
2
Complexity in Action
The market for prescription drugs in the United States is unlike most other markets for consumer products. In fact, it is unlike even other health care markets—which themselves differ from conventional markets. In conventional markets, consumers can search for available alternatives with access to information, including discounts, reviews, and ratings. Such information enables purchasers to make informed trade-offs relating to price and quality. In the case of biopharmaceutical products, such comparative information is technical and complex, and generally inaccessible. Moreover, clinicians generally do not have information about cost-sharing details and the general financial situation of their patients. As a result, the traditional process where consumers directly search for products has to a considerable degree been replaced by insurance companies that assess benefits and costs of drugs and steer patients’ choices using insurance plan design.
In contrast, the safety, efficacy, production, and distribution of prescription drugs—although not their pricing—are all highly regulated in the United States. Such regulations are intended to balance a variety of considerations, including, in particular, the needs to protect the public and to reward the risky endeavor of investing in research and development. In protecting the public, regulations are intended to ensure that drugs are both safe and effective. The severity and impact of disease need to be balanced with the possible side effects of treatment, and also with the harms that may arise from the lack of availability of those drugs. For example, lack of access to treatments for infectious diseases due to cost not only result in death of an individual, but can also lead to infection and death of others.
Regulations naturally influence the pricing of drug therapies, which are ultimately paid for by patients, their families, and society as a whole, whether through direct payments, insurance premiums, or taxes.
The processes for ensuring that various, sometimes conflicting goals are reasonably satisfied add both to the financial costs of developing drugs and to the risks and consequences of product failure. Arguably, the best interests of patients should be paramount. To begin exploring how one might approach these potential trade-offs, this chapter describes the highly complex biopharmaceutical supply chain, from drug discovery through development, distribution, financing, and the end use by patients.
RESEARCH TO RESULTS AND RETURNS
Biomedical Research
Basic biomedical research, which is usually conducted in universities and specialized research organizations, is usually the first step in a long sequence of activities that ultimately produces safe, effective, and approved drugs. Much of this research is not intended to result in directly marketable biopharmaceutical products, but rather to gain a mechanistic understanding of health, disease, and fundamental science. Historically, though, a certain amount of basic research has led to opportunities to develop new medications, at which point the applied research and development efforts commonly shift from the university or research institute setting to corporations, the latter of which bring the skills and resources necessary to develop, produce, and market prescription drugs. Almost all of these corporations operate on a for-profit basis and depend on the free market for the capital that makes them viable as developers and manufacturers of the drugs sought by patients. Researchers involved in basic research are often poorly positioned to develop their findings into a commercially viable product.
The passage through the transition from discovery to development—often termed the “valley of death”—has been (and can be further) facilitated by “incubators,” organizations that help bridge the valley between discovery and application. Such technology development facilities and related clinical trial networks have been established in many forms by research universities, private corporations, state governments, and others (IOM, 2010, 2012). These joint arrangements have served an important role in making many drugs available for the benefit of patients.
Inventions emerging from research funded by the government can be patented by the university or organization performing the research. The technology covered by the patent can then be further advanced by the patent owner or licensed to others for industrial development. This situation was created by the Bayh–Dole Act, which assigned property
rights for federally sponsored research to the inventors and their institutions rather than to the government funder (e.g., the National Institutes of Health), as had previously been the case (NRC, 2011). This major shift in how property rights were assigned led to a significant expansion in drug discovery and development within universities and other research institutions. The U.S. biopharmaceutical industry is structured as it is today in part because of the Bayh–Dole Act and the response of universities and researchers to that act (Gabriel, 2014). The annual number of patents filed and licensed from government-sponsored research is estimated to have increased by almost a factor of 10 since the passage of the Bayh–Dole Act, thereby adding billions of dollars to the U.S. gross domestic product (The Economist, 2002; Schacht, 2009). The act motivated collaboration between academia and industry, that in turn has helped enhance the transition of products from the laboratory to the public and resulted in better treatment options for patients.
Translational research and clinical development can be conducted in companies both large and small. Many “spin-off” entities have been created by universities to move basic science into more advanced stages of product development. These entities commonly receive investments from venture capital firms or individuals who gain partial ownership of the products of the entity in exchange for their infusion of capital. This investment process is fraught with risk to both the discoverer and the investor. Each step of the biopharmaceutical research and development process has a high failure rate even before a drug gets to the point where it is ready for regulatory review. As a result, the returns on investment for successful drug products may appear to be abnormally high, since the average expected return, from the manufacturer’s point of view, must also compensate for many failures. Financial markets reward those who invest in riskier ventures by providing them with higher-than-average returns. More risk leads to a higher average reward for success, thereby encouraging investments that might not otherwise occur.
Legal Exclusivity
Patent law gives an inventor exclusive right for a period of time to the use of an invention as an incentive to invent. In exchange for the legal period of exclusive use1 of the invention, patent holders must provide sufficient information in the patent (which is a public document) to allow others to use the invention once the period of exclusivity has ended. During
___________________
1 The term “legal monopoly” and related variants—including “regulated monopolies” as noted in this study’s statement of task— have also been used by some to refer to the exclusive market protection feature bestowed by U.S. patents.
the exclusivity period, patent holders have the right to prevent others from using or bringing to market products covered by the invention without the patent holder’s permission. Patents, like other forms of property, can be sold, leased, or licensed on terms mutually agreeable to the parties. Because biopharmaceutical product markets are international in scope, inventors often seek patent protection in many countries, the patent laws of which are generally coordinated through the World Intellectual Property Organization based on the Patent Law Treaty adopted in 2000. In the United States, the Leahy–Smith America Invents Act brought U.S. patent law into general alignment with these international standards.
In biopharmaceutical products, a single item often involves many patents, ranging from the chemical entity itself to the forms of delivery and sometimes even the packaging. Thus, a situation can arise where multiple patent holders mutually claim infringement by others. In such cases, agreements among the various patent holders may be necessary to bring the product to market in its final form. Patent holders are generally responsible for enforcing the exclusive use of their patent through civil court action against alleged infringement. Under 35 U.S.C. Section 284, a patent owner can claim “damages adequate to compensate for the infringement, but in no event less than a reasonable royalty.”2 The standard 25-year exclusivity period—resulting from the Hatch–Waxman Act—begins when the patent is granted, but marketing of the drug in the United States requires approval from the U.S. Food and Drug Administration (FDA). This process can take from 1 year to more than a decade depending on the drug, shortening the effective time during which the patent has economic value.
A natural tension arises in regulatory policy between safety and efficacy versus incentives for product development. One could imagine a hypothetical world wherein no FDA safety and efficacy rules existed, but only the patent exclusivity period existed. This would increase the incentives to invest in product development but could greatly reduce the safety and efficacy of drugs appearing on the market. At the other extreme, one could imagine a world with the FDA processes in place as is the case today, but shorter (or no) legal exclusivity created by patent rights. This would likely lead to less investment and a lower rate of drug discovery and development than is achieved with the current system.
___________________
2 An alternative to a patent for an inventor is to protect the invention as a “trade secret,” in which case the legal protections of patent law are not provided, but the period of exclusive use is also not limited by law. The trade secret path is not normally available to the biopharmaceutical industry because of the disclosures that the FDA requires in order for a product to be licensed for sales.
Testing and Regulation
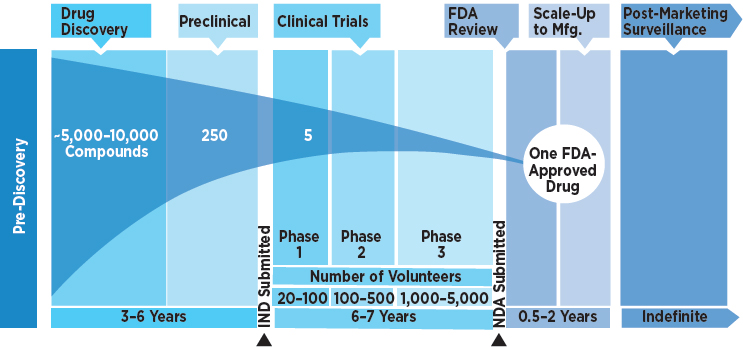
Once drugs have been developed, they must pass a rigorous regulatory review conducted by the FDA in the United States before they can be marketed—and, if they are to be sold abroad, after review by the corresponding agencies in other countries. To gain market approval, drugs must demonstrate both safety and efficacy. The very nature of this approval process demands considerable time and money and, once again, poses the risk of failure at every step of drug development. The first step in the regulatory review process involves animal testing to determine whether the drug indeed affects the intended target and to gather basic safety information concerning toxicity. Assuming that the results are satisfactory, the manufacturer will next file an investigational new drug (IND) application with the FDA, setting the formal review process in motion. For the subsequent human testing, the FDA seeks to ensure that adequate consent and human subject protection procedures are in place (Arrowsmith and Miller, 2013; FDA, 2006). Only a very small subset of new discoveries (1 out of 1,000 to 2,000 candidates) reaches the stage of IND application (AACR, 2011).3
Phase I (initial human) trials—which typically involves 20 to 80 healthy human volunteer subjects—seek to characterize drug concentrations in blood and plasma and how the drug is metabolized and to detect the most common side effects (FDA, 2016). However, in the case of severe conditions such as cancer, phase I trials may be conducted in patients with end-stage disease who have exhausted all standard therapies.
Phase II trials commonly involve several hundred human subjects and pursue several goals. First, they assess whether the drug has the potential to be effective against the target disease by testing them in patients with the disease in question, randomizing between the treatment and a placebo drug or the standard of care, or both. Researchers carrying out these trials continue to monitor side effects and safety issues. A phase II trial also seeks to determine the optimal dosing regimen (total amount, spread over a specific number of doses) and perhaps the duration of the required treatment. Approximately 70 percent of phase II trials are unsuccessful and the drug candidates are abandoned, either because the drug was no more effective than the placebo or because additional safety concerns arose during testing on the expanded number of subjects (FDA, 2017a).
___________________
3 One approach to for reducing prices is reducing the IND failure rate. In recent years, there have been a number of efforts to do so through such steps as making otherwise proprietary clinical data, especially on failed drug candidates, publicly available to all researchers. This report acknowledges that reducing IND failure rate is critical in the drug development process; however, this topic could not be considered in detail amid the competing demands of other topics presented in the study scope. Furthermore, the committee lacked the necessary expertise on this topic.
Phase III trials further expand the number of patients involved in the testing—commonly into the thousands—and continue to assess the safety and efficacy of the drug against either a placebo or the existing standard of care, depending on the status of current therapeutic options. If no current therapies exist, these studies will use placebo controls. If an existing therapy has been shown to have clinical benefit, the ethics of human subject testing normally precludes withholding that therapy. In this case the randomization compares the new drug against the existing treatment (the standard of care). Phase III studies are crucial for regulatory review, and sample sizes are predetermined to ensure that a sufficient statistical base exists to support a final FDA regulatory approval decision. About half of all the drugs that reach phase III do not proceed to market (Arrowsmith, 2011), and judging by historical trends, only about 5 to 10 percent of IND applications ever gain FDA approval (Van Norman, 2016).
The safety of human subjects is of extreme importance in these studies. Clinical trials in the United States have an independent body of experts (including clinicians and statisticians) to monitor safety by regularly reviewing interim data for the treatment being assessed and for control groups in order to spot any differences that may exist between them in either safety or efficacy. Most such trials are “double-blind”; that is, neither the researcher nor patient is aware of which specific treatment is being provided to a specific patient. Pre-specified measures are used to determine efficacy, and a review is conducted to assess the rates of adverse events, including mortality. The monitoring committees have the authority to recommend that a randomized trial be stopped at any time if major safety concerns appear or if the evidence of efficacy is so strong that there is an ethical imperative to place the therapy into general use as quickly as possible.
Upon successful completion of a phase III trial, the next step is for the sponsor of the drug to file a new drug application (NDA), submitting the results from the clinical trial to the FDA. The manufacturer also submits proposed labeling information (highly detailed data on safety, efficacy, and dosage, and the indication for which it will be approved) intended for use by clinicians. The FDA will inspect the facilities where the drug will be produced in order to assess safety and manufacturing quality standards. If everything is acceptable, the FDA will approve marketing of the new drug for the specified indication. Patients can then gain access to the new drug by prescription from a qualified professional.
Once a drug is available to consumers, it enters the phase IV safety review, known as post-market surveillance. The sponsor, typically the drug manufacturer, must submit periodic safety reports to the FDA. These data play a major role in ensuring continued drug safety, sometimes revealing adverse effects that occur too rarely to have been detected even in the large phase III trials. Per the Prescription Drug User Fee Act, all these costs are
NOTE: FDA = U.S. Food and Drug Administration.
SOURCE: Adapted from AACR, 2011.
borne by the sponsoring organization, including the cost of the FDA review process itself. The timeline for the entire process varies, but historical data indicate that it can take as long as 15 years (FDA, 2015; Ng, 2015) (see Figure 2-1). It generally takes less time to approve generics because of the data previously generated for the comparable branded drugs and also less time for those products that meet the criteria of the Orphan Drug Act.
Competitive Market Strategies Following Product Launch
“Evergreening” of Exclusivity
While evergreening is not a formal concept within patent law, it is a commonly used term that refers to various techniques for extending the legal exclusivity granted by the patent (Dwivedi et al., 2010). The practices include patenting the method of administration, patenting a minor reformulation with no therapeutic advantages, and even patenting the metabolites produced in the body after the drug is ingested (Kesselheim et al., 2006). As an example, one biopharmaceutical company filed for a patent to administer its drug after crushing it and spreading it on applesauce (Kesselheim and Mello, 2006).
Evergreening—with various versions referred to as “product hopping,” “product switching,” or “line-extension”—is frequently used when high-revenue branded drugs (i.e., “blockbusters”) reach the end of their patent life (Carrier and Shadowen, 2016; Jones et al., 2016). Under ordinary circumstances, once a branded drug reaches the end of its patent term, the
manufacturers of generic drugs are provided an opportunity to enter the market. The manufacturers of the branded drug can stall a generic competitor from entering the market by filing patents that cover not only the active ingredient, but also secondary features of the drug such as methods of formulation. A recent analysis of all drugs on the market between 2005 and 2015 found that at least 74 percent of new patents for drugs were for existing drugs. This addition of new patents and associated exclusivities was especially pronounced among blockbuster drugs. Of the roughly 100 best-selling drugs, almost 80 percent had their patent protection extended at least once, with nearly 50 percent extending patient protection more than once (Feldman and Wang, 2017).
While these patents are generally considered weaker and less novel than the original patent, they can allow the branded company to allege that the competitor will infringe these additional patents. Litigation by generic firms against branded manufacturers can help to counteract evergreening, as one analysis found that weaker patents are more likely to draw challenges from generic manufacturers (Hemphill and Sampat, 2012). Another recent analysis found that, particularly in recent years, branded firms have been less likely to win cases involving challenges to peripheral features of drugs than those involving challenges to active ingredients (Grabowski et al., 2017). However, the cost of litigation can be a deterrent to generic manufacturers, and litigation (even when ultimately unsuccessful) can be an important mechanism for extending the market exclusivity of the branded drug (Rumore, 2009).
Evergreening also sometimes refers to the creation of so-called “me too” drugs, in which minor modifications are made by manufacturers to the active ingredients in an existing pharmaceutical product. These new molecules may offer little or no additional clinical benefit compared with the existing molecules, but can nevertheless provide a substantial new stream of revenue to the branded manufacturer. From industry’s perspective, evergreening has been considered a legitimate business strategy to increase revenue. What critics may see as the exploitation of loopholes may well be seen by corporations as legitimate “product lifecycle management” and therefore an approach to maximize shareholder value.4
___________________
4 Policy developments such as the Medicare Modernization Act (MMA) have prevented companies from filing a series of patents staggered to protect specific features of biopharmaceutical products such as isomers and metabolites—patents that could otherwise result in a longer period during which manufacturers could sue for patent protection in the event they are challenged by a generic entrant. Under the Hatch–Waxman Act, a manufacturer is granted a 30-month stay during the resolution of such a challenge, during which the generic manufacturer cannot sell its product. Some observers have called for the FDA to tighten the criteria for approving minor modifications of existing drugs. For example, rather than making non-inferiority the standard for approving new applications, the FDA could in some cases (such
Delaying the Entry of Generic Products
“Pay-for-delay” reverse settlements between manufactures of branded drugs and prospective generic entrants have emerged because of the incentives for generic entry created under the Hatch–Waxman Act. Specifically, the act grants 180 days of generic market exclusivity to the first generic firm to successfully demonstrate either that a patent is not legitimate or that the generic product does not infringe the existing patent. This provision—known as “Paragraph IV”—often concerns challenges to peripheral aspects of a patented medication design. For example, the patent on the active ingredient in omeprazole (Prilosec) expired in 2001, but patents on the coating of the pill and other properties were in effect until 2007 (Kesselheim et al., 2011). In response to Paragraph IV challenges, manufacturers can countersue the generic entrant, and can delay the generic application for 30 months.
Given the uncertainty of litigation and the likelihood that a patent holder will lose its protection on a relatively weak patent, it is often mutually advantageous to the generic firm and the firm owning the patent to enter into a pay-for-delay agreement. Specifically, the firms can settle on a payment to cover the 180-day period that exceeds the amount the generic manufacturer might have earned if it were participating in the market but less than what the firm with the patent would have lost due to generic entry. Pay-for-delay keeps prices higher than they would be if a generic competitor were able to enter the market immediately.
One analysis of the economic impact of pay-for-delay settlements in response to Paragraph IV challenges found that settlements tend to
___________________
as when a generic drug is already available) require new drug applications to demonstrate superiority over existing drugs (Gagne and Choudhry, 2011).
Under recent legislative proposals, reformulations of existing drugs can attain an additional 2 years of protection from generic competition if they are shown to (as quoted from H.R.1353: PATIENT Act):
- “promote greater patient adherence to an approved treatment regime relative to the previously approved formulation or design of the drug;
- reduce the public-health risks associated with the drug relative to the previously approved formulation or design of the drug;
- reduce the manner or extent of side effects or adverse events associated with the previously approved formulation or design of the drug;
- provide systemic benefits to the health care system relative to the previously approved formulation or design of the drug; or
- provide other patient benefits that are comparable to the benefits described above.”
Such provisions highlight the challenges of effectively regulating the instances when product modifications might offer real benefits to patients, but any such benefits have to be weighed against the impact of delaying the entry of low-cost generics.
inflate prices and reduce the quantity of prescriptions for several years after the settlement—long after the protected 180-day period. For each settlement, the authors estimated a loss of $835 million in consumer benefits over 5 years. Conversely, the study found that eliminating settlements would tend to increase consumer welfare and have a minimal effect on the investment in research and development and the entry of new drugs into the market (Helland and Seabury, 2016). Pay-for-delay settlements have received substantial regulatory scrutiny, especially in those cases when they may violate antitrust law.5
Another approach by branded manufacturers uses the Risk Evaluation and Mitigation Strategies (REMS) process to delay generic entry. The FDA requires REMS as a safety strategy to manage known or potential risks (FDA, 2017b), but the Federal Trade Commission (FTC) has expressed concern about “the possibility that procedures intended to ensure the safe distribution of certain prescription drugs may be exploited by brand drug companies to thwart generic competition” (FTC, 2014a, p. 1). A recent study estimates that this could lead to about $5.4 billion in unnecessary spending on branded drugs annually (Brill, 2014). This analysis also highlighted that REMS and associated programs could also be used to impede the market entry of biosimilars.
THE MARKET STRUCTURE
The complexity and interdependence within the biopharmaceutical supply chain makes it extremely difficult to understand the motivation and behavior of its various participants, even under the best of circumstances. Nonetheless, any analysis of the availability and affordability of prescription drugs needs to take into account the frequently and extensively altered incentives, trade-offs, and constraints imposed on these markets, and the ultimate impact they have on individual patients.
___________________
5 In recent years the Federal Trade Commission has pursued antitrust cases against a number of pay-for-delay settlements. In the 2013 case Federal Trade Commission v. Actavis, the U.S. Supreme Court held that reverse payment settlements should receive antitrust scrutiny, but it did not conclude that such agreements should be presumed to be illegal; instead the court advised that they need to be evaluated on a case-by-case basis (Boumil and Curfman, 2013). After the Actavis case, lower courts have grappled with how to determine if specific cases meet the threshold for antitrust violations. Courts have searched for evidence of settlement amounts (either monetary or in-kind) that suggest an improper payoff intended to have an anticompetitive effect (Perkins Coie LLP, 2016).
A “Conventional” Market
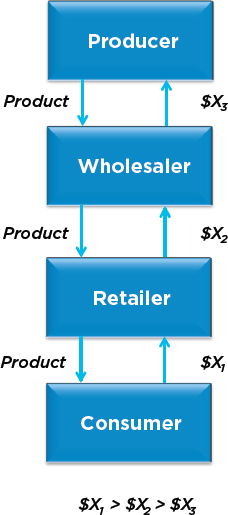
To understand the complexity of the pharmaceutical market, it is useful to first consider the characteristics of more conventional markets, such as those for automobiles, food, and consumer electronics. Each of these products involves the transformation of raw materials into final tested products. As shown in Figure 2-2, goods flow from manufacturers to consumers, and money flows from consumers back to manufacturers. Consumers collectively pay $X1 to retailers, who in turn pay $X2 to wholesalers, who in turn pay $X3 to manufacturers. The gross margin for retailers, $X1 – $X2, must cover the costs of conducting business and provide profits for their investors. Similarly, wholesalers retain $X2 – $X3 to cover their costs of operation and profits. Manufacturers receive $X3, which pays such costs as those associated with compensating its workforce, product development and manufacturing, and a return to investors. In larger firms, these “investors” are typically shareholders in corporations, bondholders, or lenders. In order of legal priority, investors (particularly, shareholders) have the last claim on assets. These residual claimants bear the greatest financial risk and in a typical financial market, demand the greatest returns on their investments.
A Simplified Standard Health Care Market Model
The U.S. health care market differs from the above standard business model in a unique way: in health care, a third party—a health insurer—intervenes in the flow of goods and services, in the flow of money, and sometimes in the choices of products available to individual consumers. People desire health insurance not only because health care is critical and intrinsically expensive, but also because the need for health care services is often highly unpredictable. This creates a significant financial risk to both individuals and to society as a whole. This risk appears in many ways, perhaps most notably in the fact that 5 percent of patients account for half of all health care spending, and the top 1 percent account for one-fifth of all medical spending. Conversely, the half with the least health care spending accounts for just 3.1 percent of medical spending (Cohen and Yu, 2012).
These data highlight how unevenly the financial burden of health care is spread and thus why it is important to have some sort of insurance against this risk—just as homeowners purchase home insurance, auto insurance, disability insurance, and life insurance in response to financial risks in those areas. However, unlike the case with these other forms of insurance, the behavior of consumers can be affected quite strongly by the simple fact that they have health insurance. Specifically, because health insurance subsidizes the cost of medical care, its presence increases the use of health care because people become less sensitive to its price (Newhouse, 1993). Simply put, if consumers pay less than they would otherwise (because of insurance) to visit a clinician or undergo a procedure or buy prescription drugs, they become more likely to do those things.
Figure 2-3 illustrates how these considerations alter the flow of services.6 Individuals, often through their employer, purchase or are provided with health insurance policies for themselves and their families, usually paying part of the annual premium (see Box 2-1 for a note on the role of employers in the control of drug prices). Then, when obtaining services such as clinician care, dental care, hospital care, or emergency room visits, or when buying products, the individual or associated party provides a copayment to the health care provider, as specified in the contract between the insurance plan and the patient. The insurance plan then pays a separate amount to the clinician or the hospital as specified in their contracts. For people without health insurance, clinicians maintain set list prices that they collect directly from the consumer (who, at this point, can appropriately be called
___________________
6 Because most health care transactions involve personal services rather than physical goods, Figure 2-3 does not include a wholesale level of the supply chain, but in some cases (e.g., durable medical goods such as wheelchairs, walking aids, or oxygen supplies) a wholesale level would appear in the medical transactions as well. This has been omitted in Figure 2-3 for simplicity.
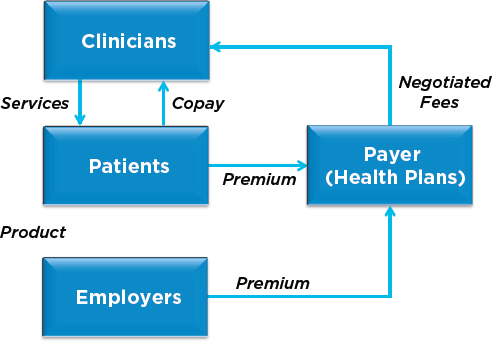
NOTES: This figure also omits two other forms of intermediaries that consolidate bargaining power. Retail pharmacies use pharmacy services administration organizations to negotiate with pharmacy benefit managers and they also use group purchasing organizations to negotiate with wholesalers.
the “patient”) or waive a part or all of such charges as charitable contributions or bad debt. However, the great majority of transactions take place not directly between the patient and provider but with the health plan as an intermediary.
For people ages 65 and over and younger adults with permanent disabilities covered by Medicare, low-income people enrolled in state Medicaid programs, or individuals in special federal programs devoted to military service members and veterans, the relevant governmental programs become the health insurance plan, and taxpayers substitute for employers as payers for some of the cost of health insurance.
A Simplified Biopharmaceutical Market
Figure 2-4 illustrates the structure of a very simplified market for biopharmaceuticals that includes many of the basic players, including patients, clinicians, drug manufacturers, health plans, and pharmacy benefit managers (PBMs). There are several distinctive characteristics of this market that are worth noting. First, individuals cannot simply choose to buy medications, but rather require a prescription from a clinician. Without such a prescription, pharmacists are not permitted to dispense prescription drugs.7
___________________
7 The same “permission” is required for some types of medical care (most obviously hospitalization), where patients cannot “admit themselves” into a hospital for (say) a surgical procedure or medical treatment.
The “manufacturer” in Figure 2-4 includes producers of branded and generic products. PBMs serve as intermediaries between both health insurance plans and retail pharmacies and the manufacturers of prescription drugs. A full portrayal of the market for biopharmaceutical products would also show the regulatory intervention of the FDA and the U.S. Patent and Trademark Office (USPTO). These additional elements are omitted from Figure 2-3 in order to focus on the product and financial flows within the prescription drug market.
As indicated in Figure 2-4, there are multiple pathways between the drug manufacturer and the patient. Before the creation of prescription drug insurance, the sole pathway operated much as displayed in Figure 2-2: drug manufacturers sold to wholesale distributors who sold to retail pharmacies who sold to patients possessing a proper prescription. This remains the primary pathway for patients without drug insurance today.
For those with drug insurance, alternative pathways exist. In one case, the PBMs (and major pharmacy chains) operate mail-order services, selling
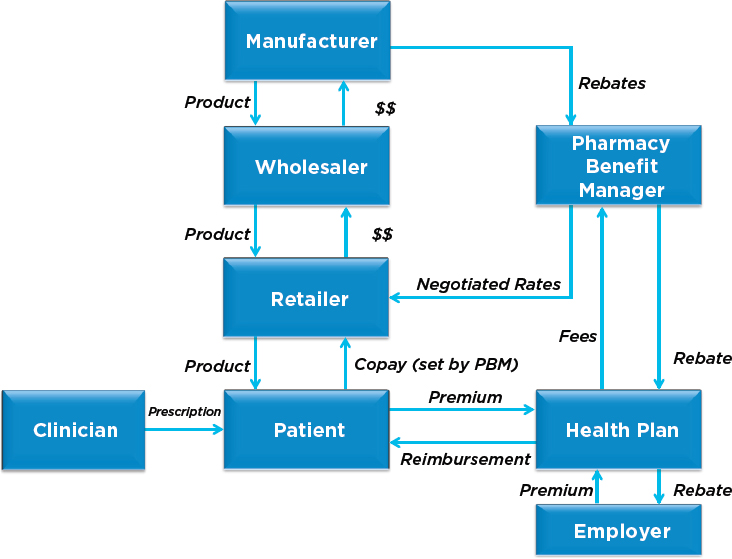
medicines directly to patients (with the usual requirement of a prescription from a clinician) and collecting copayments from the patients and payments through insurers. The other option for such patients is to go directly to a retail pharmacy and offer the copayment specified in the insurance plan. In this latter pathway, the retail pharmacy has previously purchased the drug from a wholesale distributor, and the PBM compensates the retail pharmacy (per the contract between the PBM and the pharmacy) for the cost of the drug plus a processing fee.
A More Complete Portrayal of the Biopharmaceutical Market
Figure 2-5 is a more descriptive illustration of today’s biopharmaceutical enterprise, with several additional participants beyond what is shown in Figure 2-4, but still omits elements of the distribution mechanism for certain drugs used to treat specific patient populations. In particular, many drugs are purchased by hospitals and dispensed or administered to patients cared for in the inpatient hospital and outpatient clinic setting.
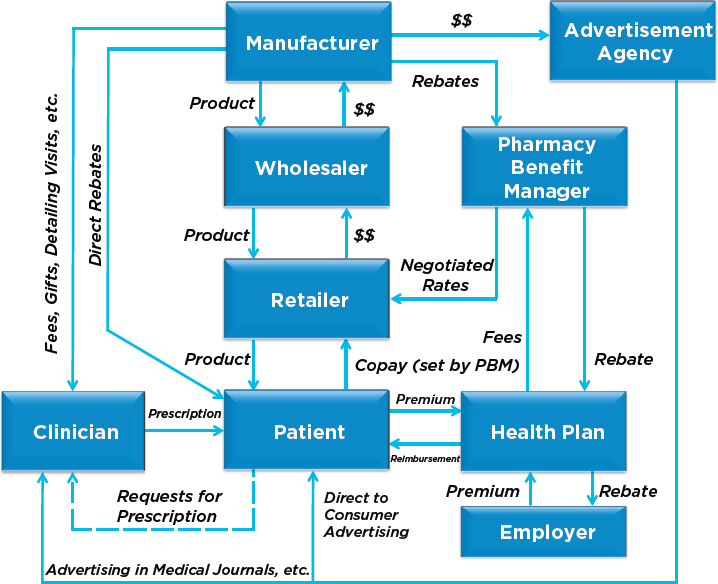
Drugs dispensed to patients in the hospital or infused or injected during an inpatient hospital stay are purchased by hospitals through wholesalers and distributors and are covered under Part A of Medicare, and inpatient benefits provided by state Medicaid programs and commercial insurers covering the non-Medicare eligible population. Use of these drugs by Medicare beneficiaries is largely covered through payments to hospitals under prospective bundled reimbursement arrangements. In 2013, Medicare spent $112 billion on payments for drugs and pharmacy services (MedPAC, 2016); 16 percent of this total (approximately $18 billion) was for drugs billed under Part A.
Standalone and hospital-based clinics purchase drugs from wholesalers and distributors and dispense or administer these drugs to patients as part of outpatient care. These drugs are covered under Part B of Medicare, and outpatient medical benefits provided by state Medicaid programs and commercial insurers covering the non-Medicare eligible population. In
2013, 15 percent of total Medicare spending on prescription drugs was for those covered under Part B (approximately $17 billion) (MedPAC, 2016). In contrast, 57 percent of total Medicare spending on prescription drugs in 2013 was on outpatient prescription drugs covered under Part D (both standalone drug plans and Medicare Advantage drug plans) (approximately $63 billion).
Drugs covered under outpatient medical insurance benefits have an unusual payment structure: clinicians, outpatient clinics, and hospital-based clinics purchase these drugs at their wholesale acquisition cost and then bill insurers and patients for their use and are paid a reimbursement price. This system is commonly called “buy and bill.” Since 2006, standalone and hospital-based clinics that administer drugs in the outpatient setting covered under Medicare Part B have been reimbursed at the average sales price of the drug plus 6 percent plus an administration fee. Commercial insurers set reimbursement rates for these drugs using average sales price, average wholesale price, or another metric, and also reimburse for administration fees.
Figure 2-5 also displays several interlinked communication channels involving pharmaceutical manufactures, clinicians, and patients. While some health care markets (and most non–health care markets) employ direct advertising to consumers, this form of marketing has several distinct features when applied to prescription drugs. Pharmaceutical companies collectively spend billions of dollars annually to “inform” and influence the choices of clinicians and patients. They do this with direct visits to clinicians (referred to as “detailing”), presentations and booths at professional medical meetings, by providing free samples to clinicians, and advertising in medical journals, and also directly providing copay coupons to patients.
BARGAINING POWER AND FORMULARY MANAGEMENT
In any market a buyer’s bargaining power is usually determined by two factors: their ability to walk away from the deal, completely or in part, and the volume of goods they are purchasing. For buyers to be able to negotiate on price, they must have credible alternatives other than purchasing from the seller (OECD, 2009). If a purchaser is always going to buy the product whatever the price, the seller can charge what price they like.
In the biopharmaceutical sector, buyers often appear to be in a weak position, with little alternative but to purchase the drug whatever the price. The drug manufacturers have a strong bargaining position, because their products are protected from competition by patents, and payers are understandably reluctant to deny patients the drugs they need. Even when payers do have credible alternatives, the fragmented nature of the U.S. health care system weakens their bargaining position.
Health care payers typically seek to gain bargaining power in drug pricing negotiations through tier-placement and through formulary design (GAO, 2007). A formulary describes which drugs a health care payer will cover for which disease indications, and at what cost. Formularies can be “open” or “closed.” An “open” formulary ostensibly covers all drugs, but typically includes mechanisms to constrain usage of drugs the payer considers too expensive, such as tiering, with higher tiers requiring greater patient cost sharing, prior authorization, or more tightly defined permissible indications. A “closed” formulary allows for drugs the payers deems very expensive or otherwise undesirable to be excluded from coverage. Formularies are used to steer patients and prescribing clinicians toward generic substitutes, biosimilars, drugs with similar therapeutic efficacy for the same disease, or other therapeutic options.
Formularies contribute to payers’ bargaining power by enabling them to restrict the volume of prescriptions in response to higher prices. Placing a drug on a higher tier triggers higher copayment from patients and therefore discourages the use of the high-priced drugs. Narrowing the indications for which a drug can be used also constrains the potential volume. Excluding the drug entirely from coverage is the most powerful lever, one most readily employed when alternatives exist to treat the same condition. For example, in 2014, Express Scripts, a PBM that covers 25 million people, negotiated a significant discount from AbbVie on its new hepatitis C drug (Viekira Pak), by making it the exclusive option in its formulary, while excluding both competitor drugs Harvoni and Sovaldi (Pollack, 2014; Wilensky, 2016a).
All of these levers work by restricting access to the drug in some way or other: if they did not, they would not contribute to the payer’s bargaining power. This points to the importance of another crucial aspect of formulary design: the basis on which these restrictions are imposed. In some contexts, such as in the United Kingdom, the British National Formulary is determined through an assessment informed by the National Institute of Clinical Evaluation. In the United States, in part due to a lack of broad agreement on how to define and assess “value,” many different approaches are used in formulary design, and transparency about decision making may be lacking (Frank and Zeckhauser, 2017).
Some other countries operate formulary systems that provide much greater ability to restrict or exclude drugs from coverage than is the case in the United States. The health systems in the United Kingdom and Australia, for example, explicitly apply cost-effectiveness and related criteria to determine whether drugs will be included in the formulary. Total exclusion from coverage is relatively rare. What happens more frequently is that approval for the most expensive drugs is only given for a tightly defined set of indications. Even if a drug is excluded from the formulary, this does not generally prohibit patients from purchasing the drug, but removes it from the realm
of insured products and services. However, the exclusion of drugs from a national formulary can generate significant controversy, since there are almost always some patients who would benefit from inclusion and who protest its removal.
By contrast, in the United States, the Centers for Medicare & Medicaid Services (CMS) makes coverage determinations for Medicare enrollees based on the language of the original legislation that created the program: that the treatment be “necessary and reasonable.” Historically, CMS and its predecessor organizations have relied on approval by the FDA for those determinations, and have not used cost as a component of coverage determinations (Neumann et al., 2005, 2008). In other areas of health care, most notably the Prospective Payment System for hospitalization in Part A and the Resource-Based Relative Value System in Part B, Medicare sets prices administratively, using a combination of historical costs and efficiencies that have been deemed achievable. The Congressional Budget Office (CBO) has not expressed confidence in this strategy as a way to produce savings on drug costs (CBO, 2014; Wilensky, 2016b), but to date, there is no CBO estimate on the effect of allowing the U.S. Department of Health and Human Services (HHS) to negotiate both drug prices and formulary placement (Shih et al., 2016). Individual states in the United States also encounter various challenges, each specific to them, in curbing prescription drug costs, with related implications on their formulary structure, a topic explored in Box 2-2.
Formulary designs in the United States typically put greater reliance on tiering than explicit exclusion, with drugs that are allocated to the higher tiers requiring higher cost sharing by patients. The logic is that higher cost sharing will simultaneously make patients more reluctant to use such drugs and, by imposing some of the costs on the individual patient, reduce the burden on the overall insurance pool and thus, control consumer premiums. However, tiering with high cost sharing can also have downsides, since it can lead to reduced adherence or the discontinuation of medications because of high out-of-pocket costs to consumers.8 Such designs may also discourage consumers with high drug expenditures from enrolling in health plans (Happe et al., 2014; Huskamp and Keating, 2005), which could further adversely affect health outcomes. But without such formulary controls within pharmacy benefit plans, insurance premiums would rise, potentially also leading to lower enrollment and similar undesired health consequences. The tiered price mechanism can be used by insurers to negotiate better prices for branded drugs (Duggan and Scott Morton, 2010).
If insurance plans do not have some cost-control mechanisms in place, increased coverage leads both to higher utilization (Newhouse et al., 1993)
___________________
8 This topic is further discussed in the insurance design section of Chapter 3.
and higher costs from providers who have any ability to set their own price. But when cost controls such as tiered prices and prior authorization enter, the net effects are theoretically ambiguous. The introduction of Part D in Medicare provided a way to estimate the net effects. One analysis tracked the prices of the most commonly prescribed branded (non-generic) drugs in Part D upon its introduction, and concluded that, on net, the extended coverage and strengthened bargaining power of the buyers (collectively, through the insurance plans) caused a reduction in many prescription drug prices (Duggan and Scott-Morton, 2010). Specifically, the analysis noted that “Part D plans have succeeded in negotiating lower price increases for Part D enrollees—approximately 20 percent lower than they otherwise would have been.”
Balancing national affordability (which translates into premium costs to consumers or to the taxpayer) and individual affordability (as reflected in copayment costs for specific drugs) is the complex task facing those designing drug insurance plans and associated formularies. But it seems
clear that from a payer’s perspective, effective bargaining cannot take place without the ability either to exclude drugs from a formulary or place them in unfavorably high tiers. Formulary management and effective bargaining go hand in hand.
In sum, significant price negotiating power entails a payer being able to refuse a deal, and a formulary that allows coverage restriction, whether through exclusion, tightening permissible indications, or tiering. The placement of drugs in a formulary needs to be based on some logic. Ideally, this would be an assessment of cost and value to both patients and society.
In addition to formulary design, the other major determinant of a buyer’s bargaining power is their scale: buying more product translates into greater bargaining power. In the world of prescription drug insurance, a commonly used measure of scale would be “covered lives.” However, this is a far from perfect measure of drug purchasing scale, because the rate of using prescription drugs varies enormously by age, among other factors. People older than age 65 spend approximately three times the amount per year on prescription drugs as adults younger than age 65, a point not captured by the “covered lives” metric.
In the United States, private payers are relatively fragmented. Currently, the largest private health insurance company (United HealthCare) has 14.1 percent of the total U.S. population, the second largest (Aetna) has 10.1 percent, and the top eight firms together have less than half of the U.S. population insured (AISHealth, 2016). Compared with other industrial sectors, this would be considered a relatively “low” level of concentration among buyers, meaning even the largest firms would have relatively weak bargaining power.
This fragmentation of purchasing power is a primary reason for the emergence of the PBMs, a form of market intermediary that appears almost unique to the United States. The PBMs negotiate prices and manage formularies on behalf of payers, whether private insurers or self-insured large employers, exploiting the fact that PBMs have achieved far greater scale and thus have greater purchasing power. Recent estimates show that the top three PBMs cover 85 percent of the individuals with prescription drug insurance (Sood et al., 2017). Increased concentration among PBMs undoubtedly enables them to have greater purchasing power versus manufacturers. However, it also enables them to have greater market power versus payers. While some PBMs act as agents for payers, receiving a fee for their services, in many case PBMs act as principals, retaining a share of the discount they have negotiated from the manufacturer. In a sense, the market concentration of PBMs can be seen as a double-edged sword from the patient and the payer perspective: it enhances the ability of the PBMs to extract bigger discounts from the manufacturer, and also the ability to pass on less of these discounts to the patients than would be the case if they
were less concentrated. These dynamics are obscured by the lack of clear information in the public domain, and the increasing integration between PBMs and insurance companies and retail pharmacies.9
Besides the private health insurance markets, the U.S. federal and state governments collectively provide health insurance coverage for a significant portion of the population, including Medicare (55 million), Medicaid (74 million), U.S. Department of Veterans Affairs hospitals and clinics (8.9 million), TRICARE for active duty and retired military and their families (4.7 million), and prisoners (2.2 million). Yet, the buying power for these different organizations is highly diffuse. Even within Medicare’s 55 million enrollees, bargaining power is highly dispersed because HHS is prohibited by statue10 from negotiating drug prices. Here again the PBMs play a central role. For virtually all of those enrolled in Part D insurance for prescription drugs dispensed through retail channels, price negotiations are delegated to the PBMs. However, for the six protected classes of drugs in Part D, for which inclusion on formularies is mandatory, PBMs may not achieve discounts because there is no real lever for negotiation. Part D retail drugs represent approximately 60 percent of the total cost of prescription drugs dispensed under Medicare. The remainder is covered under Medicare Parts A and B through sales directly to hospitals, clinics, infusion centers, and providers’ offices. These non-retail drugs—which may well constitute more than half of all prescription drug spending—come through channels that have relatively weak bargaining power, and generally lie outside the domain of PBMs, entities with the strongest bargaining power currently.
Enabling HHS to negotiate drug prices for all Medicare enrollees would increase bargaining power versus drug manufacturers if HHS also had effective formulary control. The legislative “non-interference” clause of the MMA prohibited CMS from negotiating or administratively setting prescription drug prices and instead advanced private market competition as the means of setting prices (Channick, 2006). However, in every other sphere, whether in purchasing big items like defense equipment or for infrastructure and transportation projects, or in buying more routine items like utilities, uniforms, or stationery, the government uses its scale in price negotiations to the benefit of taxpayers. Furthermore, characterizing this approach as being tantamount to price control or regulation is misleading, and the effect of not allowing HHS to negotiate prices is to tilt the balance of bargaining power further in favor of drug manufacturers.
___________________
9 In October 2017, CVS, the pharmacy chain, proposed to acquire Aetna, one of the largest health insurers in the country (Mattioli et al., 2017). Aetna had previously sought a merger with Humana, another large health insurer, a move that was abandoned after a federal judicial ruling against the proposed merger (Tracer, 2017).
10 Section 1860D-11(i) of the the Social Security Act. 42 U.S.C. § 1395w-111(i).
There are many questions regarding scope and mechanism that would need to be addressed before HHS could undertake negotiation if given the authority by the U.S. Congress to do so. For example, HHS would have to determine which drugs would be subject to negotiations, what would be negotiated (i.e., price, formulary placement), and how to implement negotiated prices in Part D plans (Shih et al., 2016). A pricing model to guide decisions would need to be defined, including how various types of evidence and other considerations would inform conclusions on drug value and price. Factors such as evidence of clinical benefit and the impact of drug pricing on future research and development would likely play a role (Shih et al., 2016). A recently proposed payment framework for negotiation focuses on drugs that are “high cost (e.g., $1,000 per month), incur high levels of Part D program spending (more than $500 million), and have few close substitutes” (Frank and Zeckhauser, 2017, p. 11). The goal of this framework is to develop a negotiated payment system that would simultaneously enable profits for manufacturers and improve health outcomes for patients. This proposal also uses quality-adjusted life-years as a key metric to assess the “value” of drugs, the subject of the following section.
While one cannot readily estimate the extent of available purchasing power that a consolidated federal agency might have, it would almost certainly exceed the largest power currently available in the private sector (85 million covered lives). If one takes the 55 million covered lives in Medicare and uses the multiplier of three times the average per-person spending that leads to 165 million effective “covered lives.” Adding other federal and state agencies would further increase the strength of bargaining power available to the government, should that power be granted through legislative authorization.
Price negotiations in biopharmaceutical markets exhibit a number of factors that differentiate them from other kinds of markets: most notably the price-setting power arising from government-granted exclusivity and the political challenges buyers face in limiting access to prescription drugs, some of which are deemed as “lifesaving.” These factors are exacerbated in the United States by the way formularies are designed, the relative fragmentation of private payers, and the diffusion of governmental purchasing power, not least through the legal restriction on CMS negotiating prices.
THE “VALUE” OF DRUGS
One common proposal to moderate the cost of prescription medicines has been to adopt the so-called “value-based pricing” for drugs, although the meaning of the term “value” varies widely among the participants in the biopharmaceutical sector. A fundamental challenge facing those who would apply value-based pricing is how to determine whether a drug intervention
is in fact of “value,” especially when sufficient evidence is lacking. It is not even straightforward to determine what “evidence” means (IOM, 2011, 2015a; Mayo-Wilson et al., 2015, 2017; NRC, 1985, 1989; Page et al., 2012; Stewart and Parmar, 1993).
Syntheses of clinical trial evidence are known to be biased in some cases, and thus may introduce additional distortion referred to as “meta-bias” (Goodman and Dickersin, 2011). Moreover, the trial and meta-analysis outcomes that are studied and reported frequently do not overlap, so the results are of little use or benefit to patients (Juthani et al., 2017; Saldanha et al., 2017). Conclusions from these studies may be more relevant to clinicians than to patients and consumers. However, the challenges of developing and identifying reliable evidence are becoming increasingly well understood, and some remedies have been proposed (IOM, 2011; NRC, 1985, 1989). At the very least, data need to be collected in a way that provides reliable evidence that can be used to inform what “value” means. In concept, if value is characterized properly and agreed upon, a value-based approach may lead to more efficient resource allocation and improved patient outcomes (Garber and Phelps, 1997; Sorenson et al., 2017). Applying such an approach could also eliminate “indication creep,” the expansion of the use of drugs and interventions that are less likely to benefit the populations.
In the United States, value assessments have been frequently conducted in the realm of oncology by evaluating changes in life expectancy and costs over time (Howard et al., 2010; Lakdawalla et al., 2010; Lichtenberg, 2009; Woodward et al., 2007). Table 2-1 describes the factors considered in various value frameworks. Despite its application in many areas and other Organisation for Economic Co-operation and Development countries (WHO, 2015), expanding the use of value assessments into the practice of actual pharmaceutical pricing and payment in the United States presents a number of challenges. The first is that, as noted earlier, there is ongoing debate about the best methods to assess value (Rubin, 2016).
Value assessments often involve cost-effectiveness analysis, in which the ratio of the added health gains from a medical intervention to the added costs of treatment is calculated, and a pre-established cutoff value is used to determine which interventions are worthy of support (Frakt, 2016; Neumann et al., 2016). The use of a quality-adjusted life-year as the health outcome measure in cost-effectiveness analyses has gained wide acceptance in many countries and is used in coverage and reimbursement decisions. The National Institute for Health and Care Excellence in the United Kingdom, for example, uses cost-effectiveness to provide advice about which drugs and treatments should be made available in its National Health Service (Sussex et al., 2013). A recent statement from the American College of Cardiology and the American Heart Association also concluded
that “it is important to consider both the cost-effectiveness and total cost of burden of performance measures before selection” (Anderson et al., 2014). However, simply using cost-effectiveness as the outcome measure leaves out other factors such as public perceptions of a disease, political interests, social justice, and other practical considerations, that need to be taken into account when making important societal decisions (Phelps and Madhavan, 2017; Phelps et al., 2017), and many frameworks do take those elements into consideration as well (Rawlins et al., 2010).
Another value-based approach would be to assess comparative effectiveness. In its simplest terms, comparative effectiveness analysis compares the therapeutic benefits of different interventions. In some randomized controlled drug trials, the comparison is a placebo treatment with no known therapeutic effects. However, even “placebo effects” can be significant, attesting to the powerful effect of the human mind on physiologic functioning. The effects may be larger when study participants are told that the drug being tested on them is very expensive (Lewitt and Kim, 2015). More importantly, despite the widespread application of cost-effectiveness criteria to coverage decisions in insurance programs in other countries (Sussex et al., 2013), federal law in the United States sharply limits the extent to which comparative effectiveness research findings can be used as the basis for coverage decisions in the Medicare program (Rosenbaum and Thorpe, 2010). Commercial insurers, however, do not face this legal restriction.
Effectiveness can, of course, have multiple dimensions. For drugs that affect life expectancy (as in the case of cancer, heart disease, strokes, and some neurological diseases), survival time is a key measure of effectiveness. But these drugs have many other relevant dimensions of effectiveness as well. Treatments (as with many chemotherapy options) often have significant adverse side effects. Some drugs may enhance the quality of life even if they do not extend it, or may even improve the quality of life for caregivers (e.g., in the case of dementia patients). Most drug therapies are effective for some patients but not for others. Side effects are also variable, affecting some patients and not others. This inconsistency in individual responses may be due to variability in disease presentation and progression, co-morbidities, differences in the patients’ biological makeup and drug dynamics in their body, and adherence to the medication regimen. The fundamental issue is “incremental effectiveness”—the additional benefit brought about in comparison to alternatives. Some newer drugs have limited incremental effectiveness compared with older drugs (including generics), but nevertheless do improve patient conditions at least to some extent.
The growing thirst for methods to measure “value” has led to the emergence of a number of proposed measures for specific disease conditions, including models for assessing the value of therapeutic options for cancer and heart diseases, as well as taxonomies to assess the strengths and
TABLE 2-1
Examples of Value Frameworks
| Organization | Factors Considered | Description |
|---|---|---|
| American College of Cardiology–American Heart Association (ACC–AHA) | Clinical benefit versus risks Magnitude of net benefit Precision of estimate based on quality of evidence Value (cost-effectiveness) | Magnitude of treatment effect ranges from class I (“benefit [greatly exceeds] risk,” “procedure or treatment is useful or effective”) to class III (“no benefit or harm,” “procedure or treatment is not useful or effective and may be harmful”). Precision of treatment effect ranges from level A (“data derived from multiple randomized trials or meta-analyses”) to level C (“only consensus opinion of experts, case studies, or standard of care”). Value corresponds to cost-effectiveness thresholds (high: less than $50,000 per QALY; intermediate: $50,000 to $100,000 per QALY; low: more than $150,000 per QALY). The framework lists the clinical benefit and value designations without combining them. |
| American Society of Clinical Oncology (ASCO) | Clinical benefit Overall survival Progression-free survival Response rate Toxicity Bonus factors Palliation Time off all treatment Cost per month | A therapy can be awarded up to 130 points. Clinical benefit (≤80 points) reflects end point and magnitude of benefit, with preference given to evidence on overall survival if available. Toxicity (±20 points) reflects the rate of grade 3 to 5 toxic effects with treatment relative to standard of care. Bonus point score reflects palliation (10 points if therapy improves symptoms) and increased time off all treatment (≤20 points). The framework does not combine each drug’s point score and cost. |
| Institute for Clinical and Economic Review (ICER) | Incremental cost-effectiveness plus care value components Comparative clinical effectiveness Other benefits and disadvantages Contextual considerations Budget impact | Cost-effectiveness ratio must not exceed a threshold ranging from $100,000 to $150,000 per QALY. Selection of final threshold is based on (a) comparative clinical effectiveness, reflecting “judgments of the health benefit magnitude” and “strength of a body of evidence”; (b) other benefits and disadvantages, including such outcomes as factors influencing adherence or return to work; and (c) contextual considerations, including “ethical, legal, or other issues” (e.g., high burden of illness, availability of alternative treatments). Budget impact is acceptable if a drug’s introduction is compatible with an annual health care budget increase of GDP growth plus 1 percent. ICER reverse-engineers a “value-based price benchmark” that independently satisfies both the cost-effectiveness and budget-impact criteria. |
| Memorial Sloan Kettering Cancer Center (DrugAbacus) | Efficacy (survival) Toxicity Novelty Research and development cost Rarity Population health burden | Framework assigns values to each domain. Efficacy is assessed as improvement in overall survival, if available. Efficacy score also reflects evidence quality. Toxicity is a drug’s impact on probability of severe side effects and treatment discontinuation. Novelty is scored as 1 (novel mechanism of action), 0.5 (“known target but different mechanism of targeting”), or 0 (“next-in-class”). Research and development cost corresponds to the “number of human subjects enrolled in the approval trials for the first indication.” Rarity is the 2015 projected disease incidence. Population health burden is the annual years of life lost to the targeted disease in the United States. Fair price is the product of the scores, each of which is scaled by a user-adjusted weight. |
| National Comprehensive Cancer Network (NCCN) | Efficacy Safety Evidence quality Evidence consistency Affordability | Each area is scored on a scale of 1 to 5, with 1 indicating least favorable and 5 most favorable. The framework presents the scores separately. There is no explicit synthesis. Stakeholders judge acceptability on the basis of their overall impression of the listed factors. |
| European Society for Medical Oncology Magnitude of Clinical Benefit Scale (ESMO-MCBS) | Toxicity Quality of life Overall survival Progression-free survival Long-term survival | The framework assesses the clinical benefit for cancer drugs using a structured approach to derive a relative ranking of the magnitude of clinically meaningful benefit that can be expected from a cancer therapy. It is developed only for solid tumors. Clinical benefits are measured on the basis of oncology-specific thresholds for overall and progression-free survival outcomes. Scores are awarded by virtue of improvements in the variables under investigation in a comparative trial or cohort study. The ESMO score ranges from 1 (worst) to 5 (best). |
NOTE: GDP = gross domestic product; QALY = quality-adjusted life-year.
SOURCE: Adapted from Neumann and Cohen, 2015.
limitations of different value frameworks (Mandelblatt et al., 2017). One approach would be to conduct post-launch surveys of therapeutic effectiveness and then adjust payments in proportion to actual (versus forecasted) success rates (Barlas, 2016). In its most extreme form, this approach would pay only for cures. Such an approach was used experimentally by the U.K. National Health Service, but no conclusions could be made about whether it was actually effective or workable in practice (Garber and McClellan, 2007). In the United States, a similar approach is being tested by Novartis, the manufacturer of a very expensive new cell-based immunotherapy for cancer, in which payment for the therapy will only be made when pediatric and young adult patients with acute lymphoblastic leukemia respond to the treatment by the end of the first month of therapy (Novartis, 2017).
Ultimately, the lack of a broad consensus on the definition of “value” is a hurdle to advancing a uniform approach to value-based purchasing. Nonetheless, health insurers are beginning to apply various approaches to foster use of treatment regimens that they consider a better value. For example, the Institute for Clinical and Economic Review is funded by nonprofit foundations to undertake value assessments within the U.S. setting, and also has membership support from pharmaceutical companies, PBMs, and insurers (ICER, 2017). PBMs use those value assessments in negotiations with manufacturers. In addition, some insurers have been experimenting with value-based insurance design, in which patient cost sharing is aligned with the value of treatments (Gibson et al., 2015; Lee et al., 2013). Some use value assessment to assign the position of drugs in formularies.
Insurers have also been defining preferred treatment pathways and incentivizing clinicians to follow them (DeMartino and Larsen, 2012; Gesme and Wiseman, 2011; Zon et al., 2016). However, one criticism of this approach is a lack of transparency on how treatment regimens are selected for pathways (IOM, 2015b). A recently announced collaboration between Optum and Merck also aims to promote a value-based form of contracting referred to as “outcomes-based risk-sharing agreements” (UnitedHealth Group, 2017). Manufacturers have also relied on value statements to justify launch prices for new drugs (McKinsey & Company, 2013).
In sum, the use of value assessments of products is not an alternative to “market forces” but they could potentially be used as tools to enhance market performance. Markets in general can work better when participants are well informed about the relative value of the goods they are trading.
OPACITY IN THE SUPPLY CHAIN
As the public concern and frustration over increasing drug prices escalate, it has become clear that information needed to directly establish the sources of these increases is lacking. Prescription drug manufacturers
blame the PBMs and insurers, saying that their price discounts are not wholly passed on to consumers (Walker, 2016). Manufacturers also say that since their rebates are commonly calculated as a fraction of the list price, reducing the manufacturer’s list prices would cause the PBMs to terminate the existing contracts because a lower list price would mean a lower discount to the PBMs (Vandervelde and Blalock, 2017). The PBMs blame both the drug manufacturers for raising prices and the insurers for not passing discounted prices on to consumers (Hopkins and Tracer, 2017).
Drug manufacturers have the ability to use a portion of their sales revenue to stimulate demand by creating incentives for various participants in the biopharmaceutical supply chain and beyond. The primary incentives they use are discounts and rebates, each of which can take a number of different forms (Eickelberg, 2015). Between 2010 and 2014, the average rebates from drug manufacturers to insurers increased by 10 percentage points, from 18 percent to 28 percent of list price, for branded drugs (QuintilesIMS, 2016), and they continue to climb.
List prices grew 9.8 percent in 2016, modestly less than the 10.8 percent increase in 2015. The resulting increases in drug prices added $8.7 billion to 2016 net income for the 28 companies analyzed in a Credit Suisse report. Rebates in 2016 were up by almost 2 percentage points from the average of 35.7 percent in 2015 (Credit Suisse, 2017), and were estimated to be around $130 billion (Goldberg, 2017). Average rebates have also risen over time in the Medicare Part D program, from 8.6 percent in 2006 to nearly 20 percent a decade later (CMS, 2016). Rebates to PBMs can actually increase out-of-pocket spending for patients who pay a percentage of their drug’s list price and those paying deductibles, as the discounts are rarely passed through to the patient at the point of sale (CMS, 2017; Dusetzina et al., 2017). The effects of such pricing strategies have been demonstrated to drive up out-of-pocket spending for patients on Medicare Part D even when net prices received by manufacturers are virtually the same (Dusetzina et al., 2017). For specialty drugs nearly all plans require enrollees to pay a percentage of their drug’s price (Dusetzina and Keating, 2015; Jung et al., 2016; Polinski et al., 2009; Yazdany et al., 2015). Recognizing the role of rebates for increasing spending by beneficiaries and the federal government, in 2016 the Medicare Payment Advisory Commission outlined recommendations for restructuring Medicare Part D to provide incentives to sponsors to improve cost protections and reduce catastrophic spending. Specifically, they would require higher cost sharing from Part D plan sponsors and eliminate patient out-of-pocket contributions in the catastrophic phase of coverage (currently set at 5 percent of drug costs with no lifetime or annual out-of-pocket maximum). They would also require manufacturer payments in the coverage gap to stop counting toward patient out-of-pocket spending (MedPAC, 2016).
The interaction between rebates and list prices can be complicated. For example, market price negotiations based on a drug’s list price can even induce drug manufacturers to further increase their drug prices (Hopkins and Tracer, 2017). Furthermore, some private insurance companies have begun to operate their own PBMs (Kirchhoff, 2015), and retail pharmacies have been merging with PBMs to provide integrated health services. These consolidations can produce conflicts of interests regarding decisions about the inclusion of drugs on a PBM’s formulary (Cook et al., 2000).
In this complex supply chain many opportunities exist to enhance various participants’ revenue and profits—often at the expense of patients. The only sources of data available to understand this system come from the participants, who release data and statements that conflict with each other and justify their positions. The relevant data needed to conclusively analyze this system do not exist at present, and, indeed, some of the participants (most notably the PBMs) argue that revealing their transactions would actually increase the drug prices paid by patients.
Arguments for and Against Transparency
The biopharmaceutical sector is rife with divergent, strongly held views regarding the concept of transparency.11 As noted earlier, various participants in the prescription drugs pricing debate offer divergent statements about who is responsible for high and steadily rising drug prices, but with little to no relevant data to support their claims. In addition, some urge greater transparency in the biopharmaceutical sector, while others assert that transparency would harm competition and thus negatively affect consumers. Proposals vary widely concerning what information should be reported, to whom, and whether this information should be linked to price control measures. No empirical studies demonstrate that transparency in the biopharmaceutical supply chain will cause harm to patients. However, the debate, including in public testimonies, continues about whether transparency would weaken the market and pose harm to patients (Balto, 2014, 2015, 2017; Shepherd, 2014).
Opponents of transparency cite a series of letters from the Federal Trade Commission (FTC) (FTC, 2004, 2006, 2009, 2011, 2014) to state officials regarding policies on the disclosure of financial transactions among
___________________
11 Public debates over transparency continue to be intense and unresolved, with numerous price transparency bills proposed at both the state and federal level. Recent legislation proposed in both chambers of the U.S. Congress would expand transparency by requiring PBMs to provide information for public posting on how much they rebate various drugs. Such price information could decrease the likelihood of excessive profiteering, particularly among PBMs—an industry mainly controlled by three firms. Several states have also introduced bills concerning drug price transparency. Much of this legislation is aimed at drug manufacturers.
the participants in the biopharmaceutical supply chain. For example, in a 2014 letter to the advisory council of the Employee Retirement Income Security Act, the FTC noted that “mandatory disclosure requirements may hinder the ability of plans to negotiate an efficient level of disclosure with PBMs” and that such disclosures, if they reveal discounts negotiated with PBMs, “may result in less aggressive pricing by, or even collusion among, pharmaceutical manufacturers” (FTC, 2014b). These concerns relate to state-by-state information and hence are more likely to reveal confidential contract information than data aggregated to the national level.
Proponents argue that lack of transparency negatively affects patients and enables the largest PBMs to engage in anticompetitive behavior, including securing kickbacks from drug manufacturers in exchange for exclusivity arrangements that may keep lower-priced drugs off the market (Balto, 2014). There is no way to test these claims in a controlled experiment. However, transparency has improved the functioning of markets in other sectors of the economy (see Box 2-3), and the potential gains from improved transparency in the biopharmaceutical sector appear to outweigh the potential risks emerging from increased transparency. The primary questions pertain to what level of information is needed and who would have access to the information. For example, the information would not need to be used directly by consumers to lead to improvements in market functioning. It could be used by other participants in the distribution chain for biopharmaceutical products, by specialists who study market behavior, and by regulators to control these markets.
One way to improve transparency in the pharmaceutical supply chain would be to require manufacturers to disclose detailed information on a drug-by-drug basis for their gross and net prices (the difference reflecting discounts given within the supply chain and discounts given directly to patients). Information about the prices paid at the end-stage of distribution in retail pharmacies or their mail-order counterparts and by hospitals, clinics, nursing homes, and other relevant organizations that purchase and directly administer drugs to patients would also need to be gathered in parallel. Logically, the difference between what the manufacturers report and what the final distributors (e.g., retail pharmacies, hospitals, doctor offices) report has been retained in the intermediary system either as costs or profits.
These data may provide clarity about the interactions—specifically the flow of funds and products—among the intermediaries of the biopharmaceutical supply chain, or they may point toward necessary regulation for additional data gathering from each participant in the biopharmaceutical supply chain. This proposed approach would involve a sequential process of first gathering information at the two ends of the supply chain—manufacturers at one end and consumer payments at the other—with the
understanding that more refined data may be needed later to completely understand how the biopharmaceutical supply chain operates. Experience from other sectors would suggest that transparency on supply chain costs tends to reduce these intermediary costs (see Box 2-3).
Profitability Across the Supply Chain
Another argument in favor of transparency is based in the importance of understanding the profitability of the participants in the biopharmaceutical supply chain. Such an understanding would, at minimum, help bring clarity into how the biopharmaceutical supply chain affects prescription drug prices in the United States compared with those in other nations. Of greatest importance in this question is how profitable the manufacturers of branded and generic drugs are, because their profitability is commonly seen as a source of funds for investing in new research and development to bring new lifesaving and life-enhancing products to the market.
To address this question, four key sources of information were reviewed. The first was a 2015 projection in Forbes showing the top 10 most profitable industries for 2016 (Chen, 2015). The listing showed generic pharmaceutical companies as the most profitable sector (with 30 percent margin), while major pharmaceutical companies appeared fourth on the list (25.5 percent margin) and biotechnology companies were sixth on the list (24.6 percent margin). Other industries on the list included investment managers (29.1 percent margin), tobacco (27.2 percent margin), Internet and software services (25 percent margin), and various banks (between 22 and 24 percent margin).
The second source was a recent study published by the University of Southern California that assessed the profit margins for various industries (Sood et al., 2017). This study found branded pharmaceuticals to be the industry with the greatest profit margins (28 percent) and generic pharmaceuticals to be fourth (26 percent), with tobacco and alcoholic beverages in between. The reported rate at which profit has been growing in the pharmaceutical sector significantly outpaced the rates of all other reported sectors of the U.S. economy (see Figure 2-6).
Third, corporate bond ratings on major pharmaceutical companies from the Morningstar bond rating agency were reviewed for insights into the financial health of companies. While some of these companies in the biopharmaceutical sector did not have bond ratings (some companies do not issue corporate bonds), of those with a rating provided by Morningstar, which accounted for 86 percent of the market capitalization considered, the bond ratings were all A– or higher, with one AAA rating (Johnson & Johnson), the highest rating given by Morningstar. The lowest bond rating among pharmaceutical companies (three firms representing 4.5 percent of the total market capitalization) was BBB–.12 For the sake of comparison, the bond ratings of some familiar corporations in other sectors of the
___________________
12 AAA = extremely low default risk; AA = very low default risk; A = low default risk; BBB = moderate default risk; BB = above average default risk; B = high default risk.
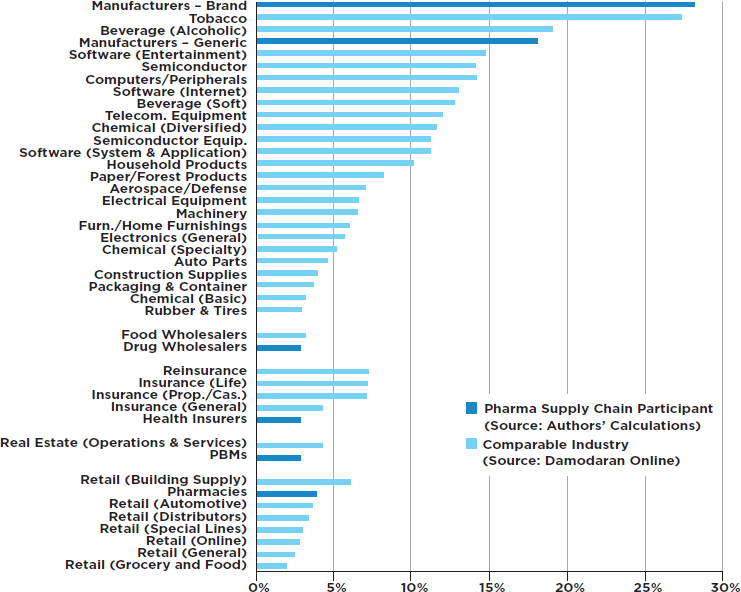
SOURCE: Sood et al., 2017, Figure 4.
U.S. economy were Microsoft: AAA; Exxon: AA+; Google: AA; Apple Computers, Chevron, Intel: AA–; Home Depot, IBM: A+; Amazon, Shell Oil (Royal Dutch), Starbucks, U.S. Steel: A; McDonalds: A–; Southwest Airlines: BBB+; Ford Motors, General Motors, Hewlett Packard, Verizon: BBB; T-Mobile, United/Continental Airlines: BB; and Sprint: B.
However, a concern about the future profitability of biopharmaceutical companies emerges from a recent analysis from Deloitte LLP (2016), which reported a steadily declining return on investment (ROI) for research and development in the biopharmaceutical sector. According to that report, the ROI fell steadily from 10.1 percent in 2010 to a low of 3.7 percent in 2016. The analysis focused on the performance of 12 large-cap companies (i.e., companies with a large market capitalization) since 2010. A group of mid-cap companies that were also newer companies had a higher average ROI, but their return also showed a decline—from a prior peak of 17.7
percent to 9.9 percent in 2016. The Deloitte report’s summary statement stated, “Costs per asset have stabilized for the original biopharma cohort, but forecast peak sales per asset continue to decline” (p. 2).
Share of Costs and Profits Within the Biopharmaceutical Supply Chain
This section presents available assessments of share of costs and profits in the biopharmaceutical supply chain. However, these assessments used different methodologies and data that differ in scope; thus, they are not directly comparable.
One recent analysis of profitability across the biopharmaceutical drug supply chain illustrates the difficulties in working with currently available data for U.S. firms (Sood et al., 2017). The U.S. analysis, reported in a white paper, used financial information that is publicly available through the Securities and Exchange Commission (SEC) for firms that are publicly traded on stock markets—a subset of the entire market that selectively eliminates both privately held firms and smaller firms. The omission of smaller firms may be more important at the front-end research and development stage of the market than further along the supply chain, but the omission is relevant to some extent at all levels of production and distribution. The approach used for this white paper also excluded nonprofit organizations. A further complication arises because some of the intermediaries in the supply chain operate in more than one segment of the supply chain, for example, both as a PBM and an insurer, as a PBM and a retail pharmacy, or other similar combinations.
While public reports generally separate these different lines of business into different areas of profit, the choice of exactly where to report profits remains somewhat arbitrary on the part of the reporting entity. For example, if a $1 million rebate is received from a drug manufacturer by a PBM, that rebate could be retained at the PBM level (where it would increase the reported PBM profit margin), or it could be passed along to an affiliated insurer or retail pharmacy. The same $1 million would have different effects on profit margins depending on the underlying level of economic activity, and it would appear smallest on a percentage of revenues if it were reported as being accrued by the largest of the component businesses.
With these considerations in mind—and emphasizing that the committee does not view these data as conclusive—the analysis does shed light on the profitability of various elements of the biopharmaceutical supply chain. The key result is presented in Figure 2-7, which shows that $41 out of every $100 spent on prescription drugs is retained in the supply chain, including wholesalers, retailers, PBMs, and insurance companies (Sood et al., 2017). This would suggest that the intermediaries in the U.S. bio-
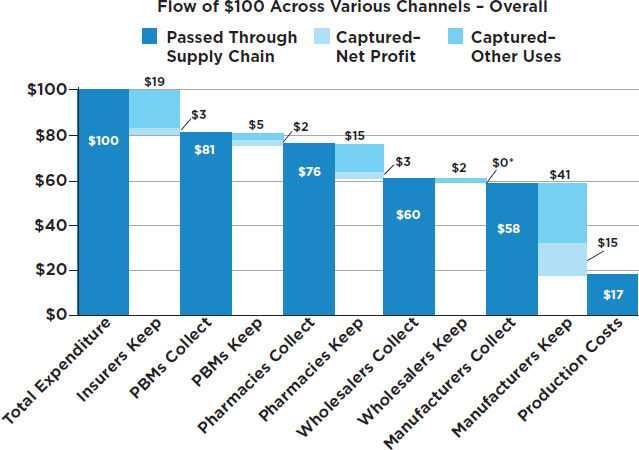
SOURCE: Sood et al., 2017, Figure 2.
pharmaceutical supply chain consume a greater fraction of total costs than is apparently the norm in other nations.13 As noted above, the distribution-related costs in six comparator countries averaged 32.7 percent of end-user price based on a simple average and 28 percent based on a population-weighted average (QuintilesIMS, 2014). The Sood analysis also estimated gross and net margins for each sector, segmented by branded drug, generic drug, and the total market, and found the following: net margins resulted: 28.1 percent for branded drug manufacturers, 18.2 percent for generic manufacturers, and 26.3 percent overall.
The intention behind reporting these international and domestic data is to emphasize that these data are unavoidably incomplete in scope and do not provide information on many key participants in the various markets. The limitations inherent in using SEC filings to garner such data also point to the need for more granular information than the SEC database can reveal.
To assess the flow of funds through the distribution system, a Barclays
___________________
13 Another study by Kanavos and colleagues (2011) found that distribution margins vary greatly at different levels of wholesale and retail segments among 27 member states of the European Union. However, none of these include distribution segments is comparable to health insurance and PBM markets in the United States.
Equity Research analysis using publicly available data from two PBMs (Express Scripts and CVS) estimated that, in 2015, an industry-wide total of $115 billion was either provided as discounts to or retained by those involved in the distribution of pharmaceutical products (i.e., those organizations between the manufacturers and the consumers). This was estimated to have been 27 percent of what gross sales would have been, based on the listed prices. The Barclays estimate of how this $115 billion is distributed is presented Figure 2-8.
In some cases, the Barclays analysis could not determine whether the funds were being retained or passed along in the supply chain because the analysts had no information from certain of the participants in this distribution system.14 For example, the analysis found that $9 to $10 billion was either “retained by distributor or passed to dispenser” at the distribution level. Manufacturers directly paid fees to PBMs ($1.5 to $2.0 billion) and additionally retained $5 billion of rebates through the commercial insurance (nongovernmental) sector, with another $23 billion passed to Medicaid programs and $21 billion passed to Medicare. The final $31 billion in the commercial sector is described as “passed to payers” with no understanding of how much of this $31 billion was passed along to consumers in terms of lower drug prices (versus being retained by payers to cover costs or as profits).
In a separate analysis of 2015 data, the Berkeley Research Group (Vandervelde and Blalock, 2017) provided estimates of the distribution of gross revenues in the pharmaceutical distribution system. They began with gross sales information from manufacturers, estimating a total of $469 billion in 2015, and then estimated the portions received by branded manufacturers (47 percent) and generic manufacturers (23 percent), which implied a total of 70 percent received by manufacturers. It was estimated that others in the supply chain receive 27 percent of the total, with 4 percent consumed in other rebates and fees (see Figure 2-9).
This calculation diverges from that of Sood and colleagues (2017) in several ways. First, the Berkeley Research Group’s method did not separate operating costs from profits. Second, its analysis began with total sales at gross list prices, whereas Sood and colleagues assessed the flow of funds only through retail sales involving commercial insurance plans. Sood and colleagues estimated that the manufacturer retained 58 percent of sales dollars, using as a base the actual amounts spent by consumers on retail sales (e.g., in pharmacies and other retail distribution outlets). Thus, the figure of 58 percent of sales calculated by Sood and colleagues (in effect, por-
___________________
14 These gaps in available data cannot be repaired using existing data sources. One of the recommendations in this report focused on new data gathering is designed to remedy this defect, providing a direct method for assessing how much is retained by the distribution system and how much passes to consumers.
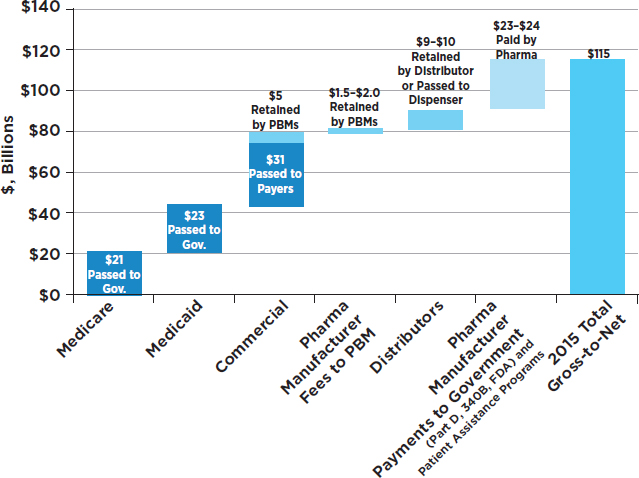
NOTE: FDA = U.S. Food and Drug Administration; PBM = pharmacy benefit manager.
SOURCE: Barclays, 2017, Figure 1.

*Includes any retrospective rebates and fees not shared with the end payer.
**Components may not sum to total due to rounding.
SOURCE: Vandervelde and Blalock, 2017, Figure 3.
traying net sales after subtracting rebates given to PBMs and others) does not directly correspond to the 70 percent figure (gross revenue) calculated by the Berkeley Research Group.
A 2014 publication reported the proportion of total pharmaceutical costs attributable to the distribution system (comprising wholesalers and retailers) in different countries (QuintilesIMS, 2014). Current estimates of the populations of these six countries were used to calculate a population-weighted average of the data. The distribution-related costs in these six nations averaged 32.7 percent of end-user price based on a simple average and averaged 28 percent when a population-weighted average was used
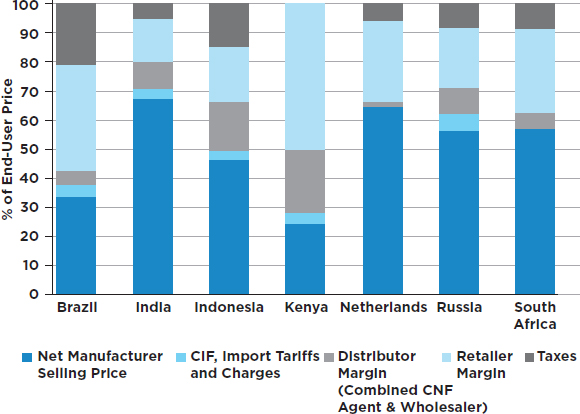
NOTES: CIF = cost, insurance, and freight charges; CNF = cost, no insurance, and freight charges.
2013 population-adjusted average. The study’s description of the countries chosen for the analysis stated:
Brazil: Upper-middle income country with large retail out-of-pocket market and a major market in the region.
India: Lower-middle income country with a large out-of-pocket market but undergoing change and implementing mechanisms to control the build-up of drug prices.
Indonesia: Lower-middle income country with little price regulation and relatively large out-of-pocket spending.
Kenya: Excluded by the committee for discussion in this report since the IMS study did not provide exact data on that country’s costs. Furthermore, the data reliability for Kenya was very small (only 30 drug prices in the sample compared to thousands in other nations).
Netherlands: High-income country with a rational approach to pricing and margins, and useful as a baseline country for comparison purposes.
Russia: High-income market with a high level of out-of-pocket, mix of regulated and unregulated market.
South Africa: Upper-middle income country with a large proportion of the medicine market funded privately, but a highly transparent pricing system in place.
SOURCE: QuintilesIMS, 2014, Exhibit 12, p. 21.
(see Figure 2-10). The data from this report data came from a sampling of drug transactions in five significant areas of pharmaceutical products: antibiotics, diabetes, epilepsy, hypertension, and respiratory diseases. However, the analysis did not include other important areas of drug spending in the United States, most notably cancer and other specialty drugs.
In summary, each analysis differs in its methodology to estimate the potential amount captured by the intermediary participants in the biopharmaceutical supply chain. Part of the challenge in making these comparisons, as noted, is that various analyses employ different approaches to defining total drug spending, part is the differing assumptions made about the pass-through of discounts and rebates, and part is whether the issue is framed in terms of the share of list price or the share of the price ultimately paid by the patient and the insurers. Similar methodological differences also bedevil cross-country comparisons, as do varying approaches to the inclusion of sales and value-added taxes.
Thus, existing data do not adequately answer a question of fundamental interest: when a pharmaceutical company provides a discounted price, how much of that discount actually reaches patients at the other end of the distribution system? Without knowing the answer to this basic question, it is impossible to determine responsibility either for the levels of prices or their rates of increase over time. This lack of clarity has led to numerous situations in which different participants in the supply chain point to other participants as the source of high and increasing prices. One cannot know with reasonable clarity how much money is retained at various levels, or how much of that which is retained is due to operational costs and how much is profit. For example, while Sood and colleagues (2017) assembled this type of information, that analysis is limited by several important omissions: (1) it included only companies that are publicly traded (SEC data), and (2) it only assessed the distribution through the retail pharmacy sector for those with insurance. Thus, public programs such as Medicaid, veterans’ benefits, TRICARE, and the 340B program, among others, are excluded, along with sales to clinicians who purchase and then directly administer prescription drugs.15
FINDINGS
Based on the material presented in this chapter, the following findings are offered:
___________________
15 This same omission accounts for the difference between estimates that prescription drugs account for 10 percent of U.S. health care costs versus other estimates of 16 percent. The former estimate includes only the retail sales portion of total sales.
Finding 2-1: The complexity, confusion, and conflicting information associated with the pricing of prescription drugs result in opaqueness of financial transactions among the participants in the biopharmaceutical supply chain.
Finding 2-2: Mandatory disclosure and public reporting of reliable information regarding financial flows and margins (which currently does not exist) would allow a fuller understanding of how participants in the biopharmaceutical supply chain operate and how they influence final prescription drug prices to the consumer. This would improve market performance and provide the basis for future policy changes as needed.
Finding 2-3: Various participants in the biopharmaceutical supply chain point to other participants as the main contributors to high and rising drug costs. The resulting discord and lack of meaningful information to evaluate those competing claims undermines the ability to confidently understand the root causes of price increases and when they are appropriate.
Finding 2-4: While some suggest that greater transparency across the biopharmaceutical supply chain could harm rather than benefit consumers, there is no compelling evidence to support this claim. Moreover, practices from other fields, such as banking, consumer loans, occupational safety, and automotive manufacturing, suggest that transparency does benefit consumers.
Finding 2-5: Most large employers self-finance their health insurance contributions for their employees and hence have a direct and significant interest in controlling health care costs.
Finding 2-6: Extensions of product exclusivity based on minor modifications to existing patents—known as “evergreening”—adversely affect consumers.
Finding 2-7: “Pay-for-delay” strategies employed by companies to keep generic competitors out of the market reduce access to reasonably priced generics.
Finding 2-8: In the United States, formulary control relies heavily on tiering, which can have mixed consequences. Placement of a drug in a higher tier can reduce adherence to the treatment plan, with potential harms to patient health, but the tiered price mechanism can also be used by insurers to negotiate better prices for branded drugs.
Finding 2-9: In the United States, the bargaining power of federal and state governments has been undercut by laws restricting what they may negotiate and exclude from coverage. Because prices tend to be lower when the purchaser has bargaining power that is at least comparable to that of the seller, the United States could achieve lower prices for prescription drugs by consolidating bargaining power and providing greater flexibility in formulary design.
Finding 2-10: Section 1861 of the Social Security Act, which requires that Medicare cover “reasonable and necessary” medical services, in conjunction with language in other statutes limiting the extent to which comparative effectiveness information can be used in Medicare coverage decisions, has precluded consideration of cost or cost-effectiveness in coverage decisions.
Finding 2-11: Value-based approaches to drug purchasing decisions and formulary control are well established in some other OECD countries where they are seen as critical to containing price pressures and ensuring affordability.
Finding 2-12: In the United States, although approaches for assessing the “value” of drugs have been developed and deployed in several clinical areas for a number of years, there remains considerable debate about the use of value-based approaches in the pricing and the purchasing of drugs, as well as for designing insurance benefits.
Finding 2-13: Rebates, discounts, and financial arrangements among biopharmaceutical companies, intermediaries, purchasers, and health care providers—coupled with tiered cost-sharing arrangements in health insurance plans—often do not benefit consumers even when intended to do so, and at times may even harm consumers and society at large.
Finding 2-14: Although the available data are not fully comparable, they indicate that intermediaries in the U.S. biopharmaceutical supply chain extract a higher fraction of total prescription drug spending than do the entities performing similar functions in other countries.
The next chapter considers in greater detail some of the main factors influencing affordability. These range from pricing policies to market exclusivity, and from patient assistance programs to the availability and the clarity of information affecting the choices available to patients and their clinicians, and also presents related findings.










































