
As noted by many workshop participants, neuroinflammation appears to play an important role in many central nervous system (CNS) diseases. The discussion that follows is not meant to provide a comprehensive review of the diseases in which neuroinflammation plays a role; rather, the examples illuminate important aspects of neuroinflammation that are relevant to the development of biomarkers and therapeutics.
MULTIPLE SCLEROSIS
MS represents the prototypic neuroinflammatory disease and is a major cause of neurological disability in young adults, although highly variable and unpredictable. Amit Bar-Or noted that in the past decade, multiple treatment options have become available, which perturb the complex pathophysiology of MS in many ways. However, selecting the treatment modality that will be best suited for individual patients remains an ongoing challenge, he said. Biomarkers that allow characterization of relevant biologies in individual patients would allow clinicians to choose the most appropriate treatment (both efficacy and safety), monitor response to treatment, and possibly change or sequence treatments, said Bar-Or.
Bar-Or described a simplified immune pathogenesis model of MS in which immune cells in the periphery are activated, upregulating a series of molecules that enable the immune cells to more efficiently cross the blood‒brain barrier (BBB), where they are reactivated. Historically, MS has been thought of as primarily a T-cell disease, but Bar-Or said that recent research shows that B cells are also important. Bar-Or said that to capture the complexity and heterogeneity of the disease, it will be necessary to measure the biologies of multiple different cell subsets. The mechanisms that underlie the aberrant balance of T, B, and myeloid cells may be a target for biomarker development, he said.
Indeed, the most recently approved treatment for MS—ocrelizumab—selectively targets B cells that express CD20, said Bar-Or. However, ocrelizumab treatment has no apparent impact on the levels of antibodies in CSF, suggesting that non-antibody functions of B cells are important (Hauser et al., 2017). Bar-Or and colleagues have shown that B cells produce different cytokines depending on the mode of activation. Compared to healthy controls, the B cells of MS patients produce increased proinflammatory cytokines and reduced levels of anti-inflammatory cytokines that regulate T-cell function and autoimmunity (Bar-Or et al.,
2010). They have also shown in patients with MS an increased number of B cells that produce granulocyte macrophage-colony stimulating factor (GM-CSF), a cytokine that activates myeloid cells to produce proinflammatory responses. B-cell depletion diminishes myeloid cell proinflammatory responses (Li et al., 2015). From a biomarker perspective, this means that measurement of these pro- and anti-inflammatory cytokines may enable definition of an individual’s functional immune profile, said Bar-Or.
Magnetic resonance imaging (MRI) is a highly sensitive tool for diagnosing MS, demonstrating that the disease is dynamic, multifocal, and diffuse, said Bar-Or, adding that gadolinium enhancement further demonstrates a breach of integrity in the BBB, which is thought to represent perivascular inflammation. However, Bar-Or said that within the CNS compartment, damage extends beyond the white-matter focal areas captured by standard T2-weighted MR images to gray-matter areas, including areas at the surface of the cortex. Indeed, cortical pathology seems to correlate better than white-matter pathology with longer-term disability, cognitive dysfunction, and social issues that people with MS experience, he said. Histopathological studies have shown that immune cells accumulate at the meninges in individuals with MS, and of interest is whether they contribute to cortical injury. Within the cortext underlying such meningeal immune cell collections, there is evidence of neuronal injury and microglial activation. This microglial activation occurs in a graded pattern from the surface inward, suggesting that a secreted factor released from meningeal immune cells contributes to neuronal damage (Magliozzi et al., 2010). Bar-Or suggested that it may be possible to develop this factor as a biomarker.
Measurement of analytes in the CSF and serum provides complementary information, said Bar-Or. For example, oligoclonal bands and immunoglobulin G (IgG) in CSF enable a diagnosis of CNS neuroinflammatory disease. Serum biomarkers that may distinguish MS from other CNS inflammatory diseases include antibodies against aquaporin 4 (AQP4), which are diagnostic for neuromyelitis optica (NMO) spectrum disease, a condition that is pathologically distinct from MS and has different therapeutic implications, said Bar-Or. There is active interest in whether the presence of serum myelin oligodendrocyte glycoprotein (MOG)-directed antibodies may reflect a subtype of MS or another entity. Other blood biomarkers that may be measured when starting treatment or considering a change in treatment include interferon beta–neutralizing antibodies, anti-JC antibodies, lymphopenia, and B-cell
counts, he said. He also mentioned many candidate biomarkers. Those that show the most promise include neurofilament in CSF and serum, chitinase 3 like 1 in CSF, and some miRNAs. The cooperativity of different cell types could also be assessed by simultaneously assessing levels of different biomarkers in those cell subsets. This would require a concerted investment in dynamic cell-based assays, but would provide a much-needed added layer to biomarker development efforts, said Bar-Or.
Indeed, despite the development of biomarkers that reveal important aspects of disease, substantial discrepancies remain among the time course of imaging markers, clinical disease, and biological progression represented by peripherally mediated and CNS-compartmentalized inflammatory injury, said Bar-Or.
Moreover, he said, the relationship between inflammation and degeneration remains unclear. He cited a recent small study of patients with poor-prognosis, early-stage, aggressive MS, which showed that complete immunoablation followed by autologous bone marrow transplantation essentially halted disease progression, suggesting that robustly removing peripheral inflammation prevented neuroinflammation and neurodegeneration (Atkins et al., 2016). Bone marrow transplantation has been unsuccessful in later-stage disease, when CNS-compartmentalized processes are acting relatively independently of peripherally mediated inflammation, said Bar-Or.
TRAUMATIC BRAIN INJURY
TBI is a major cause of death and disability that has recently attracted increased attention because of the wars in Iraq and Afghanistan and increased awareness of the negative consequences of sports injuries, said Fiona Crawford. The vast majority of military cases are mild, yet even these have significant health impacts. Chronic traumatic encephalopathy (CTE), such as that seen in boxers, football players, and other athletes, represents a subset of TBI with a distinctive presentation that may include marked behavioral changes, she said, adding that TBI is also known to be a risk factor for AD in later life or other neurodegenerative diseases.
Crawford described the complex disease processes underlying both the acute injury and the secondary consequences of TBI, including calcium dysregulation, mitochondrial dysfunction, free radical generation, and neuroinflammation. Both innate and adaptive immune responses are
invoked with the production of many cytokines and chemokines, the recruitment of T lymphocytes, and the activation of microglia, she said. Following the acute injury, these responses represent attempts to repair damage, said Crawford, and the persistence of the negative consequences of microglial activation causes chronic problems. The timing of these responses is important in order to identify the correct therapeutic window, said Crawford. Although many compounds have shown positive effects in preclinical rodent models, translation to human populations has been unsuccessful, which she said may be explained by the complexity and heterogeneity of these responses in humans.
Crawford advocated for increased attention in the preclinical space to better model the consequences of TBI. She described an array of animal models that have been developed that differ in terms of the animals used and the type of injury induced. She noted that mice have the advantage of being cheap, are available in many genetically manipulated strains, and have a life span that allows for relatively short studies. However, she also cited challenges associated with using animal models. For example, there are major differences in the brain architecture between mouse and human, and mice do not develop the hallmark pathology for CTE that is seen in humans—deposition of tau in the depths of the sulci—although she said that many other pathological features are similar across species. Crawford and colleagues have demonstrated in their mouse models similar axonal transport changes and axonal injury, accumulation of amyloid precursor protein (APP), myelin loss, astrogliosis, and inflammation microgliosis. Pigs have also been used in rotational models, she said.
Much of Crawford’s early work used a controlled cortical impact (CCI) model, which involves a craniectomy and produces a relatively severe injury. There are also blast injury, fluid percussion, and rotational models. The one common denominator in all of these models is neuroinflammation, said Crawford.
More recently, she and her colleagues have developed a closed-head-injury model of mild TBI, where they can test single or repetitive injury compared to sham injury, with no fracture or bleeding. Using this model, they showed that the mice perform poorly on neurobehavioral testing and demonstrate progressive and life-long changes in microgliosis (which they visualize histopathologically by Iba1 binding), astrocytosis (GFAP binding), and inflammation (phospho-STAT3 binding) in mice after repetitive mild TBI (r-mTBI–5 hits over 9 days) compared to sham controls (Mouzon et al., 2012, 2014). They also used this model to test whether targeting neuroinflammation with a compound called anata-
bine—which has shown efficacy in mouse models of AD, MS, and tauopathy—ameliorated the problems. In the r-mTBI model, anatabine had minimal acute effects, but dramatically improved performance on neurobehavioral testing 6 months after injury, reduced Iba1 staining in the hippocampus and corpus callosum at 9 months after injury, and reduced GFAP staining in the hippocampus, said Crawford. Anatabine reduced microgliosis and astrocytosis even when first administered 9 months after injury, suggesting a large therapeutic window, she said. However, when anatabine treatment was discontinued after 9 months, neuroinflammation appeared to reemerge, suggesting that anatabine suppressed but did not eradicate the damage (Ferguson et al., 2017). Crawford suggested that an adaptive response or epigenetic mechanism might explain this phenomenon. Indeed, she noted that posttraumatic stress disorder (PTSD) often occurs in combination with TBI, and that the sleep disturbances that characterize PTSD are thought to influence epigenetic changes.
Tau pathology persisting after the cessation of injuries has been particularly difficult to recapitulate in TBI mouse models. Using the same mild injury in a new paradigm of two hits per week over 3 months, Crawford and colleagues have demonstrated persistence of tau pathology at 3 months after injury. Crawford and colleagues suggested that the tau phenotype emerges only in the presence of concomitant neuroinflammation. This may indicate that phosphorylation of tau at the beginning of injury is a positive response, but that persistent hyperphosphorylation only happens in the inflamed environment, said Crawford.
HUNTINGTON’S DISEASE
Beth Stevens described her work to understand the mechanisms underlying neurodegenerative diseases by studying HD, the most common autosomal dominant, monogenetic neurodegenerative disease, which is characterized by disorders of movement, cognition, and behavior. The mutation that causes HD is an expansion of cytosine-adenosine-guanine (CAG) repeats in the gene for huntingtin protein, said Stevens, noting that although the mutation occurs in all cells in the body, there is a selective vulnerability to neurodegeneration of the neural circuit between the motor cortex and striatum, and medium spiny neurons in the striatum. This regional and cellular specificity and the availability of several mouse models make HD a useful model to examine the mechanisms un-
derlying synapse loss, said Stevens. Because the presence of the mutation can be used to identify individuals who will develop HD, Stevens’s lab has also been able to study synaptic loss in the earliest stages of disease in human tissue through a collaboration with Richard Faull from the University of Auckland, who operates a large HD brain bank in New Zealand. These studies show that progressive synapse loss begins in a regionally specific manner in disease and in the absence of neuroinflammation. In animal models, they have been able to follow synapse loss over time, demonstrating that it begins early in the preclinical stages of disease, even before there is overt inflammation.
Stevens’s lab has also been investigating the role of microglia and components of the complement cascade in synaptic loss. They have demonstrated a striking increase in two markers for complement—C1q and C3—in vulnerable brain regions in HD mice compared to wildtype, with deposition at the vulnerable synapses. In human tissue, they have also shown an upregulation in complement proteins in the HD brain. Using microglial stains, they have also shown a dramatic change in microglia in vulnerable regions of the mouse brain. However, to better understand functional impairments in microglia, Stevens said that novel biomarkers are needed. She has been working with Steve McCarroll to profile the proteome and RNA transcriptome of microglia in affected versus non-affected brain regions to try to create a molecular fingerprint of changes that occur. McCarroll’s approach is discussed in more detail in Chapter 6.
ALZHEIMER’S DISEASE
The brain pathology that eventually leads to dementia in people with AD starts early, when people are still cognitively normal, said Richard Perrin. He noted that the amyloid cascade hypothesis, which has been the basis of most treatments in development, posits that the deposition of amyloid plaques is the first pathological feature, followed by the accumulation of neurofibrillary tangles, with neuronal integrity declining along the way (Perrin et al., 2009). However, Perrin emphasized that there is more to AD than plaques and tangles, including neuroinflammation, synaptic and neuronal dysfunction, and cell death (Fagan and Perrin, 2012) (see Figure 4-1). Recognition of the complex pathological
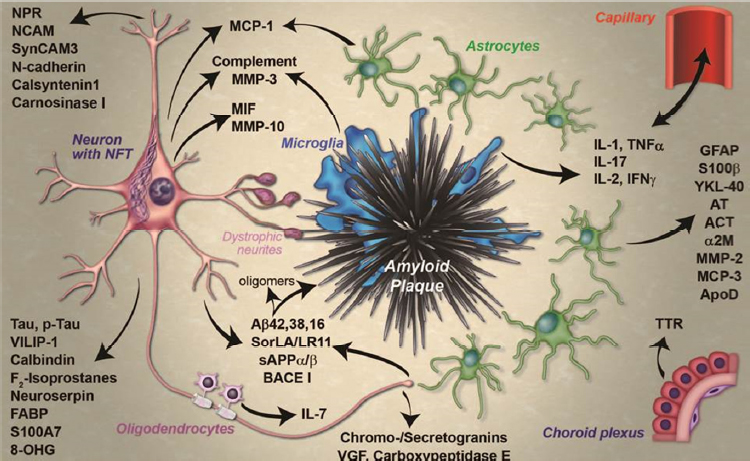
SOURCES: Presentation by Perrin, March 21, 2017; Fagan and Perrin, 2012.
processes that result in AD has given rise to a large number of promising CSF and plasma biomarker candidates. Indeed, study of these biomarkers has provided information about disease pathogenesis, said Perrin. For example, they show that brain amyloid is preceded by peripheral inflammation and low insulin signaling and that it coincides with low adiposity, low insulin signaling, and an anti-inflammatory milieu. Amyloidassociated dementia is also preceded by neuroinflammation as well as vasculopathy and BBB dysfunction and other hormonal and metabolic changes. Many of these pathophysiological changes may represent targets for therapy, said Perrin. His research to identify these novel CSF biomarkers is discussed further in Chapter 6.
IMMUNOPSYCHIATRY
Mental health disorders are the number one global cause of disability-adjusted life years, yet investment is disproportionately low, said Edward Bullmore. He suggested that innovation in drug development for neuropsychiatric disorders will require a pivot away from the focus on targets expressed in the brain, such as neurotransmitter receptors or transporters, in favor of targets in the peripheral immune system. Indeed, he noted that while 30 years ago the brain and immune system were thought to be strictly segregated from one another by the BBB, it is now recognized that there is physiological and therapeutically tractable cross-talk between the brain and immune system through the vagal inflammatory reflex (Tracey, 2002). Moreover, Bullmore said there is a strong reason to believe that immune or inflammatory mechanisms are involved in the pathogenesis of neuropsychiatric diseases. He argued that focusing on the peripheral immune system increases the potential availability of biomarkers to guide the selection of patients and assess efficacy, which would reduce the risk of expensive late-stage clinical trial failures that have plagued CNS drug development. Furthermore, because immunotherapeutics now make up a large proportion of drugs in development for oncology and other diseases, there is the potential to leverage existing expertise, facilities, and molecules, and/or repurpose drugs already on the market, said Bullmore (Bullmore and Lynall, 2014).
Bullmore noted that the role of inflammation in depression, and the potential usefulness of inflammatory biomarkers, was supported by a cumulative meta-analysis of studies over the past 20 years that showed a moderately strong and robust association between major depressive disorder (MDD) and the proinflammatory cytokines interleukin 6 (IL-6) and C-reactive protein (CRP) (Haapakoski et al., 2015). In a more recent study conducted by a consortium that Bullmore leads, which was funded by the Medical Research Council (MRC) (see Chapter 7), analysis of microarray data from earlier studies yielded a list of genes that were over- and underexpressed in depression, and showed—through an ontogeny analysis—that overexpressed genes were enriched for innate immune functions while underexpressed genes were enriched for adaptive immune functions.
Several studies have also provided evidence indicating that these markers of inflammation reflect causative mechanisms in depression, said Bullmore. For example, in a study conducted in a birth cohort population from Avon County, England, IL-6 and CRP levels obtained at 9
years of age were shown to be associated with an increased risk of developing depression at age 18 (Khandaker et al., 2014). Similarly, Bullmore cited unpublished data from the MRC Consortium showing that elevated CRP levels in non-depressed women in 2004 and 2008 were associated with approximately a threefold increased risk of becoming depressed in 2012, compared to women with no evidence of inflammation.
Identifying what the causative mechanisms are that link inflammation to depression remains a substantial gap in the literature; although a few studies have been done, said Bullmore. In one study, typhoid vaccination was used to elicit a peripheral immune response. Healthy volunteers received a placebo or a vaccination in two separate sessions, after which they completed mood questionnaires and performed an implicit emotional face perception task during functional magnetic resonance imaging (fMRI). The study showed that typhoid vaccination, but not placebo, caused an increase in circulating IL-6 that correlated with mild dysphoria as well as increased activation of the subgenual cingulate cortex in response to faces with negative-valence expressions (e.g., angry or sad), but not neutral or positive expressions (Harrison et al., 2009).
Other studies aimed at understanding molecular mechanisms of inflamed depression have demonstrated that the activation of microglia is associated with changes in monoamine metabolism, said Bullmore. For example, microglia activation is associated with activation of an enzyme called indoleamine 2,3-dioxygenase (IDO), which shunts tryptophan away from serotonin toward the formation of neurotoxic metabolites (Miller and Raison, 2016). This could help explain the relationship between peripheral inflammation and treatment-resistant depression, he said. Following up on that observation, the MRC Consortium has shown that in vitro at least, IDO activation is modulated by anti-inflammatory treatments.
Increased peripheral inflammation may also help explain the links between social and psychological stress and an increased risk of depression, said Bullmore. In one study, chronically stressed teachers were shown to have higher levels of IL-6 than more resilient teachers at baseline, and to react more strongly to the acute stress of a public speaking challenge (Bellingrath et al., 2013).
Clinical experience also suggests that non-psychiatric inflammatory disorders are often associated with depressive symptoms, said Bullmore. In a meta-analysis of anti-inflammatory trials for non-psychiatric conditions, the MRC Consortium demonstrated a reduction in depressive symptoms, although, as Bullmore noted, the trial was not designed to test
this effect. He added, however, that the anti-TNFα monoclonal antibody Remicade (infliximab) has well-known but poorly studied effects on mood, producing what clinicians commonly call the “Remicade high.” Indeed, a study of TNFα blockade in rheumatoid arthritis patients showed that even before clinical measures of disease activity were affected, subjective ratings of pain were reduced and brain regions involved in pain perception were affected, indicating that TNFα blockade causes functional CNS changes (Hess et al., 2011). Bullmore suggested that future clinical trials of anti-inflammatory drugs for non-psychiatric disorders include brain function and mental-stage changes, in addition to biomarkers of inflammation, to clarify the association between comorbid depression and non-psychiatric disorders.
TNFα blockade as a treatment for major depressive disorder was tested in a phase II clinical trial, but with disappointing results (Raison et al., 2013). However, Bullmore said that in post hoc analysis, a greater improvement in depression scores was observed for individuals with high-baseline CRP, which has led to another study in which participants are stratified according to CRP levels and suboptimal response to standard treatment. While CRP may not be the ideal biomarker, this study demonstrates the potential of biomarkers to improve clinical trials, said Bullmore. The need for better biomarkers prompted the development of BIODEP, a biomarker discovery program funded by the Wellcome Trust Consortium (see Chapter 7), which is looking deep and wide for biomarkers of the peripheral immunophenotype with imaging, cell-based assays, blood- and CSF-based cytokine analyses, and whole blood transcriptomics, and is correlating these measures with clinical and neuro-psychological assessments.
Miles Herkenham added that the adaptive immune system has also been shown to affect mood and, in animals, to improve hippocampal neurogenesis, which may be relevant in depression. He suggested that lymphocyte profiling might also represent a useful biomarker. Bullmore said that while the gene expression data suggest that innate immune-related genes from cells of the myeloid lineage were overexpressed more than adaptive immune genes from cells of the lymphoid lineage were underexpressed, current technology lacks the ability to study the adaptive immune system in great detail. The immune system is nothing if not complex and coordinated, he said, so it would be inconceivable that the two parts of the system would not be interacting. However, he said, at this point, pathophysiologically important interactions between the innate and adaptive immune systems are poorly understood.
This page intentionally left blank.












