
Appendix F
Invited Paper: Current Quality Assurance (QA) Approach: DHS BioWatch Program
Disclaimer:
This white paper was prepared for the September 18-19, 2017, workshop on Strategies for Effective Biological Detection Systems hosted by the National Academies of Sciences, Engineering, and Medicine and does not necessarily represent the views of the National Academies, the Department of Homeland Security, or the U.S. government.
Molly Isbell
Director of Quality Assurance (QA) and Data Science
Signature Science LLC
BACKGROUND
The Department of Homeland Security (DHS) BioWatch program monitors for the aerosol release of five biological threat agents through the collection of air filter samples and subsequent laboratory analysis. Air samples are collected by pulling air at approximately 100 liters per minute thorough a 3.0-micron-size filter using a portable sampling unit (PSU). For most jurisdictions, filter holders are collected approximately every 24 hours. For a small subset of sites, samples are collected more frequently. Samples are delivered to BioWatch laboratories (typically housed within a local public health laboratory facility) each morning for analysis. The number of samples collected is jurisdiction specific and was determined through population-based modeling performed by National Laboratories.
The methods and instruments used by each operational laboratory are determined by the BioWatch program and are specifically described in BioWatch program analytical standard operating procedures (SOPs). All samples are processed according to document-controlled program analytical procedures and guidance documents referenced in the BioWatch Laboratory Master Documents List. Following bead-beating and DNA extraction, analytical testing is performed in two stages, termed screening and verification, utilizing different real-time polymerase chain reaction (PCR) detection assays and reagents for each. The objective of the screening stage of analysis is to identify samples that have a significant probability of containing DNA from a specific agent or signature; Department of Defense (DoD) assays produced by DoD laboratories are used for this stage of testing. Depending on sample throughput at the laboratory, samples may be pooled 3:1, 2:1, or may be unpooled for screening analysis. Any sample pool or individual sample (whichever is applicable) producing a Ct value less than 40 is considered “reactive” and each individual sample is sent for verification analysis using the Laboratory Response Network (LRN) assay panel that corresponds to the agent for which the screening reactive was observed. In addition, for one agent group, a panel of subspeciation assays produced by the DoD labs is tested. The goal of the verification stage of analysis is to provide additional data used to assess whether the result is a BioWatch Actionable Result (BAR). The verification assays panels are agent specific and each kit contains at least three signatures; for most panels, three assays must be reactive in order for the sample to be considered positive. Operational laboratories are also required to perform the quality assurance (QA) Sample Tag assay during verification in order to assist with the interpretation of the verification results and rule out potential contamination from QA or proficiency test (PT) samples. The Viral Hemorrhagic Septicemia Virus Tag assays may be run retroactively if a verification positive is observed to rule out contamination from the verification-positive control plasmids.
On a daily basis, the laboratories are required to report their operational results to DHS and the Centers for Disease Control and Prevention (CDC) through the LRN Results Messenger reporting tool. The laboratories also provide notification to the local public health stakeholders.
BIOWATCH QA PROGRAM OVERVIEW
In 2011 DHS established a formal QA program for BioWatch that was built upon the foundational QA program developed by the Joint Program Executive Office for Chemical and Biological Defense (JPEO-CBD) in 2003 in support of fixed and mobile laboratories performing chemical, biological, radiological, nuclear, and explosives detection for DoD. Although the BioWatch QA program is fully tailored to the DHS BioWatch mission and constraints, DHS has maintained strong interagency communication and collaboration with JPEO-CBD, the National Guard Bureau Weapons of Mass Destruction Civil Support Teams, and other DoD laboratories performing biothreat analyses in the area of QA. This interagency cooperation promotes consistency, where appropriate, in the QA standards applied across these programs and allows participants to share technical and quality-related best practices and lessons learned.
The BioWatch QA program includes applicable elements from internationally recognized standards including ISO 17025:2005, Clinical Laboratory Improvement Amendments, and the College of American Pathologists. Customization of the BioWatch QA program involved stakeholder meetings to discuss objectives and approach and familiarization visits by the external QA contractor to assess QA systems already in place at the BioWatch laboratories and field collection teams and identify gaps and areas for improvement and standardization. One contractor has served as the QA contractor since the inception of the program. DoD laboratories serve as the sample standards laboratory and provide support to the QA program by preparing QA and PT samples in accordance with the plans developed by the QA contractor.
The objectives of the QA program are to:
- Implement systems, practices, and procedures to ensure that field collections and laboratory analyses will consistently generate complete, accurate, and defensible results;
- Monitor the BioWatch system to verify that the methods and assays are being implemented and are performing as intended;
- Build QA data sets that provide insight into system performance and increase local public health community confidence in operational results;
- Rapidly identify system or assay performance issues;
- Perform root-cause investigation into issues and implement corrective actions to resolve issues; and
- Foster collaboration between the program and local stakeholders.
Key elements of the BioWatch QA program include the following:
- Quality Assurance Program Plans (QAPPs). QAPPs have been developed and are maintained for both the analytical and field collection components of the BioWatch programs. These QAPPs describe the QA program elements and requirements as they pertain to the analytical and collection activities, respectively.
- Laboratory and Field Audits. Independent external audits of each field team and each laboratory are performed at least every 2 years. The audit objectives are to assess compliance with technical and quality system program requirements and to identify best practices and areas for improvement.
- External QA Samples. BioWatch laboratories are challenged daily with single-blind QA samples, prepared by the SSL by spiking either inactivated agents or plasmid constructs containing the signatures of interest onto jurisdiction-specific archived quarter filters (i.e., quarter filters for which another quarter has been analyzed by the laboratory and determined to be free of agent). These samples are analyzed following the same protocols as operational samples, except that the laboratories are not required to follow the decision algorithm or make a final call regarding the presence or absence of an agent; rather, the focus is on the assay level results. External QA samples challenge analytical performance on an ongoing basis and generate data to characterize performance, identify issues (laboratory and program wide), understand contributors to variability, and support statements of statistical confidence.
- Proficiency Tests. All laboratories participate in proficiency tests (PTs) three times annually. PTs are conducted by an ISO/IEC 17043 accredited PT provider, with PT samples prepared by the SSL. PT samples are prepared on a half filter using a common background for the entire program and laboratories are required to follow the protocols applied to operational samples, including the determination verification analyses to run based on the screening results, and the application of the decision algorithm to make a final call regarding the presence or absence of an agent. By focusing on decision-level results, PTs serve as a complement to the QA samples.
- QA Performance Reviews and Reports. The QA contractor manages all performance data and reviews QA results on an ongoing basis, providing routine feedback to DHS and the laboratories. Feedback includes weekly detailed reports to DHS and the laboratories, monthly performance snapshots to laboratory directors and laboratory leads, and semiannual reports of overall program performance, trends, changes, lessons learned, and recommendations. In addition, the QA contractor reviews Exception Report data (reports provided by the field teams to DHS via an online tool regarding maintenance, downtime, late deliver
ies, or any other unusual event associated with the collection) and provides performance summaries to DHS on at least a semiannual basis.
Since 2011, the BioWatch QA program has conducted more than 95 laboratory and 75 field audits, has tested more than 45,000 QA samples, and has conducted 16 program-wide PTs.
Quality Assurance Program Plans
Quality assurance program plans have been developed for both laboratory and field operations. The Laboratory Operations QAPP describes the details and requirements associated with the quality management systems, including document control and records management, QA associated with items and services, inventory management, corrective action and improvement, and audits and inspections. The QAPPs also describe QA requirements associated with technical systems, including training and qualification requirements; requirements related to accommodations and environmental conditions (e.g., facilities requirements, segregation of work areas and equipment, access control, environmental monitoring; contamination control, monitoring, and response; decontamination; and equipment requirements); traceability requirements; QA considerations and requirements associated with collection and analytical procedures; data review and reporting procedures; performance assessment (including quality control samples, external QA samples, proficiency tests, and metrics associate with performance assessment data); implementations of new methods, assays, and platforms; and QA reports to the DHS BioWatch program. Similar requirements are outlined in the Field Operations QAPP.
External Audits
External program audits (inspections) are conducted by the BioWatch QA contractor for each operational laboratory and field collection jurisdiction approximately once every 2 years. Additional (e.g., unannounced) external program audits may also be conducted, as required. The external BioWatch program audit addresses compliance with the applicable QAPP, established BioWatch protocols, and internal (local) SOPs specific to the BioWatch program, as well as generally accepted good practices.
All external audits performed by the BioWatch QA contractor are performed by a team of two auditors, both of whom are familiar with the BioWatch program and knowledgeable of standard auditing techniques, and at least one of whom is a current American Society for Quality Certified Quality Auditor or
equivalent.1 External program audits of the laboratories generally last 2 full days, followed by an exit briefing on the morning of the third day. Each laboratory audit begins with a formal entrance briefing, and audit activities typically include
- Reviewing documents and records, including, but not limited to, equipment use, maintenance, and repair records; temperature monitoring logs; control charts and performance data; personnel training records; internal QA records (e.g., audit records, corrective action documentation); reagent acceptance testing data and reagent preparation records; and operational sample data-related records (e.g., analytical results, instrument data files, bench logbooks, and sample analysis sheets);
- Witnessing as many in-progress laboratory or collection activities as possible;
- Conducting interviews with management and staff;
- Making note of commendable observations, deficiencies, and recommendations for continuous improvement; and
- Following up on the status of deficiencies and observations from previous audits (as applicable).
Audit deficiencies are defined as discrepancies between laboratory practices and requirements (as defined in the BioWatch program QAPP, BioWatch or LRN protocols and other program documents, and/or laboratory SOPs) or generally accepted good laboratory practices. If the auditors determine that a deficiency exists, they document the pertinent details of each deficiency, including the specific requirement that is not being met, a description of the deficiency, and the objective evidence of the deficiency. Deficiencies are classified as either critical or noncritical. Critical deficiencies include issues that will prevent the laboratory from achieving program quality goals or compromise the analytical data. If a critical deficiency is identified, it is immediately communicated to the DHS BioWatch program and the affected laboratory. Corrective action is required to be implemented for all deficiencies identified during the audit.
Observations for continuous improvement are observations made during the audit that are not linked to specific requirements or generally accepted good laboratory practices, but may improve the laboratory’s overall quality systems and practice. Laboratory action is not required for these observations, but is encouraged. In addition, the auditors may encounter discrepancies during the audit that are indicative of a program-level issue (e.g., unclear language in protocols).
___________________
1 Equivalent training or certifications may include examples such as ISO 9001 lead auditor training, NQA-1 auditor training, or Society for Quality Assurance Registered Quality Assurance Professional certification.
These discrepancies are noted as program-level observations at the exit briefing and in the audit report.
During the exit briefing, auditors discuss the audit findings in detail and obtain clarification from the laboratory, as needed. Edits or changes that clarify or correct the preliminary audit findings are made at this time. If the laboratory offers new evidence or clarification during the exit briefing that satisfactorily refutes a documented deficiency, the auditors may remove that deficiency from the summary of findings or reclassify it as an observation, if appropriate.
Following the audit, auditors prepare the draft audit report and submit it to the DHS BioWatch program for review within 2 weeks of the exit briefing. This report contains details related to deficiencies, commendable practices, observations for continuous improvement, and program-level observations identified during the audit. In addition, an assessment of the effectiveness of implemented corrective action for prior audit findings is included. The DHS BioWatch Deputy Program Manager for Quality Assurance reviews and approves (after modification, if necessary) the audit report and sends the report to laboratory management (i.e., the Laboratory Director, BioWatch Lead Biological Scientist, BioWatch QA Lead, and others as requested).
Each laboratory is required to submit a corrective action report (CAR) for all audit deficiencies. The laboratory is also requested to provide a response to observations for improvement. The CARs and observation responses must be provided to the DHS BioWatch program and QA contractor within 30 calendar days of receiving the audit report.
After the laboratory responds to the audit report, the QA contractor reviews and provides DHS with a recommendation for approval or revision. The recommendation may be to accept the corrective action as proposed, accept the corrective action with modifications, or reject the proposed corrective action. The DHS BioWatch program approves or revises the recommendations, as appropriate, and submits them to the laboratory or field team in a memorandum. If program-level observations were identified, the DHS BioWatch program is responsible for determining whether protocol modifications or other actions are necessary.
In general, the follow-up on corrective actions and responses to external program audits occurs during the next external program audit. However, if a laboratory’s response to a deficiency is considered to be unacceptable by the BioWatch program manager or if a deficiency is found to recur during follow-up audits, a phased approach is implemented to bring the laboratory into compliance. The specific approach is evaluated on a case-by-case basis. If an operational laboratory is nonresponsive to the audit report after attempts by the DHS BioWatch program to resolve any issues, the DHS BioWatch program may temporarily suspend operational laboratory operations at that site and ship samples to an alternate operational laboratory for analysis, while working with the operational laboratory to bring them into compliance. In addition, for deficiencies determined to be critical, an immediate shutdown will be considered by the DHS BioWatch program until the laboratory has shown that the discrepancy has been
corrected. For all cases, the BioWatch Deputy Program Manager for Quality Assurance discusses the laboratory situation with the DHS BioWatch program manager in real time following discussion with the DHS Director of Laboratory Operations to determine if a laboratory shutdown is necessary.
A similar audit process is followed for external audits of the field teams.
Performance Assessment Samples
There are three primary categories of performance samples used for the program: internal quality controls (QCs), external QA samples, and PT samples. Within each of these categories, there are two types of samples: (1) positive samples and controls which have been prepared with a DNA template or organism at a specified level, with the expected response of a “reactive” result, and (2) negative samples and controls (e.g., NTCs) which have not been spiked with DNA template or organism, with the expected response of a nonreactive analytical result (“undetermined”). Required QC samples are described in the BioWatch program analytical program protocols. QA samples and PTs are planned, developed, and provided to the laboratories by the QA contractor and SSL, under the direction and guidance of the DHS BioWatch program. Details associated with QA and PT samples are provided below.
QA Samples
External QA samples are prepared by the SSL in accordance with instructions provided by the QA contractor and procedures described in the SSL sample preparation SOPs. External QA samples may be either spikes or blanks, and spikes generally include one or more materials specific to the agent(s) of interest, all of which are provided to the SSL by DoD laboratories or the CDC. From 2011 to 2015, external QA samples were prepared using inactivated agents produced at DoD facilities. However, due to production issues, these materials were recalled and have become unavailable for use in the program. In the fall of 2015, the program began exploring the use of positive plasmid controls (PCP) produced by DoD labs and LRN (i.e., materials used for QC testing) and fully implemented use of these materials for QA testing in January 2016. However, after several months of QA sample data were accumulated, it became apparent through higher-than-desired false negative rates that the PCP provided by DoD labs (screening and FT subspeciation assays) was not a suitable spiking material due to its linearized format. In January 2017, the QA program transitioned to the use of Escherichia coli transformed plasmid construct material to challenge the assays provided by the DoD labs. The LRN PCP material continues to be used in addition to the plasmid constructs to challenge the LRN verification assays. DoD laboratories recently released a set of inactivated surrogates that the program plans to test and consider for use in future QA and PT samples.
The spike level used for a given material is the lowest spike level for which all assays associated with the spike material are able to be consistently detected across program laboratories. The spike levels are initially determined through range-finding studies at the SSL but are refined over time in coordination with DHS based on BioWatch laboratory results. For purposes of the spike level determination, “consistently detected across program laboratories” means mean that, over time, most laboratories, when adhering to program protocols, will be able to achieve detection rates of at least 95 percent with 95 percent statistical confidence. This is often referred to as the estimated thresholds of probable detection (TPD). The objective is to achieve spike levels that are high enough that, provided all protocols are followed correctly, all assays will be consistently detected, but low enough such that false negatives or elevated Ct values will be observed if a laboratory encounters issues associated with adherence to the protocols, background, extractions, or other elements of the analytical method. Spike level is not the only contributor to false negatives (FNs); other factors include procedural errors, background matrix effects, and assay issues. Ultimately, QA samples are intended to provide information about detection issues that could impact the detection of agent assays in operational samples. However, it should be noted that because some materials are used to test multiple assays and because a single spike level is applied for the entire program, it is possible that the spike level for one or more assays for one or more laboratories may result in a detection rate that is consistently higher than 95 percent.
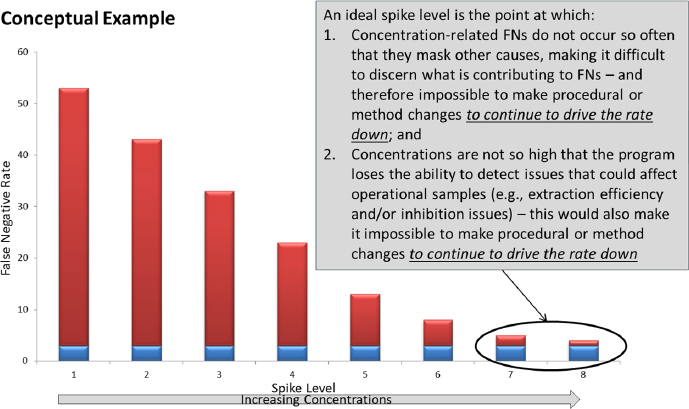
Figure F-1 provides a conceptual example of how the false-negative rate may be used to determine the optimal spike level (i.e., a spike level that is high enough such that the material is consistently detectable when the protocols are followed and reagents and materials meet the established quality requirements). In the example provided, the x axis represents a nominal spike level that increases to the right, and the y axis represents the false-negative rate at the associated spike level. The red portion of the bars represents false negatives due to the spike material concentration (e.g., extraction efficiency, low copy counts, inhibition, etc.) and the blue portion of the bars represents false negatives due to other contributors, such as analyst error. As shown in the chart, as the spike level increases, the number of false negatives due to the material concentration decreases, but the number of false negatives due to other contributors remains the same. In this example, levels 7 and 8 are considered optimal levels because they represent the level at which concentration-related false negatives do not occur so often that they mask other causes, making it difficult to discern what is contributing the errors, and the concentrations are not so high that the program loses the ability to detect issues that could affect operational samples (e.g., extraction efficiency and/or inhibition issues), both of which would make it impossible to make procedural or method changes to continue to drive the rate down.
Sample matrices are quarter filters that contain environmental backgrounds specific to each laboratory and are obtained from the archive filter inventory maintained by the SSL. For laboratories that pool samples, QA sample sets are provided (i.e., for laboratories that pool operational samples 3:1, one spike and two blank QA samples are provided to the laboratory as a set to be pooled together for screening analysis). In order to prepare the spiked QA samples, the SSL first prepares and verifies a cocktail solution that contains one or more spiking materials at a specified level, as well as QA Tag material. The purpose of the QA Tag material is to enable the laboratory to determine if detections in operational samples may be attributable to contamination from QA samples. Once the cocktail has been verified to have been prepared correctly, the cocktail is added to a quarter filter within a Petri dish. Blank QA samples are created by adding a quarter filter containing laboratory-specific background to a Petri dish. As each sample (spike or blank) is prepared, the Petri dishes are Parafilmed. Once all samples designated in a spiking scheme are prepared, they are shipped to each individual laboratory for incorporation into the daily operational analysis.
On a predetermined and documented schedule approved by DHS, external QA samples2 are analyzed by BioWatch operational laboratories (every laboratory and each shift) in accordance with the Quality Assurance Samples: Processing, Analysis, and Handling Instructions protocol. QA samples are processed and analyzed alongside (i.e., on the same plate as) operational samples and QA sample processing is rotated through all processing BSCs so that all BSCs are equally
___________________
2 For laboratories that pool, “one external QA sample” is interpreted to mean one set of samples to be pooled.
covered with QA and negative filter samples. Depending on how the sample has been spiked, some screening targets are expected to be nonreactive and others are expected to be reactive. External QA samples are not automatically carried forward to verification analyses based on the screening results. Rather, the laboratory is required to store extracted DNA refrigerated or frozen and perform verification analysis according to the provided QA Sample Analysis Scheme, which instructs the laboratory as to which verification analyses should be run on particular extracts. This approach allows the QA program to collect both false-negative and false-positive data for all assays, with a heavy emphasis on the screening assays, since those assays represent a decision gate for subsequent analyses.
Screening and verification results for all QA samples are reported into the BioWatch Laboratory Data Entry (BLaDE) tool, an online interface through which laboratories provide QA data to the QA contractor.
Proficiency Tests
Proficiency tests are performed routinely at each BioWatch operational laboratory. The QA contractor and the SSL are responsible for working together to provide up to three PTs per year for each laboratory participating in the DHS BioWatch QA program. The QA contractor is an ISO/IEC 17043:2010 accredited Proficiency Test Provider and is responsible for developing the PT design, overseeing the implementation of the PT, and reviewing and reporting all results in accordance with the ISO/IEC 17043:2010 requirements. The SSL prepares and distributes the PT test items.
Proficiency testing serves multiple objectives for the DHS BioWatch QA program. First, PTs provide information about the potential for decision-level (or final-call) errors, which serve as a complement to routine external QA samples and internal QC testing. Proficiency testing is also commonly recognized as a key element in a comprehensive QA program and helps to strengthen the overall defensibility of results from laboratories participating in the DHS BioWatch QA program.
Each proficiency test currently consists of a panel of six sample sets per operational laboratory, with identical panels sent to each laboratory (but with a different sample ID order). The samples are half filters, some of which are spiked with one or more spiking materials, and some of which are not spiked. To ensure that all laboratories receive identical panels, the SSL creates a single “pooled dirty buffer” from the archive filter inventory that represents background across all BioWatch jurisdictions. Once the pooled dirty buffer has been verified to be free of program signatures, the buffer is added to clean filters and allowed to dry. These filters serve as the dirty filters to be used to create “dirty” PT samples, which are then prepared in a manner similar to PT samples (i.e., a cocktail is prepared and verified prior to use in spiking samples and blank samples are also provided).
The laboratories are responsible for analyzing and reporting all PT samples within the specified time window. The analysis dates for each PT sample
are not specified. However, once the analysis for an individual PT sample has begun, the results must be reported as soon as possible after analysis, and within the timeframe typically allowed for operational samples. PT samples are analyzed like operational samples (i.e., the PT sample is carried through the full verification process to make a final determination within one shift). The laboratories report PT results via the BLaDE interface.
The QA contractor is reviews and evaluates the test results and provides a report to the DHS BioWatch program and the laboratories within 30 days after receipt of all sample results. This report contains a score of “pass” or “fail” for each laboratory in the areas of analytical accuracy and reporting accuracy and timeliness. The analytical accuracy score is based on the correctness of the final results and correct application of the final call algorithm. The reporting accuracy and timeliness score is based on reporting results accurately and within the specified turnaround time.
Unless otherwise specified by DHS, PT failures require the laboratory to develop and submit a CAR. In addition, in some cases, other issues or questions observed during PTs may prompt DHS to request additional responses and corrective actions. If a laboratory’s response to a proficiency test failure is considered to be unacceptable by the BioWatch program manager or if a laboratory receives two or more failures for consecutive proficiency tests, a phased approach may be implemented to bring the laboratory into compliance. The specific approach is evaluated on a case-by-case basis. If an operational laboratory is nonresponsive or continues to observe proficiency test failures after attempts by the DHS BioWatch program to resolve any issues, the DHS BioWatch program may temporarily suspend operational laboratory operations at that site and ship samples to an alternate operational laboratory for analysis, while working with the operational laboratory to bring them into compliance.
Metrics and Data Quality Expectations and Goals for QA and PT Samples
The key laboratory analysis performance parameters, or metrics, include assay-level error rates and decision-level error rates. Because the BioWatch program generates qualitative PCR data (i.e., no standard curve is included), accuracy and precision of Ct values are reviewed for information and trend purposes only. A definition for each of these key metrics is provided below, as well as an explanation of how the expectations and goals are implemented for the operational laboratories.
Assay-Level Errors
There are two types of assay-level errors: assay-level false positives and assay-level false negatives. Assay-level errors are evaluated using the results from external QA samples or PT samples. Assay-level errors are typically reviewed separately by individual assay (nucleic acid signature).
An assay-level false positive is an individual analytical result for a given nucleic acid signature that meets all SOP-defined criteria for a reactive in a sample that has not been spiked with the associated material.
An assay-level false negative is an individual analytical result for a given nucleic acid signature that does not meet all SOP-defined criteria for a reactive in a sample that has been spiked with the related spiking material. For assay-level false negatives, the level (theoretical) at which the material was spiked must be specified. Reviewing assay-level false negatives provides information about assay-level sensitivity (the likelihood of detecting an agent that is present in a sample at a given level).
Decision-Level Errors
Similar to assay-level errors, decision-level, or BAR, errors consist of two types: decision-level false positives and decision-level false negatives. Decision-level errors are typically evaluated using PT samples. Decision-level errors are computed separately by organism, and for false-negative rates, the level (theoretical) at which the material was spiked must be specified.
A decision-level false positive is a series of SOP-defined analytical results that together meet the operational criteria for a verified positive result, or BAR, for a sample that has not been spiked with materials that are expected to be reactive for the given agent. Decision-level false-positive rates are computed as the proportion of test samples that have not been spiked with an inactivated agent and/or other spiking material in which decision-level false positives are observed. Evaluation of decision-level false positives provides information about decision-level selectivity and specificity (defined in this context as the likelihood of obtaining a correct “negative,” “nonreactive,” or “undetermined” result for a sample that does not contain an agent).
A decision-level false negative is a series of SOP-defined analytical results that together do not meet the criteria for a verified positive result, or BAR, associated with a sample that contains materials specific to a given agent. Decision-level false-negative rates are computed as the proportion of test samples that have been spiked with an inactivated agent and/or other spiking material in which decision-level false negatives are observed. Evaluation of decision-level false negatives provides information about decision-level sensitivity.
Error Rate Calculations
Error rates are computed as described below. Note that the descriptions below do not provide an exhaustive description of every possible calculation that may be considered when evaluating program performance. Rather, this section describes the error rate metrics most often considered.
Error Rates by Individual Laboratory and Assay
- The laboratory/assay false-negative rate is computed as the number of assay-level false negatives for each individual assay at each individual laboratory, divided by the number of QA or PT samples analyzed by laboratory that were spiked with materials (at a designated spike level) that are expected to be reactive for the assay.
- The laboratory/assay false-positive rate is computed as the number of assay-level false positives for each individual assay at each individual laboratory, divided by the number of QA or PT samples analyzed by laboratory that were not spiked with materials that are expected to be reactive for the assay.
Error Rates by Individual Assay
- The assay-specific program-wide assay-level false-negative rate is computed as the number of assay-level false negatives for each individual assay across all laboratories, divided by the number of QA or PT samples analyzed by all laboratories that were spiked with materials (at a designated spike level) that are expected to be reactive for the assay.
- The assay-specific program-wide assay-level false-positive rate is computed as the number of assay-level false positives for each individual assay across all laboratories, divided by the number of QA or PT samples analyzed by all laboratories that were not spiked with materials that are expected to be reactive for the assay.
Error Rates by Individual Agent
Error rates by agent are typically only considered at the decision level and typically are based on PT data only.
- The agent-specific program-wide decision-level false-negative rate is computed as the number of decision-level false negatives for each individual agent across all laboratories, divided by the number of PT samples analyzed by all laboratories that were spiked with materials (at a designated spike level) that are expected to result in a final-call positive for the given agent.
- The agent-specific program-wide decision-level false-positive rate is computed as the number of decision-level false positives for each individual agent across all laboratories, divided by the number of PT samples analyzed by all laboratories that were not spiked with materials that are expected to result in a final-call positive for the given agent.
Program Goals and Expectations Associated with Error Rates
For the BioWatch program, a 2 percent false-positive rate and a 5 percent false-negative rate at the appropriate spike level have been set as the benchmarks for evaluation. These benchmarks are used as follows:
- The program minimum expectation is that the error rates are not statistically significantly greater than the benchmark. In other words, false-positive rates should not be statistically significantly greater than 2 percent, and false-negative rates should not be statistically significantly greater than 5 percent.
- The program goal is that the error rates are statistically significantly less than the benchmark. In other words, false-positive rates should be statistically significantly less than 2 percent, and false-negative rates should be significantly less than 5 percent.
Note that the goals and expectations for false-negative rates only apply to samples that are spiked at the appropriate spike level.
These expectations and goals apply to
- All assay-specific program-wide assay-level false-negative and false-positive rates,
- All agent-specific program-wide decision-level false-negative and false-positive rates,
- All laboratory-specific sample-level false-negative and false-positive rates, and
- All laboratory-specific decision-level false-negative and false-positive rates.
Expectations and goals are assessed using statistical confidence limits. Specifically, for each observed error rate, a 90 percent confidence interval is calculated. The 90 percent confidence interval describes the uncertainty in the true, long-term error rate and is influenced by the sample size used to compute the rate. The lower bound of the 90 percent confidence interval is the 95 percent lower confidence limit (LCL) and the upper bound is the 95 percent upper confidence limit (UCL).3 In order to meet the program expectation (false-positive or false-negative rate is not statistically significantly greater than 2 percent or 5 percent), the 95 percent LCL must not exceed 2 percent or 5 percent, respective-
___________________
3 Together, a 95 percent LCL and a 95 percent UCL comprise a 90 percent confidence interval. There is only a 5 percent chance that the true, long-term rate is below the 95 percent LCL, and only a 5 percent chance that the true, long-term rate exceeds the 95 percent UCL. Thus, there is 90 percent confidence that the true, long-term rate falls between the 95 percent LCL and the 95 percent UCL. Hence, the two limits form a 90 percent confidence interval.
ly. In order to meet the program goal (false-positive or false-negative rate is statistically significantly less than 2 percent or 5 percent), the 95 percent UCL should be less than 2 percent or 5 percent, respectively.
- The program expectations are applied as follows: In any proficiency test, if the program expectations are not met (i.e., if the 95 percent LCL for the false-positive error rate exceeds 2 percent or if the 95 percent LCL for the false-negative error rate exceeds 5 percent), the test will generally be classified as a failure.
- Note that for false-negative rates, the pass/fail evaluation typically only applies to samples that are spiked at or above the established TPD.
- For each operational laboratory, the cumulative data compiled in weekly QA summaries should result in the program expectations being met for all laboratory-specific sample-level false-negative and false-positive rates.
- If the expectation is not met, root-cause investigations must be undertaken by the laboratory (with support from DHS and the QA contractor, as needed), and corrective actions should be implemented to correct the identified root causes.
Metrics Related to Ct Values (Accuracy and Precision)
Accuracy refers to the nearness of a result to the true or expected value and is often measured in terms of percent recovery. Because the analytical methods for the BioWatch program do not result in an estimated concentration, analytical data are not evaluated in terms of percent recovery. Rather, the focus of accuracy is on the closeness of the average Ct value for samples that have been spiked with a given agent and spike level to the expected Ct-value range for that agent and spike level based on historical responses observed at each individual laboratory. Accuracy is evaluated separately by spiking material or nucleic acid signature.
Precision is the reproducibility of Ct values. Precision is evaluated based on the standard deviation of Ct values for samples that have been spiked with a given agent and spike level. Precision is evaluated separately by spiking material or nucleic acid signature.
Accuracy and precision of Ct values are relevant metrics because they help investigators identify trends over time, contributors to variability, and/or differences between laboratories, instruments, and reagent lots. However, because the primary focus of the BioWatch program is the ability to detect agents when they are present and to avoid false alarms when agents are not present, the evaluation of accuracy and precision is secondary to the evaluation of error rates. When the expected results with respect to analytical accuracy and precision are not met, or when systematic differences or patterns are observed (e.g., differences across instruments, reagent lot, or laboratories), the program may initiate an investigation to understand the results and the potential impact on the
ability to make correct presence/absence decisions. The extent of the investigation is tailored to the potential impact of the anomaly or pattern.
QA Data Review and Reporting
The QA contractor is responsible for obtaining key program data (including operational, QA, and PT data) from the operational laboratories and the SSL and compiling the data into the QA database, which is used to support QA-related data evaluations, summaries, and investigations. The QA database is structured to ensure that data are complete and accurate and are maintained, secured, and archived in an environment that provides appropriate levels of access to the QA contractor project team members.
QA reports to the DHS Program Office are summarized in Table F-1 below.
TABLE F-1 QA Reports to DHS BioWatch Program
| Report | Description/Frequency | Critical Elements | Interpretation |
|---|---|---|---|
| Ongoing Data Assessments | The QA contractor is responsible for reviewing external QA sample results and operational sample results (i.e., reactive rates), as well as the SSL QC data, on an ongoing basis. Updated trend charts for QA and operational data are posted to the BioWatch Portal every week. Additionally, a program-wide summary of QA performance is provided to DHS weekly, and a summary of false-positive/false-negative (FP/FN) rates in QA samples is provided monthly to the Laboratory Director and BioWatch Laboratory Lead. |
For each laboratory and assay:
|
Contributes to the evaluation of the performance capabilities and trends by laboratory, instrument, and assay. Focus is on false-positive and false-negative rates. Combined with other troubleshooting results, determines possible source of errors and identifies areas for potential improvement. |
| External Audit Reports | The auditor must provide an audit report to the DHS BioWatch program no later than 14 days following the completion of the audit. External audits are performed every 2 years for the operational laboratories, annually for the SSL, and as directed by DHS for the reagent production laboratories. |
For each external audit conducted:
|
Provides an assessment of the level to which operational laboratories adhere to the BioWatch protocols, BioWatch QAPP, and other program documents and laboratory SOPs. |
| Report | Description/Frequency | Critical Elements | Interpretation |
|---|---|---|---|
| Proficiency Test Reports | The QA contractor must provide a PT report to the DHS BioWatch program and the operational laboratories no later than 28 days after the completion of the proficiency test. Three BioWatch PTs are performed annually. |
For each proficiency test conducted:
|
Provides a periodic assessment of inter- and intralaboratory performance with respect to the potential for decision-level errors. |
| Trends Analysis Report | The QA contractor performs a comprehensive review of operational laboratory performance at least semiannually. This includes a review of external audits, summary reports from laboratories (if received), PT results, any trends or patterns observed in external QA sample results, and reported issues. |
Each report includes
|
Provides a routine summary of program laboratory performance and provides the basis for identifying and addressing program-level laboratory issues and opportunities for continuous improvement of program laboratory performance. |


















