
Proceedings of a Workshop
INTRODUCTION1
Immunotherapy is a form of cancer therapy that harnesses the body’s immune system to destroy cancer cells (Couzin-Frankel, 2013). In particular, these therapies target or modulate cells or other components of the immune system to enhance an individual’s immune response or to reduce the inhibition of an individual’s immune response. This enables the immune system to identify and subsequently eliminate cancer cells, said Samir Khleif, director of the Georgia Cancer Center at Augusta University. In recent years, immunotherapies have been developed for several cancers, including advanced melanoma, lung cancer, and kidney cancer. In some patients with metastatic cancers who have not responded well to other treatments, immunotherapy treatment has resulted in complete and durable responses. Given these promising findings, it is hoped that continued immunotherapy research and development will produce better cancer treatments that improve patient outcomes. Clinical trials are currently evaluat-
___________________
1 The planning committee’s role was limited to planning the workshop. The Proceedings of a Workshop has been prepared by the rapporteurs as a factual account of what occurred at the workshop. Statements, recommendations, and opinions expressed are those of individual presenters and participants and have not been endorsed or verified by the National Academies of Sciences, Engineering, and Medicine. They should not be construed as reflecting any group consensus.
ing immunotherapies in patients with various types of cancer, including brain, colorectal, and ovarian cancers (American Cancer Society, 2015).
Harnessing a patient’s immune system to fight disease offers potential advantages over traditional targeted therapies or chemotherapies. Under normal physiological conditions, the immune system acts with a high level of specificity to eliminate infections and generate immune cells that promote long-term protection from reinfection. In an analogous manner, immunotherapy can stimulate the immune system to identify cancer cells and to generate the same specificity and long-term protection to eliminate and prevent cancer recurrence. Because immunotherapy leverages the patient’s own immune cells, these therapies can achieve a high level of personalization that may result in fewer side effects compared with other targeted therapies or chemotherapy (Sharma et al., 2011; Yang, 2015).
There is a lot of excitement in the cancer community about the potential for immunotherapy. For example, the National Cancer Moonshot initiative describes immunotherapy as one of the key areas of investment: “This initiative will work to extend the early successes of immunotherapy for cancer treatment to virtually all solid tumors by harnessing the power of the body’s immune system by supporting basic research to increase understanding of how the immune system can be used to modify cancer cells and their activities” (The White House, 2016). With this promise, however, there is also recognition that the clinical and biological landscape for immunotherapies is novel and not yet well understood (Yang, 2015). For example, adverse events with immunotherapy treatment are quite different from those experienced with other types of cancer therapy. Similarly, immunotherapy dosing, therapeutic responses, and response time lines are also markedly different from other cancer therapies. Khleif added that combination immunotherapy regimens will likely be more promising than monotherapies. However, the sheer number of potential combinations, as well as determining which ones may be most appropriate to combine, and how they should be combined in timing and in dosing, pose incredibly complex challenges. All of these factors influence the development, regulation, and implementation of immunotherapies in clinical practice. To examine these challenges and explore strategies to overcome them, the National Cancer Policy Forum of the National Academies of Sciences, Engineering, and Medicine held the workshop “Policy Issues in the Clinical Development and Use of Immunotherapy for Cancer” on February 29 and March 1, 2016, in Washington, DC. At the workshop, researchers, clinicians, patients, and representatives from government agencies, the
pharmaceutical industry, and health care payers explored a number of topics, including
- Recent findings on how the immune system responds to tumors and how that information led to the creation of immunotherapies for cancer treatment.
- The current state of the science for cancer vaccines, immune modulator drugs, personalized cell therapies, and the use of immunotherapies in combination.
- Drug development challenges for immunotherapies, including a lack of preclinical animal models and assays; difficulty in selecting appropriate clinical trial endpoints, predicting effective doses, and anticipating, understanding, and mitigating toxicities; scarcity of validated biomarkers to identify patient populations who would benefit from immunotherapies; and the complexities of testing combination immunotherapies.
- New opportunities for collaboration and information exchange for advancing the field, including data aggregation and sharing electronic health records (EHRs) and other new sources of data, and precompetitive exchange of information.
- Clinical implementation of immunotherapies, including scaling up production of personalized cell therapies; addressing the novel adverse effects seen with the use of immunotherapies; and informing clinicians, patients, and payers about optimal use of immunotherapy.
- How to assess the value of immunotherapies and develop a sustainable economic model for clinical use of immunotherapies in cancer treatment.
These proceedings chronicle the presentations and discussions at the workshop. A broad range of views and ideas were presented, and a summary of suggestions for potential actions from individual participants is provided in Box 1. The workshop Statement of Task can be found in Appendix A and the workshop agenda can be found in Appendix B. A glossary is provided in Appendix C. The speakers’ presentations (as PDF and video files) have been archived online.2
___________________
2 See http://www.nationalacademies.org/hmd/Activities/Disease/NCPF/2016-FEB-29.aspx (accessed May 27, 2016).
BIOLOGY OF THE IMMUNE RESPONSE TO TUMORS
Khleif said that we have known for decades that the immune system continually monitors for, detects, and eliminates transformed and malignant cells, but developing therapies that use the immune system to suppress tumor growth has been very challenging (Vesely et al., 2011). However, progress in basic biomedical research has led to a better understanding of the key molecular players in the immune system, which has provided the platform for the current development and clinical use of immunotherapies in cancer treatment (Makkouk and Weiner, 2015).
Adaptive Immunity to Cancer: T-Cell Response
Several speakers—including Jay Berzofsky, chief of the vaccine branch at the National Cancer Institute’s (NCI’s) Center for Cancer Research; Malcolm Brenner, professor at the Center for Gene Therapy at Baylor College of Medicine; Naiyer Rizvi, director of thoracic oncology and director of immunotherapeutics at Columbia University Medical Center; and Steven Rosenberg, chief of the surgery branch at the NCI—discussed the current state of the science for immunotherapy in oncology. They said that tumor cells often express proteins that are not expressed under normal physiological conditions, and that these abnormal proteins (antigens) are released from the tumor cell and taken up by nearby dendritic immune cells called antigen-presenting cells. Antigen-presenting cells detect abnormal proteins using a receptor called the major histocompatibility complex (MHC). For each person, this receptor has a unique shape or cleft that selects which protein fragments can bind to the antigen-presenting cell. The antigen-presenting cell is able to capture an abnormal tumor protein fragment via the MHC receptor. This complex is recognized as “not-self ” by an inactive CD8+ T-cell (or T-lymphocyte). Other receptors on the T-cell can be costimulated, including CD28, CD27, and OX40, which activates the T-cell and triggers an expansion of different types of T-cells, including cytotoxic T-cells (CD8+) and helper T-cells (CD4+). This activated set of T-cells facilitates the immune system’s capability to recognize cells with the abnormal tumor protein. The activated cytotoxic CD8+ T-cell identifies the tumor cells and releases cytokines and toxic proteins to kill tumor cells and to recruit additional immune cells (e.g., B-cells and macrophages) to eliminate these tumor cells (see Figure 1).
Bernard Fox, chief of the Laboratory of Molecular and Tumor Immunology at the Earle Chiles Research Institute, said the immune system
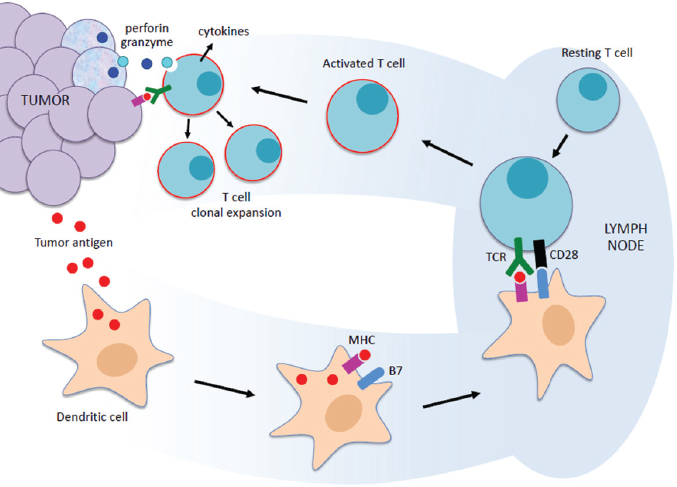
NOTES: MHC = major histocompatibility complex; TCR = T-cell receptor; perforin and granzyme are released from cytotoxic T-cells to promote cell death (Cullen et al., 2010).
SOURCES: Rizvi presentation, February 29, 2016; Scott Gettinger.
response is complex, and added that helper T-cells are necessary to achieve long-term effective immune response to tumors. He said animals that lack CD4+ T-cells are unable to stem the growth of metastatic tumors, despite injection with activated cytotoxic T-cells.
In addition to activating an immune response, the immune system also has mechanisms to curb or suppress immune responses. This prevents the production of cytotoxic T-cells once an infection or tumor has been cleared, and can help prevent an overblown immune response that can result in an autoimmune reaction that kills normal tissues. According to Berzofsky, current known T-cell suppressor mechanisms (or checkpoints) are
- Regulatory receptor/ligand pairs, such as PD-1 (programmed cell death protein-1)/PD-L1 (programmed cell death ligand 1) and CTLA-4 (cytotoxic T-lymphocyte-associated molecule-4)
- Regulatory immune cells, such as CD4+ regulatory T-cells, regulatory natural killer T-cells, myeloid derived suppressor cells, M2 macrophages, regulatory B-cells, and regulatory dendritic cells
- Regulatory cytokines, including IL (interleukin)-10, IL-13, and TGFβ (transforming growth factor beta)
All of these immune suppressors “put the brakes” on T-cell activation and can attenuate the ability of the immune system to eliminate tumor cells. “The T-cell gets turned on, and then the T-cell gets turned off,” said Rizvi. “That is just part of the normal biology that occurs when you have an immune response. You cannot have unchecked T-cell activation” (see Figure 2).
Tumor Escape from Immune Elimination
Tumors can take advantage of the naturally occurring T-cell suppressive mechanisms to escape from the immune system, a process called
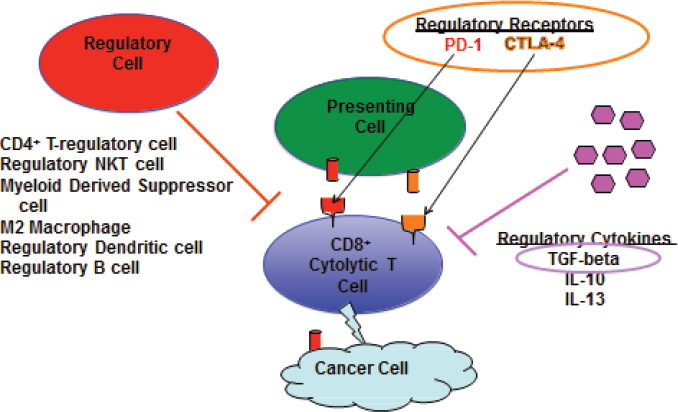
NOTES: CD4+ = cluster of differentiation 4; CD8+ = cluster of differentiation 8; CTLA-4 = cytotoxic T-lymphocyte associated molecule-4; IL = interleukin; NKT = natural killer T; PD-1 = programmed cell death protein 1; TGF = transforming growth factor.
SOURCE: Berzofsky presentation, February 29, 2016.
immunoediting, Rizvi and Fox said (Vesely et al., 2011). Chronic exposure to tumor antigens, like chronic infections, can cause the immune system to shift the balance of T-cell receptors to express a greater proportion of immune suppressing receptors, such as CTLA-4, compared to immune stimulating receptors such as CD28. Tumors can also upregulate the production of checkpoint ligands like PD-L1 on their cell surfaces to directly suppress T-cell response. The heterogeneous and continually mutating nature of cancer enables the development of these protection mechanisms via selective pressures exerted by an individual’s immune system. Only the cancer cells with excessive production of immune suppressing factors or a lack of immune stimulating factors will survive and repopulate a tumor. Rizvi said that “there is not just one pathway of immune escape, but multiple pathways, which is why single-agent immunotherapy only works in a subset of patients with cancer.”
CANCER IMMUNOTHERAPIES
Harnessing the current knowledge of immune response to tumors, researchers have developed a number of new immunotherapies for cancer, including immune modulating drugs that release the brakes on the immune system, vaccines that stimulate an antitumor immune response, and cell-based therapies that use a patient’s own T-cells, said Rizvi, Berzofsky, and Rosenberg. Berzofsky noted that unlike conventional forms of cancer therapy—such as chemotherapy, radiation, and surgery that directly target cancer cells—most immunotherapies primarily target the patient’s immune system.
Immune Modulator Drugs
A new class of drugs—immune checkpoint inhibitors—first entered the market in 2011 with the Food and Drug Administration (FDA) approval of ipilimumab for patients with melanoma, said Rizvi. The drug is a synthetic antibody that blocks the immune suppressing CTLA-4 receptor that is typically expressed on the CD4+ T-cell and CD8+ cytotoxic T-cell, in order to promote an immune system response to melanoma (Vasaturo et al., 2013). He added that in 2015, FDA approved two additional immune checkpoint inhibitor drugs, nivolumab and pembrolizumab, for use in patients with melanoma, lung cancer, and kidney cancer. Both of these drugs are also synthetic antibodies that target and inhibit the immune sup-
pressive receptor PD-1. Researchers are currently testing other agents that block CTLA-4, PD-1, or the PD-1 ligand (Topalian et al., 2015).
Rizvi reported that a pooled analysis of clinical trials of ipilimumab in 1,800 patients with advanced melanoma found nearly 20 percent of the treated patients were still alive 10 years later, suggesting an unusually durable response for a subgroup of patients (Schadendorf et al., 2015). “There are patients who, after four doses of ipilimumab, are essentially cured of metastatic melanoma, which is truly remarkable,” Rizvi said. Response rates of nearly 30 percent are seen in patients with advanced melanoma treated with anti-PD-1 drugs (Schadendorf et al., 2015). In patients with lung cancer, anti-PD-1 drugs have had a response rate of approximately 20 percent, and some lung cancer patients continue to experience a long-term benefit once active treatment is concluded (Gettinger et al., 2015). Significant response rates to approved or experimental checkpoint inhibitors have also been seen in patients with other types of solid tumors (Sunshine and Taube, 2015). “There is a huge potential impact of these drugs on the cancer landscape,” Rizvi said.
Therapeutic Cancer Vaccines
Therapeutic cancer vaccines are another immunotherapy modality under investigation. Unlike prophylactic vaccines for disease prevention, therapeutic tumor vaccines are designed to target a unique or highly overexpressed protein in an existing cancer. The vaccine aims to enhance the immune system’s targeting of the tumor cells. “Vaccines against cancer can harness the exquisite specificity of the immune system to selectively target cancer cells and avoid the kinds of side effects that one sees with less specific types of therapy,” Berzofsky said.
Two types of cancer vaccines are in development. The first is designed to stimulate the production of antibodies directed at a cell surface expressing antigen. An example of such a vaccine is one that targets the human epidermal growth factor receptor 2 (HER2), which is found in some cancers, including approximately 20 percent of breast cancers. Berzofsky said a small preliminary Phase I trial of an adeno-human HER2 vaccine is showing promising early results, including frequent objective responses or stable disease that is lasting more than 6 months in patients with advanced metastatic HER2-positive cancers—including ovarian cancer, gastro-esophageal cancer, and colon cancer—in which previous therapy has not worked. He added that this response does not require CD8+ or CD4+ T-cells, suggest-
ing the mechanism is independent of the T-cell adaptive immune response, and instead depends on induction of antibodies to HER2.
The second type of vaccine aims to elicit a more complete and possibly more durable immune response by inducing a cytotoxic T-cell response that targets tumors. “Most cancer antigens are expressed inside the cell and cannot be seen by antibodies,” Berzofsky said, making a T-cell-mediated vaccine more universally applicable. Berzofsky added that T-cells have many advantages as antitumor agents, including their ability to travel through multiple tissues, rapidly increase in number, and recruit other immune responses. Importantly, T-cells can also co-evolve with a tumor, and continue to mount an antitumor response when a tumor cell generates new antigens (known as neoantigens).
Antibody-based vaccines, in contrast, cannot alter their specificity to accommodate the production of new tumor antigens without the aid of other immune cells. “Tumors vary in single individuals not just in time, but in space so it is very difficult to come up with a universal antigen” for an antibody response, said Brenner. However, antibody-mediated tumor elimination does not rely on antigen presentation in the MHC cleft to be activated. “This is important because many tumors are MHC-negative or do not process antigens very well,” Brenner said.
Fox added that vaccine developers should not neglect B-cells that generate antibodies in favor of stimulating broader T-cell responses. He said one study found that a vaccine against the tumor antigen TRP2 prevented tumor growth, even if the mice were depleted of their CD4 and CD8 T-cells (Xu et al., 2013). In addition, when serum containing antibodies for TRP2 was transferred from immunized mice to untreated mice, Fox said that it prevented the outgrowth of an implanted tumor. “We should be thinking about not only boosting just CD4+ and CD8+ T-cell immunity, but about B-cell responses as well,” Fox said. “Cancer heterogeneity mandates we strive to get broad immunity with our cancer vaccines. If you select a limited number of antigens to go after, in terms of an antitumor response, you are giving the tumor an option to escape.”
Within the T-cell-mediated vaccines, a number of strategies are under investigation. Berzofsky described the development of a prostate cancer vaccine that targeted a protein called TARP, which is expressed in nearly all prostate cancers. The intent of the modification was to optimize an immune response by increasing the binding affinity to antigen-presenting cells and improve immunogenicity (the ability to stimulate an immune response) by inducing a T-cell response for TARP-specific T-cells (Oh et al., 2004).
This vaccine was evaluated in men whose primary prostate tumor had been removed, but whose rising levels of prostate-specific antigen (PSA) suggested microscopic recurrence of cancer. Phase I trial results showed that PSA levels were reduced in nearly three-quarters of patients 1 year after the vaccine was administered (Wood et al., 2016). Berzofsky said a Phase II trial of this vaccine was recently initiated.
Berzofsky added that common mutations in the RAS or p53 gene could be used in the development of a cancer vaccine by creating neoantigens that are recognized by T-cells and evoke an antitumor T-cell mediated immune response (Smith et al., 1997).3 In one study, patients with cancer underwent genetic analysis for mutations in RAS and p53. Researchers synthesized peptides that corresponded with the mutations and immunized patients with a cellular vaccine that included the mutated RAS or p53 peptide found in their tumors. Patients who exhibited positive T-cell responses, as measured by gamma interferon levels, had a median overall survival of 470 days, while patients who did not respond, had a median survival of 88 days (Carbone et al., 2005). Berzofsky said other studies have also found that mutant RAS can be a target neoantigen for cancer vaccines, and he noted that rapid sequencing advances may make this type of personalized vaccine more feasible.
T-cell mediated vaccines are more likely to be effective if they include components that block negative regulation of the immune system, Berzofsky said. “The vaccine is trying to induce a CD8+ T-cell that can kill a cancer cell, but there are a host of mechanisms that can inhibit this. [PD-1 and CTLA-4] are just the tip of the iceberg for immune regulation of these cells,” said Berzofsky. One cytokine with a newly discovered role in the T-cell checkpoint is TGFβ. When mice were vaccinated with an irradiated tumor vaccine combined with anti-TGFβ, there was a synergistic treatment effect, resulting in improved survival compared to monotherapy with the vaccine (Takaku et al., 2010). Researchers found that the increase in survival was mediated through CD8+ T-cells because when these T-cells were depleted, the mice had survival rates comparable to mice in the control arm.
Berzofsky described one of the first vaccine-based therapies for cancer, sipuleucel-T (Provenge), which is FDA approved for patients with metastatic prostate cancer that is resistant to hormonal therapy. This treatment involves extracting a patient’s antigen-presenting dendritic cells from a
___________________
3 The RAS gene encodes a protein important in cell signaling, and the p53 gene encodes a protein important in cell regulation. When RAS and p53 are mutated, they can drive the development of cancer.
patient’s blood. These cells are then activated using a common prostate tumor antigen and cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) at a production facility, and then are reinfused into a patient. Sipuleucel-T has been shown to extend the median survival of patients with advanced prostate cancer by 4 months (Kantoff et al., 2010).
Fox discussed the generation of neoantigens for vaccine development by using the cell degradation pathway to develop autophagosomes from degraded protein fragments (both defective ribosomal products from misfolded proteins [DRiPs] and short-lived proteins [SLiPs]). Autophagosomes can elicit a strong antitumor T-cell response, Fox said, and added that more than 100 antigens commonly overexpressed in human cancers were present in the off-the-shelf autophagosome vaccine currently in clinical trials (Page et al., 2016), including 12 of which have been prioritized for further development by the NCI (Cheever et al., 2009). Further, these autophagosome microvesicle vaccines are targeted to CLEC9A+ antigen presenting cells and contain damage-associated molecular patterns and agonist activity for 5 toll-like receptors. An initial study in mice suggested that autophagosomes were an efficient method to present tumor antigens to T-cells, and have potential for creating potent vaccines against cancer (Li et al., 2008). This method may also offer broader therapeutic potential across multiple tumor types compared with whole-cell vaccines that are only effective for the tumor for which it was designed (Twitty et al., 2011). However, Fox said that preclinical studies suggest that in order to obtain therapeutic efficacy in advanced cancer models, vaccines will need to be combined with other interventions because they are rarely effective as single agents.
Cell Therapies
Cell-based immune therapies for cancer are treatments that directly manipulate a patient’s own immune cells to attack a patient’s tumor. Several cell-based immunotherapies were discussed at the workshop, including adoptive T-cell transfer, personalized adoptive T-cell transfer, and Chimeric Antigen Receptor (CAR) T-Cell Therapy.
Adoptive T-Cell Transfer
Rosenberg provided an overview of adoptive T-cell transfer therapy for cancer treatment. The process includes removing a patient’s tumor and culturing the tumor-infiltrating lymphocytes, or TILs. The cells are
activated and their antigens are then isolated, expanded, and reinfused into the patient after the patient’s own immune cells are depleted with radiation or chemotherapy. “The T-cells can be administered [already] activated,” Rosenberg said. A patient’s immune cells are depleted prior to reinfusion of the cells in order to avoid suppression of the immune response. The time frame for adoptive T-cell transfer therapy using tumor infiltrating lymphocytes is about 6 to 8 weeks, from when the tumor is removed until the activated T-cells are reinfused.
Rosenberg presented summary data on adoptive T-cell transfer therapy from 194 patients with metastatic melanoma whose cancer had not responded to other treatments, which included data from four trials using different regimens for depletion of a patient’s immune cells. Twenty-three percent of the patients experienced a complete response, and 32 percent had partial responses. Of the 44 patients who experienced a complete response, only 2 have experienced a cancer recurrence within 4 years. “This is the hallmark of immunotherapy. If you can induce a complete response, it is very likely to be durable and curative,” Rosenberg said.
Helen Heslop, director of the Center for Cell and Gene Therapy at Baylor College of Medicine, reported on her Center’s adoptive cell transfer method for lymphoma. Similarly, this treatment involves isolating antigen-presenting cells from blood samples obtained from patients with lymphoma. These antigen-presenting cells undergo activation and incubation with five antigens prevalent on lymphoma cells. Subsequently, these antigen-presenting cells are incubated with the patient’s own T-cells along with a number of cytokines to elicit T-cell activation, and then reinfused into a patient. Because there is no well-accepted preclinical model for testing this kind of therapy, FDA required a dose-escalation design that increased the number of patients in the Phase I trial. Heslop said that thus far, three patients have exhibited T-cell activity across both incubated and non-incubated tumor antigens that correlated with clinical response (Leen et al., 2015).
Personalized Adoptive T-Cell Transfer
Expanding on the promising clinical results for adoptive cell transfer therapy, Rosenberg described the development of a technique to personalize the adoptive cell transfer method for broad application across patients and tumor types. To be recognized as an antigen, a mutant tumor protein needs to be processed intracellularly into fragments that fit into each patient’s
unique MHC cleft on his/her antigen-presenting cells. “What might be an antigen in one patient might not be an antigen in another because the peptide does not fit in the groove of the particular MHC molecules they have. This is a very important biologic point to remember as we begin to search for personalized therapies for patients with cancer,” he said.
However, Rosenberg and Harpreet Singh, managing director, founder, and chief scientific officer at Immatics Biotechnology, said methods to identify and predict which mutations within each patient’s tumor could be used as antigens are notoriously inaccurate. Therefore, Rosenberg developed an assay that evaluates the immune-stimulating ability of all the mutations within an individual’s tumor, which can range in number from a few dozen to hundreds of mutations. This high-throughput workflow starts by sequencing a patient’s tumor to identify mutations and then creates 25 amino acid peptides that correspond to each mutation (called mutated minigenes). Genes encoding these peptides are introduced to the patient’s antigen-presenting cells to identify which mutations are presented within a patient’s MHC cleft and are capable of activating a T-cell response. “There are no predictions necessary because everything is a direct measurement of the ability of a mutation to be an active antigen,” Rosenberg said. “This is a blueprint for how one might develop an immunotherapy for virtually any cancer.” Proof-of-principle testing in 25 patients whose tumors responded to adoptive cell transfer immunotherapy found that each patient harbored unique tumor antigens randomly scattered throughout their genome that were not shared by other patients. “These [antigenic mutations] are just random genes that happened to mutate and have the kind of properties that could present that mutation on an antigen-presenting cell and be recognized by the immune system,” Rosenberg said. He hypothesized that other forms of immunotherapy, such as anti-PD1 or anti-CTLA-4 drugs, also enable the immune system to target these random tumor mutations.
Rosenberg has started to apply this technique targeting personal tumor mutations in patients who have cancers other than melanoma. One patient with metastatic cancer of the bile ducts whose initial treatment failed underwent whole exome sequencing. She was found to have one mutation (out of 26 total mutations) that could be recognized by the immune system (Tran et al., 2014). Tumor-infiltrating lymphocytes that expressed this mutation were expanded in culture and reinfused into the patient, who is experiencing ongoing regression of lung and liver metastases more than 2 years after being treated. “A substantial regression for what previously was considered an untreatable cancer,” Rosenberg said. Additionally, a study of 22 patients
with epithelial cancers, including colon, breast, esophageal and rectal cancers, found 57 tumor mutations that were recognized as antigenic by T-cells, but only one (a KRAS mutation) was shared with another patient. “The lesson here is we have at least 23,000 expressed genes and any one of them can potentially become a cancer antigen,” Rosenberg said.
He said that this technique for identifying patient tumor antigens and then treating the patient with his or her own immune cells potentially could be applied to patients with any type of cancer. However, there are substantial technical requirements for successful execution. Rosenberg said the technique depends on the sequencing of mutations from tumor samples, which can be fresh or fixed in formalin and embedded in paraffin. The technique also requires the ability to grow TILs in culture, which requires fresh tumor tissue. However, fresh tumor samples are often not available or are difficult to obtain.
To overcome these challenges, Rosenberg said that his team has begun exploring use of tumor-reactive T-cells from peripheral blood rather than the tumor, because circulating T-cells will be easier to obtain. Initial research suggests that activated circulating T-cells overexpress PD-1, and could be used as a model to test whether a patient’s tumor mutations are recognized by the immune system. He said there was often comparability between recognition of tumor antigens by PD-1 expressing T-cells from tumor-infiltrating lymphocytes and from peripheral blood samples.
Rosenberg also said that this innovative treatment is challenging “because we basically develop a new drug for every patient,” which is completely counter to the way drugs are traditionally developed and regulated, and increases the cost of treatment. Nonetheless, he said that some biotechnology companies have expressed interest in personalized cell-based immunotherapy because they see the potential for its application to a wide variety of cancer types.
One participant asked what the impediments to clinical trials evaluating personalized adoptive T-cell transfer therapy are. Rosenberg said that current limitations are both biological and technical. The biological limitation is that some patients will not respond to the treatment if mutations that can stimulate an immune response cannot be found in their tumors. The technical limitation is identifying the rare T-cells that react to tumor antigens, which may comprise less than 1 percent of circulating T-cells. Researchers are currently exploring high-throughput sequencing, and Rosenberg thinks they will be able to identify and isolate those very rare tumor-reactive T-cells.
Another participant asked if clinicians should be banking viable tumor cells from cancer patients who undergo surgery in anticipation of cell-based immunotherapy. Rosenberg responded that fresh frozen tumor samples could be obtained and stored, and that peripheral blood samples could also be stored because they may contain T-cells that recognize mutations. “The storage in biorepositories will be very important,” he said. However, he said that future patients could also provide tumor and blood samples needed to develop personalized adoptive cell transfer therapy.
A third participant asked about shared tumor antigens that are found in the tumors of many melanoma patients, such as MART-1 (melanoma antigen recognized by T-cells 1) and gp100 (glycoprotein 100). He asked how they compare to the unique melanoma antigens in inciting an immune response. Rosenberg said that T-cells targeting these shared antigens tend to have weak antitumor activity and they also destroy normal melanocytes in the eye, ear, and skin, whereas unique melanoma antigens are less likely to have off-target toxicity because these mutations are only found on cancer cells. “We have tried to target those shared reactivities unsuccessfully. Cancer vaccines that have tried to target these shared relatively weak antigens have also not been effective in cancer treatment, because they are not targeting the unique mutation in that patient’s tumor,” he said.
A fourth participant questioned whether patients are more likely to respond to personalized adoptive cell transfer therapy if they have more mutations that can elicit an antitumor T-cell response. Rosenberg said that is likely true and that lung cancer patients whose tumors have more mutations are also more likely to respond to anti-PD-1 treatment. “This all fits with the hypothesis that the final common pathway of cancer immunotherapy is the recognition of cancer mutations,” Rosenberg said.
CAR T-Cell Therapy
CAR T-cell therapy is a targeted immunotherapy that combines the individualized design of adoptive T-cell therapy, the specificity of antibody therapy, and the long-term memory of vaccine therapy, said David Porter, professor and director of blood and marrow transplantation at the University of Pennsylvania Abramson Cancer Center. CAR T-cell therapy involves genetically engineering patients’ T-cells to express the antigen-binding component of an antibody on their cell surface. These T-cell-antibody hybrids, known as chimeric antigen receptors (CARs), act with the specificity of antibodies to target tumor antigens. They can stimulate immune system
activation, growth, and survival without reliance on the MHC cleft for antigen presentation. In addition, Brenner added that CAR T-cell therapy can target carbohydrates and glycolipids or non-processed surface proteins. He added that they also retain desirable characteristics of T-cells, including trafficking, expansion, persistence, and effector functions.
CAR T-cells are called “living drugs” because the chimeric receptor is propagated when T-cells multiply in the body and can last for long periods of time to provide vaccine-like activity. Similar to personalized adoptive T-cell transfer, CAR T-cell therapy takes a one-drug−one-patient approach. “Every single dose is unique to that specific patient,” Porter said. “These cells are both personalized, because every dose comes from a specific patient, and they are precise because they specifically target a protein on the tumor.”
Porter described the development of a CAR T-cell therapy directed against the CD19 receptor on B-cells, a class of immune cells responsible for producing antibodies. The CD19 receptor is expressed by most B-cell malignancies, and Porter said that research has found that antibodies against CD19 inhibit tumor cell growth. Normal B-cells and B-cell precursors also express CD19, but not the stem cells that generate blood cells, which helps minimize the risk of off-target toxicities developing with the treatment, said Porter. This CAR T-cell therapy involves isolating T-cells from a patient’s blood, introducing the CARs targeting CD19, and inducing T-cell activation and expansion. After a patient’s own immune cells have been depleted with chemotherapy, the modified CAR T-cells are reinfused into a patient. The entire process currently takes about 2 weeks, but Porter expects the process to eventually be streamlined to 5 to 7 days.
A single center pilot trial of CAR T-cell therapy was evaluated in patients with CD19 positive B-cell malignancies who had no available curative therapy options. Among the 43 patients with chronic lymphocytic leukemia, one-quarter of patients experienced a complete response and 23 percent of patients experienced a partial response, for an overall response rate of approximately 50 percent. Porter said that the treatment eradicated bulky disease, eliminating between 3 and 7.5 pounds of tumor cells in the first three patients (Kalos et al., 2011; Porter et al., 2011). “It really was quite potent,” Porter said. Biological follow-up found that the CAR T-cells expanded rapidly in patients and persisted for long periods of time (Maude et al., 2014; Porter et al., 2015). In some patients, CAR T-cells represented approximately 13 percent of the total activated T-cell population 1 year after treatment, Porter said, and are approximately 0.3 percent of the
T-cell population after 3 years. He said that by 5.5 years, CAR T-cells are still detectable in approximately 1 percent of all CD3+ cells. Preliminary findings also suggest the persisting CAR T-cells remain biologically active because patients continue to lack CD19+ B-cells 5 years after treatment.
CAR T-cell therapy was also evaluated in children and adults with relapsed acute lymphoblastic leukemia (ALL) that was resistant to other treatments, including bone marrow transplants. These patients had a poor prognosis, with expected median survival of less than 1 year. Of 30 patients who were treated, 27 had complete responses (Maude et al., 2014). “The outcomes were really quite astounding,” Porter said, although they did come with significant toxicities, such as B-cell aplasia and cytokine release syndrome4 that required some patients to be hospitalized in intensive care units. Other centers have also seen around 70 to 90 percent response rates with CAR T-cell therapies in patients with ALL, Porter said. “This really seems to be a function of the therapy, not a function of something that is unique to what we are doing in Philadelphia,” he said.
Brenner added that B-cell malignancies are especially suited for CAR T-cell therapy because most express strong, unique, and consistent antigens that are presented with ample costimulants. In addition to the findings in ALL, Brenner reported that CAR T-cell therapy directed at the CD19 receptor on B-cells has achieved 50 percent or greater complete response rates in patients with many subtypes of lymphoma. “This approach can remove even massive tumors [that are] resistant to other therapies. It is a single-dose administration that has prolonged responses,” Brenner said, adding that several biopharmaceutical companies are conducting large clinical trials of CAR T-cell therapy. Promising results are also being generated in Phase I trials using CAR T-cells directed at other types of B-cell receptors, he said.
However, unlike B-cell malignancies, Brenner said that most solid tumors have few antigens that are strong, unique and consistently expressed. More often, there is a heterogeneous pattern of antigen expression without the necessary costimulation required for T-cell activation. Furthermore, the solid tumor microenvironment contains cytokines, regulatory immune cells, and other factors that “render the environment very hostile to any immune response that might develop,” Brenner said.
___________________
4 Cytokine release syndrome occurs when patients experience inflammatory symptoms that result from rapid and large release of cytokines into the bloodstream. This can be life-threatening because it can lead to dangerously high fevers and precipitous drops in blood pressure (NCI, 2014).
To enhance costimulation of CAR T-cells for treatment of neuroblastoma, a pediatric brain cancer, Brenner described the use of a virus-specific T-cell as a platform for CAR T-cell therapy (Rossig et al., 2002; Savoldo et al., 2007). This approach uses a T-cell with a native receptor directed to a virus and a chimeric antigen receptor directed to the tumor antigen GD2, which is expressed on neuroblastoma and minimally expressed on normal cells. The rationale behind this approach was to use the virus antigen as a physiological costimulant to improve the antitumor immune response. Given that the viral-engineered T-cells recognize fragments of the virus on antigen-presenting cells “they would get all the necessary activation and would traffic the GD2-engineered T-cells to target and kill the tumor cell,” Brenner said. “This way you could turn a cold tumor, an inhibitory tumor, into something that looks like a hot virus infection to the immune system. You transfer your T-cells that target the virus and the tumor, and you develop a potent and crucially evolving immune response to solid tumor antigens.” Brenner said that this approach has demonstrated immune system expansion, persistence, and antitumor activity, but only in the presence of viral infection (Pule et al., 2008).
Combination Therapies
A number of speakers said that advances in immunotherapy treatments will require combination immunotherapies and immunotherapy provided in combination with other treatment modalities, including surgery, chemotherapy, and radiation. Berzofsky said that checkpoint inhibitors are known to be more effective in patients whose immune systems recognize a larger number of tumor antigens, presumably because more types of antigens stimulate a more potent T-cell immune response. This idea led to the hypothesis that cancer therapies (including some chemotherapies, targeted therapies, and radiation therapy) might potently activate antitumor T-cells by promoting tumor cell death and the release of multiple tumor antigens. These antitumor effects could potentially be enhanced through inhibition of immune suppression by treatment with checkpoint inhibitors, such as anti-CTLA-4 or anti-PD1 compounds, or other immune response regulators. Berzofsky said that “several research groups have found that chemotherapy’s killing of tumor cells releases antigens that act as a kind of internal vaccine. It is possible that some types of chemotherapy might synergize with something like anti-PD1 or
other types of checkpoint blockade where you allow the immune response induced by killing tumor cells to contribute to the regression or rejection of the tumor.”
Alexandra Snyder Charen, attending physician and translational researcher at Memorial Sloan Kettering Cancer Center, discussed this hypothesis in an animal model of pancreatic cancer. When researchers injected animals with pancreatic tumors followed by anti-CTLA-4 or antiPD1, as a monotherapy or in combination, there was little effect on slowing tumor growth. However, when combined with the chemotherapy drugs gemcitabine and paclitaxel with CD40, an activator of antigen-presenting cells, more than half the animals survived for 80 days after tumor injection (Winograd et al., 2015). Snyder added that in a melanoma model, the combination of radiation and anti-CTLA-4 outperformed either intervention alone. Similarly, anti-PD-1, anti-CTLA-4, and radiation treatment together provided the best outcomes (Twyman-Saint Victor et al., 2015).
Other examples of synergy with combinations of checkpoint inhibitors have also been documented. Rizvi said that one combined dose of ipilimumab and nivolumab led to a dramatic regression of a chest wall melanoma tumor in one patient (Chapman et al., 2015). “This speaks to the power of combination immunotherapy,” Rizvi said. Data also showed that the combination of nivolumab and ipilimumab caused a 58 percent response rate for melanoma patients with metastatic melanoma; FDA has since approved that combination therapy for that patient population, Rizvi added. Response rates were 72 percent in patients with melanoma who tested positive for upregulation of PD-L1 in tumors and blood (Larkin et al., 2015). However, Rizvi said that the toxicity of checkpoint inhibitors also increases when they are combined.
However, both Berzofsky and Rizvi cautioned that cancer therapies that target specific genetic defects can also inhibit the immune system, and therefore the effects of combination therapies can vary by tumor type. “For lung cancer, the patients [who] use targeted therapies are the ones least likely to respond to immunotherapy. The notion that you can use targeted therapies to convert a lung tumor to a more immunogenic environment has not been really shown yet,” Rizvi said. However, he said that there is evidence of synergy between targeted therapies and immunotherapies in melanoma.
DRUG DEVELOPMENT CHALLENGES FOR IMMUNOTHERAPIES
Several workshop speakers said there are a number of preclinical and clinical challenges that are unique to the development of immunotherapies. Immunotherapies are more complex and less well understood than other types of cancer therapies, so they require unprecedented studies and resources, said David Kaufman, associate director of vaccines clinical research at Merck. “Immuno-oncology is different than a lot of areas of cancer research in that we really do not understand how these drugs are working. We have some idea . . . but the complexity here is orders of magnitude beyond ‘here’s a pathway that you block with this drug and it works,’” he said.
Many speakers agreed that this is especially true for personalized cell-based immunotherapies. “Every actively personalized immunotherapy is different—the product is different. While you can apply certain studies, such as proof-of-principle studies, or stability and shelf-life studies on your warehouse components, it is not possible for your final drug product, which is unique for every patient,” Singh said. Peter Bross, medical review officer at FDA’s Center for Biologics Evaluation and Research (CBER), added, “When we are regulating products that are designed for a single person, we just have to develop new paradigms.” He said that given the high response rates seen in personalized cell-based immunotherapies for cancer, “we have to develop a way forward, an approach to this that will facilitate these products being available.”
In particular, Bross suggested changes to the business model for cancer immunotherapies, which some people do not view as economically feasible without some federal funding of preclinical and clinical development. Brenner added that the complexity of these treatments “means you cannot follow the standard drug development pathway from preclinical [studies to] Phase I, II, and III. Instead you are locked into an iterative phase between preclinical and Phase I where you test out your best product, go back to the lab, improve it, and go back into the clinic. It is extremely difficult for any commercial entity to entertain that open-ended time and money commitment.” He contrasted this with traditional pharmaceutical development, in which “you spend a lot of money up front developing an approved drug and manufacture it for a few cents or fractions of a cent per pill. Then you would administer it, ideally lifelong, to a patient in whom it is ameliorative and for whom you can charge the maximum possible amount.” Unlike the
pharmaceutical model, CAR T-cell therapy is ideally a one-time treatment: “They are curative given a single time. And they are very expensive to make. How do we pay for these very expensive one-off therapies?” Brenner said. He suggested one option might be to reallocate resources from developing the current forms of non-curative cancer therapies to immunotherapy development.
Kaufman added that another complexity in immunotherapy development is the need to test combination therapies because they are likely to be more potent in combination and there are numerous possible combinations. “It is going to take an enormous amount of investment in iterative clinical, preclinical, and bioinformatics research that is beyond what usually goes on for a single drug approved for a couple of indications. The research endeavor here is particularly intense,” he said. Richard Simon, chief of the NCI’s biometric research branch, agreed and suggested increased federal funding for experimental and computational initiatives is necessary to identify new antigens for adoptive T-cell therapy, as well as for integrated systems biology modeling for T-cells and their relationships to tumor biology and the immune system. “We are beset with more candidates and candidate combinations than we can really test just empirically, so we need to build a model as we go along that will collect data from studies and help us think through issues of timing and how this very complicated system works together,” he said.
Preclinical Challenges for Immunotherapy
Animal models for preclinical testing are an essential component of cancer therapy development. Whitney Helms, supervisory pharmacologist at FDA’s Office of Hematology and Oncology Products, said that FDA often requires testing in two animal species for drugs and one animal species for biologics prior to the initiation of clinical trials evaluating novel cancer therapies. However, a number of speakers noted that conducting preclinical studies with immunotherapies continues to be challenging endeavor, given a lack of appropriate animal models that can fully capture the complexity and dynamics of the human immune response to tumors.
Mouse Models
Researchers rely on a number of mouse models for establishing proof-of-principle and evaluation of on-target effects and efficacy of cancer
therapies, said Snyder. There are three main categories of mouse models used in cancer research—mice (immunodeficient or immune competent) that are implanted with tumor tissue or cell lines, mice that spontaneously develop tumors (genetically engineered and carcinogen induced), and mice that have a partially humanized immune system (Budhu et al., 2014).5 She said that immune modulator drugs that are currently being assessed in human clinical trials (e.g., drugs that modulate CTLA-4, PD-1, PDL-1, OX40, LAG3, 4-1BB, and GITR) have all been developed based on data from mouse models (Budhu et al., 2014; Pardoll, 2012). For example, to examine the function and potential toxicities following inhibition of CTLA-4, researchers generated CTLA-4-deficient mice that exhibited excessive proliferation of lymphocytes and multiple autoimmune conditions (Tivol et al., 1995). BALB/c mice injected with the B7-51BLim10 murine colon carcinoma were used to first test the effects of a CTLA-4 inhibitor on the growth of a colon cancer (Leach et al., 1996). Snyder said that these studies show that mouse models can generate useful information about drug targets and combination therapies. She reviewed some of the characteristics of commonly used mouse models, and noted that each model has different advantages and disadvantages. Common mouse models have been developed for melanoma, myeloma, ovarian, and colon cancer, but she emphasized that just because the organ of origin is the same, it does not mean that the mouse model recapitulates the human cancer.
Mouse models enable researchers to explore factors that might influence the safety and effectiveness of immunotherapies that are difficult or impossible to manipulate in humans, Snyder said. For example, researchers were able to assess the influence of the gut microbiome on checkpoint blockade by giving mice fecal implants in addition to anti-PD-L1 treatment (Sivan et al., 2015). Another study showed mice who live in germ-free facilities or are given broad-spectrum antibiotics lose their response to anti-CTLA-4 drugs compared to normally housed mice with a functional gut microbiome (Vétizou et al., 2015). These preclinical studies informed a prospective clinical study assessing the composition of gut microbiota and occurrence of colitis in CTLA-4-treated patients, said Snyder (Dubin et al., 2016).
However, mouse models also have their limitations when used to test the safety or effectiveness of cancer immunotherapies or even their mecha-
___________________
5 Snyder did not cover humanized mouse models, both because they have not yet been used to make the major discoveries in the checkpoint blockade field and due to time constraints.
nisms of action, Snyder said. A common mouse model—the implantable tumor model, or xenograft model—often uses immune-deficient mice that receive a direct tumor injection (often from human tumor cell lines). Once the tumor grows, the mouse is treated with an anticancer agent and then outcomes are assessed by measuring tumor growth and mouse survival. Because immunotherapy requires a functional immune system, immune-deficient mice models are not sufficient to evaluate hypotheses related to immunotherapies. Other syngeneic mouse models exist, in which tumor cell lines developed from the same strain are implanted into immune competent mice (for example, the B16 melanoma and ID8 ovarian cell lines in C57/BL6 mice). Advantages to their use are that they demonstrate rapid and reliable tumor growth, are available for multiple cancer types, and interventions on the tumor cell lines can be made prior to implantation. However, the drawbacks of these models are that these tumors are relatively homogeneous, consisting of a single cell line, and these models do not replicate the time course of human tumor growth because they grow at a much faster rate. “This may translate into different immune interactions taking place systemically and in the tumor microenvironment,” Snyder said. While recognizing that mice have much shorter lifespans than humans, “Undoubtedly the time course of the immune system’s knowledge of an interaction with the tumor impacts the data we get from studying the tumor and tumor microenvironment in mice,” Snyder said. She added that predicting a disease-specific application is generally not realistic with many available mouse models. Many immunotherapies require MHC matching between the immune system that is being stimulated and the tumor it needs to target. Consequently, a researcher cannot evaluate an immunotherapy in a mouse that has an implanted tumor from another strain of mouse or from a human, as is commonly done for other cancer therapies, Berzofsky added.
Mouse models that spontaneously develop tumors may recapitulate human tumor development and heterogeneity more faithfully, Snyder said. Furthermore, carcinogen-induced spontaneous tumors also model tumor immune escape mechanisms similar to humans. However, there are also challenges in using spontaneous mouse models, including the time commitment (a single experiment may take 6–12 months), expense, and challenges in interpreting results due to intermouse heterogeneity. There are also limitations with genetically engineered mouse models in which mutations are continuously present from birth, because the immune systems of these mice may develop tolerance to their tumors and certain immunotherapies may be less effective or more difficult to test in these animals.
Compared to the human immune system, the immune systems of most mouse models are also more homogeneous and demonstrate less MHC variability. Heslop and Fox added that the different immune systems of mouse models may have different target antigens and exhibit different responses. Fox described experiments designed to provoke an immune response in mouse models by genetically engineering the B16 melanoma tumor cell line to express receptors for several interleukins, interferon gamma, granulocyte-macrophage colony-stimulating factor (GM-CSF), and a number of other cytokines. While irradiated tumor cells did not stimulate significant anti-tumor activity, the investigators found that irradiated tumor cells expressing GM-CSF stimulated strong, long-lasting, and specific antitumor immunity (Dranoff et al., 1993). Snyder also said that other factors, such as environmental exposure, age, and gender variability, are also poorly modeled using mice.
Some mouse models are better suited to studying autoimmune toxicities linked to the use of checkpoint inhibitors, including NOD6 and SJL/J7 mice. The latter was used to show that injections of CTLA-4 inhibitors led to inflammation of the pituitary and circulating antipituitary antibodies. This mouse model was informative, given that hypophysitis (an inflammation/autoimmune disorder affecting the pituitary gland) is seen in 4 percent of patients treated with ipilimumab (Iwama et al., 2014). However, autoimmunity is rarely seen in mice and difficult to measure when it does occur; in addition, some autoimmune effects seen in mice, such as the cardiac toxicity following treatment with checkpoint inhibitors, are not recapitulated in humans, Snyder said (Nishimura et al., 2001; Tivol et al., 1995; Uno et al., 2006).
In addition, experiments conducted with different mouse models can lead to conflicting findings. For example, when researchers performed the same experiment in two different sarcoma models to study immunoediting, Snyder said the results were quite different (DuPage et al., 2012; Matsushita et al., 2012). “These imperfect models teach us about immunoediting, but do not tell us specifically what to do with human sarcomas. We can translate the concepts, but not the precise details from these mice models,” said Snyder. She added that different mouse models for melanoma can also result in dif-
___________________
6 NOD = non-obese diabetic.
7 SJL/J (Swiss Jim Lambert) mice display a very high incidence of reticulum cell sarcomas resembling Hodgkin’s disease by approximately 1 year of age. See https://www.jax.org/strain/000686 (accessed June 2, 2016).
fering data. When two genetically engineered mouse models with the same gene alteration but different phenotypes from two models (one developed benign noninvasive uninhibited melanocyte growth disease only while the other developed aggressive metastatic disease) were treated with an antitumor vaccine, only the first group of mice was protected from developing tumors. Because different mouse models have often generated conflicting findings, Snyder said “the evaluation of any intervention in several models is critical.”
Angela Thomas, clinical trials chair of the Biological and Vaccines Expert Advisory Group at the UK National Health Service (NHS), added that “the ultimate clinical model for these therapies is the human.” She said that a rodent model used to assess a targeted therapy did not predict the significant inflammation in the lungs that occurred when the drug was tested in humans because the model did not have the same receptors in lung cells as humans. “It is very important to realize where the pitfalls are. Nonclinical models can be very difficult and they can also be misinterpreted and lead to quite significant problems,” said Thomas.
Monkey and Other Large Animal Models
Following preclinical studies, monkeys or dogs are used to predict treatment toxicities because these animals are more evolutionarily related to humans and are considered appropriate models to assess safety concerns. However, a number of workshop speakers said that these large-animal models are limited in their ability to predict safety of cancer immunotherapy. Helms said that a major issue at FDA has been determining the relevance of the species used in animal testing for cancer immunotherapies. Helms said that studies of an anti-PD-1 drug in monkeys occasionally showed patterns of immune cell infiltrations to multiple organs, but not the autoimmunity issues experienced by some patients when these drugs are used in the clinical setting. “At exposures well above those seen clinically, there was no clear autoimmunity in the animals,” she said. “While we are not completely ready to give up on the monkeys, they seem to have a different kind of threshold for toxicity,” she said. Ramy Ibrahim, clinical vice president of Immuno-Oncology at AstraZeneca, agreed, noting that “preclinical data are not very informative in predicting toxicities and we try not to rely on preclinical models for safety.” John Connolly, scientific development advisor at Tessa Therapeutics, suggested that a limitation of monkey models is a lack of tools for characterizing their immune systems, which “will come in handy when we start looking at combinations of these biologics.”
George Weiner, director of the Holden Comprehensive Cancer Center at University of Iowa, asked if pigs or other large animals could be used as preclinical models for cancer immunotherapies. Helms said “there is no FDA guidance that says it has to be a monkey,” but because antibodies and other reagents have been developed for monkey models, FDA has more confidence in data from monkey studies. “But if you could show the target is bound in pigs and that it is a pharmacologically relevant species, I do not think you would be prevented from using that model.” She added that sometimes rabbit models are used, in addition to rodent studies, but the preference is for an animal that is further up the evolutionary tree and closer to humans for preclinical tests of toxicity.
Weiner questioned whether transgenic pigs that develop tumors could be used to test immunotherapy effectiveness. Helms suggested that these animals could be useful for proof-of-concept studies and to assess therapeutic activity, but transgenic pigs are not designed for toxicology studies. However, she said, “If you had a rigorous program, we would entertain it. We try to be flexible.” Allen Wensky, biologist at FDA’s CBER, added that research using the pig model is advancing, and “I definitely encourage that field to go forward. [There are] pig knockout models now, which are going to be amazing tools down the road, considering we [currently] have mostly mouse and rat knockout models. [Pig] models . . . could potentially be used as a safety model and they need to be developed and presented to us at FDA.” Helms added, “You just need to make your argument and make a clear case for the model. Give us your justification.”
Several speakers said that the lack of animal models for the complex interactions between the immune system and tumors creates numerous challenges when trying to identify the mechanisms of action in immunotherapies. For example, each species has a unique MHC; the human version is the human leukocyte antigen (HLA) complex, while in mice it is called the H2 complex (Neefjes et al., 2011). Because of these species-unique differences in MHC, Singh said there are no relevant toxicity-predicting animal models for the protein fragments that fit within the HLA clefts of human immune cells (or HLA-restricted peptides). “This is strictly a human-specific setting. Non-transgenic animals are not relevant because they do not carry HLA, and even HLA-transgenic animals do not express the relevant components of the human antigen processing machinery, so they may present very different peptides. If you do see a toxicity signal, it may be misleading and you may prematurely disregard a product. If you do not see a signal, this may also be misleading because you have a false sense of security,” he said.
Connelly said it is important to model the phenomenon of epitope spreading, which occurs when the immune system can target multiple sites on the same antigen or multiple antigens from a single tumor (Disis et al., 2004; Hardwick and Chain, 2011). He suggested that immunotherapies for cancer could be optimized by predicting epitope spreading in animal models. Lisa Butterfield, professor of medicine, surgery, and immunology at the University of Pittsburgh, agreed, but added that it is challenging to identify a relevant model for this biological process.
Several other workshop participants also emphasized that the lack of suitable animal models to evaluate cell-based immunotherapies can thwart progress in clinical development. For example, Heslop said that lack of appropriate animal models to evaluate the toxicity of an Epstein-Barr virus–specific T-cell therapy to prevent or treat patients with Epstein-Barr virus–related lymphoproliferative disease after receiving a bone marrow transplant led to FDA placing a hold on the clinical trial, due to concerns about cross-reactivity damaging normal healthy tissue (Heslop et al., 2010). Heslop said there was no suitable model to assess cross-reactivity.
Experimental Assays
Researchers have developed a number of experimental assays to assess the immune response to tumors following immunotherapy treatment. These assays measure a range of biological elements, including changes in immune-regulating cell abundance or release of cytokines. However, a number of presenters said that there is a need to develop new assays and to standardize and validate current assays to promote more reliable results and experimental interpretations. For example, Butterfield said it is important to determine the mechanism of action when an immunotherapy works; such as, did the treatment activate a cytotoxic T-cell or did it counter the immune suppression of regulator T-cells? “Without knowing these mechanisms of action, we do not have rational approaches to make immunotherapy combinations,” Butterfield said. She added that immune assays are costly, so researchers tend to select just a few to assess the activity of an immunotherapy, but it is unclear whether a limited set of assays will lead to misinformation about underlying mechanisms of action. Fox offered an analogy of “only looking for your keys under the streetlight,” because in immunotherapy research, “we are limited by what we can look for when we are targeting cancer with immune responses. We do not have good tools to look at the vast spectrum of targets that are potential antigens in cancer.”
Despite some progress on standardization of immune assays, such as measuring regulatory T-cells in circulation, there is mixed evidence about the biological significance of tumor infiltration of these T-cells, Butterfield said. She suggested that the mixed evidence might be due to researchers using different test methods such as immunohistochemistry and flow cytometry. In particular, there is significant variability among methods for flow cytometry. Myeloid-derived suppressor cells are thought to be potent suppressors of antitumor immune responses; in humans, there are 10 subsets of these cells and researchers are still trying to assess which type of immature myeloid cell or myeloid-derived suppressor cell affects antitumor immunity, Butterfield said. Part of the challenge is that researchers all use different parameters when using flow cytometry, called gating, even if they use the same markers. “We still need standardization in how to measure these cells,” Butterfield said.
In addition to T-cell assays, Fox described the potential use of protein arrays to assess antibody responses to cancer immunotherapies. He discussed a study of patients receiving anti-CTLA-4 treatment in combination with the cytokine GM-CSF, in which the researchers used serum from patients to run protein arrays to detect antibodies (Kwek et al., 2012). The presence of antibodies served as a surrogate marker for activated helper T-cells and possibly cytotoxic T-cells, Fox said, because activation of these T-cells is critical for B-cell-mediated antibody production. He added that a recent journal article discussing novel technologies and emerging biomarkers for personalized cancer immunotherapy included discussion of protein arrays to assess antibody responses (Yuan et al., 2016). Protein arrays to evaluate immune response in mouse models would also be useful, said Fox, but unfortunately do not yet exist.
There are also other new technologies under development, such as immune profiling, which provides in-depth characterization of the immune system through next-generation sequencing and other high-throughput analyses. Immune profiling can detect how immunotherapies alter immune cell repertoires and functional activities. But such profiling “is still in the pilot stage in a lot of settings. We have fantastic new technologies . . . but we do not yet have a robust [set] of data showing how effective they are,” Butterfield said.
Singh described the development of an immunopeptidome platform that uses high-throughput mass spectrometry to separate, identify, and sequence hundreds of thousands of tumor peptides and assess their ability to provoke immune responses using immunoassays (Walter et al., 2012; Weinschenk et al., 2002; Yadav et al., 2014b). “We would like to map as
far as possible the entirety of the immunopeptidome, not only on tumor tissues, but also on normal and healthy tissues to really understand the differences,” he said. Singh added that this platform has a target database collected from 9,000 experiments analyzing approximately 20 different tumor types and 40 different normal tissue types, resulting in a quarter million unique MHC-restricted peptides. He said that more than 1,500 of these peptides have some degree of association with cancers and might be useful for assessing personalized cancer immunotherapies. This technology has also identified neoantigens and found that most peptides containing unique mutations in cancers are not presented by MHC molecules on the cell surface, although there are rare exceptions (Yadav et al., 2014a). Singh said that tools that examine how peptides are actually presented would be more useful for predicting which cancer antigens are likely to provoke an immune response, compared to in silico prediction algorithms. Mass spectrometry immunopeptidome assays can also provide quantitative information on how many copies of a peptide are present in individual cells and assess varying copy numbers between peptides originating from the same source protein. “Every tumor is different and provides unique antigens, and ideally should be assessed in an unbiased fashion,” Singh said.
The ability of the platform to detect whether immune-stimulating peptides are shared by tumors and normal tissues can also help predict on-target toxicity, Singh said. For example, an analysis of a colorectal cancer antigen (carcinoembryonic antigen [CEA]) found that it was present in stomach cancer, non-small cell lung cancer, and colorectal cancer, but CEA was also detected on healthy colon tissue, which Singh said can help explain the toxicities experienced in the clinical trial targeting CEA in colon cancer (Parkhurst et al., 2011). Off-target toxicities can also be predicted. One analysis of the T-cell receptor that recognized the testes cancer antigen MAGE-A3 found that although the antigen is not expressed on normal tissues (with the exception of the testes), the T-cell receptor cross-reacts with another peptide derived from the protein titin, which is found in heart and muscle tissues (Cameron et al., 2013; Linette et al., 2013). “This T-cell receptor would not have moved forward because of that cross-reactivity signal you can detect early on during the discovery phase,” Singh said.
Evidence Requirements and Considerations for First-in-Human Trials
Several presenters suggested that because of the limitations of preclinical animal models and tests for cancer immunotherapies—especially
highly personalized cell-based therapies—perhaps preclinical testing in animal models is not always necessary. Instead, some workshop speakers suggested that a more thorough evaluation of the mechanisms of action and likely off- and on-target toxicities could be based on information compiled from genetic databases, computer models, and proof-of-concept studies. Once this evidence is assessed, several speakers suggested that clinical testing could initially be started with low doses that escalate slowly over time to ensure that no serious toxicities are evident before proceeding to higher doses. For example, the FDA clinical hold on the T-cell therapy clinical trial that Heslop described was lifted when investigators submitted data on structural similarities between the naturally occurring antigens and the antigens that were being used to optimize patients’ T-cell therapies. The trial was also redesigned as an antigen-escalation study, in which the first group of patients received only one antigen and the second group received two antigens, until all five antigens were provided to patients. Heslop also suggested that investigators consider options in advance to reduce clinical risk if adverse events occur, such as having the ability to ablate cells (e.g., with steroids) or to neutralize the cytokines that an immune reaction may stimulate. She added that in a CAR T-cell therapy clinical trial, researchers were able to counter the life-threatening cytokine storms in some patients by administering anti-interleukin-6 treatment.
Wensky suggested that various other sources of preclinical and clinical data could be leveraged, including any published data in peer-reviewed journals, or animal studies using an analogous product where an appropriate animal model for the intended clinical product does not exist. Other opportunities include reviewing previous clinical trial results and modifying clinical trial designs to help mitigate risk. “A lot of [what] we do is about risk mitigation so that before we enter a clinical trial, we have done what we can to make it as safe as possible, but not be too onerous about what we ask for,” Wensky said. He pointed out that FDA released a guidance document in 2013 that discusses the preclinical assessment of cellular and gene therapy products (FDA, 2013). Wensky also suggested communicating with the Office of Cellular, Tissue, and Gene Therapies within CBER early in the development process of an immunotherapy for cancer. He noted that they have an option called a pre-pre-Investigational New Drug (pre-pre-IND) consultation that may be appropriate at very early stages of development if sponsors wish to seek FDA advice on the preclinical program. “We are not there to help you develop your product and go through the research. That is not our expertise, but these consults can be extremely helpful for spon-
sors,” he said. Helms added that a thorough examination of the mechanism of the immune activity is critical throughout the development of cancer immunotherapies. This not only informs the safety of the first-in-human tests but also the continued development and post-marketing surveillance.
Helms noted that the International Council on Harmonisation (ICH) had issued a guidance for industry called, ICH S9, discussing nonclinical development of anticancer pharmaceuticals (FDA, 2010). “For biopharmaceuticals with immune agonistic properties, selection of the start dose using a minimally anticipated biologic effect level (MABEL) should be considered,” Helms said, adding that checkpoint inhibitors would fall into the class of biopharmaceuticals for which a MABEL would be appropriate. Determining a MABEL relies heavily on a variety of pharmacology studies rather than the traditional toxicology models, she said. There is no universal approach for determining a first-in-human dose based on a MABEL regardless of the indication. “It is case by case, product by product,” Wensky said. Some useful inputs have been in vitro pharmacological data from human and animal target T-cells relevant for toxicology assessments, and concentration-effect data from in vitro and in vivo studies. If animal data are used, sponsors should provide comparisons of human and animal differences in drug exposure, target expression and distribution, and affinity of binding and intrinsic efficacy, Helms said. The duration and reversibility of the biologic effects should be determined as well as the dose–exposure relationship, she added. “What we are really looking for when we receive an immunotherapeutic for review is the pharmacology of the targeted pathway—is it an agonist or is it an antagonist or is it an immune checkpoint inhibitor?” Helms said. She added that assessment of cytokine release potential is now a standard assay for immunotherapies. “If you do not have this data, it might be cause for a clinical hold,” she said. FDA also likes to see studies using human cells that take into account multiple mechanisms of action. “These are the data that are often most useful in determining a MABEL,” Helms said. It can also be useful to know receptor occupancy of an agent, although that can be problematic with some upregulated receptors, Helms noted. A MABEL based purely on receptor occupancy data can lead to extremely low doses, so it is not always used as the basis for making a MABEL determination.
Fox said that it is difficult to determine threshold doses of vaccines because they are not usually toxic and do not usually have serious side effects. When asked how FDA determines the threshold dose for other immunotherapies, Helms said that FDA generally uses the dose that is
severely toxic in 10 percent of animals tested as the highest dose to estimate an acceptable starting dose. If the MABEL assay approach is used, FDA can use in vitro functional studies, such as T-cell proliferation or production of interferon gamma, and the EC20 dose (“effective concentration” where 20 percent of maximum effect is achieved) and pharmacokinetic data from available animal studies to determine a starting dose estimate.
Khleif said that immunodulators ramp up the immune system so side effects are likely to be immune related, and to occur later and last longer than most acute toxicities in cancer treatment. Because animal models cannot often recapitulate human immune responses and side effects, he asked if FDA would consider skipping preclinical toxicology analyses for cancer immunomodulators in which there is either abundant proof-of-principle studies using the candidate or related agents or in vitro studies of the candidate agent that indicate a mechanism of action. Helms said, “The point of an animal toxicology study is not just to look for exaggerated pharmacology, which obviously we are not seeing so far with some of the immunomodulators, but to address the worry that you are going to miss something. Maybe you do not understand the target as well as you thought you did—it is expressed somewhere that we did not even consider. I do not know if there [will be] a time [when] we are going to say you do not have to do animal toxicology studies unless you have a situation where the antibody really does not bind, as for some bispecific T-cell engagers for which [there is not] a relevant animal model. In those situations, we have had times where we did not ask for a toxicology model. But if you have something that binds and it is pharmacologically relevant theoretically, it is really hard for us to say ‘don’t do it’ at this point.” Wensky added that for biologics, suitable animal models are often lacking so the investigational products are evaluated on a case-by-case, product-by-product basis. “You have to come to us and say ‘this is what is going on. This is what we have. This is the state of the art. This is what we can do.’ Oftentimes for our types of products, the best you can do is look at off-target toxicities with a bunch of cell lines in in vitro studies. It depends on the product, the indication, and the target. But I definitely think there are situations where we do not ask for studies in animals, especially nonhuman primate studies, if the results would be uninformative or masked by an overwhelming xenoresponse,” said Wensky.
Fox said there is a lot of iterative tinkering in the development of cancer immunotherapies, and suggested that instead of redoing the toxicology and other preclinical studies every time an antibody undergoes minor modifications, perhaps pilot studies could be completed in a small number of patients
in which microdoses of an agent could be injected in patients’ accessible tumors to make initial assessments of safety, effectiveness, and mechanism of action. Such testing could move combination immunotherapies into clinical practice much more quickly, he said. Singh suggested that on-target toxicity for immunotherapies can be assessed with a comprehensive study of target expression using a highly sensitive RNA sequencing method on normal, healthy tissue that could be compared to mass spectroscopy-based peptide presentation data to provide the relevant dataset “to really get as close as possible to the truth. That is what we are doing always in science—we try to approximate the truth.” To further minimize the risk of off-target toxicity of genetically engineered, T-cell receptor-based immunotherapies such as CAR T-cell therapy, he suggested determining the motif of the T-cell receptor through a peptide scan, and then checking that against genomic and immunopeptidomic databases to see if there are any cross-reactivities. Singh also suggested that clinical dose escalation studies may not be helpful in determining toxicities of cancer immunotherapies, and instead suggested assessing off-target toxicity in the preclinical setting.
Clinical Trial Design Considerations
A number of workshop participants discussed issues and challenges in the design of clinical trials for immunotherapies in cancer, including
- Inability to identify which subsets of patients are most likely to respond to specific immunotherapies. (Kaufman, Porter, Rizvi)
- Biomarker research, including issues in developing, validating, and standardizing biomarkers in immunotherapies. (Butterfield)
- Uncertainty in determining appropriate endpoints and duration of treatment, given the potential for pseudo-progression, delayed, and incomplete treatment response. (Berzofsky, Krug, Schwartzberg, Wolchok)
- Predicting effective and safe doses of immunotherapies and defining appropriate follow-up. (Bross, Porter, Simon, Sridhara)
- Ensuring quality control in immunotherapies. (Bross)
- Complexity of testing immunotherapies in combination and in sequence. (Berzofsky, Fox, Khleif, Simon)
Patient Selection
Immunotherapies often work only in a subset of cancer patients, but identifying the reasons why some patients respond is complex because each person’s immune response to his/her tumors varies both individually and over time. To find the right immunotherapy for the right patient—while avoiding therapies that are not likely to be effective and their associated side effects—a number of workshop participants discussed the important role that biomarkers play in predicting antitumor immune responses. Several workshop participants discussed the use of elevated PD-L1 expressed in tumors as an indicator for anti-PD-1/PD-L1 therapy. Rizvi said a review of checkpoint inhibitors found that elevated expression of PD-L1 on tumors correlated with greater likelihood of responding to anti-PD-L1 or anti-PD-1 therapies (Sunshine and Taube, 2015) (see Figure 3). Patients whose tumors do not show elevated expression of PD-1 and PD-L1 are less likely to respond to such therapies, but responses are seen in about 15 percent of those patients, Rizvi said. “How do we define what an acceptable cut-off is versus just giving the drug to everyone with that type of cancer?” he asked. An additional challenge with this biomarker is tumor heterogeneity; some portions of a tumor may be completely PD-L1 negative, while other areas may be strongly PD-L1 positive.
Several assays for PD-L1 are available, and could potentially help inform whether patients should receive PD-1 or PD-L1 inhibitors, or a combination of immunotherapies, Rizvi said. PD-L1 testing may also help distinguish tumor progression from pseudo-progression, with the latter being unlikely in patients who are strongly PD-L1 negative, he added. However, there is currently a lack of harmonization among all the different PD-L1 tests, each with different characteristics, assay scoring, and interpretation criteria (see the section on precompetitive collaboration for more information on PD-L1 Blueprint Project). In addition, core biopsies are not always available for PD-L1 testing, and there can be poor correlations between the PD-L1 status of biopsies versus surgical specimens, Rizvi said. Nonetheless, Kaufman said that “PD-L1 has had and may continue for a long time to have significant utility in terms of helping to triage patients receiving PD-1 directed monotherapy.” He added that most patients who respond to anti-PD-1 therapy have some degree of T-cell inflammation in their tumors at baseline that might serve as a biomarker predicting patients who are likely to respond. But he said some patients who have inflamed tumors do not respond to the treatment.
NOTE: H/N = head and neck cancer; Mel = melanoma; NSCLC = non-small cell lung cancer; RCC = renal cell carcinoma; y-axis = percentage of patients.
SOURCES: Rizvi presentation, February 29, 2016; Sunshine and Taube, 2015. Reprinted from Current Opinion in Pharmacology with permission from Elsevier.
Another proposed biomarker for identifying which patients are likely to respond to cancer immunotherapies is the mutation load of their tumors. Melanoma, lung, bladder, and other cancers that tend to have high rates of mutation (or mutation loads) are more likely to respond than prostate or thyroid cancers, which have low mutation loads (Alexandrov et al., 2013) (see Figure 4).
In addition, patients with more highly mutated melanomas, colon, or lung cancers are more likely to respond to anti-CTLA-4 or anti-PD-1 treatments than those with a lower mutation load in their tumors, Rizvi said (Alexandrov et al., 2013; Le et al., 2015; Rizvi et al., 2015). He added
NOTE: ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; CLL = chronic lymphocytic leukemia.
SOURCES: Rizvi presentation, February 29, 2016; Alexandrov et al., 2013. Reprinted with permission from Macmillan Publishers Ltd.
that mutation load combined with PD-L1 expression levels were especially robust in predicting benefit to anti-PD1 treatment in patients with lung cancer; 10 of the 11 patients (or 90 percent) who had some level of PD-L1 expression and highly mutated cancers had a durable treatment benefit (Rizvi et al., 2015). However, he said it is difficult to perform whole exome genetic sequencing of tumors in order to determine mutation load; he added that it might be possible to simplify tumor load analyses by counting the number of mutations in a targeted panel of a few hundred genes (Campesato et al., 2015). “This gives you a reasonable estimate of mutation load and can be done without doing whole exome sequencing,” Rizvi said.
Porter said an initial comparison between responders and nonresponders to CAR T-cell therapy examined patient-specific characteristics, such as age or prior therapy or genetic risk profile, but it did not discern any predictors of response. The only factor that seemed to distinguish between responders and nonresponders was that the former had rapid expansion of their T-cell in vitro (Melenhorst et al., 2010). Researchers continue to search for biomarkers for response to CAR T-cell therapy. With such biomarkers, “Not only can we select the most appropriate patients to benefit, but if we can determine why somebody may or may not respond, we may be able to alter that in the patients who previously have not responded,” Porter said.
Butterfield described a study indicating that levels of ICOS expression on CD8 and CD4 T-cells was linked to greater patient responses to
ipilimumab +/– GM-CSF therapy (Hodi et al., 2014). However, she said that this is a prognostic signal and not a validated biomarker ready to be used for prediction of patient response as part of the enrollment criteria for patients in clinical trials.
Biomarkers based on next-generation sequencing will be used to select patients for combination therapy, Kaufman said, and added that Merck is collaborating with NanoString to develop next-generation companion diagnostics. He said that for biomarkers to be useful across a broad array of combinations, they need to delineate something fundamental about the biology of the tumor—for example, if the inflamed T-cell is truly a dichotomy and not a spectrum, then that might be one potentially useful next-gen biomarker. “Probably we are going to need even more advanced tools as we start to subsegment the categories of resistance and really bring the right combinations to the right patients who are going to have unambiguous benefit, while minimizing both the clinical and financial toxicity of combinations for patients who are not going to benefit from those combinations.”
Kaufman also suggested that clinical trials be enriched with patients who have rare subsets of disease or different degrees of resistance to therapies, in order to better understand how immunotherapies perform in these patient subsets. He added that there is a need for real-time clinical genomics databases in order to identify patients who could be enrolled in clinical trials assessing immunotherapies.
Biomarker Research and Standardization
Several participants said that more standardization of biomarker assays is needed. Butterfield said that the Society for Immunotherapy of Cancer (SITC) recently convened a task force on immunotherapy biomarkers that is exploring the standardization and validation of immune monitoring assays; new developments in biomarker assays and technologies; how immune regulation and systemic modulation can be assessed with high-throughput approaches; and outcomes predictions based on baseline immunity and tumor immune environment. “We have [groups] of experts from industry, government, and academia working together to reevaluate the data and make recommendations,” she said.
There also is a need to standardize what biospecimens are analyzed for biomarkers that predict response to treatment, Butterfield said. “We do not necessarily have the right specimens [collected and stored] under standardized conditions. Everyone in this room has a slightly different immune
system, and if everyone’s peripheral blood monocytes were processed and banked slightly differently in each of our own laboratories, that is going to introduce noise that may block the identification of that immunotherapy biomarker,” she said. “We also need to know what we should be [storing] in order to find out if any of these new high-throughput immunoprofiling technologies are going to point us in the right direction,” Butterfield added.
“There is not a mechanism for [storing] a lot of different types of samples for unspecified future research to identify the biomarkers we need,” Butterfield said. She added that many tumor specimens collected in clinical trials are likely to be nonviable because they have not been prepared in the manner necessary to conduct functional assays, such as the ones needed for the personalized cancer immunotherapy that Rosenberg described. In addition, the core biopsies that are frequently preserved do not generate viable tumor cells. Different tests require different types of blood or tissue samples, and without both absolute counts and percentages of all types of immune cells, there can be misinterpretation of findings. For example, she said that tests may indicate that the number of regulatory T-cells decreased in the blood, but because the number of CD4+ T-cells also decreased, the regulatory T-cells still comprise the same percentage of T-cells.
Recognizing that there are a lot of different requirements for the different questions that can be addressed with biospecimens, Butterfield said that the International Society for Biological Therapy and SITC made a number of recommendations to improve biospecimen collection for immunotherapy development (see Butterfield et al., 2011). One of the recommendations was to store adequate biospecimens to answer current questions as well as future questions. She added that these should be collected and stored consistently by well-trained staff using standard operating procedures and full documentation of those procedures.
Mark Gorman, a patient advocate, asked if the research community has adopted standards for biomarkers or specimen collection, and Butterfield responded that most researchers conducting clinical trials seem to embrace such standardization and require standard operating procedures for biomarker assays. She added that in Europe, a program run by the Association for Cancer Immunotherapy supports the harmonization of immune monitoring tests and promotes the technical validation of in vitro assays to guide the development of innovative cancer immunotherapeutics on the basis of immunological outcomes (CIP, 2016). This initiative provides proficiency panels to investigators with samples they can test in their own laboratories to see how their results compare to the standards.
To support the development and standardization of patient response biomarkers for combination cancer immunotherapies, Kaufman suggested establishing an informatics infrastructure that is accessible to patients and community oncology practices to enable enrichment of trials with patients most likely to benefit. “We have to create an infrastructure where we can reach the patients who [are likely to benefit from therapies], and that requires really good communication with the community to enable evidence gathering. It requires not just physician engagement, but patient engagement and using some of the tools that are being developed in the informatics space that allow patients to interact with their data and with other patients and with the companies and academic centers running clinical trials,” he said.
When Khleif asked Kaufman how he would implement this concept, Kaufman said that there are information technology companies such as Flatiron, Foundation Medicine, Google, IBM, and PatientsLikeMe, “who bring pieces of this puzzle, but no one has put it all together in a way that is going to support what we need. There is both private interest and public interest in creating this space.” Kaufman also said that there is currently a convergence between immuno-oncology and conventional oncology in terms of understanding how oncogenic pathways drive the tumor immune microenvironment. “All the tools that have been developed for precision medicine and oncology are becoming relevant for immune-oncology,” he said. He suggested harnessing the unprecedented collaboration currently seen among drug companies and other stakeholders to improve immunotherapy development: “We need to bring a new corporate strategy to how we approach these issues, and it takes the input of a lot of other players in this space and it certainly takes different regulatory paradigms to support these types of changes.”
Butterfield suggested that there be more funding designated for biomarker studies because such studies are often not competitive within the large pool of NCI grants awarded for hypothesis testing. She added that exploratory biomarker research is often difficult to conduct within the National Clinical Trials Network (NCTN). One group, the ECOG-ACRIN Cancer Research Group, used to have five specialized laboratories, but Butterfield noted that current grant requirements only allow the support of a single laboratory: “one laboratory [will now have] to develop the expertise to handle all of the types of samples and do all of the banking appropriate for all of these different questions we need to ask.”
Appropriate Endpoints for Immunotherapies
Rajeshwari Sridhara, director of the division of biometrics at FDA’s Center for Drug Evaluation and Research, said that to date, FDA has based its accelerated approvals of immune modulator drugs on objective response rates and progression-free survival (PFS), whereas regular approvals have been mostly based on overall survival or PFS rates. Sridhara said traditional statistical analyses of PFS assume there is a constant treatment effect over time, which does not seem to be the case for most cancer immunotherapies because patients often have delayed responses to these treatments and may have pseudo-progression before tumor regression. She said that patient-reported outcomes are important, but they are disease dependent and have not been evaluated rigorously in cancer immunotherapies. In addition, overall response rates have been inconsistent from one cancer type to another, with melanoma patients tending to have greater responses than patients with non-small cell lung cancer, for example.
The effectiveness of cancer therapies is often assessed using the Response Evaluation Criteria In Solid Tumors (RECIST) criteria. These criteria are based on biomedical imaging results to assess changes in tumor size within a short period of time following treatment. Disease progression is assumed when tumors increase in size or if new tumors develop. Several workshop speakers said these criteria can be problematic for evaluating immunotherapies. For example, Berzofsky said it can take much longer to show a treatment effect from immunotherapy compared to conventional chemotherapy. “One cannot conclude that [an immunotherapy] has failed simply by seeing initial progress[ion] on therapy.” Berzofsky said. For example, mice treated with anti-CTLA-4 first experienced tumor growth prior to regression, said Jedd Wolchok, chief of Melanoma and Immunotherapeutics at Memorial Sloan Kettering Cancer Center (Leach et al., 1996). Wolchok compared this response to one of his patients treated with ipilimumab; the size of the tumor increased after 3 months of therapy, but then underwent a significant regression between weeks 12 and 20 (Wolchok et al., 2008). In another example, Wolchok said a patient with skin metastases that were clearly progressing at week 12 subsequently underwent complete regression. “We learned you cannot depend upon empiric time points in order to determine response. This is really important in managing expectations in patients and their families and physician colleagues, who may be calling and asking for advice as to when to change therapy,” he said.
Wolchok categorized four patterns of clinical responses to ipilimumab that investigators have seen, all of which have been linked to favorable survival: (1) shrinkage in baseline lesions, without new lesions; (2) durable stable disease (in some patients, followed by a slow, steady decline in total tumor burden); (3) response after an increase in total tumor burden; and (4) response in the presence of new lesions (Wolchok et al., 2009). The latter two novel responses make it difficult to use RECIST criteria for research purposes, especially in regard to study endpoints. “This makes the data from clinical trials really complex unless you use overall survival or landmark survival as an endpoint,” Wolchok said.
After conducting an analysis of Phase II trials of ipilimumab in melanoma patients, Wolchok found that 10 to 20 percent of patients showed tumor progression on their first set of scans, but subsequently showed a response without any additional therapy, which led to the development of immune-related response criteria as an alternative to RECIST (Wolchok et al., 2009) (see Table 1).
These response criteria try to incorporate the different response kinetics seen with checkpoint inhibitor therapies by suggesting that patients have confirmatory scans for progression at a later point if they are not symptomatically progressing. In addition, Wolchok said the appearance of new lesions can no longer be seen as a definitive indication for progression, and instead there needs to be consideration of the change in total tumor burden. “We have seen that long-term outcomes can be pretty accurately predicted with the use of these atypical response kinetic guidelines,” Wolchok said. Approximately 5 to 10 percent of patients who receive anti-PD-1 therapies also have atypical responses similar to those seen with anti-CTLA-4 therapies, Wolchok said (Wolchok et al., 2015). However, Berzofsky noted that some physicians do not yet accept the use of the immune-related response criteria, compared to the conventional RECIST criteria.
From a regulatory standpoint, Sridhara added that the challenge going forward will be determining what the primary and intermediate endpoints for immunotherapy clinical trials should be. “Overall survival is certainly a clear winner for a primary endpoint, but this may not be feasible,” she said, nor is PFS optimal, given a lack of a constant treatment effect over time. Wolchok said he originally envisioned overall survival as the primary endpoint for cancer, but acknowledged that waiting the long time periods needed to determine overall survival may not be practical, because fortunately some patients are living so much longer with these therapies. He suggested that a greater mechanistic understanding of pseudo-progressions
TABLE 1 Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Comparison Between RECIST Criteria and the Immune-Related Response Criteria
| RECIST Response Criteria | Immune-Related Response Criteria (irRC) | |
|---|---|---|
| New measurable lesions (≥5 × 5 mm) | Always represent PD | Incorporated into tumor burden |
| New non-measurable lesions (<5 × 5 mm) | Always represent PD | Do not define progression (but preclude irRC) |
| Non-target lesions | Contribute to defining BOR of CR, PR, SD, and PD | Contribute to defining irRC (complete disappearance required) |
| Complete response (CR) | Disappearance of all target lesions | Complete disappearance of all lesions (whether measurable or not, and no new lesions) |
| Partial response (PR) | ≥30 percent decrease in sum of the LD of target lesions, taking as reference the baseline sum LD | ≥50 percent decrease in tumor burden relative to baseline |
| Progressive disease (PD) | ≥20 percent increase in sum of LD target lesions, taking as reference the smallest sum LD recorded since treatment start or the appearance of one or more new lesions | ≥25 percent increase in tumor burden relative to nadir (minimum recorded tumor burden) |
| Stable disease (SD) | Neither sufficient shrinkage to qualify as PR nor sufficient increase to qualify for PD, taking as reference the smallest sum LD since treatment start | Not meeting criteria for CR or PR, in absence of PD |
NOTE: BOR = best overall response; CR = complete response; LD = longest diameter; PD = progressive disease; PR = partial response; RECIST = Response Evaluation Criteria In Solid Tumors; SD = stable disease.
SOURCES: Berzofsky presentation, February 29, 2016; Wolchok et al., 2009. Reprinted by permission from the American Association for Cancer Research.
would substantiate the importance of alternative surrogate endpoints in immunotherapy clinical trials. Wolchok said that magnetic resonance imaging (MRI) might be able to distinguish between true tumor growth due to progression and increasing tumor size due to immune infiltration. He added that positron emission tomography (PET) imaging might also provide a better mechanistic understanding of immune cell trafficking to tumors that could provide additional support for immune response criteria. “Once we can put a bit more data behind the observations, this will come more easily,” Wolchok said.
Some RECIST criteria may still be appropriate if progression is defined properly, Sridhara said. “Maybe we are defining progression wrong and for immunotherapy we should be looking at confirmed progression—that might alleviate some problems,” she said. Sridhara also suggested that alternative intermediate surrogate endpoints be explored, given the inconsistency of response rates as well as alternative ways of determining PFS.
Lee Schwartzberg, division chief of hematology and oncology at the University of Tennessee, added that surrogate markers of pseudo-progression are needed, especially to help inform decisions about whether to continue therapy. “We need to establish new technologies to detect pseudo-progression as soon as possible,” he said. Wolchok added that his colleagues are currently assessing the usefulness of a blood test (referred to as a liquid biopsy) to detect mutated BRAF DNA in circulation as a marker for melanoma patients. “If the tumors appear larger, but the circulating BRAF is dropping, then that should tell you something,” he said.
Lee Krug, Disease Area Head, Lung and Head & Neck Cancer, immuno-oncology at Bristol-Myers Squibb (BMS), said the percentage of patients who experience pseudo-progression when given immunotherapies may vary by cancer type; it may be more common in melanoma or kidney cancer, but less common for patients with lung cancer. That distinction is important, given that delaying another treatment for patients with lung cancer because there is a slight chance they might be experiencing a pseudo-progression may result in a decline in their performance status so that they are no longer eligible to receive other therapies. “There should be at least a little bit of caution about suggesting that this concept of pseudo-progression is really widespread, and we need to really work hard to try to identify if there is true tumor growth,” he said. Krug added that how patients are feeling can be another indicator of whether their tumor is progressing. “If they feel dramatically improved symptomatically, then that would be a good
indicator that maybe the scans do not really reflect what is going on with the disease,” he said.
Kaufman said that Merck is evaluating an immunotherapy for patients with colorectal cancer, and thus far, radiographic responses have been relatively slow, with patients feeling better much earlier than the changes showed on the imaging. “It is a small number of patients at this point, but it appears there may be different patterns in different disease states and we still need to flesh that out.”
Thomas added that it is hard to collect reliable data about how patients are feeling, so regulators are inclined to give more weight to a hard endpoint such as imaging. She said that often data on overall survival or PFS might show an advantage for a treatment, but there may not be patient quality-of-life data to describe how patients are feeling, so assessing patient-reported outcomes (PROs) with immunotherapy is important.
Lee Newcomer, senior vice president of oncology, genetics, and women’s health at UnitedHealthcare, asked whether improvements in symptoms is enough of a signal to suggest waiting a longer period of time before searching for imaging evidence of tumor regression. Wolchok said it is infrequent that imaging does not reflect how a patient is feeling, but added that it is “very unusual to see imaging [completed] with any decision attached to it before 12 weeks. . . . In the context of the life expectancies of some of these patients, [that time period] is unfortunately a reasonable fraction of that. We do need the imaging, but we need to be careful about very early time points. That is where we can see a higher rate of discordance between potential response and enlargement of lesions on imaging,” Wolchok said. He suggested that patients in clinical trials be offered the possibility of continuing on an immunotherapy beyond the point when progression is detected by imaging, as long as their performance status is maintained, but added “we are all learning about this together.”
Ibrahim said the FDA guidance document for vaccine development acknowledges the need for new response criteria and suggests that the studies with immunotherapies should have an exploratory endpoint that tries to characterize the immune response rate observed. Wolchok said that researchers can start validating the immune response criteria using data from exploratory analyses. “We have to standardize our definition of an immune responder and generate data that will be convincing to regulators that this is an endpoint that can be used in clinical trials when we are assessing the clinical activity. We have advanced our understanding
to the point where we have to standardize and validate those endpoints,” he said.
Determining Dose, Toxicity, and Follow-Up Time
There are also challenges in determining the appropriate dose and expected toxicity for immunotherapy clinical trials, especially for vaccines and cell-based immunotherapies in which toxicity does not appear to be dose related. Simon said a literature review of a large number of vaccine clinical trials found that grade 3 or 4 toxicities were exceedingly low—generally less than 1 percent of patients or less than a tenth of 1 percent of vaccine administrations. In those patients who did experience grade 3 or 4 toxicities, there was little evidence that it was dose related, Simon added (Rahma et al., 2014).
Porter said that there was also no obvious dose−response effect in a dose−optimization study of a CAR T-cell therapy, although a few more patients who received the higher dose experienced a tumor response than those who received the lower dose. He added that responses were more likely to depend on the degree of T-cell proliferation that occurred when CAR T-cell therapy was administered to patients, rather than the quantity of T-cells that were initially administered. “It has more to do with how many cells you end up with rather than how many cells you put in, so we really did not expect to see a dramatic dose−response effect,” Porter said. He also said that there was not a dose−toxicity relationship in that study; however, the patients who responded to the therapy developed a lack of B-cells. Because the treatment targets B-cells, this finding was not surprising and he said it is an indicator that the treatment was working. Another indication of the treatment response was that patients developed tumor lysis syndrome (although this was found to be delayed, reversible, and manageable)8: “While this is a measure of toxicity, it is also a testament to the potency of these cells,” Porter said.
In addition, nearly all responding patients also developed cytokine release syndrome that could be countered with anti-IL-6 therapy. “Although we have a good handle on how to intervene for this cytokine release syn-
___________________
8 “A condition that can occur after treatment of a fast-growing cancer, especially certain leukemias and lymphomas. . . . As tumor cells die, they break apart and release their contents into the blood. This causes a change in certain chemicals in the blood, which may cause damage to organs, including the kidneys, heart, and liver.” (NCI, 2016)
drome, we do not know exactly when to intervene. There is some fear that if we abrogate this response too early, we may prevent the antitumor response,” Porter said.
“The toxicity with this living drug is unique because cells expand, so the dose administered is very different than the final dose these patients see, and the cells persist. This drug may continue to be active for years after initial therapy. The safety and toxicity grades that we are all used to thinking of were not designed for immunotherapies and need to be redesigned,” Porter said. To evaluate the CAR T-cell therapy, Porter said that he and his colleagues had to design their own novel toxicity grading criteria (Porter et al., 2015)
Bross agreed that is challenging to determine the appropriate doses of cell-based therapies because “these cells are expanding in the patients and last forever.” He added that although these therapies can result in a cure for some patients, they can also cause serious and sometimes fatal toxicities. Furthermore, he said the long-term effects of genetically modified cells that induce a prolonged lack of B-cells is unknown.
Determining the off-target effects of treatments that use a patients’ own cells is also challenging, Bross added. “How do you determine whether it is off-target activity when the target is different in every case?” he asked. It is also difficult to determine the appropriate follow-up time, with several participants suggesting that longer follow-up times are needed to assess the toxicities and responses to immunotherapies because these often occur later than they do for standard chemotherapy. “Beyond treatment, follow-up is necessary and we should think about making sure that this is included in trial designs,” Sridhara said. She added that there should also be critical consideration of the duration of treatment.
Quality Control
There are numerous challenges to ensuring quality control of personalized cell-based immunotherapies, Bross said. Given that each of these products is unique, it can be difficult to determine consistency and potency, as well as proper oversight of their stability, transportation, and tracking so that the right patient receives the right product. There are also manufacturing challenges, such as ensuring identity comparability and sterility of products that can affect the statistical interpretation of clinical trials, Bross said. But perhaps the ultimate challenge, he suggested, is “How are we supposed to regulate a product that is different for every patient?”
Testing Combinations
Khleif said that when cancer patients receive immunotherapy as a monotherapy, only a subset of patients experience a tumor response, and some patients experience recurrences after an initial response. Some preclinical and clinical studies of combination immunotherapies have shown better and more durable response rates, suggesting “combination is going to be the name of the game for immunotherapies,” said Khleif. In 2007, a study identified 20 potential immunotherapies that could be evaluated for combination cancer therapy; Khleif said that the number of potential immunotherapies that could be assessed in combination today is probably three times as many (NCI, 2007). These agents could be tested not only in combination with each other, but in combination with chemotherapies, targeted cancer therapies, or radiation therapies. “One statistician calculated the possibility of putting together all of these in combination and calculated it would take 300 years to clinically test them,” Khleif said. “What are the clinical trial designs that would be necessary to move this field further so we could do it in 10 or 15 years rather than waiting 300 years?” he asked. Simon added, “It is likely that to really improve [cancer treatment], we will have [therapies] with even more components than what we are seeing today—numerous candidate immunomodulating agents—and they will need to be evaluated in combinations with current regimens.”
Helms said FDA traditionally is asked to review drug combination therapies in which there has been abundant clinical experience using each agent in the combination singly. For these regimens, combination toxicology studies are not usually required. Instead, the drugs are tested clinically in combination by initially reducing their doses and proceeding based on the clinical experience. For example, a sponsor who was developing a combination therapy that included a kinase inhibitor and an anti-PD1 antibody was not required to submit pharmacology or toxicology studies on the combination, because there was already significant clinical experience with both products used in the combination regimen. Because previous preclinical toxicology findings in animals showed that the kinase inhibitor could cause a serious inflammation of the heart (although this toxicity had not been seen clinically), FDA was concerned that its use in combination with a checkpoint inhibitor might increase the likelihood of this occurrence, or exacerbate this effect. Consequently, FDA requested that the sponsor lower the starting dose of the kinase inhibitor to approximately 20 percent of the dose causing the cardiac inflammation in the preclinical studies.
If there is no clinical experience with either product used in a combination, then a combination toxicology study might be warranted, Helms said. In cases where there is limited clinical experience with one or both products, then combination pharmacology studies are often recommended, including in vitro and rodent studies. For example, FDA placed a clinical hold on a sponsor who wanted to conduct clinical tests of a combination regimen in which there was no clinical information for either agent in the regimen. FDA asked for in vitro pharmacology studies to help determine a reasonable combination dose to be used in the combination therapy study. The hold was lifted and clinical tests of the combination regimen proceeded after the sponsor completed clinical dosing studies for each of the monotherapies in at least one patient cohort. Occasionally, a sponsor will add a combination arm to one of their general toxicology studies of a new agent.
Clinical trials of combination cancer immunotherapies are complex because investigators need to consider which agents to combine and how to sequence these agents to maximize efficacy and minimize toxicity, several speakers said. When the immune stimulant OX40 was given concurrently with anti-PD1 treatment in mice, the animals did not survive as long as they did when given OX40 alone, Fox said. But when the researchers delayed anti-PD1 treatment a day after OX40 treatment, there was a synergistic effect—the mice lived much longer than when they received the treatments simultaneously, or either treatment singly. “Adding the dimension of time makes designing clinical trials much more difficult because not only do you have to test every possible combination, but you have to figure out the right order and how far apart they need to be spaced,” Berzofsky said. Sridhara added that there are challenges involved in labeling sequenced combination therapies. “You have a new product followed by another product and another product. What do you put on the labels of those drugs?” she asked.
Simon suggested that there should be more computational systems biology modeling of T-cell effectors and their relationship to tumors and the immune system that can help assess which combinations of immunotherapies should be tested in patients. “We are beset with more candidates and more candidate combinations than we can test empirically, so we need something to go along with all of these empirical studies,” he said. One idea is to use available data to “build a model as we go along that will help us think through issues of timing and how this very complicated system works together,” he said.
Simon also said there are access and licensing challenges in testing combination therapies and suggested drug companies make immune
modulating agents available to academics for clinical discovery studies on novel combinations. He said that acquiring agents from different companies can be a long, drawn-out process. “The difficulty of having different immune modulating agents from different companies is one of the serious roadblocks that somehow needs to be tackled if we are really going to make maximum progress,” Simon added. Reiterating that clinical trials to evaluate combination immunotherapies can be complex, Sridhara said improved collaboration and more resources are necessary to conduct these studies.
Innovations in Clinical Trial Designs
Individual workshop participants discussed innovative trial designs—including patient enrichment and adaptive designs—as opportunities to improve the clinical development and evaluation of cancer immunotherapies. Changing the regulatory paradigm to better support innovative clinical trial designs and registration pathways could also be helpful, Kaufman said. Simon suggested that a clinical trial could be designed to include a brief treatment period followed by an assessment of an intermediary biomarker, such as interferon gamma levels or T-cell quantity. This information could be used to enrich a clinical trial with likely responders to immunotherapies, based on the biomarker data, because patients who are less likely to experience a tumor response would be taken off the trial. Patients who respond would be randomized to either continue or discontinue immunotherapy treatment. “You essentially are using a post-treatment marker as a predictive biomarker,” he said, which works in situations where there are known predictive biomarkers for a treatment. When such predictive markers are not known, he suggested treating patients for a short period of time and then measuring several candidate biomarkers, whose predictive abilities could be evaluated at the end of the clinical trial. Patients would be randomized to continue or discontinue the treatment independent of their biomarker results, so that subset analyses could be performed with enough statistical power, despite small sample sizes, Simon said.
He also recommended defining upfront the duration of therapy in clinical trials, independently of progression of disease for the treatment arm containing the immunotherapy, to overcome the challenge of pseudo-progression in analyses. For example, clinical endpoints for Phase IIb trials could be response status after 6 months of treatment, and the clinical endpoints for Phase III studies could be overall survival, he said.
Simon proposed a new Phase I design for a cancer vaccine in which the Phase II starting dose is the minimal active dose of the vaccine and its adjuvant. To determine that dose, one patient per tested dose is treated until an immune response is induced. Then the dose level is increased, one patient at a time, until achieving an additional immune response. If no additional immune response is achieved in seven patients, researchers stop adding patients and continue to escalate the dose in one patient at a time (Rahma et al., 2014). For vaccines used in combination with immunomodulator drugs, Simon suggested starting off with a biologically active dose of the vaccine and its adjuvant in combination with the immunomodulator, and any dose modification that is made be done using the vaccine in combination and not as a single agent. “Finding the optimal dose as a single agent will not get you where you want to go,” he said. Bross added that “Everybody wants to combine their favorite checkpoint inhibitor with a cancer vaccine du jour and even if preclinical tests are relevant, we take it on a case-by-case basis.”
Simon proposed that instead of conducting traditional large clinical studies of combination immunotherapies, researchers should first conduct smaller clinical pilot studies, which he called discovery Phase II studies. He said that large treatment effects can be detected with smaller patient sample sizes and cautioned against having what he called a Type III error—the error made by not studying an intervention. He said there is an opportunity cost in doing a large clinical trial on just one combination, because those same funds could be allocated to smaller trials assessing a variety of combinations. Simon showed several examples of study designs for Phase II clinical trials of combination therapies. One is to first treat patients with a single immunotherapy, and then for those patients who do not respond, investigators could provide an additional immune modulator. Alternatively, patients who do not initially respond to an immunotherapy, such as an anti-PD1 checkpoint inhibitor, could be randomized into three different treatment arms, each with the anti-PD1 treatment combined with a different immune modulator. An advantage of such a trial is that the combination treatments are more comparable than if they were tested in separate trials, although it is more complicated to run a three-armed study, Simon added.
Simon also suggested a factorial study design of a treatment used singly or with one or two additional treatments in all possible combinations, that is, an anti-PD1 treatment versus an anti-PD1 treatment plus X, versus an anti-PD1 treatment plus Y, versus an anti-PD1 treatment plus X plus Y. Although this would be a four-armed study, it could be analyzed as separate
two group comparisons: anti-PD1 treatment plus or minus Y versus antiPD1 treatment plus or minus X. “If you want to evaluate X and its contribution to the anti-PD1 antibody, you compare the two arms containing X, some of which contained Y and some of which do not, to the two arms that do not contain X, some of which are anti-PD1 alone and some that are anti-PD1 with Y,” Simon added. Such a design is often not pursued when testing combination chemotherapies that are highly toxic because the dose of X precludes delivering the full dose of Y and vice versa. But this design may be possible for testing combination immunotherapies if they do not cause dose-limiting toxicity in this context.
The Phase II discovery studies of combination therapies are not intended to be used for regulatory approval, Simon said. “These are small screening designs that may not be appropriate in a regulatory context, but would tell us what we want to study more fully,” he said. Bruce Chabner, director of clinical research at the Cancer Center at Massachusetts General Hospital, noted that small discovery Phase II studies and even Phase I studies have led to accelerated approvals of individual checkpoint inhibitors because they generated such high response rates and durable responses. “It all depends on whether you can select patients appropriately for the trial and have a very high response rate,” Chabner said, adding that discovery trials for combination therapies could quickly become trials used to garner drug approvals if the right combinations go to the right patients. “The problem now with immunotherapy is the inability to select patients, but once we get to that point, things could change dramatically,” Chabner said. Simon responded, “Vince DeVita [a former NCI director] used to say that the best prognostic factor is treatment, and what he meant by that was what we really want to do is improve outcome, and I personally think that is as important today as finding the correct predictive biomarker.”
Ibrahim noted that “the way we are designing and running clinical trials right now is cost prohibitive,” with late-phase trials of immunotherapies having as many as 2,000 patients and as many as six arms. The large size of these Phase III combination studies is due in part to assessing many patient response biomarkers and combination immunotherapies, and also partly due to inadequate prior assessment of the monotherapies used in the combination. He suggested conducting more adaptive trials in which various combination treatment arms can be dropped if an initial small cohort of patients does not respond to them. These patients can then be put on a different arm of the study. “We need to pause and think about what we
can do before we reach Phase III studies so we do not end up with six-arm randomized Phase III studies,” he said.
Singh described a Phase I clinical trial supported by a European Union consortium that is testing a patient-specific vaccine in 20 patients with glioblastoma. To personalize the vaccine, researchers conduct an analysis of each patient’s tumor antigens and assess whether any of 71 premanufactured peptide fragments that are commonly found in glioblastoma are prevalent in the patient’s tumor and stimulate an immune response. They also use next-generation sequencing to identify neoantigens specific to each patient’s tumor. Following these steps, a patient is vaccinated with up to 10 of the premanufactured peptides within 3 months of surgery and 2 peptides derived from the patient-specific tumor antigens within 6 months of surgery (see Figure 5). “Each patient is receiving his or her individual peptide vaccine cocktail,” Singh said.
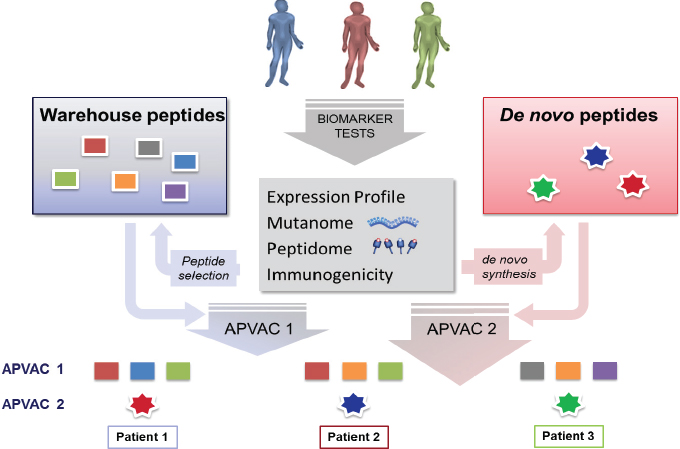
NOTES: Warehouse peptides = premanufactured peptides commonly found in glioblastoma. De novo peptides are created from next generation sequencing-identified neoantigens. APVAC1 = premanufactured peptide-based vaccine; APVAC2 = neoantigen-based vaccine.
SOURCES: Singh presentation, February 29, 2016; Immatics Biotechnologies.
This approach can also be used for other immunotherapies, and is currently being applied to a clinical trial evaluating a T-cell immunotherapy for patients with solid tumors, Singh said. Each patient will receive an adoptive cellular therapy based on the subset of his/her own T-cells that target up to 4 of 10 different tumor antigens.
Porter said that in the pilot clinical trial of CAR T-cell therapy, they did not start with a traditional Phase I dose escalation study “because this is a living drug and while we knew a reasonable dose to start with from some animal model studies, we did not really think a traditional Phase I study was appropriate.” However, the researchers conducted a follow-up study—essentially a randomized Phase II dose-optimization trial—that tested two dose levels, both of which had induced complete remissions in the pilot trial.
Most new immunotherapies for cancer have entered the market via the accelerated approval pathway based on objective response rates, Sridhara said (see Box 2 for a summary of the different FDA approval pathways and designations and Box 3 for an overview of European regulation of drugs and biologics). She added that the large response rates seen with recently approved immunotherapies, such as checkpoint inhibitors, raises the bar on what can be considered for accelerated approval or given a breakthrough therapy designation; new therapies will now have to show improvement over the response rates of therapies already on the market and not of traditional chemotherapies. “We could be reaching a ceiling effect where response rates are so high that unless you find the right biomarker and the right patient subgroup, you may not be able to substantially surpass it,” Sridhara said.
Marc Theoret, lead medical officer in the Division of Oncology Products at FDA, asked Thomas to describe the difference between FDA’s accelerated approval pathway versus adaptive licensing. Thomas responded that with FDA’s accelerated approval, a license continues under the condition that confirmatory data are provided within a certain time frame, at which point the license may be revoked. With adaptive licensing, early in drug testing there are meetings with stakeholders to determine the best trial designs and in which patient populations a drug should be tested. A drug that is approved for adaptive licensing is considered licensed, but it can have a very restricted indication, such as a third-line therapy indication. “But if it is used and shows its worth and safety profile with the extra information being favorable, then the indication may roll out to second or even first-line. That is where the adaptive licensing should be helpful.” Because FDA’s
breakthrough therapy designation enables intensive guidance during the drug testing stage, Theoret added that it is somewhat similar to the European Medicines Agency adaptive licensing approach.
COLLABORATION AND INFORMATION EXCHANGE
Several speakers discussed how clinical data collection, sharing, and analysis—enabled by EHRs, data warehouses, and data analytic approaches—can help advance progress in immunotherapy development
and clinical implementation. Individual workshop participants also discussed opportunities for greater precompetitive collaborations to foster the exchange of information and development of tools that can benefit all stakeholders.
Clinical Practice Data Collection, Sharing, and Analysis
Eric Perakslis, senior vice president of informatics at Takeda Pharmaceuticals, said that faster and cheaper computations are fueling a new data paradigm in health. He added that there are many readily available sources of data, new technological innovations under development, and the potential to answer research and clinical care questions in real time. A patient’s health information can be stored in a variety of places: community health care settings, academic and government medical centers, registries of clinical trials, omics-based databases such as The Cancer Genome Atlas, as well as in patient-powered databases like PatientsLikeMe9 and PCORnet.10 “Today there is so much data out there that you could use. What people can now do with all these data is getting more impressive and interesting every day,” Perakslis said. He encouraged researchers and clinicians to take advantage of these technological innovations to rapidly assess large sets of information.
Perakslis and Amy Abernethy, chief medical officer and senior vice president of oncology at Flatiron Health, said that data warehouses—central repositories of integrated data from multiple sources—enable new opportunities for research. Data warehouses can link patient information from processed EHR data to other datasets, including immunology, mortality, genomic, health insurance claims, and patient-reported outcomes data. By leveraging these linkages, EHRs can “start to serve a critical purpose of creating an information backbone for our future,” Abernethy said.
Analysis of Clinical Practice Data
Information in EHRs includes both structured and unstructured data, Abernethy said (see Figure 6). Structured data, such as demographic information and diagnostic testing results, can be easily digitized in a structured format. However, even structured information in EHRs can be “sloppy,” if
___________________
9 See https://www.patientslikeme.com (accessed May 18, 2016).
10 See http://www.pcornet.org (accessed May 18, 2016).
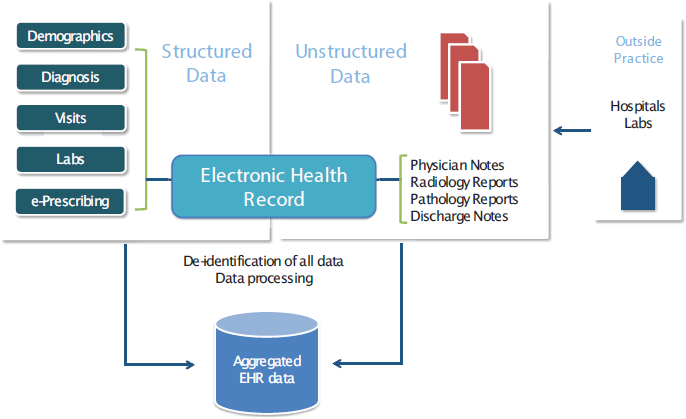
SOURCES: Abernethy presentation, February 29, 2016; Flatiron Health.
it uses nonspecific and nonstandardized terms. For example, a diagnostic test report may not clearly indicate whether an albumin test is serum albumin or albumin derived from urine. Other critical information, such as the genetic mutation status of a patient’s tumor, is often buried in the verbiage of laboratory reports and difficult to extract, she said.
Most information in EHRs exists as unstructured data (e.g., clinician notes, radiology reports, or pathology reports) that have to be processed before they can be merged with other data in a database and analyzed, Abernethy said. Each speaker—Abernethy, Perakslis, and Mary Horowitz, chief of the division of hematology and oncology at the Medical College of Wisconsin—said that current technologies are insufficient to translate unstructured data into quantitative variables. While technology is facilitating the translation of unstructured data, Horowitz noted “You have to have people who understand the disease and the patients” to accurately translate this information. Perakslis added that collaborations among individuals who understand the technologies and those who have expertise in cancer are critical.
Abernethy said that although EHRs may have a plethora of information, there are “only a series of critical variables that are going to be needed for the overall dataset.” For example, a clinician’s assessment that a patient’s
disease has progressed can be distilled into a number of anatomic variables that are consistent with progression. This assessment requires understanding the clinical activity and how that translates into variables: “There is going to be an entirely new vocabulary about how people think about variables that will come from abstracted data,” she said.
Horwitz said that a major challenge in using EHRs in research is the variability in data completeness and accuracy. She said estimates for EHR data incompleteness range from 24–86 percent, estimates of errors in computerized provider order entry are 51–91 percent, and other data inaccuracies and errors are estimated at approximately 4 percent (Balas, 2015). “Someone has to note [in the EHR], for example, that 30 percent of the skin was involved with a graft-versus-host disease,” she added. Administrative claims data can present similar challenges, Horowitz said. She and her colleagues used a claims database that compiled information from more than 115 million patients and more than 100 payers to compare the 1-year costs of hematopoietic cell transplantation (HCT) or chemotherapy treatment in patients with acute myeloid leukemia. Although researchers could identify more than 29,000 patients with this diagnosis, data from only 985 patients (3 percent) were considered sufficiently reliable for inclusion, based on the researchers’ confidence in the diagnosis and treatment that patients received. Perakslis said that data completeness will be a persistent challenge, in part because data standards are continually evolving. “No matter what, [EHRs are] never complete and that is okay—it just means you cannot rely on them for everything. You are always going to have to be creative and say, ‘these are the standards that I have today and I am going to use them to solve the problem I have. Tomorrow they will be better, but I am not going to wait.’”
Abernethy said that at Flatiron, there is an audit trail for all data entered, including the date, time, and person who entered the data. Flatiron also provides indicators of the quality of all data entered, and how likely the variable entered approximates reality. A quality rating assigned to each data variable is based on completeness, accuracy of the data abstractor at the last testing, and agreement among data abstractors on the variable. For example, there is greater agreement on entries for bone metastases, which are easier to extract from reports, than there is for lung metastases, which can be confused with lung infiltrates. She added that researchers need to understand how variables perform and the degrees of confidence associated with specific variables in order to draw appropriate conclusions. “You should be able to understand every variable in your dataset with that kind of quality and that is what datasets going forward are going to look like,” Abernethy said.
“We need to recognize that unstructured data processing is costly, and that is something we are going to have to deal with. It is kind of like oil—it has to be refined to be useful,” Abernethy said. “We need a workforce that is facile in dealing with these datasets and doctors who are ready to be a part of this in the future,” Abernethy said. Horwitz added that “If we are going to rely on providers to give us the information we [need], we have to make it easy for them.” She suggested that all clinicians receive improved education regarding the importance of clinical documentation in EHRs. In addition, individuals who were previously involved in transferring records manually could be employed to help ensure that all necessary data are entered accurately when medical records are transferred electronically.
Challenges and Opportunities for Clinical Data Aggregation
Once EHR data are structured, they can be used “to populate registries of the future,” Abernethy said. She described a registry as an “an aggregation of single patient stories pulled together in order to create the overarching story,” and suggested that datasets are organized around these patient stories (see Figure 7). “Ultimately you get up-to-date datasets that are EHR based, organized, and ready for use,” Abernethy said. Being up to date is critical given the rapidly changing landscape of cancer immunotherapies, she said. For example, nationwide PD-L1 checkpoint inhibitors made up only about 1 percent of all treatments for patients with non-small cell lung cancer in November 2014. By December 2015, these therapies represented 22 percent of treatment. “We need real-time datasets that show us what the overall story looks like in order to inform our future,” she said.
Kaufman asked Abernethy how information from patients who are treated in the community can be used by researchers to inform enrichment studies aimed at understanding the biology of patients who are likely to benefit from specific treatments. Abernethy suggested integrating currently available clinical genomics registries with processed EHR data. She added that EHR data could also be combined with prospective data capture, and noted that that several organizations are working to develop such capabilities in their data warehouses. Greater access to biospecimens collected from patients as part of routine care could also be leveraged: “We need the whole community coming together for this—the scientists, oncologists, patients, and tech community have to try to coordinate this in a thoughtful way,” she said.
Linking practice-based clinical and claims data can also inform clini-
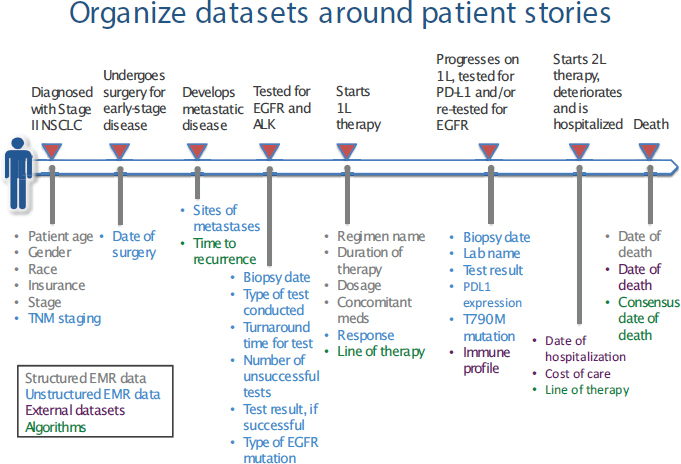
NOTES: ALK = anaplastic lymphoma kinase; EGFR = epidermal growth factor receptor; NSCLC = non-small cell lung cancer.
SOURCES: Abernethy presentation, February 29, 2016; Flatiron Health.
cians and patients about the effectiveness and costs of different cancer therapies, Newcomer said. UnitedHealthcare uses an online prior authorization process, in which practices input patient information in order to display National Comprehensive Cancer Network–recommended treatments. Recently UnitedHealthcare linked clinical and claims data in this program, and beginning in 2017, clinicians will be able to see the performance data of each regimen in specific patient populations. Newcomer said the database is live and continually updated, and UnitedHealthcare plans to make the data available to researchers and other interested parties outside of its system. “It is my hope that the data will begin to influence selection of regimens . . . [when] patients and clinicians could start to see how well some of these drugs perform in situations exactly like theirs,” Newcomer said.
There are a number of challenges to integrating and analyzing clinical data from multiple sources for use in immunotherapy research, said Abernethy, Horwitz, and Perakslis. Some of the challenges discussed
included a lack of interoperability of health care information systems, fragmentation in health care services and the long duration of follow-up needed to assess immunotherapies, infrastructure and analysis costs, and concerns about patient data protections.
Critical to aggregating patient data is interoperability, which Perakslis defined as “the ability of different information technology systems and software applications to communicate, exchange data, and use the information that has been exchanged” (Healthcare Information and Management Systems Society, 2013). Interoperability can be viewed as a linguistics problem, Perakslis said, but added that it is broader than considering the language of different datasets and computer programs that have to be harmonized. Abernethy said that policy makers should demand interoperability: “We should be able to move our documents back and forth in order to take care of our patients every day,” she said. Horowitz suggested that meaningful use requirements for EHRs include the ability to export data for research in order to be reimbursed by the Centers for Medicare & Medicaid Services (CMS).
The fragmented health care system in the United States makes it difficult to locate all the data needed, Horowitz noted. “Patients move from one provider to the next without assurance that the data flows with them,” she said. For example, she reviewed quality of care data derived from health claims that suggested a high proportion of women treated at her hospital did not have a needle biopsy prior to surgery for breast cancer, which is a required procedure. When Horowitz’s team reviewed patient records, they found that all of these women had a biopsy, but in a different health care system before referral to surgery. Horowitz suggested that there should be a mechanism to conduct long-term follow-up of patients, especially for cellular immunotherapies that persist in the body for long durations. But she added that the fragmentation of the health care system makes such long-term follow-up difficult. For example, when she was asked to review the records of a patient who had died, she found that the patient was still registered as being alive because he had died in a different health care system. She added that cancer mortality rates at a specific site can appear exceptionally low because they do not capture mortality data from other sites. “Follow-up is really hard and with these cellular immune therapies, we are going to have to do 15 years of follow-up, which is going to require innovation in how we maintain patient contact,” Horowitz said. For example, if a patient received CAR T-cell therapy at one institution after another immunotherapy failed at a different institution, and then went on to have a bone marrow trans-
plant at a third institution, “How do we integrate all that data? Who reports what to where?” she asked.
Horowitz said that integrating and analyzing data from multiple datasets can be expensive and time consuming. As an example, she described the bone marrow transplant database AGNIS (A Growable Network Information Service), which is an open-source messaging system designed to exchange patient blood and bone marrow transplant data using a secure, standards-based system (CIBMTR, 2016). A goal of AGNIS was to link data from transplant centers in the United States, the Asia-Pacific transplant registry, the European transplant registry, the umbilical cord blood transplant registry, and cord blood banks, said Horowitz. While AGNIS has had a number of successes, she said that establishing the data linkages has been challenging, expensive, and slow; for example, it took 12 years for the European registry to enter their data into the electronic network. Abernethy added that it is critical to develop the business models to support these mechanisms for data linkage. Horowitz said that “once you make it easier to take clinical data and make it available for research, it frees up your resources to add in other things. The easier you can make it to transfer the data from point of care into a research database, the more things you can think about doing.”
Data integration and sharing also requires consideration of data confidentiality and security, and patient consent. Abernethy recommended exploring new approaches to data security and confidentiality, including data-clean zones, to prevent or mitigate the potential for reidentification of de-identified patient data. Horowitz added that researchers need to ensure that they receive appropriate patient consent for biospecimen research. Gorman said that patients usually want to share their data and are frustrated when they cannot. “Patients have the perspective that these data are about them and belong to them and, therefore, they should have the ability to send them forth to do good, and the walls that have been built that prevent the free flow of data” can be very frustrating, he said.
Precompetitive Collaboration
Several speakers said precompetitive collaboration among industry, academia, patients, payers, and clinicians will be needed to improve the development and implementation of cancer immunotherapies in clinical practice. Brenner called for a culture of collaboration and transparency among academic institutions and pharmaceutical firms. He said that aca-
demics tend to have a publish-or-perish attitude, whereas industry tends to have a perish-if-published perspective, and those two points of view need to be bridged for progress in immunotherapy development.
Immunotherapy development is also hampered by a lack of cross-licensing of products across companies, Brenner said. It is often difficult to acquire licenses from more than one company to assess immunotherapies from different companies in combination. “That is something that needs to be considered and addressed,” Brenner said. One participant added that from a public health perspective, it makes sense to develop an infrastructure that would provide access to many immunotherapy agents early in the drug development pipelines from a variety of companies to advance the pace of research on combination immunotherapies. He added that the NCI already has an agreement with several companies to access immunotherapy agents for combination therapy studies without the need for separate agreements between companies.
Brenner said that academic researchers who are involved in Phase I immunotherapy clinical trials are more likely to have or be perceived as having conflicts of interest. He said that these trials have major financial implications—the valuation of a company could change significantly based on the results of single patients enrolled in a Phase I study. “This makes it difficult for any academic institution or investigator who is involved with one of these studies to really be clear that they have no conflict of interest,” Brenner said. “It is manageable, but it is an important consideration.”
He said that typically during the drug development process, academic involvement tends to diminish over time “because the costs increase logarithmically and you really have to get involved with an entity that has far more financial muscle than an academic [institution].” But he said that complex immunotherapies often require longer academic involvement, which makes it more likely that new agents may “end up in the valley of death,” in which academic funding has been depleted but industry funding that has yet to emerge. To prevent this funding cliff, Brenner described the mission of the Center for Cell and Gene Therapy, which conducts basic and translational research necessary for a cell therapy to advance into Phase II clinical trials. “We have de-risked the product by doing those time-consuming iterative Phase I studies so we can form partnerships with biopharma to take them forward,” Brenner said. Phase II trials are then completed as broad-based collaborations with an industry sponsor or they are completed by the Center individually.
Precompetitive collaboration can also be leveraged to address diag-
nostic testing issues, said Steven Averbuch, vice president of Development, Oncology and Pharmacodiagnostics at BMS. In partnership with diagnostic companies, AstraZeneca, BMS, Merck, and Roche-Genentech had each developed their own biomarker diagnostic to predict patient responsiveness to their own anti-PD-1 agents. Because the assays were developed for use with a specific checkpoint inhibitor, each diagnostic had a different design and different criteria for scoring and interpretation, said Averbuch. The different assays were found to be highly discordant, and one study recommended a multicenter international standardization effort to address significant concerns about testing complexity (Kerr et al., 2015) (see Table 2). “You had four different drugs, four different antibodies, all against the same target and there was a lot of concern about the confusion this was going to cause in the community practices as these drugs became available,” he said. Each assay had a different platform, different reagent system, and used different machinery and software, and each agent could potentially have a different clinical response based on its biology, chemistry, and mechanism of action. Averbuch said that running a different test for each drug would be impractical given patients’ limited tumor tissue and the turnaround time needed for multiple tests. But using one test that included the biomarkers for all the drugs would be equally impractical because all tests could not be run on the same platforms and the scoring and interpretation guidelines were not harmonized. FDA recognized that patients could be harmed if clinicians did not prescribe the right drug with the right test.
Recognizing all these challenges, before any of the anti-PD-1/PD-L1 agents or assays received FDA approval, the leadership of the pharmaceutical and diagnostic companies explored the potential for precompetitive collaboration and formed a consortium called Blueprint, which was facilitated by the American Association for Cancer Research. The consortium enlisted what Averbuch called “an independent honest broker,” the International Association for the Study of Lung Cancer, to compare the analytical performance of the different diagnostic assays and pave the way for postmarket standardization. All participating companies provided data and other information needed to conduct the study, which initially focused on non-small cell lung cancer. The goal of the study was to lay the groundwork for post-approval studies that will help inform patients, clinicians, pathologists, and others on how best to use the test results to determine treatment decisions once the tests are approved (AACR, 2016).
“Blueprint is a successful precompetitive collaboration that is a good
TABLE 2 Summary of Published Findings for PD-L1 Immunohistochemistry in Therapeutic Trials
| Drug | Biomarker Antibody | Rx Line | Definition of “Positive”a (%) | N Positive (%) | Positive Predictive Outcome | ORR % IHC pos. Cases | ORR % IHC neg. Cases | Ref. |
|---|---|---|---|---|---|---|---|---|
| Nivolumab | Dako 28-8 | 1st | ≥5 in >100 cells | 59 | Yes | 31b | 10 | 7, 8f |
| Nivolumab | Dako 28-8 | ≥2nd | ≥5, ≥1 | 49, 56 | No | 15, 13 | 14, 17 | 9, 10 |
| Nivolumab + Ipilimumab | Dako 28-8 | 1st | ≥5 in >100 cells | 42 | No | 19 | 14 | 11 |
| Nivolumab | Dako 28-8 | ≥2nd | ≥5 | 33c | Yes | 24 | 14 | 12f |
| Nivolumab | 5H1d | ≥2nd | ≥5, also studied TIICs | 67 | Yes | No data for lung | No data for lung | 13 |
| Prembrolizumab | Dako 22C3 | Any | “Strong” ≥50, “Weak” 1-49 | 25, 70 | Yes, Yes | 37, 17 | 9 | 14 |
| Pembrolizumab | Dako 22C3 | 1st | ≥50, ≥1 | ? | Yes | 47, 26 | ? | 15 |
| MPDL3280A | Roche Ventana, SP142 | ≥2nd | ≥10,e ≥5, ≥1 TIICs | 13, 28, 56 | Yes | 83, 46, 31 | 18, 18, 20 | 16–18 |
| MEDI-4736 | Roche Ventana, SP263 | ≥2nd | Data not available | 41 | Yes | 25 | 3 | 19, 20 |
NOTES: IHC = immunohistochemsitry; ORR = Overall Response Rate (Response Evaluation Criteria in Solid Tumors [RECIST]); TIIC = tumor infiltrating immune cell.
a Expression in tumor cells unless otherwise stated.
b The 31% figure is for all tumors. The ORR was 37% in nonsquamous tumors and 12% in squamous cases. In PDL-1 negative cases, ORR was 14% in nonsquamous tumors and 0% in squamous tumors.
c This study concerned squamous cell carcinomas only.
d These authors also used the anti-PD-1 monoclonal M3 in their immunohistochemical analysis.
e ICH score 3, ≥10% TIICs positive; IHC score 2–3, ≥5% TIICs positive; IHC score 1-2-3, ≥1% TIICs positive.
f ORR quoted are those actually presented, as opposed to those published in the abstract.
SOURCES: Averbuch presentation, February 29, 2016; Kerr et al. 2015. Reprinted from Journal of Thoracic Oncology with permission from Elsevier.
example of how we can move the field forward,” Averbuch said. “Blueprint provides a springboard for other professional societies, such as the American Society of Clinical Oncologists (ASCO) or the Association for Molecular Pathology (AMP), to take this forward for other types of cancers. It is the professional societies that should set the standards,” he added. Rasika Kalamegham, associate group director of U.S. Regulatory Policy at Genentech, said that Blueprint was successful because all the PD-1/PD-L1 agents and assays were at the same stage of development, rather than one being further along in the drug development pipeline. Blueprint also worked well, she added, because it clearly defined a narrow scope of non-small cell lung cancer and did not include tests developed in small academic laboratories, “while keeping in mind the focus of doing the right thing for the patient. It was always about the patient. That is what kept everybody together.”
CLINICAL IMPLEMENTATION
Many workshop presenters and discussants said there are a number of challenges in implementing cancer immunotherapies in clinical practice, including scaling up complex cell-based therapies, detecting and managing adverse side effects that are often non-specific, and educating clinicians and patients on what responses to expect from immunotherapies, which can differ markedly from standard cancer treatments.
Bringing Cell Therapies into Clinical Practice
Mark Dudley, director of Cell Process Development, Cell and Gene Therapies at Novartis, described the challenges of scaling up CAR T-cell therapy developed in an academic setting. He added that manufacturing processes for cell-based therapies that are developed in an academic setting are not capable of fulfilling global demand because they are often not standardized or validated, usually involve manual processes requiring numerous people to execute, and have complicated logistics. Whereas an experimental treatment process can be flexible in an academic center—researchers may vary their reagents, processes, or personnel—in a commercial setting, “the process has to work every time and you cannot change anything,” Dudley said. The advantage of an inflexible process is that each step can be optimized “as a principally science-based development approach moves into a principally operations approach,” Dudley said.
Prior to the transfer of CAR T-cell therapy to Novartis, the treatment required an immense amount of personnel and resources. “At University of Pennsylvania we have now treated over 200 patients with our CAR T-cell [therapy], but to do that we essentially had to develop a brand-new organization from scratch,” Porter said. The transfer of the treatment to Novartis required collaboration of a diverse team, including members from academia, and experts in good manufacturing practices, technical development, quality assurance, and regulatory issues, Dudley said.
According to Dudley, the initial steps to develop a commercial process for producing a therapy are to understand its scientific basis and key technologies. Novartis is currently focused on these two steps with CAR T-cell therapy. The next steps will be to automate production of the therapy, and then to design manufacturing and operations to enable supply-chain efficiencies, which will help with the cost of goods and scalability, Dudley said.
Pretransfer steps involved the commercial team watching the process, making a process map, conducting a risk assessment, completing a data-mining exercise, and determining the industrial process of production, Dudley said (see Figure 8). Once the process was transferred into the commercial facility, Novartis conducted test runs, trained the manufacturing staff, and completed split engineering runs at Novartis and the University of Pennsylvania using the same patient samples.
Once Novartis was satisfied that it had adequately duplicated the cell therapy, it started manufacturing the T-cell products for use in clinical practice and conducted a review of its process capabilities. Factors of particular concern when scaling up production include having better control of and streamlining the process by improving unit operations, and having better product characterization. Attention to patient safety and efficacy is a priority in every step of the scale-up process, Dudley said.
Dudley also described the complex chain of custody for products involved in CAR T-cell therapy because once blood products are removed from patients, they have to be transported to a central manufacturing facility where the T-cells are genetically engineered and expanded in number, and then transported back to the patient while being kept at a precise temperature by a certified transporter. Each step in that chain is overseen by careful labeling and quality assurance measures, and was improved by more stringent sterility procedures and other enhancement efforts that Novartis instituted. “We used to manufacture a product and write on the bag the expiration date, which was an hour after we made it, and then we would walk it over to the patient and infuse it. You cannot do that in a commer-
NOTE: GMP = good manufacturing practice; Penn = University of Pennsylvania.
SOURCE: Dudley presentation, March 1, 2016. Copyright owned by Novartis Pharmaceuticals Corporation. All rights reserved.
cial setting. Whatever process that is established has to be appropriate for multiple regulatory environments if you are going to have a global product. It is quite an endeavor,” Dudley said. Novartis was able to generate a T-cell expansion rate within a tighter range, but comparable to that seen at the University of Pennsylvania, Dudley reported, and the pharmaceutical company is currently conducting a global Phase II trial of the CAR T-cell therapy in patients with acute lymphocytic leukemia. “We started by making one product at a time, treating a couple patients a month. We are now at a point where this can be operationalized and standardized, but to make this really practical, we have to have smarter production so that this can treat not tens or hundreds, but thousands of patients around the world,” Porter said.
Novartis is not the only company involved in scaling up cellular therapies for commercial production, Porter said. “This is something many people are trying to operationalize for large-volume therapies,” he said (see Table 3).
TABLE 3 Examples of Pharmaceutical and Biotechnology Companies Working on Adoptive Cell Therapy
| Company | Technology/Cell Type | Indication |
|---|---|---|
| Lion Biotechnologies | TIL (autologous) | Metastatic melanoma |
| Autolus | CAR (autologous) | Unspecified |
| Novartis | CAR (autologous) targeting CD19 | Pediatric and adult ALL, diffuse large B-cell lymphoma, NHL |
| Juno Therapeutics | CAR (autologous) targeting CD19, TCR targeting Wilms tumor protein-1 |
Adult and pediatric ALL, NHL, AML, NSCLC |
| Cardio3 Biosciences | CARs targeting NKp30; NKG2D; B7H6 | Range of hematological malignancies and solid tumors |
| Cellular Biomedicine Group (China) | CARs targeting CD19, CD20, CD30, and EGFR | Range of hematological malignancies and solid tumors |
| CARsgen | CARs targeting GPC-3 | Hepatocellular carcinoma |
| Celgene/Bluebird | CAR (autologous) | Range of hematological malignancies and solid tumors |
| Kite Pharma/Amgen | CAR (autologous) targeting CD19, TCR | Relapsed or refractory ALL |
| Cellectis/Servier/Pfizer | CAR (allogeneic, UCART 19) | CLL, ALL, and AML in preclinical stage, Phase I for B-cell leukemia to be initiated in 2015 |
| GSK/Adaptimmune | TCR (autologous) targeting the cancer testis antigen NY-ESO-1 and other targets | Trials in MM, melanoma, sarcoma, and ovarian cancer |
| Janssen/Transposagen | CAR (allogeneic) | Unspecified |
| Company | Technology/Cell Type | Indication |
|---|---|---|
| Unum Therapeutics/Sanofi-Genzyme | Antibody-coupled TCR (autologous) | Unspecified |
| Ziopharm Oncology/Intrexon | CAR | Unspecified |
| Opus Bio | CAR (autologous) targeting CD22 | Pediatric and adult ALL and NHL, CD22 licensed to Juno |
| Takara Bio (Japan) | CAR (autologous) targeting CD19, TCR, MAGE-A4 | NHL, esophageal cancer |
| Bellicum Pharmaceuticals | CAR (autologous) targeting CD19 with a proprietary safety switch to mute unwanted adverse events, such as cytokine release syndrome | Potential hematological malignancies and solid tumors |
| Cellular Therapeutics Ltd. (UK) | CAR (autologous) | Metastatic melanoma, esophago-gastric cancer |
| Cell Medica (UK) | Virus-specific T-cell (allogeneic) targeting Epstein-Barr virus antigen | Advanced NK/T cell lymphoma |
| Celdara Medical | CAR (autologous) targeting NKG2D | AML, advanced myelodysplastic syndrome (MDS), MM |
| Catapult Cell Therapy (UK) | TCR (autologous) targeting WT-1—overexpressing cells | AML, MDS |
| Medigene (Germany) | TCR (autologous) | Hematological malignancies |
| TheraVectys (France) | CARs (autologous) targeting CD19, CD33, and CD123 | ALL, CLL, AML |
| Company | Technology/Cell Type | Indication |
|---|---|---|
| CARsgen (China) | CAR (autologous) targeting GPC-3 expressed in hepatocellular carcinoma; other CARs | Liver, lung, and brain cancers |
| FF CanVac | Virus-specific T cells (autologous) | Head and neck cancer |
| Apceth | Genetically engineered mesenchymal stem cells (MSCs) (autologous) | Advanced, recurrent, or metastatic gastrointestinal cancer |
| Pocastem | Genetically engineered MSCs | Solid tumors (head and neck, brain) |
| TVAX Biomedical | Antigen-specific T cells (autologous) | Solid tumors (brain, kidney) |
| TC Biopharm (Scotland) | γ/δ T cells (autologous) | Melanoma |
| Immunovative Therapies (Israel) | Activated T cells (allogeneic) | Hematological malignancy, prostate cancer, breast cancer, glioblastoma, colorectal cancer with liver metastases, kidney cancer, NSCLC |
| CytoVac (Denmark) | Activated T cells/NK cells (autologous) | Glioblastoma, prostate cancer, pancreatic cancer |
| Conkwest | CAR NK cell line | AML |
| Coronado Biosciences | Activated NK cells (autologous) | AML |
NOTES: Adoptive cell therapy applications are shown for cancers, infections, and graft versus host disease. ALL = acute lymphoblastic leukemia; AML = adult acute myeloid leukemia; B7H6 = B7 homolog 6; CAR = chimeric antigen receptor; CD = cluster of differentiation; EGFR = epidermal growth factor receptor; GPC-3 = glypican-3; MM = multiple myeloma; NHL = non-Hodgkin’s lymphoma; NK= natural killer; NKG2D = NK group 2, member D; NKp30 = NK cell p30-related protein; NSCLC = non-small cell lung cancer; TCR = T-cell receptor; TIL = tumor infiltrating lymphocytes.
SOURCES: Porter presentation, February 29, 2016; adapted from June et al., 2015. Reprinted with permission from the American Association for the Advancement of Science.
Addressing Adverse Effects
Wolchok reiterated that “the toxicities for immunotherapy are unlike what we are used to seeing from other standard anticancer therapies.” Ibrahim added that these side effects are often non-specific, such as inflammation, and can occur in any organ. But the safety profile of these agents can be explained by their mechanism of action, or the induction of nonspecific activation of the immune system, he said. Some organs are more prone to developing toxic outcomes from non-specific inflammation. For example, the gastrointestinal tract is especially susceptible and patients can present with diarrhea or colitis. The skin and the liver are also prone to inflammatory side effects, as well as the pancreas and the endocrine system. But “the key message about these adverse events is that they can occur in any organ, not just those organs that are commonly affected,” Ibrahim said.
He said there is also variability in how soon after treatment the adverse events are seen and that appears to depend on the organ affected as well as the molecule being tested (see Figure 9). Adverse events in the skin following anti-CTLA-4 therapies usually are seen early, whereas adverse events involving the liver or endocrine system have more of a delayed onset.
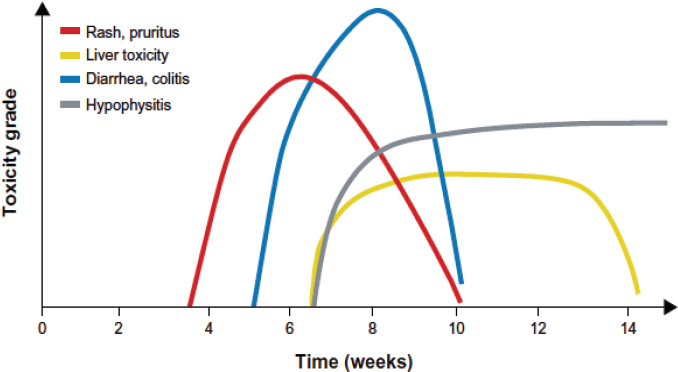
NOTE: Hypophysitis = inflammation of the pituitary gland, the primary gland of the endocrine system.
SOURCES: Wolchok presentation, March 1, 2016; Weber et al., 2012. Reprinted with permission. © 2012 American Society of Clinical Oncology. All rights reserved.
Most adverse events are low grade in nature, and in many patients can be managed and reversed with the aid of management guidelines that were established using data generated from clinical trials, Ibrahim said. For example, these guidelines specify that clinicians should prescribe an immune-suppressing drug to patients who develop a serious non-specific inflammation. Low-grade events are usually managed with treatments aimed at relieving symptoms or with topical agents, but if the condition persists or worsens, patients are usually prescribed systemic corticosteroids, he said.
Education and vigilance are key to managing adverse events in patients who receive cancer immunotherapies because the earlier the recognition of the inflammation and its treatment, the less likely the patient will develop serious complications, Ibrahim said. These complications, which can include bowel perforation in patients who develop colitis, can be life threatening. For that reason, diarrhea that develops in these patients should be taken seriously, Ibrahim said. “It is all about vigilance and early recognition of the signs and symptoms. We have to really rely on patient education. The patients are our partners in trying to identify and manage those adverse events early on,” Ibrahim said.
Despite available guidelines and treatments, there are still challenges in addressing the side effects that patients develop with cancer immunotherapies, including variations in how these adverse events are defined, difficulties in detecting them, and a lack of clinician experience and understanding of cancer immunotherapies and their side effects, according to Ibrahim.
Ibrahim said that an immune-related adverse event can be defined as any inflammatory event for which there is no other possible cause that could explain it. But others define this type of side effect as any toxicity managed with corticosteroids. “We do not have a consistent way of defining those adverse events, and even the terminology we are using to describe them is not consistent,” Ibrahim said. He pointed out that some of these events are reported as immune-related adverse events, immune-mediated adverse events, or events of special interest. “Do we rely on the investigator’s assessment for causality or do we need to have an independent review of those adverse events?” he asked.
Identification of adverse events can also be challenging because many cannot be detected in a physical exam. For example, a patient may report to her clinician that she has fatigue, but the clinician cannot detect the underlying endocrine inflammation that may be causing fatigue in a routine
physical exam. “It is not necessarily a simple matter to decide whether the toxicity is coming from the therapy or coming from the disease. Each individual clinician outside of a clinical trial has to make this decision on his/her own, which is a challenge,” Schwartzberg said. In addition, patients may be reluctant to report adverse events because they worry their treatment will be stopped and they might try over-the-counter remedies first to attempt to control their side effects. If patients develop acute or severe toxicities, they may not see their medical oncologist, but rather an emergency room clinician or internist, Ibrahim added. Some toxicities to ipilimumab and pembrolizumab did not occur until more than 200 days after treatment initiation, Schwartzberg said. By the time patients developed adverse reactions, they may no longer be seeing their oncologist frequently “so they need very clear instructions,” said Thomas.
Ibrahim said Phase I trials may not identify dose-limiting toxicities because of delayed onset of adverse events that are not captured in the limited observation periods for toxicity in these dose-setting trials. “When making dose-selection decisions, it is very important when we are trying to characterize the safety of these agents early on to look at the totality of the data, not just the few weeks that we use them in the dose-limiting-toxicity observation period,” Ibrahim said.
Another challenge is clinician inexperience with immunotherapies. While investigators involved in clinical trials of immunotherapies know how to identify and manage adverse events, this “might not be the case once the drugs get marketed or investigators who have less experience start treating patients with these agents,” Ibrahim said. He added that clinical trials occur in closely monitored environments and there are very strict criteria about when to stop treatment when adverse events occur, unlike in community oncology settings. Oncologists who have minimal experience with cancer immunotherapies may also be reluctant to treat patients experiencing inflammation with an immunosuppressive drug for fear of reducing the effectiveness of the cancer immunotherapy. In addition, oncologists who are not familiar with immunotherapies may try to manage adverse events symptomatically rather than mechanistically (providing patients with colitis an antidiarrheal agent rather than treating the inflammation causing the colitis). In addition, patients’ adverse events are often managed by internists, gastroenterologists, or endocrinologists who may be unfamiliar with cancer immunotherapies.
Ibrahim suggested several policy measures to encourage better management of patients’ side effects of immunotherapies, including standardizing
how these adverse events are defined. He suggested having input from regulators, academics, and industry representatives for this endeavor, and considering including a separate term for immune-adverse events in the Common Terminology Criteria for Adverse Events that are used to characterize the adverse events of agents in clinical trials.
Ibrahim also suggested developing novel ways of designing Phase I studies to characterize the safety of agents, as well as using expanded access to collect peri-approval data once Phase III studies are completed, but before the agent receives approval for marketing, as well as collecting safety data in the postmarketing setting. He also suggested including more community hospitals and oncology practices in clinical trials to gain “real-world” clinical experience early in drug testing.
Schwartzberg said the ability to manage patients’ adverse events varies according to the size of the practice providing cancer immunotherapies. Small- to medium-sized community practices, which provide approximately 80 percent of cancer care in the United States, may have challenges addressing the side effects of immunotherapies, given that many may be reported by patients during the weekend when practice clinicians are not available. Such off-hours reporting may necessitate a hospital admission and care by someone not familiar with immunotherapies. Academic centers tend to have more after-hours coverage of their patients, in contrast, and more expertise providing cancer immunotherapies, said Schwartzberg.
Ibrahim said patients should be better educated so they are aware of how important it is to report adverse events early. He also suggested leveraging patient-reported outcomes and quality-of-life assessments, rather than just considering investigators’ assessments of adverse events. “You need to talk with and listen to your patients and understand that these toxicities need to be managed mechanistically and not symptomatically,” Wolchok added. In addition, clinicians outside of the medical oncology field need to be educated on adverse events linked to cancer immunotherapies, especially emergency room clinicians, specialists, and internists, Ibrahim said. Wolchok and Schwartzberg suggested the development of patient materials to explain how immunotherapies work, expected side effects, and when those side effects might occur.
Schwartzberg described an electronic PRO system called Patient Care Monitor, which is intended “to keep patients on treatment and out of the hospital,” Schwartzberg said. This system collects information from patients on physical symptoms, functional status, and psychological status. It takes approximately 7 minutes to complete, has been validated and used by more
than 100,000 patients, and can be incorporated directly into an EHR. It is also available as a Web-enabled, cloud-based system that works on mobile devices. The system documents a patient’s symptom history, highlights severe problems, triggers appropriate information to be sent to patients (e.g., rash management), and tailors notifications to the care team.
Schwartzberg said electronic PROs are a way to have patients report their side effects of immunotherapies in a timely fashion that might alleviate some of the more serious complications of such treatments. He said that one study found that quality of life was better in those patients assessed routinely with an electronic PRO system and that those patients were less likely to have emergency room visits (Basch et al., 2016).
Educating Clinicians, Patients, and Payers
Schwartzberg described the rapid progress seen in immunotherapies for cancer, with nine approvals of five different drugs or drug combinations within the course of 1 year:
- March 2015: Nivolumab approved for squamous cell cancers following platinum-based therapy
- September 2015: Nivolumab and ipilimumab approved for BRAF V600 wild type metastatic melanoma
- October 2015: Pembrolizumab approved for PD-L1 positive non-small cell lung cancer following platinum-based therapy (companion diagnostic)
- October 2015: Nivolumab indication expanded to non-squamous non-small cell lung cancer (complementary diagnostic)
- October 2015: Imlygic (talimogene laherparepvec or T-VEC) approved for recurrent melanoma with injectable lesions
- October 2015: Ipilimumab approved for adjuvant treatment of lymph node-positive melanoma
- November 2015: Nivolumab approved for renal cell carcinoma after prior antiangiogenic therapy
- December 2015: Pembrolizumab approved for metastatic melanoma
- January 2016: Ipilimumab plus nivolumab approved for advanced BRAF wild type or mutated melanoma
This rapid progress requires educational initiatives to ensure that clinicians are up to speed with the new treatments, Schwartzberg said, in addi-
tion to opportunities for patient education (see Box 4). “There is an overwhelming amount of knowledge that needs to be integrated quickly in order to get these life-saving drugs to the patients who need them,” Schwartzberg said, including clinician familiarity with the types of therapies available, determining appropriate use, and assessing patient responses. In addition, Wolchok emphasized that a critically important aspect of education is early recognition and proper management of the side effects of immunotherapies.
There is a great need for immunotherapy training among clinicians, Schwartzberg said. A 2014 survey of the Association of Community Cancer Centers (ACCC) members found that more than half reported they were only somewhat, slightly, or not at all familiar with the concept of cancer immunotherapies. Clinicians said that the main challenges to using these therapies in clinical practice were a lack of information about specific therapies, few experts to consult with, and a lack of immunotherapy education and training opportunities for their staff and patients. Recognizing this need, Schwartzberg said that the ACCC established the Institute for Clinical Immuno-Oncology (ICLIO) to provide information about cancer
immunotherapies to the full multidisciplinary team taking care of patients with cancer, including physicians, nurses, practice managers, financial counselors, patient navigators, and social workers. ICLIO’s educational initiatives have five domains: (1) management best practices, (2) clinical optimization, (3) training and development, (4) patient access and advocacy, and (5) coverage and reimbursement.
Wolchok added that that the SITC provides immunotherapy training sessions at their annual meetings, conducts an immunotherapy primer session for clinicians who do not have experience with cancer immunotherapies, and has developed online and mobile device-based educational activities. SITC also developed a traveling course, Advances in Cancer Immunotherapy, to make education on cancer immunotherapies more convenient for clinicians. ASCO also has integrated cancer immunotherapy topics into its ongoing online classes and other education programs for oncologists, and has collaborative sessions with SITC at both the ASCO and SITC annual meetings. The two organizations are offering a clinical immuno-oncology symposium in 2017.11
Wolchok added that the Cancer Research Institute is working collaboratively with the Oncology Nursing Society to develop educational programs and materials for nurses and patients about cancer immunotherapies. There also is a policy working group convened by Friends of Cancer Research with a cancer immunotherapy education initiative. “There are many new agents rolling out and there is a flood of information for people to integrate,” Wolchok said. “I cannot stress enough how important it is to have these other organizations that have made a commitment to educating clinicians, nurses, and other treating colleagues in this important and growing area.”
VALUE OF IMMUNOTHERAPY
A number of workshop speakers highlighted a fundamental tension with immunotherapy treatment for cancer: these therapies have great promise to achieve durable treatment responses for some patients with cancer; however, they are also very expensive. Krug described the dramatic shift in the therapeutic options for patients with lung cancer: “When I finished my fellowship training, really the only treatment available for patients with advanced lung cancer was chemotherapy, [which] has a limited effect on patients’ survival, and, of course, has [significant] toxicities associated with
___________________
11 See http://immunosym.org (accessed May 26, 2016).
it,” Krug said. When the data on checkpoint inhibitors found improved survival for some patients with lung cancer, “it changed the paradigm and I think what was most impressive was the fact that these responses were so durable and patients were having much more long-term benefit. And that is really where the value is,” said Krug. At the same time, there are concerns about the high costs of cancer immunotherapies—both currently approved therapies, as well as the expected high costs of future cell- and vaccine-based therapies, and combination therapies.
Escalating Cancer Drug Costs
Many workshop speakers noted that discussion of the value of immunotherapies is occurring against the backdrop of escalating prices for many cancer drugs (Howard et al., 2015). “The pricing model for oncology drugs is badly broken and it is hurting us as a society,” said Scott Ramsey, director at the Fred Hutchinson Cancer Research Center. He added that the price of the cancer drug Gleevec (imatinib) has tripled since it was introduced about 20 years ago, even though manufacturing costs have likely gone down, and its effectiveness has not improved.12 “These prices are not being set based on what health economists recognize as value, but on what the manufacturers’ think the market will bear. It is not a supportable model and until we start facing that reality, I am worried that access to these promising immunotherapies is going to be curtailed,” Ramsey said.
“There is no obvious reason why something like Gleevec should keep going up in price when in fact the cost of its development was years ago,” Berzofsky said. Newcomer responded that although that would be true of a free-market system, such a free market does not exist in health care, as federal and state laws and regulations require coverage of most FDA-approved cancer drugs, and CMS cannot consider cost when deciding what drugs to cover (Bach, 2009).
Woodman said that the value of a drug can also change over time: When Gleevec was first approved for patients with chronic myelogenous leukemia, he said that impact on overall survival and cure was not fully appreciated. In addition, Gleevec has subsequently been approved for eight other cancers, for which it often is curative, he said. Newcomer responded that pricing a product based on the years of life given to patients is not
___________________
12 Gleevec’s patent protection ended in 2016, which is expected to result in a lower price (Kodjak, 2016).
financially sustainable. Ramsey added that the second-generation tyrosine kinase inhibitors are priced at triple that of Gleevec, which was the first such inhibitor to enter the market, “yet we do not yet have evidence that they are clearly superior. They have better response rates, but if your argument is that when we see prolonged survival then we can charge more, why are you charging so much more for new drugs before the evidence is there?”
Ramsey added that although it is expensive to develop drugs, the cost of developing drugs is not going up commensurate with the price increases seen in cancer therapies. Many of these drugs underwent accelerated review, which reduced their cost of development because sponsors did not have to conduct large and expensive Phase III trials, he said. But Ramsey did note that the cost of manufacturing many immunotherapies, especially cell-based therapies, is much higher than it is for traditional drugs.
Due to the extremely high costs of new cancer therapies, the financial burden to payers and patients has to be considered, Ramsey said. He added that patients are particularly vulnerable to the high costs of new treatments, because insurers are increasingly shifting the higher costs of care to patients and families through higher co-payments and co-insurance rates. Newcomer added that in about 10 years, one estimate found that the amount of money households will spend on insurance premiums and out-of-pocket health care expenses will be equivalent to the projected average U.S. household income (Young and DeVoe, 2012). Newcomer said, “It is naïve to think we are not going to have to address costs. That is magical thinking.”
“There are things we are going to have to give up in order to pay for these more exciting treatments,” he said. Newcomer listed several examples of wasteful health care spending that could be averted, including forgoing PET scans for women with early-stage breast cancers who are at low risk of recurrence. Newcomer also suggested that drug companies be prevented from packaging expensive infused medications in quantities that are larger than patients need. He noted that a recent analysis suggested billions of dollars could be saved, because leftover medicine has to be discarded for safety reasons (Bach, 2016).
Patient-Centered Definition of Value
Gwen Darien, executive vice president for Patient Advocacy at the National Patient Advocate Foundation, said that any assessment of value for immunotherapies should consider the patient perspective. “If we talk about patient-centered care and talk about putting the patient in the center, we have
to ask the patient what she or he wants, values, and considers to be a cost,” she said. “The idea that all patients want access to all drugs no matter what the cost is false. Most patients do not have conversations with their clinicians about cost, and people make different decisions based on different values. For some people, a benefit of a few months at a huge cost either to their family, to their society, or their quality of life is not worth it,” said Darien.
The patient survey described in Box 4 asked respondents how they define value when describing their cancer experience. Darien noted that most responses focused broadly on answering questions such as, “What do I value in my life and how does that impact my decision making? What do I value in terms of what I am going to be able to do and how does that impact decision making?” Darien said that none of the answers really focused on the cost and she added that cost is broader than purely financial cost and financial impact. When patients focused on describing value related to their health care, what they valued the most was communication with their health care team. “This was paramount to how they felt about their treatment across all modalities,” Darien said.
But unfortunately, clinicians are not reimbursed for taking more time to explain cancer immunotherapies to their patients, said Patricia Ganz, distinguished professor at the University of California, Los Angeles, School of Medicine. “One of the big challenges is that our payment models do not allow clinicians and patients to have the time to communicate about this. We need to make sure these discussions are happening so patients are offered [meaningful] choices,” Ganz said. She said that discussion can be focused both on access to immunotherapy treatment, as well as opportunities to be involved in collecting information about these new therapies, such as participation in expanded access programs or registries. Ganz asked if CMS might be able to reimburse for the time clinicians take to have these discussions with their Medicare patients. James Rollins, director of the Division of Items and Devices in the Coverage and Analysis group at CMS, said the agency is currently working on promoting shared decision making by establishing reimbursement for this activity (see Box 5 for more information about CMS coverage policies). Such shared decision making would give patients and clinicians an opportunity to communicate when deciding on a treatment plan, he said.
Pricing and Payment Models for Immunotherapies That Incorporate Value
The escalating cost of health care suggests the need to prioritize high-value treatments and ensure value-based pricing, several workshop participants said. Newcomer suggested prioritizing how health dollars are spent on cancer care, noting that it may be more appropriate to focus on first-line immunotherapies, rather than developing it as a fourth-line or fifth-line option. For example, if CAR T-cell therapy works well as a first-line therapy for patients with leukemia, it may be less costly because it could potentially avoid several rounds of chemotherapy or a bone marrow transplant, Newcomer said. Several opportunities to ensure that immunotherapy pricing and payment emphasize value were discussed, including immunotherapy-specific assessment of incremental value and outcomes-based pricing.
Immunotherapy-Specific Assessment of Incremental Value
Ramsey suggested that when determining a price for new treatments, an assessment of the incremental value of the treatment is needed—that is, the additional health gain for the additional cost of the treatment. Ramsey suggested using a new economic model for assessing the incremental value of cancer immunotherapies that incorporates the probability that a patient will experience a durable response to treatment. He said the minority of patients who have durable responses may live for decades, whereas for those whose cancers do not respond, the immunotherapy may not look much better than standard treatment. “If there is a cure fraction, then I think accounting for this with this mixture model improves things,” Ramsey said.
He said that people whose cancers respond to the treatment will increase the total treatment costs if the immunotherapy is given to them over a longer period of time. But Ramsey added “that it is okay because they are [receiving] additional survival and quality of life.” Using this model can substantially affect the incremental cost-effectiveness evaluation, he said. When he applied the model to data on patients with advanced melanoma who received ipilimumab treatment, the incremental cost-effectiveness ratio (ICER) was $113,000/quality-adjusted life year (QALY) versus $324,000/QALY using the Standard Weibull model (see Table 4 for analysis parameters) (Lin et al, 1997; Seidler et al., 2010; Tromme et al., 2014). “As we further refine the patient populations and identify markers for response so we do not have to give full courses of drugs for people who are not going to benefit, it could be possible to push the ICER down into territory that
TABLE 4 Parameters for Ipilimumab Case Study
| gp100 | Ipilimumab | |
|---|---|---|
| Weibull analysis without cure modeling | ||
| Mean overall survival (years) | 0.90 | 1.60 |
| Mixture cure model analysis | ||
| Mean overall survival of cured patients (years) | 26 | 26 |
| Mean overall survival of uncured patients (years) | 0.75 | 0.83 |
| Cure proportion (percent) | 6 | 21 |
NOTE: Mean overall survival of cured patients was greater than mean overall survival of uncured patients, and 15 percent more patients were cured by ipilimumab than gp100 (glycoprotein 100).
SOURCE: Ramsey presentation, March 1, 2016.
might be considered reasonable value,” he said. “We need to tweak our economic models to consider a new approach that accounts for patients who may have durable responses,” he added.
Krug agreed, saying, “The paradigm has shifted now for these immunotherapies. They are not providing the incremental benefits we saw in the past when patients may be reluctant to pay for a therapy that improves their survival by a month or two. Now therapies can potentially extend patient’s lives much longer than that and these responses are much more durable. So we need to make sure that models that involve pricing take that type of information into consideration.”
Woodman also agreed that one should consider durable responses when calculating the value of immunotherapy, but raised the issue of how to define such responses, noting that some patients with durable responses still have residual disease but no symptoms. Greg Rossi, oncology business unit director at AstraZeneca, suggested that traditionally when cancer patients achieve a life expectancy similar to that of their age- and gender-matched peers, they are considered to have a durable response, if not a cure.
Outcomes-Based Pricing and Bundled Models
Rossi suggested use of value-based pricing of cancer immunotherapies for each indication and use of outcomes-based agreements with drug sponsors. “Instead of having a pharma product-centric or regulator population-centric or payer system-centric or clinician-centric view of the world, we need to have an integrated view with the patient at the center of it,” he said. Rossi said Italy and other European countries have a pay-for-response model for the majority of therapies that enter the market, with the benefit for cancer patients assessed by how long the treatment is continued “because oncologists are not going to withdraw therapy that is benefiting patients.” However, in the absence of a biomarker that predicts response to therapy, continuing therapy for a certain period of time can be a poor surrogate for effectiveness of the therapy, and payers can “claw back or rebate either partial or full funds associated with anyone who progresses within that period of time,” he said.
Ramsey added that currently fewer than one-third of patients have meaningful responses to cancer immunotherapies. In a pay-for-performance model, the payer would pay for the treatment, but if there is no response, then they receive a rebate for that patient. “That allows manufacturers to get paid when the product works and paid less when it does not work,” he said.
Bundled or episode-of-care payment models are another way to limit treatment costs and encourage use of the most cost-effective therapies, said Rossi and Ron Kline, medical officer at CMS. With this approach, a single amount is paid for a treatment episode, so that clinicians have an incentive to choose high-value therapies. But because both the response and treatment duration will be unknown for many cancer immunotherapies, Rossi said calculating their value is difficult. “How do we understand what we are getting for the money we are spending? Uncertainty in the immuno-oncology setting is a conversation we need to have,” Rossi said, noting the difficulties of doing value-based pricing when the evidence continues to evolve for immunotherapies.
Improving the Evidence Base to Inform Treatment and Value
Newcomer said that health care resources are often squandered by spending them on new treatments for which there is incomplete data on who is likely to benefit from them. “We still have to find out who are the right patients for these therapies, as exciting as the technology is, because if we give it to folks who are not going to benefit, we are wasting precious resources,” Newcomer said.
However, Richard Schilsky, chief medical officer of ASCO, noted that providing therapy that is anticipated to only benefit some patients substantially is already standard in many adjuvant treatments settings for early-stage cancers (e.g., chemotherapy or radiation therapy after a lumpectomy for early-stage breast cancers). “We accept that these adjuvant treatments are high-value therapies despite the fact that many people who get them do not need them and many who need them do not benefit from them. We cannot tell who those people are because the patients have no evidence of disease when they begin the treatment. We accept the notion that it is okay to treat everybody in the relevant population for what is generally a small percentage of individuals who will have long-term benefit from adjuvant treatment,” said Schilsky. He added that in the adjuvant setting, there are also efforts—using biomarker tests and gene expression panels—to better determine which patients may be at low risk of recurrence who could safely forego additional therapy. These same issues are emerging in the immunotherapy setting, in which only a small subset of those who are treated with immunotherapies will derive long-term benefit: “But the majority of patients will not [benefit] and we cannot yet decide who is in which group. We are confronted with the notion of treating everybody to try to get that
long-term benefit for the few,” said Schilsky. Rossi agreed that the adjuvant setting is somewhat analogous to immunotherapy, and highlighted the critical role that biomarkers could play in identifying who is most likely to benefit from immunotherapy treatment.
In addition to considering different approaches to assessing value, several workshop participants suggested improving the evidence base through clinical trials research. “Let’s make [immunotherapy] trials accessible so we can get the answers we need. My moonshot challenge to Vice President Biden is let’s get 100,000 people [enrolled in] trials in the next 3 years. If we did, we could have a much more informed debate 3 years from now because we would have the data we need,” Newcomer said. Rossi added that because many immunotherapies are entering the market based on surrogate endpoints from early clinical trials, the generalizability of that early data is also unknown. He suggested considering how early access programs for immunotherapies could be used to collect information on patients’ long-term outcomes.
Newcomer noted that patients often have to travel long distances to enter clinical trials and many are not able to do so. “Let’s get the trials to them,” he said. Krug agreed and said that the Immuno-Oncology-Integrated Community Oncology Network, a network of community cancer centers, is currently collaborating with BMS so more community cancer centers are incorporated into clinical trials and novel treatments can be made available to more patients. Effort is also being made to make eligibility criteria for clinical trials broader so more patients can be enrolled, and to simplify and streamline the clinical trial process so data can be collected more quickly, Krug said. Khleif added that no one expected that progress in immunotherapy development and clinical use “would happen so fast and be so impactful; so now it is important for us to start thinking of the different ways the field needs to go for the [next] 5, 10, or 15 years.”
REFERENCES
AACR (American Association for Cancer Research). 2016. A blueprint proposal for companion diagnostic comparability. http://www.aacr.org/AdvocacyPolicy/GovernmentAffairs/Pages/industry-working-group-blueprint-proposal.aspx#.VxUrczYrJmA (accessed May 12, 2016).
Alexandrov, L. B., S. Nik-Zainal, D. C. Wedge, S. A. J. R. Aparicio, S. Behjati, A. V. Biankin, G. R. Bignell, N. Bolli, A. Borg, A.-L. Borresen-Dale, S. Boyault, B. Burkhardt, A. P. Butler, C. Caldas, H. R. Davies, C. Desmedt, R. Eils, J. E. Eyfjord, J. A. Foekens, M. Greaves, F. Hosoda, B. Hutter, T. Ilicic, S. Imbeaud, M. Imielinsk, N. Jager, D. T. W. Jones, D. Jones, S. Knappskog, M. Kool, S. R. Lakhani, C. Lopez-Otin, S. Martin, N. C. Munshi, H. Nakamura, P. A. Northcott, M. Pajic, E. Papaemmanuil, A. Paradiso, J. V. Pearson, X. S. Puente, K. Raine, M. Ramakrishna, A. L. Richardson, J. Richter, P. Rosenstiel, M. Schlesner, T. N. Schumacher, P. N. Span, J. W. Teague, Y. Totoki, A. N. J. Tutt, R. Valdes-Mas, M. M. van Buuren, L. van’t Veer, A. Vincent-Salomon, N. Waddell, L. R. Yates, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, J. ZucmanRossi, P. Andrew Futreal, U. McDermott, P. Lichter, M. Meyerson, S. M. Grimmond, R. Siebert, E. Campo, T. Shibata, S. M. Pfister, P. J. Campbell, and M. R. Stratton. 2013. Signatures of mutational processes in human cancer. Nature 500(7463):415-421.
American Cancer Society. 2015. What’s new in cancer immunotherapy research?http://www.cancer.org/treatment/treatmentsandsideeffects/treatmenttypes/immunotherapy/ immunotherapy-whats-new-immuno-res (accessed April 8, 2016).
Bach, P. B. 2009. Limits on Medicare’s ability to control rising spending on cancer drugs. New England Journal of Medicine 360(6):626-633.
Bach, P. B., R. M. Conti, R. J. Muller, G. C. Schnorr, and L. B. Saltz. 2016. Overspending driven by oversized single dose vials of cancer drugs. BMJ 352:i788
Balas, E. A. 2015. Big data clinical research: Validity, ethics, and regulation. Studies in Health Technology and Informatics 216:448-452.
Basch, E., A. M. Deal, M. G. Kris, H. I. Scher, C. A. Hudis, P. Sabbatini, L. Rogak, A. V. Bennett, A. C. Dueck, T. M. Atkinson, J. F. Chou, D. Dulko, L. Sit, A. Barz, P. Novotny, M. Fruscione, J. A. Sloan, and D. Schrag. 2016. Symptom monitoring with patient-reported outcomes during routine cancer treatment: A randomized controlled trial. Journal of Clinical Oncology 34(6):557-565.
Budhu, S., J. Wolchok, and T. Merghoub. 2014. The importance of animal models in tumor immunity and immunotherapy. Current Opinion in Genetics & Development 24:46-51.
Butterfield, L., A. K. Palucka, C. M. Britten, M. V. Dhodapkar, L. Hakansson, S. Janetzki, Y. Kawakami, T.-O. Kleen, P. P. Lee, C. Maccalli, H. T. Maecker, V. C. Maino, M. Maio, A. Malyguine, G. Masucci, G. Pawelec, D. M. Potter, L. Rivoltini, L. G. Salazar, D. J. Schendel, C. L. Slingluff, W. Song, D. F. Stroncek, H. Tahara, M. Thurin, G. Trinchieri, S. H. van Der Burg, T. L. Whiteside, J. M. Wigginton, F. Marincola, S. Khleif, B. A. Fox, and M. L. Disis. 2011. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clinical Cancer Research 17(10):3064-3076.
Cameron, B. J., A. B. Gerry, J. Dukes, J. V. Harper, V. Kannan, F. C. Bianchi, F. Grand, J. E. Brewer, M. Gupta, G. Plesa, G. Bossi, A. Vuidepot, A. S. Powlesland, A. Legg, K. J. Adams, A. D. Bennett, N. J. Pumphrey, D. D. Williams, G. Binder-Scholl, I. Kulikovskaya, B. L. Levine, J. L. Riley, A. Varela-Rohena, E. A. Stadtmauer, A. P. Rapoport, G. P. Linette, C. H. June, N. J. Hassan, M. Kalos, and B. K. Jakobsen. 2013. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Science Translational Medicine 5(197):197ra103.
Campesato, L. F., R. Barroso-Sousa, L. Jimenez, B. R. Correa, J. Sabbaga, P. M. Hoff, L. F. L. Reis, P. A. F. Galante, and A. A. Camargo. 2015. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice Oncotarget 6(33):34221-34227.
Carbone, D. P., I. F. Ciernik, M. J. Kelley, M. C. Smith, S. Nadaf, D. Kavanaugh, V. E. Maher, M. Stipanov, D. Contois, B. E. Johnson, C. D. Pendleton, B. Seifert, C. Carter, E. J. Read, J. Greenblatt, L. E. Top, M. I. Kelsey, J. D. Minna, and J. A. Berzofsky. 2005. Immunization with mutant p53- and k-RAS-derived peptides in cancer patients: Immune response and clinical outcome. Journal of Clinical Oncology 23(22):5099-5107.
Chapman, P. B., S. P. D’Angelo, and J. D. Wolchok. 2015. Rapid eradication of a bulky melanoma mass with one dose of immunotherapy. New England Journal of Medicine 372(21):2073-2074.
Cheever, M. A., J. P. Allison, A. S. Ferris, O. J. Finn, B. M. Hastings, T. T. Hecht, I. Mellman, S. A. Prindiville, J. L. Viner, L. M. Weiner, and L. M. Matrisian. 2009. The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clinical Cancer Research 15(17):5323-5337.
CIBMTR (Center for International Blood & Marrow Transplant Research). 2016. AGNIS. https://www.cibmtr.org/DataManagement/TrainingReference/AGNIS/Pages/index.aspx (accessed May 23, 2016).
CIP (CIMT Immunoguiding Program). 2016. About CIMT immunoguiding program. http://www.cimt.eu/workgroups/cip/#anchor_about (accessed May 13, 2016).
Couzin-Frankel, J. 2013. Breakthrough of the year 2013. Cancer immunotherapy. Science 342(6165):1432-1433.
Cullen, S. P., M. Brunet, and S. J. Martin. 2010. Granzymes in cancer and immunity. Cell Death & Differentiation 17(4):616-623.
Disis, M. L., V. Goodell, K. Schiffman, and K. L. Knutson. 2004. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. Journal of Clinical Immunology 24(5):571-578.
Dranoff, G., E. Jaffee, A. Lazenby, P. Golumbek, H. Levitsky, K. Brose, V. Jackson, H. Hamada, D. Pardoll, and R. C. Mulligan. 1993. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proceedings of the National Academy of Sciences of the United States of America 90(8):3539-3543.
Dubin, K., M. K. Callahan, B. Ren, R. Khanin, A. Viale, L. Ling, D. No, A. Gobourne, E. Littmann, C. Huttenhower, E. G. Pamer, and J. D. Wolchok. 2016. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nature Communications 7:10391.
DuPage, M., C. Mazumdar, L. M. Schmidt, A. F. Cheung, and T. Jacks. 2012. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482(7385):405-409.
FDA (Food and Drug Administration). 2010. Guidance for industry: S9 nonclinical evaluation for anticancer pharmaceuticals. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm085389.pdf (accessed May 17, 2016).
FDA. 2013. Guidance for industry: Preclinical assessment of investigational cellular and gene therapy products. http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/ucm376136.htm (accessed May 18, 2016).
Gettinger, S. N., L. Horn, L. Gandhi, D. R. Spigel, S. J. Antonia, N. A. Rizvi, J. D. Powderly, R. S. Heist, R. D. Carvajal, D. M. Jackman, L. V. Sequist, D. C. Smith, P. Leming, D. P. Carbone, M. C. Pinder-Schenck, S. L. Topalian, F. S. Hodi, J. A. Sosman, M. Sznol, D. F. McDermott, D. M. Pardoll, V. Sankar, C. M. Ahlers, M. Salvati, J. M. Wigginton, M. D. Hellmann, G. D. Kollia, A. K. Gupta, and J. R. Brahmer. 2015. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, bms-936558, ono-4538) in patients with previously treated advanced non-small-cell lung cancer. Journal of Clinical Oncology 33(18):2004-2012.
Hardwick, N., and B. Chain. 2011. Epitope spreading contributes to effective immunotherapy in metastatic melanoma patients. Immunotherapy 3(6):731-733.
Healthcare Information and Management Systems Society. 2013. What is interoperability?http://www.himss.org/library/interoperability-standards/what-is-interoperability (accessed May 18, 2016).
Heslop, H.E., K. S. Slobod, M. A. Pule, G. A. Hale, A. Rousseau, C. A. Smith, C. M. Bollard, H. Liu, M.-F. Wu, R. J. Rochester, P. J. Amrolia, J. L. Hurwitz, M. K. Brenner, and C. M. Rooney. 2010. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 115(5):L925-L935.
Hodi, F. S., S. Lee, D. F. McDermott, U. N. Rao, L. H. Butterfield, and A. A. Tarhini. 2014. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: A randomized clinical trial. JAMA 312(17):1744-1753.
Howard, D. H., P. B. Bach, E. R. Berndt, and R. M. Conti. 2015. Pricing in the market for anticancer drugs. Journal of Economic Perspectives 29(1):139-162.
Iwama, S., A. De Remigis, M. K. Callahan, S. F. Slovin, J. D. Wolchok, and P. Caturegli. 2014. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Science Translational Medicine 6(230):230ra245.
June, C. H., S. R. Riddell, and T. N. Schumacher. 2015. Adoptive cellular therapy: A race to the finish line. Science Translational Medicine 7(280):280ps287.
Kalos, M., B. L. Levine, D. L. Porter, S. Katz, S. A. Grupp, A. Bagg, and C. H. June. 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Translational Medicine 3(95):95ra73.
Kantoff, P. W., C. S. Higano, N. D. Shore, E. R. Berger, E. J. Small, D. F. Penson, C. H. Redfern, A. C. Ferrari, R. Dreicer, R. B. Sims, Y. Xu, M. W. Frohlich, P. F. Schellhammer, and IMPACT Study Investigators. 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. England Journal of Medicine 363(5):411-422.
Kerr, K. M., M.-S. Tsao, A. G. Nicholson, Y. Yatabe, I. I. Wistuba, and F. R. Hirsch. 2015. Programmed death-ligand 1 immunohistochemistry in lung cancer: In what state is this art? Journal of Thoracic Oncology 10(7):985-989.
Kodjak, A. 2016. Modest price cut expected for generic version of cancer pill gleevec. http://www.npr.org/sections/health-shots/2016/02/01/465139901/generic-gleevec-imatinib-savings-will-be-modest (accessed May 26, 2016).
Kwek, S. S., V. Dao, R. Roy, Y. Hou, D. Alajajian, J. P. Simko, E. J. Small, and L. Fong. 2012. Diversity of antigen-specific responses induced in vivo with CTLA-4 blockade in prostate cancer patients. Journal of Immunology 189(7):3759-3766.
Larkin, J., V. Chiarion-Sileni, R. Gonzalez, J. J. Grob, C. L. Cowey, C. D. Lao, D. Schadendorf, R. Dummer, M. Smylie, P. Rutkowski, P. F. Ferrucci, A. Hill, J. Wagstaff, M. S. Carlino, J. B. Haanen, M. Maio, I. Marquez-Rodas, G. A. McArthur, P. A. Ascierto, G. V. Long, M. K. Callahan, M. A. Postow, K. Grossmann, M. Sznol, B. Dreno, L. Bastholt, A. Yang, L. M. Rollin, C. Horak, F. S. Hodi, and J. D. Wolchok. 2015. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New England Journal of Medicine 373(1):23-34.
Le, D. T., J. N. Uram, H. Wang, B. R. Bartlett, H. Kemberling, and A. D. Eyring. 2015. PD-1 blockade in tumors with mismatch-repair deficiency. New England Journal of Medicine 372(26):2509-2520.
Leach, D. R., M. F. Krummel, and J. P. Allison. 1996. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271(5256):1734-1736.
Leen, A., I. Tzannou, M. Bilgi, H. Liu, J. F. Vera, U. Gerdemann, R. T. Kamble, C. A. Ramos, B. Grilley, A. P. Gee, C. M. Bollard, M. K. Brenner, H. E. Heslop, C. M. Rooney, and G. Carrum. 2015. Immunotherapy for lymphoma using T cells targeting multiple tumor associated antigens. Paper presented at American Society of Hematology 57th Annual Meeting and Exposition, Orlando, FL.
Li, Y., L. X. Wang, G. Yang, F. Hao, W. J. Urba, and H. M. Hu. 2008. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Research 68(17):6889-6895.
Lin, D. Y., E. J. Feuer, R. Etzioni, and Y. Wax. 1997. Estimating medical costs from incomplete follow-up data. Biometrics 53(2):419-434.
Linette, G. P., E. A. Stadtmauer, M. V. Maus, A. P. Rapoport, B. L. Levine, L. Emery, L. Litzky, A. Bagg, B. M. Carreno, P. J. Cimino, G. K. Binder-Scholl, D. P. Smethurst, A. B. Gerry, N. J. Pumphrey, A. D. Bennett, J. E. Brewer, J. Dukes, J. Harper, H. K. Tayton-Martin, B. K. Jakobsen, N. J. Hassan, M. Kalos, and C. H. June. 2013. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122(6):863-871.
Makkouk, A., and G. J. Weiner. 2015. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Research 75(1):5-10.
Matsushita, H., M. D. Vesely, D. C. Koboldt, C. G. Rickert, R. Uppaluri, V. J. Magrini, C. D. Arthur, J. M. White, Y.-S. Chen, L. K. Shea, J. Hundal, M. C. Wendl, R. Demeter, T. Wylie, J. P. Allison, M. J. Smyth, L. J. Old, E. R. Mardis, and R. D. Schreiber. 2012. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482(7385):400-404.
Maude, S. L., N. Frey, P. A. Shaw, R. Aplenc, D. M. Barrett, N. J. Bunin, A. Chew, V. E. Gonzalez, Z. Zheng, S. F. Lacey, Y. D. Mahnke, J. J. Melenhorst, S. R. Rheingold, A. Shen, D. T. Teachey, B. L. Levine, C. H. June, D. L. Porter, and S. A. Grupp. 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine 371(16):1507-1517.
Melenhorst, J. J., A. M. Leen, C. M. Bollard, M. F. Quigley, D. A. Price, C. M. Rooney, M. K. Brenner, A. J. Barrett, and H. E. Heslop. 2010. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood 116(22):4700-4702.
NCI (National Cancer Institute). 2007. National Cancer Institute immunotherapy agent workshop, July 12, 2007.https://dcb.nci.nih.gov/Reports/Documents/immunotherapyagentworkshop/Final_NCI_Workshop_Proceedings_23Oct07.pdf (accessed May 26, 2016).
NCI. 2014. CAR T-cell therapy: Engineering patients’ immune cells to treat their cancers.http://www.cancer.gov/about-cancer/treatment/research/car-t-cells (accessed June 21, 2016).
NCI. 2016. NCI dictionary of cancer terms.http://www.cancer.gov/publications/dictionaries/cancer-terms (accessed May 23, 2016).
Neefjes, J., M. L. Jongsma, P. Paul, and O. Bakke. 2011. Towards a systems understanding of MHC Class I and MHC Class II antigen presentation. Nature Reviews Immunology 11(12):823-836.
Nishimura, H., T. Okazaki, Y. Tanaka, K. Nakatani, M. Hara, A. Matsumori, S. Sasayama, A. Mizoguchi, H. Hiai, N. Minato, and T. Honjo. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291(5502):319-322.
Oh, S., M. Terabe, C. D. Pendleton, A. Bhattacharyya, T. K. Bera, M. Epel, Y. Reiter, J. Phillips, W. M. Linehan, C. Kasten-Sportes, I. Pastan, and J. A. Berzofsky. 2004. Human CTLS to wild-type and enhanced epitopes of a novel prostate and breast tumor-associated protein, TARP, lyse human breast cancer cells. Cancer Research 64(7):2610-2618.
Page, D. B., T. W. Hulett, T. L. Hilton, H.-M. Hu, W. J. Urba, and B. A. Fox. 2016. Glimpse into the future: Harnessing autophagy to promote anti-tumor immunity with the dribbles vaccine. Journal for ImmunoTherapy of Cancer 4(1):1-5.
Pardoll, D. M. 2012. The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer 12(4):252-264.
Parkhurst, M. R., J. C. Yang, R. C. Langan, M. E. Dudley, D. A. Nathan, S. A. Feldman, J. L. Davis, R. A. Morgan, M. J. Merino, R. M. Sherry, M. S. Hughes, U. S. Kammula, G. Q. Phan, R. M. Lim, S. A. Wank, N. P. Restifo, P. F. Robbins, C. M. Laurencot, and S. A. Rosenberg. 2011. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Molecular Therapy 19(3):620-626.
Porter, D. L., B. L. Levine, M. Kalos, A. Bagg, and C. H. June. 2011. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New England Journal of Medicine 365(8):725-733.
Porter, D. L., W. T. Hwang, N. V. Frey, S. F. Lacey, P. A. Shaw, A. W. Loren, A. Bagg, K. T. Marcucci, A. Shen, V. Gonzalez, D. Ambrose, S. A. Grupp, A. Chew, Z. Zheng, M. C. Milone, B. L. Levine, J. J. Melenhorst, and C. H. June. 2015. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science Translational Medicine 7(303):303ra139.
Pule, M. A., B. Savoldo, G. D. Myers, C. Rossig, H. V. Russell, G. Dotti, M. H. Huls, E. Liu, A. P. Gee, Z. Mei, E. Yvon, H. L. Weiss, H. Liu, C. M. Rooney, H. E. Heslop, and M. K. Brenner. 2008. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine 14(11):1264-1270.
Rahma, O. E., E. Gammoh, R. M. Simon, and S. N. Khleif. 2014. Is the “3+3” dose-escalation Phase I clinical trial design suitable for therapeutic cancer vaccine development? A recommendation for alternative design. Clinical Cancer Research 20(18):4758-4767.
Rizvi, N. A., M. D. Hellmann, A. Snyder, P. Kvistborg, V. Makarov, and J. J. Havel. 2015. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124-128.
Rossig, C., C. M. Bollard, J. G. Nuchtern, C. M. Rooney, and M. K. Brenner. 2002. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: Potential for improved immunotherapy. Blood 99(6):2009-2016.
Savoldo, B., C. M. Rooney, A. Di Stasi, H. Abken, A. Hombach, A. E. Foster, L. Zhang, H. E. Heslop, M. K. Brenner, and G. Dotti. 2007. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood 110(7):2620-2630.
Schadendorf, D., F. S. Hodi, C. Robert, J. S. Weber, K. Margolin, O. Hamid, D. Patt, T.-T. Chen, D. M. Berman, and J. D. Wolchok. 2015. Pooled analysis of long-term survival data from Phase II and Phase III trials of ipilimumab in unresectable or metastatic melanoma. Journal of Clinical Oncology 33(17):1889-1894.
Seidler, M. L. Pennie, E. Veledar, S. D. Culler, and S. C. Chen. 2010. Economic burden of melanoma in the elderly population: Population-based analysis of the Surveillance, Epidemiology, and End Results (SEER)—Medicare data. Archives of Dermatology 146(3):249-256.
Sharma, P., K. Wagner, J. D. Wolchok, and J. P. Allison. 2011. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nature Reviews Cancer 11(11):805-812.
Sivan, A., L. Corrales, N. Hubert, J. B. Williams, K. Aquino-Michaels, Z. M. Earley, F. W. Benyamin, Y. Man Lei, B. Jabri, M.-L. Alegre, E. B. Chang, and T. F. Gajewski. 2015. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350(6264):1084-1089.
Smith, M. C., C. D. Pendleton, V. E. Maher, M. J. Kelley, D. P. Carbone, and J. A. Berzofsky. 1997. Oncogenic mutations in RAS create HLA-a2.1 binding peptides but affect their extracellular antigen processing. International Immunology 9(8):1085-1093.
Sunshine, J., and J. M. Taube. 2015. PD-1/PD-L1 inhibitors. Current Opinion in Pharmacology 23:32-38.
Takaku, S., M. Terabe, E. Ambrosino, J. Peng, S. Lonning, J. M. McPherson, and J. A. Berzofsky. 2010. Blockade of TGF-beta enhances tumor vaccine efficacy mediated by CD8(+) T cells. International Journal of Cancer 126(7):1666-1674.
Tivol, E. A., F. Borriello, A. N. Schweitzer, W. P. Lynch, J. A. Bluestone, and A. H. Sharpe. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3(5):541-547.
Topalian, S. L., C. G. Drake, and D. M. Pardoll. 2015. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 27(4):450-461.
Tran, E., S. Turcotte, A. Gros, P. F. Robbins, Y.-C. Lu, M. E. Dudley, J. R. Wunderlich, R. P. Somerville, K. Hogan, C. S. Hinrichs, M. R. Parkhurst, J. C. Yang, and S. A. Rosenberg. 2014. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344(6184):641-645.
Tromme, I., B. Devleesschauwer, P. Beutels, P. Richez, A. Leroy, J. F. Baurain, F. Cornelis, C. Bertrand, N. Legrand, J. Degueldre, L. Thomas, C. Legrand, J. Lambert, J. Haagsma, and N. Speybroeck. 2014. Health-related quality of life in patients with melanoma expressed as utilities and disability weights. British Journal of Dermatology 171(6):1443-1450.
Twitty, C. G., S. M. Jensen, H. M. Hu, and B. A. Fox. 2011. Tumor-derived autophagosome vaccine: Induction of cross-protective immune responses against short-lived proteins through a p62-dependent mechanism. Clinical Cancer Research 17(20):6467-6481.
Twyman-Saint Victor, C., A. J. Rech, A. Maity, R. Rengan, K. E. Pauken, E. Stelekati, J. L. Benci, B. Xu, H. Dada, P. M. Odorizzi, R. S. Herati, K. D. Mansfield, D. Patsch, R. K. Amaravadi, L. M. Schuchter, H. Ishwaran, R. Mick, D. A. Pryma, X. Xu, M. D. Feldman, T. C. Gangadhar, S. M. Hahn, E. J. Wherry, R. H. Vonderheide, and A. J. Minn. 2015. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520(7547):373-377.
Uno, T., K. Takeda, Y. Kojima, H. Yoshizawa, H. Akiba, R. S. Mittler, F. Gejyo, K. Okumura, H. Yagita, and M. J. Smyth. 2006. Eradication of established tumors in mice by a combination antibody-based therapy. Nature Medicine 12(6):693-698.
Vasaturo, A., S. Di Blasio, D. G. A. Peeters, C. C. H. de Koning, J. M. de Vries, C. G. Figdor, and S. V. Hato. 2013. Clinical implications of co-inhibitory molecule expression in the tumor microenvironment for DC vaccination: A game of stop and go. Frontiers in Immunology 4:417.
Vesely, M. D., M. H. Kershaw, R. D. Schreiber, and M. J. Smyth. 2011. Natural innate and adaptive immunity to cancer. Annual Review of Immunology 29(1):235-271.
Vétizou, M., J. M. Pitt, R. Daillère, P. Lepage, N. Waldschmitt, C. Flament, S. Rusakiewicz, B. Routy, M. P. Roberti, C. P. M. Duong, V. Poirier-Colame, A. Roux, S. Becharef, S. Formenti, E. Golden, S. Cording, G. Eberl, A. Schlitzer, F. Ginhoux, S. Mani, T. Yamazaki, N. Jacquelot, D. P. Enot, M. Bérard, J. Nigou, P. Opolon, A. Eggermont, P.-L. Woerther, E. Chachaty, N. Chaput, C. Robert, C. Mateus, G. Kroemer, D. Raoult, I. G. Boneca, F. Carbonnel, M. Chamaillard, and L. Zitvogel. 2015. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350(6264):1079-1084.
Walter, S., T. Weinschenk, A. Stenzl, R. Zdrojowy, A. Pluzanska, C. Szczylik, M. Staehler, W. Brugger, P.-Y. Dietrich, R. Mendrzyk, N. Hilf, O. Schoor, J. Fritsche, A. Mahr, D. Maurer, V. Vass, C. Trautwein, P. Lewandrowski, C. Flohr, H. Pohla, J. J. Stanczak, V. Bronte, S. Mandruzzato, T. Biedermann, G. Pawelec, E. Derhovanessian, H. Yamagishi, T. Miki, F. Hongo, N. Takaha, K. Hirakawa, H. Tanaka, S. Stevanovic, J. Frisch, A. Mayer-Mokler, A. Kirner, H.-G. Rammensee, C. Reinhardt, and H. Singh-Jasuja. 2012. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nature Medicine 18(8):1254-1261.
Weber, J. S., K. C. Kahler, and A. Hauschild. 2012. Management of immune-related adverse events and kinetics of response with ipilimumab. Journal of Clinical Oncology 30(21):2691-2697.
Weinschenk, T., C. Gouttefangeas, M. Schirle, F. Obermayr, S. Walter, O. Schoor, R. Kurek, W. Loeser, K.-H. Bichler, D. Wernet, S. Stevanović, and H.-G. Rammensee. 2002. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Research 62(20):5818-5827.
The White House. 2016. Fact sheet: Investing in the National Cancer Moonshot. https://www.whitehouse.gov/the-press-office/2016/02/01/fact-sheet-investing-national-cancer-moonshot (accessed May 13, 2016).
Winograd, R., K. T. Byrne, R. A. Evans, P. M. Odorizzi, A. R. L. Meyer, D. L. Bajor, C. Clendenin, B. Z. Stanger, E. E. Furth, E. J. Wherry, and R. H. Vonderheide. 2015. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunology Research 3(4):399-411.
Wolchok, J., R. Ibrahim, V. DePril, M. Maio, P. Queirolo, K. Harmankaya, L. Lundgren, A. Hoos, R. Humphrey, and O. Hamid. 2008. Antitumor response and new lesions in advanced melanoma patients on ipilimumab treatment. In ASCO Annual Meeting Proceedings 26(15):3020.
Wolchok, J. D., A. Hoos, S. O’Day, J. S. Weber, O. Hamid, C. Lebbé, M. Maio, M. Binder, O. Bohnsack, G. Nichol, R. Humphrey, and F. S. Hodi. 2009. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clinical Cancer Research 15(23):7412-7420.
Wolchok, J. D., O. Hamid, A. Ribas, C. Robert, R. Kefford, W.-J. Hwu, J. S. Weber, A. M. Joshua, T. C. Gangadhar, R. S. Dronca, A. Daud, A. Patnaik, R, W. Joseph, H. M. Zarour, X. N. Li, D. Xue, S. Ebbinghaus, S. P. Kang, A. M. P., and F. S. Hodi. 2015. Atypical patterns of response in patients (PTS) with metastatic melanoma treated with pembrolizumab (MK-3475) in keynote-001.http://meetinglibrary.asco.org/content/149728-156 (accessed May 18, 2016).
Wood, L. V., A. Fojo, B. D. Roberson, M. S. B. Hughes, W. Dahut, J. L. Gulley, R. A. Madan, P. M. Arlen, M. Sabatino, D. F. Stroncek, L. Castiello, J. B. Trepel, M. Lee, H. L. Parnes, S. M. Steinberg, M. Terabe, J. Wilkerson, I. Pastan, and J. A. Berzofsky. 2016. TARP vaccination is associated with slowing in PSA velocity and decreasing tumor growth rates in patients with stage D0 prostate cancer. OncoImmunology doi: 10.1080/2162402X.2016.1197459.
Xu, G., T. Smith, F. Grey, and A. B. Hill. 2013. Cytomegalovirus-based cancer vaccines expressing TRP2 induce rejection of melanoma in mice. Biochemical and Biophysical Research Communications 437(2):287-291.
Yadav, M., S. Jhunjhunwala, Q. T. Phung, P. Lupardus, J. Tanguay, and S. Bumbaca. 2014a. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515(7528):572-576.
Yadav, M., S. Jhunjhunwala, Q. T. Phung, P. Lupardus, J. Tanguay, S. Bumbaca, C. Franci, T. K. Cheung, J. Fritsche, T. Weinschenk, Z. Modrusan, I. Mellman, J. R. Lill, and L. Delamarre. 2014b. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515(7528):572-576.
Yang, Y. 2015. Cancer immunotherapy: Harnessing the immune system to battle cancer. Journal of Clinical Investigation 125(9):3335-3337.
Young, R. A., and J. E. DeVoe. 2012. Who will have health insurance in the future? An updated projection. Annals of Family Medicine 10(2):156-162.
Yuan, J., P. S. Hegde, R. Clynes, P. G. Foukas, A. Harari, T. O. Kleen, P. Kvistborg, C. Maccalli, H. T. Maecker, D. B. Page, H. Robins, W. Song, E. C. Stack, E. Wang, T. L. Whiteside, Y. Zhao, H. Zwierzina, L. H. Butterfield, and B. A. Fox. 2016. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. Journal for ImmunoTherapy of Cancer 4:3.






































































































