
Methods and processes used to identify and measure CQAs for raw materials and regenerative medicine products were examined during this session. The three panelists—Anne Plant, the chief of the biosystems and biomaterials division at NIST; Linda Kelley, the director of the cell therapies processing facility at the Moffitt Cancer Center; and Robert Deans, the chief technology officer at BlueRock Therapeutics—discussed measurement methodology and approaches to ensure that measurements are accurate and reproducible. They also described future needs and new technologies for measuring CQAs.
ACHIEVING CONFIDENCE IN MEASUREMENTS FOR REGENERATIVE MEDICINE PRODUCTS
The process of defining and measuring the critical characteristics of regenerative medicine products is often listed as one of the fundamental challenges in the field, Anne Plant told the workshop participants. “Figuring out what the quality attributes are that define a product is not a straightforward thing to do,” she said. The quality attributes of regenerative medicine products can include identity, quantity, purity, sterility, viability, and markers of biological activity, Plant said. While FDA defines a number of quality attributes that must be provided with any regulatory submission, there are many different types of products in development, each with its own set of unique characteristics, making it challenging to develop common assays for product characterization. Furthermore, Plant said, for many regenerative medicine products that are in development there is not yet a complete understanding of their mechanisms of action. As a result, it can be unclear what needs to be measured in order to assess biological activity or identity.
Furthermore, it is often unclear which in vitro metrics will predict in vivo activity, Plant said, which can be challenging when the goal is to
develop products that are both safe and effective at treating disease. Gaps in fundamental knowledge about biology are partly to blame for this situation, she said, but so too is the fact that there are many variables in the analytical assays and in the differences between the samples harvested from the patients who will receive these therapies. Because there are so many assay and sample variables, it is difficult to design experiments and measurement methods that can get a handle on all of these things simultaneously, Plant said. To complicate the situation even more, the cells and tissues under study are dynamic, living entities that are growing and possibly even changing.
Defining CQAs for a particular product is challenging without accurately measuring endpoints. It is important to ensure correct measurements, Plant said, but it is also important to take measurements that are meaningful to the clinical outcome. Ensuring the comparability of measurements is another major challenge for researchers and product manufacturers in this area, she said. Given the complexity of the measurements needed to characterize these products, she continued, it is important to have confidence that measurements can be compared. Comparability is especially important because there may be many changes to how a material is handled as a process moves from the research laboratory to the manufacturing setting, Plant said. Changes during the scale-up process can include new raw materials, new suppliers, new storage conditions, and even new technicians. Therefore, she added, product characterization is needed as the manufacturing process changes to make sure that the product generated in the manufacturing pipeline has the desired characteristics. If manufacturers are unable to demonstrate that a product has the same characteristics as it did in the past, they may be asked to perform additional experiments or even perform another clinical trial, Plant said. She noted that there is FDA guidance about how to qualify and validate measurements.1
Characterizing an assay’s precision, reproducibility, accuracy, robustness, sensitivity, specificity, dynamic range, response function, and limit of detection can lead to confidence that measurements are yielding data that can support good decision making, Plant said. Knowing these variables can help manufacturers draw comparisons during the process of scaling up and allows them to have confidence that when they get an answer that is unexpected, it is due to changes in the product and not the assay.
FDA requires validated assays for registration-enabling clinical trials. However, Plant said, validating assays as early as possible leads to better decision making at each step along the translation process and to more
___________________
1 The FDA guidance document Potency Tests for Cellular and Gene Therapy Products is available here: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm243392.pdf (accessed August 11, 2017).
confidence that an observed effect is real. Developing confidence in an assay and collecting accurate measurements may be easier in the pre-clinical stage, she said, because there will be less variability in the samples being tested.
In Plant’s experience, academic researchers will often not have a problem getting reproducible results from an assay in their laboratories, but when results of that assay are compared across laboratories, the results can vary considerably. One approach to addressing this issue is to go back and re-examine the parameter space and where variability in an assay can occur; by doing so, it is possible to create an assay protocol that generates comparable inter-laboratory results. “That gives you confidence that you know what you are measuring and that your measurement process is under control,” Plant said.
In some cases, the availability of reference materials can lead to measurement assurance. For example, NIST is using a standard reference material to calibrate flow cytometry beads made by different manufacturers.2 Calibrating each manufacturer’s beads to the NIST reference material will mean that each manufacturer’s beads will be normalized to one another, Plant said. In other cases, when the measurement is too complex for a reference material composed of cells, it may be possible to build rigorous statistical models based on fundamental principles that researchers can use to evaluate relative precision and accuracy, as NIST has done for cell counting, she added.
Plant noted that a NIST-led consortium, in partnership with the Standards Coordinating Body, is working to improve measurement assurance in gene editing.3 This project will compare existing assays, define minimum metadata to report, design benchmark materials, and compare informatics platforms. A number of elements can contribute to more robust assay development, Plant said, including
- inter-laboratory studies
- experimental design
- testing assumptions
- traceability to a reference material
- statistical methods
- assay qualification
- consistent reporting
___________________
2 Background information on the Flow Cytometry Quantitation Consortium can be found here: https://www.federalregister.gov/documents/2016/07/15/2016-16761/flow-cytometry-quantitation-consortium (accessed August 16, 2017).
3 More information about the NIST-led consortium on measurements in gene editing can be found here: https://www.nist.gov/programs-projects/measurements-targeted-genome-editing (accessed August 15, 2017).
While working through this list can be time consuming and requires a great deal of effort, doing so may reduce the risk of making decisions based on wrong answers and potentially slowing down a development project, Plant said. Community efforts can mitigate the challenge of working through this list, she added, and standards development organizations can be good partners in such an effort. These organizations can codify best practices and define reliable protocols. Workshops, white papers, and data sharing can then spread these best practices and reliable protocols.
In summary, Plant said, developing assays that generate comparable data and allow for a better understanding of the important characteristics of a given product or system will allow the researchers and manufacturers in the field to learn from one another’s experiences, share those data, and perhaps develop a better understanding of mechanisms of action and complex biology. NIST is trying to help the regenerative medicine field through hosting a number of workshops, many held in collaboration with FDA,4 she added.
POSSIBLE OPPORTUNITIES AND CHALLENGES FOR DETERMINING CRITICAL QUALITY ATTRIBUTES IN EARLY-PHASE CLINICAL TRIALS
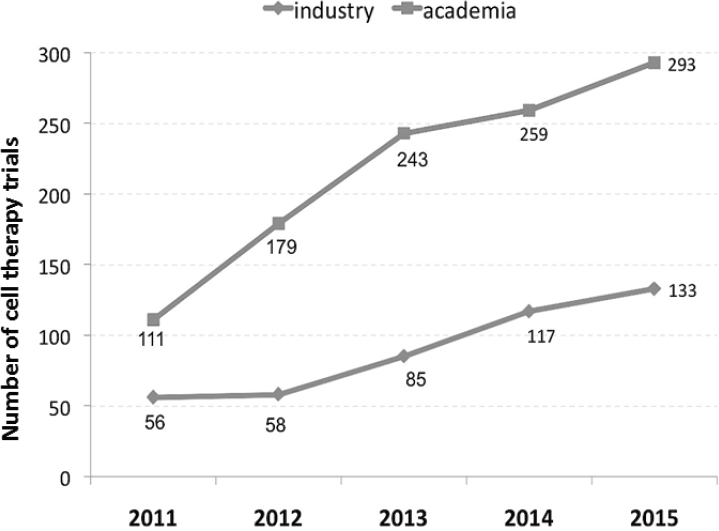
The comparability testing of CQAs could be carried out more efficiently by tapping into global collaborations as a way of accessing a larger number of patients and managing costs, Linda Kelley said. As of April 2017, there were 55 approved cell-based therapeutic products (Bersenev, 2017), and only one-third of those had received regulatory approval in the United States. This difference is due in part to the geographic location of the patients who participated in clinical trials, Kelley said, but it is also a result of new infrastructures that are being implemented in other countries. The number of industry-sponsored and academia-sponsored clinical trials continues to increase, she said, making partnerships between industry and academia more common as a means of leveraging the science, know-how, and cost-sharing (see Figure 3-1).
When such collaborations first started, there was a very steep learning curve on how to make them work, Kelley said, but that learning curve is starting to plateau. Valuable experience has been gained on how to make these collaborations effective, she said. Nonetheless, global collaborations still have constraints, particularly in navigating the different regulatory frameworks that exist worldwide. In the United States and Europe, for
___________________
4 Additional information from NIST on regenerative medicine biomanufacturing can be found here: https://www.nist.gov/programs-projects/regenerative-medicine-biomanufacturing (accessed August 11, 2017).
SOURCES: Linda Kelley, National Academies of Sciences, Engineering, and Medicine workshop presentation, June 26, 2017. Originally from A. Bersenev. 2016. Number of academic versus industry-sponsored cell therapy trials, listed in databases. https://doi.org/10.6084/m9.figshare.1504124.v4 (accessed August 11, 2017).
example, select cell and tissue products require pre-market approval, while in Japan many of the products are considered on a decision-to-treat basis. With regard to CQAs, the U.S. Pharmacopeia (USP) and the European Pharmacopeia play similar roles.
As an example of an international, multi-centered trial, Kelley discussed her experience carrying out Phase I and Phase II clinical trials with autologous CAR T cells. The trial originated in Brussels and included her institution in Tampa, Florida. Part of the purpose of the collaboration was to treat patients in both countries and to manufacture the cell products in two places. The idea, she explained, was not for her facility to manufacture cells for U.S. patients and for her collaborators in Brussels to be manufacturing cells for European patients, but rather for the products created at the two
sites to be interchangeable. By creating redundancy in their manufacturing capacity, the hope was that this would allow the team to mitigate issues that arose at one site by potentially allowing manufacturing to continue at the other site, she said. Another reason for the dual site manufacturing was to create flexibility in the production process to accommodate the European regulatory agency’s regulations regarding audits that can require facilities to shut down completely.
The challenge for Kelley and her collaborators was to demonstrate that the two manufacturing sites could produce comparable products so that the data from both sites could contribute to the results of the clinical trial. The process of creating autologous CAR T cells is lengthy and can take approximately 3 to 4 weeks, Kelley explained. The first step involves collecting T cells from a patient, and this is followed by transduction, expansion, and cryopreservation of the cells. Next the cells are subjected to a variety of quality control assays and release tests before the final product is shipped for treatment of the patient.5 Kelley and her colleagues hypothesized that by making sure that these steps were executed in the same way in both locations, they could create the same product in both facilities.
Ideally, Kelley said, the time to address CQA testing is during the original product development and also during technology transfer and whenever the manufacturing process changes. In the case of this particular collaboration, both she and her collaborators started with well-validated analytical methods with well-developed standard operating procedures, although at first there was no consensus between the two groups as to whose methods were the best. Where possible, the collaborators used known reference samples, and they used actual patient samples when doing their comparability testing. The collaborators also identified critical reagents, and in their initial studies the two groups shared exactly the same reagents, not just reagents from the same vendor or with the same lot number. The two groups established acceptance criteria, a process that required having technical experts in the same place at the same time. Ultimately, the team from Brussels traveled to Kelley’s laboratory and spent 1 month working on this approach.
Kelley and her collaborators went through the process of establishing acceptance criteria before they started tackling the manufacturing process. Given the desire of the stakeholders in the collaboration to begin the clinical trial quickly, Kelley said, it was challenging to convince some of them that this time-consuming approach was necessary, but with all the planning for logistics and resources it would have been difficult to do it all at the same time. By agreeing on CQAs, analytical methods, and acceptance criteria,
___________________
5 Additional information on how CAR T cells are made and used can be found here: https://www.cancer.gov/about-cancer/treatment/research/car-t-cells (accessed August 15, 2017).
Kelley and her team gained confidence in the products they plan to manufacture in this collaboration moving forward (see Table 3-1).
Implementing timely and cost-effective CQA rapid release assays has been particularly challenging, Kelley said. One example concerned sterility testing, which has been a subject of conversation with regulators and stakeholders in the field for a long time. Currently, FDA requires that all parenteral biological products undergo sterility testing to ensure that the products are sterile when they reach the patient. Previously, the gold standard assay for sterility testing was the compendial sterility method, which takes 2 weeks, but FDA has now sanctioned validated and automated systems for sterility testing such as BacT/ALERT and BACTEC. Historically, the automated systems utilized culture media that is only FDA approved for testing blood products, and therefore they are being used off-label for cell therapy products. New media for sterility testing has recently been introduced to the market with enhanced characteristics that make them more appropriate for cell therapy products (e.g., “industry bottles”), Kelley said. There is good reason to move forward with using this type of technology, she said, but it will require lengthy revalidation. Revalidation can be a time- and cost-intensive process, but Kelley said she is planning on moving forward with a new study with the help of consultants to demonstrate the value of using industry bottles in her laboratory. In thinking about testing for every
TABLE 3-1 Critical Quality Attributes, Analytical Methods, and Acceptance Criteria
| Criteria | Methods | Limits |
|---|---|---|
| Viability | Flow cytometry | ≥ 70% |
| Purity (CD3+ cells) | Flow cytometry | ≥ 80% |
| Cell count | Flow cytometry | Specified dose ± 25% |
| Identity (NKG2D+ cells) | Flow cytometry | ≥ 50% |
| Microbiological tests | ||
|
Ph. Eur. 2.6.27 | No growth |
|
Ph. Eur. 2.6.14 | ≤ 8.67 EU/mL |
|
Ph. Eur. 2.6.7 | No mycoplasma |
| Safety tests | ||
|
Ph. Eur. 2.6.21 | < 5.0 copies/cell |
|
Ph. Eur. 2.6.21 | No detection |
NOTE: CD3 = cluster of differentiation 3; EU/mL = Endotoxin units per milliliter; NKG2D = natural killer group 2, member D; Ph.Eur = European Pharmacopoeia.
SOURCE: Linda Kelley, National Academies of Sciences, Engineering, and Medicine workshop presentation, June 26, 2017.
organism on the U.S. and European Pharmacopeia lists, Kelley held discussions with both organizations about reasonable expectations, and she was able to narrow the list based on the likely contaminants.
Another challenge Kelley mentioned was related to testing for replication competent retroviruses (RCRs), which, if present, can result in multiple integrations within host cell genomes and potentially lead to oncogenesis. RCR testing is currently required by FDA if a cell product is in culture for more than 4 days after transduction. However, a 2012 study found no RCR-positive samples across 29 ongoing gene therapy trials, Kelley said (Bear et al., 2012). Based on their findings, the study authors proposed that master cell banks used for production of infectious virus should continue to undergo rigorous RCR testing but argued that final T cell products that incorporate retroviral vectors and patient peripheral blood samples do not need to undergo active screening at defined time intervals. The reason, Kelley said, is that the original RCR testing method is costly and lengthy. Despite the findings from the 2012 study, FDA requirements have not changed, she said. Therefore, most groups are migrating to using quantitative polymerase chain reaction assays, a faster and more cost-effective option, she said. Doing so requires each laboratory to validate their assays, which can be time-consuming and costly. In closing Kelley asked, “Are we requiring too much for validation and not embracing the recommendations of scientists in the field?”
A COMMERCIAL PERSPECTIVE ON BUILDING PRODUCT ATTRIBUTES
In the early phase of translating products into the clinic, investigators often worry about the core biological hypothesis and demonstrating that the therapeutic approach is safe, said Robert Deans, the third presenter in the session. Regulatory agencies encourage that focus by asking questions of investigators about proof points and whether their methods and assays are providing statistical confidence regarding pre-clinical and early clinical results. On the commercial side, however, there are other attributes that become important, such as whether the product will meet effectiveness endpoints, if it can be produced at a reasonable cost that payers will reimburse, and whether physicians will adopt the new product.
To address these concerns as early as possible in the commercial phase of development, Deans said, companies build what is called a target product profile to guide decisions over the course of the development process (see Box 3-1). The target product profile is an extensive document that covers both the biological properties of the product and its safety and quality
attributes.6 It also addresses many of the product’s financial and commercial considerations, such as whether the ease and frequency of delivery is linked to compliance problems and whether patients can afford the product.
The wave of new products resulting from CAR T cell technology shows how effective product development can be when there is a major clinical effect tied to a well-understood mechanism of action, Deans said. Also important from a commercial perspective is the ability to forecast how technological advances, such as the development of gene editing and gene therapy, might affect how competitive a product will be in the future against the current standard of care approach.
To demonstrate how a company assesses important product attributes, Deans discussed two examples. The first concerned an assay his company developed to demonstrate potency for a mesenchymal stem/stromal cell (MSC)7 product in the cardiovascular area. Once industry scientists had identified what Deans called the black box benefit they observed when their cells were injected into an in vivo animal model, they developed an in vitro surrogate assay that had comparable benefits to those seen in the animal model. The company next worked on identifying a negative control by using a cell line that did not stimulate angiogenesis, Deans said, and it used
___________________
6 The FDA Guidance for Industry and Review Staff on the target product profile can be found here: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm080593.pdf (accessed September 19, 2017).
7 The term “mesenchymal stem cell” is often used interchangeably with “mesenchymal stromal cell” or “multipotent stromal cell.” MSCs are a diverse population of multipotent cells that include osteoblasts, chondrocytes, myocytes, and adipocytes.
that cell line to compare proteomic and transcriptional profiles and identify key parameters that differed between the successful product and the unsuccessful product. The company also used genetic knockdown and antibody inhibition to find the core angiogenic factors involved in the product’s beneficial effects. The knockdown models enabled the company to add those factors back in order to get an understanding of the minimal threshold for biological activity. This approach gave Deans and his colleagues a statistical basis for defining pass/fail criteria. Using the information gained from these studies, company researchers were able to quantify how its production lots varied in terms of their functional performance and the measurement of these parameters (Lehman et al., 2012). This effort resulted in these assays being implemented in a lot release assay panel and also in the development of intellectual property around cell composition.
Overarching Challenges to Product Identity Testing
On the subject of product identity testing, Deans said that he sees many potential challenges with using individual markers or cell surface markers as a distinguishing mark if they do not correlate to a specific cellular function. Instead, his company has used a unique approach to identify a functional distinction between its product and another product and then defines a molecular surrogate that correlates with that distinction that could be used reproducibly as an assay for in-process development or lot release. Deans and his colleagues took a reductionist approach to characterizing the cell type using a number of different epigenetics-based methods, including transcriptional profiling, gene methylation, miRNA profiling, and proteomic surveys. Next, they analyzed many donors, bioprocess variants, and a competitor’s product to understand the principal components that defined their product. This analysis identified a block of microRNAs that can regulate a cascade of genes that reflect functions within their cell population, and further studies across the manufacturing process resulted in a fingerprint profile for miRNA patterning that serves as a rapid process and lot release assay (Crabbe et al., 2016). miRNA-mRNA interactions and cell phenotypes were studied and Deans and his colleagues concluded that miRNA markers could be linked to cell differentiation and cycle cell regulation in human MSCs and multipotent adult progenitor cells.
With regard to the attributes of living medicines such as CAR T cells or MSC products, Deans said, it is important to understand how cells distribute, amplify, and persist in vivo, and currently there are only poor surrogates for these biological attributes. For example, the patient response to CAR T cell therapies is poorly linked to cell production numbers. Circulatory distribution and tissue penetration are critical therapeutic attributes, he said, yet those do not have sufficient in vitro surrogates.
Another area that needs further study, Deans said, is the development of standardized attributes for safety, such as the formation of ectopic tissue and teratogenicity. These measurements have not been adequately developed and are major rate-limiting factors in the clinical development pipeline. The current teratogenicity assay, in Deans’s experience, takes about 9 to 12 months in immuno-compromised mice and can cost upward of $1 million. The time and expense of this assay is precluding the field from doing comparability studies between embryonic stem cells and iPSCs, he said, and is hindering the development of personalized iPSC therapeutics. A challenge for the field, he said, will be to develop efficient and effective assays linked to key safety attributes so that the development cycle can accelerate. One potential solution may involve implanting cells into pre-existing tumors, which would provide a tumor microenvironment that might speed up the development of teratomas, if they were going to develop at all.
It may be challenging for the community to come together to define quality attributes or standards, Deans said. One reason is that therapeutic sponsors do not want to reveal upfront their cell sourcing or gene manufacturing trade secrets. As a result, most co-development opportunities focus on downstream processing. Analytical testing and assay development are two areas that are ripe for co-development, Deans said, and the field could benefit from pursuing those opportunities. However, he added, tool providers and contract manufacturers are not enthusiastic about leveling the playing field and making all processes common.
Therapeutic societies have focused on assay harmonization and have constructed assays for key product class attributes. However, these organizations do not have much leverage for encouraging widespread adoption of those assays across the field. Societies are often unaware of standards-setting organizations and the processes involved in having assays accepted as standards by those organizations, Deans said. He commended NIST for working with the Alliance for Regenerative Medicine to build a standards coordinating body.
Regulatory Pathways and Commercial Development
In closing, Deans said that regulatory science is changing industry’s development strategies. Regulatory approval pathways have become critically important levers in commercial development, he said, particularly with regard to securing capital to continue development. Those pathways are also having a significant effect on regional economic development in the life sciences. The regulatory pathway in Japan has an accelerated approval option that is aimed at measuring safety and efficacy, Deans said. Allowing more rapid market approval and cost recovery for clinical trials has
resulted in products reaching the market faster, and post-market studies have allowed for the collection of additional data and statistical depth, which helps to stratify patients and further refine product indications. Australia’s regulatory process features two distinct pathways. One of them, the Clinical Trial Notification path,8 allows for the accelerated entry of emerging therapeutics, Deans said. Companies are required only to have an institutional review board approval regarding the safety of a manufactured product before beginning studies looking at early patient experience. This has led an increasing number of companies to perform first-in-human trials in Australia followed by Phase I or Phase II trials back in the United States or Western Europe, he said. It is very important to figure out how to gain early human clinical experience in a safe and responsible way, Deans concluded.
DISCUSSION
Novel Technologies for Improving Cell-Based Manufacturing
Synthetic biology may offer new avenues for programming cells to grow and differentiate as part of the manufacturing process, but safety testing standards associated with gene editing and gene modification have yet to be defined. One of the exciting things about this field is the merging of synthetic biology and genome engineering to create products, Plant said. There are still many details to work out, she said, which is why she is currently speaking with experts about the state of the science and the issues associated with using gene editing for therapeutic purposes.
With regard to whole-genome sequencing, Plant said that NIST formed a public–private academic consortium called Genome in a Bottle that is establishing important benchmarking data on a limited number of human genomes from de-identified individuals.9 A number of groups using different platforms are sequencing these genomes and analyzing the results using unique bioinformatics tools. The goals are to compare methods, learn how they differ from one another, and perhaps identify those areas of the genome that are either easier or more difficult to sequence with confidence. “I think that those kinds of benchmarking data will help us understand how to interpret whole-genome sequencing and some of the slight modifications that might show up, whether they are real or not, and whether they are meaningful or not,” Plant said.
___________________
8 For more information on the Clinical Trial Notification pathway and the Australian scheme for clinical trials, see https://www.tga.gov.au/clinical-trials-glance (accessed August 14, 2017).
9 More information about the Genome in a Bottle consortium is available here: http://jimb.stanford.edu/giab (accessed August 14, 2017).
The pace of technology may be outstripping the ability to establish and implement an assay, Deans said. “We are going to get way in front of ourselves with potentially very effective new therapeutics and an inability to understand the development path.”
One workshop participant described an effort in his laboratory to use artificial intelligence to define the quality of cell therapy products as part of ongoing work to develop an autologous iPSC-derived retinal pigment epithelium patch for retinal regeneration. His team is developing an imaging-based convolutional neural network, which is a computing system inspired by physiological neural networks that is used to analyze visual imagery. The research goal is to use the convolutional neural network to effectively predict measurable cell functions. According to the workshop participant, the neural network was powerful enough that with one training session it could differentiate among the quality of cells and could potentially be implemented during the manufacturing process across different centers with no raw materials required for the assay. He said that his concern was how to validate this type of system for regulatory purposes.
NIST is very interested in this type of approach, Plant said, adding that she and her colleagues are learning what would be required to validate it for regulatory purposes. The challenge, she said, is deciding who the experts are that develop a training set, given that one expert is likely to produce a different training set than another one would. The key will be to validate the algorithm and training sets against a thoroughly validated functional assay.
Sharing Data in the Pre-Competitive Space
Both the issue of assay variability and the challenges associated with getting people to share assays were raised by speakers in this session. Martha Somerman, the director of the National Institute of Dental and Craniofacial Research of the National Institutes of Health, asked panelists if they had any suggestions on how to improve data sharing efforts in the field. One suggestion was to start in the pre-competitive space. It may be challenging to share information about characteristics or CQAs of a particular product, Plant said. An easier approach might be to encourage sharing of information about analytical methods or tools that everybody uses, such as approaches for counting cells reliably. Regardless of where the process starts, she said, it has to be a grassroots effort. The Standards Coordinating Body is putting together several projects in the pre-competitive space, said workshop co-chair Claudia Zylberberg.
Deans said that the problem he sees in co-development is the high expense of generating raw materials for testing. “One of the barriers to getting standardized testing or getting group assessment of common mate-
rials is deciding who is going to pay for the process,” he said. There may be opportunities for the clinical academic community to develop and use CQA assays related to function much earlier in the development process (i.e., before researchers turn a product over to the commercial sector). CQA assays should be used as early as possible, Deans said, and he predicted that as the field gets feedback from some of the epigenetic analyses from patient follow-up assays, academic laboratories will start using those data in their work. “We are really just beginning the cycle of going from bedside back to bench,” he said. “I think we are now positioned to apply those early, apply them to mode of action, and then have improved processes going forward.”
Regarding early phase clinical data, there is an opportunity for the collection and dissemination of more epigenetic analyses and clinical trial data including that of patients treated with placebos, Deans said. Sponsors are logically and justifiably conservative about revealing patient response data, he said, but there is information on placebo or control groups that, if deposited into a public database, would be extremely helpful. Making that happen, however, will require an edict from a reputable organization, he said.
Potential Opportunities for Developing Reference Materials
The MSC research community has debated the value of a gold standard reference MSC line and the idea of agreeing on standards that adequately define an MSC product, Deans said. It can be frustrating that the community has not been able to agree on a consensus identity definition or to work collectively together to harmonize cell characterization, he said, noting that there currently a few hundred different names being used to describe very similar cell products. The field should define the cell type by the bioprocess used to make it, not by phenotypic comparisons or epigenetic traits, he said, in addition to creating functional profiles and assays that correspond to phenotypic or epigenetic traits.
Together with the World Health Organization (WHO), international reference laboratories such as the National Institute for Biological Standards and Control in the United Kingdom have played a critical role in providing standards and assays as well as in funding that work, one workshop participant said. WHO has shown interest in the cell therapy area and NIST often collaborates with it, Plant said, though its way of generating standards differs slightly from NIST’s approach.
Ongoing Financial, Logistical, and Technical Challenges
The inter-laboratory comparison that Kelley and her colleagues undertook was expensive and required having people from the laboratory in
Brussels come to Tampa and live there for 1 month, something that will happen again when the researchers start the transfer of manufacturing technology. In the end, though, the results were valuable, and, perhaps more importantly, a consensus developed within the team and team members gained the confidence to move forward as true collaborators who are on the same page.
A unique challenge to certain cell-based regenerative medicine products is that part of the manufacturing process occurs in the patient, where the cells expand, proliferate, and change phenotype after injection. A workshop participant asked the panelists for their thoughts on the responsibilities that developers have to understand that part of the process and what type of assays might be needed to understand what is happening to the product. It depends on the cell type and the purpose of using that cell type, Kelley answered. One approach that she is taking is to address the specificity of the cell types her team is manufacturing using real-time assays that look at appropriate function over time. Currently, she said, her group is in the process of collecting enough data to determine if there are CQAs that are more meaningful and that can be incorporated into the manufacturing process cost-efficiently. There are many new technologies becoming available for quickly screening for cytotoxicity, phenotype, and other important attributes that the field could explore, she said.
Biomedical imaging with novel tracers—an approach that needs to be further developed—might provide information on cell persistence, Deans said. The field needs model systems to predict what will happen inside a patient, Plant said. “Everything we are going to be measuring is in vitro, and we are assuming the environment we are creating in our in vitro assays has something to do with the in vivo environment,” she said.
When conducting an evaluation of comparability based entirely on in vitro assays, it is important to know what the CQAs are, but in many cases, a workshop participant said, they are not known. It may be helpful to identify an experimental in vivo situation that can distinguish in a meaningful way between a product that works and one that does not as well as determining what magnitude change in an attribute results in a product that does not work. The true population variance of many of these parameters may make it difficult to conduct a study with sufficient statistical power to know if the difference between two products is real. Getting all the critical process parameters worked out early in development can save many headaches down the line, the participant noted.
















