
4
Understanding Risks Related to Future Biotechnology Products
There is a history of risk assessments and regulatory determinations for biotechnology products through the Coordinated Framework. However, the scope, scale, complexity, and tempo of products to be developed in the next 5–10 years (outlined in Chapter 2) will likely be substantially different from the scope, scale, complexity, and tempo of products developed between the 1980s and 2016. Under these new conditions, regulators will have to assess whether the risks of future products are different from products that have come before and whether the risk-analysis approaches that have been used (outlined in Chapter 3) are sufficient. If those approaches need to be revised for future products, then regulators will need to have the appropriate scientific capabilities, tools, and expertise to support oversight of those products.
RISKS FROM FUTURE BIOTECHNOLOGY PRODUCTS: SIMILARITIES TO THE PAST AND GAPS GOING FORWARD
As discussed in Chapter 3 (see Box 3-1), risks are comprised of undesirable outcomes (what), the possibility of occurrence (how likely), and state of reality (ways the risk occurs in pathways) (Renn, 1992). Risk-assessment endpoints are societal, human health, or environmental values that need to be managed or protected (NASEM, 2016a). There can be many pathways by which those risk-assessment endpoints are reached, and risk assessments provide a quantitative or qualitative evaluation of the endpoints.
The committee’s statement of task posed two questions related to risk:
- Could future biotechnology products pose different types of risks relative to existing products and organisms?
- Are there areas in which risks or lack of risks of biotechnology products are well understood?
The first question in the committee’s charge was interpreted as a request to reflect on the degree to which regulatory human health and ecological endpoints used in risk assessments for existing
biotechnology products are likely to be similar to or different from the endpoints that would be selected when assessing risks for future biotechnology products. For ecological risk-assessment endpoints, the committee considered the potential similarities and differences between explicitly defined ecological entities and their attributes within the ecosystems possibly at risk (see EPA, 1998) for existing biotechnology products and future biotechnology products. For human health risk-assessment endpoints, the similarities and differences considered for existing and future products were those associated with responses of individuals at the subcellular, cellular, tissue, and individual levels of biological organization; such responses typically serve as endpoints within specified human subpopulations (for example, see NRC, 2007, 2009). To compare specific risk-assessment endpoints between existing and future products, the committee was also asked to evaluate whether the exposure and effect pathways under which endpoints can be expressed differed between the two. That is, will the ways humans or an environment may be exposed to or the degree to which they may be affected by a future biotechnology product differ from the ways exposure and effect occur for existing biotechnology products? The U.S. Environmental Protection Agency (EPA, 1998), Suter (2007), and a 2009 National Research Council report (NRC, 2009) discuss the need to specify the spatial and temporal dimensionality of an assessment during its problem-formulation phase. Consideration of dimensionality also incorporates concepts of toxicity (NRC, 2007) and adverse outcome pathways (Ankley et al., 2010), which link subcellular and cellular perturbations to potential adverse outcomes at the tissue, organ, individual, population, or community level of biological organization. The committee incorporated the perspectives on dimensionality by EPA, Suter, and the 2007 and 2009 National Research Council reports when it evaluated what may be different or similar for risk-assessment endpoints associated with existing versus future biotechnology products.
The term “well understood” in the statement of task was interpreted to mean that the degree of uncertainty in estimates of risk does not preclude a formulation of risk-management options, consistent with the goals and objectives established during the problem-formulation phase of a risk assessment (see EPA, 1998; NRC, 2009; Box 4-1). Although the committee interpreted “well understood” in a risk-analysis context, it noted that “well understood” is a value judgment that in some instances can be informed, at least in part, by the statutory definitions of “safety.” The committee also was aware that although a risk analysis for a given product may be “well understood” in one context, it may not be “well understood” in another. As described in this section, the committee examined whether future biotechnology products could pose types of risks different from those associated with existing products (including organisms). It also reviewed risks that are “well understood” and those that may not be in human health and environmental risk assessments.
The Extent to Which Future Products Could Pose Different Types of Risks: Scenarios of Different Use Patterns of Future Biotechnology Products
In 1987, the National Academy of Sciences published a report that stated “[t]he risks associated with the introduction of [recombinant] DNA-engineered organisms are the same in kind as those associated with the introduction of unmodified organisms and organisms modified by other methods” and there is “no evidence that unique hazards exist either in the use of [recombinant] DNA techniques or in the movement of genes between unrelated organisms” (NAS, 1987). What is meant by “same in kind”? The present committee hypothesized that this phrase referred to the final risk-assessment endpoints identified in human health and ecological risk assessments. As these are the kind of assessments that have most commonly been conducted under the auspices of the Coordinated Framework, the committee interpreted its charge to identify “different types of risks” to mean that it should assess the degree to which risk-assessment endpoints identified in human health and ecological risk assessments for existing biotechnology products are likely to be similar to or different from the endpoints that would be selected when assessing risks for future biotechnology
products. The question in the statement of task about different types of risks was also interpreted by the committee as a request to compare the risk hypotheses linking assumed routes of exposure to possible effects and the spatial and temporal scales used in existing risk assessments with those that may be needed for future products.
In the 2017 update to the Coordinated Framework (EOP, 2017), the regulatory agencies used a number of case studies to illustrate how the updated Coordinated Framework would be applied to products. To a large extent, the case studies review how a particular product would navigate the Coordinated Framework. For all the products reviewed, the route through the Coordinated Framework was relatively well defined. In its evaluation, the committee attempted to articulate scenarios for biotechnology products that could emerge over the next 5–10 years for which the path through the regulatory system would be less clear than for the case-study products. The committee also took into consideration whether risk-assessment endpoints for future biotechnology products would be different from existing biotechnology products.
To organize the scenarios, the committee considered different ways that future biotechnology products might be used or manufactured because exposure to a product or any potential effects on human health or an environment are connected to how a product is used or manufactured. The scenarios include products (including living organisms) that are designed to be released into an
open environment and products that are manufactured in contained systems (albeit with limited environmental releases). The committee also examined products with intended or unintended reversible effects as well as products with intended or unintended irreversible effects. It considered exposure of biotechnology products to people or the environment but did not consider human or environmental exposure to compounds produced from biotechnology products.1 The committee did not attempt to review all available risk assessments and risk-management decisions for existing biotechnology products available in the public domain or prepare problem formulations for possible future biotechnology products. Rather, the committee evaluated several scenarios in which future biotechnology products may be used or manufactured to illustrate key issues and concepts that are required to address the statement of task’s question about different types of risk. Some of the scenarios are drawn from two National Academies of Sciences, Engineering, and Medicine reports (NASEM, 2016a,b), while others are drawn from Drinkwater et al. (2014) and presentations made to this committee. The scenarios provided below are intended to be illustrative of the issues that need to be considered to determine similarity in risks of existing biotechnology products with those risks that may be associated with future biotechnology products.
Scenario 1: Contained Products Used in Commercial Manufacturing Facilities That Generate Waste Streams
Future microbial biotechnology products that are used in indoor, contained manufacturing processes and regulated under the Toxic Substances Control Act (TSCA) are likely to be similar in terms of risk-assessment endpoints and the nature of the risk-assessment dimensionality to existing microbial biotechnology products that have already been permitted for manufacturing. Even though such products are intended for use in contained environments, problem formulation would need to include risk-assessment endpoints for the possibility of accidental or intended open release into the environment. For example, if not properly treated, the waste streams from manufacturing processes (for existing and future biotechnology products) may contain engineered biological elements, ranging from genetically engineered organisms to microbial consortia to synthetic DNA. The waste streams themselves may involve either local or interstate activities, depending on how and where the waste streams are treated and distributed.
Under TSCA, EPA has responsibility to address the human health and environmental risks of products released into waste streams. To the extent that control measures in the manufacturing process have a high probability of preventing the release of living organisms into wastewater or solid waste, EPA assumes the risk to humans (that is, adverse health effects) or to the environment (for example, effects on microbial community structure and function) is negligible. EPA also has a list of “pre-approved” microbes that biotechnology-product developers can use; this list is based on risk assessments for these specific microbial species. The human health and ecological risk-assessment endpoints would therefore likely be the same for existing and future products, but risk assessments for a future product—for example, a microbial consortium intended for use in a contained system—may not be as “simple” as current risk assessments for a single, engineered microbe. For example, if there was an accidental release of a living consortium into a waste stream, what is the potential
___________________
1 The committee observed that if compounds produced by future biotechnology products were already regulated by EPA (for example, industrial chemicals regulated under the Toxic Substances Control Act) or the U.S. Food and Drug Administration (FDA; for example, cosmetics or food additives under the Food, Drug, and Cosmetic Act that are already sold, distributed, or marketed across state lines) or the compounds (or their proposed uses) are new, existing EPA or FDA processes to assess risks are not different between existing and future compounds. (Of course, there may be unique new chemical compounds created by a biotechnology organism, but the committee concluded the risk analysis of the new chemical compound is outside the report’s scope.) However, the committee also observed that the domestic manufacture and use of compounds derived from biotechnology products may not fall under a federal agency’s oversight process.
survival and reproduction of the consortium or of each individual microbe? Do the community effects of the consortium affect the ability of individual species within the consortium to survive? How can the consortium be mimicked in laboratory or field studies under a range of environmental conditions that may affect survival and reproduction? Could the consortium or its individual microbes be of concern if consumed by humans? Could the consortium be of concern if consumed by humans even if consumption of its individual species is not a concern? What is the likelihood that the escaped microbes would affect native microbial communities, and is that likelihood different for the consortium versus the individual species? The dimensionality of the risk assessments is thus likely to be more complex than current assessments.
There are also potential regulatory gaps and redundancies. For example, it was not clear to the committee how state regulatory authorities issue national pollutant discharge elimination system permits for potential release of biotechnology products to publicly owned treatment works or to receiving bodies. To what extent would EPA coordinate activities across TSCA, the Clean Water Act, the Resource Conservation and Recovery Act, and the activities of the states (which are, in part, delegated implementation of water and solid waste laws) to minimize redundancy and maximize efficiency in any post-market monitoring? How would potential effects on the environment be monitored, regardless of who has the responsibility to do it? This lack of clarity does not necessarily suggest that there is a risk of concern or that new risks of concern may arise with future biotechnology products; however, the extent to which any adverse effects may be expected by intentional or accidental releases to U.S. waters is not clear.
Scenario 2: Products Manufactured Within Homes for Use by Household Members
Personal or domestic products (such as probiotics, cosmetics, cleaning agents, and antimicrobial pesticides) can be made in traditional factories, but increasingly such products may also be made and used within a home. For example, a purchased kit could allow consumers to engineer organisms to produce a desired chemical, probiotic, or microbe. Combinations of organisms, whether engineered or not, might also be combined in a home environment for use as a nutraceutical or fertilizer.
These types of products may not fall under federal regulatory oversight because presumably they would not be marketed, sold, or distributed and would not cross state boundaries. Risk-assessment dimensionality (time and space), if not risk-assessment endpoints themselves, for future biotechnology products that are manufactured within a home and intended to be used indoors by household members is likely more complex than it is for future products associated with Scenario 1. The nature of exposure pathways and means to estimate environmental releases are less certain as compared to the dimensionality of risk assessments associated with contained manufacturing processes traditionally regulated under TSCA, the Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA), or the Food, Drug, and Cosmetic Act (FDCA). For example, there is likely to be greater variability in geography, use patterns, and disposal in household use than in manufacturing plants and that variability will be difficult for regulators to assess. In addition, the number of children and adults potentially exposed to personal care products or other products used in domestic settings, the number and nature of the settings under which they may be exposed, and the potential adverse effects of the biotechnology products or compounds derived from the products may be less certain as compared to products regulated under TSCA, FIFRA, or FDCA.
As in Scenario 1, the nature and extent of point-source releases of biotechnology products to publicly owned treatment works or non–point source releases to receiving bodies requires a more extensive exposure analysis. Monitoring by the U.S. Geological Survey (USGS) has documented detection of organic wastewater contaminants and pharmaceuticals in public and private drinking-water sources, surface-water receiving bodies, and septic systems (see, for example, Focazio et al.,
2008; Writer et al., 2013; Schaider et al., 2014; Phillips et al., 2015), so it may be reasonable to assume that future biotechnology products manufactured in domestic settings (or the compounds derived from such products) will be released into surface-water or groundwater sources. The results from USGS monitoring studies could provide insights on the dimensionality of the risk hypotheses for future human health and ecological risk assessments. The potential effects of released organisms to the microbial systems in publicly owned treatment works or receiving bodies would be less certain if the potential effects had not been previously characterized. Finally, as with Scenario 1, the dimensionality of risk hypotheses for the disposal scenarios of future biotechnology products will likely be higher and the information to support risk characterization less certain.
Scenario 3: Open-Release, Next-Generation Biotechnology Plants for Agricultural and Other Uses
At the time the committee was writing its report, biotechnology plants consisted of genetically engineered (GE) varieties of a few widely grown crops, such as corn, soybean, and cotton. In total, GE varieties of 10 crop species were grown in the United States in 2015 (NASEM, 2016b). However, the committee anticipated that the number of crop species modified by biotechnology will increase substantially over the next 5–10 years. For example, in the United States, citrus trees engineered to resist citrus greening disease (huanglongbing), which is fatal to the tree, were already in confined trials, and transgenic research was under way to fight Pierce disease in grapes and bacterial spot disease in tomatoes (Ricroch and Hénard-Damave, 2016). Outside the United States, examples of future biotechnology products include GE banana and cassava, for which trials were being conducted in varieties with improved insect and disease resistance and increased nutrient content (Ricroch and Hénard-Damave, 2016). The committee expected that, along with the greater number of GE crop species, the number of engineered genes in a crop would also increase as multiple or more complex traits are targeted and genome-editing techniques such as CRISPR-Cas9 enable certain genetic manipulations to be more readily accomplished. Indeed, a number of crops engineered with genome editing had already been brought to the U.S. Department of Agriculture (USDA) for an “Am I Regulated?” determination (see Table 9-3 in NASEM, 2016b). The committee did not anticipate that the risk-assessment endpoints associated with these new crops would be different from those associated with crops that had already gone through the regulatory process. However, dimensionality, pathways to risk, and the magnitude of risk might change as the synergistic effects of multiple genetic changes could lead to unintended effects in the biochemistry of crops (affecting nutrients, immunogens, phytohormones, or toxicants) or in the phenotypic characteristics of crops due to more complicated epigenetic effects. Off-target effects in genes from base-pair insertions and deletions via genome editing should also be considered.
The committee also expected the use of biotechnology plants to spread beyond agricultural fields. One example would be a novelty product like the glowing plant, discussed in Box 3-4. However, a more important type of plant that the committee thought would be likely to become increasingly common is endangered or locally extinct plants that have been engineered to be able to thrive in natural ecosystems in which they once were widespread. An example discussed in Chapter 2 is the American chestnut, a hardwood tree species native to the U.S. eastern seaboard that has been decimated by a fungal blight introduced in the early 1900s. Genetically engineered resistance to the blight could allow this tree species to grow to maturity and reclaim some of its native range. Ecological risk assessments will be needed for likely inadvertent release of novelty plants into natural ecosystems as well as for the intentional release of plants such as blight-resistant American chestnut.
There may also be regulatory gaps associated with these types of products. For example, if USDA determines that a product is not regulated by virtue of the mechanism used to insert the
genetic modification or the source of the genetic material, and that product may be a plant pest or weedy species, there would not be oversight when oversight is warranted.
Scenario 4: Open-Release Microorganisms and Microbial Consortia
Engineered microbial consortia is a potential area of rapid growth in new biotechnology products for open release in the environment for a broad range of markets including mining, bioremediation, and nutrition. As described in Chapter 2, researchers have worked to establish stable synthetic consortia of microorganisms—and the biological principles behind their establishment and maintenance—that could be used as the bases of a wide variety of future applications.
The committee concluded that open release of engineered, naturally occurring or artificial microbial consortia with multiple modifications—some or all of which may be orthogonal—should have a similar suite of risk-assessment endpoints as those used to assess the risks of nonengineered microorganisms, but, as in Scenario 3, the pathways to risk and the magnitude or dimensionality of risk could change. Ecological risk assessments concerning the use of engineered or nonengineered microbial consortia used in bioremediation or biomining would likely address perturbations of native microbial communities including effects on energy flow, horizontal gene transfer, and evolution. The dimensionality of these risk assessments may be more complex with engineered microbes as compared to nonengineered microorganisms, depending on such variables as the use pattern, taxonomic relationships, use of orthogonal genes, environmental conditions within and outside a release site (for example, pH), and use of engineered “kill switches” that terminate the organism when the energy or nutritional sources fall below a certain level. For example, native or artificial consortia designed to alter earthworm digestion of cellulose, change honey bee behavior by manipulating levels of neurotransmitters, or confer nitrogen-fixation properties to nonlegume crops may require a new or expanded suite of risk-assessment endpoints and pathways. If endosymbiotic microorganisms were used to confer nitrogen-fixation properties to nonlegume crops, a human health risk assessment might be needed if the endosymbionts were present in the edible parts of the crop.
Scenario 5: Open-Release Products Designed to Suppress, Eradicate, or Enhance a Target Species Population
Consistent with the National Academies report Gene Drives on the Horizon: Advancing Science, Navigating Uncertainty, and Aligning Research with Public Values (NASEM, 2016a) and examples of risk-assessment methods for non-GE biocontrol agents and the release of non-native organisms (Fairbrother et al., 1999; Orr, 2003; Landis, 2004),2 risk assessments for future biotechnology products that are designed to introduce a new species or suppress or enhance an existing species reflect a high degree of dimensionality and entail a diversity of endpoints at varying levels of biological organization. These assessments will generally require spatially and temporally explicit assessments that address direct and indirect ecological effects and evolutionary effects. Given that some biotechnology products could suppress or enhance a species population at a rate that is faster than natural ecological processes or evolutionary rates, these new products may require the definition of a new suite of risk-assessment pathways.
As an example of the complex pathways that might arise in this type of scenario, a possible risk pathway could be through horizontal gene transfer of the kill-switch mechanism in a gene drive
___________________
2 See also the Framework for Assessment described by the U.S. Fish and Wildlife Service learning module on managing invasive plants. Available at https://www.fws.gov/invasives/staffTrainingModule/assessing/introduction.html#part2. Accessed September 13, 2016.
to other species, perhaps to important species in the ecosystem that are beneficial or desired. For example, the possible transfer of a kill switch from a GE organism with a gene drive to that organism’s non-GE predator and the detrimental effects of such a transfer would be intermediate risk-assessment endpoints of concern. If the nontargeted predator were to disappear from the ecosystem, its decline could leave a niche open for an invasive species or another pest species. The presence of the harmful species would then be another intermediate endpoint of concern. The results of the harmful species on human health, agriculture, or ecosystems would be ultimate risk-assessment endpoints. Such complex and multilayered risk pathways have been dealt with in other risk analyses for population suppression of mosquitoes (for example, with engineered Wolbachia) in fault tree and Bayesian-analysis approaches (Murphy et al., 2010; Murray et al., 2016). Risk assessments for these types of complex pathways may also be able to take advantage from approaches used to assess the risks of non-GE biocontrol agents and the release of non-native organisms (for example, Fairbrother et al., 1999; Orr, 2003; Landis, 2004).3
Given the rapid pace of technological change and the ways and environments in which resulting products can be used, it will be important to create and regularly update scenarios such as the ones above to explore emerging risks and the adequacy of the regulatory framework. A recent set of recommendations by the National Academy of Public Administration called for the U.S. government to “systematically integrate foresight into policy development,” with an emphasis on the use of scenarios “to consider how different trends and developments may come together in unexpected ways to put policy objectives at risk or create opportunities for more effective action on these objectives” (NAPA, 2016:9,11). Such scenarios should reflect and integrate changes in technologies, capabilities, actors, business models, and risk pathways. Scenarios can be incorporated into a portfolio of approaches, including horizon scanning, to create an early warning system for emerging risks, an approach being explored by the European Union (SEP, 2016).
Risks or Lack of Risks That Are Well Understood
As noted in the introduction of the chapter, the committee interpreted the first question in this part of the statement of task as a request to reflect on the degree to which regulatory human health and ecological risk-assessment endpoints used in risk assessments for existing biotechnology products are likely to be similar to or different from the endpoints that would be selected when assessing risks for future biotechnology products. The second question concerns risks or lack of risks of biotechnology products that are well understood. The term “well understood” was interpreted to mean situations when uncertainty in estimates of risk does not preclude a description of the possible risks consistent with the goals and objectives established during the problem-formulation phase of a risk assessment (see EPA, 1998; NRC, 2009; Box 4-1). However, even with that interpretation, the committee’s statement of task was difficult to address given the ambiguity of the phrase “risks or lack of risk.” Terms such as “lack of risks,” “low risks,” “minimal risks,” or “acceptable risks” contribute to linguistic ambiguity (NASEM, 2016a) and do not provide a meaningful framework from which to distinguish between scenarios of a product’s use in which there are “risks” versus situations where there is a “lack of risks.”
Because risk assessments available for existing biotechnology products do not, in general, employ probabilistic estimates of risk, but rather use deterministic expressions of risks (Box 4-2), the committee could not quantitatively address the extent to which risks or the lack of risks are well understood. Existing risk assessments typically characterize risks in a qualitative or deterministic
___________________
3 See also the Framework for Assessment described by the U.S. Fish and Wildlife Service learning module on managing invasive plants. Available at https://www.fws.gov/invasives/staffTrainingModule/assessing/introduction.html#part2. Accessed September 13, 2016.
manner, which precludes the means to quantitatively compare risks of existing and future biotechnology products and of biotechnology products to nonbiotechnology products designed for similar use patterns. Advancing quantitative risk assessments will be useful generally, given the characteristics of future open-release products (for example, the mechanisms of action, degree of reversibility and recovery, or movement within ecosystems). There is a need to advance risk-assessment techniques for potential adverse outcomes that have not been rigorously addressed for both biotechnology products and environmental stressors4 in general (NRC, 2013; NASEM, 2016a). Given the nature of the use patterns for future open-release products, spatially and temporally explicit risk assessments will also facilitate a more insightful identification of risk patterns.
Risks That Are Well Understood
Although it was not possible to quantitatively determine risks that are well understood, a future biotechnology product that is based on a similar genetic modification and has a similar use pattern as an existing biotechnology product with a safe-use record likely has a risk profile similar to that of the existing product.
The scenarios in the above section could be used for pilot projects to develop probabilistic estimates of risks for existing biotechnology products and thereby provide the means to compare the likelihood of adverse effects from future biotechnology products to the likelihood of adverse effects from existing biotechnology and nonbiotechnology products, assuming risk assessments for future products incorporate probabilistic methods. Such analyses would help identify high-priority information needs to reduce uncertainty in risk estimates and inform the classification of comparable products based on the nature of risk-assessment endpoints, dimensionality of risk assessments, and the probabilities of adverse effects. Pilots would be particularly helpful for products intended for wide-area environmental release in low-management environments (for example, open-release organisms with gene drives or genetically engineered bacteria for bioremediation or fuel production). Estimated probabilities of immediate, medium, and long-term environmental and human health risks would be appropriate (Suter, 2007; Warren-Hicks and Hart, 2010; NRC, 2013; NASEM, 2016a).
For new biotechnology products without comparators, risk estimates could be lower than, similar to, or higher than such estimates for existing biotechnology or nonbiotechnology products based on the design of the future products and their use patterns.
Risks That Are Not Well Understood
Although risk-assessment endpoints for human health and environmental effects for existing and future biotechnology products will likely be similar (assuming, for example, similar manufacturing controls, use patterns, and properties of the products), the magnitude of the risks may change, the pathways could be more complex and multidimensional, and existing risk assessments have limitations. It is not always clear in existing risk assessments how assessment endpoints were selected or how the dimensionalities of the risk assessments were considered. Furthermore, existing risk assessments are generally comparative risk assessments—that is, they rely in large part on the comparison of a new biotechnology product to an existing nonbiotechnology product. Future products of biotechnology will involve more complex comparisons that, for example, potentially encompass multigene traits in consortia deployed in novel environments and management scenarios, and the new products may not have a counterpart or precedent to allow a ready means for an “as safe as” comparison to a product that already exists. The comparative risk-assessment
___________________
4 A stressor is any agent or actor with the potential to alter a component of the ecosystem (NASEM, 2016a).
process may, however, be more easily applied to human health risk assessments than to ecological risk assessments. Finally, it is difficult and not typical to incorporate social and cultural factors into existing risk assessments, yet these factors may ultimately affect human health and ecological risk (for example, social systems and variability in use of products). In addition, social and cultural values, including social and cultural risks, are not usually included in regulatory risk assessments and are difficult to assess.
Products Without Comparators. Existing biotechnology risk assessments are guided by comparative risk approaches and are informed by comparisons to nonbiotechnology counterparts that help establish the “as safe as” criteria used by regulatory decision makers. As described in Chapter 2, existing biotechnology products have typically involved a host (the organism into which new material is introduced) and a source (the organism from which the new material was taken). For example, for a GE insect-resistant variety of corn, the corn is the host organism and the bacterium Bacillus thuringiensis (Bt) is the source organism. In the comparative risk-assessment paradigm, the GE corn variety is compared against a genetically similar variety of corn that does not contain the GE
trait. The two varieties are compared in terms of nutrient and chemical composition to ascertain if there are unintended differences (the GE trait is an intended difference). This comparison includes toxicity testing and allergenicity screening to help identify any potential human health issues related to intended or unintended effects from the GE trait.
With the advancement of biotechnology from recombinant-DNA technology to genome engineering, the use of comparators is becoming more challenging, even for GE crops (EFSA, 2011). Furthermore, transformations can be made in host organisms that are less well characterized than corn. For example, genome-editing technologies allow product developers to make changes in genomes of nearly any host organism for which there is a genome sequence available, from microbes to insects to mammals (Reardon, 2016). There may not be baseline data on the nontransformed counterpart host. Furthermore, novel gene sequences—including synthetic ones—can be introduced into host organisms; there may be no nonbiotechnology product to which they can be compared.
If potential off-target effects of new technologies, such as genome editing, are similar to those that occur naturally—for example, point mutations or epigenetic changes—and the probability of
off-target effects is not substantially different from the background rate of such changes, then any additional risk (that is, beyond that associated with the intended target of genome-editing technology) is low. On the other hand, the range of genome-editing techniques is rapidly expanding, and if certain new technologies cause off-target effects that do not typically occur naturally or increase the number of such changes, then this may pose an additional challenge for comparative risk assessment. Note that conventional-breeding techniques such as mutation breeding in plants could increase the extent and the range of types of off-target effects to a much greater extent than existing genome-editing techniques (NASEM, 2016b). With rapid changes in and lowering costs of genome sequencing and genome-interrogating technologies (see Chapter 2), genome-wide identification of off-target effects is increasingly straightforward. However, what remains a challenge is evaluating whether any off-target effects pose a risk because of the difficulty in obtaining proper comparators. Finally, for organisms made by inserting entirely new pathways of genes derived from multiple unrelated sources or consortia of organisms engineered with multiple genes, how a product developer or regulator would conduct protein toxicity testing is complicated. Synergistic effects of multiple genes and organisms compound such testing, and it might not be accurate to test each organism or gene separately. In these cases, basic biology and ecology studies may be necessary to develop baseline environmental behaviors, as in new knowledge about gene transmissibility, persistence in the environment, and toxin production before small-scale environmental release.
On a related note, products without comparators do not have a clear path already charted through the U.S. regulatory system. The degree to which risks associated with a new type of product are “well understood” is a subjective determination, but formal uncertainty analyses can help determine when a reduced level of regulatory scrutiny may be warranted. Use of a biological component or system that has a history of safe use provides an existing regulatory path with known nonbiotechnology comparators and clear risk-assessment endpoints, whereas using an unfamiliar component or system does not. Future biotechnology products may be unique and therefore lack adequate precedents. For example, the GE mosquito developed by the company Oxitec was a first-of-its-kind product; it was genetically engineered to carry a gene that would render its offspring sterile with the goal of effectively eradicating the mosquito population (Aedes aegypti) that, among other things, carries dengue, chikungunya, and Zika viruses. The developer and the regulatory agencies both shared uncertainty on the front end as to whether the GE mosquito should be regulated by EPA under FIFRA as an insecticide (because it kills mosquitoes) or by the U.S. Food and Drug Administration (FDA) as a new animal drug (the pathway that was used for genetically engineered salmon). After a number of years of agency deliberations, in August 2016 FDA ultimately released a final environmental assessment and a finding of no significant impact, agreeing with Oxitec’s environmental assessment that its proposed field trial would not have significant impacts on the environment. However, FDA’s assessment was not a quantitative, probabilistic one and was not based on field data looking for harm to nontarget species or to ecosystems; field data from other countries only looked at the efficacy of population suppression (FDA, 2016). The committee heard a similar concern about a lack of clarity with regards to the regulatory path for engineered microbes. The microbes were designed for open-environmental release to extract gold and copper from low-grade ore. An invited speaker emphasized that there was ambiguity about which regulatory agency would be responsible for the product, what data would be required by the agency, and what nontransformed host would serve as the comparator in a risk assessment (DaCunha, 2016).
Social and Cultural Effects. Methods to incorporate social and cultural values into risk analysis are limited because they often cannot be put on the same scale as health risks, environmental externalities, and monetized costs and benefits. However, some risk-analysis frameworks have been developed to incorporate values of various publics during the problem-formulation or risk-management stage, when other options are compared to the proposed action of releasing a techno-
logical product (Nelson et al., 2004). Previous National Academies reports have also emphasized the importance of public participation in the risk-analysis process, particularly in risk characterization (which involves complex, value-laden judgments) and problem formulation, where public involvement can improve acceptance of the analysis (Box 4-3) and improve the analysis for the purposes of risk management (NRC, 1996, 2008, 2009; NASEM, 2016a).
Future products of biotechnology will be more complex in terms of their interactions with their environment and society, and more research may be needed to develop methods for governance systems that integrate ethical, cultural, and social implications into formulation of risk-assessment endpoints and risk characterization in ways that are meaningful. At the same time, it may not be feasible or even justified for all new biotechnology products—for instance, products with which there is already familiarity or products that will not be released into the environment. Genetically engineered organisms used in the research laboratory to develop new chemical synthesis methods are not likely to require the same level of public dialogue according to decision criteria for public engagement proposed by several scholars and think-tanks as will products that have more uncertainty associated with them, such as organisms with gene drives (Funtowicz and Ravetz, 1995; NRC, 1996; Renn, 2005; NASEM, 2016a).
EXISTING FEDERAL CAPABILITIES, EXPERTISE, AND CAPACITY
To address risks that are not “well understood” and to oversee the profusion of products anticipated from the discussion in Chapter 2, federal agencies need to be prepared with the appropriate
scientific capabilities, expertise, and capacity to conduct regulatory science. On the basis of definitions provided by FDA5 and the Society for Risk Analysis, the committee understood regulatory science to involve developing and implementing risk-analysis methods and maximizing the utility of risk analyses6 to inform regulatory decisions for biotechnology products, consistent with human health and environmental risk–benefit standards provided in relevant statutes. Regulatory science includes establishment of information and data quality standards, study guidelines, and generation of data and information to support risk analyses. It can also include the development of risk-mitigation measures and the development and implementation of safety training and certification programs to help ensure the intended benefits of products are realized and risks to workers, users, and the environment are minimized. Individuals in government, industry, academia, and nongovernmental organizations that contribute to the advancement of regulatory science have degrees across disciplines in the natural, socioeconomic, and computational sciences, engineering, and public policy.
Federal capacity is not limited to the agencies that participate in the Coordinated Framework. To assess the capacity of the federal government to regulate future biotechnology products, the committee looked at the existing capabilities in the workforce, the available external resources that could be drawn upon by the agencies, and the present investment in key tools for biotechnology-product evaluation. The committee also noted current opportunities to enhance capability and capacity through interactions across the federal agencies and through partnerships with developers, nongovernmental organizations, and academia.
Existing Scientific Capabilities in the Federal Workforce
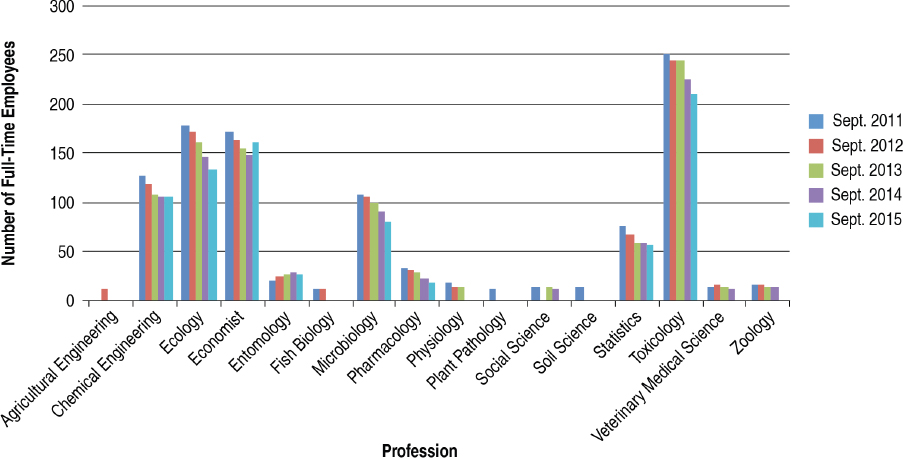
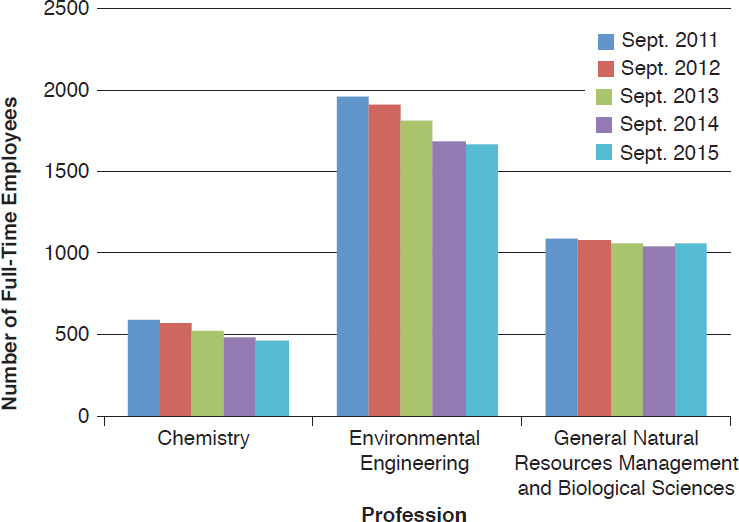
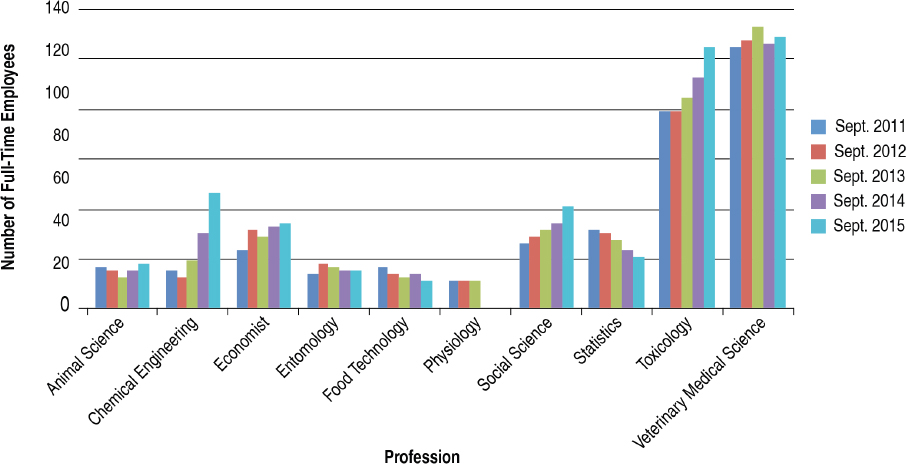
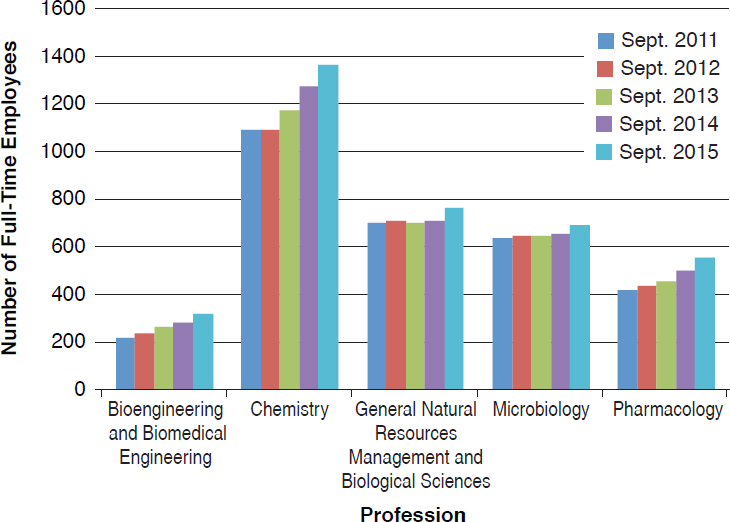
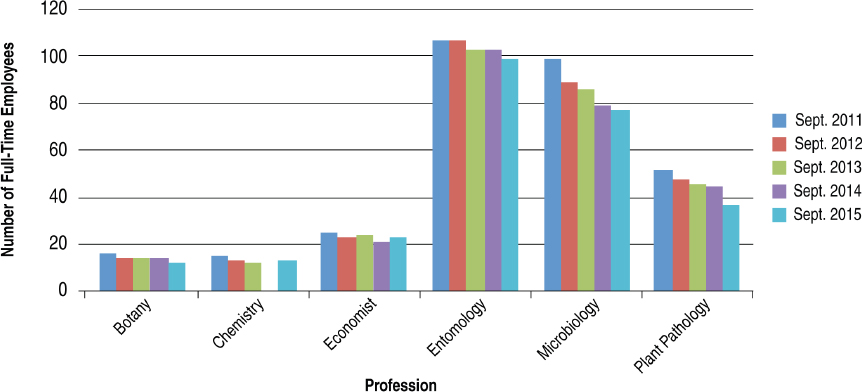
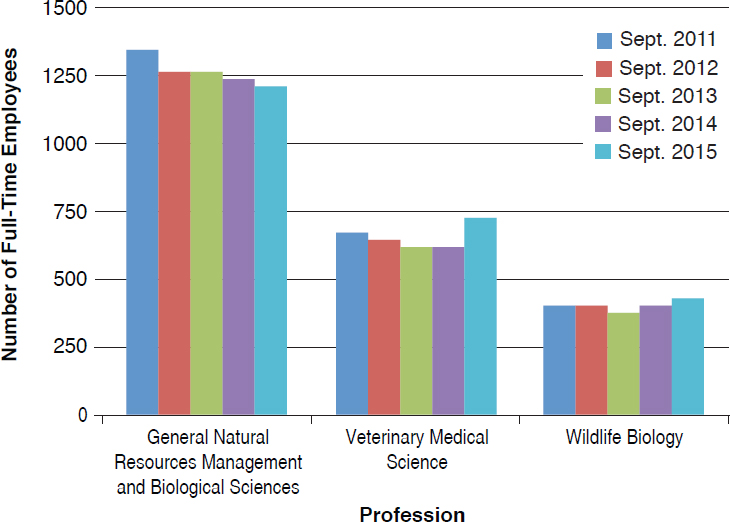
The committee made use of the FedScope database within the U.S. Office of Personnel Management to ascertain the types of expertise within the regulatory agencies. The database provided information about the trends in number of staff with each type of expertise for the fiscal years 2011–2015.7 From the list of professional occupations provided in the database, the committee selected 33 that it surmised would be necessary to the regulatory responsibilities of EPA, FDA, or USDA–Animal and Plant Health Inspection Service (APHIS) personnel; the search returned results for at least one agency in 25 of the occupations.8Figures 4-1 through 4-6 show the employment trends for the occupations in each agency. There are two figures per agency to address issues of scale. Professions or years which returned a value of “NA” in the database are not included in the figures.
The committee recognizes that the data provided in Figures 4-1 through 4-6 are not as specific as would be desired. The committee was primarily interested in the expertise available with FDA’s Center for Food Safety and Applied Nutrition and Center for Veterinary Medicine, EPA’s Office
___________________
5 U.S. Food and Drug Administration. Advancing Regulatory Science: Moving Regulatory Science into the 21st Century. Available at http://www.fda.gov/ScienceResearch/SpecialTopics/RegulatoryScience/default.htm?utm_campaign=Goo. Accessed December 13, 2016.
6 According to the Society for Risk Analysis, risk analysis defined broadly includes “risk assessment, risk characterization, risk communication, risk management, and policy relating to risk, in the context of risks of concern to individuals, to public and private sector organizations, and to society at a local, regional, national, or global level” (SRA, 2015).
7 FedScope employment trends are available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
8 The professions listed in the FedScope database correspond to the qualification standards for federal positions used by the Office of Personnel Management. The committee searched the following professions: general natural resources management and biological sciences, microbiology, pharmacology, ecology, zoology, physiology, entomology, toxicology, botany, plant pathology, plant physiology, horticulture, genetics, soil conservation, forestry, soil science, agronomy, fish and wildlife administration, fish biology, wildlife biology, animal science, environmental engineering, bioengineering and biomedical engineering, agricultural engineering, chemical engineering, chemistry, forest products technology, food technology statistics, social science, sociology, economist, and veterinary medical science. No results were returned for the professions plant physiology, horticulture, genetics, soil conservation, forestry, agronomy, sociology, and fish and wildlife administration.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
NOTE: General Natural Resources Management and Biological Sciences includes employees hired with expertise in biological sciences, agriculture, natural resource management, chemistry, or related disciplines appropriate to the position.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
NOTE: General Natural Resources Management and Biological Sciences includes employees hired with expertise in biological sciences, agriculture, natural resource management, chemistry, or related disciplines appropriate to the position.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
NOTE: This figure has been updated since the prepublication release. The number of full-time employees in the prepublication version of the report was incorrect.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
NOTE: General Natural Resources Management and Biological Sciences includes employees hired with expertise in biological sciences, agriculture, natural resource management, chemistry, or related disciplines appropriate to the position.
DATA SOURCE: U.S. Office of Personnel Management. FedScope database, employment trend cubes. Available at https://www.fedscope.opm.gov/etrend.asp. Accessed December 12, 2016.
of Chemical Safety and Pollution Prevention, and USDA–APHIS’s Biotechnology Regulatory Services. This level of resolution was not available; as a result, the number of full-time employees (FTEs) in the regulatory agencies whose activities are most directly relevant to nonmedical biotechnology products is likely well below those depicted in the figures. Nevertheless, the information returned through the committee’s search of the FedScope database shows that, for much of the expertise relevant to regulation of biotechnology, the number of employees has fallen over the period of time covered in the database. For some applied disciplines important to understanding the use and deployment of biotechnology products, the decrease in numbers is a cause for concern (for example, the numbers of plant pathologists, soil scientists, fisheries biologists, and agricultural engineers within EPA) given the committee’s prediction that the number and novelty of future biotechnology products will increase. Because the FTE data available to the committee likely overestimate the breadth and depth of regulatory agency staff, the trends in the federal workforce raise the possibility there may not be sufficient capacity to meet future regulatory demands. There is the possibility of using contract scientists to manage the day-to-day workload of review, analysis, and summarization of regulatory submissions. Agencies within the Coordinated Framework have and do maintain contract personnel and have this built into agency budgets. Proper utilization of such outside expertise will require appropriate training and oversight to ensure the quality of regulatory reviews is maintained.
Enhancing Federal Expertise Through External Scientific Advisory Committees
The agencies can and have supplemented their internal expertise by making use of external advisory committees. Independent federal advisory committees can not only assist with scientific peer review on issues of human health and ecological risk assessment but can also provide input on issues such as risk-assessment methodology, environmental modeling, life-cycle analyses, sustainability, and environmental justice, among others, which may become more relevant in assessing risks and benefits of future products if the conception of risk is broadened (see Box 3-1). EPA has used such committees to provide independent, external peer review, with public comment, on specific, well-defined risk-assessment methodology and assessment issues for specific biotechnology products.9 In the context of pesticidal biotechnology products, EPA has used its science advisory panels (SAPs) to review risk assessments and resistance management plans for Bt plant-incorporated protectants since the mid-2000s.10 In 2014, the FIFRA SAP reviewed EPA’s proposed problem-formulation approach for RNA interference (RNAi) technology in anticipation of future human health and ecological risk assessments of specific RNAi products (EPA, 2014a). Consistent with an iterative process in advancing risk-assessment approaches for a new technology, EPA convened a SAP in September 2016 to review a draft assessment for a specific RNAi plant-incorporated protectant (EPA, 2016c).
Other agencies acting within the Coordinated Framework have utilized programmatic environmental impact statements to elicit public input as a means to consider need for future regulation for biotechnology products. For instance, USDA–APHIS undertook programmatic environmental
___________________
9 See, for example, EPA FIFRA Scientific Advisory Panel for pesticides, including biotechnology products (available at https://www.epa.gov/sap, accessed January 14, 2017); the EPA Science Advisory Committee on Chemicals, including biotechnology products regulated under TSCA (TSCA Scientific Peer Review Committees, available at https://www.epa.gov/tsca-peer-review, accessed January 14, 2017); and USDA’s Advisory Committee on Biotechnology & 21st Century Agriculture (available at http://www.usda.gov/wps/portal/usda/usdahome?contentidonly=true&contentid=AC21Main.xml, accessed January 14, 2017).
10 See Scientific Advisory Panel Meetings on Issues Related to PIPs (External Peer Review). Available at https://www.epa.gov/regulation-biotechnology-under-tsca-and-fifra/overview-plant-incorporated-protectants#scientific. Accessed January 14, 2017.
impact statement activities in 2004 for the purpose of rulemaking to determine if their regulatory remit for biotechnology regulation could be clarified and expanded (USDA–APHIS, 2004), and it was involved in a similar process at the time the committee was writing its report (USDA–APHIS, 2016). In addition, USDA–APHIS has sponsored third-party activities to gain expert input that was subsequently reflected in views and approaches used in biotechnology risk assessment (Traynor and Westwood, 1999). USDA–APHIS has also directly sponsored expert workshops (Rose et al., 2006). There also have been expert meetings hosted by the former interagency Agricultural Biotechnology Risk Analysis Task Group to understand research priorities (NSTC, 2007). Agencies responsible for regulating future biotechnology products can also build from experience to proactively gain advice and input on proposed risk-assessment approaches for other future products and assessment techniques in areas like nanotechnology (EPA, 2009), computational toxicology (EPA, 2011), spatially explicit ecological assessments (EPA, 2015), and pollinator protection (EPA, 2012).
EPA, FDA, and USDA–APHIS have used independent external scientific input and review several times in the past. For example, EPA sought input from the National Research Council (NRC, 2012) to provide advice on future environmental science and engineering challenges and technological advances and to assess the overall capabilities of the agency to meet emerging and future mission challenges. One of the recommendations was to “[e]ngage in a deliberate and systematic ‘scanning’ capability involving staff from [EPA’s] ORD [Office of Research and Development], other program offices, and the [EPA regional offices]. Such a dedicated and sustained ‘futures network’ (as EPA has called groups in the past with a similar function), with time and modest resources, would be able to interact with other federal agencies, academe, and industry to identify emerging issues and bring the newest scientific approaches into EPA” (NRC, 2012:11). Consistent with this horizon-scanning recommendation, in 2016 EPA shared a preliminary view of emerging and potential issues, which included the inevitability of transformational biotechnology products (Greenblott et al., 2016).
The benefit of this input from external experts is reflected in guidance that lays the groundwork for implementation of strengthened approaches for risk assessment. For instance, guidance for identifying and selecting ecological risk-assessment endpoints (including those that address ecological goods and services) across biological, spatial, and temporal scales have been developed (for example, EPA, 2003, 2016g), and EPA has summarized experiences gained in several case studies that assessed the human health and ecological risks of exposure to combinations of disparate chemical, biological, and physical stressors. These case studies also highlight the importance of engaging stakeholders throughout the risk-assessment phases and risk management (Gallagher et al., 2015; see also Box 4-3). The U.S. Fish and Wildlife Service (FWS), the National Marine Fisheries Service (NMFS), USDA, and EPA were in the process of implementing recommendations from the 2013 National Research Council report Assessing Risks to Endangered and Threatened Species from Pesticides at the time the committee was writing its report.11 EPA’s Risk Assessment Forum has developed peer-reviewed white papers on the use of probabilistic human health and ecological risk assessments (EPA, 2014b) in response to the 2013 Institute of Medicine report on uncertainty in environmental decision making (IOM, 2013). Probabilistic human health risk assessments are performed on a routine basis for food-use pesticides (EPA, 2016a) based on advice from the FIFRA SAP. Probabilistic methods have been developed for some ecological risk-assessment scenarios (NRC, 2013; EPA, 2016e) and have been employed in a limited number of cases. Ongoing research to advance computational toxicology techniques, including high-throughput screening, illustrates EPA’s commitment to advance 21st-century approaches to assess chemical stressors (NRC, 2007)
___________________
11 Implementing NAS Report Recommendations on Ecological Risk Assessment for Endangered and Threatened Species. Available at https://www.epa.gov/endangered-species/implementing-nas-report-recommendations-ecological-risk-assessment-endangered-and. Accessed September 13, 2016.
and develop and employ the information-technology infrastructure needed to manage and analyze large data sets (for example, see EPA, 2016f).
EPA, USDA, and USGS already maintain and employ large geospatial data sets to support human health and ecological risk assessments12 and agriculture and natural resource research and management within the governance of the Federal Geographical Data Committee (NRC, 2013). Research is ongoing to develop the data, models, and tools to expand community stakeholders’ capabilities to consider the social, economic, and environmental impacts of decision alternatives on community well-being; develop the causal relationships between human well-being and environmental conditions; develop and implement monitoring designs and indicators to support national, regional, and state reports of environmental condition; and advance tools and metrics to support life-cycle analyses (Yeardley et al., 2011; EPA, 2016d). The agencies also have established processes to address cross-cutting research and scientific issues, including dialogue with stakeholders,13 which could be expanded to complement cross-cutting issues relevant to the Coordinated Framework and future research and risk-assessment needs.
Another example of an external advisory group is the EPA Pesticide Program Dialogue Committee (PPDC),14 which was established in 1995 as a forum for a diverse group of stakeholders—environmental and public-interest groups, pesticide manufacturers, trade associations, commodity groups, public health and academic institutions, federal (including USDA, FDA, FWS, NMFS, and the Centers for Disease Control and Prevention) and state agencies, and the general public—to provide feedback to EPA on various pesticide regulatory, policy, and program implementation issues. The PPDC has provided advice to EPA in implementing far-reaching changes in risk-assessment and risk-management approaches mandated with passage of the Food Quality Protection Act in 1996 and provided input on issues including implementation of 21st-century toxicology testing and nonanimal testing alternatives, endangered species and pollinator protection options, classification systems for reduced-risk pesticides, and approaches for documenting label claims, among others. The PPDC also provided feedback on EPA’s development and implementation of public review and comment processes for proposed new pesticide registration decisions (EPA, 2016b) and the reevaluation of registered pesticides (including problem formulation, draft risk assessments, and proposed regulatory decision steps15). This EPA advisory committee could be employed to help guide the development of a governance approach for pesticidal biotechnology products in conjunction with the Coordinated Framework.
Independent external advice and input can help expand the ability of a federal agency to meet its future scientific challenges. Successful implementation of recommendations from an advisory committee is contingent on the breadth and depth of an agency’s workforce. Downward trends in the staffing of certain areas of expertise at regulatory agencies (summarized in Figures 4-1 through 4-6) raise concerns that the staff may not have sufficient time or skills to take advantage of external advice and prepare for the future.
___________________
12 EnviroAtlas. Available at https://www.epa.gov/enviroatlas. Accessed October 11, 2016.
13 See, for example, Computational Toxicology Communities of Practice, available at https://www.epa.gov/chemical-research/computational-toxicology-communities-practice, and Pesticide Environmental Modeling Public Meeting–Information, available at https://www.epa.gov/pesticide-science-and-assessing-pesticide-risks/environmental-modeling-public-meeting-information. Both accessed January 10, 2017.
14 Pesticide Program Dialogue Committee. Available at https://www.epa.gov/pesticide-advisory-committees-and-regulatory-partners/pesticide-program-dialogue-committee-ppdc. Accessed January 14, 2017.
15 Registration Review Process. Available at https://www.epa.gov/pesticide-reevaluation/registration-review-process. Accessed January 14, 2017.
Federal Research Funding to Advance Risk Analysis on Future Biotechnology Products
In an attempt to better ascertain the nature and extent of federal research designed to support risk analyses of biotechnology products, the committee solicited input from relevant agencies through a request for information (RFI) keyed to programmatic work addressing risk analysis for products of biotechnology. The questions posed in the RFI (see Appendix C) were derived, in part, from the report Creating a Research Agenda for Ecological Implications of Synthetic Biology, published in 2014 following two workshops organized by the Massachusetts Institute of Technology Program on Emerging Technologies and the Woodrow Wilson Center’s Synthetic Biology Project (Drinkwater et al., 2014) and from a workshop and Delphi study on synthetic-biology governance funded by the Sloan Foundation and hosted by North Carolina State University’s Genetic Engineering and Society Center (Roberts et al., 2015). The committee was interested in programmatic work related to fundamental and applied research efforts that can inform human, animal, and ecological risk assessments and socioeconomic costs and benefits. Research related to potential risks of future human drugs or medical devices was not included in the committee’s statement of task and therefore was not part of this RFI, except to the extent such research may be broadly applicable to other biotechnology products.
The committee sent the RFI to 28 federal offices and received responses from 17. Twelve of the 17 had information to share that was relevant to the committee’s request. The RFI recipients are listed in Appendix C. The RFI specifically asked about the ongoing research the agencies had with regards to
- The nature and extent to which future biotechnology products were similar to or different from nontransformed (nonbiotechnology) products serving as comparators.
- Off-target gene effects16 and phenotype characterization of future biotechnology products.
- Impacts of future biotechnology products on nontarget organisms.
- Gene fitness, gene stability, and propensity for horizontal gene transfer in future biotechnology products.
- Measures designed to control organismal traits and mitigate risk in the event of intentional or accidental release of future biotechnology products.
- Life-cycle analysis of future biotechnology products.
- Monitoring and surveillance of future biotechnology products.
- Modeling to inform risk-based hypotheses, collect data to reduce uncertainties, and provide findings or predictions in risk characterization with regards to future biotechnology products.
- Economic costs and benefits of future biotechnology products.
- Social costs and benefits of future biotechnology products.
The committee also included an “other” category to catch any other areas of research not described above in the event any of the agencies receiving the RFI had additional information to share. Information falling into this category is not included in the following figures but is described within the text.
EPA reported it had no ongoing research directly tied to any one area of the RFI. However, the agency did provide the committee with information regarding efforts within its Office of Research
___________________
16 It is important to distinguish between nontarget effects and off-target effects. Nontarget effects are unintended, short- or long-term consequences for one or more organisms other than the organism intended to be affected by an action or intervention. Concern about nontarget effects typically centers around unforeseen harms to other species or environments, but nontarget effects can also be neutral or beneficial. Off-target effects are unintended, short- or long-term consequences of an intervention on the genome of the organism in which the intended effect was incorporated. See also NASEM (2016a).
and Development that would enhance risk-assessment capabilities and could be applied to biotechnology. It is worth noting that EPA previously funded intramural and extramural biotechnology research programs, but these activities were discontinued in 2012. At the time the committee was writing its report, EPA possessed some capacity and capability for implementing probabilistic ecological risk assessments (as noted above) but had done so only on a limited basis for nonbiotechnology pesticides; models appropriate for ecological risk assessments of biotechnology products had not been developed.
USDA reported a total of approximately $13.23 million invested during 2012–2015 across 10 research areas (Figure 4-7). The response from USDA reflects investments through its Biotechnology Risk Assessment Grants (BRAG) program, which is jointly administered by USDA’s Agricultural Research Service (ARS) and National Institute of Food and Agriculture. The BRAG program receives input regarding its program priorities through multiple regulatory agencies that have an interest in the environmental risk related to the introduction of GE organisms, including USDA–APHIS, EPA, and FDA. About 75 percent of BRAG award recipients for the years included were scientists with land-grant universities or USDA–ARS.
The committee also contacted and received feedback in response to the RFI from several federal agencies that are not primary agencies within the Coordinated Framework. Of the seven
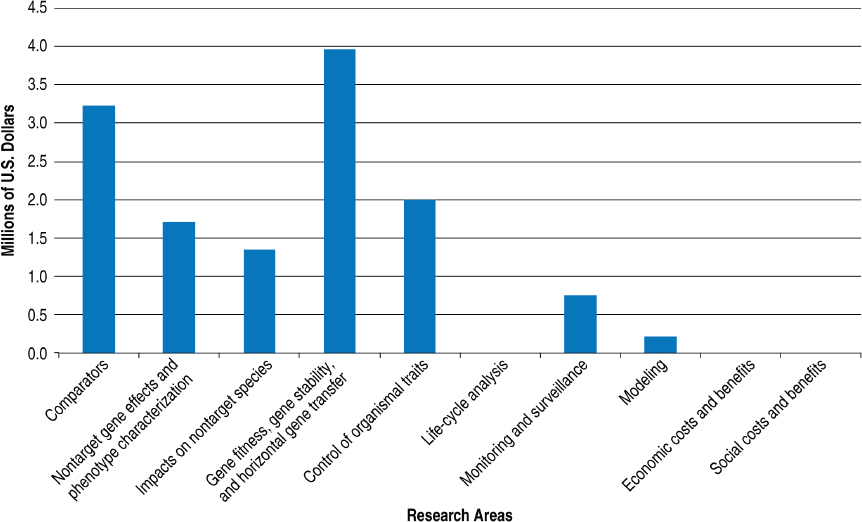
NOTES: Life-cycle analysis, economic costs and benefits, and social costs and benefits are outside the authorization of the BRAG program. USDA provided information regarding the awards and which of the research areas an award belongs to. The value entered for projects that fit into more than one research area was obtained by dividing the total value of the award by the number of research areas it fit into.
SOURCE: Information provided by the U.S. Department of Agriculture. Available upon request from the National Academies’ Public Access Records Office at PARO@nas.edu.
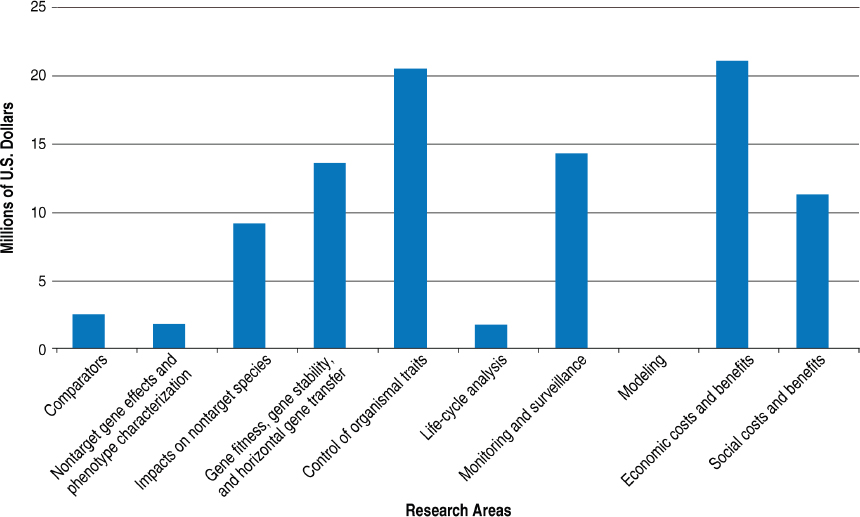
NOTES: SES and CBET submitted information for fiscal years 2012–2015. IIP submitted information for fiscal years 2012–2016. This figure has been updated since the prepublication release. The amount of money directed toward control of organismal traits has been corrected.
SOURCES: Information provided by the National Science Foundation’s Division of Industrial Innovation and Partnerships (IIP), Division of Social and Economic Sciences (SES), and Division of Chemical, Bioengineering, Environmental and Transport Systems (CBET). Available upon request from the National Academies’ Public Access Records Office at PARO@nas.edu.
offices at the National Science Foundation (NSF) that were sent the RFI, the committee received responses from three of them: the Division of Industrial Innovation and Partnerships, the Division of Social and Economic Sciences, and the Division of Chemical, Bioengineering, Environmental and Transport Systems. These divisions reported total investments of approximately $95.8 million during 2012–2016 (Figure 4-8); additionally, they reported approximately $44.8 million in investments fitting under “other,” which primarily involved the product-development research and not research pertaining to the risk assessment of those products. The U.S. Army Corps of Engineers invested $2.08 million during the same time period ($1.24 million in fitness, gene stability, and horizontal gene transfer; and $0.84 million in monitoring and surveillance).
The Office of Naval Research (ONR), the Defense Threat Reduction Agency (DTRA), and the U.S. Army Research Laboratory (ARL) provided the committee with information pertaining to their investments in 2012–2016; most of this research was on biotechnology products (falling under the “other” category) and not directly related to risk-analysis research areas outlined in the RFI. ONR reported approximately $32.9 million, DTRA reported approximately $149.8 million, and ARL reported approximately $88 million in awards pertaining to research and development of biotechnology products.
The Intelligence Advanced Research Projects Agency (IARPA) and the Defense Advanced Research Projects Agency (DARPA) indicated that they were initiating efforts in the areas outline by the RFI as well as research into future products of biotechnology. IARPA indicated to the committee (Julias, 2016) that a 2016 research initiative addressing several of the topics in the RFI was under way and will address comparators; computational modeling; off-target gene effects and phenotypic characterization; fitness, genetic stability, and horizontal gene transfer; control of organismal traits; and impact on nontarget organisms. Although the total level of future investments was not provided, IARPA anticipated the majority of funding would initially address comparators and computational modeling. At the time the committee was writing its report, DARPA was launching a “Safe Genes Program” to support responsible innovation while mitigating the risk of unintended consequences of genome editing and derivative technologies, including gene drives.17 The level of future research investments for this program was not publicly available.
The Office of Biological and Environmental Research within the U.S. Department of Energy (DOE) indicated investments of approximately $15 million per year between 2012 and 2016 made by the Genomic Science program. The committee was unable to get resolution as to the specific areas (as outlined by the RFI), but it appeared that awards had gone toward the control of organismal traits or future products of biotechnology (the latter falling under the “other” category). Other agencies that responded to the RFI (U.S. Air Force Office of Scientific Research and FDA) indicated that their intramural and extramural programs do not currently address the topics identified by the committee. The committee also heard from the National Invasive Species Council (NISC) Secretariat, which provides support to the NISC to coordinate control of invasive species across the federal government. The council has interest in and has supported work in this area in the past but was not directly funding the areas outlined in the RFI at the time the committee was writing its report.
In summary, for those federal agencies that responded to the RFI, risk-analysis research resources cumulatively totaled to $111.08 million over the 2012–2016 period (Table 4-1). Of note is the lack of any research funding by FDA to address risk analyses for future biotechnology products. Although EPA did not have direct investments for risk analyses of future biotechnology products, its efforts in risk-analysis research in other areas can be applied toward biotechnology products.
A 2015 Woodrow Wilson Center report estimated that in 2008–2014 the U.S. government invested $820 million in synthetic-biology research (with a significant increase in 2010–2014) with DARPA, the U.S. Department of Defense (excluding DARPA), DOE, NSF, and the National Institutes of Health investing the majority resources (Wilson Center, 2015). Of the total investment, the Wilson Center estimated less than 1 percent was invested in risk research and approximately 1 percent was invested in ethical, legal, and social issues. The results of the committee’s RFI indicated approximately $6.98 million was invested in social research in 2012–2015, which is fairly consistent with the Wilson Center findings. However, assuming a flat budget for total synthetic-biology research in 2015 as compared to 2014 (that is, approximately $220 million in 2015 or an estimated total of $1.04 billion in 2008–2015), the results of the RFI indicate approximately 9 percent of the total ($89.92 million when excluding social research) was invested in risk–benefit research. Excluding research on economic costs and benefits during the period 2012–2015 ($20.93 million), risk research during that period (approximately $68.98 million) would represent approximately 7 percent of the total synthetic-biology research portfolio.
The results of the Wilson Center study and the committee’s RFI indicate that from the “outside looking in” it is difficult to ascertain the level of research funding to support risk analyses, and it appears it may also be a challenge for the U.S. government to aggregate investment totals across agencies. Consequently, the committee acknowledges uncertainty in its estimates of risk-analysis
___________________
17 DARPA Safe Genes Proposers Day. Available at https://www.eiseverywhere.com/ehome/196223/443234. Accessed October 11, 2016.
TABLE 4-1 Federal Investment in Risk-Analysis Research, 2012–2015
| Type of Risk Research | Amount (in millions) |
|---|---|
| Comparators | $5.70 |
| Off-target gene effects and phenotypic characterization | $3.02 |
| Impacts on nontarget species | $8.86 |
| Fitness, gene stability, and horizontal gene transfer | $15.44 |
| Control of organismal traits | $19.36 |
| Life-cycle analyses | $1.72 |
| Monitoring and surveillance | $14.67 |
| Modeling | $0.22 |
| Economic costs and benefits | $20.93 |
| Social costs and benefits | $6.98 |
| TOTAL | $96.90 |
NOTES: This table does not reflect the investments made by IARPA, DARPA, or DOE as there was not sufficient resolution or figures given to be categorized under a specific type of research. A total of $251.27 million was reported to be invested in research that was considered to fall under the “other” category by the committee or the agencies themselves. For consistency, this table represents the amounts reported through 2015, even though some agencies reported figures for 2016. The U.S. Army Corps of Engineers only reported numbers for 2012 and 2015, which are also included here.
SOURCE: Responses to the committee’s request for information, available upon request from the National Academies’ Public Access Records Office at PARO@nas.edu.
research investments, due the level of resolution different agencies provide for their yearly budgets. The committee also realizes there may be related research efforts outside of synthetic biology that can support future risk-analysis methods (for example, research undertaken by USDA’s Agricultural Research Service and the Agricultural Experiment Stations at public universities). However, the estimates suggest that research in specific areas such as modeling and life-cycle analyses, which are critical for supporting premarket risk assessments and socioeconomic cost–benefit analyses, represent 0.02 percent and 0.17 percent, respectively, of the total synthetic-biology research investment. Monitoring and surveillance research, which is critical to supporting post-market assessments and implementing risk-mitigation measures, represents approximately 1.4 percent of the total research portfolio. Research concerning comparators; off-target gene effects and phenotypic characterization; impacts on nontarget species; gene fitness, stability, and horizontal gene transfer; and control of organisms (containment and confinement) combined to represent approximately 5 percent of the total synthetic-biology research investment. These research areas can support both premarket and post-market risk analyses. It is difficult to determine the appropriate level of investment for risk research to support oversight of future biotechnology products; however, the sense of the committee is that the current level is insufficient.
The committee is also concerned that the current U.S. government risk-analysis research portfolio may not be planned in a manner that can maximize its return on investment. It is encouraging that USDA’s BRAG program includes other federal agencies, including EPA and FDA, in identifying research priorities; however, there is no indication that these interactions include regulatory risk assessors, risk managers, and researchers working together to vet and adapt research products for use in risk assessments and socioeconomic cost–benefit analyses. Based on the responses to the RFI, there does not appear to be significant interaction between DARPA and other agencies in the U.S. Department of Defense, DOE, and NSF with USDA, EPA, and FDA regulators in research
planning or in envisioning a new paradigm for advancing risk-analysis approaches for future biotechnology products.
While the reported research portfolio may be relevant to the risk-analysis needs for future biotechnology products, it is not clear that it is responsive to the nature and extent of future challenges facing public- and private-sector risk assessors, risk managers, and other interested and affected parties. Finally, the committee notes that the financial resources needed to establish an adequate research portfolio for the United States need not fall solely on the U.S. government and the nation’s tax payers. The U.S. government may want to explore establishing open and transparent approaches to integrate and optimize public investments, private investments, and public–private partnerships to realize the needed resources to support development of a responsive, nimble, and robust risk-analysis paradigm.
The consequences of current levels of intramural and extramural funding in research and to support future risk-assessment needs suggest the number of products poised to enter the marketplace in the coming 5–10 years may outpace the means and capacity for voluntary- or regulatory-based risk–benefit assessments to inform premarket decision making or post-market oversight, including environmental monitoring.
SUMMARY AND CONCLUSIONS
In this chapter, the committee examined whether product risks associated with future biotechnology will be similar to or different from those of existing biotechnology products and how well understood those risks are. It also reviewed the extent to which the current capabilities of the regulatory system are appropriately aligned with the likely needs in oversight of those future products. The committee reached the following conclusions.
Conclusion 4-1: The risk-assessment endpoints for future biotechnology products are not new compared with those that have been identified for existing biotechnology products, but the pathways to those endpoints have the potential to be very different in terms of complexity.
The biotechnology products emerging in the next 5–10 years pose a diverse array of environmental, health, and safety risks that vary widely in terms of their potential impacts, likelihood of occurrence, spatial and temporal dimensions, and the appropriate regulatory policies for their assessment. Although the nature of human health and ecological risk-assessment endpoints that will need consideration are similar to those identified with existing products, the pathways to these endpoints will differ in complexity. To the extent future indoor manufacturing will occur in domestic settings, the types of risk-assessment endpoints of future biotechnology products that need to be considered will likely be similar to those used with existing indoor manufacturing; however, the dimensionality of these risk assessments will be more complex and the risk estimates will be more uncertain. Open-release microbial consortia may have more complexity in the dimensionality of their associated risk assessments, but the degree to which that complexity differs from nonengineered microorganisms is dependent upon on a number of variables, such as use pattern and environmental conditions. Such products as well as those designed to suppress, eradicate, or enhance a target species will have new risk pathways and increased dimensionality in the risk assessments.
Transparently elaborated regulatory decisions that provide precedents or comparative examples may shape development strategies for future products. The degree to which risks associated with a product are “well understood” is a subjective determination and formal uncertainty analyses will be needed to determine where a reduced level of regulatory scrutiny may be warranted.
The committee concludes that it is reasonable to assume that existing and future biotechnol-
ogy products that are similar in terms of their properties, mechanisms, and use patterns may have similar risk profiles, but methods for quantifying similarity between products need to be developed.
Conclusion 4-2: Gaps in the risk-analysis capability of the regulatory system can create a real or perceived impression that some products are entering the marketplace without any government oversight, which can undercut public confidence.
Regulators have the two-fold concern of maintaining and building public confidence in the regulatory process and of engaging in continual improvement of risk analyses to ensure human health and environmental safety. Public confidence in government oversight of emerging technologies may be eroded to the extent there is a lack of transparency and clarity as to how regulatory authorities are undertaking risk assessments, including identifying societal values in addition to taking input from biotechnology developers in formulating regulatory decisions. Given the nature and diversity of future biotechnology products, increased public and developer participation may improve both the understanding and quality of risk-analysis approaches. With better understanding of real or perceived gaps in the risk-analysis process, regulators would be better equipped with the capabilities needed to strengthen risk analysis.
Conclusion 4-3: The expertise and capacity of EPA, FDA, USDA, and other agencies that have interests related to future biotechnology products may not be sufficient to handle the expected scope and scale of future biotechnology products.
Although the regulatory agencies have access to a number of external advisory committees, the number of in-house experts and the responses to the RFI indicate that there may not be sufficient scientific capability, capacity, and tools within and across the agencies to address the risk-assessment challenges for future biotechnology products. The number of products poised to enter the marketplace in the coming years may outpace the means and capacity for voluntary- or regulatory-based assessment processes to inform decision making. This imbalance, if not addressed in the near term, could impede the development of new biotechnology products in the long term. In addition, based on the expected scope and complexity of products of biotechnology that are likely in the next 5–10 years, advances in regulatory science will be needed for effective and appropriate evaluation. Clearly, the profusion of future biotechnology products poses a significant potential stress to the existing regulatory system. Regulatory agencies are likely not prepared with sufficient staff, appropriate risk-analysis approaches, and corresponding guidance for development and evaluation of data packages submitted by product developers.
REFERENCES
Ankley, G.T., R.S. Bennett, R.J. Erickson, D.J. Hoff, M.W. Hornung, R.D. Johnson, D.R. Mount, J.W. Nichols, C.L. Russom, P.K. Schmieder, J.A. Serrano, J.E. Tietge, and D.L. Villeneuve. 2010. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry 29(3):730–741.
DaCunha, C. 2016. Universal Bio Mining: Transforming Mining with Synthetic Biology. Presentation to the National Academies of Sciences, Engineering, and Medicine Committee on Future Biotechnology Products and Opportunities to Enhance Capabilities of the Biotechnology Regulatory System, June 1, Washington, DC.
Drinkwater, K., T. Kuiken, S. Lightfoot, J. McNamara, and K. Oye. 2014. Creating a Research Agenda for the Ecological Implications of Synthetic Biology. Joint Workshops by the MIT Program on Emerging Technologies and the Wilson Center’s Synthetic Biology Project. Available at https://www.wilsoncenter.org/sites/default/files/SYNBIO_create%20an%20agenda_v4.pdf. Accessed August 10, 2016.
EFSA (European Food Safety Authority). 2011. Guidance on selection of comparators for the risk assessment of genetically modified plants and derived food and feed. EFSA Journal 9:2149.
EOP (Executive Office of the President). 2017. Modernizing the Regulatory System for Biotechnology Products: An Update to the Coordinated Framework for the Regulation of Biotechnology. Available at https://obamawhitehouse.archives.gov/sites/default/files/microsites/ostp/2017_coordinated_framework_update.pdf. Accessed January 30, 2017.
EPA (U.S. Environmental Protection Agency). 1998. Guidelines for Ecological Risk Assessment. Federal Register 63:26846–26924.
EPA. 2003. Generic Ecological Assessment Endpoints (GEAEs) for Ecological Risk Assessment. EPA/630/P-002/004F. Risk Assessment Forum, Washington, DC. Available at https://www.epa.gov/sites/production/files/2014-11/documents/generic_endpoinsts_2004.pdf. Accessed January 30, 2017.
EPA. 2009. Assessment of Hazard and Exposure Associated with Nanosilver and Other Nanometal Pesticides. FIFRA Scientific Advisory Panel Meeting, November 3–6. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2009-0683. Accessed January 14, 2017.
EPA. 2011. Integrated Approaches to Testing and Assessment Strategies (IATA): Use of New Computational and Molecular Tools. FIFRA SAP Meeting May 24-26, 2011. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2011-0284. Accessed October 25, 2016.
EPA. 2012. FIFRA SAP Meeting on Pollinator Risk Assessment Framework, September 11–16. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2012-0543. Accessed January 14, 2017.
EPA. 2014a. Notice of FIFRA SAP Meeting; RNAi Technology as a Pesticide: Problem Formulation for Human Health and Ecological Risk Assessment, January 28. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2013-0485. Accessed on August 10, 2016.
EPA. 2014b. Risk Assessment Forum White Paper: Probabilistic Risk Assessment Methods and Case Studies. EPA/100/R-14/004. Risk Assessment Forum, Washington, DC. Available at https://www.epa.gov/sites/production/files/2014-12/documents/raf-pra-white-paper-final.pdf. Accessed on November 20, 2016.
EPA. 2015. Development of a Spatial Aquatic Model (SAM) for Pesticide Risk Assessment. FIFRA Scientific Advisory Panel Meeting, September 15–18. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2015-0424. Accessed January 14, 2017.
EPA. 2016a. DEEM-FCID/Calendex Software Installer. Available at https://www.epa.gov/pesticide-science-and-assessing-pesticide-risks/deem-fcidcalendex-software-installer. Accessed on August 10, 2016.
EPA. 2016b. Public Participation Process for Registration Actions. Available at https://www.epa.gov/pesticide-registration/public-participation-process-registration-actions. Accessed on August 10, 2016.
EPA. 2016c. FIFRA Scientific Advisory Panel; Notice of Public Meeting. Available at https://www.regulations.gov/docket?D=EPA-HQ-OPP-2013-0485. Accessed on August 10, 2016.
EPA. 2016d. EPA Sustainable and Healthy Communities National Research Program: 2015 Accomplishments. EPA 601/R-16/004. Office of Research and Development, EPA. Available at https://www.epa.gov/sites/production/files/2016-08/documents/shc2015finalepa601-r16-004web-508_metadata.pdf. Accessed September 13, 2016.
EPA. 2016e. TIM Version 3.0 Beta–Technical Description and User’s Guidance. Available at https://www.epa.gov/pesticide-science-and-assessing-pesticide-risks/tim-version-30-beta-technical-description-and-users. Accessed August 10, 2016.
EPA. 2016f. Research on Analyzing the Life Cycle of Chemicals. Available at https://www.epa.gov/chemical-research/research-analyzing-life-cycle-chemicals. Accessed August 10, 2016.
EPA. 2016g. Generic Ecological Assessment Endpoints (GEAEs) for Ecological Risk Assessment: Second Edition with Generic Ecosystem Services Endpoints Added. EPA/100/F15/005. Risk Assessment Forum, Washington, DC. Available at https://www.epa.gov/sites/production/files/2016-08/documents/geae_2nd_edition.pdf. Accessed November 20, 2016.
Fairbrother, A., J. Gentile, C. Menzie, and W. Munns. 1999. Report on the Shrimp Virus Peer Review and Risk Assessment Workshop: Developing a Qualitative Risk Assessment. Washington, DC: EPA.
FDA (U.S. Food and Drug Administration). 2016. Preliminary Finding of No Significant Impact in Support of an Investigational Field Trial of OX513A Aedes aegypti Mosquitoes. March. Available at http://www.fda.gov/downloads/AnimalVeterinary/DevelopmentApprovalProcess/GeneticEngineering/GeneticallyEngineeredAnimals/UCM487379.pdf. Accessed September 30, 2016.
Focazio, M.J., D.W. Kolpin, K.K. Barnes, E.T. Furlong, M.T. Meyer, S.D. Zaugg, L.B. Barber, and M.E. Thurman. 2008. A national reconnaissance for pharmaceuticals and other organic wastewater contaminants in the United States — II. Untreated drinking water sources. Science of the Total Environment 402(2-3):201–216.
Funtowicz, S.O., and J.R. Ravetz. 1995. Science for the post normal age. Pp. 146–161 in Perspectives on Ecological Integrity, L. Westra and J. Lemons, eds. Dordrecht, Netherlands: Springer.
Gallagher, S.G., G.E. Rice, L.J. Scarano, L.K. Teuschler, G. Bollweg, and L. Martin. 2015. Cumulative risk assessment lessons learned: A review of case studies and issue papers. Chemosphere 120:697–705.
Greenblott, J.M., T. O’Farrell, and B. Burchard. 2016. EPA’s strategic foresight pilot project. Presentation at the Institute for Alternative Futures’ Public Sector Foresight Network and Federal Foresight Community of Interest Foresight Day, July 22, Washington, DC.
IOM (Institute of Medicine). 2013. Environmental Decisions in the Face of Uncertainty. Washington, DC: The National Academies Press.
Julias, J. 2016. IARPA Overview of NAS RFI. Webinar presentation to the National Academies of Sciences, Engineering, and Medicine Committee on Future Biotechnology Products and Opportunities to Enhance Capabilities of the Biotechnology Regulatory System, July 25.
Kuzma, J., J. Larson, and P. Najmaie. 2009a. Evaluating oversight systems for emerging technologies: A case study of genetically engineered organisms. Journal of Law, Medicine, and Ethics 37(4):546–586.
Kuzma, J., A. Kuzhabekova, and K. Wilder. 2009b. Improving oversight of genetically engineered organisms. Policy and Society 28(4):279–299.
Kuzma, J., A. Kuzhabekova, S. Priest, and L. Yerhot. 2010. Expert opinion of emerging technologies oversight: Lessons for nanotechnology from biotechnology. Pp. 133–156 in Understanding Nanotechnology: Philosophy, Policy, and Publics, U. Fiedeler, C. Coenen, S.R. Davies, and A. Ferrari, eds. Amsterdam: IOS Press.
Landis, W. 2004. Ecological risk assessment conceptual model formulation for nonindigenous species. Risk Analysis 24(4):847–858.
Murphy, B., C. Jansen, J. Murray, and P. De Barro. 2010. Risk Analysis on the Australian Release of Aedes aegypti (L.) (Diptera: Culicidae) Containing Wolbachia. Indooroopilly, Australia: CSIRO.
Murray, J.V., C.C. Jansen, and P. De Barro. 2016. Risk associated with the release of Wolbachia-infected Aedes aegypti mosquitoes into the environment in an effort to control dengue. Frontiers in Public Health 4:43.
NAPA (National Academy of Public Administration). 2016. Strategic foresight. Pp. 7–11 in Transition 2016: Equipping the Government for Success in 2016 and Beyond. Available at http://www.napat16.org/images/7.8.2016_T16_Final_Report.pdf. Accessed February 12, 2017.
NAS (National Academy of Sciences). 1987. Introduction of Recombinant DNA-Engineered Organisms into the Environment: Key Issues. Washington, DC: National Academy Press.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2016a. Gene Drives on the Horizon: Advancing Science, Navigating Uncertainty, and Aligning Research with Public Values. Washington, DC: The National Academies Press.
NASEM. 2016b. Genetically Engineered Crops: Experiences and Prospects. Washington, DC: The National Academies Press.
Nelson, K.C., G. Kibata, L. Muhammad, J.O. Okuro, F. Muyekho, M. Odindo, A. Ely, and J.M. Waquil. 2004. Problem formulation and options assessment (PFOA) for genetically modified organisms: The Kenya case study. Pp. 57–82 in Environmental Risk Assessment of Genetically Modified Organisms, Vol. 1. A Case Study of Bt Maize in Kenya, A. Hilbeck and D. Andow, eds. Cambridge, MA: CABI Publishing.
NRC (National Research Council). 1996. Understanding Risk: Informing Decisions in a Democratic Society. Washington, DC: National Academy Press.
NRC. 2000. Genetically Modified Pest-Protected Plants: Science and Regulation. Washington, DC: National Academy Press.
NRC. 2007. Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington, DC: The National Academies Press.
NRC. 2008. Public Participation in Environmental Assessment and Decision Making. Washington, DC: The National Academies Press.
NRC. 2009. Science and Decisions: Advancing Risk Assessment. Washington, DC: The National Academies Press.
NRC. 2012. Science for Environmental Protection: The Road Ahead. Washington, DC: The National Academies Press.
NRC. 2013. Assessing Risks to Endangered and Threatened Species from Pesticides. Washington, DC: The National Academies Press.
NSTC (National Science and Technology Council). 2007. Agricultural Biotechnology Risk Analysis Research in the Federal Government: Cross Agency Cooperation. Available at https://www.nsf.gov/pubs/2007/nsf07208/nsf07208.pdf. Accessed October 26, 2016.
Orr, R. 2003. Generic nonindigenous aquatic organism risk analysis. Pp. 415–438 in Invasive Species: Vectors and Management Practices, G.M. Ruiz and J.T. Carlton, eds. Washington, DC: Island Press.
Phillips, P.J., C. Schubert, D. Argue, I. Fisher, E.T. Furlong, W. Foreman, J. Gray, and A. Chalmers. 2015. Concentrations of hormones, pharmaceuticals and other micropollutants in groundwater affected by septic systems in New England and New York. Science of the Total Environment 512–513:43–54.
Reardon, S. 2016. Welcome to the CRISPR zoo. Nature 431(7593):160–163.
Renn, O. 1992. Concepts of risk: A classification. Pp. 53–79 in Social Theories of Risk, S. Krimsky and D. Golding, eds. Westport, CT: Praeger.
Renn, O. 2005. Risk Governance: Towards an Integrative Approach. Geneva: International Risk Governance Council.
Ricroch, A.E., and M.-C. Hénard-Damave. 2016. Next biotech plants: New traits, crops, developers and technologies for addressing global challenges. Critical Reviews in Biotechnology 36(4):675–690.
Roberts, J.P., S. Stauffer, C. Cummings, and J. Kuzma. 2015. Synthetic Biology Governance: Delphi Study Workshop Report. GES Center Report No. 2015.2. Available at https://research.ncsu.edu/ges/files/2014/04/Sloan-Workshop-Report-final-ss-081315-1.pdf. Accessed October 10, 2016.
Rose, R., S. McCammon, and S. Lively. 2006. Proceedings: Workshop on Confinement of Genetically Engineered Crops During Field Testing. U.S. Department of Agriculture. Available at https://www.aphis.usda.gov/brs/pdf/conf_ws_proc2.pdf. Accessed October 4, 2016.
Schaider, L.A., R.A. Rudel, J.M. Ackerman, S.C. Dunagan, and J.G. Brody. 2014. Pharmaceuticals, perfluorosurfactants, and other organic wasterwater compounds in public drinking water wells in a shallow sand and gravel aquifer. Science of the Total Environment 468–469:384–393.
Selin, C., K.C. Rawlings, K. de Ridder-Vignone, J. Sadowski, C.A. Allende, G. Gano, S.R. Davies, and D.H. Guston. 2016. Experiments in engagement: Designing public engagement with science and technology for capacity building. Public Understanding of Science [online]. Available at http://pus.sagepub.com/content/early/2016/01/13/0963662515620970.full.pdf+html. Accessed October 25, 2016.
SEP (Science for Environment Policy). 2016. Future Brief: Identifying Emerging Risks for Environmental Policies. Issue 13. Bristol, UK: European Commission.
SRA (Society for Risk Analysis). 2015. SRA glossary. June 22. Available at http://sra.org/sites/default/files/pdf/SRA-glossary-approved22june2015-x.pdf. Accessed January 14, 2017.
Sunstein, C.R. 2005. Laws of Fear: Beyond the Precautionary Principle. Cambridge, UK: Cambridge University Press.
Suter, G.W. 2007. Ecological Risk Assessment. Boca Raton, FL: CRC Press.
Traynor, P.L., and J.H. Westwood, eds. 1999. Proceedings of a Workshop on Ecological Effects of Pest Resistance Genes in Managed Ecosystems, January 31–February 3, Bethesda, MD. Available at http://www.isb.vt.edu/documents/proceedings.pdf. Accessed October 4, 2016.
USDA–APHIS (U.S. Department of Agriculture–Animal and Plant Health Inspection Service). 2004. Environmental Impact Statement; Introduction of Genetically Engineered Organisms. Federal Register 69:3271–3271.
USDA–APHIS. 2016. Environmental Impact Statement; Introduction of the Products of Biotechnology. Federal Register 81:6225–6229.
Warren-Hicks, W.J., and A. Hart, eds. 2010. Application of Uncertainty Analysis to Ecological Risks of Pesticides. Boca Raton, FL: CRC Press.
Wilson Center. 2015. U.S. Trends in Synthetic Biology Research Funding. Synthetic Biology Project. Washington, DC: Woodrow Wilson Center for International Scholars.
Wolt, J.D., and R.K.D. Peterson. 2010. Prospective formulation of environmental risk assessments: Probabilistic screening for Cry1A(b) maize risk to aquatic insects. Ecotoxicology and Environmental Safety 73:1182–1188.
Writer, J.H., I. Ferrer, L.B. Barber, and E.M. Thurman. 2013. Widespread occurrence of neuro-active pharmaceuticals and metabolites in 24 Minnesota rivers and wastewaters. Science of the Total Environment 461–462:519–527.
Yeardley, R.B., Jr., B. Dyson, and M. Tenbrink. 2011. EPA Growing DASEES (Decision Analysis System for a Sustainable Environment, Economy and Society) To Aid in Making Decisions on Complex Environmental Issues. EPA/600/F-11/023. Washington, DC: EPA. Available at https://cfpub.epa.gov/si/si_public_record_report.cfm?dirEntryId=238232. Accessed September 24, 2016.






























