
3
Laboratory Quality Systems for Research Testing of Human Biospecimens
Many expert groups have agreed that if individual research results are to be returned to participants, the test results should have a high level of validity (Bookman et al., 2006; Green et al., 2013; Jarvik et al., 2014; PCSBI, 2013). The validity of a result depends on the test used and the laboratory environment in which the test is conducted. Research using human biospecimens is conducted in a broad range of laboratory types where, due to the nature of the research and regulatory requirements, the quality management systems (QMSs)1 in place vary significantly. For example, certain types of research settings and questions, such as field-based studies, may require less documentation with respect to pre-analytic, analytic, and post-analytic quality measures and reporting capabilities (see Figure 3-1 for details on what is included in these analytic phases) than required in clinical laboratories. This is because the Clinical Laboratory Improvement Amendments of 1988 (CLIA) regulates clinical laboratories (discussed in more detail later in this chapter). Consequently, determining whether the individual results generated over the course of research studies are valid may be challenging because documented evidence supporting result accuracy may not be available. While the committee does not expect all research laboratories to operate under a single quality standard, the lack of certainty about the research result validity is a barrier to the return of research results. Using appropriate quality processes in scientific research will be important to ensure the validity of individual research
___________________
1 Quality management systems (QMSs) are defined by the World Health Organization (WHO), the International Organization for Standardization (ISO), and the Clinical and Laboratory Standards Institute (CLSI) as “coordinated activities to direct and control a laboratory with regard to result validity and reliability” (WHO, 2011).
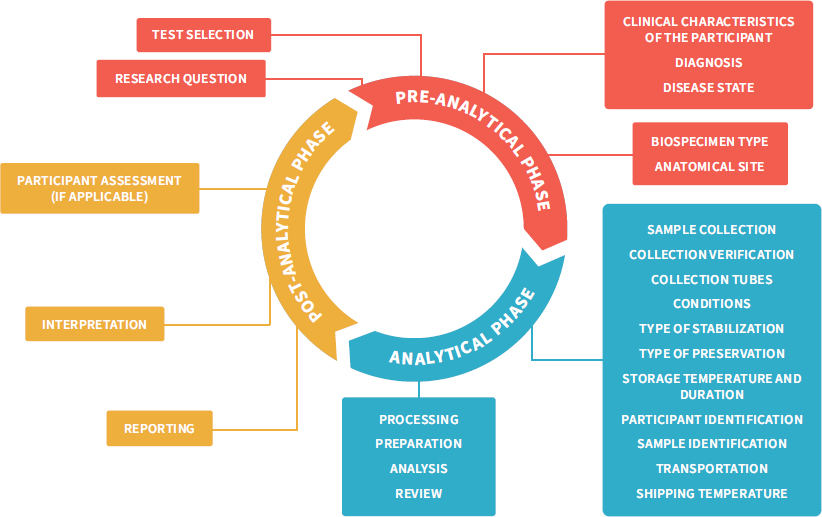
NOTES: This figure adapts the total testing process in clinical research, traditionally separated into the three phases pre-analytical, analytical, and post-analytical (Hawkins, 2012), to the broader range of research settings and questions. It highlights the total research testing process, starting with the identification of a research question followed by an investigator selecting the tests best suited to address the hypothesis; then sample collection, transport, and processing; analysis and interpretation; and finally, showing how the result can influence the development of new hypotheses or even participant care. Rigorous control measures are required throughout the pre-analytical phase to avoid errors associated with specimen handling and identification, which would result in additional errors downstream in the analytical and post-analytic phases (LabCE, 2018).
SOURCE: Figure adapted from Reisner, 2015. © McGraw-Hill Education.
results returned to participants and the integrity and quality of science more broadly. This chapter discusses the need for confidence in the validity of research results that may be returned to participants and describes the associated infrastructure needed to implement the processes to generate high-quality research results.
THE SPECTRUM OF TRANSLATIONAL RESEARCH
Translational research using human biospecimens occurs across a spectrum, ranging from the discovery of basic mechanisms of human physiology and epidemiological associations to first-in-human studies, phase I clinical trials, phase II–IV clinical trials, and implementation studies (IOM, 2013a). There are a variety of laboratory methods used for the research on this spectrum, depending on the samples analyzed and the research questions being addressed. The test methods used and the regulations with which investigators must comply, including the quality processes in place, may vary according to the particular translational phase of the research.
In seeking new physiological pathways or novel methodologies, hypothesis-driven basic research is inherently more exploratory than clinical testing and, as such, requires greater flexibility in the standard operating procedures used in the laboratory. In the conduct of such research, investigators may make frequent modifications to the test protocols in order to identify the optimal procedures, and these modifications necessitate frequent revalidations of test performance. As a result, these studies are generally not conducted under a regulated QMS. Procedurally this makes sense; however, it has implications for the reproducibility, interpretability, and validity of test results. Furthermore, as research moves from bench research on participants’ biospecimens and into more controlled clinical settings—for example, into clinical trials—human participants play a more involved role. In fact, the testing done in clinical research can affect clinical decision making or generate evidence used to gain Food and Drug Administration (FDA) approval of new drugs or devices, and therefore this testing is often performed in the regulated environment of the clinical laboratory, where protocols are more fixed and laboratory quality assurance processes are more stringent. In some cases, validated clinical tests already used in health care may be conducted during the course of a study—for example, to determine inclusion eligibility for study enrollment.
The progression to more fixed protocols and the prospect of high-quality results generated in the clinical research setting may imply that studies further along the translational spectrum are more amenable to the return of individual research results—or may simply lead to the assumption that the closer that research and test results seem to clinical application, the more reasonable it is to return research results. This assumption may also result in part from the belief that investigators conducting clinical research (and some forms of public health
research) have several or even ongoing interactions with the participants, perhaps creating a more robust investigator–participant relationship. However, this is not always the case. Some research will generate long-term relationships with participants through recurring contact and communication with the participants. The contact may occur through additional specimen collection, for instance, or follow-up visits with the trial team (i.e., in longitudinal studies). Some studies, however, may have little or no investigator–participant interaction or communication outside of the collection of a biospecimen (e.g., a one-time specimen collection for an environmental exposure study). In some situations, samples are acquired from a biobank, and, as a result, investigators may have never interacted with the participants who donated the biospecimen. In situations where the biospecimen is anonymized, return will not be possible. Further discussion of anonymized biospecimens or biospecimens acquired from a biobank is beyond the scope of this study.
THE IMPORTANCE OF ANALYTIC AND CLINICAL VALIDITY OF RESEARCH RESULTS
A core concern with the return of research results to participants relates to the potential risks of harm (physical or psychosocial) to the individual if the results are not accurate or were mislabeled and hence are not actually the results for that specific participant. Specimen labeling and handling are critically important quality issues in clinical laboratories, as errors can lead to patient injury from an incorrect diagnosis or treatment (Nakhleh et al., 2011). In research, sample mislabeling can lead to invalid comparisons and problems with reproducibility (Toker et al., 2016). Such sample mislabeling may not affect the overall aggregate results from a research study, as long as the study is powered correctly, but it can have a critical effect in the case of individual research results; if results from mislabeled specimens are provided to the incorrect individual participants, a variety of harms can follow. For example, this can result in unnecessary clinical consultations, medical procedures, and testing that can lead to participant harm. In addition, returning results to the incorrect individual can cause the investigator to miss key information that could inform participant care (Epner et al., 2013). As a result, possible errors in specimen labeling and handling are a central concern in debates on returning individual research results.
This type of risk is ameliorated, although not completely avoided, in the case of clinical test results because of processes in clinical laboratories that are in place to reduce the likelihood of mislabeling or other errors and to otherwise ensure the validity and quality of individual clinical test results (Agarwal, 2014). Specifically, clinical laboratories have requirements for personnel training and ongoing competency, equipment validation and maintenance, testing facility operations (e.g., written document control logs and standard operating procedures), establishing and verifying performance, the specifications of tests across different patient
groups, the retention of records and reports, specimen transport and management, result reporting requirements, and personnel safety.2 Clinical laboratories must assess and document test precision, reliability, accuracy, sensitivity, and specificity as well as reference ranges and other test characteristics relevant to each test. Together, these requirements form the basis of a quality system that supports the analytic validity of the tests performed in the laboratory.
These quality standards required for clinical laboratories were established by the Centers for Medicare & Medicaid Services (CMS) to protect patients. CMS stipulated that laboratories that report patient-specific test results that will be made available for “the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings” are required to meet the CLIA3 quality standards or an equivalent or superior standard of an approved accrediting organization (Yost, 2003), as discussed further in Chapter 6. These standards help ensure the validity of laboratory test results. The more limited quality-assurance processes in place in many research laboratories pose a problem for the return of research results to participants because the validity of an individual test result may be difficult to assess. The fact that science as a whole is examining its own performance in the area of research rigor and reproducibility and finding itself falling short of its expectations and goals (Baker, 2016; Begley et al., 2015; McNutt, 2014) (as discussed in more detail later in this chapter) lends credence to concerns over the effectiveness of quality procedures in research laboratories in general. Certainly, the relative lack of rigor is not characteristic of all research laboratories. However, the variability that exists from laboratory to laboratory—thanks to the absence of a minimum QMS requirement akin to the requirement that clinical laboratories must meet CLIA standards—creates uncertainty concerning the validity of test results from research laboratories.
A test’s analytic validity (AV) and clinical validity (CV) are key considerations in deciding whether to return a research result based on that test (Bookman et al., 2006; Ravitsky and Wilfond, 2006) and may also inform the framing of information that accompanies the research results when they are returned (discussed in more detail in Chapter 5). AV refers to the ability of a test to measure what it is designed to measure (NASEM, 2017a). Results from tests lacking AV may be inaccurate and therefore misleading. Consequently, most expert groups have agreed that it is not appropriate to return results that lack credible evidence demonstrating AV (Fabsitz et al., 2010; Jarvik et al., 2014). CV is a measure of a test’s ability to identify or predict accurately and reliably the clinically defined disorder or final health or medical outcomes of interest in an individual (NASEM, 2017a). CV describes, for example, whether a biomarker being tested is associated with a disease, outcome, or response to treatment, and it takes into account clinical sensitivity (the ability to identify those with the disease), clinical specificity (the
___________________
2 42 C.F.R. § 493.
3 42 C.F.R. § 493.
ability to identify those without the disease), and the positive and negative predictive value of the test (NASEM, 2017a). In the context of genetic testing, the National Institutes of Health (NIH) defines CV as “how well the genetic variant being analyzed is related to the presence, absence, or risk of a specific disease” (NIH, 2018). CMS does not require a demonstration of CV for a laboratory to meet CLIA requirements (Willmarth, 2015). However, FDA, which is concerned with the safety and effectiveness of diagnostic tests, may review the CV of a test to make sure that it identifies, measures, or predicts the presence or absence of a clinical condition or predisposition toward disease in a patient (FDA, 2018; Gottlieb, 2018). If the CV of a test is not established, the meaning of any result from that test for an individual will not be clear, raising questions about how, or whether, the result should be returned. One challenge with research results is that the AV and CV of the tests are not always known, even when the tests are performed under a QMS; indeed, establishing AV or CV may in fact be one of the purposes of a given research study.
Establishing Analytic Validity
Methods for establishing the AV of a test may include (Jennings et al., 2009; NASEM, 2017a)
- comparison to another test measuring the same analyte;4
- the use of controls that contain the analyte or biomarker (possibly using controls that contain a specific amount of the analyte if the test is quantitative);
- the use of human samples known to contain the analyte or biomarker; or
- the use of samples which are admixed with a known amount of analyte to simulate a patient sample that contains the analyte.
The demonstration of AV also requires assessing a range of specimens that are known not to contain the analyte—especially specimens from patients with similar diseases who may also be tested in a clinical setting to reach a diagnosis—in order to evaluate the incidence of false-positive results (Jennings et al., 2009). However, obtaining the controls and samples of varying concentrations of analyte for establishing AV can be challenging (IOM, 2012; Mattocks et al., 2010). Some analytes, such as glucose or blood gases, are unstable and can only be accurately measured under strictly controlled specimen collection and handling conditions (Nichols, 2011). Collection, storage, and even the type of preservative used to collect the sample can affect the stability of analytes or the test accuracy. Some
___________________
4 Analyte: any substance or constituent being subjected to analysis or on which the laboratory conducts testing (Segen’s Medical Dictionary, 2011).
rare diseases make finding positive samples with the relevant analyte a challenge (Jennings et al., 2009; Maddalena et al., 2005). AV must take into account all specimen types that will eventually be used for testing, the cost of analysis, automation, and labor. In addition, assessing the reproducibility of test results that are performed over time, by different technologists, or at different test sites using different instruments is part of the analytic validation of a test (IOM, 2012). Some methods that are highly automated and inexpensive, such as testing for sodium or glucose levels in blood, may be validated using hundreds of samples. However, for tests with more labor-intensive or computationally complex methods, it may be necessary to validate with fewer samples (Jennings et al., 2009). The extent of AV determines how each performance characteristic of the test is known, including its accuracy, precision, linear range, limit of detection, and interference potential (Jennings et al., 2009; Magnusson and Ornemark, 2014). If AV has not been established or if only limited validation studies have been performed, the reliability of the individual results is affected.
The final step of a validation process is to write a standard operating procedure (SOP) that describes all aspects of performing the test (IOM, 2012). This includes detailing the controls to be run with each test or batch; the steps in the testing process, including the exact quantities of reagents and the timing of incubations or other steps; the validation of new lots of reagents; the documentation of testing steps each time that the test is run; and reporting requirements. Other aspects of a QMS ensure that the test SOPs are being followed each time testing is performed (WHO, 2011).
Establishing Clinical Validity
When evaluating the CV of a test, AV is assumed to have been established. As a result, the clinical scenario becomes the important consideration (Jennings et al., 2009; NASEM, 2017a). For example, the CV of a given test can depend on the purpose of the test, such as whether it is diagnostic, prognostic, or predictive. CV is generally established through the testing of positive and negative control samples as well as through testing specimens from the population being studied with and without the disease, biomarker, or analyte that the test detects (Chen et al., 2009). Case control studies and longitudinal cohort studies may be used to establish CV in the diagnostic setting as long as the number of cases is sufficient (NASEM, 2017a). For prognostic and predictive tests, CV can be established with, respectively, longitudinal observational studies and clinical trials. Prognostic CV also can be established with the control arm of a clinical trial. CV can evolve over time, with additional use of the test for purposes or diseases other than those initially assessed during test validation, especially as the test is used in clinical practice and further characterized through additional research (NASEM, 2016).
Implications of Pretest Probability for Test Result Interpretation
The prevalence of a disease in the population being tested determines the predictive value of a positive test (Flyn, 1996; Jennings et al., 2009). The same test may have very good clinical performance in a high-risk population but perform poorly in predicting disease in a low-risk population. For example, a positive HIV test result is more likely to be a true positive for patients in a high-risk sexually transmitted disease clinic than for those in a low-risk population (e.g., a population that is not sexually active), where the same positive HIV test is more likely a false positive (Irwig et al., 2008). Thus, for proper context the interpretation of a test result must consider pretest probability, the patient history, and symptomology. Test results in isolation of a patient’s history have a higher probability of being misinterpreted (Flyn, 1996). In the context of clinical care, clinicians order tests because a patient presents with symptoms consistent with or indicative of a particular disease. In the research context, testing is often conducted to prove a hypothesis or to support a study. Testing is not necessarily conducted, or ordered, based on a patient’s pretest probability of disease and likelihood of treatment (because actions and treatment are often dictated as part of the trial protocol). Thus, pre-test probability may affect whether a research result is likely to be a true positive for an individual research participant.
LABORATORY QUALITY SYSTEMS TO INCREASE CONFIDENCE IN THE VALIDITY OF RESEARCH RESULTS
Research and clinical laboratories are held to different regulatory standards because of their different recognized purposes (Burke et al., 2014; Clayton and McGuire, 2012). Therefore, research and clinical tests are often conducted in vastly different laboratory environments—although clinical laboratories can also perform research tests.
Many research laboratories are designed to train new researchers, including graduate students and postdoctoral fellows (Bosch and Casadevall, 2017; NASEM, 2017b). They also allow for creativity and flexibility in laboratory protocols in order to foster an environment that can make cutting-edge discoveries. To this end, research laboratories have a more innovative culture with regards to assays and testing protocols. Investigators may make frequent modifications to a testing procedure in order to optimize methodologies (e.g., a novel assay may be developed for more rapid or sensitive detection of a disease) or to answer a specific scientific question. These practices are distinct from those of clinical laboratories, which do not experiment with testing methods and are required to train their staff in quality essentials, process control, documentation, and common sources of pre-analytic and analytic error in order to help maintain specimen control, assay
validity, and reproducibility (American Academy of Family Physicians, 2018).5 Clinical laboratories are regulated to ensure adherence to more stringent quality standards because their results are designed to be used for clinical decision making. By contrast, the needed flexibility in many research laboratories means that their practices may not align with those required of clinical laboratories.
Research laboratories lacking CLIA certification may still maintain high standards for quality, and in some cases their quality assurance and control processes may even exceed the quality requirements established by CLIA. For example, the committee heard at its public workshop in September 2017 that some genome sequencing results generated by the Clinical Sequencing Evidence-Generating Research (CSER) consortium were less prone to error than results from CLIA-certified laboratories. This was because the CSER laboratory was highly automated and less prone to human error than the CLIA laboratory that was conducting the confirmation testing and not using the same automation.6 While this laboratory was highly automated with set SOPs, other research laboratories may not have established internal operational standards or may not meet any formal recognized quality standards. The lack of adherence to a formal standard limits investigators’ ability to demonstrate the validity of their results.
CLIA
The CLIA requirements for clinical laboratories ensure the quality and integrity of clinical testing, accurate documentation of test validation and test performance, and the comparability of test results regardless of the personnel conducting the testing or the test location.7 To achieve CLIA certification, laboratories are required to have various systems in place to meet the standards for AV (American Academy of Family Physicians, 2018),8 but the regulations do not prescribe the design or implementation of those systems. They do not, for example, define specific methods or standards for how to demonstrate the performance characteristics of a test. The laboratory director is required to meet all CLIA regulatory standards for quality and safety and is held accountable to CLIA inspectors who perform on-site assessments of regulatory compliance of
___________________
5 42 C.F.R. § 493.
6 Testimony of Rex Chisholm of Northwestern University, eMERGE, at the public meeting of the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on September 6, 2017.
7 The enactment of CLIA 1988 and the regulation of clinical laboratories on a national scale followed public outcry in response to scandals reported in The Wall Street Journal involving commercial laboratories that had inaccurately analyzed Pap smears resulting in the deaths of several women (Yost, 2003).
8 42 C.F.R. § 493.
non-waived testing every 2 years.9 The laboratory director is also accountable to the physicians and patients who rely on the quality of the laboratory services. This accountability and partnership between the laboratory director and the clinician is critical to protecting patient safety and strengthens confidence in the reliability of any test results used for clinical care.
A laboratory seeking CLIA certification or accreditation must apply for a certificate, pay a biennial fee (which can be as low as $150, but depends on the type of laboratory and tests performed there) (American College of Physicians, 2014), comply with the regulatory requirements of CLIA or another accrediting agency recognized by CLIA, and agree to be inspected at least every 2 years. Depending on the complexity of the test methods used in a laboratory, those checking compliance with the regulatory standards will examine such things as analytic validation of test performance; written SOPs with documentation showing that the SOPs are being followed; staff qualifications, training and ongoing educational requirements, and regular ongoing competency assessments; instrument validation and maintenance; the proper handling and verification of reagents; general laboratory safety; and a QMS to control the handling and processing of samples from test order through collection, transportation to the laboratory, processing, analysis, reporting of test results, and investigation of issues of non-conformance with laboratory SOPs or other errors.10 In addition, CLIA-certified laboratories performing non-waived testing are required to participate in proficiency testing on a regular basis using specimens sent from an external source approved by CMS (CMS, 2014b). Alternative methods for proficiency testing can be used by clinical laboratories when external proficiency testing is not available. Proficiency testing is required a minimum of twice per year, using multiple samples for each assessment, and must be conducted for each test performed by the laboratory (CMS, 2014b).
Although CLIA has significantly improved the quality of clinical laboratory results used in medical decision making (Ehrmeyer and Laessig, 2004), its requirements are not always appropriate for the kinds of testing performed in the research context, such as tests relevant to biomonitoring for environmental contaminants (NRC, 2006; Ohayon et al., 2017). One investigator noted that “CLIA certification does not cover lab work for the majority of chemicals measured in biological or environmental media samples, but rather is primarily relevant for diagnostic and treatment-related tests such as genetic screening and cholesterol measurements” (Ohayon et al., 2017, p. 144). While laboratories may meet the quality controls and assurance that CLIA mandates, “the lack of well-validated
___________________
9 On-site testing every 2 years is required for laboratory sites performing non-waived testing. Certificate of waiver (COW) laboratories and provider performed microscopy facilities are not routinely inspected or surveyed (CMS, 2014a). More than half of CLIA certificates are for COWs (184,298), sites limited to performing only waived tests. Approximately 16,000 sites have a certificate of accreditation for more complex testing (CMS, 2018).
10 42 C.F.R. § 493.
methods for measuring some cutting-edge biomonitoring analytes precludes their ability to be accredited” (Ohayon et al., 2017, p. 144).
Moreover, with the rapid pace of technological innovation (e.g., DNA sequencing technologies), CLIA regulatory requirements are outdated (Ferreira-Gonzalez et al., 2008). For example, CLIA requirements are “inadequate to ensure the overall quality of genetic testing because they are not specifically designed for genetic tests and because they do not give sufficient emphasis to pre- and post-analytic phases of testing” (Task Force on Genetic Testing, 1997). One particular concern is that current CLIA requirements do not address the complexity of the informatics analyses, interpretation, and reporting that are required for next-generation sequencing (NGS) technologies or other omics testing (Gargis et al., 2012). Addressing these gaps has been a focus of several U.S. and international workgroups (Aziz et al., 2014; Euformatics, 2017; Gargis et al., 2012; Rehm et al., 2013; Task Force on Genetic Testing, 1997). However, NGS testing is just one example of how technology, including newer “omics” technology, is rapidly influencing research and health care more broadly. The challenge will be for regulators and their requirements for quality systems to keep pace with the rapidly changing clinical testing environment.
Some organizations that CMS has approved to issue CLIA accreditation have quality standards for NGS tests and other more complex testing methods. CLIA allows CMS-approved organizations to inspect and otherwise ensure that CLIA requirements are met by the clinical laboratories when the requirements of the accrediting organizations are equal to or more stringent than CLIA requirements (CMS, n.d.; Yost, 2003). Accrediting organizations with the authority to certify clinical laboratories under CLIA include the College of American Pathologists, American Association for Laboratory Accreditation, COLA, and others (CMS, n.d.). The College of American Pathologists and the New York State Department of Public Health are two examples of accrediting organizations that have quality standards for NGS tests and other more complex testing methods (Aziz et al., 2014; New York State Department of Health, 2016). Thus, laboratories conducting cutting-edge research and considering pursuing CLIA certification may find more value in certification through an accreditation organization with standards that align with the testing performed in research laboratories rather than with CLIA.
CLIA and the Return of Individual-Specific Research Results
Under the current CMS interpretation of CLIA, if research laboratories return individual research results to participants, the laboratory must be CLIA certified. While the direct cost of CLIA certification is not prohibitory, meeting the requirements to obtain the certification by compliance with all of the regulatory standards would come with significant costs for most research laboratories (Barnes et al., 2015), although the extent of the burden would depend on the
infrastructure and processes already in place in the laboratory.11 Most research laboratories operate under the direction of a single principal investigator (PI), and many investigators have never worked in a clinical laboratory setting and may not be familiar with the quality procedures, proficiency testing, and software required for CLIA certification. To begin the process of becoming CLIA certified, each PI would likely need to hire a consultant to provide guidance, pre-inspection evaluations, competency evaluations, and laboratory management plans as well as to obtain proper software for logging laboratory samples, reagents, and other processes (Riedl and Dunn, 2013; Robins et al., 2006). To obtain this type of guidance and to assemble the necessary infrastructure for meeting CLIA standards, including laboratory personnel requirements, would require funding and institutional support. This could divert research resources from the conduct of research to the process for CLIA certification. The Secretary’s Advisory Committee on Human Research Protections (SACHRP) considered the challenges that would be associated with research laboratories wishing to return results becoming CLIA certified and concluded that it would not be realistic, “as it would impose tremendous, new transaction costs on research and could even lead to the elimination of some research laboratories and the consolidation of others, which would reduce research opportunities overall” (SACHRP, 2015).
Research tests used in clinical decision making, such as some tests conducted in the course of a clinical trial, must be performed in a CLIA-certified laboratory. As alternatives to pursuing CLIA certification, investigators can outsource testing to a CLIA-certified laboratory, can have only those results that will be returned retested in a CLIA-certified laboratory for verification prior to disclosure, or can build or modify an existing laboratory or core facility to make it CLIA compliant. Not all clinical laboratories will have the resources to validate and perform research tests, and sometimes no equivalent test exists, in which case other clinical tests may not be available to assess the significance of the research result. In the latter situation, creating a CLIA-compliant core facility would not be without challenges. For example, the University of Maryland School of Medicine worked to make a genomics core facility CLIA-compliant. A report on that experience concluded that it was “not without difficulty” and offered a list of challenges that anyone taking on such a task could expect to face, including the need for
(1) a CLIA-qualified director as well as qualified key personnel; (2) appropriate space to allow for a unidirectional work flow separating pre- and post-amplification processes; (3) developing a validation study and implementation for each assay offered and participating in a CLIA-approved proficiency test or sample exchange program; (4) having an experienced quality program manager to oversee the quality program and document management system; (5) having
___________________
11 Testimony of Karen Dyer of CMS at an open session of Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on July 19, 2017.
the financial resources to invest in developing and operating this unique regulatory environment. (Ambulos, 2013, p. S21)
Given the diversity of research activities that use human biospecimens, it may not be reasonable to expect that all research laboratories performing testing on human biospecimens meet CLIA standards in order to return research results, particularly given the operational challenges of becoming CLIA certified discussed above. If a laboratory plans to return results that are not intended for clinical decision making in a study protocol, the laboratory should consider the use of other quality systems (see Box 3-1 on the committee definition of results not intended for clinical decision making in a study protocol). For the purposes of this report, results not intended for clinical decision making in a study protocol include results that have no known or established clinical implications as well as those with potential medical value but which require confirmation prior to a clinical response.
While other recognized laboratory quality standards, such as those from the ISO,12 allow flexibility in approaches while still supporting technical rigor (Thelen et al., 2015), they generally have requirements that are similar to or even more stringent than those of CLIA (see Table 3-2 at the end of this chapter).13 Consequently, adopting such standards may present hurdles similar to CLIA certification for investigators who want to return research results. For an investigator or institution considering implementing a recognized laboratory quality standard, many factors should be considered, such as legal obligations to obey state and federal laws, the type of laboratory, the type of testing, cost, institutional support, training, and other variables. Put simply, “one-size quality program does not fit all” (NCI, 2016). In fact, given the variety of laboratory tests, including the use and development of cutting-edge tests and the assessment of novel analytes, the quality systems used by clinical laboratories may not be the most appropriate for research laboratories; however, they may serve as guidance for laboratories considering the adoption of quality practices.
While CLIA is especially critical when results will be used in clinical decision making, many research laboratories are generating results that are not for use in clinical decision making. In these cases, the best way to ensure laboratory quality controls are in place may be through the adoption of a QMS designed specifically with research laboratories in mind and tailored to the nature of the research being conducted. There is a great deal of momentum in this area and immense interest in improving research laboratory rigor and quality, although an alternative quality management system for research laboratories has not yet been established.
___________________
12 ISO accreditation is not a legally permitted alternative to CLIA certification for laboratories conducting clinical testing in the United States.
13 Testimony of Randy Querry of the American Association for Laboratory Accreditation at a public webinar of the at the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on December 7, 2017.
CONCLUSION: When individual research results are intended for use in clinical decision making, tests must be performed in laboratories that are CLIA certified.
CONCLUSION: When individual research results are not intended for use in clinical decision making in a study protocol, CLIA certification may not be an appropriate or necessary mechanism to ensure that research test results are of sufficient quality to permit their return. However, no alternative accepted quality standard exists for such research laboratories.
Establishing Quality Management Systems for Biomedical Research Laboratories
Human biospecimens in research
are subject to a number of different collection, processing, and storage factors that can significantly alter their molecular composition and consistency. These biospecimen pre-analytical factors, in turn, influence experimental outcomes and the ability to reproduce scientific results. Currently, the extent and type of information specific to the biospecimen pre-analytical conditions reported in scientific publications and regulatory submissions vary widely. To improve the quality of research utilizing human tissues, it is critical that information
regarding the handling of biospecimens be reported in a thorough, accurate, and standardized manner. (Moore et al., 2011, p. 57)
These pre-analytical procedures are especially critical when individual research results will be returned to participants, as it documents sample handling, which contributes to confidence that the sample was processed appropriately in a way that preserved the analyte being tested and ensured that the result belongs to a specific individual participant.
In academic biomedical research laboratories, laboratories are not centrally regulated, and the PI sets requirements for quality and monitors compliance with those requirements (Bosch and Casadevall, 2017), although research sponsors or scientific journals may mandate quality standards as part of funding or publication requirements, respectively.14 The lack of common regulation is not a flaw in the system. Rather, the validation of research results is expected to occur through integrity in the scientific process. In this system, results are verified by peer review and confirmed by other scientists who replicate the results. “In other words, there is no official seal of approval—quality assurance comes from the expert judgment of communities of scientists, who are supposed to be able to filter out the good from the bad on their own” (White, 2015).
However, the widely reported concerns regarding the lack of reproducibility in science may drive changes in the requirements for research laboratories and motivate the development of quality standards or the training of PIs in basic quality management (Begley et al., 2015; Calabrese and Palm, 2008; Collins and Tabak, 2014; Davies et al., 2017; Loew et al., 2015; McNutt, 2014; NIH, 2017b; Titus and Bosch, 2010). The reasons behind the reported reproducibility problems are varied and may include increased scrutiny, the complexity of experiments and statistical methods, pressures on investigators to publish leading to inadequate repetition or even falsified data (Baker, 2016), and a failure to embrace best practices in preclinical study design (Grens, 2017; Vahidy et al., 2016). The lack of reproducibility in biomedical research is concerning because it impedes the translation of research discoveries into clinical practice (Perry and Lawrence, 2017). Some contributing factors can be controlled by implementing quality measures or adopting a QMS.
Although most research laboratories are not formally regulated, some nascent efforts are encouraging the adoption of voluntary quality assurance processes to improve the integrity of the science and to address issues with reproducibility (Calabrese and Palm, 2008; Freedman and Inglese, 2014; Glick and Shamoo, 1993; Herman and Usher, 1994; Scientific Working Group on Quality Practices in Basic Biomedical Research, 2001; Volsen et al., 2004). NIH, for example, has
___________________
14 Testimony of Rebecca Davies of the University of Minnesota at a public webinar of the Committee on the Return of Individual-Specific Research Results Generated in Research Laboratories on December 7, 2017.
acknowledged that reproducibility is an issue and is taking steps to address quality in pre-clinical research as well as in research on biospecimens (Collins and Tabak, 2014; Engel et al., 2014; NCI, 2011). In fact, a workshop held at the National Cancer Institute (NCI) that discussed biospecimen reporting standards resulted in the development of the Biospecimen Reporting for Improved Study Quality (BRISQ) guidelines. These reporting requirements detail the elements that should be reported in order to improve the evaluation and quality of the data generated from biospecimens. The elements that were identified are tiered and
prioritized according to the relative importance of their being reported. The first tier, items recommended to report, includes information such as the organ(s) or the anatomical site from which the biospecimens were derived and the manner in which the biospecimens were collected, stabilized, and preserved. . . . Each reporting element included in [the] guidelines is backed by evidence that the factor could have an effect on the integrity and molecular characteristics of the biospecimen or on the ability to perform certain assays on the biospecimen and obtain reliable results. (Moore et al., 2011, p. 59)
Tier 1 BRISQ reporting requirements are shown in Table 3-1. While this list does not include all of the biospecimen quality requirements that should be met for the return of research results, it does provide an example foundation of simple-to-implement procedures that laboratories conducting research on biospecimens can begin executing now in order to improve pre-analytic quality.
Several other organizations, including many outside the United States, are also working in this area. In 2013, for example, the Global Biological Standards Institute published The Case for Standards, which emphasizes the benefits of adopting widespread quality standards in order to improve the quality of the biological sciences (Global Biological Standards Institute, 2013). WHO issued a handbook on quality practices in basic biomedical research outlining for
institutions and researchers the necessary tools for the implementation and monitoring of quality practices in their research, thus promoting the credibility and acceptability of their work. The handbook highlights non-regulatory practices that can be easily instituted with very little extra expense. (Scientific Working Group on Quality Practices in Basic Biomedical Research, 2001, p. 2)
Several ongoing initiatives in Europe are aimed at producing guidance and recommendations to assist investigators in meeting quality management essentials (EQIPD, 2017; PAASP, 2018b). Box 3-2 briefly describes several European initiatives that have been set up to establish standardized quality practices and improve data quality in research.
In the United States there is currently no standardized QMS for biomedical research laboratories that accommodates the wide variation in laboratory procedures, that can be adopted or modified to suit the study context, and that
| DATA ELEMENTS | EX AMPLES |
|---|---|
|
Serum, urine |
| Solid tissue, whole blood, or another product derived from a human being | |
|
Liver, antecubital area of the arm |
| Organ of origin or site of blood draw | |
|
Diabetic, healthy control |
| Controls or individuals with the disease of interest | |
|
Premenopausal breast cancer patients |
| Available medical information known or believed to be pertinent to the condition of the biospecimens | |
|
Postmortem |
| Alive or deceased patient when biospecimens were obtained | |
|
Breast cancer |
| Patient clinical diagnoses (determined by medical history, physical examination, and analyses of the biospecimen) pertinent to the study | |
|
HER2-negative intraductal carcinoma |
| Patient pathology diagnoses (determined by prior to research use) pertinent to the study | acro- and/or microscopic evaluation of the biospecimen at the time of diagnosis and/or |
|
Fine-needle aspiration, preoperative blood draw |
| How the biospecimens were obtained | |
|
Heparin, on ice |
| The initial process by which biospecimens were stabilized during collection | |
|
Formalin fixation, freezing |
| The process by which the biospecimens were | ustained after collection |
|
10 percent neutral-buffered formalin, 10 U.S. Pharmacopeia heparin units/mL |
| The make-up of any formulation used to maintain the biospecimens in a nonreactive state | |
|
−80°C, 20°C to 25°C |
| The temperature or range thereof at which the biospecimens were kept until distribution/analysis. | |
|
8 days, 5–7 years |
| The time or range thereof between biospecimen acquisition and distribution or analysis | |
|
−170°C to −190°C |
| The temperature or range thereof at which biospecimens were kept during shipment or relocation | |
|
Minimum 80 percent tumor nuclei and maximum 50 percent necrosis |
| Parameters used to choose biospecimens for the study | |
SOURCE: Moore et al., 2011.
documents the data elements that affect result validity. The development of such a system would allow investigators, journal editors, research sponsors, and regulators to better evaluate, compare, and reproduce experimental results, thereby bolstering confidence in the validity of research results that may be returned to participants. The potential benefits to the research enterprise extend beyond the issue of return of results and address a broader need for improved reproducibility and rigor (as discussed in this section above). But while this would improve research, it is important to note that any results that were to be used in clinical decision making in a study protocol would still need to be generated in a CLIA-certified laboratory in order to protect patient safety.
As detailed above, support is growing among investigators, sponsors, and regulators in the United States for the development of a standardized QMS for biomedical research laboratories. However, given the myriad of interested and invested parties, a coordinated effort will be needed with all stakeholders at the table to provide input on the quality system elements and key implementation processes. Many government agencies, private institutions, pharmaceutical companies, and patient organizations are sponsoring and participating in research that
would benefit from the development and use of a QMS for biomedical research laboratories. A joint effort by these stakeholders would increase efficiency and avoid waste, redundancy, and confusion on the part of investigators from different quality standards across sponsors and funding agencies. Investigators funded by both an NIH and a Centers for Disease Control and Prevention (CDC) grant, for example, would not need to implement different quality practices because both agencies would have agreed upon and would require the same QMS.
NIH, as the predominant sponsor of biomedical research in the United States, would be an obvious choice to lead this effort, especially given the groundwork already begun by NCI (Moore et al., 2011; NCI, 2011). The engagement of the relevant federal agencies, including CMS, FDA, and CDC, as well as nongovernmental stakeholders such as the Patient-Centered Outcomes Research Institute, private-sector research sponsors, scientific professional societies, and participant and patient advocacy groups will be critical to ensuring that diverse perspectives are taken into account and thus to ensure that the QMS will be broadly applicable across all types of biomedical research. The involvement of the relevant stakeholders at the earliest steps and throughout the development and into the implementation of the new QMS may help to mitigate the kinds of concerns about lack of flexibility and applicability that arose in 2014 when NIH released new guidelines for preclinical research intended to address rigor in research and scientific publishing (Baker, 2015; Haywood, 2015; NIH, 2017a).
Recognizing that the purposes and methodologies of laboratories engaged in biomedical research are highly variable, the committee does not expect that the required quality practices for laboratories across the translational spectrum should be the same. The committee stresses the importance of a tiered system, akin to that recommended for the BRISQ reporting requirements (discussed above, see Table 3-1). One could envision rubrics for quality being developed that could be adjusted based on the nature of the research and on whether results will involve human biospecimens or be returned to research participants. Attention will need to be given to allowing flexibility in the biomedical research QMS so that it can evolve and be updated over time, as the system is adopted and used by more laboratories and as new technologies are developed. An ongoing advisory committee to review and provide guidance on needed updates, analogous to the Clinical Laboratory Improvement Advisory Committee (CLIAC)15 (CDC, 2018a), may be considered. It is not just technologies that will evolve, however, but science as well, and as scientific knowledge progresses, interpretations and terminologies will regularly need to be verified and harmonized across disciplines. NCI’s Thesaurus
___________________
15 “CLIAC, managed by the Centers for Disease Control and Prevention (CDC), provides scientific and technical advice and guidance to the Department of Health and Human Services (HHS) related to improvement in clinical laboratory quality and laboratory medicine practice, as well as revision of the CLIA standards. The Committee includes diverse membership across laboratory specialties, professional roles (laboratory management, technical, physicians, nurses), and practice settings (academic, clinical, public health), and includes a consumer representative” (CDC, 2018b).
initiative, for example, conducts an ongoing review of the literature to provide reference terminology for research and clinical care that stakeholders can reference and use. Efforts to achieve consensus on definitions, terminology, and standards are also systematically occurring in the genetics field (Caudle et al., 2017; NCI, 2018; Ritter et al., 2016), and to aid in the effective implementation of the QMS across the range of biomedical research, standard terminology for research outside of the field of genetics may need to be developed (as has been done by the Clinical Data Interchange Standards Consortium [CDISC] and its data standards for Alzheimer’s disease research) (CDISC, 2011a,b). This will be particularly important if the results generated in laboratories might be used to inform clinical decision making, as clinicians may need to translate research terms to terms more commonly used in clinical practice, making data and terminology standards for interoperability particularly important. However, it should be noted that while standardization will be important, it will not ensure data quality (IOM, 2013b).
A central element of established quality management systems, such as CLIA, is a method for evaluation and external accountability to demonstrate that quality standards are being met by a laboratory. In the absence of a system for independent verification (i.e., inspection by external experts without conflict of interest or intractable bias toward any one investigator or, perhaps even bias toward the institution), determining which laboratories are adhering to quality essentials is challenging. In the development of the NIH-led QMS, stakeholders will need to consider a system of accountability. Several models could be considered, but it will be important to have a body independent of the laboratory that will perform ongoing (e.g., annual or biennial) assessments to verify that the defined standards are being met. The external monitoring function could be housed within the research institution, as is currently the case for institutional biosafety committees; it could be done through an accreditation model similar to that used for clinical laboratories; or the NIH-led stakeholder group could establish a review workgroup to conduct the assessment. Ultimately, the monitoring process will be up to the NIH-led stakeholder group, but external accountability will be critical if research results are to be returned to participants.
With proper representation, an externally accountable QMS that details best practices for laboratories across the biomedical research spectrum has the potential to improve the conduct of research, address current gaps in training and practice, and benefit the whole of the research enterprise. While potentially all research laboratories would benefit from a QMS to improve the reproducibility of their science, the committee was asked to focus on research using human biospecimens. Initiating the development and implementation of a QMS for research that tests human biospecimens would be more limited in scope and more easily tailored than a research-wide QMS. Such a program would also acknowledge the value and potential scarcity of human biospecimens and the participants who contribute to the success of the medical research enterprise.
Review of Quality Practices for the Return of Research Results
The committee recognizes that the proposed NIH-led QMS discussed above will not be immediately developed or implemented. In fact, it is likely that once such a standard is developed, the implementation of the system would take several years as investigators, institutions, and other stakeholders become familiar with the requirements and begin to establish the infrastructure, training, and other required support (infrastructure requirements are discussed in more detail below; see section “Addressing Resource and Infrastructure Needs in Research Laboratories to Enable Return of High-Quality Individual Research Results”). Given the extended time-frame necessary for implementation, institutions would benefit from the development of interim processes to assess the quality of research testing conducted by investigators planning to return results from human biospecimens. For example, an institutional review board (IRB) may still approve the disclosure of research results generated in a laboratory without CLIA certification or another recognized QMS when the quality of the laboratory analysis is deemed sufficient and the risks of return are considered low, as long as information is provided regarding the limits of the test’s validity (see Recommendation 3c). The limits of test validity will not be a standard definition and will vary based on the test used and the extent of knowledge—i.e., what is currently known about the analyte of interest, the extent to which it has been researched and published on, and what may need to be experimentally completed to provide the test in routine practice.
Implementing this type of review process will likely require training and funding for IRBs and their institutions as IRBs may not have the necessary expertise to review laboratory quality. When expertise in quality essentials is lacking, institutions will need to hire staff with the appropriate expertise, solicit training in quality management practices for their current staff, consult with an external advisor with expertise in quality practices, or work with scientific review committees so that they are able to review laboratory practices. With the proper expertise, this review could be performed through the use of a central IRB, and decisions could be expedited, when appropriate. For research laboratories at academic medical centers, the pathologists and laboratory scientists who oversee the clinical laboratories at the academic hospital may be an excellent resource for the expertise needed to review laboratory quality practices, the quality of the laboratory
tests,16 and for the implementation and oversight of a QMS in the research setting, whether it is the NIH-led QMS (see Recommendation 2) or another.
In addition to the quality practices of a laboratory, the characteristics of the tests being used will be important for IRBs to consider. Tests in the development phase (intended for either research use or clinical use) may not generate results that are appropriate for return because their validity may still be in question—once validity testing is performed, results could be returned to participants; however, the test would have to be run in the proper laboratory environment following proper protocols. Some specific test performance characteristics as described in CLIA will be important to consider. These include, but are not limited to, precision, analytic measurement range, accuracy (either diagnostic or correlation with other well-characterized assays), analytic reportable range (i.e., sensitivity at the low end or the ability to dilute high-concentration samples above the linear range and what diluents have been validated), interferences, carry-over, and clinical validity. Enabling return from novel, validated tests would require institutional oversight to ensure scientific integrity and proper research and reporting practices.17
As part of the NIH-led QMS development process, stakeholders could define a more exact role for the IRB and perhaps develop a checklist to aid in IRB review of study protocols and design. Initially this guidance could be based on the experiences of IRBs or oversight boards already involved in making decisions around the return of research results. For example, some IRBs are already playing a role in the return of research results and provide guidance for the disclosure of research results to participants from non-CLIA-certified laboratories on a case-by-case basis (Office of Ethics and Compliance, 2017). Additionally, informed cohort oversight boards (ICOBs) have been formed in response to the need of IRBs to handle the return of results—specifically, how they will be returned if they are to be returned (see Box 3-3). ICOBs are most often, but not always, associated with a biobank and may provide insight and guidance to IRBs as the return of research results becomes more expected and routine.
Regardless of their approach to the review, however, IRBs will need to assess the critical elements needed for quality assurance. A list developed by NCI of the
___________________
16 The quality of the laboratory tests could be assessed, for example, by knowing the quality systems used in the research laboratory that generated the result as well as by a review of the documentation of the handling and testing of the specific participant’s sample, similar to how it is done with CLIA.
17 There are many research results derived from research with human participants that do not entail removing a biospecimen from the body. The development of an algorithm for insulin delivery, for example, might involve measuring glucose by a glucometer that is worn continuously; efficacy of a medication to treat Parkinson’s disease may involve measuring initiation of, or frequency of, movement through a smart phone application; mobile health technologies may be important “research tests” in the future. The development of algorithms generally, and the application of valid, quality artificial intelligence tools, are important research measures for discussion; however, they were out of scope of this committee.
key elements needed for the implementation and auditing of quality assurance and quality control processes is provided in Box 3-4. It would also benefit the NIH-led QMS development process if it capitalized on the existing guidance for best laboratory practices already established for some tests, such as the American College of Medical Genetics practice guidelines for next-generation sequencing (Rehm et al., 2013). In addition, numerous documents exist that may provide guidance to laboratories considering the adoption of quality practices. In addition to CMS, NIH and CDC have issued key resources for proper laboratory procedures for management and quality (CDC, 2017, 2018a; Ned-Sykes et al., 2015; NIH, 2013), as have consensus groups such as the CLSI and international
organizations such as ISO (CLSI, 2018; ISO, 2018). These existing guidance documents may help inform investigators and institutions interested in improving the quality of research through an interim voluntary quality management system as well as IRBs and other review committees seeking guidelines for use in assessing the quality of laboratory testing.
In addition to IRBs assessing the laboratory quality other issues will need to be considered; these include determining the relative value of return—this value may be from personal and not just clinical benefits—and the relative risk (discussed in more detail in Chapter 4).
If investigators do not adhere to quality practices in the conduct of their research, it would be inadvisable for them to return research results. However, with the appropriate laboratory quality practices in place for the given research and proper institutional oversight, return is appropriate. Recommendation 2 focuses on the development of a research-grade QMS to mirror the clinical QMS that CLIA provides. A research QMS will ensure that the results returned to participants are valid and that any interpretation of the results is backed scientifically by method validation and ongoing control processes. For example, in the case of genomics this would include attention to variant interpretation as well as to laboratory procedures. The committee recommends three pathways for ensuring the appropriate return of high-quality research results; these are detailed in Recommendation 3 and shown schematically in Figure 3-2. The pathways presented in Recommendation 3 are applicable if the research results are originally planned to be returned or if they are later returned upon request of the participant. As noted in Chapters 2 and 4, participants have the right to request their results, and when individual research results are offered, participants have the right to decide whether to receive or to share their results.
CONCLUSION: For investigators conducting research testing on human biospecimens, the adoption of an externally accountable quality management system would improve confidence in result validity and help ensure that the results returned to participants are of high quality.
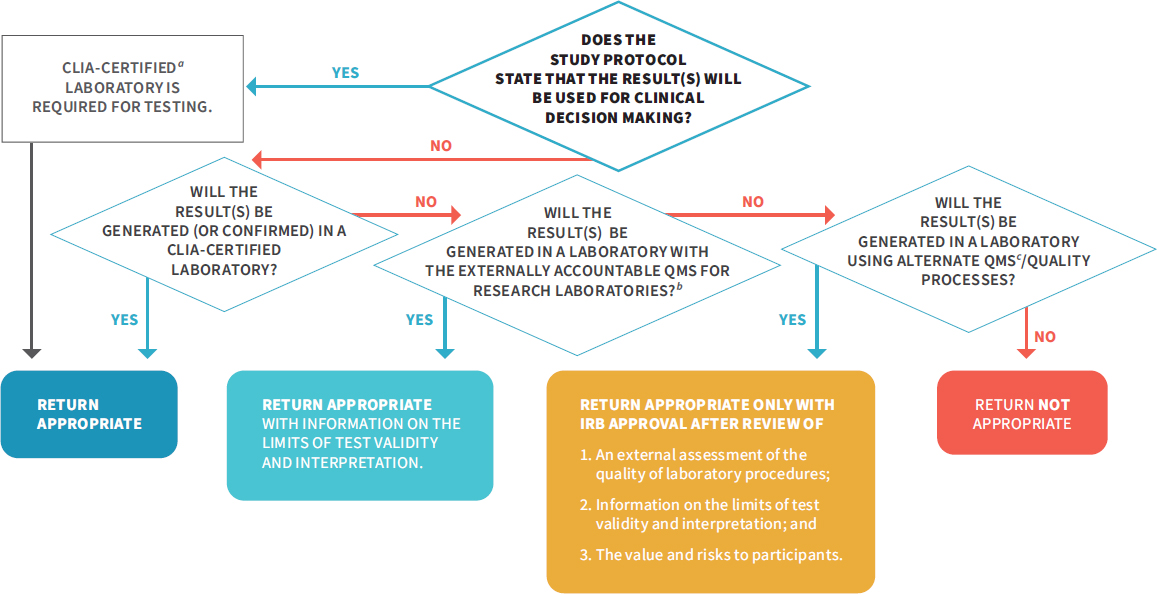
NOTES: The above flowchart details two critical aspects in the return of individual research: (1) the use of the result in research protocols and (2) the result validity. If results will be used for clinical decision making in the study protocol, they must be generated in a CLIA-certified laboratory, and return is appropriate. If results will not be used for clinical decision making in the study protocol (see Box 3-1), there are additional pathways for the return of individual research results, as long as the result is accompanied with information on the limits of the test validity and interpretation. For IRBs to have confidence in the quality of a research result and determine that it is appropriate for return, investigators should do one of the following: (1) perform their testing or get confirmation in a CLIA-certified laboratory, (2) use the NIH-led research QMS once it has been developed (see Recommendation 2), or (3) use an alternate QMS or quality processes that a review process independent of the laboratory determines is sufficient. This flowchart is also applicable to a situation in which an investigator has an unanticipated result and is considering whether to return it to a participant. CLIA = Clinical Laboratory Improvement Amendments of 1988; DRS = designated record set; HIPAA = Health Insurance Portability and Accountability Act of 1996; NIH = National Institutes of Health; QMS = quality management system.
In Recommendation 3 above, the committee details situations where the return of individual research results to participants is appropriate; however, the committee acknowledges that given the current interpretation of CLIA by CMS, this will create a potential legal conflict between investigators and CMS. This is because CMS currently interprets CLIA regulations as meaning that any laboratory returning individual research results to participants must be CLIA certified (discussed in more detail in Chapter 6). Therefore, to fully implement Recommendation 3, CMS will need to change the CLIA regulations or else the CMS interpretation of the CLIA regulations to enable investigators to return research results to participants. Allowing research laboratories to return results will not affect the conduct of clinical laboratories or investigators who will generate results for clinical decision making in the study protocol, as the committee emphasizes that it is of the utmost importance that tests used for clinical decision making need to be performed in a CLIA-certified laboratory to protect participant safety.
ADDRESSING RESOURCE AND INFRASTRUCTURE NEEDS IN RESEARCH LABORATORIES TO ENABLE RETURN OF HIGH-QUALITY INDIVIDUAL RESEARCH RESULTS
Given that few research laboratories currently operate under a QMS with external accountability, significant investment in infrastructure will be needed in order to substantially increase the number of laboratories that meet quality standards necessary for the return of individual research results to participants. Investigators will likely need both guidance and assistance from their institutions and research sponsors. The initial training, cost, and time commitment will likely be high, but the value added will be considerable, both for participants and for biomedical research overall.
Challenges and Costs Associated with Implementing a Quality Management System
Despite the need for systems to be fit for purpose and to take into account the nature of the research experiences, there are a number of commonalities in the requirements for implementing a QMS at university laboratories, in departments in academic medical centers, and in research and development laboratories in industry (Hooper et al., 2018; Mathews et al., 2017; Volsen et al., 2004; Zapata-García et al., 2007). Specifically, the development of such systems requires commitment and investment on the part of the investigators, buy-in from staff and faculty across all levels of the organization, extensive training in quality practices, and the commitment and support of general management, the department, or the institution (Vermaercke, 2000).
QMSs can be met with skepticism because they are viewed as rigid, bureaucratic, and impinging on the freedom of research (Vermaercke, 2000). Nevertheless, investigators have responded positively to the implementation of quality measures despite the extra effort entailed because, generally speaking, investigators take great pride in their work and are passionate about delivering high-quality research (Volsen et al., 2004). Obtaining the necessary buy-in from investigators tasked with implementing the standards is easier when they can see how it adds value to their work (Robins et al., 2006) and when the QMS is developed through a bottom-up approach. This enables the investigators, with the support of management, to identify where their critical quality challenges in the laboratory are and to develop quality processes to address these quality gaps (Volsen et al., 2004). The necessary changes in practice and culture will only be sustainably embraced if there are proper incentives and leadership commitment from the outset (see Box 3-5).
While adopting a QMS provides many benefits to a research enterprise, implementing a quality system is not without its challenges. Laboratories and institutions will need to consider the resources necessary to institute a QMS, as the process comes with costs that are not always transparent (Vermaercke, 2000). Figure 3-3 depicts some of the costs, infrastructure, and resources needed to implement a QMS.
Personnel costs are a key expenditure. These costs include training or bringing on additional trained staff, such as quality assurance coordinators, project leaders, and several task leads. However, it is important to note that the cost of staff time drops dramatically after the system has been implemented and becomes more mature (Vermaercke, 2000). Time is another key consideration. A group in Barcelona found that the implementation of the system took a total of 18 months, even having started with part of a previous system in place (Zapata-García et al., 2007). This highlights the point that change will not be immediate. The rollout of a high-quality system will take time and concerted effort on the part of all players in order to be successful. These are just a few of the factors that must be taken
into account when making decisions about adopting a QMS. When approaching the development and implementation of a QMS, the challenges of addressing the key laboratory and institutional gaps must be balanced with the need for quality improvement (see Figure 3-4). Once these challenges are overcome the gains are substantial. Quality management systems have been shown to make work more efficient, facilitate the training of new staff, improve reproducibility, increase patient safety, and enhance data integrity (Davies, 2013; Global Biological Standards Institute, 2013).
The committee acknowledges that the return of research results to participants may not sufficiently motivate research laboratories to adopt a QMS. However, this ought not be the only consideration for laboratories deciding whether to
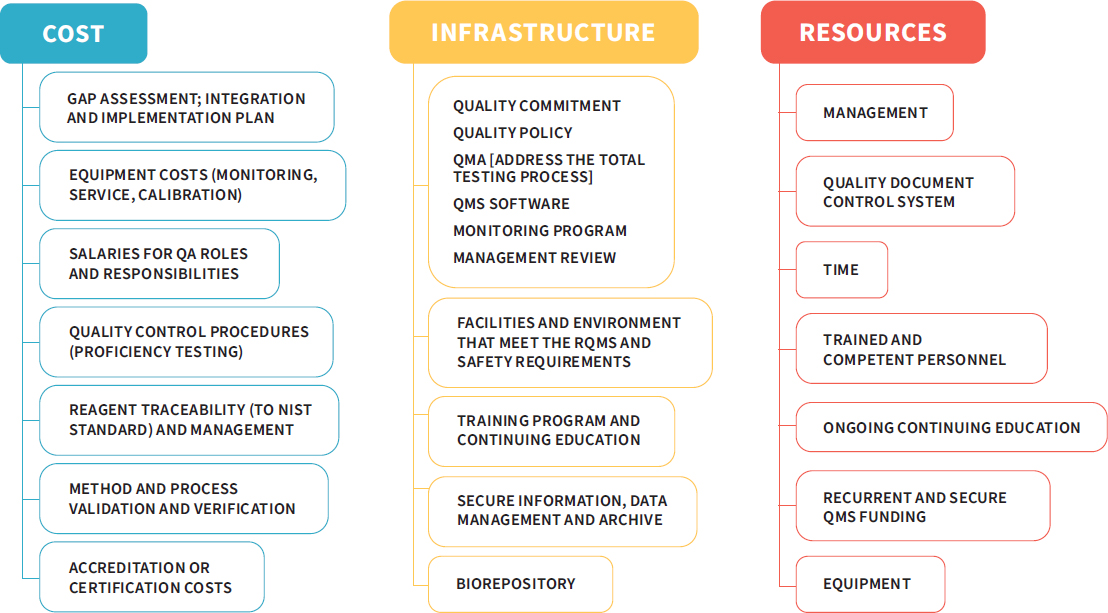
NOTE: NIST = National Institute of Standards and Technology; QA = quality assurance; QMA = quality management approach; QMS = quality management system; RQMS = roll quality management system.
SOURCE: Adapted from a presentation by Rebecca Davies, December 7, 2017.
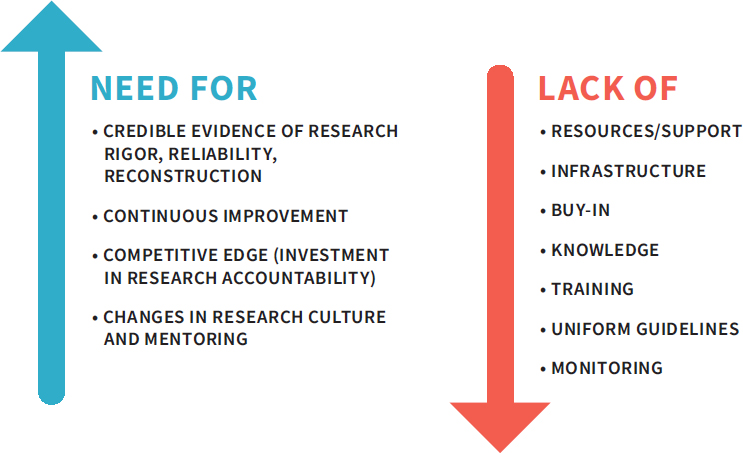
NOTES: This figure highlights the opposing factors that investigators and their institutions will encounter when considering the implementation of a quality management system (QMS). The challenges to implementation may be significant, but the need for quality is also great.
SOURCE: Adapted from a presentation by Rebecca Davies, December 7, 2017.
do so. Rather, investigators should consider how the use of a quality management system could benefit their research overall, including how it can contribute to reproducibility in science and the return on investment from research funding. See Figure 3-5 for how the implementation of standards can benefit all research stakeholders.
Institutional Support for the Generation of High-Quality Research Results
As discussed above, returning results will require assurance concerning result validity and laboratory processes and will create additional demands on investigators for quality practices beyond those required for laboratories that will not return results. As a result, institutions will need to assist investigators in tackling these additional demands. In anticipation of the NIH-led research QMS (see Recommendation 2), it would be prudent for institutions to begin initial groundwork intended to support investigators as they work to adopt the laboratory quality practices necessary for return of individual research results. This may include providing training programs in quality management for investigators, assisting

NOTE: This figure, developed by the Global Biological Standards Institute, highlights the fact that “research standards can be a unifying driver of quality improvement efforts and have the potential to benefit all stakeholders.”
SOURCE: Global Biological Standards Institute, 2013, p. 30.
investigators in an initial gap assessment to identify risks to data quality and integrity, and facilitating the acquisition and adoption of quality management software (for example, electronic laboratory notebooks or quality and data management software). Other institutional responsibilities may include
- educating laboratory leadership and other laboratory staff about QMS expectations;
- establishing platforms, templates, and access to expert consultants to evaluate and assess investigators’ current level of quality management practices, advise the IRB, and implement quality standards;
- developing a system for laboratory inspection to support compliance with the external review of quality standards; and
- advising the investigators, IRBs, and institution as to the required compliance with the QMS, which has implications for the potential validity of research results and the ability to return research results to participants.
Numerous guidance documents discussing research laboratory quality practices are available to support institutions in these efforts, including those published by NIH and WHO (Moore et al., 2011; NCI, 2016; NIH, 2017a; Scientific Working Group on Quality Practices in Basic Biomedical Research, 2001).
Many institutions may already have resources in place that could be used to assist investigators with the generation of high-quality research results and facilitate the process of returning results to participants before a QMS is adopted. These include expert review committees that can provide guidance on necessary quality measures and core laboratory facilities that already operate under a quality management system, such as that required by CLIA. The use of core facilities may also help achieve economies of scale and serve as resources for those laboratories without adequate infrastructure to implement a QMS. For institutions without core facilities, one option would be to develop partnerships with institutions that already have established core facilities. This sharing of institutional resources could be further encouraged by NIH, which could call on institutions with federally funded core facilities—Clinical and Translational Science Awards hubs, for example—to make these accessible beyond their parent institution. Additionally, third parties may serve as resources for investigators and laboratories wishing to return results but who do not have the appropriate on-site resources. Identifying and partnering with third-party organizations for research testing performed under a QMS may require institutions to develop a working list of partners for their investigators to consider when research test results on human biospecimens will be returned to participants. Ideally, to help keep costs lower for research budgets, research pricing structures would be available or negotiated by the institution on a contract basis rather than by individual investigators.
Guidance will be needed to assist investigators and laboratories in determining whether they are best suited to pursue CLIA certification, adopt the proposed
NIH-led QMS, or leverage external resources. These decisions should be made based on the type of research that investigators conduct, the types of samples they test, and the intended use of the results as well as the potential value and risks of returning those results to participants. Adopting the NIH-led QMS will not be instantaneous, and some better-resourced institutions will be more able to take on this task. In less well-resourced institutions, there will likely be delayed implementation, but these institutions will benefit from the work of early adopters. The early-adopter institutions can create helpful tools, lessons learned, and best practices that can be shared with other institutions to facilitate their adoption of high-quality practices.
Generating valid results is only one piece of the required infrastructure for the return of individual research results to participants. The other considerations are how to assess the value and risk of returning results and the feasibility of return (discussed in Chapter 4) and how investigators untrained in lay communication and communication of test results can appropriately return results. The process of communication will require additional institutional mechanisms and financial support, as discussed in Chapter 5.
CONCLUSION: Investigators, institutions, and research sponsors will need to anticipate the needed time, costs, and resources required for adopting a quality management system to both enable the return of research results and improve research overall.
The research enterprise is facing growing expectations—from participants and even research sponsors, as in the case of NIH’s Precision Medicine Initiative (All of Us Research Program, 2017; Precision Medicine Initiative Working Group, 2015)—concerning the return of research results. The quality of the research results will be a crucial factor to be weighed as investigators and institutions consider whether to return results to participants, but it is only one element in the decision-making process. The next chapter addresses two additional dimensions, value and feasibility, that will help investigators make decisions on what to return, and it describes the need for a plan and process that includes engaging participants and communities in making these determinations.
TABLE 3-2 Common Elements of Quality Systems
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| External Quality Control | |||||
| External inspections required | ✓ | ✓ | ✓ | ||
| External inspections voluntary | ✓ | ||||
| Participation in proficiency testing from an approved tester required for regulated analytes | ✓ | ✓ | ✓ | ✓ | |
| Alternative performance assessment required for unregulated analytes | ✓ | ✓ | ✓ | ✓ |
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| Internal Quality Control | |||||
| Internal quality control checks required | ✓ | ✓ | ✓ | ✓ | ✓ |
| Documentation describing policies and procedures necessary to assure the quality of test results | ✓ | ✓ | ✓ | ✓ | ✓ |
| Methods, supplies, equipment, and testing are appropriate to provide results within stated performance specifications and type and volume of testing | ✓ | ✓ | ✓ | ✓ | ✓ |
| Requirements for specimen collection | ✓ | ✓ | ✓ | ✓ | ✓ |
| Established and reviewed procedures for specimen transport to and from reference laboratories (timing, record-keeping, environmental conditions, communications) | ✓ | ✓ | ✓ | ✓ | ✓ |
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| Guidelines for specimen acceptance and rejection | ✓ | ✓ | ✓ | ✓ | ✓ |
| System for storage and use of supplies | ✓ | ✓ | ✓ | ✓ | ✓ |
| System of equipment checks, calibration, and maintenance (with records) | ✓ | ✓ | ✓ | ✓ | ✓ |
| Defined system for specimen identification, storage, handling, and tracking through all phases of testing | ✓ | ✓ | ✓ | ✓ | ✓ |
| Reportable range is established for all procedures before implementation | ✓ | ✓ | ✓ | ✓ | |
| System for reporting results | ✓ | ✓ | ✓ | ✓ | ✓ |
| Information management and record storage systems (tests requested, tests run, results, etc.) | ✓ | ✓ | ✓ | ✓ | ✓ |
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| Established process for documenting and fixing failed conditions | ✓b | ✓ | ✓ | ✓ | ✓ |
| System for ensuring the privacy of the client (information management protocols) through all phases of testing | ✓ | ✓ | ✓ | ✓ | ✓ |
| Communication system in place to handle internal and external complaints and problems reported to the laboratory | ✓ | ✓ | ✓ | ✓ | |
| Verification for unmodified tests | ✓ | ✓ | ✓ | ✓ | ✓ |
| Verification for modified tests | ✓ | ✓ | ✓ | ✓ | ✓ |
| Personnel and Facility | |||||
| Hierarchy and personnel responsibilities are clear | ✓ | ✓c | ✓ | ✓ | ✓ |
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| Personnel are trained, qualified, and there is sufficient staff for the work required | ✓ | ✓ | ✓ | ✓ | ✓ |
| Qualified laboratory director (a role with responsibility over the administrative oversight of the laboratory) | ✓ | ✓ | ✓ | ✓d | ✓ |
| Qualified laboratory manager/supervisor/assistant director | ✓e | ✓ | ✓ | ✓ | |
| Qualified technical consultant or supervisor (a role with responsibility over the technical oversight of the laboratory) | ✓ | ✓e | ✓ | ✓ | |
| Qualified clinical consultant (a role that acts as a liaison between the laboratory and the client and is responsible for matters related to the reporting and interpretation of results) | ✓ | ✓e | ✓ | ||
| Quality Systems Manager | ✓ | ✓ |
| ✓= REQUIRED | CLIA NON-WAIVED (HIGHLY COMPLEX LABORATORIES) | CLIA ACCREDITATION ORGANIZ ATIONS | ISO 15189 | CLIA EXEMPT STATE STANDARD (NY) | VOLUNTA RY S TA NDA RDa |
|---|---|---|---|---|---|
| Qualified laboratory testing personnel (testing of specimens) | ✓ | ✓ | ✓ | ✓ | ✓ |
| Steps taken to prevent environmental fluctuations (power or humidity fluctuations) | ✓ | ✓ | ✓ | ✓ | ✓ |
| Environment is safety-focused and has adequate space and utilities for all phases of the testing process | ✓ | ✓ | ✓ | ✓ | ✓ |
NOTE: The table lists common elements of quality systems. A check (✓) indicates that the element is required; if there is no check, the element is not required. This table does not capture nuanced differences (for example, the number of external inspections); rather the aim of the table is to convey the common key elements in quality management systems that laboratories could consider in implementing their own quality management system.
*This table is an example of some elements present in laboratory quality systems.
a The voluntary standards captured in this table are based on recommendations from the WHO and Research Quality Association (RQA) guidelines. A research laboratory voluntarily adopts a research quality management system that applies to research activity throughout the research life cycle.
b For CLIA this includes a “Quality assessment system to monitor, assess, and correct problems, review the effectiveness of corrective actions, revise SOPs to prevent reoccurrence, and discuss QA reviews with appropriate staff.”
c This includes the need for written, clear, and explicit delegation of responsibilities by the laboratory director to individuals qualified to assume those duties.
d New York State (NYS) regulations require that there be a qualified laboratory director with responsibility over the administrative and technical oversight of the laboratory. The pertinent NYS regulations are the following:
- Section 19.1(a) of Title 10 of the New York State Codes, Rules and Regulations (10 NYCRR) defines the clinical laboratory director as the individual responsible for administration of the technical and scientific operation of a clinical laboratory or blood bank.
- 10 NYCRR Section 58-1.2(a) states that the director is responsible for direction and technical operation of the laboratory.
e One individual may fulfill more than one position (e.g., a laboratory director may also serve as a clinical consultant).
SOURCES: American College of Physicians, 2014; COLA, 2017; College of American Pathologists, 2017; ISO, 2007; New York State Department of Health, 2017; UN Industrial Development Organization, 2009; WHO, 2006.
REFERENCES
Agarwal, R. 2014. Quality-improvement measures as effective ways of preventing laboratory errors. Laboratory Medicine 45(2):e80–e88.
All of Us Research Program. 2017. Return of genetic results in the All of Us research program—Agenda, March 6–7, 2017. https://www.nih.gov/sites/default/files/research-training/initiatives/pmi/return-of-results-agenda-0306-0717.pdf (accessed March 13, 2018).
Ambulos, N. P. 2013. The good, the bad and the ugly: Getting CLIA certification for a basic research lab. Journal of Biomolecular Techniques 24(Suppl):S21.
American Academy of Family Physicians. 2018. Quality assurance, CLIA, and your lab. https://www.aafp.org/practice-management/regulatory/clia/quality-assurance.html (accessed March 13, 2018).
American College of Physicians. 2014. CLIA & your laboratory: A guide for physicians and their staff.https://www.acponline.org/system/files/documents/running_practice/mle/clia-and-your-lab.pdf (accessed May 21, 2018).
Aziz, N., Q. Zhao, L. Bry, D. K. Driscoll, B. Funke, J. S. Gibson, W. W. Grody, M. R. Hegde, G. A. Hoeltge, D. G. B. Leonard, J. D. Merker, R. Nagarajan, L. A. Palicki, R. S. Robetorye, I. Schrijver, K. E. Weck, and K. V. Voelkerding. 2014. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Archives of Pathology & Laboratory Medicine 139(4):481–493.
Baker, M. 2015. U.S. societies push back against NIH reproducibility guidelines. Nature, April 17. https://www.nature.com/news/us-societies-push-back-against-nih-reproducibility-guide-lines-1.17354 (accessed March 13, 2018).
Baker, M. 2016. 1,500 scientists lift the lid on reproducibility. Nature 533(7604):452–454.
Barnes, M., S. Stayin, D. Forster, M. Russell-Einhorn, D. Peloquin, and A. Medina-Jordan. 2015. The CLIA/HIPAA conundrum of returning test results to research participants. Medical Research Law & Policy Report, July 15. https://www.ropesgray.com/~/media/Files/articles/2015/July/2015-07-15-Bloomberg-BNA.ashx (accessed May 18, 2018).
Begley, C. G., and J. P. Ioannidis. 2015. Reproducibility in science: Improving the standard for basic and preclinical research. Circulation Research 116(1):116–126.
Begley, C. G., A. M. Buchan, and U. Dirnagl. 2015. Robust research: Institutions must do their part for reproducibility. Nature 525(7567):25–27.
Berlin Institute of Health. 2017. Opening of the Quest Center for Transforming Biomedical Research. https://www.bihealth.org/en/quest-center/quest-opening (accessed March 29, 2018).
Berlin Institute of Health. 2018. Premier. https://www.bihealth.org/de/quest-center/projekte/premier (accessed February 27, 2018).
Bookman, E. B., A. A. Langehorne, J. H. Eckfeldt, K. C. Glass, G. P. Jarvik, M. Klag, G. Koski, A. Motulsky, B. Wilfond, T. A. Manolio, R. R. Fabsitz, and R. V. Luepker. 2006. Reporting genetic results in research studies: Summary and recommendations of an NHLBI working group. American Journal of Medical Genetics, Part A 140(10):1033–1040.
Bosch, G., and A. Casadevall. 2017. Graduate biomedical science education needs a new philosophy. mBio 8(6):e01539-17.
Burke, W., B. J. Evans, and G. P. Jarvik. 2014. Return of results: Ethical and legal distinctions between research and clinical care. American Journal of Medical Genetics 166C(1):105–111.
Calabrese, R., and U. Palm. 2008. Proposal for a national quality standard for biomedical research. Quality Digest, October 2. https://www.qualitydigest.com/inside/health-care-article/proposal-national-quality-standard-biomedical-research.html (accessed May 21, 2018).
Caudle, K. E., H. M. Dunnenberger, R. R. Freimuth, J. F. Peterson, J. D. Burlison, M. Whirl-Carrillo, S. A. Scott, H. L. Rehm, M. S. Williams, T. E. Klein, M. V. Relling, and J. M. Hoffman. 2017. Standardizing terms for clinical pharmacogenetic test results: Consensus terms from the clinical pharmacogenetics implementation consortium (CPIC). Genetics in Medicine 19(2):215–223.
CDC (Centers for Disease Control and Prevention). 2017. Laboratory quality assurance and standardization programs. https://www.cdc.gov/labstandards/overview.html (accessed May 10, 2018).
CDC. 2018a. About CLIAC. https://wwwn.cdc.gov/cliac/About.aspx (accessed March 13, 2018).
CDC. 2018b. Clinical Laboratory Improvement Advisory Committee (CLIAC). https://wwwn.cdc.gov/cliac (accessed March 29, 2018).
CDISC (Clinical Data Interchange Standards Consortium). 2011a. Alzheimer’s therapeutic area user guide v1.0. https://www.cdisc.org/standards/therapeutic-areas/alzheimers/alzheimers-therapeutic-area-user-guide-v10 (accessed May 11, 2018).
CDISC. 2011b. Critical Path Institute and Clinical Data Interchange Standards Consortium announce release of data standard for Alzheimer’s disease research. October 17. https://c-path.org/critical-path-institute-and-clinical-data-interchange-standards-consortium-announce-release-of-data-standards-for-alzheimers-disease-research (accessed May 21, 2018).
Chen, B., M. Gagnon, S. Shahangian, N. L. Anderson, D. A. Howerton, and D. J. Boone. 2009. Good laboratory practices for molecular genetic testing for heritable diseases and conditions. Morbidity and Mortality Weekly Report 58(RR-6):1–29.
Clayton, E. W., and A. L. McGuire. 2012. The legal risks of returning results of genomics research. Genetics in Medicine 14(4):473–477.
CLSI (Clinical and Laboratory Standards Institute). 2018. CLSI standards: Guidelines for health care excellence. https://clsi.org/standards (accessed May 10, 2018).
CMS (Centers for Medicare & Medicaid Services). 2014a. Certificate of waiver laboratory project. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Certificate_of_-Waiver_Laboratory_Project.html (accessed May 10, 2018).
CMS. 2014b. Proficiency testing and PT referral: Dos and don’ts. https://www.cms.gov/Regulations-andGuidance/Legislation/CLIA/Downloads/CLIAbrochure8.pdf (accessed May 21, 2018).
CMS. 2018. CLIA update—January 2018. U.S. Centers for Medicare and Medicaid Services. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/statupda.pdf (accessed May 21, 2018).
CMS. n.d. List of approved accredidation organizations under the Clinical Laboratory Improvement Amendments (CLIA). https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/AOList.pdf (accessed March 13, 2018).
COLA (formerly the Commission on Office Laboratory Accreditation). 2017. COLA laboratory accreditation manual.https://www.colacentral.com/files/pdf/COLA%20Laboratory%20Accreditation%20Manual.pdf (accessed May 21, 2018).
College of American Pathologists. 2017. All common checklist. Northfield, IL: College of American Pathologists. http://www.cap.org/ShowProperty?nodePath=/UCMCon/Contribution%20Folders/DctmContent/education/OnlineCourseContent/2016/LAP-TLTM/resources/AC-all-common.pdf (accessed May 21, 2018).
Collins, F. S., and L. A. Tabak. 2014. Policy: NIH plans to enhance reproducibility. Nature 505(7485):612–613.
Davies, R. 2013. Good research practice: It is time to do what others think we do. Quasar July 13:21–23.
Davies, R., C. London, B. Lascelles, and M. Conzemius. 2017. Quality assurance and best research practices for non-regulated veterinary clinical studies. BMC Veterinary Research 13(1):242.
Ehrmeyer, S. S., and R. H. Laessig. 2004. Has compliance with CLIA requirements really improved quality in U.S. clinical laboratories? Clinica Chimica Acta 346(1):37–43.
Engel, K. B., J. Vaught, and H. M. Moore. 2014. National Cancer Institute biospecimen evidence-based practices: A novel approach to pre-analytical standardization. Biopreservation and Biobanking 12(2):148–150.
Epner, P. L., J. E. Gans, and M. L. Graber. 2013. When diagnostic testing leads to harm: A new outcomes-based approach for laboratory medicine. BMJ Quality & Safety 22(Suppl 2):ii6.
EQIPD (European Quality in Preclinical Data). 2017. European Quality in Preclinical Data. http://www.dcn.ed.ac.uk/camarades/eqipd (accessed March 13, 2018).
Euformatics. 2017. Evolving standards in clinical NGS. http://euformatics.com/evolving-standards-in-clinical-ngs (accessed February 5, 2018).
Fabsitz, R. R., A. McGuire, R. R. Sharp, M. Puggal, L. M. Beskow, L. G. Biesecker, E. Bookman, W. Burke, E. G. Burchard, G. Church, E. W. Clayton, J. H. Eckfeldt, C. V. Fernandez, R. Fisher, S. M. Fullerton, S. Gabriel, F. Gachupin, C. James, G. P. Jarvik, R. Kittles, J. R. Leib, C. O’Donnell, P. P. O’Rourke, L. L. Rodriguez, S. D. Schully, A. R. Shuldiner, R. K. Sze, J. V. Thakuria, S. M. Wolf, G. L. Burke, and the National Heart, Lung, and Blood Institute Working Group. 2010. Ethical and practical guidelines for reporting genetic research results to study participants: Updated guidelines from a National Heart, Lung, and Blood Institute Working Group. Circulation: Cardiovascular Genetics 3(6):574–580.
FDA (Food and Drug Administration). 2018. Use of public human genetic variant databases to support clinical validity for genetic and genomic-based in vitro diagnostics: Guidance for stakeholders and Food and Drug Administration staff.https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM509837.pdf (accessed May 21, 2018).
Ferreira-Gonzalez, A., S. Teutsch, M. S. Williams, S. M. Au, K. T. FitzGerald, P. S. Miller, and C. Fomous, on behalf of the members of the Secretary’s Advisory Committee on Genetics, Health, and Society. 2008. U.S. system of oversight for genetic testing: A report from the Secretary’s Advisory Committee on Genetics, Health, and Society. Personalized Medicine 5(5):521–528.
Flyn, K. 1996. Assessing diagnostic technologies. Boston, MA: Veterans Health Administration’s Office of Research an Development.
Freedman, L. P., and J. Inglese. 2014. The increasing urgency for standards in basic biological research. Cancer Research 74(15):4024–4029.
Gargis, A. S., L. Kalman, M. W. Berry, D. P. Bick, D. P. Dimmock, T. Hambuch, F. Lu, E. Lyon, K. V. Voelkerding, B. A. Zehnbauer, R. Agarwala, S. F. Bennett, B. Chen, E. L. H. Chin, J. G. Compton, S. Das, D. H. Farkas, M. J. Ferber, B. H. Funke, M. R. Furtado, L. M. Ganova-Raeva, U. Geigenmüller, S. J. Gunselman, M. R. Hegde, P. L. F. Johnson, A. Kasarskis, S. Kulkarni, T. Lenk, C. S. J. Liu, M. Manion, T. A. Manolio, E. R. Mardis, J. D. Merker, M. S. Rajeevan, M. G. Reese, H. L. Rehm, B. B. Simen, J. M. Yeakley, J. M. Zook, and I. M. Lubin. 2012. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nature Biotechnology 30(11):10.1038/nbt.2403.
Glick, J. L., and A. E. Shamoo. 1993. A call for the development of “good research practices” (GRP) guidelines. Accountability in Research 2(4):231–235.
Global Biological Standards Institute. 2013. The case for standards in life science research: Seizing opportunities at a time of critical need. Washington, DC: Global Biological Standards Institute.
Gottlieb, S. 2018. Remarks by Dr. Gottlieb at the American Clinical Laboratory Association annual meeting. https://www.fda.gov/NewsEvents/Speeches/ucm599551.htm (accessed May 10, 2018).
Green, R. C., J. S. Berg, W. W. Grody, S. S. Kalia, B. R. Korf, C. L. Martin, A. L. McGuire, R. L. Nussbaum, J. M. O’Daniel, K. E. Ormond, H. L. Rehm, M. S. Watson, M. S. Williams, and L. G. Biesecker. 2013. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genetics in Medicine 15(7):565–574.
Grens, K. 2017. Preclinical studies don’t regularly adhere to best practices. https://www.the-scientist.com/?articles.view/articleNo/49375/title/Preclinical-Studies-Don-t-Regularly-Adhere-to-Best-Practices (accessed April 5, 2018).
Hawkins, R. 2012. Managing the pre- and post-analytical phases of the total testing process. Annals of Laboratory Medicine 32(1):5–16.
Haywood, J. R. FASEB response to NIH principles and guidelines for reporting preclinical research, e-mail message to Lawrence A. Tabak, April 8, 2015. =https://www.faseb.org/Portals/2/PDFs/opa/2015/4.8.15%20FASEB%20Response%20to%20NIH%20Preclin%20Guidelines.pdf (accessed May 29, 2018).
Herman, D. R., and R. W. Usher. 1994. Good research practices: A commonsense approach to ensuring quality in research facilities. Quality Assurance (San Diego, Calif.) 3(4):355–359.
Holm, I. A., S. K. Savage, R. C. Green, E. Juengst, A. McGuire, S. Kornetsky, S. J. Brewster, S. Joffe, and P. Taylor. 2014. Guidelines for return of research results from pediatric genomic studies: Deliberations of the Boston Children’s Hospital Gene Partnership Informed Cohort Oversight Board. Genetics in Medicine 16(7):547–552.
Hooper, J. E., H. Richardson, A. W. Maters, K. C. Carroll, and P. J. Pronovost. 2018. The association of departmental quality infrastructure and positive change: A pathology department illustration. Academic Pathology 5. doi: 10.1177/2374289517744753.
IOM (Institute of Medicine). 2012. Evolution of translational omics: Lessons learned and the path forward. Washington, DC: The National Academies Press.
IOM. 2013a. The CTSA program at NIH: Opportunities for advancing clinical and translational research. Washington, DC: The National Academies Press.
IOM. 2013b. Sharing clinical research data: Workshop summary. Washington, DC: The National Academies Press.
Irwig, L., J. Irwig, L. Trevena, and M. Sweet. 2008. Smart health choices: Making sense of health advice. London: Hammersmith Press.
ISO (International Organization for Standardization). 2007. Medical laboratories—Particular requirements for quality and competence.https://www.iso.org/standard/56115.html (accessed May 21, 2018).
ISO. 2018. We’re ISO: We develop and publish international standards. https://www.iso.org/standards.html (accessed May 10, 2018).
Jarvik, G. P., L. M. Amendola, J. S. Berg, K. Brothers, E. W. Clayton, W. Chung, B. J. Evans, J. P. Evans, S. M. Fullerton, C. J. Gallego, N. A. Garrison, S. W. Gray, I. A. Holm, I. J. Kullo, L. S. Lehmann, C. McCarty, C. A. Prows, H. L. Rehm, R. R. Sharp, J. Salama, S. Sanderson, S. L. Van Driest, M. S. Williams, S. M. Wolf, W. A. Wolf, Electronic Medical Records and Genomics Act–Return of Results (ROR) Committee, Consent, Education, Regulation, and Consultation Committee, Clinical Sequencing Exploratory Research Act–ROR Working Group, and W. Burke. 2014. Return of genomic results to research participants: The floor, the ceiling, and the choices in between. American Journal of Human Genetics 94(6):818–826.
Jennings, L., V. M. V. Deerlin, and M. L. Gulley. 2009. Recommended principles and practices for validating clinical molecular pathology tests. Archives of Pathology & Laboratory Medicine 133(5):743–755.
Kohane, I. S., K. D. Mandl, P. L. Taylor, I. A. Holm, D. J. Nigrin, and L. M. Kunkel. 2007. Re-establishing the researcher-patient compact. Science 316:836–837.
LabCE. 2018. Pre-analytical, analytical, and post-analytical phases of testing. https://www.labce.com/spg650097_pre_analytical_analytical_and_post_analytical_phas.aspx (accessed March 30, 2018).
Loew, L., D. Beckett, E. H. Egelman, and S. Scarlata. 2015. Reproducibility of research in biophysics. Biophysical Journal 108:E01.
Maddalena, A., S. Bale, S. Das, W. Grody, and S. Richards. 2005. Technical standards and guidelines: Molecular genetic testing for ultra-rare disorders. Genetics in Medicine 7:571–583.
Magnusson, B., and U. Ornemark (eds.). 2014. Eurachem guide: The fitness for purpose of analytical methods—A laboratory guide to method validation and related topics. 2nd ed. https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf (accessed May 21, 2018).
Mathews, S. C., R. Demski, J. E. Hooper, L. D. Biddison, S. A. Berry, B. G. Petty, A. R. Chen, P. M. Hill, M. R. Miller, F. R. Witter, L. Allen, E. C. Wick, T. S. Stierer, L. Paine, H. A. Puttgen, R. J. Tamargo, and P. J. Pronovost. 2017. A model for the departmental quality management infrastructure within an academic health system. Academic Medicine 92(5):608–613.
Mattocks, C. J., M. A. Morris, G. Matthijs, E. Swinnen, A. Corveleyn, E. Dequeker, C. R. Müller, V. Pratt, and A. Wallace. 2010. A standardized framework for the validation and verification of clinical molecular genetic tests. European Journal of Human Genetics 18(12):1276–1288.
Mayo Clinic. 2018. Mayo clinic biobank—Governance and oversight. http://www.mayo.edu/research/centers-programs/mayo-clinic-biobank/about/governance-oversight (accessed May 11, 2018).
McCarty, C. A., R. L. Chisholm, C. G. Chute, I. J. Kullo, G. P. Jarvik, E. B. Larson, R. Li, D. R. Masys, M. D. Ritchie, D. M. Roden, J. P. Struewing, and W. A. Wolf. 2011. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Medical Genomics 4:13.
McNutt, M. 2014. Journals unite for reproducibility. Science 346(6210):679.
Moore, H. M., A. Kelly, S. D. Jewell, L. M. McShane, D. P. Clark, R. Greenspan, P. Hainaut, D. F. Hayes, P. Kim, E. Mansfield, O. Potapova, P. Riegman, Y. Rubinstein, E. Seijo, S. Somiari, P. Watson, H.-U. Weier, C. Zhu, and J. Vaught. 2011. Biospecimen reporting for improved study quality. Biopreservation and Biobanking 9(1):57–70.
Nakhleh, R. E., M. O. Idowu, R. J. Souers, F. A. Meier, and L. G. Bekeris. 2011. Mislabeling of cases, specimens, blocks, and slides: A College of American Pathologists study of 136 institutions. Archives of Pathology & Laboratory Medicine 135(8):969–974.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2016. Biomarker tests for molecularly targeted therapies: Key to unlocking precision medicine. Washington, DC: The National Academies Press.
NASEM. 2017a. An evidence framework for genetic testing. Washington, DC: The National Academies Press.
NASEM. 2017b. Fostering integrity in research. Washington, DC: The National Academies Press.
NCI (National Cancer Institute). 2011. National Cancer Institute best practices for biospecimen resources.https://biospecimens.cancer.gov/bestpractices/2011-ncibestpractices.pdf (accessed May 21, 2018).
NCI. 2016. Quality management. https://biospecimens.cancer.gov/bestpractices/to/qac.asp (accessed March 13, 2018).
NCI. 2018. NCI Thesaurus. https://ncit.nci.nih.gov/ncitbrowser (accessed May 11, 2018).
Ned-Sykes, R., C. Johnson, J. C. Ridderhof, E. Perlman, A. Pollock, and J. M. DeBoy. 2015. Competency guidelines for publich health laboratory professionals: CDC and the Association of Public Health Laboratories. Morbidity and Mortality Weekly Report Supplement 64(1):1–81.
New York State Department of Health. 2016. Validation of next generation sequencing (NGS)-based methods of identification and/or characterization of infectious agents (isolates only).https://www.wadsworth.org/sites/default/files/WebDoc/ID%20WGS%20NGS%20Molecular%20Guidelines%20for%20Isolates_0.pdf (accessed May 21, 2018).
New York State Department of Health. 2017. Clinical laboratory standards of practice, part 1—General systems. Albany, NY: New York State Department of Health. https://pingpdf.com/pdf-clinicallaboratory-standards-of-practice-wadsworth-center.html (accessed May 21, 2018).
Nichols, J. H. 2011. Laboratory quality control based on risk management. Annals of Saudi Medicine 31(3):223–228.
NIH (National Institutes of Health). 2013. DAIDS guidelines for good clinical laboratory practice standards.https://www.niaid.nih.gov/sites/default/files/gclp.pdf (accessed May 21, 2018).
NIH. 2017a. Principles and guidelines for reporting preclinical research. https://www.nih.gov/researchtraining/rigor-reproducibility/principles-guidelines-reporting-preclinical-research (accessed March 13, 2018).
NIH. 2017b. Rigor and reproducibility. https://www.nih.gov/research-training/rigor-reproducibility (accessed December 15, 2017).
NIH. 2018. How can consumers be sure a genetic test is valid and useful?https://ghr.nlm.nih.gov/primer/testing/validtest (accessed March 13, 2018).
NRC (National Research Council). 2006. Human biomonitoring for environmental chemicals. Washington, DC: The National Academies Press.
Office of Ethics and Compliance. 2017. CLIA compliance and lab test results. University of California, San Francisco. https://irb.ucsf.edu/clia-compliance-and-lab-test-results (accessed September 12, 2017).
Ohayon, J. L., E. Cousins, P. Brown, R. Morello-Frosch, and J. G. Brody. 2017. Researcher and institutional review board perspectives on the benefits and challenges of reporting back biomonitoring and environmental exposure results. Environmental Research 153:140–149.
PAASP (Partnership for Assessment and Accreditation of Scientific Practice). 2018a. EQIPD. http://paasp.net/about-us/eqipd (accessed May 11, 2018).
PAASP. 2018b. Our mission. http://paasp.net/our-mission (accessed March 13, 2018).
PCSBI (Presidential Commission for the Study of Bioethical Issues). 2013. Anticipate and communicate ethical management of incidental and secondary findings in the clinical, research, and direct to consumer contexts. Washington, DC: Presidential Commission for the Study of Bioethical Issues.
Perry, C. J., and A. J. Lawrence. 2017. Hurdles in basic science translation. Frontiers in Pharmacology 8:478.
Precision Medicine Initiative Working Group. 2015. The Precision Medicine Initiative cohort program: Building a research foundation for 21st century medicine.https://acd.od.nih.gov/documents/reports/DRAFT-PMI-WG-Report-9-11-2015-508.pdf (accessed May 21, 2018).
Ravitsky, V., and B. S. Wilfond. 2006. Disclosing individual genetic results to research participants. American Journal of Bioethics 6(6):8–17.
Rehm, H. L., S. J. Bale, P. Bayrak-Toydemir, J. S. Berg, K. K. Brown, J. L. Deignan, M. J. Friez, B. H. Funke, M. R. Hegde, and E. Lyon. 2013. ACMG clinical laboratory standards for next-generation sequencing. Genetics in Medicine 15:733–747.
Reisner, H. M. 2015. Pathology: A modern case study. New York: McGraw-Hill Education.
Riedl, D. H., and M. K. Dunn. 2013. Quality assurance mechanisms for the unregulated research environment. Trends in Biotechnology 31(10):552–554.
Ritter, D. I., S. Roychowdhury, A. Roy, S. Rao, M. J. Landrum, D. Sonkin, M. Shekar, C. F. Davis, R. K. Hart, C. Micheel, M. Weaver, E. M. Van Allen, D. W. Parsons, H. L. McLeod, M. S. Watson, S. E. Plon, S. Kulkarni, S. Madhavan, and the ClinGen Somatic Cancer Working Group. 2016. Somatic cancer variant curation and harmonization through consensus minimum variant level data. Genome Medicine 8(1):117.
Robins, M. M., S. J. Scarll, and P. E. Key. 2006. Quality assurance in research laboratories. Accreditation and Quality Assurance 11(5):214–223.
SACHRP (Secretary’s Advisory Committee on Human Research Protections). 2015. Attachment C: Return of individual results and special considerations of issues arising from amendments of HIPAA and CLIA.https://www.hhs.gov/ohrp/sachrp-committee/recommendations/2015-september28-attachment-c/index.html (accessed May 21, 2018).
Scientific Working Group on Quality Practices in Basic Biomedical Research. 2001. Handbook: Quality practices in basic biomedical research. Geneva, Switzerland: World Health Organization.
Segen’s Medical Dictionary. 2011. Analyte.https://medical-dictionary.thefreedictionary.com/analyte (accessed August 3, 2018).
Stack, C. B., N. Gharani, E. S. Gordon, T. Schmidlen, M. F. Christman, and M. A. Keller. 2011. Genetic risk estimation in the Coriell Personalized Medicine Collaborative. Genetics in Medicine 13:131–139.
Task Force on Genetic Testing. 1997. Promoting safe and effective genetic testing in the United States: Final report of the Task Force on Genetic Testing. Baltimore, MD: Johns Hopkins University Press.
Thelen, M. H., F. J. Vanstapel, C. Kroupis, I. Vukasovic, G. Boursier, E. Barrett, B. Andreu Francisco, M. Brguljan Pika, D. Brugnoni, M. Lohmander, L. Sprongl, T. Vodnik, I. Ghita, M. Vaubourdolle, W. Huisman, and the Working Group Accreditation ISO/CEN Standards of EFLM. 2015. Flexible scope for ISO 15189 accreditation: A guidance prepared by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Working Group Accreditation and ISO/CEN standards (WG-A/ISO). Clinical Chemistry and Laboratory Medicine 53(8):1173–1180.
Titus, S., and X. Bosch. 2010. Tie funding to research integrity. Nature 466(7305):436–437.
Toker, L., M. Feng, and P. Pavlidis. 2016. Whose sample is it anyway? Widespread misannotation of samples in transcriptomics studies. F1000Research, August 30. https://f1000research.com/articles/5-2103/v1 (accessed May 21, 2018).
United Nations Industrial Development Organization. 2009. Complying with ISO 17025: A practical guidebook for meeting the requirements of laboratory accreditation schemes based on ISO 17025:2005 or equivalent national standards. Vienna, Austria: United Nations Industrial Development Organization.
Vahidy, F., W. R. Schäbitz, M. Fisher, and J. Aronowski. 2016. Reporting standards for preclinical studies of stroke therapy. Stroke 47(10):2435–2438.
Vermaercke, P. 2000. Sense and nonsense of quality assurance in an R&D environment. Accreditation and Quality Assurance 5(1):11–15.
Volsen, S. G., J. M. Kent, and M. Masson. 2004. Quality: An old solution to new discovery dilemmas? Drug Discovery Today 9(21):903–905.
White, M. 2015. Do we need formal quality standards for science? Pacific Standard, May 7. https://psmag.com/environment/do-we-need-formal-quality-standards-for-science (accessed March 13, 2018).
WHO (World Health Organization). 2006. Handbook: Quality practices in basic biomedical research. Geneva, Switzerland: World Health Organization.
WHO. 2011. Laboratory quality managment system: Handbook. Geneva, Switzerland: World Health Organization.
Willmarth, K. 2015. The FDA and genetic testing: Improper tools for a difficult problem. Journal of Law and the Biosciences 2(1):158–166.
Wolf, S. M., B. N. Crock, B. Van Ness, F. Lawrenz, J. P. Kahn, L. M. Beskow, M. K. Cho, M. F. Christman, R. C. Green, R. Hall, J. Illes, M. Keane, B. M. Knoppers, B. A. Koenig, I. S. Kohane, B. Leroy, K. J. Maschke, W. McGeveran, P. Ossorio, L. S. Parker, G. M. Petersen, H. S. Richardson, J. A. Scott, S. F. Terry, B. S. Wilfond, and W. A. Wolf. 2012. Managing incidental findings and research results in genomic research involving biobanks and archived data sets. Genetics in Medicine 14(4):361–384.
Yost, J. A. 2003. Laboratory inspection: The view from CMS. Laboratory Medicine 34(2):136–140.
Zapata-García, D., M. Llauradó, and G. Rauret. 2007. Experience of implementing ISO 17025 for the accreditation of a university testing laboratory. Accreditation and Quality Assurance 12(6):317–322.






















































