
C
Analysis of Legal and Regulatory Landscape Relevant to Return of Individual Results Generated from Biospecimens in Research1
III. State Laws Equivalent to CLIA
A. Overview of Legal Obligations Related to Access
B. Potential Conflicts Between CLIA and HIPAA
___________________
1 A white paper commissioned by the National Academies of Sciences, Engineering, and Medicine’s Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories, written by Christi J. Guerrini, J.D., M.P.H., Baylor College of Medicine.
EXECUTIVE SUMMARY
This memorandum describes the U.S. legal and regulatory landscape relevant to the return of individual results generated from biospecimens in research.
The Clinical Laboratory Improvement Amendments of 1988 (CLIA) requires certification (or waiver of certification) of all laboratories, which are defined as facilities where human specimens are examined for the purpose of providing information for diagnosis, prevention, treatment, or health assessment. However, research laboratories need not become certified so long as they do not report results to tested individuals, their physicians, or researchers, where the results could be used for diagnostic, preventative, treatment, or health assessment purposes.
Since 2000, the Health Insurance Portability and Accountability Act (HIPAA) has provided individuals with a right of access to inspect and obtain a copy of their protected health information that is contained within a designated record set, which is defined broadly as any record used by covered entities to make any kind of decision about individuals. So defined, the designated record set may include laboratory test reports and related information. Research laboratories are HIPAA-covered entities if they electronically conduct at least one billing-related transaction or function as part of a larger covered entity, such as a hospital or academic medical center.
Before 2014, all laboratories, including research laboratories, were not only exempted by HIPAA from compliance with the right of access, but also were prohibited by CLIA from returning laboratory test results directly to tested individuals unless explicitly authorized to do so by state law. In 2014, however, both CLIA and HIPAA were amended to require all HIPAA-covered laboratories, including HIPAA-covered research laboratories, to comply with the right of access.
It is generally recognized that these amendments have created a dilemma for research laboratories that are covered by HIPAA but not certified by CLIA. To comply with the expanded access rules, these laboratories must now return test reports and related information contained within designated record sets when individuals request them to do so, but research laboratories cannot do so without becoming CLIA-certified. Yet, CLIA certification is time consuming and expensive, and it may be unrealistic to require all research laboratories to become CLIA-certified in order to comply with HIPAA. Some institutions have responded to this dilemma by adopting policies that interpret the designated record set to exclude some research-related information or by making case-by-case determinations to return certain research results even if generated by laboratories that are not CLIA-certified.
The return of research results is also relevant to regulations for the protection of research participants. These include the Federal Policy for the Protection of Human Subjects (also known as the Common Rule) and relevant regulations adopted by the Food and Drug Administration (FDA). Neither set of regulations explicitly allows or prohibits the return of results to study participants. However, they both require that, where appropriate, research participants be informed of significant findings that may relate to participants’ willingness to continue participation. Moreover, pending revisions to the Common Rule will require that plans to return results be provided as an element of informed consent in some circumstances. In practice, when a study protocol includes a plan to return results, an institutional review board (IRB) will review the plan to ensure its benefits outweigh its risks. While IRBs can prohibit investigators from
returning results, however, they cannot block access when study participants request results under HIPAA.
The return of individual research results is relevant to other FDA regulations related to the agency’s responsibility to protect and promote public health by ensuring the safety and effectiveness of medical drugs and devices, which include laboratory tests. First, the return and subsequent use of results generated by laboratory-developed tests (LDTs) have factored into FDA interest in abandoning its policy of enforcement discretion of LDTs. The return of results directly to consumers also has played a role in FDA regulation of specific genetic tests. Further, FDA regulation of investigational devices, including laboratory tests, depends in part on whether and how results from such devices will be returned. Finally, the communication of interpreted research results in some cases may constitute prohibited promotion of devices.
In general, state courts have not viewed research results, including data generated from genetic tests, as legal property belonging to research participants. However, in the context of genetics, some states, including Colorado and Alaska, have enacted statutes that explicitly recognize property rights of individuals in their test results. Individuals can also privately agree to allocate rights in test results that are different from default legal rules.
The return of research results may give rise to tort liability under state law for researchers and laboratories. Tort liability associated with the return of research results can generally be categorized as non-disclosure liability or disclosure liability; the most probable cause of action for both is negligence. In general, individuals owe a duty of reasonable care under the circumstances, but tort law imposes no affirmative duties to act for another’s benefit and individuals are not required to warn others of impending harm. A number of factors can overcome this general tort law notion that individuals do not owe others affirmative duties, however, including the existence of a fiduciary relationship or other “special relationship,” as well as contractual obligations. While physicians are held to be fiduciaries of their patients, researchers are generally not viewed as fiduciaries of their research participants. Nevertheless, in some cases, researchers have been held to have a “special relationship” with their research participants giving rise to affirmative duties. Whether those duties include the return of certain test results depends on the prevailing standard of care.
Researchers who return results must do so consistent with the standard of care and regulatory requirements. Many kinds of actions associated with the return of research results may give rise to tort liability, including disclosure of incorrect results as a result of, e.g., improper test administration. Meanwhile, disclosure of results to individuals who are not authorized to receive them may give rise to negligence claims where, among other things, the tested individual suffered discrimination as a result.
There is a complex web of federal and state laws that address unwanted access to and discriminatory use of health information. Two major federal statutes are the Genetic Information Nondiscrimination Act (GINA), which limits access to and use of genetic information in health insurance and employment contexts, and the Americans with Disabilities Act (ADA), which limits discrimination against individuals with disabilities in employment, public services, and public accommodations contexts. However, GINA and the ADA do not preempt state laws that provide equal or greater protection, and over the years, many state anti-discrimination statutes have been enacted that vary widely in scope and applicability. The majority of states have enacted laws that regulate employment and/or insurance discrimination based upon genetic test results or genetic status, and some also regulate genetic discrimination by life, disability, or long-term care insurers.
INTRODUCTION
I. Background
A. Legal Landscape
This memorandum describes the U.S. legal and regulatory landscape relevant to the return of individual results generated from biospecimens in research. Black’s Law Dictionary defines “law” broadly as “[t]he body of authoritative grounds of judicial and administrative action.”1 The legal landscape of a particular issue therefore encompasses the collective legal rules and practices that are followed when deciding controversies relevant to that issue.
The legal landscape consists of: federal and state constitutions (constitutional law); federal and state statutes (statutory law); federal and state regulations and administrative practices (administrative law); and laws and principles derived from federal and state judicial decisions (common law).
B. Regulatory Landscape
The legal landscape relevant to a particular issue necessarily includes its regulatory landscape. The regulatory landscape refers to the regulations adopted and practices followed by administrative agencies, such as the Department of Health and Human Services (HHS).
Within the regulatory landscape, agency action can be classified as rulemaking or adjudication.2 Focusing on rulemaking, many agencies are authorized to issue what are known as legislative rules that grant legal rights to or impose legally binding obligations on regulated parties.3 Legislative rules must be issued in accordance with notice-and-comment procedures.4 Examples of legislative rules include regulations implementing the CLIA and the Health Insurance Portability and Accountability Act (HIPAA).
Final legislative rules are codified in the Code of Federal Regulations.5 They are also published in the Federal Register and are typically preceded by a preamble that describes the regulatory changes taking effect.6 Although a preamble cannot control the meaning of a regulation and so does not itself have the force of law,7 courts have recognized that a preamble may serve as evidence of “contemporaneous agency intent” regarding the meaning and operation of the regulation.8
___________________
1 BLACK’S LAW DICTIONARY (10th ed. 2014) (“law”).
2 CHARLES H. KOCH, JR. & RICHARD MURPHY, 3 ADMIN. L. & PRAC. § 2.10 (3d ed. 2017).
3 JAMES T. O’REILLY, ADMINISTRATIVE RULEMAKING § 2:3 (2017 ed.).
4See KOCH & MURPHY, supra note 2, at § 4.10.
5See id. at § 1:21.
6See O’REILLY, supra note 3, at §§ 10.1, 12.1.
7See id. § 10.2. However, an agency’s own procedural rules may give a Federal Register preamble more authority. See, e.g., 21 C.F.R. § 10.85(d)-(e) (2017) (providing that a Federal Register preamble to a final Food and Drug Administration rule constitutes an advisory opinion that FDA is obligated to follow until it is amended or revoked).
8 Wyo. Outdoor Council v. U.S. Forest Serv., 165 F.3d 43, 53 (D.C. Cir. 1999) (“Although the preamble does not ‘control’ the meaning of the regulation, it may serve as a source of evidence concerning contemporaneous agency intent.”); see also City of Las Vegas, Nev. v. Fed. Aviation Admin. 570 F.3d 1109, 1117 (9th Cir. 2009) (“When a regulation is ambiguous, we consult the preamble of the final rule as evidence of context or intent of the agency promulgating the regulations.”).
In addition to legislative rules, agencies may adopt procedural rules directed at organizing and improving their operations and interpretive rules that interpret a statute or another rule.9 Because both procedural and interpretive rules do not create new duties, rights, or obligations, they may be issued without following notice-and-comment procedures.10 Finally, and similar to interpretive rules, general policy statements (sometimes set forth in or labeled as guidance documents, guidelines, or manuals) are announcements to advise the public prospectively of the manner in which an agency proposes to exercise its discretionary powers.11 Like an interpretive rule, a general policy statement does not purport to establish a binding norm and so does not have the force of law.12 Nevertheless, courts hold that it is prudent to give deference to interpretive rules and policy statements.13
C. Legal Hierarchy
The U.S. legal system functions as a hierarchy that dictates how different categories of law rank in authority. The U.S. Constitution is the supreme law of the land.14 Because no federal or state law may contradict it, federal constitutional law represents the highest legal authority.15 Second in rank is federal statutory law, which is enacted by Congress and must be followed by the states, and third is federal regulations that interpret federal statutes.16 The lowest legal authority in the federal system is federal common law.17
In the event of a conflict between a federal law and state law, the federal law preempts the state law.18 However, states can generally offer greater protections than federal law, and when this occurs, there is no conflict and state law controls.19 Moreover, state laws generally can address issues that are not addressed by federal law so long as they do not violate the U.S. Constitution or the state’s constitution.20
At the state level, the highest legal authority is the state’s constitution, followed by state statutes, state regulations, and, finally, state common law.21
D. Conceptual Distinctions
At the outset, it is important to acknowledge certain conceptual distinctions that are relevant as a legal, practical, or technical matter to this analysis. First, there is a generally
___________________
9See O’REILLY, supra note 3, at §§ 2.4-2.5.
10See id.
11See id. § 2.6.
12See id.
13See KOCH & MURPHY, supra note 2, at § 10:22.
14See 16 C.J.S. Constitutional Law § 9 (2017).
15See id.
16See 2 AM. JUR. 2D Administrative Law § 218 (2d ed. 2017).
17 Kent Greenawalt, The Rule of Recognition and the Constitution, 85 MICH. L. REV. 621, 628 (1987) (describing legal hierarchy); Michael J. Glennon, Raising the Paquete Habana: Is Violation of Customary International Law by the Executive Unconstitutional?, 80 NW. U. L. REV. 321, 334 (1985) (same).
18See 16A AM. JUR. 2D Constitutional Law § 232.
19See id. § 231.
20See id. § 11.
21See Greenawalt, supra note 17, at 628.
recognized distinction between research and clinical care.22 Research is focused on the production of generalizable knowledge, where the responsibility of researchers is to preserve the integrity of the research process.23 While researchers are obligated to minimize harms to participants, they do not have a duty to optimize participants’ health.24 By contrast, the responsibility of clinicians is to provide care directed to the best interests of patients.25
The distinction between research and clinical care is central to laws and responsibilities relevant to the conduct of research and medical practice.26 In addition, the distinction is used for practical purposes to classify, e.g., results of laboratory tests of biospecimens as research results or clinical results and laboratories that perform such tests as research or clinical laboratories.
Distinctions can also be made between the kinds of information generated by laboratory tests. These include uninterpreted raw data and interpreted findings. In the context of a genetic test, uninterpreted raw data are sequencing data, whereas an interpreted finding might be information that the test identified a genetic variant that increases one’s risk of developing a particular disease or condition.27 For the sake of simplicity, this analysis will refer to the spectrum of information generated by laboratory tests of biospecimens generally as “results” except where finer distinctions are required.
In a research context, test results may be relevant to primary study aims or they may describe incidental or additional findings.28 Research results can further be distinguished based on whether they pertain to individual research participants or are aggregated and reported as general study results,29 as well as when the results are generated in research—at baseline, while the research is in process, or at study end.30 Further, test results may be those that are originally generated (and possibly also reported), or they may be results that are later revised to correct errors or reflect new knowledge.31
Test results may be linked (or not) to research participants according to different standards. Thus, de-identified results can be linked to specific individuals but information that would identify those individuals with the results has been removed in accordance with HIPAA
___________________
22See, e.g., Susan M. Wolf, The Role of Law in the Debate Over Return of Research Results and Incidental Findings: The Challenge of Developing Law for Translational Science, 13 MINN. J. L. SCI. & TECH. 435, 443-44 (2012) (noting that the traditional architecture of health law and bioethics has “largely accepted and built upon a dichotomy between the two spheres” of research and clinical care).
23See Wylie Burke, Barbara J. Evans & Gail P. Jarvik, Return of Results: Ethical and Legal Distinctions Between Research and Clinical Care, AM. J. MED. GENETICS SEMINARS MED. GENETICS 105, 106 (2014).
24See id.
25Id.
26See Wolf, supra note 22, at 443-44. However, Prof. Wolf explains that the traditional “wall” between research and clinical care is starting to resemble a membrane as research insights increasingly move into clinical practice. Id. at 443-45. For a discussion of the relevance of the clinical care-research distinction to tort liability, see Part IX, infra.
27See Adrian Thorogood et al., APPLaUD: Success for Patients and Participants to Individual Level Uninterpreted Genomic Data, 12 HUMAN GENOMICS 7, 7-8 (2018) (distinguishing uninterpreted raw data from interpreted results); Anna Middleton et al., Potential Research Participants Support the Return of Raw Sequence Data, 52 J. MED. GENETICS 571, 571 (2015) (same).
28See Barbara J. Evans, The First Amendment Right to Speak About the Human Genome, 16 U. PA. J. CONST. L. 549, 555-56 (2014).
29See SACHRP, JULY 21, 2016 SACHRP LETTER TO THE HHS SECRETARY, ATTACHMENT B: RETURN OF INDIVIDUAL RESEARCH RESULTS (passed May 19, 2016).
30See id.
31See generally Part IX.B, infra (discussing tort liability for failing to update or correct previously disclosed results).
standards.32Non-identified results can also be linked to known individuals but identifying information has been removed in accordance with the Federal Policy for the Protection of Human Subjects (also known as the Common Rule), which prescribes standards that are different than HIPAA standards.33Re-identified results are de-identified or non-identified results whose links to known individuals have been restored.
Finally, distinctions can be made regarding to whom test results are returned. Depending on applicable laws, results can be returned to the individuals whom they describe, their relatives, or other authorized persons.34 Distinctions also can be made between returning results when the individuals to whom they pertain are alive versus deceased, as well as when the individuals, if alive, are capacitated versus incapacitated.35
REGULATORY LANDSCAPE
II. CLIA
A. Scope
The Centers for Medicare & Medicaid Services (CMS) is responsible for administering the regulatory standards governing laboratories known as CLIA.36 CLIA establishes quality standards for laboratories to ensure the accuracy, reliability, and timeliness of individual test results.
CLIA defines regulated “laboratories” as any:
[F]acility for the . . . examination of materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings.37
___________________
32See 45 C.F.R. § 164.514(b) (2017) (prescribing standards for de-identification under HIPAA).
33See id. §§ 46.101(b)(4), 102(f)(2) (defining research regulated by the Common Rule as involving “identifiable private information” and describing a regulatory exemption for research involving existing non-identified data and biospecimens); OHRP, CODED PRIVATE INFORMATION OR SPECIMENS USE IN RESEARCH, GUIDANCE (Oct. 16, 2008), https://www.hhs.gov/ohrp/regulations-and-policy/guidance/research-involving-coded-privateinformation/index.html (describing standards for non-identification by coding). The convention of referring to data and biospecimens that are not identifiable according to Common Rule standards as “non-identified” is explained in the Federal Policy for the Protection of Human Subjects: Notice of Proposed Rulemaking, 80 Fed. Reg. 53,933, 53,942-43 (Sept. 8, 2015) (“Consistent with historical interpretation of identifiable private information under the Common Rule, the terms ‘non-identified’ or ‘non-identifiable’ are used throughout this [notice] to signify biospecimens or data that have been stripped of identifiers such that an investigator cannot readily ascertain a human subject’s identity.”).
34See generally Susan M. Wolf et al., Returning a Research Participant’s Genomic Results to Relatives: Analysis and Recommendations, 43 J. L. MED. ETHICS 440 (2015).
35See id. at 453-59.
36 FDA and the Centers for Disease Control and Prevention also have responsibilities related to CLIA. See FDA, Clinical Laboratory Improvement Amendments (CLIA), FDA.GOV, https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/IVDRegulatoryAssistance/ucm124105.htm (last updated March 22, 2018).
37 42 C.F.R. § 493.2 (2018).
CLIA requires certification (or waiver of certification) of all laboratories, so defined, except “CLIA exempt” laboratories,38 which have been licensed by a state that has enacted laws relating to CMS-approved laboratory requirements “that are equal to or more stringent than CLIA requirements.”39 As discussed in Part III, infra, CMS has approved the licensure programs of Washington and New York. Licensed laboratories in these states therefore qualify as “CLIA exempt.”
CLIA further provides that its rules do not apply to “components or functions” of certain laboratories that are referred to as “exceptions.”40 For purposes of this analysis, the most important CLIA exception covers:
Research laboratories that test humans but do not report patient specific results for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of the health of individual patients.41
CMS has interpreted this provision to mean that “only those facilities performing research testing on human specimens that do not report patient-specific results may qualify to be excepted from CLIA certification.”42 If a research laboratory intends to report individual-level results, and those results “will be or could be” used to diagnose, treat, prevent, or assess human health, the laboratory must first obtain CLIA certification.43 In practice, CMS has taken the position that a research laboratory may not report individual-level research results to any person or entity, where “[t]he results are available to be used for health care for individual patients,” unless the laboratory is CLIA-certified.44 Thus, a research laboratory may not report individual-level test results to tested individuals or their clinicians unless it is CLIA-certified.45 Further, a research laboratory may not report individual-level test results to investigators where those results could be used in the treatment of research participants, which includes assignment of participants to control and treatment arms.46
___________________
38Id. § 493.3(a).
39Id. § 493.2.
40Id. § 493.3(b).
41Id. § 493.3(b)(2).
42 CMS, Research Testing and Clinical Laboratory Improvement Amendments of 1988 (CLIA) Regulations, CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/Research-Testing-and-CLIA.pdf (last updated Dec. 10, 2014) (emphasis in original).
43Id.
44See Penelope Meyers, CLIA and Research Results, Presentation to the Secretary’s Advisory Committee on Human Research Protections (Mar. 8, 2011), available athttp://wayback.archive-it.org/org745/20150824191143/http://www.hhs.gov/ohrp/sachrp/mtgings/mtg03-11/rirr_by_p_meyers.pdf [hereinafter Meyers, SACHRP Presentation]. Moreover, it is CMS’s position that research laboratories returning results cannot avoid the requirement of CLIA certification by including disclaimers that, e.g., the testing was conducted in a research setting and/or the clinical meaning of the results is unknown. Telephone communication with Penelope Meyers, Technical Director, Division of Laboratory Services, CMS (Nov. 16, 2017). Accord David H. Ledbetter & W. Andrew Faucett, Issues in Genetic Testing for Ultra-Rare Diseases: Background and Introduction, 10 GENETICS MED. 309, 310 (2008) (noting the misconception that CLIA allows research laboratories to return results that might be used to impact diagnosis, management, or decision making by patients or their physicians if they are “simply qualif[ied] with statements (verbal or written) that testing was done on a research basis”).
45 Telephone communication with Penelope Meyers, supra note 44.
46See id.; Meyers, SACHRP Presentation, supra note 44.
Table C-1 summarizes these three categories of laboratories: laboratories regulated by CLIA and requiring CLIA certification; “CLIA-exempt” laboratories; and research laboratories that are “exceptions” to CLIA.
TABLE C-1 CLIA Categories of Laboratories
| CLIA Definition | CLIA Certification Required? | |
|---|---|---|
| Laboratories regulated by CLIA | “[F]acilit[ies] for the . . . examination of materials derived from the human body for the purpose of providing information for the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings” | Yes |
| CLIA-exempt laboratories | Laboratories licensed by states that have “enacted laws relating to CMS-approved laboratory requirements that are equal to or more stringent than CLIA requirements” | No, but subject to, CMS-approved, state regulations |
| Research laboratories | Facilities “that test humans but do not report patient-specific results for the diagnosis, prevention or treatment of any disease or impairment of, or the assessment of, the health of individual patients” | No |
Laboratories may obtain waivers from CLIA to the extent that they perform only tests that: are so simple and accurate that the likelihood of error is negligible; pose no reasonable risk of harm if performed incorrectly; or are cleared by the FDA for home use.47
With respect to genetic testing, there has been a trend in recent years of unbundling collection of biospecimens and test administration from data interpretation.48 Because CLIA is limited to regulation of laboratories, legal scholars have noted that it should not extend to firms offering only interpretation services.49 CMS is currently considering its position on this issue.50
B. Original Access Rule
Until 2014, CLIA restricted the disclosure of laboratory test results as follows:
[T]est results must be released only to authorized persons and, if applicable, the individual responsible for using the test results and the laboratory that initially requested the test.51
CLIA defines an “authorized person” as “an individual authorized under State law to order tests or receive test results, or both.”52 Thus, until 2014, laboratories were legally permitted to release
___________________
47See 42 C.F.R. § 493.15.
48See Margaret A. Curnutte, Karen L. Frumovitz, Juli M. Bollinger, Amy L. McGuire & David L. Kaufman, Development of the Clinical Next-Generation Sequencing Industry in a Shifting Policy Climate, 32 NATURE BIOTECH. 980, 981-82 (2014).
49See Gail H. Javitt & Katherine Strong Carner, Regulation of Next Generation Sequencing, 42 J. L. MED. & ETHICS 9, 15-16 (2014 supp.).
50 Telephone communication with Karen Dyer, Director, Division of Laboratory Services, CMS (Dec. 14, 2017).
51 42 C.F.R. § 493.1291(f) (effective to April 6, 2014).
52Id. § 493.2 (effective to July 10, 2014).
results only to health care providers, ordering laboratories, and persons authorized by state law to order tests or receive test results. In states that did not provide for direct access to laboratory test results, individuals were required to request and obtain their results through their ordering providers.53
C. New Access Rule
Seeking to harmonize the existing CLIA access rule with revisions to the HIPAA access rule (see Part IV, infra), in 2014, HHS amended CLIA to expand individuals’ access to their laboratory test results. HHS did so by retaining the original CLIA access rule and adopting a new provision that:
Upon request by a patient (or the patient’s personal representative), the laboratory may provide patients, their personal representatives, and those persons specified under [the HIPAA access rule], as applicable, with access to completed test reports. . . .54
The new rule does not define a “completed test report,” although HHS explained in the Federal Register preamble to the new access rule that it considers test results to be “complete” when “all results associated with an ordered test are finalized and ready for release.”55 HHS further clarified that laboratories are not required to provide any interpretation of the test reports that they provide upon request.56
The new rule provides that the return of completed test reports is discretionary (“may”) in the identified circumstances. Thus, to the extent that the return of completed test reports to individuals would conflict with a state law that prohibits disclosure without provider consent, the state law controls unless it is preempted by another federal law, such as HIPAA (see Part IV, infra).
Today, both the original and new CLIA access rules apply to all requests for access to results of tests performed by CLIA-regulated laboratories. Table C-2 describes key distinctions between the rules.
TABLE C-2 Original Versus New CLIA Access Rule*
| Original Access Rule | New Access Rule | |
|---|---|---|
| Who may request access? | N/A |
|
| Who may obtain access? |
|
|
___________________
53See CMS, Memorandum from Thomas Hamilton, Director, Survey and Certification Group, CMS, to State Survey Agency Directors, Ref: S&C:14-11-CLIA (Feb. 7, 2014), available athttps://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/Survey-and-Cert-Letter-14-11.pdf.
54 42 C.F.R. § 493.1291(l) (2017).
55 CLIA Program and HIPAA Privacy Rule, 79 Fed. Reg. 7290, 7295 (Feb. 6, 2014).
56Id. at 7293.
|
||
| What may be obtained? | “Test results” | “Completed test reports” |
| NOTE: * Both the original and new CLIA access rules are legally in effect. | ||
D. Enforcement
CMS is authorized to enforce CLIA. Its principal enforcement mechanism is the suspension, limitation, or revocation of a laboratory’s CLIA certificate, which also can result in cancellation of a laboratory’s approval to receive Medicare payments for its services.57 For research laboratories that are not CLIA-certified, CMS generally has two enforcement options: (1) impose a civil money penalty of $50–$10,000 per day of noncompliance or per violation, depending on whether the deficiency poses an “immediate jeopardy”; or (2) file a civil lawsuit to enjoin continuation of any activity that CMS has reason to believe constitutes a “significant hazard to the public health.”58
CMS publishes a Laboratory Registry every year identifying laboratories and individuals that have been sanctioned for CLIA violations.59 Based on these registries, there do not appear to have been any actions taken against laboratories that involved the return of research results. Further, a search of CMS’s website did not identify any published hearing decisions involving research laboratories.60
Otherwise, there are few known instances in which CMS has used less formal mechanisms to enforce CLIA against research laboratories that returned or planned to return individual-level test results. The most recent such instance involved ORIG3N, a direct-to-consumer (DTC) genetic testing firm that offers genetic tests purporting to identify genetic variants associated with intelligence, athleticism, and metabolism.61 After ORIG3N announced plans to give away tests at a Baltimore Ravens game in September 2017, CMS intervened to examine whether those tests are subject to CLIA.62 ORIG3N claimed to be outside the scope of CLIA as a “research laboratory that does not provide patient specific results,” but instead provides results to “customers.”63 CMS rejected this characterization, however, and concluded that ORIG3N is subject to CLIA because it provides information for health assessment purposes, and CMS directed ORIG3N to apply for certification.64
___________________
57 42 U.S.C. § 263a(i)(1); 42 C.F.R. §§ 493.1806(a)-(b), 493.1840(a)-(b), 493.1842(a).
58 42 U.S.C. §§ 263a(h)(1)-(2), § 263a(j); 42 C.F.R. §§ 493.1806(c)(3)-(d), 493.1834(c)-(d), 493.1846; see also telephone communication with Karen Dyer, supra note 50.
59See CMS, Laboratory Registry, CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Laboratory_Registry.html (last modified Apr. 28, 2017).
60See CMS, CLIA-Related Hearing Decisions, CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/Hearing-Index-August-14-2017.pdf (current through Aug. 14, 2017).
61 ORIG3N, https://orig3n.com/?gclid=Cj0KCQiA0vnQBRDmARIsAEL0M1nAUesRebAYPjT6btWL3udxNIvQ2X u8OjtyVe2AqFJadSfYqpLR6f0aAqUwEALw_wcB (last visited Jan. 11, 2018).
62See Jeff Barker, “DNA Day” Planned for Ravens’ Game Undergoes Federal and State Scrutiny, BALT. SUN, Sept. 18, 2017, http://www.baltimoresun.com/business/bs-bz-ravens-dna-day-20170918-story.html.
63 Letter from Karen Dyer, Director, Division of Laboratory Services, CMS, to Kate Blanchard, Chief Operating Office, ORIG3N (Oct. 30, 2017) (on file with author) (summarizing ORIG3N’s asserted position).
64Id.
III. State Laws Equivalent to CLIA
CMS has determined that the laboratory licensure programs of Washington and New York are equivalent to CLIA requirements and so laboratories in these states can qualify as “CLIA exempt.”65
A. Washington
Washington law regulates “medical test sites,” defined as any facility or site “which analyzes materials derived from the human body for the purposes of health care, treatment, or screening.”66 Washington provides exceptions for two kinds of facilities, neither of which is relevant to this analysis.67 When asked whether research laboratories are considered medical test sites that require certification, an official with the Washington State Department of Health explained that if a research laboratory “is giving out results that get to patients and/or providers,” the testing will be considered clinical testing by a medical test site subject to state regulation.68 In this respect, Washington’s rule prohibiting the return of research results generated by unlicensed laboratories is identical to the CLIA prohibition.69
The default access rule in Washington requires that “test reports” be released to “authorized persons or designees,” defined as individuals allowed by state law to order tests or receive test results.70 After the new CLIA access rule was enacted, Washington adopted a similar provision that test reports may be released to patients and their personal representatives.71
B. New York
New York law regulates “clinical laboratories” located in New York or that accept specimens from New York.72 New York’s definition of clinical laboratories is similar to CLIA’s definition of laboratories except that New York’s regulations also encompass laboratory testing for forensic and identification purposes.73
___________________
65See CMS, List of Exempt States Under the Clinical Laboratory Improvement Amendments (CLIA), CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/ExemptStatesList.pdf (last visited Jan. 12, 2018).
66 WA. REV. CODE ANN. §§ 70.42.005, 70.42.010(8) (West 2017); WASH. ADMIN. CODE §§ 246-338-001, 246-338-010(25) (2017).
67See WASH. ADMIN. CODE § 246-338-010(25).
68 E-mail from Susan Walker, Program Manager, Laboratory Quality Assurance, Washington State Department of Health, to author (Sept. 14, 2017) (on file with author).
69 Telephone communication with Susan Walker, Program Manager, Laboratory Quality Assurance, Washington State Department of Health (Nov. 2, 2017).
70 WASH. ADMIN. CODE §§ 246-338-010(2), 246-338-070(3)(b).
71See id. § 246-338-070(3)(c).
72See N.Y. PUB. HEALTH LAW §§ 570, 571(1), 572 (McKinney 2017).
73See id. § 571(1) (defining “clinical laboratory” to include examination for the purpose of “obtaining information” for health assessment and identification purposes); NY STATE DEP’T OF HEALTH WADSWORTH CENTER, CLINICAL LABORATORY EVALUATION PROGRAM: A GUIDE TO PROGRAM REQUIREMENTS AND SERVICES 3 (rev. Jan. 2017) [hereinafter NY STATE PROGRAM GUIDE] (providing that “[c]linical laboratories located in New York State, and laboratories conducting clinical or forensic testing on specimens originating in New York State regardless of location, must hold a New York State Department of Health clinical laboratory permit”).
Like CLIA, New York provides an exception for facilities that “perform laboratory tests solely for research purposes.”74 In its Clinical Laboratory Evaluation Program’s Guide to Program Requirements and Services, the New York Department of Health clarified the clinical-research laboratory distinction as follows:
Research testing is considered clinical in nature if a patient-identified result is generated. This would include results used to make clinical decisions for patient management under an IRB-approved research protocol or clinical trial.75
If a result is obtained during the course of research testing that a laboratory feels ethically compelled to report to a clinician or research participant, the laboratory must obtain a New York clinical laboratory permit prior to reporting.76 In practice, New York’s rule prohibiting the return of research results generated by unlicensed laboratories is comparable to the CLIA prohibition.77
The default access rule in New York restricts the reporting of test results of specimens “submitted for evidence of human disease or medical condition” to three categories of individuals: physicians, their agents, and persons legally authorized “to employ the results thereof in the conduct of [their] practice or in the fulfillment of [their] official duties.”78 After the new CLIA access rule was enacted, New York adopted a similar provision that test reports may be released to patients.79
IV. HIPAA
A. Scope
HIPAA applies to three categories of individuals and entities: health plans, health care clearinghouses, and health care providers who transmit “any health information in electronic form” to carry out certain activities related to furnishing, billing, or receiving payment for health care.80 Such covered transactions include sending claims to health plans to inquire about eligibility to receive health care or to request payment for medical services.81 The privacy and
___________________
74See N.Y. PUB. HEALTH LAW § 580(2).
75 NY STATE PROGRAM GUIDE, supra note 73, at 4; see also NY State Dep’t of Health, Test Approval: LDTs used in Clinical Trials, WADSWORTH.ORG, https://www.wadsworth.org/regulatory/clep/clinical-labs/obtain-permit/test-approval (last visited Jan. 12, 2018) (“Examples of testing performed for participant management include those that influence enrollment (exclusion or inclusion), safety, or dosing.”).
76 NY State Dep’t of Health, Test Approval: LDTs used in Research Testing, WADSWORTH.ORG, https://www.wadsworth.org/regulatory/clep/clinical-labs/obtain-permit/test-approval (last visited Jan. 12, 2018).
77 Telephone communication with Stephanie Shulman, Director, New York Clinical Laboratory Program (Nov. 14, 2017).
78 N.Y. COMP. CODES R. & REGS. tit. 10, § 58-1.8 (2018).
79Id.
80 42 U.S.C. §§ 1320d-1(a), 1320d-2(a) (2018); 45 C.F.R. § 160.103 (2018); see also CMS, Administrative Simplification: Covered Entity Guidance, CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/Administrative-Simplification/HIPAA-ACA/Downloads/CoveredEntitiesChart20160617.pdf (last visited Jan. 16, 2018) [hereinafter CMS, Administrative Simplification] (summarizing covered transactions).
81See 42 U.S.C. § 1320d-2(a)(2) (listing covered transactions); 45 C.F.R. §§ 162.1101, 162.1201 (defining transactions relevant to “health care claims or equivalent encounter information” and “eligibility for a health plan”). See also CMS, Administrative Simplification, supra note 80; CMS, Transactions Overview, CMS.GOV, https://www.cms.gov/Regulations-and-Guidance/AdministrativeSimplification/Transactions/TransactionsOverview.html (last modified July 26, 2017).
security regulations that implement HIPAA also extend to “business associates” of covered entities.82 A business associate is any person who creates, receives, maintains, or transmits protected health information (PHI) on behalf of a covered entity or provides services to a covered entity that includes disclosure of PHI.83 PHI is defined as individually identifiable health information, which is any information (including genetic information) that: (1) is created or received by a covered entity or employer; (2) “relates to the past, present, or future physical or mental health or condition of an individual; the provision of health care to an individual; or the past, present, or future payment for the provision of health care to an individual”; and (3) identifies or could be used to identify the individual.84
Research laboratories are HIPAA-covered entities in two situations. The first is when they electronically conduct a covered transaction.85 HHS has emphasized that the conduct of a single covered transaction will transform a laboratory into a covered entity “with respect to all protected health information that it creates or maintains,” not just the individuals or information associated with the covered transaction.86
The second situation in which research laboratories are covered entities is when they function as part of larger covered entities. Thus, research laboratories that operate within HIPAA-covered hospitals, medical centers, or medical schools may also be covered by HIPAA by virtue of these relationships.87 However, a covered entity may elect to become a “hybrid entity,” which is defined as a covered entity whose business activities include both covered and non-covered functions.88 When a covered entity elects to become a hybrid entity, it must ensure that its designated health care components that perform covered functions do not disclose PHI to other components except as permitted by HIPAA.89 This becomes difficult in the case of a clinician-investigator who is an employee of and performs duties for both a health care component and a non-covered component of a hybrid entity.90 In the end, hospitals, medical centers, and medical schools often do not elect hybrid entity status and designate their research laboratories as non-covered components because of the operational complexities and high transaction costs associated with doing so successfully.91
___________________
82See 45 C.F.R. §§ 162.923(c), 164.302, 164.500(c).
83Id. § 160.103.
84Id.
85See supra notes 80-81 and accompanying text.
86 CLIA Program and HIPAA Privacy Rule, 79 Fed. Reg. 7290, 7291 (Feb. 6, 2014).
87See Barbara J. Evans, Michael O. Dorschner, Wylie Burke & Gail P. Jarvik, Regulatory Changes Raise Troubling Questions for Genomic Testing, 16 GENETICS MED. 799, 801 (2014) (explaining that a research laboratory may “fall under HIPAA because of its business organizational arrangements (for example, if it is part of a HIPAA-covered academic medical center)”).
88See 45 C.F.R. §§ 164.103, 164.105(a)(2)(iii)(D).
89See id. § 164.105(a)(2)(ii). Health care components include every component that “would meet the definition of a covered entity or business associate if it were a separate legal entity.” Id. § 164.105(a)(2)(iii)(D).
90See id. § 164.105(a)(2)(ii)(C); telephone communication with Mark Barnes, Partner, Ropes & Gray LLP (Nov. 15, 2017).
91 Telephone communication with Mark Barnes, supra note 90. Further, if a research laboratory functions as a business associate to a hospital, medical center, or medical school that has elected hybrid entity status, it must be designated a covered health care component. See 45 C.F.R. § 164.105(a)(2)(iii)(D); see also Modifications to the HIPAA Privacy, Security, Enforcement, and Breach Notification Rules Under the Health Information Technology for Economic and Clinical Health Act and the Genetic Information Nondiscrimination Act, 78 Fed. Reg. 5566, 5588 (Jan. 25, 2013) (explaining that the final rule “requires that the health care component of a hybrid entity include all business associate functions within the entity”); telephone communication with David Peloquin, Associate, Ropes & Gray LLP (Nov. 22, 2017).
B. Original Access Rule
Since 2000, HIPAA regulations have included a rule that individuals have a “right of access” to inspect and obtain a copy of their PHI that is maintained within a “designated record set” for as long as the PHI is maintained in the designated record set.92 A designated record set is defined as a “group of records” maintained by or for a covered entity that includes medical, claims, and billing records, as well as any other record “[u]sed, in whole or in part, by or for the covered entity to make decisions about individuals.”93 HHS has interpreted this definition broadly to mean that the designated record set includes all “records that are used to make decisions about any individuals, whether or not the records have been used to make a decision about the particular individual requesting access.”94 Further, qualifying “decisions” include but are not limited to health care decisions “because other decisions by covered entities can also affect individuals’ interests.”95
Before 2014, the HIPAA access rule provided an exception for HIPAA-covered laboratories. Specifically, two provisions excluded from access any PHI maintained by:
- Laboratories “[s]ubject to [CLIA], to the extent the provision of access to the individual would be prohibited by law”; and
- Laboratories “[e]xempt from [CLIA], pursuant to 42 C.F.R. § 493.3(a)(2)” (which refers to “CLIA-exempt” laboratories).96
The first provision excluded CLIA-regulated laboratories because at that time CLIA prohibited the return of results to individuals except in states that explicitly authorized such returns (see Part II, supra). The second provision excluded “CLIA-exempt” laboratories regulated by New York and Washington. Importantly, however, in the preamble to the original access rule, HHS interpreted the second provision excluding laboratories “[e]xempt from [CLIA], pursuant to 42 C.F.R. § 493.3(a)(2)” to also include research laboratories—even though research laboratories are excluded from CLIA under a different regulatory section.97 HHS explained that this interpretation was necessary because if research laboratories are “subject to the access requirements of this regulation, such entities would be forced to meet the requirements of CLIA from which they are currently exempt.”98 “To eliminate this additional regulatory burden,” HHS viewed research laboratories as excluded from the HIPAA access requirement.99
___________________
92 45 C.F.R. § 164.524(a).
93Id. § 164.501. A “record,” in turn, is defined as any “item, collection, or grouping of information” that includes PHI and is “maintained, collected, used, or disseminated by or for a covered entity.” Id.
94 Standards for Privacy of Individually Identifiable Health Information, 65 Fed. Reg. 82,462, 82,606 (Dec. 28, 2000).
95Id.
96 45 C.F.R. § 164.524(a)(1)(iii) (effective to April 7, 2014).
97 Standards for Privacy of Individually Identifiable Health Information, 65 Fed. Reg. at 82,485. Dr. Evans and colleagues argue that CMS forgot this history when it eliminated the exception that kept “CLIA-exempt” laboratories from having to comply with the HIPAA access rule, thereby inadvertently putting “HIPAA-covered, non-CLIA laboratories squarely in the crosshairs of individuals’ new § 164.524 access right.” Evans et al., supra note 87, at 801.
98 Standards for Privacy of Individually Identifiable Health Information, 65 Fed. Reg. at 82,485 (referring to the CLIA “exemption” for research laboratories under 42 C.F.R. § 493.3(a)(2)).
99Id. Research laboratories might also have been indirectly excluded by the terms of the first provision. In the view of CMS, research laboratories cannot return results unless they are CLIA-certified, so a research laboratory’s return of results would trigger the need for CLIA certification, but at that time, CLIA prohibited certified laboratories from returning results.
C. Revised Access Rule
In 2014, following three years of deliberation, HHS announced its elimination of the laboratory exclusion from the HIPAA access rule and the CLIA prohibition on the return of laboratory test results to individuals.100 HHS was motivated by concerns that these rules impeded individuals’ access to their records, thereby preventing them from having a more active role in personal health care decisions.101 HHS also stated that removing these impediments would support its commitments to personalized medicine and the widespread adoption of electronic health records.102
Focusing on HIPAA, the revisions eliminated the original HIPAA access rule’s carve-out for laboratories.103 In the Federal Register preamble, HHS explained that the purpose of this change was to require all HIPAA-covered laboratories to comply with the access rule regardless of their status under CLIA:
Even if CLIA does not apply to the conduct of certain types of laboratory tests, HIPAA may still apply to require access to certain test reports to the extent the laboratory is a HIPAA covered entity and the information to which an individual is requesting access is protected health information under HIPAA.104
Elsewhere in the preamble, HHS further explained that under the proposed rule, which was adopted with only minor clarifications and conforming changes, “HIPAA covered entities that are laboratories subject to CLIA, as well as those that are exempt from CLIA, would have the same obligations as other types of covered health care providers” with respect to providing individuals access to their PHI.105 That “exempt” in this context encompasses not only “CLIA-exempt” (i.e., New York and Washington) laboratories but also research laboratories is reinforced by the preamble’s repeated reference to the expanded access obligations of all HIPAA-covered laboratories.106
Table C-3 compares the original and revised HIPAA access rules.
TABLE C-3 Original Versus Revised HIPAA Access Rule*
| Original Access Rule | Revised Access Rule | |
|---|---|---|
| Who may request access? | Individuals who are the subject of PHI | Individuals who are the subject of PHI |
| Who may obtain access? | Individuals who are the subject of PHI and other persons as directed by individuals | Individuals who are the subject of PHI and other persons as directed by individuals |
___________________
100See CLIA Program and HIPAA Privacy Rule, 79 Fed. Reg. 7290 (Feb. 6, 2014).
101See id.
102See id.
103See 45 C.F.R. § 164.524(a)(1) (2018) (effective beginning April 7, 2014).
104 CLIA Program and HIPAA Privacy Rule, 79 Fed. Reg. at 7296-97.
105Id. at 7292.
106 This interpretation was also confirmed by OCR. Telephone communication with Deven McGraw, Deputy Director (former), Health Information Privacy, OCR (Jan. 5, 2018).
| What may be obtained? | PHI about an individual maintained within a designated record set | PHI about an individual maintained within a designated record set |
| From whom may information be obtained? | HIPAA-covered entities, but not CLIA-regulated labs, “CLIA-exempt” labs, and research labs | HIPAA-covered entities |
| NOTE: * Only the revised HIPAA access rule is legally in effect. | ||
In sum, the revised HIPAA access rule provides individuals with a broad right of access to their PHI contained within designated record sets maintained by HIPAA-covered laboratories.107 A designated record set includes at least laboratory test reports, but as noted above, it also includes all other PHI maintained by a laboratory that is used to make any kind of decision about any person.108
In 2016, the Office for Civil Rights (OCR), the HHS office responsible for enforcing HIPAA, published guidance explaining the kinds of information that may fall within the designated record set maintained by laboratories.109 The guidance states that in the context of a genetic test conducted by a clinical laboratory, the designated record set includes: the “completed test report”; the “full gene variant information generated by the test”; the “underlying data used to generate the report”; “as well as any other information in the designated record set concerning the test.”110
There are two limits to the HIPAA access rule that are relevant to this analysis. First, the rule provides for a temporary suspension of access related to clinical research activities. Specifically, it provides that an individual’s access to PHI created or obtained “in the course of research that includes treatment may be temporarily suspended for as long as the research is in progress” provided that the individual has consented to this temporary denial of access.111 However, the right of access must be reinstated upon completion of the research.112
The second limit to the access rule is set forth in HIPAA’s authorizing statute. It provides that HIPAA standards “shall not require disclosure of trade secrets or confidential commercial information” by covered entities.113 Thus, a covered entity may legally refuse to provide
___________________
107 Dr. Evans argues that, as applied to genetic information, the access rule is a federal civil rights regulation compelled by the understanding that “access to one’s own genomic data is a foundational civil right that empowers people to protect all their other civil rights.” Barbara J. Evans, HIPAA’s Individual Right of Access to Genomic Data: Reconciling Safety and Civil Rights, 102 AM. J. HUM. GENETICS 5, 6-7 (2018).
108See id. at 7295; see also notes 94-95 and accompanying text.
109See HHS, Individuals’ Right Under HIPAA to Access Their Health Information 45 C.F.R. § 164.524, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/privacy/guidance/access/index.html (last visited Jan. 16, 2017) [hereinafter HHS, Individuals’ Right Under HIPAA]. Relevant FAQs include “Does an Individual Have a Right Under HIPAA to Access from a Clinical Laboratory the Genomic Information the Laboratory Has Generated About the Individual?” and “Does an Individual Have a Right Under HIPAA to Access More Than Just Test Results from a Clinical Laboratory?” Both FAQs were last reviewed on June 24, 2016.
110Id. The guidance refers only to access to genomic information “maintained by or for a clinical laboratory that is a covered entity”; it does not address the access obligations of research laboratories. Id. However, earlier guidance states that research participants shall have access to “any research records or results that are actually maintained by the covered entity as part of a designated record set.” See HHS, What Does the HIPAA Privacy Rule Say About a Research Participant’s Right of Access to Research Records or Results?, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/faq/311/what-does-hipaa-say-about-research-participants-right-of-access/index.html (last reviewed July 26, 2013). This earlier guidance was created in 2002 and last reviewed in 2013.
111 45 C.F.R. § 164.524(a)(2)(iii) (2018).
112Id.
113 42 U.S.C. § 1320d-1(e) (2018).
individuals any requested PHI contained within a designated record set that the entity views as a trade secret or confidential commercial information.114 This limit on the HIPAA access rule may have special significance in the context of laboratories that maintain proprietary databases of test data and associated algorithms.115
If these limits do not apply, the covered entity must provide individuals “the access requested by the individuals,” including inspection or obtaining a copy of the requested PHI, within 30 days of receipt of the request.116 Alternatively, the covered entity may provide a summary or explanation of the PHI if agreed upon by the requesting individual.117 The PHI must be provided in the form and format requested by the individual to the extent that it is readily producible in that form and format; otherwise, the PHI must be provided in “readable hard copy form” or any other agreed-upon form and format.118 Finally, the covered entity may charge the requestor a reasonable, cost-based fee covering its labor, supplies, and postage expenses associated with responding to requests for copies.119
The 2016 guidance makes clear that an individual’s reasons for requesting access to his or her PHI maintained in a designated record set are irrelevant to a covered entity’s obligation to respond to that request:
[A] covered entity may not require an individual to provide a reason for requesting access, and the individual’s rationale for requesting access, if voluntarily offered or known by the covered entity or business associate, is not a permitted reason to deny access.120
Finally, the revised HIPAA access rule preempts any contrary provisions of state law.121 Thus, state laws that prohibit an individual’s direct access to test results are void to the extent they conflict with HIPAA.122 However, states may provide greater rights of access than those set forth in HIPAA.123
___________________
114 In at least one instance, HHS has explicitly authorized a covered entity’s refusal to disclose in these circumstances. See Robin Feldman & John Newman, Copyright at the Bedside: Should We Stop the Spread?, 16 STAN. TECH. L. REV. 623, 644 & n.105 (2013) (explaining that HHS informed a developer of proprietary cognitive test materials that it need not disclose any test results under HIPAA, even if the results include PHI, “to the extent that doing so would result in a disclosure of trade secrets”).
115See Christi J. Guerrini, Amy L. McGuire & Mary A. Majumder, Myriad Take Two: Can Genomic Databases Remain Secret?, 356 SCIENCE 586 (2017) (describing the application of the HIPAA access rule to proprietary genomic databases). HIPAA’s restriction on access to trade secret information is consistent with the trend in this country toward enhanced protection of trade secrets. See, e.g., Defend Trade Secrets Act of 2016, Pub. L. No. 114-153 (May 11, 2016) (creating a federal civil cause of action for trade secret misappropriation).
116 45 C.F.R. §§ 164.524(b)(2)(i), 164.524(c)(1).
117Id. § 164.524(c)(2)(iii).
118Id. § 164.524(c)(2)(i).
119Id. § 164.524(c)(4).
120 HHS, Individuals’ Right Under HIPAA, supra note 109 (emphasis in original).
121 45 C.F.R. § 160.203.
122See, e.g., CONN. AGENCIES REGS. § 19a-36-D32 (2018) (providing that laboratory findings on specimens may be reported to “lay persons” only upon the written request of their health care providers).
123 45 C.F.R. §§ 160.202, 160.203(b).
D. Enforcement
OCR enforces HIPAA by investigating complaints of HIPAA violations filed by individuals and conducting compliance reviews of covered entities.124 Since April 2003, OCR has received over 169,000 HIPAA complaints and initiated over 860 compliance reviews.125
Following the filing of a complaint by an individual, OCR will investigate if the complaint is timely and alleges a violation against a HIPAA-covered entity.126 If OCR concludes that a violation has occurred, it will attempt to resolve the case by obtaining voluntary compliance, corrective action, or a signed resolution agreement, and most investigations are concluded through these mechanisms.127 However, if the covered entity does not take action to resolve the matter in a way that is satisfactory to OCR, OCR can impose civil money penalties upwards of $50,000 for each violation (but not more than $1,500,000 for identical violations per calendar year).128
Individuals’ lack of access to their health information is among the top five issues that OCR investigates every year.129 OCR’s website identifies several examples of access-related complaints that it has investigated and resolved.130 None of these appear to involve research laboratories.
An example of an ongoing OCR investigation alleging a clinical laboratory’s denial of access was initiated against Myriad Genetics by four individuals for whom Myriad had performed genetic testing.131 The individuals claim that HIPAA entitles them to four categories of information specific to those tests: (1) raw and assembled genetic sequence data; (2) a list of all variants identified, including benign variants; (3) results of large-scale analyses; and (4)
___________________
124See id. §§ 160.306, 164.524(d)(2)(iii) (individual complaints); id. § 160.308 (compliance reviews).
125See OCR, Enforcement Results as of November 30, 2017, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/data/enforcement-highlights/index.html (last reviewed Jan. 9, 2018) [hereinafter OCR, Enforcement Results].
126See OCR, What OCR Considers During Intake & Review, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/examples/what-OCR-considers-during-intake-and-review/index.html? language=en (last reviewed June 7, 2017).
127See 45 C.F.R. § 160.312(a)(1) (authorizing resolution by informal means); OCR, How OCR Enforces the HIPAA Privacy & Security Rules, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/examples/how-OCR-enforces-the-HIPAA-privacy-and-security-rules/index.html (last reviewed June 7, 2017). “A resolution agreement is a settlement agreement signed by HHS and a covered entity or business associate in which the covered entity or business associate agrees to perform certain obligations and make reports to HHS, generally for a period of three years,” and may also agree to pay a resolution amount. OCR, Resolution Agreements and Civil Money Penalties, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/agreements/index.html (last reviewed Dec. 28, 2017) [hereinafter, OCR, Resolution Agreements].
128See 45 C.F.R. §§ 160.312(a)(3)(ii), 160.402(a), 160.404(b)(2), 160.410(c); OCR, Resolution Agreements, supra note 127. In the case of a continuing violation, a separate violation occurs each day the covered entity or business associate is in violation. Id. § 160.406.
129See OCR, Enforcement Results, supra note 125; OCR, Top Five Issues in Investigated Cases Closed with Corrective Action, by Calendar Year, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/data/top-five-issues-investigated-cases-closed-corrective-action-calendar-year/index.html?language=es (last reviewed June 7, 2017).
130See OCR, All Case Examples, HHS.GOV, https://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/examples/all-cases/index.html#case6 (last reviewed June 7, 2017). Based on the website descriptions, it is unclear whether civil monetary penalties were imposed in any of these cases.
131See Health Information Privacy Complaint (filed with OCR on May 19, 2016), available athttps://www.aclu.org/sites/default/files/field_document/2016.5.19_hipaa_complaint.pdf.
“records relating to clinical interpretation” of identified variants.132 While Myriad initially refused to provide this information, it eventually disclosed to each complainant a list of identified variants and raw data from Myriad’s large rearrangement test.133 Myriad stated that it does not retain and so cannot disclose any other requested sequence information.134 Nevertheless, OCR opened an investigation, which is ongoing.135
V. CLIA-HIPAA Interactions
A. Overview of Legal Obligations Related to Access
Table C-4 summarizes laboratories’ current legal obligations regarding individual access under CLIA and HIPAA. Boxes A and C describe access obligations of HIPAA-covered laboratories with additional detail in Box C for laboratories not certified by CLIA before and after the 2014 regulatory changes. Boxes B and D describe access obligations of laboratories not covered by HIPAA.
TABLE C-4 Legal Obligations Related to Individual Access, by Type of Laboratory
| HIPAA-Covered Laboratory | Not HIPAA-Covered Laboratory | |
|---|---|---|
CLIA-certified laboratory |
A Federal law (HIPAA): Mandatory access State law: Preempted unless provide greater access Example: Clinical laboratory |
B Federal law (CLIA): Permissive access State law: Not preempted; can mandate, permit, or prohibit access Example: Independent clinical laboratory that does not seek third-party reimbursement |
| Not CLIA-certified laboratory | C1 (pre-2014) Federal law (CLIA): Prohibited access unless authorized by state law State law: Not preempted; could mandate, permit, or prohibit access C2 (current) Federal law (HIPAA): Mandatory access (but disclosure requires laboratory to become CLIA-certified according to CMS) State law: Preempted unless provide greater access Example: Research laboratory that is part of a covered entity |
D Federal law: N/A State law: Not preempted; can mandate, permit, or prohibit access Example: Independent research laboratory |
___________________
132Id. at Ex. 1.
133See id. at Exs. 2-3.
134See id.
135 E-mail from Thomas Dresslar, Media Relations Associate, American Civil Liberties Union, to author (Dec. 1, 2017) (on file with author).
B. Potential Conflicts Between CLIA and HIPAA
As explained above, CMS has interpreted the CLIA exception for research laboratories to apply only where laboratories do not return individual-specific results or otherwise use those results to make clinical decisions. If laboratories return results to individuals or their clinicians for any reason, CMS’s position is that they must become CLIA-certified.136 Further, if laboratories return results to investigators and those results could be used in the treatment of research participants, they must become CLIA-certified.137
It is generally recognized that the 2014 changes to CLIA and HIPAA have created a dilemma for research laboratories that are not certified by CLIA but are covered by HIPAA because they conduct at least one electronic covered transaction or by virtue of their relationships with HIPAA-covered entities.138 To comply with the expanded access rules, HIPAA-covered research laboratories must now return PHI contained within designated record sets (including but not limited to test results) when individuals request them to do so, but these laboratories cannot do so without becoming CLIA-certified (see Table C-4, Box C2).
Yet, the Secretary’s Advisory Committee on Human Research Protections (SACHRP) has stated that it would be unrealistic to require all research laboratories to become CLIA-certified in order to comply with HIPAA.139 That is because the process of CLIA certification is expensive and time consuming.140 A National Heart, Lung, and Blood Institute Working Group has noted that many research laboratories are not CLIA-certified and many existing biobanks and current studies do not use CLIA-certified laboratories.141
Relying on principles of statutory interpretation, some scholars argue that, contrary to CMS’s interpretation, the return of results by research laboratories should not trigger a requirement to obtain CLIA certification.142 Focusing on the provision in CLIA that certification is not required if research laboratories do not return individual results “for the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of individual patients,”143 they understand that the need for CLIA compliance is dependent on the purpose for which a laboratory reports results.144 Although CLIA provides no guidance on how to assess the purpose of returning results, these scholars argue that returning results to an individual along with a suggestion that the individual seek confirmatory testing or consult a
___________________
136See supra notes 42-45 and accompanying text.
137See supra note 46.
138See, e.g., Evans et al., supra note 87, at 801; Mark Barnes, Susan Stayn, David Forster, Michele Russell-Einhorn, David Peloquin & Andres Medina-Jordan, The CLIA-HIPAA Conundrum of Returning Test Results to Research Participants, BNA MED. RES. L. & POL’Y REP. at 5 (July 15, 2015), available athttps://www.ropesgray.com/~/media/Files/articles/2015/July/2015-07-15-Bloomberg-BNA.ashx. This dilemma has been considered by several national committees and working groups, but none has made recommendations regarding how to reconcile the regulations. See Gail P. Jarvik et al., Return of Genomic Results to Research Participants: The Floor, the Ceiling, and the Choices in Between, 94 AM. J. HUM. GENETICS 818, 819 (2014).
139See SACHRP, SEPT. 28, 2015 SACHRP LETTER TO THE HHS SECRETARY, ATTACHMENT C: RETURN OF INDIVIDUAL RESULTS AND SPECIAL CONSIDERATION OF ISSUES ARISING FROM AMENDMENTS OF HIPAA AND CLIA (passed July 22, 2015).
140 Barnes et al., supra note 138, at 6.
141See Richard R. Fasbitz et al., Ethical and Practical Guidelines for Reporting Genetic Research Results to Study Participants: Updated Guidelines from a National Heart, Lung, and Blood Institute Working Group, 3 CIRCULATION: CARDIOVASCULAR GENETICS 574, 576-77 (2010).
142See, e.g., Burke et al., supra note 23, at 107-08; Evans, supra note 28, at 562-63.
143 42 C.F.R. § 493.3(b)(2) (2018).
144 Burke et al., supra note 23, at 107-08; Evans, supra note 28, at 562-65.
physician constitutes informational communication and does not amount to reporting for diagnostic, preventative, treatment, or health assessment purposes.145 Thus, the return of results by research laboratories in such circumstances should not trigger the requirement to obtain CLIA certification.146 Moreover, one of these scholars has separately argued that there may be a First Amendment right for a willing researcher to share results generated by a research laboratory with a willing participant.147
Finally, some practitioners have noted that even if a conflict exists between CLIA and HIPAA, it is unclear whether OCR will require research laboratories to comply with the new access rule.148 In this regard, it may be notable that OCR’s 2016 guidance on access to genetic test information refers only to information maintained by or for clinical laboratories and does not also address the access obligations of research laboratories.149
C. Institutional Responses
Institutions have responded to the perceived CLIA-HIPAA conflict in different ways. Some institutions may be minimizing the conflict through policies that interpret the institution’s designated record set to exclude some research-related information. For example, the policy of Johns Hopkins Medicine (JHM) is that researchers may not disclose results of research tests to subjects, patients, families, or caregivers “when such tests have been performed in laboratories that have not been CLIA-certified and do not have a state laboratory license.”150 More generally, JHM has taken the position that a “research record” is categorically not part of any designated record set and so is not subject to the HIPAA access rule.151 Rather, “only information that is entered into an individual’s medical record during the course of research would be part of the ‘designated record set.’”152 However, the policy recognizes that if the research involves treatment of a patient, and there is only one “record,” the research and medical record could be the same.153 The policy concludes: “[T]his is not a settled area of the law. Different experts have different opinions. But until there is further clarification, this is our position on this issue.”154
Similarly, NYU Langone Health System’s policy is that results of tests performed at laboratories not certified by CLIA to perform such tests are categorically not part of any designated record set and so are not subject to the HIPAA access rule.155 The designated record
___________________
145 Burke et al., supra note 23, at 108.
146Id. However, these scholars note that where the research is a clinical trial occurring in a health care setting, the distinction between research and clinical care may be so fine that the “prudent course” is for investigators to presume that the requirements of clinical care will apply and return only those results generated or confirmed in CLIA-certified laboratories. See id. at 109.
147 Evans, supra note 28.
148 Barnes et al., supra note 138, at 3.
149See notes 109-110 and accompanying text; telephone communication with David Peloquin, supra note 91.
150 Johns Hopkins Medicine, Organization Policy 101.2: Research Laboratory Testing Results (Aug. 2013), available athttp://www.hopkinsmedicine.org/institutional_review_board/guidelines_policies/organization_policies/101_2.html.
151 Johns Hopkins Medicine, HIPAA Questions and Answers Relating to Research, http://www.hopkinsmedicine.org/institutional_review_board/hipaa_research/faq_research.html (Feb. 2015).
152Id.
153Id.
154Id.
155 NYU Langone Health System, Policy: Designated Record Set (last rev. Nov. 1, 2017), available athttps://nyulangone.org/files/policy-designated-record-set-nov-17.pdf.
set is further interpreted to exclude research records that are not used or available to treating providers to make health care decisions about patients.156
However, there is anecdotal evidence that institutional policies prohibiting the return of results generated by research laboratories are being overruled in some instances. For example, a qualitative interview study of 31 IRB professionals at six sites across the United States reported two cases in which research test results that could not be confirmed in CLIA-certified laboratories were nevertheless reported to individual research participants.157 In one of these cases, the researcher had identified several genes associated with hyper-coagulability in a participant, and the IRB recommended returning this result after concluding that doing so posed a low risk of harm but high anticipated benefit to the participant.158 Although additional instances have been noted in the literature,159 the frequency with which these decisions are being made in practice is unclear.
VI. Federal Research Participant Protections
The return of results generated from biospecimens in research is relevant to federal regulations for the protection of research participants. These include the Common Rule160 and regulations adopted by FDA.161 Although this analysis is limited to federal protections, it is noted that several states also have adopted protections for research participants.162
A. Common Rule
1. Current The Common Rule applies to all “research” in which data or biospecimens are obtained through “intervention or interaction” with a “human subject,” where the research is federally funded, federally supported, or conducted by institutions that have voluntarily agreed (through federal-wide assurances) to comply with the Common Rule for both covered and non-covered research.163 However, several categories of research are excluded from the Common Rule’s scope, including secondary studies involving only data or biospecimens that cannot be identified as originating from specific individuals.164 For covered studies, the Common Rule requires IRBs to ensure that the risks of participation are minimized and reasonable in relation to
___________________
156Id.
157 Lynn G. Dressler et al., IRB Perspectives on the Return of Individual Results from Genomic Research, 14 GENETICS MED. 215, 216-17 (2012).
158Id. at 217.
159See, e.g., Anya E.R. Prince, John M. Conley, Arlene M. Davis, Gabriel Lázaro-Muñoz & R. Jean Cadigan, Automatic Placement of Genomic Research Results in Medical Records: Do Researchers Have a Duty? Should Participants Have a Choice?, 43 J. L. MED. & ETHICS 827, 837 (2015) (describing the practice of the Familial Dilated Cardiomyopathy Research Project to notify participants of “suspected meaningful results” generated by a research laboratory).
160See 45 C.F.R. pt. 46 (2018).
161See 21 C.F.R. pts. 50, 56 (2018).
162See CAL. HEALTH & SAFETY CODE §§ 24170-24179.5 (West 2018); MD. CODE. ANN. HEALTH-GEN. § 13-2001 to -2004 (West 2018); N.Y. PUB. HEALTH LAW §§ 2440-2446 (McKinney 2018); VA. CODE ANN. § 32.1-162.16 to 162.20 (2017).
163See 45 C.F.R. §§ 46.101(a), 46.102(f). Research, in turn, is defined as “a systematic investigation . . . designed to develop or contribute to generalizable knowledge.” Id. § 46.102(d).
164See id. § 46.102(f).
the anticipated benefits and also that participation is conditioned on informed consent.165
The Common Rule neither explicitly allows nor prohibits the return of results to study participants. Although it requires that potential participants be notified of certain study features for consent to be valid, these features do not include the study’s plan (or not) to return results. Still, legal scholars have noted, the Common Rule requires that, when appropriate, a research participant be informed of “significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation.”166 They note that this requirement, at a minimum, may obligate investigators “to disclose the fact that significant findings might be discovered during the course of research and whether or not those will be offered to subjects and/or their physicians.”167
In practice, when a study protocol includes a plan to return results, the IRB will review the plan to ensure its benefits outweigh its risks.168 While IRBs can prohibit investigators from returning results, however, they cannot block access when study participants request results under HIPAA.169
2. Pending revisions In January 2017, following many years of deliberation, HHS announced its adoption of revisions to the Common Rule.170 Most of the revisions, including all of those mentioned in this analysis, were scheduled to go into effect on January 19, 2018.171 However, HHS postponed the effective date by six months to July 19, 2018.172
The revisions continue to exempt secondary studies involving only non-identifiable data or biospecimens. They also identify new categories of exempt studies, including secondary research involving only collection and analysis of identifiable health information originally collected for other purposes if that use already is regulated by HIPAA.173 This exemption was adopted on grounds that HIPAA protections already in place for this kind of research are sufficiently “adequate” and it is “unduly burdensome and confusing” to require such research to also be subject to Common Rule protections.174
In addition to exempting new categories of research, the Common Rule revisions specify new categories of research eligible for limited IRB review. These include secondary studies of identifiable data and biospecimens where the investigator “does not include returning individual
___________________
165Id. §§ 46.109, 46.111(a), 46.116.
166Id. § 46.116(b)(5).
167 Amy L. McGuire, Bartha Maria Knoppers, Ma’n H. Zawati & Ellen Wright Clayton, Can I Be Sued for That? Liability Risk and the Disclosure of Clinically Significant Genetic Research Findings, 24 GENOME RES. 719, 720 (2014).
168See Stephanie A. Alessi, The Return of Results in Genetic Testing: Who Owes What to Whom, When, and Why?, 64 HASTINGS L. J. 1697, 1702-03 (2013).
169See Evans et al., supra note 87, at 800 (“Under the Privacy Rule, institutional review boards overseeing human-subjects research have no power to block § 164.524 access.”).
170 Federal Policy for the Protection of Human Subjects, 82 Fed. Reg. 7149 (Jan. 19, 2017).
171Id. at 7259 (revised § 101(l)). The requirement for one IRB to review cooperative research projects conducted in the U.S. will go into effect in January 2020. Id.
172See Federal Policy for the Protection of Human Subjects: Delay of the Revisions to the Federal Policy for the Protection of Human Subjects, 83 Fed. Reg. 2885 (Jan. 22, 2018).
173See Federal Policy for the Protection of Human Subjects, 82 Fed. Reg. at 7262 (revised § 104(d)(4)(iii)); see also id. at 7191-92 (explaining that the information and biospecimens covered by this exclusion “would generally be found by the investigator in some type of records (in the case of information) or some type of tissue repository (such as a hospital’s department for storing clinical pathology specimens)”).
174Id. at 7194 (noting HIPAA’s requirement that researchers obtain an individual’s authorization for certain research uses of protected health information or a waiver of that authorization by an IRB or HIPAA privacy board).
research results to subjects as part of the study plan.”175 The regulations make clear, however, that an investigator of a study that falls within this category may still return results if required by law to do so.176
Otherwise, the changes require, for the first time, that investigators disclose their plans regarding return of results in some circumstances. Specifically, the revised Common Rule sets forth a new element of information that, when appropriate, must be provided to research participants.177 That element is:
A statement regarding whether clinically relevant research results, including individual research results, will be disclosed to subjects, and if so, under what conditions.178
Furthermore, with respect to the storage, maintenance, and secondary research use of identifiable data and biospecimens, a research participant may provide “broad consent,” which must be conditioned on disclosure of the following information:
Unless it is known that clinically relevant research results, including individual research results, will be disclosed to the subject in all circumstances, a statement that such results may not be disclosed to the subject.179
Under the new rules, it is therefore possible for a study investigator to have no plans to return research results, and to inform (and be required to inform) study participants that individual research results will not be returned, yet be required by HIPAA to return results to participants upon their request according to procedures outside of IRB review.
B. FDA Protections
FDA research participant protections apply to all “clinical investigations,” regardless of funding source, that are regulated by FDA or that support applications for research or marketing permits for products regulated by FDA.180 A clinical investigation is defined as an experiment involving a test article and one or more human participants.181 Because the reach of FDA protections partially overlaps with the Common Rule, some research studies must comply only with FDA protections, some must comply only with the Common Rule, and some must comply with both.
FDA protections and the Common Rule have different regulatory purposes and so their substance is not identical. Still, many FDA protections are the same as or similar to provisions of the Common Rule.182 Thus, for covered investigations, FDA regulations (like the Common Rule)
___________________
175Id. at 7263 (revised § 104(d)(8)).
176See id. (revised § 104(d)(8)(iv)).
177Id. at 7266 (revised § 116(c)(8)).
178Id.
179Id. at 7266-67 (revised § 116(d)(6)).
180 21 C.F.R. §§ 50.1(a), 56.101(a) (2018).
181Id. §§ 50.3(c), 56.102(c).
182See Bonnie M. Lee, Comparison of FDA and HHS Human Subject Protection Regulations, FDA.GOV, https://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/EducationalMaterials/ucm112910.htm (last updated Mar. 10, 2009).
require IRBs to ensure that the risks of participation are minimized and reasonable in relation to the anticipated benefits and also that participation is conditioned on informed consent.183 Also like the Common Rule, FDA regulations neither explicitly allow nor prohibit the return of results to study participants. Although they require that potential participants be notified of certain study features for consent to be valid, they do not include notification of the study’s plan (or not) to return results.184 However, FDA protections (like the Common Rule) include the requirement that, when appropriate, a research participant be informed of “significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation.”185 In practice, an IRB overseeing an FDA-regulated study will review the study’s return-of-results plan to ensure that adequate protections accompany any return and prospective participants are informed through the consent process and provided an opportunity to opt-out of receiving results if not essential to the study.186
The pending revisions to the Common Rule have no effect on FDA research participant protections.187 However, in the Federal Register preamble to the revisions, HHS stated its intention to “consider the need for updates to FDA regulations and other relevant federal departmental or agency regulations with overlapping scope.”188 Further, the 21st Century Cures Act, enacted in 2016, requires HHS to harmonize differences between the Common Rule and FDA research participant regulations “to the extent practicable.”189 Therefore, it should be expected that many Common Rule revisions will be incorporated into FDA regulations.
VII. FDA Regulations
A. Scope
Under the authority of the Federal Food, Drug, and Cosmetic Act (FDCA), FDA is responsible for protecting and promoting public health by ensuring the safety and effectiveness of medical drugs and devices.190 Devices regulated by FDA are defined broadly to include articles “intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals.”191 So defined, FDA-regulated devices cover many laboratory tests, including in vitro diagnostic tests.
FDA classifies each device intended for human use on the basis of risk to consumers.192 The greater the risk posed by a device, and the more control presumed necessary to ensure its safety and effectiveness, the higher will be its classification. Thus, Class I devices are subject only to general controls, including registration and labeling requirements, whereas most Class II devices require pre-market notification and “special controls,” including stricter labeling
___________________
183See 21 C.F.R. §§ 50.20, 50.25, 56.103, 56.109, 56.111.
184See id. § 50.25.
185Id. § 50.25(b)(5).
186 Telephone communication with Abram Barth, Associate, Ropes & Gray LLP (Nov. 24, 2017).
187See James E. Valentine & David B. Clissold, The Final Common Rule: Much Either Retained or Removed, But Not Much New Added, FDA LAW BLOG (Feb. 17, 2017), http://www.fdalawblog.net/2017/02/the-final-common-rule-much-either-retained-or-removed-but-not-much-new-added.
188 Federal Policy for the Protection of Human Subjects, 82 Fed. Reg. 7149, 7151 (Jan. 19, 2017).
189 21st Century Cures Act, Pub. L. No. 114-255 § 3023(a)-(b) (2016).
190See 21 U.S.C. ch. 9 (2018).
191Id. § 321(h).
192See id. § 360c; 21 C.F.R. pt. 860.
requirements.193 Class III devices are subject to the most stringent standards and require premarket approval before marketing.194 In determining the safety and effectiveness of a device for purposes of its classification, FDA considers four factors: (1) the individuals who are intended or represented as the users of the device; (2) conditions of use of the device; (3) the “probable benefit to health from the use of the device weighed against any probable injury or illness from such use”; and (4) the device’s reliability.195 FDA has classified over 1,700 distinct types of medical devices.196
B. Regulation of LDTs and DTC Genetic Tests
FDA has discretion in its enforcement of regulations, and historically the agency has followed a policy of enforcement discretion with respect to laboratory-developed tests (LDTs).197 An LDT is an in vitro diagnostic device that is designed, manufactured, and used within a single laboratory.198 In 2010, responding to concerns about the increasing complexity, reach, and risk of LDTs, as well as the use of results from faulty LDTs to direct major treatment decisions, FDA announced its intent to reconsider its policy of enforcement discretion with respect to LDTs.199 Although some scholars questioned FDA’s legal authority to regulate LDTs,200 these concerns became moot in 2017 when FDA announced that it would not issue a final guidance to allow for further public discussion and to give Congress the opportunity to develop a legislative solution.201
Nevertheless, FDA generally does not exercise enforcement discretion against firms providing direct-to-consumer genetic tests, whether or not they constitute LDTs,202 in part due to
___________________
193See 21 U.S.C. § 360c(a)(1)(A)-(B); 21 C.F.R. § 860.3(c)(1)-(2).
194See 21 U.S.C. § 360c(a)(1)(C); 21 C.F.R. § 860.3(c)(3).
195See 21 C.F.R. § 860.7(b).
196 FDA, Device Classification Panels, FDA.GOV, https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/ucm051530.htm (last updated June 26, 2014).
197See FDA, Laboratory Developed Tests, FDA.GOV, https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/LaboratoryDevelopedTests/default.htm (last updated Dec. 26, 2017).
198See id.
199See id.
200See, e.g., Paul D. Clement & Laurence H. Tribe, Laboratory Testing Services, as the Practice of Medicine, Cannot be Regulated as Medical Devices (Jan. 6, 2015), http://www.acla.com/wp-content/uploads/2015/01/Tribe-Clement-White-Paper-1-6-15.pdf. Accord Gail Javitt, FDA’s Legally-Suspect Shift of Clinical Lab Test Regulation Through Guidance Documents, THE WLF LEGAL PULSE (Aug. 20, 2014), https://wlflegalpulse.com/2014/08/20/fdas-legally-suspect-shift-of-clinical-lab-test-regulation-through-guidance-documents (explaining that FDA’s “legal authority to regulate LDTs, which are used to provide a medical service and are not distributed in interstate commerce as freestanding products, remains a subject of debate”).
201 FDA, Discussion Paper on Laboratory Developed Tests (LDTs), at 1, FDA.GOV (Jan. 13, 2017), https://www.fda.gov/downloads/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/LaboratoryDevelopedTests/UCM536965.pdf.
202 FDA, Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories: Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs), at 5, n.4, FDA.GOV (Oct. 3, 2014), https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416685.pdf (explaining that “FDA generally does not exercise enforcement discretion for direct-to-consumer (DTC) tests regardless of whether they meet the definition of an LDT”).
concerns associated with returning results directly to customers.203 In 2013, for example, FDA sent 23andMe a Warning Letter that its marketing of the Personal Genome Service (PGS) Test without FDA clearance or approval violated the FDCA, citing concerns that customers might self-manage their treatments based on false positive or false negative test outcomes.204 In 2017, after 23andMe submitted a request for de novo classification of the PGS Test, FDA granted permission to market the test for certain genetic health risks as a Class II device.205 However, based on concerns that included customers’ potentially incorrect interpretation of results, FDA imposed special controls that include providing customers information to help them interpret results and requiring their opt-in to receive results related to risk of life-threatening but unpreventable or untreatable conditions.206
C. IDE Requirements
The FDCA and implementing regulations describe a path of regulatory exemption for the conduct of clinical investigations to determine the safety or effectiveness of “investigational devices.”207 Procedures for this exemption, known as an investigational device exemption (IDE), are detailed in Part 812 of FDA regulations and summarized in Figure C-1. Devices subject to an approved IDE are exempt from regulatory requirements related to, among other things, performance standards and premarket notification and approval.208
___________________
203 Telephone communication with Alberto Gutierrez, Director (retired), Office of In Vitro Diagnostics and Radiological Health, FDA (Nov. 21, 2017).
204 Warning Letter from Alberto Gutierrez, Director, Office of In Vitro Diagnostics and Radiological Health, FDA, to Ann Wojcicki, CEO, 23andMe, Inc. (Nov. 22, 2013).
205 FDA, Evaluation of Automatic Class III Designation for the 23andMe Personal Genome Service (PGS) Genetic Health Risk Test for Hereditary Thrombophilia, Alpha-1 Antitrypsin Deficiency, Alzheimer’s Disease, Parkinson’s Disease, Gaucher Disease Type 1, Factor XI Deficiency, Celiac Disease, G6PD Deficiency, Hereditary Hemochromatosis and Early-Onset Primary Dystonia, Decision Summary, DEN160026 (correction date Nov. 2, 2017), available athttps://www.accessdata.fda.gov/cdrh_docs/reviews/DEN160026.pdf.
206Id. Accord Kayte Spector-Bagdady, “The Google of Healthcare”: Enabling the Privatization of Genetic Bio/Databanking, 26 ANNALS EPIDEMIOLOGY 515, 518 (2016) (explaining that “FDA’s current risk assessment of the 23andMe service is based entirely on the data and information that are returned to the customer” (emphasis in original)).
207See 21 U.S.C. § 360j(g) (2018); 21 C.F.R. pt. 812.
208 21 C.F.R. § 812.1(a). However, they may still be subject to requirements of IRB review. See id. §§ 50.1, 56.101, 812.62.
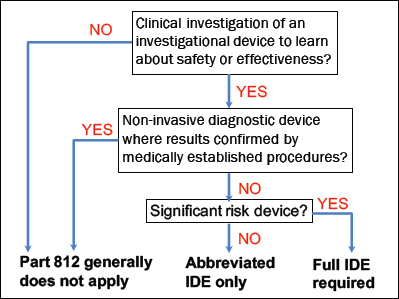
NOTE: IDE = investigational device exemption.
Part 812 defines an “investigational device” as one “that is the object of an investigation,” where an “investigation” is defined as “a clinical investigation or research involving one or more subjects to determine the safety or effectiveness of a device.”209 An approved or cleared device that is used in a study “in accordance with the approved or cleared labeling is not investigational and, therefore, is not subject to the IDE regulation.”210
FDA has stressed that Part 812 is limited to investigations conducted for purposes of determining safety or effectiveness, and not for other purposes.211 Nevertheless, FDA has viewed Part 812 as applying to all investigations involving devices where investigators expect to learn about the safety or effectiveness of the device, regardless of whether that is the primary research purpose.212
If investigators do expect to learn about the safety or effectiveness of an investigational device, the question becomes whether it poses a “significant risk,” meaning it “presents a potential for serious risk to the health, safety, or welfare of a subject.”213 FDA has explained that risk depends not on the nature of the device, but rather on how the information that it generates will be used in a specific study, and also that risk is evaluated on a case-by-case basis according to the worst-case scenario.214 Factors that FDA considers in determining whether an investigational device poses significant risk include the health status of the study population and
___________________
209 21 C.F.R. § 812.3(g)-(h); see also id. § 812.2(a) (providing that “[t]his part applies to all clinical investigations of devices to determine safety and effectiveness”).
210 FDA, Guidance for Industry and FDA Staff: In Vitro Diagnostic (IVD) Device Studies-Frequently Asked Questions, at 9, FDA.GOV (June 25, 2010) [hereinafter FDA, IVD FAQs]. A device may be an investigational device subject to Part 812 regardless of whether it is used in a clinical or research lab. Telephone communication with Alberto Gutierrez, supra note 203.
211See Procedures for Investigational Device Exemptions, 45 Fed. Reg. 3732, 3735 (Jan. 18, 1980).
212See NHGRI, Investigational Device Exemptions (IDEs) and Genomics Workshop: Meeting Report, GENOME.GOV (June 10, 2016), https://www.genome.gov/multimedia/slides/ideworkshop/ide_workshop_meeting_report.pdf [hereinafter NHGRI, Meeting Report].
213 21 C.F.R. § 812.3(m).
214See NHGRI, Meeting Report, supra note 212.
the manner in which results will be returned.215 Factors that FDA does not consider in determining whether an investigational device poses significant risk include the size of the cohort and potential benefits to participants.216
Investigational devices that do not pose significant risk are subject only to abbreviated IDE requirements, including proper labeling and IRB approval.217 When a device satisfies these abbreviated requirements, FDA considers it to have an approved IDE application.218
Further, Part 812 does not apply to a diagnostic device if it is properly labeled, “noninvasive,” and not used for diagnostic purposes without confirmation of the diagnosis by a “medically established” diagnostic product or procedure.219 The regulations define blood sampling that involves simple venipuncture, as well as the use of surplus body fluids and tissues originally taken for non-investigational purposes, as non-invasive.220
There is ambiguity, however, with respect to the kinds of devices that qualify as medically established. Devices that are used for purposes for which they already have been approved or cleared qualify as medically established.221 But it is unclear whether unapproved or uncleared LDTs used in laboratories can qualify as medically established and whether and how CLIA certification of the laboratories might affect that determination.222 In the context of genetic testing, FDA has stated that Sanger sequencing will sometimes constitute a medically established procedure.223
Recently, the IDE regulations were an issue for four studies funded by NIH as part of the Newborn Sequencing in Genomic Medicine and Public Health (NSIGHT) program.224 In 2013, FDA asked the investigators of those studies, which include genetic testing of newborns, to participate in the IDE process.225 FDA considered whether the genetic tests used in the studies were significant risk given that the results might be used to influence medical decision making for newborns.226 FDA also expressed concern that results might not be confirmed in every case by medically established procedures before their return to participants.227 Ultimately, all of the
___________________
215 Telephone communication with Alberto Gutierrez, supra note 203; see also NHGRI, Points to Consider Regarding the Food and Drug Administration’s Investigational Device Exemption Regulations in the Context of Genomics Research, GENOME.GOV, https://www.genome.gov/27561291/points-to-consider-in-assessing-when-an-investigational-device-exemption-ide-might-be-needed (last updated July 27, 2017) [hereinafter, NHGRI, Points to Consider] (providing as an example of a significant risk study one that involves genome sequencing of healthy participants with an intent to return variants of unknown significance because the test results might lead healthy individuals to pursue unnecessary treatments that could expose them to harm).
216See NHGRI, Points to Consider, supra note 215.
217See 21 C.F.R. § 812.2(b)(1).
218See id.
219See id. §§ 812.2(a), 812.2(c)(3). However, it is still subject to regulations related to disqualification of investigators. See id. § 812.119.
220Id. § 812.3(k).
221See FDA, IVD FAQs, supra note 210, at 11.
222 Telephone communication with Abram Barth, supra note 186.
223See NHGRI, Meeting Report, supra note 212 (noting that “although Sanger is analytically valid, it is not clinically valid,” whereas “medically established” procedures are, by definition, clinically valid).
224See Julia Karow, First Newborn Sequencing Study Gets FDA Green Light While Others Still Await Approval, GENOMEWEB (Dec. 17, 2014), https://www.genomeweb.com/sequencing-technology/first-newborn-sequencing-study-gets-fda-green-light-while-others-still-await.
225See id.
226 Personal communication with Amy McGuire, Leon Jaworski Professor of Biomedical Ethics, Baylor College of Medicine (Oct. 3, 2017).
227See id.
studies filed a “pre-IDE” to determine whether they needed to obtain an IDE, but only the North Carolina study was deemed to pose significant risk and required to submit a full IDE application.228 Although FDA eventually approved the application, the process of obtaining approval set back the study at least a year.229
In 2016, the National Human Genome Research Institute (NHGRI) held a workshop to discuss the IDE regulations as they apply to clinical studies that use genomic technologies.230 To develop the content of this event, NHGRI collaborated with FDA’s Center for Devices and Radiological Health, which reviews IDE submissions and is responsible for FDA’s medical device regulations.231 Outstanding questions identified at the workshop’s conclusion that are relevant to the return of results included whether FDA considers professional guidelines recommending the return of results to represent a standard of care, and if so, how those guidelines factor into FDA risk assessments.232
D. Unlawful Promotion
Although FDA regulates statements that device manufacturers can make about the clinical significance of test results, the agency’s jurisdiction over the practice of medicine is limited.233 According to its authorizing statute, FDA may not “limit or interfere with the authority of a health care practitioner to prescribe or administer any legally marketed device to a patient for any condition or disease within a legitimate health care practitioner-patient relationship.”234 However, the statute makes clear that the exclusion does “not change any existing prohibition on the promotion of unapproved uses of legally marketed devices.”235 Moreover, with respect to investigational devices, FDA regulations state that sponsors, investigators, and persons acting on their behalf may not promote or test market an investigational device before FDA has approved it for commercial distribution or “represent” that the device “is safe or effective for the purposes for which it is being investigated.”236
According to legal scholars, these rules mean that FDA likely cannot prohibit statements that physicians make to patients about their laboratory test results, including explanations of the clinical significance of the results, if they are made during the course of medical practice.237 However, it is unclear whether there are circumstances in which an investigator’s communication to participants of interpreted results generated from an investigational device
___________________
228See Karow, supra note 224. The “pre-IDE” process is briefly described in FDA, IVD FAQs, supra note 210, at 19-20.
229 Telephone communication with Jonathan Berg, Assistant Professor, Department of Genetics, University of North Carolina at Chapel Hill (Dec. 20, 2017).
230See NHGRI, Meeting Report, supra note 212.
231See id.
232See id.
233But see Patricia J. Zettler, Toward Coherent Federal Oversight of Medicine, 52 SAN DIEGO L. REV. 427, 460-64 (2015) (identifying examples of indirect FDA regulation of the practice of medicine).
234 21 U.S.C. § 396 (2018).
235Id.
236 21 C.F.R. §§ 812.7(a), 812.7(d).
237See Evans at 273; Sarah Y. Kwon, Regulating Personalized Medicine, 31 BERKELEY TECH. L. J. 931, 951 (2016); see also telephone communication with Alberto Gutierrez, supra note 203. However, if physicians’ claims about test results violate the standard of care, they may be subject to tort lawsuits or disciplinary actions by state medical practice boards. See Evans at 272-73.
would constitute unauthorized activity, and where, precisely, the FDA draws the line between permissible communications and impermissible promotion.238
OTHER LEGAL ISSUES
VIII. Property Rights
The return of research results is also relevant to the issue of property rights, which is generally governed by the states. In general, state courts have not viewed research results, including data generated from genetic tests, as legal property belonging to research participants. This conclusion has been reached by several courts in lawsuits where individuals who provided biospecimens for research claimed an ownership interest in results of tests performed on those biospecimens or, more generally, information and discoveries obtained through analysis of the biospecimens.239 In Greenberg v. Miami Children’s Hospital Research Institute, for example, a federal district court in Florida held that donors’ property rights in their biospecimens “evaporates once the sample is voluntarily given to a third party” and declined to recognize a “continuing right for donors to possess the results of any research.”240 Nevertheless, legal scholars describe a widespread belief that individuals “own” their personal data.241
Moreover, in the context of genetics, some state legislatures have explicitly recognized property rights of individuals in their test results.242 A Colorado statute, for example, provides that “[g]enetic information is the unique property of the individual to whom the information pertains,”243 although it permits the research use of information derived from genetic testing so long as the identity of any individual to whom the information pertains is not disclosed to third parties.244 An Alaska statute similarly provides that “a DNA sample and the results of a DNA analysis performed on the sample are the exclusive property of the person sampled or analyzed.”245 The statute further authorizes civil and criminal liability against any person who
___________________
238 Telephone communication with Patricia Zettler, Associate Professor, Georgia State University College of Law (Nov. 2, 2017).
239See, e.g., Moore v. Regents of the Univ. of Cal., 51 Cal. 3d 120, 136-41 (Cal. 1990), cert. denied, 111 S. Ct. 1388 (1991) (declining to find that a patient retained ownership rights in his excised cells); Ande v. Rock, 256 Wis. 2d 365, 382-83 (Wis. Ct. App. 2002) (rejecting plaintiffs’ claim that, under Wisconsin law, they had a property interest in diagnostic results that had not been returned to them). Accord Washington Univ. v. Catalona, 490 F.3d 667, 675 (8th Cir. 2007), cert. denied, 128 S. Ct. 1122 (2008) (holding that, under Missouri law, individuals who donated their biological materials voluntarily to a research institution did not retain an ownership interest allowing them to direct the transfer of the materials to third parties).
240 264 F. Supp. 2d 1064, 1075-76 (S.D. Fl. 2003) (applying Florida law, dismissing a claim of conversion of body tissue and genetic information voluntarily given to researchers).
241See, e.g. Mark A. Rothstein, Ethical Issues in Big Data Health Research, 43 J. L. MED. & ETHICS 425, 427 (2015) (noting that “many individuals strongly believe that their biological specimens and health records ‘belong to them’” so that they at least “ought to be consulted and asked for permission before their specimens and data are collected, analyzed, stored, and used for research”). Whether the propertization of data and biospecimens is socially desirable or not is the subject of ongoing academic debate. See, e.g., Jorge L. Contreras, Genetic Property, 105 GEO. L. J. 1 (2016); Gail H. Javitt, Take Another Little Piece of My Heart: Regulating the Research Use of Human Biospecimens, 41 J. L. MED. & ETHICS 424 (2013).
242See, e.g., ALASKA STAT. ANN. § 18.13.010(a)(2) (West 2017); COLO. REV. STAT. ANN. § 10-3-1104.7(1)(a) (West 2017); FLA. STAT. ANN. § 760.40(2)(a) (West 2017).
243 COLO. REV. STAT. ANN. § 10-3-1104.7(1)(a).
244See id. § 10-3-1104.7(5).
245 ALASKA STAT. ANN. § 18.13.010(a)(2).
retains a DNA sample or the results of a DNA analysis without informed consent.246 A lawsuit alleging unlawful disclosure of test results in violation of these provisions is currently pending before the federal district court in Alaska.247 The court has not yet interpreted the substantive provisions of the statute, although the test provider filed notice that it is challenging the statute as unconstitutionally vague because it does not allow reasonable people to understand what behavior would run afoul of it.248
Finally, parties can privately agree to allocate rights in test results that are different from default legal rules.249 This is done, for example, by the Personal Genome Project (PGP), which conducts genetic sequencing on biospecimens donated by participants, collects results of health surveys and previous genetic tests, and publishes all individual-level data on the internet for use by researchers.250 PGP’s informed consent document provides that donors retain ownership of the data they provide, although by participating in PGP, they license it to use that data without restriction.251 On the other hand, any information created or prepared by PGP from donated biospecimens and data, including the results of any research or analysis performed by or in collaboration with PGP, are “the property of and owned by the PGP and not by [participants].”252 Although PGP will attempt to make this information available to participants and the public, it states that it “is unable to guarantee if, when or in what form [participants] will receive access to any information, data or materials as part of [their] participation.”253
IX. Tort Liability
The return of research results may give rise to tort liability under state law for researchers and laboratories. A tort is a civil wrong (other than breach of contract) for which a remedy may be obtained, usually in the form of damages.254 Tort liability associated with the return of research results can generally be categorized as non-disclosure liability or disclosure liability.
A. Non-Disclosure Liability
There are several legal theories under which a researcher can be sued for failure to return results, although negligence has been identified as the most probable cause of action.255 An individual is liable for negligence where the individual owed a duty to another person, but breached that duty, and the person was harmed as a result.256
___________________
246Id. §§ 18.13.020, 18.13.030.
247See Complaint, Cole v. Gene by Gene, Ltd., Case No. 1:14-cv-00004-SLG (D. Alaska) (filed May 13, 2014).
248See Jennifer K. Wagner, A Constitutional Challenge to Alaska’s Genetic Privacy Statute, GENOMICS L. REP. (July 18, 2017), https://www.genomicslawreport.com/index.php/2017/07/18/a-constitutional-challenge-to-alaskas-genetic-privacy-statute.
249 Individuals also can contractually agree to give broad rights to use their test results. Telephone communication with David Peloquin, supra note 91.
250 HARVARD PERSONAL GENOME PROJECT, https://pgp.med.harvard.edu/about (last visited Jan. 29, 2018).
251See Personal Genome Project, Consent Form § 8.2 (rev. May 5, 2015), available athttps://my.pgphms.org/static/PGP_Consent_2015-05-05_online_stamped.pdf (last visited Jan. 29, 2018).
252Id. § 8.3.
253Id.
254 BLACK’S LAW DICTIONARY (10th ed. 2014) (“tort”).
255See McGuire et al., supra note 167, at 720.
256See id.
Whether one owes a legal duty to another depends on the nature of their relationship and is highly context specific.257 In general, individuals owe a duty of reasonable care under the circumstances, but tort law imposes no affirmative duties to act for another’s benefit and individuals are not required to warn others of impending harm.258 A number of factors can overcome this general tort law notion that individuals do not owe others affirmative duties, including the existence of a fiduciary relationship or other “special relationship,” as well as contractual obligations.259
1. Fiduciary relationships Fiduciary relationships are two-way relationships based on trust: “[t]he principal must have placed trust in the fiduciary and the fiduciary must have accepted that trust.”260 The fiduciary duty has been described as “extremely high,” where the fiduciary has “a duty to act with undivided loyalty in the best interests of the principal.”261 Physicians are held to be fiduciaries of their patients and are obligated to use their specialized knowledge and skill to act primarily in their patients’ best interests.262
Researchers, on the other hand, are generally not viewed as fiduciaries because their obligation is to produce generalizable knowledge, which may require acting in ways that are not primarily for the benefit of research participants.263 In several notable cases, courts have declined to view researchers as fiduciaries of participants in their studies where the researchers were not also the participants’ treating physicians.264
There is a question, however, whether a researcher’s act of returning clinically relevant results to a research participant, including interpretation of the clinical relevance of those results, by itself constitutes the practice of medicine that transforms their relationship into one of physician and patient. If so, the researcher assumes the fiduciary duties of a physician. No court apparently has addressed this issue in a precedential opinion, although legal scholars have argued that communication of the need to seek care is not necessarily the practice of medicine:
___________________
257See Elizabeth R. Pike, Karen H. Rothenberg & Benjamin E. Berkman, Finding Fault? Exploring Legal Duties to Return Incidental Findings in Genomic Research, 102 GEO. L. J. 795, 816-17 (2014); Prince et al., supra note 159, at 835.
258See Pike et al., supra note 257, at 816; Stacey A. Tovino, Incidental Findings: A Common Law Approach, 15 ACCOUNTABILITY RES. 242, 248 (2008) (“Under general principles of tort law, individuals do not have a duty to warn of risks they did not create absent a special relationship or other exceptional circumstances.”)
259See Pike et al., supra note 257, at 816-17.
260 Tovino, supra note 258, at 250-51.
261Id. at 251.
262See McGuire et al., supra note 167, at 720.
263See id. at 720-21; see also Pike et al., supra note 257, at 820 (explaining that, “[a]s a general rule, researchers are not fiduciaries of participants, so they do not owe fiduciary duties to act in a participant’s best interest”); Leslie A. Meltzer, Undesirable Implications of Disclosing Individual Genetic Results to Research Participants, 6 AM. J. BIOETHICS 28, 29 (2006) (stating that, in contrast to the clinical care setting, “in the research setting, investigators are not fiduciaries of participants” because their primary goal “is not, and indeed cannot be, to benefit any one participant”).
264See Moore v. Regents of the Univ. of Cal., 51 Cal. 3d 120, 133 (Cal. 1990) (rejecting the claim that a researcher who was not a physician had a fiduciary relationship with the complainant); Greenberg v. Miami Children’s Hosp. Research Inst., Inc., 264 F. Supp. 2d 1064, 1070-72 (S.D. Fl. 2003) (explaining that there is “no automatic fiduciary relationship that attaches when a researcher accepts medical donations” and finding that the plaintiffs’ failure to sufficiently allege that the defendant researchers had accepted the plaintiffs’ trust was fatal to their claim of breach of fiduciary duty); Ande v. Rock, 256 Wis. 2d 365, 377-79 (Wis. Ct. App. 2002) (finding no legally cognizable allegations in the complaint that there existed a physician-patient relationship between tested children and non-treating researchers).
Law recognizes a distinction between informing a person of the need to seek medical care and actually rendering medical care. . . . Return of research results lacks the treatment step that is necessary to create a [physician-patient relationship] and transform research into medical practice.265
Another question is whether a laboratory is a health care provider, which is presented in a pending federal lawsuit in South Carolina. The Williams v. Quest Diagnostics lawsuit is based on allegations that a clinical laboratory returned erroneous genetic test results and did not provide corrected test results until almost eight years later.266 Bringing suit on behalf of her son, who died as a result of receiving the wrong treatment, the plaintiff alleges negligence and other claims based on the laboratory’s failure to correctly classify her son’s genetic variant consistent with then-known scientific information.267 However, the laboratory seeks to reframe the case as a medical malpractice suit, which would be barred under the state’s statutes of limitations and repose.268 To resolve this issue, the trial judge asked the South Carolina Supreme Court to determine (“certify” the question) whether a clinical laboratory is a licensed healthcare provider under South Carolina law.269 Although the federal court’s decision in this case will not be binding on subsequent cases except in limited circumstances,270 it nevertheless may have practical significance to the extent that judges presiding over similar cases choose to follow its reasoning.271
2. Special relationships In addition to fiduciary relationships, “special relationships” between researchers and research participants can give rise to affirmative duties to disclose certain findings.272 Two courts have recognized the potential existence of such a relationship in the research context in the absence of any physician-patient relationship. First, in Blaz v. Michael Reese Hospital Foundation, a federal court in Illinois found a special relationship existed
___________________
265 Burke et al., supra note 23, at 107. However, where the testing is conducted at the request of a physician, some scholars have argued that the “tests are merely an extension of the doctor’s favored methods for evaluating a patient and diagnosing the problem” and so the testing service is “part and parcel of the doctor’s practice of medicine.” Clement & Tribe, supra note 200, at 12 (emphasis in original) (arguing that FDA cannot regulate LDTs because doing so would constitute unlawful interference with the practice of medicine).
266 Complaint, Williams v. Quest Diagnostics, Inc., Case No. 2016-CP-40-01166 (S.C. Ct. Com. Pl.) (filed Feb. 24, 2016).
267Id.
268See Laurel Coons, Williams v. Athena Motion to Dismiss Hearing—SC Supreme Court May Be Asked to Decide Whether a Diagnostic Laboratory Qualifies as a Healthcare Provider, GENOMICS L. REP. (Jan. 26, 2017), https://www.genomicslawreport.com/index.php/2017/01/26/williams-v-athena-motion-to-dismiss-hearing-sc-supreme-court-may-be-asked-to-decide-whether-a-diagnostic-laboratory-qualifies-as-a-healthcare-provider.
269See Turna Ray, Wrongful Death Suit Awaits Input from South Carolina Supreme Court, GENOMEWEB (Apr. 4, 2017), https://www.genomeweb.com/molecular-diagnostics/wrongful-death-suit-awaits-input-south-carolina-supreme-court.
270 Deborah Levenson, Lawsuit Raises Questions about Variant Interpretation and Communication, 173 AM. J. MED. GENETICS SEQUENCE 838, 839 (2017) (summarizing commentary by John Conley, William Rand Kenan Jr. Professor of Law, UNC School of Law). Specifically, if the decision is appealed to the U.S. Court of Appeals for the Fourth Circuit, that appellate court’s decision would be binding on federal courts within the Fourth Circuit applying the same South Carolina laws. See id.
271See Coons, supra note 268 (afterword by John Conley). Moreover, the South Carolina Supreme Court’s ruling on the narrow question whether clinical laboratories are licensed healthcare providers in South Carolina, which it has certified, will be binding on all courts applying South Carolina law. See id.
272See McGuire et al., supra note 167, at 721.
between research participants and a physician in charge of follow-up to the research program by virtue of his specialized knowledge and communication with participants.273 Even though the participants were not the physician’s patients, the court held that the special relationship created a duty on the part of the physician to warn of risks.274
Two years later, in Grimes v. Kennedy Krieger Institute, Maryland’s highest court took a more expansive view and suggested that a special relationship between researchers and participants can exist by virtue of the “very nature of nontherapeutic scientific research” and also that informed consent documents can give rise to duties to warn.275 At issue in Grimes was a nontherapeutic study led by Kennedy Krieger Institute (KKI) to investigate the effectiveness of lead-based paint abatement strategies on local housing rented to families with small children. The study included treatment groups of homes that received varying levels of less-than-comprehensive lead abatement and control groups of homes that received comprehensive lead abatement or were recently constructed and presumed not to have lead-based paint.276 Effectiveness of the abatement strategies was determined by comparing lead levels in the blood of the children with lead samples taken from the homes, exterior soil, and drinking water.277
The Grimes opinion is directed to the research experiences of two participants who were children. Participant Ericka Grimes lived with her family in a home where initial lead dust testing revealed “hot spots” of lead, although her mother was not informed of these results until nine months after sample collection.278 In the meantime, Miss Grimes was tested three times for lead in her blood; the second and third tests detected elevated lead levels.279 Her family sued for failure to timely disclose the property’s elevated lead dust levels.280 Participant Myron Higgins lived with his mother in a home that tested positive for lead but had received partial abatement.281 After they moved in, lead dust samples were taken using two different methods; his mother was not informed of the elevated lead dust levels detected by one of the methods.282 Mr. Higgins was tested three times for lead in his blood and all tests detected elevated lead levels.283 His family sued for failure to timely disclose the property’s original lead test results and failure to ever disclose the elevated lead dust levels detected after they moved in.284
The lower court granted summary judgment for KKI in both cases on grounds that KKI had no legal duty to warn the research participants of potential harms, and the rulings were appealed to the Court of Special Appeals.285 The Court of Appeals of Maryland then granted certiorari to consider the relevant issues and concluded that summary judgment was incorrectly granted because such a duty to warn may exist as a matter of law.286
___________________
273 74 F. Supp. 2d 803, 806-807 (N.D. Ill. 1999).
274See id.
275 Grimes v. Kennedy Krieger Inst., Inc., 782 A.2d 807, 834-35, 843-46 (Md. 2001).
276See id. at 820-23.
277See id. at 822.
278See id. at 824-25.
279See id. at 825.
280See id. at 825-26, 844-45.
281See id. at 826.
282See id. at 827-28, 845.
283See id. at 828-29.
284See id. at 829-31, 845.
285See id. at 818.
286See id. at 818-19.
The court held that “the very nature of nontherapeutic research on human subjects can, and normally will, create special relationships out of which duties arise.”287 The creation of such a relationship is especially likely where researchers “recruit people, especially children, whose consent is furnished indirectly,” to participate in potentially dangerous research.288 Alternatively, the court held that the informed consent documents signed by the participants’ family members created bilateral contracts that obliged KKI to provide “full, detailed, prompt, and continuing warnings as to all the potential risks and hazards inherent in the research or that [arose] during the research.”289 In its subsequent denial of a motion for reconsideration, however, the court clarified that its opinion was limited to the finding that summary judgment was improperly granted and that “[e]very issue bearing on liability or damages remains open for further factual development.”290 Further, the opinion has been widely criticized and generally not followed.291
3. Scope of duties If researchers owe legal duties to participants with whom they interact, the scope of that duty and whether it includes an obligation to return certain results depends on the prevailing standard of care. The standard of care can be established by guidance and recommendations to return results, recognition by scholars and the research community of an ethical obligation to return results, and a common practice of returning results.292 Legal scholars have explained that the “more encompassing guidelines and practices are with regard to return of results, the more sweeping the potential ethical and legal obligation” to do so will be.293 Furthermore, if the practice of returning results becomes routine, researchers “will be legally required” to do so because “[t]his is the way tort law has worked for decades.”294 Finally, in the case of physician-investigators, legal scholars have suggested that if the research itself generates individualized and identifiable data, medical obligations may trump research obligations and support a “higher duty to disclose relevant and significant findings.”295
B. Disclosure Liability
Many kinds of actions associated with the return of research results may give rise to tort liability. These include: (1) disclosure of correct results to the wrong individual as a result of, e.g., improper labeling; (2) disclosure of incorrect results to the right individual as a result of, e.g., improper test administration; (3) disclosure of results to individuals who are not authorized
___________________
287Id. at 834-35.
288Id. at 845-46.
289Id. at 843-44.
290 Grimes v. Kennedy Krieger Inst., No. 128 Sept. Term 2000 (Md. 2001) (per curiam) (denying motion for reconsideration).
291See, e.g., Pike et al., supra note 257, at 820 n.132 (noting that “courts that have considered similar fact patterns have generally refused to extend the holding of Grimes, and Grimes has been the subject of significant scholarly criticism”); Diane E. Hoffmann & Karen H. Rothenberg, Whose Duty is it Anyway? The Kennedy Krieger Opinions and its Implications for Public Health Research, 6 J. HEALTH CARE L. & POL’Y 109 (2002) (criticizing the duty implied by the court as unclear and potentially overbroad).
292See Pike et al., supra note 257, at 798, 822-23; Tovino, supra note 258, at 255. But see Wolf, supra note 22, at 440-41 (2012) (describing numerous ethics recommendations on return of results that have been promulgated over the years, none of which apparently has been cited in support of legal liability).
293 Ellen Wright Clayton & Amy L. McGuire, The Legal Risks of Returning Results of Genomics Research, 14 GENETICS MED. 473, 475 (2012).
294Id. (emphasis in original).
295 McGuire et al., supra note 167, at 721.
to receive them; and (4) failure to update previously disclosed results and return the updated results.
Researchers who return results must do so consistent with the standard of care and regulatory requirements.296 Thus, laboratories that deviate from good laboratory practices and standards regarding interpretation may be exposed to tort liability if they return erroneous results to individuals who are harmed as a result.297 In fields like genomics, however, the standard of care regarding interpretation is rapidly evolving.298 “[G]iven that there is still fervent debate about how to interpret variants,” legal scholars have argued, “it will be extremely difficult to prove what the standard of care is or that it has clearly been breached by a researcher acting in good faith” who provides interpreted genetic results.299
Nevertheless, researchers are generally required to comply with standards that seek to maximize the analytic and clinical validity of findings.300 Legal scholars have concluded that if researchers return erroneous results generated by a research laboratory and not validated in a CLIA-certified lab, and they do not make clear that the results need to be repeated before any clinical interventions are undertaken, the researchers may be liable in tort.301 However, the legal effect of such disclaimers, and whether they will absolve researchers of tort liability, remains unclear.
Meanwhile, disclosure of results to individuals who are not authorized to receive them may give rise to negligence claims where, among other things, the tested individual suffered discrimination as a result. These negligence claims would be in addition to any privacy claims that might be available to the tested individual. On the other hand, a laboratory’s obligation to update previously returned results in response to, for example, new scientific evidence or consensus, or following the laboratory’s adoption of a different classification scheme, remains unsettled. This question is raised in Williams v. Quest Diagnostics but has not yet been resolved.302
X. Anti-Discrimination Statutes
There is a complex web of federal and state laws that address unwanted access to and discriminatory use of health information. Whereas unwanted access is the domain of privacy laws, which are based on ethical principles of autonomy, discriminatory use is the domain of
___________________
296See id. at 721-22.
297See id. at 722.
298See id.
299Id.
300Id. at 721.
301See id. at 722.
302See Complaint, Williams v. Quest Diagnostics, Case No. 2016-CP-40-01166 (S.C. Ct. Com. Pl.) (filed Feb. 24, 2016). In Williams, the clinical laboratory allegedly made an error in the original variant classification, which did not reflect the then-existing literature, and it did not correct this classification in a revised report until almost eight years later. See id. The plaintiff’s position is that the revised report “corrected an error in the original classification rather than provid[ed] an update or reinterpretation.” Jennifer K. Wagner, Litigating the Accountability of Clinical Genomics Laboratories, GENOMICS L. REP. (May 31, 2016), https://www.genomicslawreport.com/index.php/2016/05/31/litigating-the-accountability-of-genomics-laboratories (emphasis in original). The analogous question regarding the potential duty of physicians to provide patients new information that might be relevant to their ongoing medical care is addressed in Mark A. Rothstein & Gil Siegal, Health Information Technology and Physicians’ Duty to Notify Patients of New Medical Developments, 12 HOU. J. HEALTH L. & POL’Y 93 (2012).
anti-discrimination laws, which are animated by concerns with equality and fairness.303 As noted by legal scholars, however, privacy laws can “do the work” of preventing discrimination by blocking access to information that might be the basis for discriminatory conduct.304 Therefore, provisions relevant to both types of activities are sometimes included in the same statute.
A. Federal Statutes
The major federal statutes that address problematic downstream uses of health information, whether generated in research or clinical contexts, are the Genetic Information Nondiscrimination Act (GINA), which is focused on genetic information, and the Americans with Disabilities Act (ADA), which is directed toward actual and perceived disabilities.
1. Genetic Information Nondiscrimination Act Passed in 2008, GINA limits access to and use of genetic information in health insurance and employment contexts, where an individual’s genetic information is generally defined to encompass information about his or her genetic tests, the genetic tests of family members, the manifestation of a disease or disorder in family members, the request or receipt of genetic services by the individual or family members, and participation in clinical research by the individual or family members that includes genetic services.305 The legislative purpose of GINA is to promote genetic testing for personal health and research purposes by allaying concerns with the potential misuse of information learned from genetic tests.306
Focusing on its application to health insurance, prior to GINA, HIPAA prohibited group health insurers from using genetic information to determine eligibility or set premiums for individuals or from treating genetic information as the basis of any pre-existing condition exclusion.307 GINA extended these protections to individual health insurers and further prohibits group health insurers from using genetic information about an individual to determine coverage or set premiums for a group.308 Moreover, under GINA, health insurers cannot request or require genetic testing prior to an individual’s enrollment and cannot request, require, or purchase genetic information for underwriting purposes.309 While health insurers cannot deny coverage on the basis of genetic information, however, GINA permits them to do so based on already expressed genetic conditions.310 This loophole was closed in 2011 by the Patient Protection and Affordable Care Act (ACA), which prohibits health insurers from denying coverage based on any pre-existing condition.311
___________________
303See Jessica L. Roberts, Protecting Privacy to Prevent Discrimination, 56 WM. & MARY L. REV. 2097, 2105-07, 2109-12 (2015).
304See id. at 2121-22 (describing a privacy-nondiscrimination “symbiosis”).
305 Genetic Information Nondiscrimination Act (GINA) of 2008, Pub. L. No. 110-233 (2008) (codified as amended in scattered sections of 26, 29, and 42 U.S.C.).
306See id. § 2.
307See 29 U.S.C. § 1181 (effective to Feb.16, 2009); 29 U.S.C. § 1182 (effective to May 20, 2008).
308See Amanda K. Sarata & Jody Feder, The Genetic Information Nondiscrimination Act of 2008 (GINA), Cong. Res. Serv. Report No. RL34584, at 5, 10 (Aug. 6, 2015) (explaining the legal effect of GINA).
309See 42 U.S.C. §§ 300gg-4(c)(1), 300gg-4(d)(1) (2018).
310See Robert C. Green, Denise Lautenbach & Amy L. McGuire, GINA, Genetic Discrimination, and Genomic Medicine, 372 N. ENGL. J. MED. 397, 397-98 (2015).
311See Patient Protection and Affordable Care Act (ACA), Pub. L. No. 111-148 § 1201 (2010) (codified at 42 U.S.C. § 300gg-3(a)).
GINA similarly limits both access to and use of genetic information by employers. Thus, employers are prohibited from requesting, requiring, or purchasing genetic information about employees or their family members.312 Employers also may not use genetic information to make employment decisions related to, e.g., hiring, firing, promotion, and compensation, or to deprive employees of employment opportunities.313 Finally, employers must treat the genetic information of their employees as confidential medical records that generally may not be disclosed.314
Yet GINA, even as amended by the ACA, has important limits. First, GINA applies only to health insurers and employers; it does not apply to life, disability, or long-term care insurers or to other contexts in which discrimination may occur, such as housing and education.315 Further, with respect to its prohibitions on employers, GINA does not protect against discrimination based on non-genetic health information or manifested disease.316 For these and other reasons, GINA has been widely criticized.317
From fiscal years 2010 to 2017, the Equal Employment Opportunity Commission (EEOC), which is responsible for enforcing GINA, received between 201 and 333 GINA-related complaints each year.318
2. Americans with Disabilities Act While GINA prohibits discrimination on the basis of genetic information, the ADA prohibits discrimination against individuals with disabilities in employment, public services, and public accommodations contexts.319 The threshold issue in every ADA case is whether the individual alleging discrimination has a disability. The ADA, as amended by the ADA Amendments Act of 2008, defines a disability as: (1) a physical or mental impairment that substantially limits one or more “major life activities” of an individual; (2) a record of such an impairment; or (3) being regarded as having such an impairment.320 A major life activity is defined to include activities such as concentrating, communicating, caring for oneself, and performing manual tasks, as well as the operation of major bodily functions.321
Although the ADA instructs that the definition of disability should “be construed in favor of broad coverage of individuals . . . to the maximum extent permitted,”322 it is unclear whether an asymptomatic individual, such as an individual who has a genetic predisposition for a not-yet-manifested condition, can have a disability recognized by the ADA.323 This question is presented
___________________
312See 42 U.S.C. § 2000ff-1(b).
313See id. § 2000ff-1(a).
314See id. § 2000ff-5(a)-(b).
315See Mark A. Rothstein, GINA, the ADA, and Genetic Discrimination in Employment, 36 J. L. MED. & ETHICS 837, 837 (2008).
316See id.
317See, e.g., Mark A. Rothstein, Putting the Genetic Information Nondiscrimination Act in Context, 10 GENETICS MED. 655 (2008).
318See EEOC, Genetic Information Non-Discrimination Act Charges, EEOC.GOV, https://www.eeoc.gov/eeoc/statistics/enforcement/genetic.cfm (last visited Feb. 2, 2018).
319 Americans with Disabilities Act of 1990 (ADA), Pub. L. No. 101-336 (1990) (codified at 42 U.S.C. ch. 126 and amended by the ADA Amendments Act of 2008, Pub. L. No. 110-325 (2008)).
320 42 U.S.C. § 12102(1).
321See id. § 12102(2).
322Id. § 12102(4)(A).
323Compare 1 DISABILITY DISCRIMINATION IN THE WORKPLACE § 13:8 (last updated Oct. 2017) (stating that asymptomatic individuals might be covered if they are “regarded as” or perceived to be disabled), with Rothstein, supra note 315, at 839 (stating that, “[i]n the context of genetic discrimination in employment, asymptomatic individuals are unlikely to be covered by the ADA”), and Anya E.R. Prince & Benjamin E. Berkman, When Does an Illness Begin: Genetic Discrimination and Disease Manifestation, 40 J. L. MED. & ETHICS 655, 657 (2012) (stating that “[t]here is arguably no protection for individuals who have manifested some symptoms, but whose symptoms have not risen to the level of substantial limitations”).
in Chadam v. Palo Alto Unified School District, a lawsuit filed on behalf of a California student who was asked to transfer middle schools on the basis of his genetic status as a carrier of a variant associated with cystic fibrosis.324 The student, who has not exhibited symptoms of the disease, alleged that the school district’s actions violated the ADA, and in 2016, the U.S. Court of Appeals for the Ninth Circuit upheld his claim on appeal of its dismissal.325 The case remains pending.326
In employment contexts, the ADA prohibits employers from discriminating against individuals who, with or without reasonable accommodation, can perform the essential functions of employment positions.327 Employers are further prohibited from conducting medical examinations or making inquiries of job applicants and employees as to whether they have a disability or the nature or severity of such disability unless the examinations or inquiries are job-related.328 However, employers may conduct examinations and make inquiries after a job offer has been made (but before employment has begun) for any reason, and they may condition the offer on the results so long as any exclusionary criteria that are applied are job-related and consistent with business necessity.329
From fiscal years 2010 to 2017, the EEOC, which also is responsible for enforcing alleged violations of the employment discrimination provisions of the ADA, received approximately 25,000 ADA-related complaints each year.330
B. State Statutes
GINA and the ADA establish a floor of minimum protection against health-related discrimination and do not preempt state laws that provide equal or greater protection.331 Over the years, many state anti-discrimination statutes have been enacted that vary widely in scope and applicability.
Focusing on genetic discrimination, some states have enacted anti-discrimination statutes limited to specific genetic conditions. For example, in 1975, North Carolina became the first state to prohibit employment discrimination against individuals with sickle cell trait or
___________________
324See Second Amended Complaint for Damages, Case No. 4:13-cv-04129-CW (N.D. Cal.) (filed Feb. 28, 2014).
325See Memorandum, Chadam v. Palo Alto Unified School District, No. 14-17349, D.C. No. 4:13-cv-04129-CW (9th Cir. 2016).
326 A 5-day jury trial has been scheduled for September 2018. See Jennifer K. Wagner, Keeping an Eye on “Perceived Disability” Litigation in California: Chadam v. Palo Alto Unified School District, GENOMICS L. REPORT (May 2, 2017), https://www.genomicslawreport.com/index.php/2017/05/02/keeping-an-eye-on-perceived-disability-litigation-in-california-chadam-v-palo-alto-unified-school-district.
327See 42 U.S.C. §§ 12111(8), 12112(a)-(b).
328See id. §§ 12112(d)(1)-(2), 12112(d)(4).
329See id. § 12112(d)(3). The ADA’s regulation of employer medical examinations and inquiries based on stage of employment are described in Mark. A. Rothstein, Innovations of the Americans with Disabilities Act: Confronting Disability Discrimination in Employment, 313 J. AM. MED. ASS’N 2221 (2015).
330See EEOC, Americans with Disabilities Act of 1990 (ADA) Charges, EEOC.GOV, https://www.eeoc.gov/eeoc/statistics/enforcement/ada-charges.cfm (last visited Feb. 2, 2018).
331See 42 U.S.C. § 2000ff-8(a)(1) (GINA employment provisions); 42 U.S.C. § 12201(b) (ADA).
hemoglobin C trait.332 States like New Jersey have expanded this list to also include thalassemia trait, Tay-Sachs trait, and cystic fibrosis trait.333
Most states, however, have enacted general laws that prohibit employment and/or insurance discrimination based upon genetic test results or “genetic status.”334 According to NHGRI, 35 states and the District of Columbia have enacted statutes that limit unwanted access to and/or discriminatory use of genetic information in employment contexts, and 48 states and the District of Columbia have enacted similar statutes limiting these activities in health insurance contexts.335 Moreover, 24 states have passed laws regulating genetic discrimination by life, disability, or long-term care insurers.336 In 2011, California passed the most comprehensive anti-discrimination statute to date, the California Genetic Information Nondiscrimination Act (CalGINA), which prohibits genetic discrimination in emergency medical services, housing, mortgage lending, and state-funded programs, including public education.337
___________________
332See Karen Rothenberg et al., Genetic Information and the Workplace: Legislative Approaches and Policy Challenges, 275 SCIENCE 1755, 1755 (1997) (citing N.C. GEN. STAT. ANN. § 95-28.1 (West 2018)).
333See N.J. STAT. ANN. §§ 10:5-5(x), 10:5-12(a) (2017).
334See, e.g., ARIZ. REV. STAT. ANN. § 41-1463(B)(3) (2017) (prohibiting employment discrimination against individuals based on the results of genetic tests received by employers); FL. STAT. ANN. § 627.4301(2)(a) (West 2017) (prohibiting health insurers from canceling, limiting, or denying coverage, or establishing differentials in premium rates, based solely on genetic information).
335See NHGRI, Table of State Statutes Related to Genomics. GENOME.GOV, https://www.genome.gov/27552194/table-of-state-statutes-related-to-genomics (last updated Oct. 11, 2017).
336See id.
337See California Genetic Information Nondiscrimination Act, 2011 Cal. Legis. Serv. ch. 261 (West 2011) (Senate Bill No. 559); see also Jennifer K. Wagner, Genetic Discrimination Case Against School District is Appealed to Ninth Circuit, GENOMICS L. REPORT (Feb. 2, 2016), https://www.genomicslawreport.com/index.php/2016/02/02/genetic-discrimination-case-against-school-district-is-appealed-to-ninth-circuit (explaining CalGINA’s applicability to educational contexts).
This page intentionally left blank.












































