
9
Reticulation, Divergence, and the Phylogeography–Phylogenetics Continuum
SCOTT V. EDWARDS,*§SALLY POTTER,†‡C. JONATHAN SCHMITT,* JASON G. BRAGG,†‡ AND CRAIG MORITZ†‡
Phylogeography, and its extensions into comparative phylogeography, have their roots in the layering of gene trees across geography, a paradigm that was greatly facilitated by the nonrecombining, fast evolution provided by animal mtDNA. As phylogeography moves into the era of next-generation sequencing, the specter of reticulation at several levels—within loci and genomes in the form of recombination and across populations and species in the form of introgression—has raised its head with a prominence even greater than glimpsed during the nuclear gene PCR era. Here we explore the theme of reticulation in comparative phylogeography, speciation analysis, and phylogenomics, and ask how the centrality of gene trees has fared in the next-generation era. To frame these issues, we first provide a snapshot of multilocus phylogeographic studies across the Carpentarian Barrier, a prominent biogeographic barrier dividing faunas spanning the monsoon tropics in northern Australia. We find that divergence across this barrier is evident in most species, but is heterogeneous in time and demographic history, often reflecting the taxonomic distinctness of lineages spanning it. We then discuss a variety of forces generating reticulate patterns in phylogeography, including
__________________
* Department of Organismic and Evolutionary Biology and Museum of Comparative Zoology, Harvard University, Cambridge, MA 02138; †Research School of Biology, The Australian National University, Acton, ACT 2601, Australia; and ‡Centre for Biodiversity Analysis, Acton, ACT 2601, Australia. §To whom correspondence should be addressed. Email: sedwards@fas.harvard.edu.
introgression, contact zones, and the potential selection-driven outliers on next-generation molecular markers. We emphasize the continued need for demographic models incorporating reticulation at the level of genomes and populations, and conclude that gene trees, whether explicit or implicit, should continue to play a role in the future of phylogeography.
Phylogeography is being revolutionized by a whole-genome perspective driven by next-generation sequencing (NGS) in combination with development of coalescent-based methods of analysis within and among species. The classical phylogeographic foundation from which genome-scale phylogeography has grown was established in the decades spanning the early 1980s’ emphasis on animal mtDNA (Avise et al., 1987; Moritz et al., 1987) to the mid-2000s, just before the first genomewide surveys of genetic variation in humans (Novembre et al., 2008; Novembre and Di Rienzo, 2009). By the early 2000s, phylogeographic surveys of nonmodel species typically included a handful of loci, mostly using methods that facilitated a locus-by-locus phylogeographic analysis (Dolman and Moritz, 2006; Brito and Edwards, 2009). There are now a growing number of studies realizing a distant goal of phylogeography, geographically informed whole-genome resequencing (Jones et al., 2012; Wallberg et al., 2014), as well as many more sampling subgenomes through varied approaches (Potter et al., 2016; McCormack et al., 2012; Smith et al., 2014a; Harvey and Brumfield, 2015). With the expansion to genomewide analyses afforded by NGS, phylogeographic analysis has necessarily expanded its analytical toolkit.
The increasingly routine analysis of genome-scale data has blurred the disciplinary boundaries between phylogeography and its sister discipline, population genetics, and has allowed phylogeography to contribute to endeavors such as scans for selection and association mapping (Edwards et al., 2015). Indeed, with burgeoning data and increasing applications of related analytical tools, such as site-frequency spectra and coalescent simulations, we can ask whether and how phylogeography is now distinct from population genetics (Edwards et al., 2015). We contend that there is still value in the original conception of phylogeography as a bridge between population biology and phylogenetics (Avise et al., 1987) (Fig. 9.1). This bridge can be thought of across geography and time, as is often the case with practitioners, or across gradients of migration rates and linkage disequilibrium (Slatkin, 2008), with the former decreasing and the latter increasing from the population to phylogenetic scale. That phylogeography sits centrally in this process-oriented space emphasizes the importance of understanding interactions between reticulation (gene flow/introgression and recombination), drift, and protracted isolation.
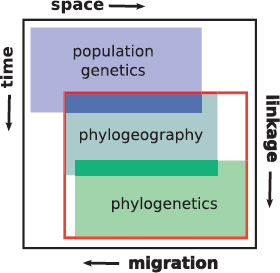
This combination of processes sets phylogeography apart from traditional population genetics and phylogenetics.
Scanning entire genomes of closely related organisms has unleashed a level of heterogeneity of signals that was largely of theoretical interest in the PCR era. This genomic heterogeneity is profoundly influencing our basic concepts of phylogeography and phylogenetics, and indeed our views of speciation processes. It is now routine to encounter a diversity of gene trees across the genome that is often as large as the number of loci surveyed (Song et al., 2012; Fontaine et al., 2015). Aside from variation induced by the coalescent process within and across species, we are only beginning to understand how such gene tree heterogeneity arises (Fontaine et al., 2015; Nater et al., 2015). Recognition of this heterogeneity has driven the development of phylogenetic methods for accommodating such conditional independence of gene trees, so-called “species tree”
methods (Liu et al., 2009a, 2015; Knowles and Kubatko, 2010; Edwards, 2016). For phylogeographic analyses, at the transition from population structure to phylogenetic divergence, incomplete lineage sorting (ILS) is prevalent where populations have been separated for less than 4Ne generations, where Ne is the effective population size (Neigel and Avise, 1986; Rosenberg, 2002). Another increasingly evident source of heterogeneity is introgression among species (Fontaine et al., 2015) (Fig. 9.2), the converse of the deep phylogeographic structure often observed in low-dispersal taxa. Such reticulation has long been recognized in plants, or in microbial systems, where horizontal gene transfer is an established paradigm. Increasingly, zoologists are also finding evidence for extensive movement of genes between phenotypically divergent taxa (Novick et al., 2009; Lamichhaney et al., 2015), including nonsister species. Such observations have increased attention to models of “speciation-with-gene-flow” (Feder et al., 2012). The new genome-scale analyses are causing evolutionary biologists to reevaluate the very nature of species (Mallet, 2008; Mallet et al., 2016), which, in some cases, appear to maintain phenotypic distinctiveness despite extensive gene flow across most of the genome (Mavárez et al., 2006; Heliconius Genome Consortium, 2012; Soria-Carrasco et al., 2014; Malinsky et al., 2015), and to recognize introgression as an important source of adaptive traits in a variety of study systems (Nosil and Crespi, 2004; Rheindt and Edwards, 2011; Nosil and Feder, 2012). Analytically, evidence of introgression among species is driving the emergence of network models of diversification (Nakhleh, 2013). Clearly genome-scale biology and the abundant reticulations across the “Tree of Life” are turning much of evolutionary biology, including phylogeography, on its head.
In this chapter, we explore the themes of reticulation and the genomics of speciation as key processes across the phylogeography–phylogenetics continuum. Reticulation presents challenges to many concepts and methods in phylogeography and speciation that were not as evident when we were locus-poor in the PCR era. Reticulation in the form of recombination causes gene trees to depart from a strictly bifurcating pattern, hence posing challenges for some methods of reconstructing evolutionary history (Lanier and Knowles, 2012). Recombination has also long been known to play a central role in speciation (Butlin, 2005), and the suppression of recombination, such as occurs in chromosomal inversions, can dramatically reduce the local genomic rate of gene flow (Joron et al., 2011; Nishikawa et al., 2015). Reticulation via gene flow is to be expected among intraspecific lineages that are the classical domain of phylogeography (Fig. 9.2). Finally, reticulation is becoming increasingly conspicuous at the level of diverging species and adaptive radiations (Rheindt and Edwards, 2011; Ellegren et al., 2012; Yeaman, 2013; Hermansen et al., 2014; Delmore et al., 2015;
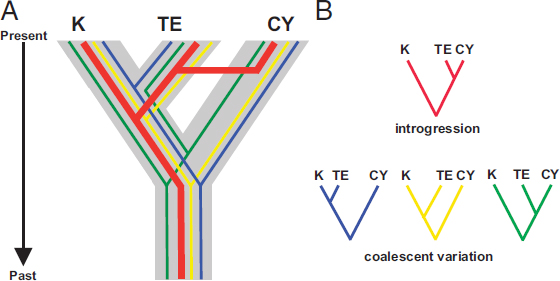
Lamichhaney et al., 2015), and is causing biologists to consider “ephemeral species,” with frequent lineage mergers, not only among sister taxa but between lineages with any geographic co-occurrence (Rosenblum et al., 2012). The goal, however imperfect it is now realized, is to develop and apply models that integrate phylogeography, demography, and genome evolution in ways that will allow more nuanced interpretation of myriad interacting evolutionary processes from patterns of genomic diversity (Cutter, 2013). Acknowledging and modeling reticulation at various levels in the hierarchy of life will be an important part of reaching this goal.
KEY PROCESSES OF DIVERGENCE AND RETICULATION IN NATURE
Comparative Phylogeography Across the Australian Monsoonal Tropics
In essence, comparative phylogeography is about establishing commonalities of spatial patterns of genetic and gene tree diversity across codistributed species (Bermingham and Moritz, 1998; Avise, 2000). Combined with population genetic (coalescent) and spatial modeling (Knowles, 2009b), this effort has yielded insights into biogeographic history, such as locations of refugia and expansion areas (Hewitt, 2011) and the varying effects of ecological or physical dispersal barriers. In a comparative setting, such studies can identify how landscape features and regional climatic variation have interacted with the varying ecologies of species to shape current diversity (Carnaval et al., 2014) and how these interactions can influence speciation processes (Moritz et al., 2009; Ellegren et al., 2012).
To explore divergence vs. reticulation processes in a comparative context, we focus on phylogeographic data for ecologically diverse species from northern Australia, a vast stretch of monsoonal savanna and woodlands with interspersed ancient sandstone plateaus (Bowman et al., 2010) (Fig. 9.3). That this rich tropical fauna is biogeographically structured has long been known (Keast, 1961) and formalized using cladistic biogeography by Cracraft (1986), who recognized a basal dichotomy across the treeless “Carpentarian Barrier” (CB) separating the Kimberley (KIM) and Top End (TE) faunas from the faunas in Cape York (CY) and the eastern Australian forest (EF), as well as New Guinea. KIM and TE are, in turn, separated by hot, low-relief, and relatively dry regions associated with several smaller barriers (Eldridge et al., 2011), collectively referred to here as the Kimberley-Top End Barrier (KTEB). Early sequence-based phylogeographic studies in babblers [Pomatostomus (Edwards, 1993)] reported deep divergence across the CB relative to divergence within the eastern and western regions of the continent. Subsequent multilocus analyses of several avian systems revealed mostly Pleistocene divergences across the CB for congeners (Jennings and Edwards, 2005; Balakrishnan et al., 2010; Toon et al., 2010), as well as within species (Lee and Edwards, 2008; Kearns et al., 2011). Some studies examining divergence across the region have discovered clines (Rollins et al., 2012) or complex reticulate patterns in the form of introgression; for example, in butcherbirds (Cracticus), populations east of the KTEB are introgressed with mtDNA from populations of arid-adapted species to the south that expanded during the Last Glacial Maximum, whereas populations west of the KTEB are not introgressed (Kearns et al., 2014).
Fewer multilocus phylogeographic studies have been conducted for mammals across the monsoonal tropics, yet some common themes
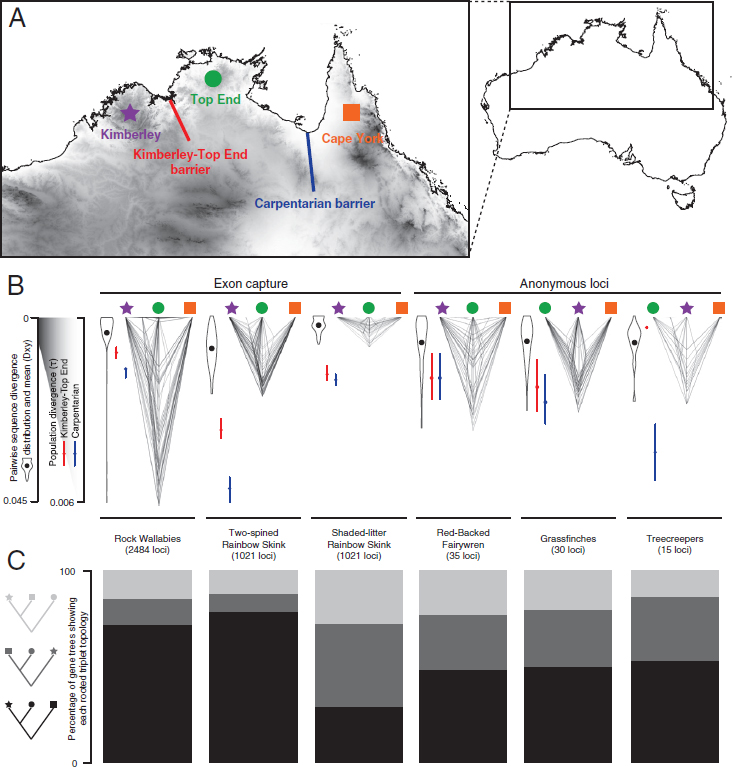
emerge. Rock-wallabies (Petrogale), specialists of rocky habitats as the name implies, are strongly structured across the disjunct sandstone plateaus of the region, with deeper divergences across the CB than across the KTEB (Potter et al., 2012a,b) (Fig. 9.3). Other macropod species have different degrees of geographic and genetic discontinuity across the CB, suggesting the species’ ecology has played a key role in their ability to adapt and persist across this region. The antilopine wallaroo (Macropus antilopinus), a savanna-woodland specialist, has a disjunct distribution but shallow divergence on either side of the CB grasslands, suggesting recent gene flow or range expansion. By comparison, a more ecologically generalized and widespread congener, the common wallaroo (Macropus robustus), has a more continuous distribution but substantial divergence across the CB (Eldridge et al., 2014). Preliminary analyses of other mammals suggest deep divergences across the CB, but require more expansive molecular study (Malekian et al., 2010; Aplin et al., 2015).
Low-dispersal species, such as lizards and frogs, have been the subject of a burst of recent multilocus phylogeographic studies across the region, including early applications of NGS in this context. Relative to birds and mammals, these taxa exhibit phylogeographic structure at a finer spatial scale, often with cryptic species and greater phylogenetic depth among regions, possibly reflecting a combination of lower dispersal and higher localized persistence through cycles of harsh climate. Deep structure across the CB, and often also the KTEB, is observed across phylogenetically and ecologically diverse reptiles, including species complexes of agamid lizards (Melville et al., 2011), rainbow skinks (Dolman and Hugall, 2008; Potter et al., 2016), several species complexes of geckos (Oliver et al., 2014; Moritz et al., 2016), and toadlet frogs (Catullo et al., 2014). In many cases, the divergence across the CB appears at deep phylogenetic scales rather than within species. For example, Carlia rainbow skinks have radiated across the KIM and TE, yet these taxa diverged from the eastern species of Carlia in the mid-Miocene (Dolman and Hugall, 2008). Analyses of ~2,000 exons for the two-spined rainbow skink (Carlia amax) inferred recent population expansion from western KIM across the KTEB to the western TE, emphasizing that the KTEB is a more porous filter than the CB (Potter et al., 2016). Studies of low-dispersal taxa (Catullo et al., 2014; Moritz et al., 2016; Potter et al., 2016) are also revealing congruent patterns at a finer scale than the major barriers envisioned by Cracraft (1986). These congruent patterns include deep structuring between offshore islands and mainland populations and an unexpected north-south split from the TE to the northern desert region (Catullo et al., 2014; Moritz et al., 2016; Potter et al., 2016). Closely related northern desert taxa often have ranges that are more widespread than those ranges across the savannas and sandstones to the north, and sometimes with evidence of broad-scale introgression
(Catullo et al., 2014; Moritz et al., 2016; Potter et al., 2016). For example, in contrast to strong phylogeographic structuring within C. amax, an arid-adapted congener, Carlia munda (shaded-litter rainbow skink) includes a single widespread clade from the west coast across the northern desert to the east coast (Fig. 9.3).
Gene Tree Heterogeneity Across the CB
The complex landscapes and dynamic climate history across this region have resulted in a combination of often strongly vicariant processes across the CB and a mix of divergence and dispersal or introgression across the KTEB. Given that gene tree heterogeneity arises from both ILS and gene flow between populations (Fig. 9.2), we can expect to see a more dominant phylogenetic signal across the CB in which the deepest split for a majority of gene trees spans the CB, with fewer loci having their deepest split across the KTEB, or between the CY/EF and TE (Fig. 9.3). We explored this hypothesis for exemplar avian, mammal, and lizard taxa for which we had multilocus sequence data spanning these geographic regions (Fig. 9.3 and Tables S1 and S21). As expected, among four-tip gene trees (one allele sampled for the KIM, TE, and CY/EF plus outgroup), we found diverse gene tree distributions across the region, with gene trees exhibiting deeper divergence times across the CB than the KTEB being the most frequent (Fig. 9.3 and Table S3). An exception to this pattern is C. munda, the more arid-adapted lizard, in which the dominant gene tree is one in which the TE and CY alleles are sisters, implying a more isolation-by-distance than vicariance model (Cracraft, 1986). Analyzing the larger datasets in which these simple gene trees are embedded with coalescent models (Rannala and Yang, 2003) uniformly suggests deeper population divergence and speciation across the CB than across the KTEB, although these divergences are quite close temporally in several cases (Fig. 9.3 and Fig. S1). Although our sample sizes are small, the analysis also suggests that the highest genetic diversity currently segregating within each complex varies among regions; in fairy wrens and wallabies, the highest diversity is in the CY/EF, whereas diversity is similar among regions in C. amax. Finally, the analysis does not support a key prediction of the simplest vicariance scenario: that the effective population sizes of descendant lineages are smaller than the sizes of the ancestral populations inhabiting the area before the vicariant event. Our estimates of ancestral Ne for at least four of the six species groups are smaller than for contemporary lineages
__________________
1 Supporting information for this chapter, which includes additional text, Figs. S1 and S2, and Tables S1, S2, S3, and S4, is available online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1601066113/-/DCSupplemental.
in the KIM, TE, or CY. A challenge with our brief analysis of comparative phylogeography across the monsoon tropics of Australia is the diversity of markers, which prevents easy comparison across groups due to differences in substitution rate. Use of a common set of markers, such as provided by various forms of target capture (Brumfield et al., 2003; Lemmon and Lemmon, 2012; Jones and Good, 2016), whether coding or noncoding, will be an important focus of future research.
Reticulation Driven by Ecology and Introgression
Our comparative phylogeographic analysis for northern Australia highlights the complex mix of divergence and reticulation and diverse spatial and temporal scales of phylogeographic structure that can emerge. Much of this heterogeneity appears to relate to differences among species in their capacity to persist or disperse across the landscape as climates oscillated over the Quaternary. Whereas it is convenient to focus on common patterns of divergence, in a classic vicariance mindset, closer attention to differing outcomes of reticulation, such as we have seen when comparing the results for C. munda with the results for other taxa spanning the CB, will yield more insight into speciation processes (Harrison and Larson, 2014).
Following secondary contact, genetically distinct populations can form “tension zones,” maintained over time by a balance between dispersal and selection against hybrids (Barton and Hewitt, 1985), progressively merge via introgression [i.e., ephemeral taxa (Cutter, 2013)], or overlap while maintaining their integrity (Fig. 9.4). A special case of introgression occurs when an expanding lineage overrides a static (relictual) one, but is itself invaded by genes from the resident population due to sequential founder events during the spatial expansion (Ray and Excoffier, 2009). Over time, introgressed chromosome segments will recombine between lineages, leading to a mosaic of coalescent histories within and across loci. These reticulation events can manifest at two scales: in genetic clines, for single-nucleotide polymorphisms (SNPs) at the contact zone(s) themselves, and in lineage-scale migration, as estimated from allopatric populations using isolation-migration (IM) models (Fig. 9.4). Genome-scale data are enabling new approaches (reviewed in Payseur and Rieseberg, 2016), including genomic clines (Gompert and Buerkle, 2009) and analyses of lengths of introgressed haplotype blocks (Pool and Nielsen, 2009). Given estimates of recombination rate, the length of immigrant haplotypes can, in principle, be used to estimate the timing of recent introgression events at a lineage scale, a parameter that has proved difficult to infer from IM models (Sousa et al., 2011).
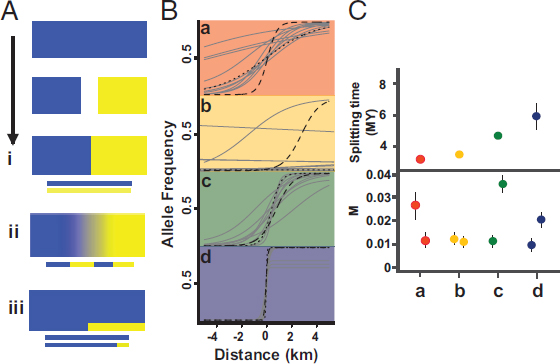
In the context of comparative phylogeography, insights into reticulation processes can be gleaned by comparing outcomes for taxa with varying ecologies and lineage divergence times across a common geographic and paleoenvironmental setting. Suture zones can be useful for this purpose, where multiple taxa have co-occurring contact zones (Dasmahapatra et al., 2010). The fauna endemic to the rainforest of northeastern Australia are a case in point. Climate-driven fluctuations of rainforest-dependent taxa on mountaintops have resulted in spatially concentrated contact zones between morphologically indistinguishable but genetically distinct lineages (Moritz et al., 2009). A comparative analysis of clines and genetic disequilibria across different contact zones (Singhal and Moritz, 2013) revealed less introgression and stronger genetic disequilibrium between more divergent lineage pairs, showing that reproductive isolation between these phenotypically cryptic lineages scales with divergence
time (Fig. 9.4). However, at the lineage scale, levels of gene flow inferred from IM analyses of comparative transcriptomes are generally low and do not scale with divergence time (Fig. 9.4). These contrasting patterns remind us that estimates of gene flow are often averaged over the entire divergence history.
The Nexus of Comparative Phylogeography and Speciation Genomics
So how will a fully genomic perspective enrich our understanding of the nexus between comparative phylogeography and speciation? A plethora of recent whole-genome comparisons among sister taxa reveal fascinating, but complex, heterogeneity of divergence across the genome (reviewed in Payseur and Rieseberg, 2016). The most common outcome among recently diverged taxa is stronger differentiation on X and Z sex chromosomes than autosomes and scattered “islands” of high divergence against a background of low divergence. Islands of divergence were initially taken as suggesting locations of incompatible genes in the context of ongoing gene flow (Wu, 2001). However, it is also possible that they reflect varying levels of background selection in the absence of gene flow (Payseur and Nachman, 2005; Cruickshank and Hahn, 2014), leading to reinterpretation of some high-profile examples (Burri et al., 2015).
A key factor emerging from these studies and earlier scans of intraspecific diversity is the strong effect of recombination rate variation on the spatial patterning of genomic diversity, mediated most strongly by hitchhiking (Charlesworth et al., 1993). Thus, we expect to see reduced within-lineage diversity in regions of low recombination, with a corresponding increase in divergence using measures that are sensitive to levels of within-lineage diversity [e.g., Wright’s fixation index Fst (Nachman and Payseur, 2012)]. Paradoxically, it has also been proposed that low-recombination regions, as might occur within chromosomal inversions or near centromeres, will accumulate locally adapted alleles, thereby contributing to genetic incompatibility between lineages (Kirkpatrick, 2010). Empirical evidence for this proposal is mixed, but there are some positive examples (Lemmon et al., 2012; Lemmon and Lemmon, 2013; Zhou et al., 2014). Finally, genome comparisons among closely related taxa have also highlighted introgression of adaptive alleles from one lineage to another (Rheindt and Edwards, 2011; Hedrick, 2013), an old concept reborn (Lewontin and Birch, 1966). Such alleles can readily flow across contact zones even if there is strong hybrid breakdown. Analytical challenges aside, such cases point to the exciting prospect of understanding how adaptive evolution influences divergence and reticulation among lineages.
One limitation of many of the above analyses of genomic divergence during speciation is that the historical biogeographic and environmental setting of isolation and reconnection of diverging lineages over time is often not well established (Payseur and Rieseberg, 2016). Understanding these processes is the core business of phylogeography, and closer interaction between analyses of historical biogeography and speciation genomics can be expected to bear fruit. Conversely, in the genomic era, comparative phylogeographers will not just have to master details of environmental history, species’ ecology, and the plethora of methods for NGS and demographic inference but will also have to comprehend effects of selection and recombination rate variation across the genome. This challenge is exciting and will serve to strengthen further the link between population genomics and phylogenetics.
RECONSTRUCTING PROCESSES OF DIVERGENCE AND RETICULATION
Evolution of Molecular Markers in the Next-Generation Era
Having outlined some of the key processes of divergence and reticulation observable with genomic data, we now ask: How do we reconstruct phylogeographic history in the era of NGS? What new practical and analytical challenges do the increased detail afforded by NGS bring to phylogeographic reconstruction? The glimpse of comparative phylogeography across the CB in northern Australia makes clear the implications of one of the key components of any effort at reconstructing demographic history, namely, how we select molecular markers and the need for easy comparison across datasets. As a start, we may ask: Has NGS finally liberated phylogeographers from the constraints of marker choice, allowing unfettered access to the most appropriate markers for the questions being asked? Which combinations of markers may promote the further integration of phylogeography and phylogenetics? In these still-early days of next-generation phylogeography, marker choice is still constrained somewhat by technical and resource considerations, and will remain so until whole-genome sequencing of individuals or at least exemplars of the clades being studied becomes routine. The emergence of several widely used NGS platforms and marker suites in the past few years illustrates this point. For example, the flanking regions of ultraconserved elements (UCEs) have been promoted as suitable for phylogeographic questions, with the advantage that they are variable and their presence in can be predicted in uncharacterized genomes (McCormack et al., 2013). Another comparison (Brandley et al., 2015) found similar phylogeographic resolution between exons (drawn randomly from transcriptomes) vs. anchored
hybrid enrichment (AHE) loci, which mostly target conserved exons (Lemmon and Lemmon, 2013; Lemmon et al., 2012). Exon capture has been effectively used to study diverging lineages of both vertebrates and invertebrates (e.g., Zhou et al., 2014; Hugall et al., 2016; Potter et al., 2016) and is particularly appropriate for retrieving genomic data from museum specimens (Rowe et al., 2011; Bi et al., 2013; Jones and Good, 2016).
Arguably, most UCE, AHE, or exon capture loci that have been used thus far for next-generation phylogeography are under mild or even strong purifying selection. Such selection is not necessarily a problem; after all, much of the animal mitochondrial genome, despite its high variability, is under purifying selection. However, purifying selection will likely reduce variation and bias the site-frequency spectrum toward low-frequency variants in a manner similar to, but less extreme than, selective sweeps, making gene trees compressed toward the tips (Nielsen, 2005). There is also clear evidence that loci in the vicinity of exons exhibit reduced levels of ILS compared with anonymous genomic regions (Scally et al., 2012). So long as researchers frame their findings within the context of the diversity of loci found throughout the genome, exons and UCEs are likely to remain a powerful force in phylogeography. The pervasiveness of natural selection, particularly for species with large effective population sizes (Corbett-Detig et al., 2015), is, however, a force with which phylogeographers have not yet fully come to grips. One wonders whether any of the loci used in phylogeography in the next-generation era are genuinely neutral.
The approach using restriction-site-associated DNA sequences, or RAD-seq, is a popular application of NGS to phylogeography, and yields large but sometimes patchy matrices of relatively short and mostly noncoding loci (Andrews et al., 2014a), which are often analyzed in the form of SNPs. Such markers can be powerful measures of phylogeographic structure and, in some cases, seem relatively free of strong selection (Dierickx et al., 2015). Within-locus recombination is irrelevant to SNPs, whereas recombination may pose challenges for analysis of the longer loci such as are generated by target capture and AHE. RAD-seq loci are less amenable to the type of gene tree building that has characterized phylogeography (Hare, 2001; Brumfield et al., 2003; Knowles, 2009a), but few NGS loci of any kind yield highly resolved gene trees when applied on a phylogeographic scale. The power of next-generation methods lies primarily in generating more independent loci, although the phylogenetic informativeness of individual loci also plays a role, especially in species tree reconstruction (Liu L et al., 2015). Going forward, it will be important to compare the behavior and informativeness of different types of markers and genomic compartments explicitly in phylogeographic settings (Fontaine et al., 2015; Nater et al., 2015).
The ideal phylogeographic marker in the next-generation era presumably depends on the questions being asked and the temporal and taxonomic scales over which comparisons are made. Whereas introns and anonymous loci were popular sequence-based markers in the PCR era (Hare et al., 1996), and continue to be captured by various NGS approaches (Lemmon and Lemmon, 2012; Barrow et al., 2015; Perez et al., 2016), targeting of such unconstrained sequence-based markers has made few inroads in the next-generation era, presumably because compared with exons (Bragg et al., 2016), such loci are difficult to predict, and therefore capture, in unknown genomes using probes from other species. Herein lies a conflict between the ease of retrieving markers and their variability within species: Until whole-genome sequencing of phylogeographic exemplars allows us to design probes that are optimal for a given species, and consistent across taxa, the practicalities of easily and cheaply capturing large numbers of loci may tend the field toward conserved loci. Ultimately, phylogeographers should embrace a diversity of marker types even within individual studies, not only to allow the phylogeographic history of different marker types to illuminate each other but also to study genomic diversity and history in an unbiased way that facilitates the discovery of genomic loci underlying adaptation.
Insight into Processes of Reticulation from Gene Tree Outliers
Gene tree outliers, like Fst outliers, may be important indicators of nonneutral or locus-specific processes in the genome. We used a newly proposed gene tree outlier approach, KDEtrees (Weyenberg et al., 2014), to explore the behavior of gene tree distributions in empirical phylogeographic and low-level phylogenetic datasets of several marker types (Fig. S2 and Table S4). KDEtrees appears effective at identifying loci that result from horizontal gene transfer or are clear outliers, such as gene trees generated by a species tree different from the majority. However, it is unclear how KDEtrees behaves when confronted with loci influenced largely by demographic processes, or how the number of outliers varies by marker type. Although our sample size is small, our analysis of eight datasets (Fig. S2 and Table S2) suggests that many phylogeographic and transcriptome datasets harbor surprisingly few gene tree outliers, and so conform well to expected distribution based on overall patterns and levels of divergence. For example, given our chosen level of sensitivity (λ = 1.5), for which we expect roughly 5% of gene trees to exhibit outlier behavior simply by chance, none of the datasets we analyzed contains a significant number of outliers. In the future, the KDEtrees approach, and other methods (Gruenstaeudl et al., 2015), should be a useful tool to explore gene tree heterogeneity within and between datasets.
METHODS FOR DETECTING RETICULATION: RECOMBINATION AND INTROGRESSION
Reticulation and Phylogenetic Networks
As we have seen, as phylogeography and speciation studies begin to probe the genomes of diverse species on a large scale, reticulation, in the forms of introgression and recombination, appears much more common than previously supposed. Accordingly, a major challenge going forward is to incorporate reticulation as a standard component of phylogeographic analysis. Many computational methods targeted at the phylogeography–phylogenetics continuum necessarily ignore some kinds of reticulation. Key examples include models to estimate species trees from multiple unlinked loci using the multispecies coalescent (MSC) model (Edwards, 2016). MSC methods ignore two fundamental aspects of reticulation: recombination within loci and postspeciation hybridization. Some MSC methods (Liu et al., 2009b) are known via simulations and theoretical arguments to be robust to reticulations, such as introgression, particularly when datasets are large and when introgression is confined to a subset of loci. However, other MSC methods are not robust to such model violations (Edwards, 2016). It is not surprising that phylogeographers are among the most comfortable working with MSC methods because of the similarity in assumptions they apply to multilocus datasets. At the same time, due to their familiarity with reticulating lineages, phylogeographers are the most likely to identify shortcomings arising from the inherent simplifications of standard MSC models.
Conceptually, networks subsume trees; networks are trees with reticulation (figure 1 of Edwards et al., 2016). Genome-scale evidence for introgression is renewing enthusiasm for coalescent phylogenetic models that allow for hybridization between diverging lineages. Several recent phylogenomic datasets, including those datasets analyzing human populations as well as distantly related lineages of birds or mammals (Hallström and Janke, 2010; Jarvis et al., 2014), have noted signals for reticulation in the form of ancient interlineage hybridization. Phylogenomic network models based on the MultiSpecies Network Coalescent (MSNC) (Park and Nakhleh, 2012; Nakhleh, 2013; Stenz et al., 2015) are likely to be an important new tool for phylogeneticists in general and phylogeographers in particular. Early studies suggest that application of the MSNC to genome sequences from diverging species will yield new insights into complex evolutionary histories of divergence and reticulation (Wen et al., 2016).
Better insight into the presence of reticulation at the level of populations need not involve computationally intensive algorithms. For example, application of simpler SNP-based tests of introgression and admixture [e.g., the “ABBA-BABA” test (Durand et al., 2011)] will help flag
phylogeographic scenarios that may be more complex than originally envisioned. Although they have yet to make inroads into the phylogeography of nonmodel species, a suite of recently developed drift (F) statistics, related to but distinct from Wright’s F-statistics, provide simple and powerful metrics to test various models of population history, such as whether populations are related in a treelike fashion [Reich et al., 2009; Patterson et al., 2012; reviewed by Peter (2016)]. Tools for model selection in phylogeography (Hickerson et al., 2010; Ray et al., 2010; Carstens et al., 2013; Excoffier et al., 2013; Tsai and Carstens, 2013; Chan et al., 2014; Xue and Hickerson, 2015) will also be critical for determining whether reticulation at the population level is an important part of the demographic history under study.
Capturing Heterogeneity with the Sequentially Markovian Coalescent
Recombination within loci violates assumptions of most phylogenomic analyses, whether informed by the MSC or not. The departure from the assumptions of the MSC could be particularly acute for datasets consisting of sequences from long loci relative to the distance over which linkage disequilibrium decays, which can be <1 kb in many organisms. The one simulation study exploring effects of intralocus recombination (without introgression) on the performance of species tree methods (Lanier and Knowles, 2012) found little effect, and then only on very short trees, as is typical of phylogeographic datasets. However, Potter et al. (2016) observed incongruent and less resolved species trees among lineages of C. amax (as in Fig. 9.3) when using full-length exons compared with the longest nonrecombining segments of these loci. Still, phylogeography is no stranger to intralocus recombination. Several phylogeographic models have been adapted to incorporate recombination (Becquet and Przeworski, 2007; Naduvilezhath et al., 2011), using information from the joint site frequency spectrum among loci and other data. If justified, such models could be adapted to MSC and MSNC methods to allow for intralocus recombination. Additionally, several postgenome phylogeographic models have emerged that incorporate recombination via the sequentially Markovian coalescent, a new approach that models the coalescent site-by-site along the genome, exploiting the variation in site patterns among linked SNPs (Marjoram and Wall, 2006; Li and Durbin, 2011; Wang et al., 2014). Sequentially Markovian coalescent models have obvious applications in traditional species tree methods and may alleviate lingering concerns about recombination. A final means of addressing the issue of intralocus recombination in phylogeography is by using SNP data, which obviates intralocus recombination. Phylogeographic models using SNP data have been available for a number of years (e.g., Beerli and Felsenstein,
1999; Beerli and Palczewski, 2010), and several MSC methods (Bryant et al., 2012; Chifman and Kubatko, 2014) now use linked or unlinked SNP data to estimate phylogenetic trees without explicitly estimating constituent gene trees. It remains to be seen whether the limited genealogical information in SNPs is compensated for by the large number of SNPs that can be collected in typical phylogenomic datasets.
CONCLUSION
Phylogeography has come a long way from its origins of analyzing single gene trees across geography (Avise et al., 1987). Sophisticated statistical inference, integration with spatial modeling, model choice, parameter estimation, and now access to sequence or SNP data for thousands of loci have all enriched the field tremendously. It will be interesting to see how closely future phylogeographers adhere to its conceptual roots, the “mitochondrial DNA bridge,” as mirrored in gene trees empirically derived from nuclear sequence data. On the one hand, extensive reticulation in the form of recombination and the convenience of analyzing large numbers of unlinked SNPs with rapid parametric tests may be ushering in an era of phylogeography beyond gene trees, or, at the very least, an era that acknowledges them only implicitly, via connections with coalescent theory (Degnan and Rosenberg, 2009; McVean, 2009). On the other hand, some of the currently popular methods of locus capture are showing promise for capturing genetic diversity in the form of gene trees, even if weakly resolved at lower taxonomic levels. What seems clear is that next-generation approaches are pushing phylogeography toward a future dominated by SNPs or sequence data for thousands of loci, a positive development that we believe will help bridge the phylogeography–phylogenetics continuum as envisaged by Avise et al. (1987).
In the future, we can expect integration of phylogeography with increased understanding of genome organization and parameters, such as variation in recombination rate across the genome (Cutter, 2013). We can also expect continuing integration of phylogeography with speciation biology and with analyses of adaptive variation, including phenotype-genotype associations (Edwards et al., 2015). Almost by definition, phylogeography will retain its distinctions from sister disciplines like population genetics by its emphasis on broad geographic sampling and the natural history origin of the questions it seeks to answer. One of the thrills of phylogeographic research is the ability of researchers to absorb cutting-edge technologies that now put whole-genome variation within our grasp, yet also retain the exploratory field spirit that has motivated the discipline since its inception. With such a detailed view of genomic variation across geography, reticulation is likely to be omnipresent, push-
ing phylogeography to reinvent itself, question its foundations, and strive for new syntheses.
ACKNOWLEDGMENTS
We thank Mark Eldridge (Australian Museum Research Institute) for access to tissues and data of rock-wallabies; Ke Bi and Sonal Singhal for assistance with Carlia data collection and preparation of Fig. 9.4; Brant Faircloth, John McCormack, and Robb Brumfield for assistance in acquisition of online datasets; and John Wakeley, Rudy Yoshida, and Alan Lemmon for helpful discussion. S.V.E.’s research is supported by the U.S. National Science Foundation and Harvard University. S.P., J.G.B., and C.M. are supported by grants from the Australian Research Council.




















