Activities and must implement stipulated guidelines for biosafety known as the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH 2016a). When certain kinds of novel experiments are proposed to local IBCs, these must be referred to the Office of Biotechnology Activities, and its advisory body, the Recombinant DNA Advisory Committee (RAC) (NIH, 2016b), for consideration and recommendations. A 2014 Institute of Medicine report, Oversight and Review of Clinical Gene Transfer Protocols: Assessing the Role of the Recombinant DNA Advisory Committee, recommends that the kinds of protocols the RAC assesses should be restricted, particularly if an assessment can be adequately performed by another regulatory and oversight process such as an IBC (IOM, 2014, p. 4). However, these recommendations were developed before the first gene drive proof-of-concept studies were published, and may need to be reconsidered in light of potential gene drive technologies. Indeed, a new National Academies of Sciences, Engineering, and Medicine study is under way that will identify near term biotechnology products, such as gene drive technologies, and provide advice on “the scientific capabilities, tools, and expertise that may be necessary to regulate those forthcoming products.”2
IBCs and other government policies reinforce a system of professional best practices in research. Best practices standards in research consist of both technical and ethical considerations and are essential for the research enterprise. If a laboratory conducts research that involves recombinant DNA, the principal investigator must register the research project with the university and the IBC assigns the project a biosafety level at which the work must be carried out. IBCs are authorized to conduct periodic safety audits to document compliance with the requirements for the project’s laboratory biosafety level, biosafety work practices, and training requirements (HHS, 2009). These laboratory inspections entail a discussion of documentation of lab-specific training and standard operating procedures to ensure that records are up-to-date and reflect the types of experiments being carried out in the laboratory. For example, a typical university laboratory audit might note how microbes, chemicals, compressed gas, and hazardous waste are stored and handled; the state of the current equipment in the laboratory, and the laboratory itself, and whether the conditions impact safety; the presence of required emergency equipment (e.g., chemical spill kits, eyewash, safety shower); whether documentation on personnel training is up to date and if the laboratory possesses a chemical hygiene plan that includes a chemical inventory and standard operating procedures; the presence of relevant personal protective equipment; a risk plan that details experimental purpose, protocols used, types of infectious agents and route of infection, if necessary; annual biosafety cabinet inspections and certifications; a list of where all agents are stored; and whether appropriate signage is present in the laboratory (e.g., laboratory caution, emergency and waste guidelines).
If vertebrate animals are being used in the research, the project’s principal investigator must develop a clearly articulated protocol to be filed with the Institutional Animal Care and Use Com-
___________________
2The project website “Future Biotechnology Products and Opportunities to Enhance Capabilities of the Biotechnology Regulatory System”: http://nas-sites.org/biotech (accessed April 4, 2016).
mittee (IACUC). Protocols must be submitted to the IACUC for scientific and ethical review, and must be approved, prior to the initiation of any animal research. These protocols contain information regarding: experimental design (e.g., number of animals needed, how they will be treated, experiments to be performed and endpoints, pain category); personnel qualifications and training; justification for breeding, breeding methodology, and genotyping; emergency treatment and care (including euthanasia methods); and hazardous agents and how they will be used. In addition, annual updates on the approved protocol must be provided to the IACUC. These updates contain such information as the number of animals (living or dead), whether the protocol will remain active or will be terminated (and why), and if the research objectives have been met or changed. The National Research Council’s Guidance for the Care and Use of Laboratory Animals, Eighth Edition, is an important science-based resource that scientists may draw upon as the develop protocols and carry out their research (NRC, 2011). In addition, research must be conducted in accordance with the Animal Welfare Act, which regulates research on a number of live or dead “warm-blooded” animals, excluding birds, rats (Rattus species), mice (Mus species), and food animals. As of May 2016, the committee is unaware of formal gene drive research proposals on animals that fall within the regulatory jurisdiction of the Animal Welfare Act.
Certain laboratory work on genetically modified plant species and “plant pests” is subject to federal regulations under the Biotechnology Regulatory Services of the Animal and Plant Health Inspection Agency (APHIS) of USDA. This body maintains jurisdiction over certain genetically modified organisms, particularly plant pests, including the transport of seeds or plants intended for laboratory use. The regulations are intended to help ensure that regulated genetically modified organisms are not harmful to plants or plant products by controlling the importation, interstate movement, or release into the environment of regulated organisms. Unauthorized (including accidental) importation, interstate movement, or release of a regulated article is a violation of the APHIS regulations (Plant Protection Act of 2000).
In sum, existing systems to govern biotechnology research in the laboratory include professional guidelines, institutional oversight committees that, in most cases, are accountable to federal agencies, and a process through which novel and controversial research can be considered by federal authorities before it proceeds. These systems are likely to have the flexibility to adapt well to gene drive technologies.
Governance Mechanisms for Phase 2 (Field Based Research) and Phase 3 (Staged Environmental Release)
Because US governance and regulatory considerations for Phase 2 and Phase 3 are similar, the following discussion applies to both phases, unless otherwise noted.
As noted above, regulatory authority for gene drive technology will likely be dictated by the Coordinated Framework for the Regulation of Biotechnology. Ideally, the standards and regulations appropriate for field testing or environmental release of gene drive technologies would be commensurate with potential harms, and take into account the extent to which a gene is expected to spread throughout the target population (e.g., Oye et al., 2014). However, as described below, the current US regulatory system does not particularly account for the intentional spread of genetically modified organisms or their potential persistence in the environment. In addition, it is not clear how existing biotechnology regulations apply to gene drive technologies.
Through its regulatory programs, APHIS has used its “plant pest authority” under the Plant Protection Act as the major tool for regulating biotechnology and releases into both contained and open areas. The Plant Protection Act also gives APHIS authority to regulate “noxious weed.” APHIS is actively considering revising its rules to incorporate this additional authority into regulation, but to date has not done so (Pearson, 2015). Whether USDA can or will regulate a gene drive technology such as the gene-drive modified Palmer amaranth in Case Study 6 (see Table 8-2) is unclear, because the noxious weed authority has not yet been translated into regulation.
TABLE 8-2 Potential US Regulatory Mechanisms to Oversee Environmental Release: Analysis of Selected Gene Drive Case Studies
| Case Study 1 (mosquito) | Case Study 3 (mosquito) | Case Study 4 (mouse) | Case Study 6 (plant) | |
| Application of the gene-drive modified organism | Reduce or eliminate the spread of dengue from mosquitoes to humans | Reduce the spread of avian malaria to threatened and endangered birds in the Hawaiian islands | Reduce or eliminate invasive mouse species from islands | Reduce or eliminate Palmer amaranth on agricultural fields in the southern United States |
| Regulatory authority under the current Coordinated Framework | FDA is likely to regulate genetic constructs within a gene-drive modified mosquitoes as “new animal drugs†as the agency has with the Oxitec genetically engineered mosquito; however it is unclear from the Coordinated Framework and guidance documents how that authority was determined | Regulation of a gene-drive modified mouse could fall under any one of three agencies if mice are considered a plant pest (USDA), if the gene drive is considered a new animal drug (FDA), or if it is considered a pesticide/rodenticide (EPA) | The Plant Protection Act gives USDA the authority to regulate noxious weeds. The agency has not yet revised its rules to incorporate noxious weeds into their biotechnology regulatory authority | |
| Agency-specific assessment under the current Coordinated Framework | Impact assessment under National Environmental Protection Act. If FDA assumes regulatory control, then they develop set of tailored assessment questions for each potential product | Without clarity of regulatory authority, assessment would be based on voluntary actions of research partnerships involved in development of the gene-drive modified mouse | If USDA assumes regulatory control field tests and environmental release impact assessments would be conducted under that National Environmental Protection Act | |
| Select regulatory uncertainties |
Differences in agency approach to assessing harms, public consultation, and other components of decision making. In Case Study 4, for example, regulation of a gene-drive modified mouse as a rodenticide under the EPA, would likely trigger agency policies for ecological risk assessment, a quantitative and much more rigorous assessment than environmental impact assessments that FDA and USDA might carry out under the auspices of NEPA.
The role of agencies with authority over sites where gene-drive modified organisms might be released or over species that may be affected by the release. In Case Study 3, for example, what is the role of US Fish and Wildlife, which has authority over endangered and threatened honeycreeper birds and USDA Forest Service, which has authority over much of the Hawaiian forests where endangered birds reside? The role of tribal governments in the decision making process for the field testing or release of gene-drive modified organisms on or near tribal lands. It is uncertain how institutional decisions regarding gene drives will be integrated with tribal governance frameworks to ensure justice and respect. In Case Study 1, for example, if dengue moves into the southern United States, who has authority to determine whether gene-drive modified mosquitoes could be released in or near tribal lands? Mechanisms in place for international considerations and coordination for field testing or release of gene-drive modified organisms near national borders. In Case Study 6, for example, Palmer amaranth is a weed in the southern United States that can interbreed with related plant species that are cultivated as vegetable crops in Mexico. What mechanisms are in place for dialogue with the Mexican national government? How will any concerns raised by the Mexican government be incorporated into US decision-making processes? |
|||
For technologies that qualify, the APHIS-Biotechnology Regulatory Services system specifies permit conditions for field trials. These conditions are customized to the organism, trait, and release locations, in order to maximize confinement. Supplemental permit conditions can include a minimum separation distance to wild relatives and post-harvest monitoring requirements, among others. In 2014, USDA authorized close to 11,000 field trials of more than 12,000 types of genetically modified organisms (Pearson, 2015). These organisms include insect plant pests, such as the pink bollworm and the diamondback moth, which have been engineered to suppress pest populations. APHIS draws a distinction between “containment procedures,” which are used to prevent exposure of modified organisms to the environment, e.g., in laboratories, greenhouses, and during transport, and “confinement procedures” used during field trials to ensure the modified organism does not persist in the environment. The latter include reproductive isolation and post-harvest monitoring. For “contained” settings, the probability of release should be near zero; for “confined” settings, the probability of persistence in the environment should be near zero. Because some gene drive technologies will be intended to persist in the environment, there is a clear mismatch with the current regulatory goal to prevent environmental persistence.
New engineering techniques are likely to lead to a higher number of genetically modified plants that will not be subject to USDA review (Carter et al., 2014). This is because APHIS’s authority to regulate engineered plants relies on its “plant pest” authority. Even if APHIS were to add “noxious weed” authority to its biotechnology regulations, the limits are still likely to apply. This regulatory gap could mean that an increasing number of genetically modified plants may eventually be cultivated “for field trials and commercial production without prior regulatory review for possible environmental or safety concerns” (Carter et al., 2014). This result could also occur if the modifications are made using gene drive technologies, although this is perhaps less likely because gene drive applications are more likely to be aimed at the control of plant pests.
It is likely, but not certain, that FDA has the authority under the Federal Food Drug and Cosmetics Act (FFDCA) to regulate gene-drive modified organisms. The trigger for FDA oversight of gene drive technologies would be the operable term “drug,” defined in part as “articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals” or as “articles (other than food) intended to affect the structure or any function of the body of man or other animals” (Rudenko, 2015). The FDA’s Center for Veterinary Medicine (CVM) is currently treats the genetic construct within an organism as a “new animal drug,” requiring both premarket approval and post-approval oversight. The CVM states that “the [heritable] rDNA construct in a genetically engineered animal that is intended to affect the structure or function of the body of the genetically engineered animal, regardless of the intended use of products that may be produced by the genetically engineered animal, meets the FFDCA drug definition.” In other words, it is the rDNA construct itself, and not the animal into which it has been inserted, that is considered a “drug” (FDA, 2015a). Commercial entities wishing to market “regulated articles” under FDA’s authorities over genetically modified animals must demonstrate that they are safe and effective.
However, the FDA has recently specified a definition of genetically engineered organisms that does not encompass modified insect disease vectors, modified invasive species, or many of the other types of applications likely to be relevant to gene drives. In its Guidance for Industry 187: Regulation of Genetically Engineered Animals Containing Heritable Recombinant DNA Constructs (FDA, 2015b), FDA defines genetically engineered animals “as those animals modified by rDNA techniques, including the entire lineage of animals that contain the modification.” The guidance document also enumerates six classes of animals “based on the intended purpose of the genetic modification,” as follows:
- to enhance production or food quality traits (e.g., pigs with less environmentally deleterious wastes, faster growing fish);
- to improve animal health (e.g., disease resistance);
- to produce products intended for human therapeutic use (e.g., pharmaceutical products or tissues for transplantation; these GE animals are sometimes referred to as “biopharm” animals);
- to enrich or enhance the animals’ interactions with humans (e.g., hypo-allergenic pets);
- to develop animal models for human diseases (e.g., pigs as models for cardiovascular diseases); and
- to produce industrial or consumer products (e.g., fibers for multiple uses).
The six criteria create some uncertainty as to whether the FDA has the regulatory authority to consider gene-drive modified organisms such as mosquitoes designed to prevent the spread of infectious disease in humans or animals (Case Studies 1, 2, and 3), or a mouse designed to reduce or eliminate nonindigenous mice on islands (Case Study 4). Despite the lack of clarity in the guidance, FDA is reviewing an Investigational New Animal Drug (INAD) application for a genetically engineered mosquito developed by the company Oxitec Limited more than 10 years ago. The mosquito is designed to suppress wild populations of Aedes aegypti, a species that transmits a variety of human infectious diseases including dengue, chikungunya, Zika, and yellow fever. Since 2008, Oxitec pursued discussions with the USDA and other regulatory agencies concerning the proper oversight of a field trial in Florida (Waltz, 2015). Oxitec seeks to conduct a field trial in Key Haven, Florida. In March 2016, the FDA released for public comment the draft environmental assessment submitted by Oxitec (FDA, 2016).
State and local laws, regulations and ordinances also contribute to the complex regulatory environment for outdoor research with gene drive constructs in animals. Of greatest import may be the state-level environmental laws (e.g., the California Environmental Quality Act), and state and local notification requirements for the release of genetically modified organisms (e.g., Virginia Biotechnology Research Act Sec. 2.2-5500-5509).3
Gene-drive modified organisms released into the environment have the potential for transboundary movement. Governance will require communication and coordination between adjacent countries or states with separate regulatory jurisdiction. Both regional and national rules and regulations would apply. Laws and regulations at the country and local levels (nation, state, province, county, or lesser levels of jurisdiction control, such as a village) are also likely to play a significant role in the governance of the release of gene-drive modified organisms and their potential transboundary movement. The phase of staged environmental release, in particular, will have direct effects and implications for communities near and adjacent to the location of release, animating the issue of community participation in research governance.
Environmental Assessment and Public Consultation Under the National Environmental Policy Act
Like all other federal agencies, FDA and USDA/APHIS are subject to the National Environmental Policy Act (NEPA). NEPA requires agencies to determine if an environmental analysis is needed for a proposed action, and to assess impacts of those actions that have the potential to harm the environment (see Chapter 6 for additional discussion of the NEPA process). In the context of the Coordinated Framework, NEPA requires an environmental assessment (EA) to determine whether the introduction (field test of environmental release) of a specific biotechnology or related product has the potential to cause significant environmental effects, and inform federal government decisions whether to allow such an introduction. Federal agencies must prepare an Environmental Impact Statement (EIS) if a proposed major federal action is determined to significantly affect the quality of the human environment. The procedural requirements for an EIS are more detailed and rigorous than the requirements for an EA (40 CFR Part 1502).
___________________
3See http://law.lis.virginia.gov/vacodepopularnames/virginia-biotechnology-research-act.
Federal agencies can develop their own guidance for developing and evaluating environmental assessments. For example, APHIS performs EAs before providing permits for the release of modified organisms. The hazards of interest in such assessments include the potential for (1) a modified plant to become a weed in agricultural settings or to be invasive in natural habitats; (2) gene flow from the modified plant to sexually compatible plants whose hybrid offspring may become more weedy or more invasive; (3) the modified plant to become a plant pest; or (4) the modified plant to have an impact on non-target species. As is discussed in Chapter 6, EAs require supporting data to estimate impacts, but often the anticipated effects are not quantified as they would be in a risk assessment. Once a genetically modified organism is shown to lack hazardous traits and enters the commercial marketplace, it is no longer regulated by APHIS (Pearson, 2015).
Applications for products that are genetically modified animals are evaluated by FDA using what the agency calls a “risk-based approach.” FDA develops a specific set of questions about potential harms and benefits using a case-by-case approach for each product under evaluation. The intended application of the product drives the environmental assessment based on product definition, conditions of use, and other factors.
Two critical points need to be made in describing the potential role of NEPA and associated environmental assessments in the analysis of environmental effects of gene-drive modified organisms. First, to recap an important point from Chapter 6, while the preparation of an EA requires the assessment of potential impacts of the research activity, an EA does not require an ecological risk assessment. Thus, the necessary evidence to quantitatively estimate risk may not be gathered for environmental assessment procedures normally performed under NEPA.
Second, NEPA includes provisions for some public engagement. For environmental assessments, agencies sometimes take into account public views in the form of a public hearing or comment period. The INAD that Oxitec submitted for its genetically engineered mosquito includes an environmental assessment. FDA issued a preliminary finding of no significant impact (FONSI) that agrees with the draft EA’s conclusion. However, FDA has said it will review public comments on the EA before issuing either a final EA and FONSI, or an EIS (FDA, 2016). NEPA explicitly requires public consultation for an EIS. Through mandatory public hearings and comment periods, members of the public can express their views about the relative value of potential benefits and harms, and concerns about assumptions built into the environmental impact statement Thus, through provisions requiring transparent decision making and public input of various kinds, NEPA affords stakeholders and the general public the opportunity to participate directly in governance.
Some warn that the EA and EIS process “can be quite costly and time-consuming for the product developer” (Carter et al., 2014). NEPA has also been a tool for those who would use the courts to challenge an EA and FONSI and force a full EIS, which can delay matters for years and fundamentally alter the economics of a proposed innovation. Nevertheless, given the desirability of creating space for public engagement, NEPA would seem to be an important regulatory resource for the integration of public values into the governance processes.
Examining US Regulation of Gene-Drive Modified Organisms Through Case Studies
Which federal agency has the jurisdiction to approve field tests or environmental release of gene-drive modified organisms in the United States? Table 8-2 illustrates how the Coordinated Framework might apply to select case studies: Case Study 1 (gene-drive modified mosquito to combat dengue); Case Study 3 (using the house mosquito to combat avian malaria); Case Study 4 (controlling populations of nonindigenous house mice to protect biodiversity on islands); and Case Study 6 (controlling Palmer amaranth to increase agricultural productivity).
Notably, in all four cases, how gene-drive modified organisms fit within regulatory jurisdiction of FDA, USDA, and EPA is unclear, and their processes for assessing risks may differ from one another. In addition, there are many regulatory uncertainties, some of which have been
listed in Table 8-2. For one example, if a gene-drive modified organism, such as the Culex mosquito (Case Study 3) has the potential to effect an endangered species such as honeycreeper birds (for good or for ill), what is the role of the Endangered Species Act and the US Fish and Wildlife service, which has regulatory authority over actions that may affect the birds? A second example, is determining oversight for gene-drive modified organisms where there may be regulatory overlaps among the USDA, FDA, or EPA. In the case of a gene-drive modified mouse (Case Study 4), USDA could be considered the regulatory authority under the Animal Health Protection Act (7 U.S.C. § 8301) if the mouse is considered a threat to animal health, or under the Plant Protection Act (7 U.S.C. § 7701) if the mouse poses a threat to plants. The FDA could also be considered the regulatory authority for the mouse because the genetic construct (the T complex) used to develop a gene drive in the mouse might be considered an animal drug, because the T complex would be used to influence fertility. Although, it is clear that suppressing or eradicating a species population is not encompassed by FDA’s six classes of animals “based on the intended purpose of the genetic modification.” Finally, the EPA could be considered the appropriate regulatory authority under the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA) (7 U.S.C. § 136 et seq.) if the wild-type mice are considered a pest, and the gene-drive modified mouse or the gene drive construct within the mouse is considered a “substance or mixture of substances intended for preventing, destroying, repelling, or mitigating” the wild-type pest (FIFRA § 2[u], 7 U.S.C. § 136[u]). A real world example of this confusion occurred with the genetically engineered mosquito developed by Oxitec. “The question of FDA versus USDA jurisdiction circled for years, until finally an understanding was reached: The FDA Center for Veterinary Medicine would be the lead agency coordinating other federal and state agencies, [but] by then, Oxitec had begun trials in South America and the Caribbean” (Charo and Greely, 2015). This Oxitec case demonstrates both a major challenge (years of delay) when there are overlaps in regulatory jurisdiction and a potential solution, creating a process to quickly develop memorandums of understanding between federal agencies when regulatory jurisdiction is uncertain. As a third example, the mechanisms to solicit input from communities that live in or near potential sites for field testing or environmental release of a gene-drive modified organism are unknown. NEPA mandates public involvement to include, at a minimum, “reasonable public notice” of environmental assessments, but it is not clear which mechanisms each agency would use in the case of a gene-drive modified organism or how public input would be incorporated in the decision making process is not clear. In addition “reasonable public notice” falls short of the engagement that is needed for gene-drive modified organisms (see Chapter 7).
BIOSECURITY CONSIDERATIONS
An area that will need continual discussion and evaluation is the biosecurity and related uncertainties of gene drive research. It is assumed that efforts to introduce a gene drive into an organism are performed with good intent, that ethical and regulatory standards will be followed, and that the necessary review and approval by oversight committees will be sought. However, concerns related to gene drive technology include not only unintended or unanticipated effects, but also the potential for the unethical, intentional creation of an organism with the capacity to spread undesirable traits into a population. As an illustrative case study, in late 2011, manuscripts by two independent research groups describing research on the highly-pathogenic avian influenza H5N1 that increased the transmissibility of the virus (Herfst et al., 2012; Imai and Kawaoka, 2012; Russel et al., 2012) gained the attention of the US National Science Advisory Board for Biosecurity; the concern that the studies could turn H5N1 into a bioweapon resulted in a worldwide moratorium on the research and legal battles to get manuscripts published.
A US government policy for oversight of dual use research of concern in the life sciences developed in 2012 was modified in 2014 to include new requirements for oversight and training (S3, 2014). Research involving any of 15 agents or toxins must be reviewed in the context of dual use potential, and it is possible that gene drive research could fall under one of the seven
categories of experiments listed in the policy. As described in Chapter 5, planning research that involves genetically engineering mosquitoes requires multiple steps with associated guidelines (including for physical and biological containment), regulations, and laws that determine progress from concept to release. However, engineering that includes the introduction of a gene drive may require modification of current governance and perhaps the implementation of review criteria that to date have not yet been applicable to the field. Although they seem unlikely, examples of possible scenarios where dual use might apply are described below.
Gene drives are likely to raise similar biosecurity concerns as those raised in the discussion of genetic modification and synthetic biology techniques. In these cases, state-sponsored terrorism is considered to be the most serious threat to biosecurity and also to be the most difficult to pre-empt. Reports dealing with the governance of synthetic biology have cited a range of precautionary measures to address biosecurity threats (Lowrie and Tait, 2010; Presidential Commission for the Study of Bioethical Issues, 2010; IRGC, 2011). These reports have also pointed out that the most effective means to deal with such threats should they materialize is to use the relevant scientific expertise to develop rapid diagnostic techniques and synthesis methods for vaccines and antibiotics to enable a fast response to a threat (Presidential Commission, 2010; IRGC, 2011). The availability of rapid diagnostic and synthesis technologies will also enable states to respond rapidly to the much more likely threat of a naturally occurring emerging disease or a future pandemic.
There are several types of concerns related to safe, ethical, and secure research:
- Unintended and unforeseen consequences of release;
- Unintended releases due to negligence or natural disasters;
- Release of information that could be used for intentional misuse; and
- Intentional release or misuse of a gene-drive modified organism.
Unintended consequences or releases are the domain of biosafety. In general, scientific norms and institutional guidelines on biosafety adequately address these issues. The potential for misuse of research, however, is the domain of biosecurity. As noted by the International Academy Panel (IAP), it is difficult to predict the outcome and consequences of research; nonetheless the potential for misuse must be “anticipated and minimized to the extent possible in the planning, performance, and dissemination of research” (IAP, 2016). The IAP emphasizes that scientists have a responsibility “to participate in discussions about the possible consequences of their work, including harmful consequences, in planning research projects.”
Intentional Misuse
Gene drive research has advanced considerably for mosquitoes (see Chapter 2). The impetus for genetically engineering mosquitoes is to control mosquito-borne diseases, either by suppressing mosquito populations or by replacing existing wild populations with mosquitoes that have a reduced capacity to be infected with or transmit a pathogen, such as dengue viruses or Plasmodium species that cause malaria. As described in preceding chapters, a number of excellent guides are available to ensure that researchers working to genetically engineer mosquitoes follow ethical steps from concept to application and are performing these experiments in situations and facilities that protect the public and minimize the risk of accidental release into the environment. Although the committee firmly believes that members of legitimate research community working on gene drives do so ethically and work with the intent to benefit society, for the sake of completeness, the possibility that there may be researchers (or regimes who control research agendas) whose motivation is to cause harm needs to be considered. Given the current understanding of the genetics of vector competence, using gene drives in mosquitoes for malicious intent would seem to be extremely difficult from a technical standpoint, making gene drive research an unattractive proposition compared with other options for causing harm. Yet, with a
better understanding of the basis of mosquito—pathogen interactions, it is not inconceivable that rather than developing a resistant mosquito, one could develop a more susceptible mosquito capable of transmitting a specific pathogen more efficiently than wild-type mosquitoes. It might even be possible to develop mosquitoes that could transmit a pathogen that is not normally vector-borne, or that could even be able to deliver a toxin. The latter might be accomplished by engineering a gene encoding a toxin with a secretion signal under the control of a salivary gland gene. Unlikely as this may sound, early discussions on applications for genetically engineered mosquitoes included expression of heterologous proteins to vaccinate the humans on whom they fed (Crampton et al., 1999) and a patent was issued to protect this technology.4 As a proof of principle, Kamrud and colleagues (1997) infected mosquitoes with a viral expression vector and were able to detect a marker in the mosquito saliva. Other researchers used a similar approach to express a toxin gene in mosquitoes, although the location of the protein in specific tissues was not attempted because toxin expression at very low levels rapidly killed the mosquitoes (Higgs et al., 1995).
The actual and potential use of insects as weapons has been discussed; for example, by releasing insects infected with human pathogens or releasing agricultural pests (Lockwood, 2012). However, the availability of a gene drive provides a new opportunity for malicious use because its self-sustaining nature poses a perhaps more significant threat. In the context of such research being performed in an academic setting, such experiments would be subject to scrutiny via the IBC review process. Since September 2015, if the reviewed research meets certain criteria, the research institution is required by the US Department of Health and Human Services (HHS) government policy (S3 2015) to determine whether the proposed research should be designated as Dual Use Research of Concern.
Research to introduce a gene drive into mosquitoes could conceivably be interpreted as meeting experimental criteria included in the HHS dual use policy. Such criteria apply to research that, for example, could disrupt immunity or effectiveness of immunization, could increase the transmissibility or ability to disseminate an agent or toxin, or could alter the host range or tropism of an agent or toxin. Mosquito salivary gland proteins can influence immune responses of the vertebrates on which they feed and can influence pathogen establishment and diseases development (Schneider and Higgs, 2008). Moreover, since it may be possible to engineer mosquitoes to be more efficient vectors, which in effect increases the transmissibility of pathogens, it is probable that some approaches to genetically modifying mosquitoes may constitute dual use research of concern. As stated above, this discussion applies in the context of developing a gene-drive modified mosquito with good intent; however, just as there are inadequacies associated with the Cartagena Protocol with regards to oversight and jurisdiction, those who would deliberately create modified mosquitoes with malicious intent will likely operate outside of the purview of ethics, biosafety, and other review committees. In the 2016 Worldwide Threat Assessment of the US Intelligence Community, the US Director of National Intelligence classified genome editing as a weapon of mass destruction and proliferation (Clapper, 2016). The assessment states “given the broad distribution, low cost, and accelerated pace of development of this dual-use [genome editing] technology, its deliberate or unintentional misuse might lead to far-reaching economic and national security implications” (p. 9). The impact the Worldwide Threat Assessment may have on gene drive research is not yet known.
There are well-developed resources and guidelines adequate to enable safe and secure research with appropriate oversight in, for example, academic environments in which research is being performed. Manipulation of mosquitoes should be performed in arthropod containment level two (ACL-2) insectaries at a minimum, which fulfill facility design criteria, with appropriate standard operating procedures and adequately trained personnel. In addition, the NRC publi-
___________________
4Delivery system US 20030192067 A1, Inventors Robert Sinden and Julian Crampton. For more information, see http://www.google.com/patents/US20030192067.
cation Understanding Biosecurity: Protecting Against the Misuse of Science in Today’s World details the role of the scientific community and governments in preventing misuse (NRC, 2010). Box 8-4 summarizes key concepts in the report (NRC, 2010) that are relevant for gene drives.
GOVERNANCE OF GENE DRIVES IN GLOBAL CONTEXTS
International sources of governance that may apply to gene drive research have as much impact on whether and how science develops, as do national the United States’ sources of domestic governance. As gene drive research advances, the scientific community and regulators will need to consider mechanisms and policies for global engagement for two main reasons. First, gene drive science is a global endeavor. The early stages of gene drive research (e.g., phase 1 and phase 2; see Figure 5-1) are predominantly taking places in high income countries like the United States, the United Kingdom, and Australia. However, later phases of research—contained and open field tests—are likely to take places in other parts of the world. One example is the use of gene-drive modified mosquitoes to combat human malaria (Case Study 2), a disease that disproportionately impacts the tropics and the southern hemisphere, particularly in low- and middle-income countries. Field trials are most likely to take place in countries where malaria is endemic just as related research on the use of Wolbachia and the RIDL mosquito to combat dengue has been concentrated in these countries. However, some of the jurisdictions targeted for field testing or releases may lack the capacity to assess safety of experiments in a scientifically and socially robust fashion.
Second, the unique qualities of gene-drive modified organisms to spread and persist in the environment will require any nation planning field tests or environmental releases to consider whether and how gene-drive modified organisms will cross national borders. As noted previously, for example, Palmer amaranth is a damaging weed in the United States (Case Study 5), but a related Amaranthus species, with which Palmer amaranth can interbreed, is cultivated for food in in Mexico, South America, India, China, and Africa. The escape of a suppression drive in Palmer amaranth could affect non-targeted species and negatively impact valued Amaranthus vegetable crops. There are currently no national regulatory mechanisms worldwide that adequately address field testing and environmental releases of gene-drive modified organisms. Scholars of governance warn that the regulation of new technologies with societal implications will require evidence-based policy processes, with deliberate and participatory engagement in policy making by the people who will be impacted by these innovations (Lyall and Tait, 2005).
Gene drive research will require international collaborations, and attention should be given to the research capacity and biosafety regulations in other parts of the world, particularly in low- and middle-income countries. Reconciling differences between preventative and permissive regulatory schemes, as described earlier in this chapter, will likely be a considerable challenge for the international development and testing of gene-drive modified organisms. The difficulties introduced by widely divergent regulatory systems are compounded by the potential for these organisms to spread across state and national borders. Careful consideration will need to be given to whether national differences in approaches to governance will create gaps in the ability to protect human health and the environment, or whether such differences could impede basic research that does not yet have clear benefit for society. For these reasons, responsible systems of governance will need to incorporate clear mechanisms for international dialogue among governing authorities, and perhaps, formal or informal agreements about the use of potential gene drive technologies and comparable standards for biosafety.
International cooperation and attempts to harmonize research standards for science and technology is not a new endeavor. Policy tools that span guidelines for research to legally enforceable treaties have been considered and developed for many areas of science, such as stem cells research, climate change, and nanotechnology. In general, there are three commonly used governance mechanisms for international agreements: policy, international coordination and cooperation, and formal treaties (Breggin et al., 2009; see Table 8-3). In the 2015 International Summit on Human Gene Editing (NASEM, 2016), Gary Marchant, Professor of Emerging Technologies, Law and Ethics at Arizona State University, laid out a number of disadvantages and advantages to international systems of governance (Marchant, 2015). Marchant observed that it is difficult to integrate social, political, and ethical norms of different countries into a single policy, and that developing international systems of governance may require substantial resources that may take away from developing strong national-level oversight. On the other hand, the benefits of internationalization include standards that provide consistent requirements for scientists and their research institutions, and such standards could ensure equal protection for citizens of all nations. Marchant noted that it is difficult to develop international harmonization of governance when some countries lack national regulations; nonetheless developing national regulations in every country before putting harmonization mechanisms in place may unduly delay international agreements and be more difficult in the face of entrenched and inconsistent national regulations, such as those on genetically modified organisms.
Two relevant sources of international governance of gene drive research are the United Nations Convention on Biological Diversity (CBD) and the World Health Organization’s (WHO’s) Guidance Framework for Testing Genetically Modified Mosquitoes. Neither CBD nor the WHO explicitly addresses gene drive research, although discussions are under way.
TABLE 8-3a Types of International Agreements
| Policy Tool | Definition | Example |
| Policy | Informal communications and policy learning between regulators | US–UK Agreement for Scientific and Technological Cooperation |
| International coordination and cooperation | Formal or informal congruent approaches without large-scale adjustment of domestic law and regulation | World Health Organization Guidance Framework for Testing Genetically Modified Mosquitoes |
| Treaties | Formal negotiated agreements on common rules and standards for domestic regulation | Convention on Biological Diversity |
aBased on Breggin et al., 2009.
The Convention on Biological Diversity and Its Protocols
The United Nations Convention on Biological Diversity is the main international regulatory instrument governing the development and use of genetically modified organisms. The CBD is a multilateral treaty focused on the global conservation of biological diversity. To date, 193 states are parties to the Convention. The objectives of the Convention are threefold:
- conservation of biological diversity;
- sustainable use of the components of biological diversity; and
- fair and equitable sharing of the benefits arising out of the utilization of genetic resources.
Parties to the Convention5 are required to “establish or maintain means to regulate, manage, or control the risks associated with the use and release of living modified organisms resulting from biotechnology which are likely to have adverse environmental impacts that could affect the conservation and sustainable use of biological diversity, taking also into account the risks to human health” (UN, 1992).
The Convention on Biological Diversity is implemented through its two protocols (international agreements), the Cartagena Protocol on Biosafety and the Nagoya Protocol (NP) on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization. Although the Convention itself does not strictly police compliance, many of the Parties have regulatory systems, developed under the Convention and its protocols, that are based on a strong precautionary, near preventative approach, and implement its provisions in a way that is seen by some to be overly restrictive of these technologies, the EU being the most prominent example (Strauss et al., 2009; Freeman and Swidicki, 2013).
The Cartagena Protocol was developed primarily because of concerns related to genetically modified crops, with the purpose of addressing potential risks posed by releases of genetically modified organisms into the environment (CBD, 2016). However, potential extensions of the powers of the Convention in the governance of gene drive research, and, relatedly, synthetic biology, are being explored. A 2012 report of an ad hoc technical group on risk assessment includes discussion on mosquitoes modified with a gene drive (CBD, 2010). In addition, the 2012 “Guidance Document on Risk Assessment of Living Modified Mosquitoes” (CBD, 2012), recognizes that “In cases where living modified mosquitoes are modified with gene drives, containment may not be possible even when efforts are made to reduce long-distance dispersal due to anthropogenic activities.” In 2015, the Open-ended Online Forum on Synthetic biology was
___________________
5To become a Party to the CBD and its protocols, a nation must first have gone through a process of ratification, acceptance, and approval or accession, after which it can take part in decision making processes, and is also obliged to pass national laws implementing CBD provisions.
held to inform work of Ad Hoc Technical Expert Group. For the Forum, the Conference of the Parties (COP) invited Parties, other governments, relevant international organizations, indigenous and local communities, and other relevant stakeholders to submit information on seven topics related to synthetic biology to the Executive Secretary. The Forum demonstrated that there is a broad range of differing opinions, between and within nations, about the operation of the Convention, and about how its current provisions would relate to the governance of synthetic biology (CBD, 2015), and by extension to gene drives.
At the time of the adoption of the Convention on Biological Diversity and the Cartagena Protocol, there were good reasons to take a cautious approach to the potential harms to biodiversity that might arise from the development of genetic engineering technologies, particularly in agriculture. However, there is a growing body of evidence that genetically engineered crops deliver many significant benefits for agriculture, particularly for resource-poor farmers, as well as for biodiversity and ecosystem services (ISAAA, 2015). The apprehension about the extension of the powers of the Convention and its protocols to cover synthetic biology, expressed in the consultations referenced above, relates to the lack of adaptation of the strong precautionary approach in light of the evidence we now have for safety and benefits. Indeed, within these consultations, there are continuing calls for additional enhanced levels of restraint for use of genetically engineered crops themselves and particularly for synthetic biology, including calls for a moratorium on all forms of synthetic biology research, even in contained use.
The challenges for gene drive research arising from the Convention and the Protocols lie mainly in the way in which individual countries choose to implement their provisions, rather than in the provisions themselves. Concerns about the impacts of these regulatory provisions on future innovative developments are based on assumptions that there will be no future downwards adaptation of regulatory provisions based on experience in use of new technologies. Indeed, these discussions and consultations are being seen by some as an opportunity to reinforce and extend a preventative emphasis.
Many low- and middle-income countries are signatories to the Cartagena Protocol, which has guided the development of their national regulatory frameworks for governance of living modified organisms, which it defines as any living organism that possesses a novel combination of genetic material obtained through the use of modern biotechnology. Under the Cartagena Protocol, countries are obligated to notify one of the United Nation’s International Biosafety Clearing-Houses and any affected nations about activities that may lead to movement of living modified organisms with potential adverse effects on biological diversity or human health. However, some countries do not have sufficient resources to enforce such legislation. As a result, capacity building and public awareness activities in low-income countries have largely been top down, with governments playing a passive role and non-governmental organizations taking up the brokering role (Kingiri and Hall, 2012).
Some countries have developed regional regulation in order to assist individual countries with adoption of the Cartagena Protocol. For example, the African Model Law on Biosafety (2011) aims to help countries that are members of the Cartagena Protocol on Biosafety to implement the provisions of the Protocol at the national level (see Figure 8-1). Paarlberg (2012) argues that African countries in particular have largely followed the European Union’s precautionary approach, which limits the deployment of biotechnology. Furthermore, politics can delay deployment of promising innovations as has occurred in the Philippines (Brooks, 2010; Kupferschmidt, 2013), Kenya (Paarlberg, 2001; Paarlberg, 2009; Brooks, 2010; Zhu, 2014) and India (Herring, 2008; Jayaraman, 2010) where field trials of genetically engineered crops have been destroyed or disrupted.
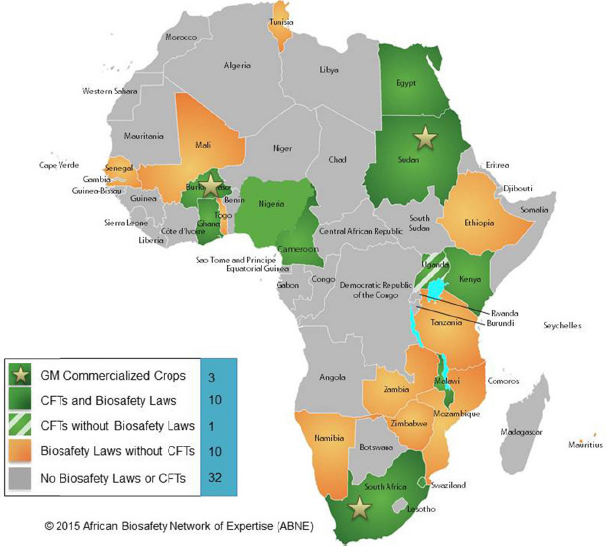
These experiences point toward the need for governance mechanisms that recognize the urgent need to improve poor countries’ ability to exploit new scientific and technological advances to address social and economic challenges affecting the poor (Watkins and Ehst, 2008; Juma, 2011).
Transnational Governance Through Standards and Guidelines
There is currently no overall framework that policy makers and standards bodies can use to assess what mix of standards, guidelines, and regulations is appropriate for transnational governance of any particular technology. In his presentation at the 2015 International Summit on Hu-
man Gene Editing, Gary Marchant discussed the idea of “transnational new governance.” Transnational new governance originates from a “soft law” concept in international law. It entails substantive obligations and requirements created by instruments that are not directly legally enforceable. These instruments have an international scope, focus, and participation and can broaden oversight from top-down government requirements to include a much broader range of decision makers, for example, companies, researchers, nongovernmental organizations (NGOs), public—private partnerships, and other third parties. Their advantages include the fact that they are voluntary, cooperative and reflexive; can be adopted or revised relatively quickly; allow many different approaches to be tried simultaneously: and can be gradually “hardened” into more formal regulatory oversight (Allen and Sriram, 2000; Langlois and Savage, 2001). They do, however, have limitations. For example, their norms and standards are not directly enforceable; they are not always as flexible and adaptable as hoped; there is potential for confusion and overlap; and they have less legitimacy. Examples of transnational non regulatory and non-legislative governing tools are provided in Table 8-4. In transnational new governance, a number of respected, non-regulatory authorities, such as the International Council for Science, InterAcademy Partnership,6 Academy of Sciences for the Developing World, and the WHO, may have important roles to play in shaping responsible practices for gene drive research internationally.
TABLE 8-4 Examples of Transnational Governance Tools in Science That Are Non Regulatory and Non-Legislative
| Policy Tool | Example |
| Transnational regulatory dialogue and networks | Working groups of the Organisation for Economic Co-operation and Development |
| International harmonization committees | International Conference on Harmonization |
| United Nations declarations | International Declaration on Human Genetic Data |
| International principles | World Medical Association, Helsinki Principles |
| International scientific assessment | Intergovernmental Panel on Climate Change |
| Research guidelines developed by international professional scientific societies or other non-regulatory science authorities | International Society for Stem Cell Research Guidelines for Embryonic Stem Cell Research |
| World Health Organization’s Guidance Framework for Testing Genetically Modified Mosquitoes | |
| World Organisation for Animal Health (OIE)—Ensuring | |
| Good Governance to Address Emerging and Re-emerging | |
| Animal Disease Threats Food and Agriculture | |
| Organisation (FAO)—Biosafety of Genetically Modified | |
| Organisms: Basic concepts, methods and issues | |
| International Committee of Medical Journal Editors | |
| International statements of policy | Human Genome Organization’s statement on the patenting of DNA sequences |
| Private/industry standards | International Standards Organisation |
| International Gene Synthesis Consortium’s Harmonized | |
| Screening Protocol |
Source: Adapted from Marchant, 2015.
___________________
6InterAcademy Partnership (IAP) is a global network of the world’s science academies, launched in 1993: http://www.interacademies.net.
There is also an increasingly well recognized capacity for guidelines and standards to supplement and contribute to the implementation of regulations in order to support the speedy and effective delivery of science that safely meets the public’s needs and desires; and, where appropriate, contribute to the development of a supportive innovation environment (SBLC, 2016). In other words, such guidelines and standards can have a formative role in the very early development of radically innovative technologies to guide future regulatory decisions (see Figure 8-2). Thus, an innovation such as a gene drive, in early stages of development (phase 1), could most appropriately be governed through the adoption of a of consensus standard designed to set an agreed level of good practice or quality or establish trust in an innovative product or service. This agreement would be a preliminary step to better informed development of more formal regulatory guidelines (phase 2 in Figure 8-2) and then, as understanding of the new technology and its properties increases, to legally based regulations (Phase 3). At phase 4, once the properties of a novel technology have been fully codified and understood, standards will continue to serve their traditional subsidiary role to regulations, for example, to ensure quality and safety of products and good manufacturing practice (see Table 8-4).
Although not a regulatory body, the WHO serves as the de facto authority to its member states (194 countries worldwide), regarding which interventions have demonstrated public health value and therefore should be considered for adoption by Ministries of Health. The WHO has been instrumental in facilitating the generation of normative guidance and standards for research, biosafety, and risk assessment on vector control paradigms. A series of guidelines with outlined testing protocols and pre-defined efficacy standards are available at no cost to investigators, industry, and other stakeholders, their purpose being to ensure that proven, effective products are used in the field. Such resources are particularly valuable to low- and middle-income countries where regulatory infrastructure may be lacking but in-country decision makers/authorities recognize the need to comply with the Cartagena Protocol. Many countries, particularly those who lack national or regional governance mechanisms, look to these guidelines to develop their biosafety governance systems. The WHO also manages expert panels to encourage self-governance. For example, the Vector Control Advisory Group (VCAG) was established in 2012 to provide expert advice to investigators on the evaluation and development of vector control products and strategies. The VCAG serves as a venue to vet scientific concepts, appropriate study designs, efficacy needs, and considerations on decisions points for advancing evaluation from one phase of testing to the next. The expert advice facilitates the generation of rigorous findings and provides a foundation for science-based decisions points. All of VCAG’s critiques and comments are posted in the public domain for full transparency to enhance VCAG’s engagement with a broad range of stakeholders that may be impacted by the research and/or implementation of specific strategies.
Most relevant to this report, the WHO Special Programme for Research and Training in Tropical Diseases (WHO/TDR) has coordinated specific efforts to develop internationally accepted guidelines for the testing of Genetically Modified Mosquitoes (GMM). The Guidance Framework for Testing of Genetically Modified Mosquitoes (WHO, 20147) highlights the need for a staged-approach to the evaluation of GMM to ensure evidence-based decision points are utilized for further development of the strategy. A complementary training manual, Biosafety for Human Health and the Environment in the Context of the Potential use of Genetically Modified Mosquitoes (GMMs) (WHO, 2015), provides investigators of GMMs a tool for governing their research as it relates to mitigating risk of accidental releases during field-based trials or open environmental release.
Many countries, particularly those that lack national or regional governance mechanisms, look to the WHO’s GMM arthropod containment guidelines to develop their biosafety governance systems for mosquitoes. It is expected that many of the WHO’s normative principles for evaluating GMMs will also apply to gene-drive modified mosquitoes; however, the challenge with emerging gene drive technology, is that not all aspects of the WHO GMM principles may apply. This may lead to challenges for considering gene-drive modified mosquitoes and other gene drive modified organisms.
The WHO’s Special Programme in Research and Training in Tropical Diseases (WHO/TDR) also funded a project to produce Best Practice Guidance for Deployment of Genetic Control Methods against Mosquito Vectors in Disease Endemic Countries (MosqGuide) (Mumford et al., 2009). This initiative is already engaging Panama, Brazil, Mexico, Thailand, Kenya, and India.
In some of the jurisdictions targeted for experimentation, there may be no governance system in place or only one that lacks the capacity to assess experiments in a scientifically and socially robust fashion. International organizations like the WHO are attempting to address this problem by promoting ethical codes and best practices that might be used in these situations.
A number of challenging questions for governance in global contexts remain. Is local review by authorized committees in host countries required for an experiment to proceed ethically? Is prior consent at the national level necessary? International standards raise the potential problem of legitimacy gaps. If international organizations are filling the governance gap at the international level, what procedures are being used to produce those standards, and who should participate in that process?
CONCLUSIONS AND RECOMMENDATIONS
The governance of research begins with the personal responsibility of the investigator, is formalized in professional guidelines, and often extends to legally binding policies and enforceable regulations. In the United States, it is clear that gene drive activities will trigger a variety of governance mechanisms. However, some of these mechanisms may be inadequate for identifying immediate and long-term potential environmental and public health implications of individual gene drive applications because they lack clarity in their jurisdiction, they are challenged by the novel characteristics of gene drives, or they provide insufficient structures for public engagement.
Currently, institutions, funders, and professional societies work in concert to encourage professional best practices in research, and this cooperation will be key to maintaining high standards. Professional codes of conduct that address technical and ethical considerations in research are an important source of governance that helps both to promote awareness among researchers and encourage them to take responsibility for their science. Approaches currently in use to incentivize and refine good practices are to provide resources for education (conceptual) and training (practical) in the responsible conduct of research and to publicly acknowledge re-
___________________
7WHO/TDR annual reports can be found at http://www.who.int/tdr/publications/about-tdr/annual-reports/en.
searchers for their standards of practice. These will be important for reinforcing responsible practices in gene drive research.
Laboratory-based research conducted at an institution that receives funding from the NIH is subject to NIH’s guidelines on biosafety and oversight by IBCs. These guidelines, although international in nature, are adapted to specific institutional contexts and are complemented by good laboratory practices. Moreover, the NIH guidelines stipulate that all research at NIH-funded institutions may be regulated by laws established at the local, state, and federal levels, even in the absence of NIH funding for a specific project (e.g., other federal agencies, private foundations).
Over the last few decades, IBCs have provided a robust system of health and environmental protection for laboratory research. Part of the advantage of an IBC is its flexibility: reflected in the use of guidelines that can be modulated as technology and experience develop, a delegated system of oversight that operates at the local level but is accountable to a governmental body, and a process through which novel and controversial research can be considered at a level higher than the research laboratory.
Although these features of IBCs will be useful as gene drive research moves forward, IBCs also have important limitations. Due to the novel characteristics of gene drives, capacity issues, and an absence of clearly defined guidelines for gene drive research, current IBCs may not have the expertise or resources to evaluate the biosafety of gene drives effectively. IBCs are also not equipped to examine biosecurity or willful misuse issues. However, there is potential to learn from IBCs at institutions where gene drive research has been ongoing.
At the institutional level, it is essential that gene drive research continue to be governed by good professional practices, strict adherence to standard operating procedures, and comprehensive training of research personnel.
Recommendation 8-1: Institutions, funders, and professional societies should provide face-to-face instruction and online, open access resources for education and training on the responsible practices in gene drive research.
Recommendation 8-2: Due to the novel characteristics of gene drives, funding agencies and research institutions should take responsibility to ensure the development of the necessary expertise to assess safety within Institutional Biosafety Committees and their equivalents.
Each phase of research activity—from developing a research plan to post-release surveillance—raises different levels of concern depending on the organism being modified and the type of gene drive being developed. A one-size-fits-all approach to governance is not likely to be appropriate. Governance and regulation of gene drive research will need to be proportionate to the hazards posed by the specific activity. In addition, governance will need to be responsive to changes in scientific best practices and ethical considerations as gene drive technologies develop.
Recommendation 8-3: Researchers and funders should take measures to review the study design and implementation on an ongoing basis to ensure that harms and benefits remain reasonably distributed and balanced.
In the United States, regulation of gene-drive modified organisms will most likely fall under the Coordinated Framework for the Regulation of Biotechnolgy. However, the US Food and Drug Administration (FDA), the US Department of Agriculture (USDA), and the US Environmental Protection Agency (EPA), the federal agencies included in the current Coordinated Framework, do not have clear lines of authority over the potential applications of gene drive research. The diversity of potential gene-drive modified organisms and contexts in which they might be used reveal a number of regulatory overlaps and gaps. For example, regulatory practices for the assessment of potential ecological and public health effects of field experiments or
planned releases are inadequate for gene drive research due to these policies’ predication on containment. For some potential applications of gene drive technologies, regulatory jurisdiction may overlap, which suggests the need for a process to quickly determine which agency should coordinate governance of that technology.
Recommendation 8-4: The US government should clarify the assignment of regulatory responsibilities for field releases of gene-drive modified organisms, including the roles of relevant agencies that are not currently included in the Coordinated Framework for the Regulation of Biotechnology.
The introduction of novel genetic constructs intended to modify ecosystems increases the uncertainties that gene drives raise in ways that make robust assessment of their risks more critical, but also more difficult. Regulation will be needed that facilitates fundamental, applied, and translational research so that the potential harms and benefits of gene drives can be responsibly explored in laboratory and field studies.
Recommendation 8-5: Relevant agencies and decision making bodies will need to develop the capacity for robust assessment of a gene-drive modified organism’s risks and uncertainties on a case-by-case basis that looks at the organism’s intended function as well as the biological construct.
Recommendation 8-6: Regulatory agencies with oversight authority over genetic modification research should review risk assessment models and procedures to ensure that they capture the characteristics of gene drives, drawing upon multiple models and integrating experts’ comprehensive knowledge of practical conditions for gene drive research.
There is broad agreement on the importance of engaging affected communities and broader publics in decision making about activities involving gene drives. Mechanisms for public engagement and deliberation already exist within the relevant authorized agencies, but there is generally little clarity on how public engagement should feed into governance and a lack of consensus about best practices in this regard. This is due to at least two factors: first, because regulatory authority remains unclear, the availability of particular formal and customary mechanisms for public engagement also remain unclear; second, although the National Environmental Protection Act will in some cases require public input and afford opportunity for public comment, these mechanisms are an inadequate platform for the more robust forms of engagement discussed in Chapter 7.
The scientific community, including individual researchers, institutions, and funders, have an obligation to engage in conversations with policy makers about best practices to safeguard against unintentional or intentional misuse of gene-drive modified organisms. Safeguards will be aided by rigorous attention to confinement and containment protocols in laboratory and field tests; active awareness about the potential for misuse; and participation in education and training programs about the dual use potential of gene drive research. Governance mechanisms need to be in place to address questions about the biosecurity implications of gene drive research and consider develop mitigation strategies that are not dependent on the underlying technology.
Recommendation 8-7: Researchers’ institutions, regulators, and funders should collaborate to develop oversight structures to regularly review the state of gene drive science and its potential for misuse. Such reviews should also recommend or develop educational programs for researchers and members of the public about biosecurity concerns, the potential for dual-use research, responsible practices, and the funding of gene drive science.
Research on gene drives is global and likely to become even more so in the future. Responsible governance will need to be international and inclusive, with clearly defined global reg-
ulatory frameworks, policies, and best practice standards for implementation. Low- and middle-income countries where gene-drive modified organisms may be employed will need to be involved in relevant governance, recognizing that many countries lack the capacity to develop a comprehensive regulatory scheme for gene drives from scratch. To cope with the unique aspects of gene drives, existing approaches to governance need to be adapted and combined for broad international use. Integrating new policy and law for gene drives into existing international governance frameworks will require attention to the values, experiences, and perspectives of people in many disparate nations. It is unlikely that a successful one-size-fits-all approach or a single mechanism, such as regulation, policy, or professional codes alone, will be sufficient for appropriate international governance of gene drive technology.
The most broad-ranging and widely accepted international governance system is the United Nations Convention on Biological Diversity, as implemented through the Cartagena and Nagoya Protocols. Many countries are now developing regulatory systems in response to the Cartagena Protocol. Many such systems are predicated on a strong precautionary, nearly preventative approach, which may restrict further gene drive research out of concern about gene drives’ intrinsic ability to spread and persist in the environment. Given that the United States is not a Party to the Cartagena Protocol, it is a major gap in international governance that the United States does not have a clear policy for collaborating with other countries with divergent systems of governance, especially when such countries may, in fact, lack the capacity to assess the safety of gene drive research, undertake public engagement and societal dialogue, and maintain regulatory institutions. This gap is also significant because many sites for field testing, and ultimately environmental release of gene-drive modified organisms are likely to be outside of the United States.
Recommendation 8-8: If field testing or environmental releases are expected to be conducted in other countries, United States funders and researchers should give careful consideration to the regulatory systems in place in those countries, their adequacy to control the development and release of gene-drive modified organisms, and the relevant community and other voices that will need to be considered in related governance.
In practice, a significant amount of field research on genetically-modified mosquitos operates under guidelines established by international organizations, such as the WHO, and by the research community itself. Although these guidelines provide a useful foundation for the establishment of guidelines for gene-drive modified organisms they have important gaps and may not address all of the unique aspects of gene drives or the range of potential organisms to be used. For example, guidelines may need to be adapted to align to local contexts in order to be implemented. Moreover, most guidelines are not tied explicitly to public oversight and implementation.
There is a need to reach international agreement on the adaptation of existing governance approaches in the United States and other countries to cope with the distinguishing features of gene drives, particularly their intentional persistence upon release to the environment.
Recommendation 8-9: To ensure the long-term safety of human health and the environment, decision makers should consider a large toolbox of policies, including regulatory and non-regulatory mechanisms, for the rapidly developing field of gene drive research.
Recommendation 8-10: Research institutions, regulators, and funders should revist international regulatory frameworks, national laws, non-governmental policy, and professional codes of conduct on research and the release of genetically modified organisms to determine whether and how they may be applied to the specific context of gene drive research, particularly with regard to site selection issues, capacity building for responsible and inclusive governance systems, scientific and post release surveillance, and stakeholder engagement.
REFERENCES
Allen, R.H., and R.D. Sriram. 2000. The role of standards in innovation. Technol. Forecast. Soc. 64(2-3):171-181.
Annas, G.J., and M.A. Grodin. 1992. The Nazi Doctors and the Nuremberg Code: Human Rights in Human Experimentation. New York: Oxford University Press.
ASM (American Society of Microbiology). 2005. Code of Ethics [online]. Available: http://www.asm.org/index.php/governance/code-of-ethics [accessed April 27, 2016].
Baltimore D., Berg P., Carroll D., Charo R.A., Church G., Corn J.E., Daley G.Q., Doudna J.A., Fenner M, Greely H.T., Jinek M., Martin G.S., Penhoet E., Puck J., Stenberg S.H., Weissman J.S., Yamamoto K.R. 2015. A prudent path forward for genomic engineering and germline gene modification. Science 348(6230):36-38.
Breggin, L., R. Falkner, N. Jaspers, J. Pendergrass, and R. Porter. 2009. Securing the Promise of Nanotechnologies: Towards Transatlantic Regulatory Cooperation. London: Chatham House [online]. Available: https://www.chathamhouse.org/sites/files/chathamhouse/public/Research/Energy,%20Environment%20and%20Development/r0909_nanotechnologies.pdf [accessed April 27, 2016].
Brooks, S. 2010. Biosafety Regulation: Lessons from Kenya and the Philippines. STEPS Centre, Institute of Development Studies, University of Sussex, Brighton, UK [online]. Available at: http://steps-centre.org/wp-content/uploads/STEPSsumBiosafety.pdf [accessed November 4, 2015].
Carroll, D., and R.A. Charo. 2015. The societal opportunities and challenges of genome editing. Genome Biol. 16:242.
Carter, S.R., M. Rodemeyer, M.S. Garfinkel, and R.M. Friedman. 2014. Synthetic Biology and the U.S. Biotechnology Regulatory System: Challenges and Options. J. Craig Venter Institute [online]. Available at: http://www.jcvi.org/cms/fileadmin/site/research/projects/synthetic-biology-and-the-us-regulatory-system/full-report.pdf [accessed April 27, 2016].
CBD (Convention on Biological Diversity). 2010. Final Report of the Ad Hoc Technical Expert Group on Risk Assessment and Risk Management under the Cartagena Protocol on Biosafety. UNEP/CBD/BS/ AHTEG-RA&RM/2/5. UNEP [online]. Available at https://www.cbd.int/doc/meetings/bs/bsrarm02/official/bsrarm-02-05-en.pdf [accessed April 28, 2016].
CBD. 2012. Guidance on Risk Assessment of Living Modified Organisms: Risk Assessment of Living Modified Mosquitoes [online]. Available: http://bch.cbd.int/onlineconferences/guidancedoc_ra_mosquitoes.shtml [accessed April 28, 2016].
CBD. 2015. Report of the Ad Hoc Technical Expert Group on Synthetic Biology. UNEP/CBD/SYNBIO/ AHTEG/2015/1/3 [online]. Available: https://www.cbd.int/doc/?meeting=SYNBIOAHTEG-2015-01 [accessed April 28, 2016].
CBD. 2016. The Cartagena Protocol on Biosafety [online]. Available at: http://bch.cbd.int/protocol/ [accessed April 27, 2016].
Charo, A. 2015. Comparative approaches to biotechnology regulation. Presentation at the International Summit on Human Gene Editing. December 1-3, 2015. Washington, D.C [online]. Available at: https://vimeo.com/album/3703972/video/149182567 [accessed April 27, 2016].
Clapper, J.R. 2016. Statement for the Record Worldwide Threat Assessment of the U.S. Intelligence Community. Senate Armed Service Committee, February 9, 2016 [online]. Available: https://www.dni.gov/files/documents/SASC_Unclassified_2016_ATA_SFR_FINAL.pdf [accessed April 27, 2016].
Crampton, J.M., S.L. Stowell, M. Karras, and R.E. Sinden. 1999. Model systems to evaluate the use of transgenic haematophagous insects to deliver protective vaccines. Parassitologia 41(1-3):473-477.
Dubnick, M.J., and H.G. Frederickson. 2010. Accountable agents: Federal performance measurement and third-party government. J. Publ. Adm. Res. Theor. 20(suppl. 1):i143-i159.
El-Zahabi-Bekdash, L, and J.V. Lavery. 2010. Precaution through effective community engagement in research with modified mosquitoes. AsPac. J. Mol. Biol. Biotechnol. 18(2):247-250.
Emanuel, E.J., D. Wendler, and C. Grady. 2000. What makes clinical research ethical? JAMA 283(20):2701-2711.
FDA (US Food and Drug Administration). 2015a. Genetically Engineered Animals: General Q&A [online]. Available: http://www.fda.gov/AnimalVeterinary/DevelopmentApprovalProcess/GeneticEngineering/GeneticallyEngineeredAnimals/ucm113605.htm [accessed April 27, 2016].
FDA. 2015b. Guidance for Industry 187: Regulation of Genetically Engineered Animals Containing Heritable Recombinant DNA Constructs [online]. Available: http://www.fda.gov/downloads/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/ucm113903.pdf [accessed April 27, 2016].
FDA. 2016. FDA Announces Comment Period for Draft Environmental Assessment for Genetically Engineered Mosquito [online]. Available: http://www.fda.gov/AnimalVeterinary/NewsEvents/CVMUpdates/ucm490246.htm [accessed March 11, 2016].
Freeman, G., and P. Swidicki. 2013. EU Impact on Life Sciences. UK Government Fresh Start Project. Available at: http://www.eufreshstart.org/downloads/lifesciences2.pdf [accessed November 4, 2015].
Herfst, S., E.J. Schrauwen, M. Linster, S. Chutinimitkul, E. de Wit, V.J. Munster, E.M. Sorrell, T.M. Bestebroer, D.F. Burke, D.J. Smith, G.F. Rimmelzwaan, A.D. Osterhaus, and R.A. Fouchier. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336(6088):1534-1541.
Herring, R.J. 2008. Opposition to transgenic technologies: Ideology, interests and collective action frames. Nat. Rev. Genet. 9(6):458-463.
HHS (US Department of Health and Human Services). 2009. Biosafety in Microbiological and Biomedical Laboratories, 5th Ed. HHS(CDC) 21-1112 U.S. Department of Health and Human Services, Public Health Service-Centers for Disease Control and Prevention-National Institutes of Health [online]. Available: http://www.cdc.gov/biosafety/publications/bmbl5 [accessed April 27, 2016].
Higgs, S., K.E. Olson, L. Klimowski, A.M. Powers, J.O. Carlson, R.D. Possee, and B.J. Beaty. 1995. Mosquito sensitivity to a scorpion neurotoxin expressed using an infectious Sindbis virus vector. Insect Mol. Biol. 4(2):97-103.
Holdren, J.P., H. Shelanski, D. Vetter, and C. Goldfuss. 2015. Modernizing the Regulatory System for Biotechnology Products. Memorandum for Heads of Food and Drug Administration, Environmental Protection Agency, and Department of Agriculture, from Executive Office of the President of the United States, Washington, DC. July 2, 2015 [online]. Available at: https://www.whitehouse.gov/sites/default/files/microsites/ostp/modernizing_the_reg_system_for_biotech_products_memo_final.pdf [accessed March 17, 2016].
IAP (InterAcademy Parternship). 2016. Doing Global Science: A Guide to Responsible Conduct in the Global Research Enterprise. Princeton, NJ: Princeton University Press [online]. Available at: http://www.interacademycouncil.net/File.aspx?id=29431 [accessed April 27, 2016].
Imai, M., and Y. Kawaoka. 2012. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2(2):160-167.
IOM (Institute of Medicine). 2014. Oversight and Review of Clinical Gene Transfer Protocols: Assessing the Role of the Recombinant DNA Advisory Committee. Washington, DC: The National Academies Press.
IRGC (International Risk Governance Council). 2011. Reports on Special Issues: Synthetic Biology [online]. Available: http://www.irgc.org/publications/reports-on-special-issues/ [accessed April 27, 2016].
ISAAA (International Service for the Acquisition of Agri-biotech Applications). 2015. Top Ten Facts. ISAAA Brief 49-2014 [online]. Available at: http://www.isaaa.org/resources/publications/briefs/49/toptenfacts/default.asp [accessed April 27, 2016].
Jayaraman, K. 2010. Bt brindjal splits Indian cabinet. Nat. Biotechnol. 28(4):296.
Juma, C. 2011. The New Harvest: Agricultural Innovation in Africa. New York: Oxford University Press.
Kamrud, K.I., K.E. Olson, S. Higgs, A.M. Powers, J.O. Carlson, and B.J. Beaty. 1997. Detection of chloramphenicol acetyltransferase in the saliva of Culex pipiens mosquitoes. Insect Biochem. Mol. Biol. 27(5):423-429.
Kingiri, A.N., and A. Hall. 2012. The role of policy brokers: The case of biotechnology in Kenya. Rev. Pol. Res. 29(4):492-522.
Krimsky, S. 1982. Genetic Alchemy: The Social History of the Recombinant DNA Controversy. Cambridge, MA: The MIT Press.
Kupferschmidt, K. 2013. Activists destroy ‘golden rice’ field trial. Science Magazine, August 9, 2013 [online]. Available at: http://www.sciencemag.org/news/2013/08/activists-destroy-golden-rice-field-trial [accessed March 31, 2016].
Langlois, R.N., and D.A. Savage. 2001. Standards, modularity, and innovation: The case of medical practice. Pp. 149-168 in Path Dependence and Path Creation, R. Garud, and P. Karnøe, eds. Abingdon, UK: Psychology Press.
Lockwood, JA. 2012. Insects as weapons of war, terror, and torture. Annu. Rev. Entomol. 57:2015-2027.
Lowrie, H., and J. Tait. 2010. Guidelines for Appropriate Risk Governance of Synthetic Biology. Policy Brief. Geneva, Switzerland: International Risk Governance Council. [online]. Available: http://www.irgc.org/IMG/pdf/irgc_SB_final_07jan_web.pdf [accessed April 27, 2016].
Lyall, C., and J. Tait. 2005. New Modes of Governance: Developing an Integrated Policy Approach to Science, Technology, Risk and the Environment. Surrey, UK: Ashgate Publishing Ltd.
Marchant, G.E. 2015. Human Gene Editing: International Governance. Presentation at the International Summit on Human Gene Editing, December 1-3, 2015. Washington, DC [online]. Available at: https://vimeo.com/album/3704161/video/149197332 [accessed April 26, 2016].
Mumford, J., M.M. Quinlan, C. Beech, L. Alphey, V. Bayard, M.L. Capurro, P. Kittayapong, J.D. Knight, M.T. Marrelli, K. Ombongi, J.M. Ramsey, and R. Reuben. 2009. MosqGuide: A project to develop best practice guidance for the deployment of innovative genetic vector control strategies for malaria and dengue. AsPac. J. Mol. Biol. Biotechnol. 17(3):93-95.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2016. International Summit on Human Gene Editing: Global Discussion. Washington, DC: The National Academies Press.
NIH (National Institute of Health). 2016a. NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines). April 2016 [online]. Available: http://osp.od.nih.gov/office-biotechnology-activities/biosafety/nih-guidelines [accessed April 27, 2016].
NIH. 2016b. Biomedical Technology Assessment. Recombinant DNA Advisory Committee. Office of Biotechnology Activities [online]. Available: http://osp.od.nih.gov/office-biotechnology-activities/biomedical-technology-assessment/hgt/rac [accessed April 27, 2016].
NRC (National Research Council). 1996. Understanding Risk: Informing Decisions in a Democratic Society. Washington, DC: National Academy Press.
NRC. 2010. Understanding Biosecurity: Protecting Against Misuse of Science in Today’s World. Washington, DC: The National Academies Press.
NRC. 2011. Guidance for the Care and Use of Laboratory Animals, 8th Ed. Washington, DC: The National Academies Press.
NRC. 2015. The Industrialization of Biology: A Roadmap to Accelerate the Advanced Manufacturing of Chemicals. Washington, DC: The National Academies Press.
Oye, K.A., K. Esvelt, E. Appleton, F. Catteruccia, G. Church, T. Kuiken, S.B. Lightfoot, J. McNamara, A. Smidler, and J.P. Collins. 2014. Biotechnology. Regulating gene drives. Science 345(6197):626-628.
Paarlberg, R.L. 2001. The Politics of Precaution: Genetically Modified Crops in Developing Countries. International Food Policy Research Institute (IFPRI). Baltimore, MD: Johns Hopkins University Press.
Paarlberg, R.L. 2009. Starved for Science: How Biotechnology is Being Kept out of Africa. Cambridge, MA: Harvard Press.
Paarlberg, R.L. 2012. Governing the dietary transition: Linking agriculture, nutrition and health. Pp. 191-199 in Reshaping Agriculture for Nutrition and Health, R. Pandaya-Lorch, and S. Fan, eds. Washington, DC: International Food Policy Research Institute (IFPRI).
Pearson, A. 2015. Regulation of Agricultural Biotechnology by USDA-APHIS: APHIS Protects Plant Health through Rigorous Regulation of GE Organisms. Webinar, December 8, 2015. Available: http://nas-sites.org/gene-drives/2015/11/14/webinar-us-regulations/ [accessed March 17, 2016].
Presidential Commission for the Study of Bioethical Issues. 2010. New Directions: The Ethics of Synthetic Biology and Emerging Technologies. Washington, DC: Presidential Commission for the Study of Bioethical Issues.
Rudenko, L. 2015. Regulation of GE Animals at FDA: FD&C Act and NEPA. Webinar, December 9, 2015. Available: http://nas-sites.org/gene-drives/2015/11/14/webinar-us-regulations/ [accessed March 17, 2016].
S3 (Science Safety Security). 2014. U.S. Government Policy for Institutional Oversight of Life Science Dual Use Research of Concern, March 2014 [online]. Available: http://www.phe.gov/s3/dualuse/Pages/InstitutionalOversight.aspx [accessed April 27, 2015].
S3. 2015. U.S. Government Policy for Institutional Oversight of Life Science Dual Use Research of Concern, November 2, 2015 [online]. Available: http://www.phe.gov/s3/Documents/life-sci-dual-use.pdf [accessed April 27, 2015].
SBLC (Synthetic Biology Leadership Council). 2016. Biodesign for the Bioeconomy: UK Synthetic Biology Strategic Plan 2016 [online]. Available: https://connect.innovateuk.org/documents/2826135/31405930/BioDesign+for+the+Bioeconomy+2016+-+DIGITAL.pdf/0a4feff9-c359-40a2-bc93-b653c21c1586 [accessed March 17, 2016].
Schneider, B.S., and S. Higgs. 2008. The enhancement of arbovirus transmission and disease by mosquito saliva is associated with modulation of the host immune response. Trans. R. Soc. Trop. Med. Hyg. 102(5):400-408.
Slovic, P. 1987. Perception of risk. Science 236(4799):280-285.
Strauss, S.H., H. Tan, W. Boerjan, and R. Sedjo. 2009. Strangled at birth? Forest biotech and the Convention on Biological Diversity. Nat. Biotechnol. 27:519-527.
Sunstein, C.R. 2009. Laws of Fear: Beyond the Precautionary Principle. Cambridge: Cambridge University Press.
Tait, J. 2008. Risk governance of genetically modified crops: European and American perspectives. Pp. 133-153 in Global Risk Governance: Concept and Practice Using the IRGC Framework, O. Renn, and K. Walker, eds. Dordrecht, NL: Springer.
Tait, J. 2014. Bringing it all together. Pp. 129-136 in Innovation: Managing Risk not Avoiding It. Evidence and Case Studies, Annual Report of the Government Chief Scientific Adviser [online]. Available at: https://www.gov.uk/government/publications/innovation-managing-risk-not-avoiding-it [accessed April 27, 2015].
Tait, J., and G. Banda. In press. Proportionate Governance of Innovative Technologies: The role of regulations, guidelines and standards. Report to the British Standards Institution.
UN (United Nations). 1992. Convention on Biological Diversity [online]. Available at https://www.cbd.int/doc/legal/cbd-en.pdf [accessed April 27, 2016].
Waltz, E. 2015. A face-lift for biotech rules begins. Nat. Biotechnol. 33(12):1221-1222.
Watkins, A., and M. Ehst. 2008. Science, Technology, and Innovation: Capacity Building for Sustainable Growth and Poverty Reduction. Washington DC: The World Bank.
WHO (World Health Organization). 2014. The Guidance Framework for Testing Genetically Modified Mosquitoes. Geneva, Switzerland: World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR).
WHO. 2015. Training Manual: Biosafety for Human Health and Environment in The Context of the Potential Use of Genetically Modified Mosquitoes (GMM). Geneva, Switzerland: World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR) [online]. Available: http://apps.who.int/iris/bitstream/10665/180388/1/9789241549271_eng.pdf?ua=1 [accessed May 2, 2016].
Zhu, A. 2014. Kenya’s GM ban and the future of GM policy in Africa. The Huffington Post [online]. Available at: http://www.huffingtonpost.com/april-zhu/can-we-be-rational-kenyas_b_5434687.html [accessed March 31, 2016].






























