
5
Nutrigenomics: Regulatory, Ethical, and Science Policy Considerations
In session 3, moderated by Patsy Brannon, speakers considered a range of policy and ethical issues in personalized nutrition. Their presentations took a close look at the nature and strength of nutrigenomic evidence in terms of both what it needs to be and what it is, and at consumer perspectives, behaviors, and ethics. This chapter summarizes the session 3 presentations and discussion, with highlights provided in Box 5-1.
SCIENTIFIC BASIS OF GENETICALLY PERSONALIZED NUTRITION:
ETHICAL IMPLICATIONS OF METHODOLOGICAL LIMITATIONS
To begin her discussion of the ethical implications of the methodological limitations of the scientific evidence for personalized medicine, Cecile Janssens, Emory University, remarked that she had presented a similar lecture at the 2008 Evaluation of Genomic Applications in Practice and Prevention (EGAPP) meeting in Atlanta, Georgia. The title of that talk was “A Critical Appraisal of the Scientific Basis of Commercial Genomic Profiles Used to Assess Health Risks and Personalize Diet and Lifestyle Interventions.” At the time, she recalled, there were many companies selling personalized diet DNA tests via the Internet, with many people believing what Janssens referred to as the “myths about weight loss.” For example, Genotrim was among the first companies to introduce a DNA-customized “solution” for weight, telling consumers that the advice being provided would last a lifetime because, the company claimed, “your genes are not a
fad.” Some of the genomic profiles being sold by those companies, Janssens noted, focused on specific diseases or disease categories, such as heart health, bone health, or inflammation health, while others were what she characterized as a potpourri of gene variants supposedly statistically associated with some kind of health outcome. She added that the companies would then provide dietary recommendations based on having detected, for
example, an increased risk for heart disease. Her presentation began with a review of the evidence behind these claims.
Reviewing the Evidence Behind Company Claims That Gene Variants Are Associated with Disease Risk
In an article published in the American Journal of Human Genetics, Janssens and colleagues (2008) review the scientific literature for evidence of any association between the gene variants being tested by those early companies and disease risk. Janssens explained that she and her colleagues did not limit the outcomes of their search to certain classifications of disease. For example, if a company was using a gene to provide consumers with information about heart health, they searched for evidence of an association between that gene and any disease, not just heart disease. Additionally, they required that the evidence be robust, so they reviewed only meta-analyses, not single studies. Specifically, they investigated the claims of seven different companies, pooling all patients tested for a total of 69 polymorphisms in 56 genes. Of those 56 genes, they found no evidence at all for almost half (24); that is, none of those genes had been included in a meta-analysis. Another 7 of the 56 genes had been analyzed in meta-analyses but showed no statistically significant association with any disease. The remaining 25 genes also had been included in meta-analyses and had been shown to be significantly associated with disease, but with 28 different diseases, and most of the reported effects were small. What Janssens characterized as the two most amazing findings of this review were, first, that the genes being used to construct consumers’ cardiogenomic profiles were more frequently associated with noncardiovascular than with cardiovascular diseases; and, second, that two of the five genes used to construct consumers’ osteogenomic profiles (i.e., risk for bone diseases) were associated not with bone disease but with Alzheimer’s disease, asthma, non-Hodgkin’s lymphoma, obesity, psoriasis, and systemic lupus erythematosus.
While this review was conducted 10 years ago, Jannsens continued, “I’m not really very positive that the situation at this moment is very much different.” In a recent commentary, Janssens and colleagues (2017) describe their observation of two nutrigenomic studies in which the investigators had tested all participants for the APOE gene in order to tailor recommendations on saturated fat intake, but without telling them that one of the APOE alleles is a major risk factor for Alzheimer’s disease. Thus now, she explained, all of the people tested in one of these studies know whether they carry either one or two copies of the risk allele, and all of the people tested in the other study know whether they carry at least one copy. And, she added, as soon as they search for and read about APOE on the Internet, they will know they carry an allele that is associated with a risk
for Alzheimer’s. For Janssens, that they were not told about this risk is an indication that too few clinical genetics experts were involved in the studies. In her opinion, even a clinical geneticist in training would have recognized the association between APOE and an increased risk for Alzheimer’s disease. She believes this example of the APOE allele illustrates many ethical issues, such as those of informed consent, privacy, data sharing, and return of results.
Janssens went on to describe another study similar to Janssens et al. (2008), conducted by a team of researchers in Greece (Pavlidis et al., 2015). She explained how they identified several nutrigenomics companies, examined whether the genes included in the companies’ profiles were associated with any disease or pathological condition, and found no single statistically significant association for any of the 38 genes of interest. In cases in which a weak association was demonstrated, she noted, the evidence was based on only a limited number of studies. These authors concluded, “As solid scientific evidence is lacking, commercially available nutrigenomics tests cannot be presently recommended.”
Janssens remarked that, as a critical reviewer of scientific literature, she is always aware of confirmation bias. She acknowledged that the results of the Pavlidis et al. (2015) review accord with her skepticism about nutrigenomics, but she also expressed the view that the review was not very well conducted. The investigators conducted their search of the literature using the following combination of terms: “nutrigenomics,” “[gene name],” and “[disease name].” Thus, the only articles that appeared in their search results were articles with “nutrigenomics” in their title or abstract. But Janssens pointed out that many of the possible associations of relevance to the 38 genes of interest could have been studied by researchers who were not interested in nutrigenomics, in which case that term probably would not have appeared anywhere in their papers. She said she was unsure whether the results of the Pavlidis et al. (2015) meta-analysis would have been any different if the investigators had conducted a more thorough search.
Regarding what is argued by researchers working in the field, Janssens quoted the Academy of Nutrition and Dietetics’ position paper on nutritional genomics (Camp and Trujillo, 2014): “Although the discipline of nutritional genomics holds promise for tailoring diet to a person’s genotype and influencing chronic disease development, the science is still developing.” She noted that this paper was being updated with new evidence, but did not know whether the updates would alter this conclusion. She also cited another paper, written by Görman and colleagues (2013), the principal investigators of the Food4Me study, a large nutrigenomic trial in Europe, who concluded “There is convincing evidence that common diet-related diseases are influenced by genetic factors, but knowledge in this area is fragmentary and few relationships have been tested for causality.
The evidence that genotype-based dietary advice will motivate appropriate behavior changes is also limited.” Janssens interpreted these papers to mean that the field is “not ready for prime time” yet. “It was premature in 2008,” she said, and “it’s still premature in 2017.”
Company Claims Versus Disclaimers
Despite this lack of what she characterizes as robust evidence, Janssens continued, companies advertising genetic testing for consumers continue to claim that insights from their DNA can help them eat a healthy diet best suited to their genetic makeup, metabolism, and lifestyle, or simplify dieting with a personalized nutrition plan based on their DNA. One company claims that people’s DNA plays a large role in determining how their body interacts with food, affecting preferences, sensitivities, and metabolism. But does DNA really play such a large role?, Janssens asked. Her answer was no. These companies, she asserted, are not providing recommendations—they are promising genetically personalized eating plans. In her opinion, their claims are too optimistic and can be misleading.
In contrast to their claims, Janssens continued, these companies’ disclaimers are very transparent with regard to the limitations of the testing they provide and the marginal role of genetics in how the body responds to diet. As an example she cited Helix, an online marketplace that sells nutrition-related DNA testing applications, whose disclaimer states, “Genetic variants related to nutrition are connected by the way that your body processes food, but they do not guarantee that you will or will not be successful with any given diet plan. Your DNA may help you narrow in on new diet plans that you might prefer or find more successful than others, or even just a better understanding of your existing preferences. Everyone, regardless of their genetics, will benefit from a well-balanced diet.”1
Janssens went on to point out that all of these companies provide legal disclaimers, although consumers must scroll down to the bottom of their terms of service pages to find this information, and when they do, the information is in legal language. To illustrate, she showed a screenshot of a disclaimer that reads,
This site and the information, services and materials contained on this site are provided on an “as is” basis and your use of this site is at your own risk. . . . Neither Vitagene nor its affiliates warrant that the information on this site is accurate, reliable or current. . . . Neither Vitagene nor its affiliates nor any third party supplier can be assured that the user, in using this site, has selected an appropriate service provider. Again, you should
___________________
1 See https://www.helix.com/shop/dnafit-mealplanner (accessed April 17, 2018).
use this site for general informational and educational purposes only and should direct any further inquiries to a professional health care provider.2
The point is, Janssens said, “if you put this on the bottom of your site, you can get away with any test.”
For Janssens, the content of these claims and the way these disclaimers are being communicated is where ethical principles come into play. The ethical norms of both medicine and marketing, she stressed, basically say the same thing: do good with the best intentions, and treat your patient or customer with honesty, responsibility, and transparency. In her opinion, the companies whose claims and disclaimers she had described are not meeting these criteria. She elaborated on two ethical principles in particular, as summarized below: doing good (beneficence) and autonomy.
Beneficence
Janssens explained that beneficence, which is the intent of doing good, involves, first, developing and maintaining skills and knowledge and continuously updating them to reflect the best of what is available; and, second, considering the individual circumstances of all patients. She expressed uncertainty as to how the latter criterion should be applied on the Internet, but asserted that at least the first criterion can be met. But an important question for her is whether, given that nutrigenomics research is still so young, the commercial offers of these companies are in the best interest of customers or the best interest of the companies. She highlighted as another important question whether there is any evidence on how to “compensate” genetic effects with diet, to which, in her opinion, the answer is no. “The knowledge that we have does not provide enough evidence for those kinds of tests,” she argued.
To explore further the concept of beneficence in personalized nutrition, Janssens described the analytic framework used by the U.S. Preventive Services Task Force (USPSTF) to investigate whether a screening program provides a benefit to persons at risk—for example, whether a glucose test used to screen obese individuals for prediabetes results in improved health after a dietary or exercise intervention (Melnyk et al., 2012) (see Figure 5-1a). She remarked that, while many studies link diet and genes to final outcomes—which in the glucose/prediabetes example include reduced diabetes, cardiovascular disease, and mortality—clear evidence linking diet and genes to an intermediate outcome—which in the glucose/prediabetes example would be weight loss—should be adequate for personalized nutrition (see Figure 5-1b). The problem, she emphasized, is that there is no
___________________
2 See https://vitagene.com/consent/terms_of_use (accessed April 17, 2018).
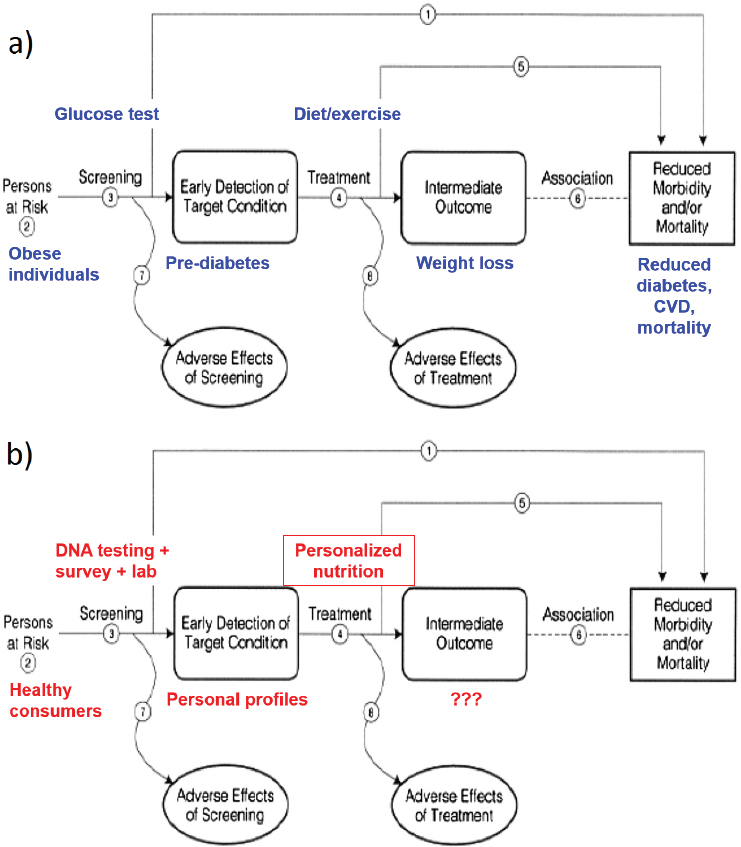
NOTE: CVD = cardiovascular disease.
SOURCES: Presented by Cecile Janssens on December 5, 2017, adapted from USPSTF, 2017.
evidence showing that a genetic disadvantage can be compensated by changing one’s diet. In other words, as represented by the question marks in Figure 5-1b, there is no evidence showing that personalized nutrition is associated with any beneficial intermediate outcomes, let alone final outcomes. “I think that’s where the science is lacking,” Janssens asserted—not with gene–disease associations, but with the solution that is being offered by these companies to break these associations.
Janssens reiterated, “I think there is not enough evidence yet to offer those tests online.” She believes that if people want to purchase these tests, they should be able to do so, but they should also be provided with the information necessary to make informed decisions about whether to make such a purchase.
Autonomy
In addition to beneficence, Janssens views autonomy as an ethical problem with current nutrigenomics companies. She explained that many websites use proprietary algorithms that provide consumers with no insight into how the company makes its recommendations based on a consumer’s DNA profile. This means, she stated, that neither consumers nor scientists can verify whether the information being provided makes sense, nor can anyone verify what the company is doing with the algorithm. For Janssens, this raises the question of whether a company is using an advanced algorithm to develop a personalized diet based on multiple SNPs with small effects, or using a simple list of specific recommendations, or rules (e.g., the rule for one SNP might be “eat more broccoli,” and for another, “consume more vitamin D”), and then combining them into a personalized combination of recommendations. Janssens argued that to build trust, it is essential to provide some insight into how advanced a company’s algorithm is.
If the companies are not using advanced algorithms, Janssens continued, they are providing essentially the same services that were offered 10 years ago, when, for example, it was recommended that an individual with variation in MTHFR, MTRR, MTR, or CBS add a supplement containing 800 mcg folic acid, 15 mg vitamin B6, and 20 mcg B12 (Arkadianos et al., 2007). Basically, she elaborated, there was a specific recommendation for every gene, and one’s personalized diet was the combination of all of these specific recommendations. “But that was 2008,” she said, adding that, by 2017, one would have expected the field to have advanced further.
Janssens called for companies to be straightforward when evidence is lacking, as she said 23andMe has been. She described how she had her DNA tested by 23andMe in 2009 and how the information she received from the company indicated that, while her risk for diabetes, for example, was what it was at the time of the testing, evidence was accumulating
that could alter her risk. Additionally, the company provided her with exact information about the volume of patients used to predict her risk for diabetes and how her risk would increase or decrease if she had other genotypes. In her opinion, that was enough information to verify what the company was doing and to draw a conclusion about whether the information being provided made sense.
Final Remarks
In closing, Janssens called for better and more relevant scientific studies, especially those showing whether personalized nutrition improves intermediate outcomes. Additionally and more important, in her opinion, she called for companies to show more respect to consumers. She cautioned against “spoiling the field” before personalized nutrition matures, noting that it is lacking an appropriate scientific basis.
VETTING PERSONALIZED AND GENOMICALLY GUIDED NUTRITION: ISSUES AND STRATEGIES
Delving more deeply into the vetting of personalized, genomically guided nutrition, Nicholas Schork, J. Craig Venter Institute,3 began by listing the three themes he would be covering during his presentation: (1) how to leverage trends in the biomedical sciences in nutrition-based health care, touching on themes addressed by previous speakers; (2) how to identify, verify, and vet nutrition strategies for individuals, borrowing strategies used in cancer and chronic disease management; and (3) how to apply N-of-1, aggregated N-of-1, and personal threshold–based trials.
Leveraging Trends in Biomedical Science in Nutrition-Based Health Care
Schork identified four trends in today’s biomedical science that he believes could be leveraged in nutrition-based health care: (1) personalized health care; (2) a number of emerging technologies that could facilitate personalized health care, such as DNA sequencing, proteomics, wireless technologies, and novel imaging technologies; (3) big data and the use of information technology to identify patterns in the massive amounts of data that are being collected at the population level; and (4) emerging strategies in artificial intelligence. He identified as the goal for nutrition-based health
___________________
3 Schork was chair of the planning committee for the 2006 National Academies workshop on nutrigenomics (IOM, 2007). The workshop summary is available at https://www.nap.edu/catalog/11845 (accessed March 23, 2018).
care combining these trends such that something compelling can be said about the nutritional needs of individuals.
However, Schork continued, these trends also raise some questions, beginning with what he termed the “garbage in, garbage out principle”: that unless data are of sufficient quality, an analysis of those data will not yield reliable results. In the context of vetting nutritional strategies, he elaborated, a number of questions need to addressed, including how to develop these strategies in the first place, how to test them in humans, and how to deploy them at the population level. Additionally, he raised the question of what these nutritional strategies are trying to optimize—individual outcomes; cost savings for the community as a whole, such as by reducing the incidence of disease in the population at large; or quality of life. He encouraged the workshop participants to keep these questions mind as he proceeded and suggested that they could shed light on some of the controversies he would be describing.
In addition to these emerging trends in biomedical science, Schork mentioned recent, relevant changes at the U.S. Food and Drug Administration (FDA). He predicted that some of these changes will bear on the claims one can make in the future about nutritional interventions. Specifically, he was referring to the 21st Century Cures Act, signed into law by President Obama in December 2016, which under certain conditions allows companies to provide “data summaries” and “real-world evidence,” such as results of observational studies, insurance claims data, patient input, and anecdotal data, rather than full clinical trial results (FDA, 2017a). The data must be compelling and collected in a sophisticated way, although definitions of “compelling” have yet to be proposed. Nonetheless, Schork characterized this as “a complete game changer.” He noted that FDA has issued a number of white papers on various aspects of this new legislation that he thinks may be worthy of consideration by nutritional scientists.
Identifying, Verifying, and Vetting Nutrition Strategies for Individuals
For Schork, a key question to consider when thinking about how to leverage this new legislation and new technologies to identify, verify, and vet nutrition strategies for individuals is what is actually being tailored to what. Is a gross diet, such as the Atkins or Mediterranean diet, being tailored to an individual’s genetics? Or are refined nutrient recommendations being tailored to an individual based on the collection of many different types of data?
Schork described four levels of nutrition strategies for individuals. First is the traditional, one-size-fits-all strategy, which involves simply providing everyone with the same diet. Second is stratified nutrition, which may involve using a couple of biomarkers of relevance to nutritional response
to place people in homogeneous categories and then providing each category, or subgroup, with a specific diet. Third, taking that strategy one step further, multiple biomarkers could be used to refine the subgroups, an approach Schork referred to as “precision nutrition.” Finally, at the individualized, or personalized, nutrition level, every individual is provided a uniquely nuanced diet based on his or her genetic or biochemical profile. But, again, Schork suggested, these possibilities raise questions: Which of these four levels works best? How does one define “best” (e.g., economics, patient benefit, scientific understanding)? And how does one prove that one or another approach is best?
As an example of work in this area, Schork referenced a study by Zeevi and colleagues (2015), who developed a strategy for collecting a large amount of data on a group of individuals and then matching those profiles to certain dietary recommendations in an attempt to identify subgroups of individuals who would respond best, or optimally, to particular dietary interventions. Additionally, the authors took their study one step further to pursue what Schork said amounted to a small randomized controlled trial that showed that in fact, the strategy had value.
Schork went on to state that this stratified approach has an analogy in the cancer space. The cancer community knows, he explained, that certain drugs can overcome the defects induced by certain genetic perturbations (i.e., mutations) often found in tumors (Simon and Roychowdhury, 2013). But to test, or vet, each drug–perturbation match would require what Schork described as “a zillion small clinical trials,” which no one is likely to pay for or pursue. So, he said, the cancer community has developed a few strategies for vetting drug–mutation matches. As an example, he cited the “basket trial,” whereby patients who are enrolled in a trial are steered toward whatever treatment “basket” is most relevant, or most likely to counteract the defect(s) caused by their mutation profile, based on an a priori scheme, or algorithm, for matching patients to drugs. He emphasized that it is not the individual drugs that are being tested in these basket trials, but the a priori scheme for matching a drug to a mutation. Based on his conversations with FDA, he remarked that this same vetting strategy is applicable not just to cancer but to all diseases, and that people can be profiled not just at the genomic level but on the transcriptomic and proteomic levels as well. He predicted that in the future of nutrition, it will be these algorithms that will be tested, not individual nutrients versus individual profile characteristics.
Schork noted that in his discussions with FDA, another issue that arose was that this type of study often does not take into account insights derived outside of a trial. According to historical FDA standards, he elaborated, someone initiating a trial to test a particular matching scheme would not be able to incorporate any new information with bearing on the drug or
nutrient being tested that emerged over the course of the trial. According to Schork, this potentially could result in a disservice to the individuals participating in the trial. In the future, he argued, some discussion will need to take place around how to make these trials more adaptive such that they can incorporate data external to a trial into whatever strategy is being vetted.
Schork stated that in his opinion, it should not be obligatory to test the algorithms being used to develop nutrition strategies for individuals (e.g., via legislation or regulatory oversight at some level)—that is, to show that people who are provided therapies based on an algorithm have better outcomes than those resulting from the standard of care or experienced by some comparator group. However, he suggested further that if a company is not curious enough about or confident enough in its technologies to want to see if its algorithm works, that company should probably be approached “with major caution.”
N-of-1, Aggregated N-of-1, and Personal Threshold–Based Trials
Finally, Schork considered several emerging trial designs that focus on the well-being of an individual rather than that of the population at large (Schork, 2015). He characterized the basic idea behind these trials as fairly simple. He explained that one may want to measure an individual’s phenotype and then modify it through intervention, but not know what intervention would be useful. So one could measure the phenotype; subject the individual to a particular intervention, or diet; take the individual off the diet; then subject him or her to a comparator diet; and finally measure the phenotype over the course of these alternating applications. Then on this basis, Schork stated, objective claims could be made about which intervention worked best for that particular individual. He added that the same N-of-1 study on a different individual might yield a different result; that is, the second individual might respond better to a different intervention.
Schork noted that although N-of-1 studies have been pursued in the literature (Lillie et al., 2011), only some have dealt with dietary manipulations. But he envisioned the approach being used more in nutrition in the future, leveraging wireless technology to collect phenotypic information continuously.
There are several different N-of-1 study designs, Schork continued, including the sequential design (i.e., making decisions in real time), as well as aggregated N-of-1 studies (Schork and Goetz, 2017). He explained that the latter design involves aggregating results from multiple N-of-1 studies so that patterns can be detected, and subsets of individuals with the same kind of response can be identified. Then the next question would be, he said,
What is it about one subset that differentiates it from another? It could be, for example, that individuals in that subset possess a particular genotype, or perhaps they were exposed to something that was not accounted for when the study was started. Schork suggested thinking about N-of-1 studies as a way to identify phenotypes for later detailed study.
Schork cited as an example of an N-of-1 study the work of David and colleagues (2014), who collected information on one of the investigator’s own microbiomes, as well as information on his diet, every day for more than 1 year. At the end of the study, they found a number of compelling associations—such as that between fiber in the diet and changes in the microbiome—that they believed might provide insights into how one can optimize one’s diet. Schork himself conducted an N-of-1 study on the effects of interventions on blood pressure. In this study, he and his colleagues found that one of two drugs had a greater effect, but they were unable to discern whether the individual’s drop in blood pressure was due to the effect of that one drug or to weight loss, because over the course of the study, the individual had become more health conscious and had lost 10 pounds. Schork also was involved in another N-of-1 study that helped identify an optimal strategy for treating a genetically mediated sleep disorder.
In closing, Schork differentiated between population and personal thresholds. He defined population thresholds are those defined on the basis of epidemiological studies; if a person’s biomarker level exceeds the population threshold, he or she is at risk. Personal thresholds, in contrast, are based on an individual’s personal average (with error bars), obtained from historical or legacy measures of the biomarker in that individual, with any deviation over time being an indication of a health status change even if the person’s biomarker level remains below the population threshold. Schork concluded by observing that, as demonstrated by Drescher and colleagues (2013), using a personal as opposed to a population threshold can minimize the amount of time a person might have latent disease.
POTENTIAL REGULATORY POLICY CONSIDERATIONS PRESENTED BY NUTRIGENOMICS IN THE COMMERCIAL CONTEXT
The third and final speaker of this session, Sarah Roller, Kelly Drye & Warren, LLP, shared her thoughts on key regulatory issues she believes merit further consideration as nutrigenomics moves forward commercially. She began by providing an outline of her talk. First, she would provide an overview of the current federal legal framework that governs genetic testing and health benefit claims for the types of foods that might be used in the context of nutrigenomics. She noted that she would not have time to cover state law, but emphasized that states have regulatory authority that is
comparable to federal law and in some cases is even more stringent. Next, she would be highlighting recent legal developments related to commercial direct-to-consumer genetic testing. Finally, she would be highlighting some key regulatory considerations for the commercialization of nutrigenomics moving forward.
Federal Legal Framework for Health-Related Genetic Testing
Roller explained that all laboratories that perform health-related testing, including genetic testing, are subject to federal regulatory standards administered by the Centers for Medicare & Medicaid Services (CMS) under the Clinical Laboratory Improvement Amendments (CLIA). These standards, she noted, govern how tests are performed, the qualification of laboratory personnel, and quality control procedures for each laboratory. She added that they are designed to ensure the analytical validity, but not the clinical validity, of genetic testing in laboratories that perform health-related testing.
In contrast, Roller continued, FDA regulates genetic testing kits and components that are sold to clinical laboratories or other persons under the Federal Food, Drug and Cosmetic Act (FDCA), which requires that products be cleared by FDA before marketing. In accordance with the definition of “device” in the statute, FDA’s authority to regulate medical devices covers in vitro reagents and any genetic testing kit or related article that is intended for use in the diagnosis of disease or other conditions or in the cure, mitigation, treatment, or prevention of disease. Roller noted that, while there is some controversy concerning the precise scope of FDA’s authority to regulate tests that are developed in house by laboratories (i.e., laboratory-developed tests, or LDTs), FDA has taken the legal position that laboratories offering LDTs are subject to both the CLIA and the FDCA, yet FDA has generally exercised enforcement discretion and refrained from enforcing the pre-market clearance requirements for LDTs.
Roller went on to point out that in 2015, FDA issued a report on its findings from an evaluation of 20 publicly available case studies involving what it called “problematic LDTs,” including some genetic tests (FDA, 2015). According to Roller, none of the problematic LDTs appeared to have been related to nutrigenomics, but among those 20 cases, FDA identified a number of critical issues posed by LDTs that had not been cleared by the agency, including lack of evidence supporting clinical validity, lack of pre-market review of performance data, and unsubstantiated product claims. In the specific context of genetic tests, she reported, FDA found that some tests yielded too many false positives and others too many false negatives, some detected factors that had no clear relevance to the disease at issue, some linked to treatments that were based on disproven scientific
concepts, and others had problems with lack of validation. She pointed out that all of these 20 problematic LDTs had met the minimum CLIA standards.
Roller emphasized that only health-related genetic tests qualify as medical devices. So, for example, Helix’s many applications, which she noted that Janssens had also mentioned during her presentation (in the context of her discussion of the contrast between companies’ claims and disclaimers about the genetic tests they are selling), are classified as entertainment applications of genetic testing. To illustrate this point, Roller observed that through Helix, one can order genetic testing to gain insight into the types of wine one is likely to prefer based on taste preference, how much “the Neanderthal genome” is reflected in one’s own genome, or whether one’s metabolism is more farmer or hunter based. She added that consumers can even purchase socks, scarves, or tartans that are color-coded to reflect their personal genetic profile. Because these are not health applications, she reiterated, they do not qualify as medical devices, so FDA does not have jurisdiction to regulate the tests involved under the medical device provisions of the FDCA.
Direct-to-Consumer (DTC) Genetic Testing
Roller went on to point out that, while a number of personal test kits have been cleared by FDA for DTC marketing, the very first DTC genetic test—23andMe’s Genetic Health Risks (GHR) test(s)—was not cleared until early 2017. She noted that the initial set of approved GHR tests was intended for use in determining an individual’s genetic predisposition to 10 diseases, and as far as she was aware, that initial set had already been expanded by the time of the workshop. She explained that the tests analyze DNA from a saliva sample and provide results intended to help individuals make decisions about lifestyle choices or inform discussions with their health care providers.
FDA cleared the 23andMe tests through the de novo premarket review pathway, which Roller explained is available for devices that are novel, that is, not substantially equivalent to an already legally marketed device, and that present low to moderate risk. The conditions of approval were designed to provide reasonable assurance of the safety and effectiveness of both the initially approved and similar GHR tests produced by 23andMe. According to Roller, the agency intends to exempt additional 23andMe GHR tests from pre-market review and may also exempt GHR tests of other makers after they submit their first pre-market notification. Because of the higher risks associated with diagnostic tests, she added, FDA approval excluded diagnostic tests from its scope.
Regulatory Considerations for the Commercialization of Nutrigenomics: Health Benefit Claims for Foods
Under the FDCA, FDA also regulates the safety and labeling of food, Roller continued. “Food” is a broad category, she noted, defined as “articles used for food or drink,” “chewing gum,” and “components” of these articles. “Food” encompasses conventional foods and beverages, including nutritionally fortified and enriched foods; dietary supplements, which are foods that are not in conventional food form and are consumed to supplement the diet with an essential nutrient or other dietary ingredient; foods for special dietary use, which Roller noted is a very old category, one that includes foods designed to serve particular dietary needs due to a physical, physiological, pathological, or other condition, such as disease convalescence, pregnancy, lactation, food allergy, underweight, or overweight; and medical foods, which are foods that are formulated to be consumed or administered orally under the supervision of a physician and are intended for the specific dietary management of a disease or condition for which distinctive nutritional requirements have been established that cannot be satisfied by dietary modification alone (FDA, 2017b). Roller cited Lofenalac4 for people with phenylketonuria as an example of a medical food.
Roller stressed that, regardless of category, a food may also be a drug because the FDCA defines a drug as “(B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals [excludes FDA-cleared ‘health claim’].” Thus, she elaborated, if a vendor makes a claim for a food product suggesting that the food has benefits with respect to diagnosing, curing, mitigating, treating, or preventing a disease, FDA may regulate that product as a drug unless the particular claim has been cleared by the agency as a health claim. If the product is approved on the basis of a health claim instead—that is, if the claim suggests that a food has health maintenance or health promotion benefits rather than disease prevention benefits—the marketer must instead substantiate the claim under what Roller described as the structure/function claim carve-out from the drug definition.
According to Roller, while FDA historically has interpreted the drug provisions of the FDCA very broadly so as to limit the range of disease-related claims that can be made for food products without triggering drug status, the agency also has broad authority to interpret and enforce the statute and regulations flexibly. She offered a few examples in which FDA has relied on this authority to exercise enforcement discretion and refrain from enforcing the letter of the statute or agency rules. She cited as a
___________________
4 Lofenalac is an oral powder prescribed to replace milk in the diets of infants and children with phenylketonuria; it has been regulated as a food since 1972.
recent example FDA’s guidance allowing “healthy” claims for higher-fat foods that contain a healthy balance of fatty acids, even though the rule in the Code of Federal Regulations (CFR) does not include this provision, adding that this guidance was partly in response to the most recent dietary guidelines. She referred to qualified “health claims”—truthful health claims substantiated by credible evidence that does not meet the “significant scientific agreement” evidence standard—as a second example. Finally, as a third example she pointed to FDA currently allowing medical device data systems (MDDSs) and many medical apps to be marketed without meeting the requirements for medical devices, even though their makers call their products medical devices. This latter policy, she remarked, is intended to avoid overregulation of promising lower-risk health information technology (HIT) applications.
Regulatory Considerations for the Commercialization of Nutrigenomics: Federal Trade Commission (FTC) Authority
Finally, in addition to CLIA and FDA standards and regulations, Roller discussed FTC’s broad authority under the Federal Trade Commission Act (FTCA) to prohibit unfair and deceptive acts and practices, including false advertising. In contrast with FDA, she elaborated, the scope of FTC’s authority does not cover products, only acts and practices. In the context of nutrigenomics, she explained, FTC authority encompasses advertising claims for genetic testing and food products and services, as well as data security practices of companies that produce and store personal consumer information (e.g., social security numbers and personal genetic test results).
Roller went on to point out that based on a large body of FTC false advertising case law, FTC has developed detailed policies and guidance under the FTCA concerning requirements for claim substantiation. She then highlighted a few of these policies.
First, Roller noted, both expressed and implied claims must be accurate and substantiated before they are used. Second, Roller continued, a claim must be supported by evidence that provides a reasonable basis for the claim. The basis need not be complete, she clarified, just reasonable, and the reasonable basis standard is flexible. She explained that FTC takes several factors into account when determining the nature and amount of evidence required, including the type of product and claim, the benefits of a truthful claim, the consequences of a false claim, the cost and feasibility of developing claim substantiation, and the nature and amount of evidence that experts in the relevant field believe is reasonable to support this kind of claim. For Roller, the last criterion is especially important. “It’s what people like you believe would be necessary for there to be a reasonable basis,” she said.
Roller identified as a third FTC policy concerning claim substantiation that “competent and reliable scientific evidence” is generally required for health-related claims. This means, she clarified, that studies must be conducted objectively by qualified persons using investigative procedures generally accepted in the relevant scientific community, such as randomized controlled trials. She added that even in the context of this “competent and reliable scientific evidence” standard, there is flexibility regarding the amount and type of evidence, quoting FTC guidance (FTC, 2001):
A guiding principle for determining the amount and type of evidence that will be sufficient is what experts in the relevant area of study would generally consider to be adequate. The FTC will consider all forms of competent and reliable scientific research when evaluating substantiation. As a general rule, well-controlled human clinical studies are the most reliable form of evidence. Results obtained in animal and in vitro studies will also be examined, particularly where they are widely considered to be acceptable substitutes for human research, or where human research is infeasible. . . . Although there is no requirement that a . . . claim be supported by a specific number of studies, the replication of results in an independently conducted study adds to the weight of the evidence. In most situations, the quality of studies will be more important than quantity. When a clinical trial is not possible (e.g., in the case of a relationship between a nutrient and a condition that may take decades to develop), epidemiologic evidence may be an acceptable substitute for clinical data, especially when supported by other evidence.
As an example of recent FTC enforcement, Roller reported that in 2013, FTC took an enforcement action against Genelink, Inc., and foruTM International Corporation, alleging that both companies had violated the FTCA by making false and unsubstantiated health benefit claims about their genetic tests and genetically customized dietary supplements, and by using inadequate data security practices, putting consumers at risk of identity theft. Roller did not delve into the details of the allegations, but pointed out that they addressed both the claims and the characterizations of the evidence, as well as the companies’ data security and data sharing practices falling short of requirements to protect private consumer information. The cases were settled in 2014.
Roller cited as another example that, although 23andMe’s GHR tests were recently cleared through FDA, it sent the company a warning letter in 2013 because 23andMe was marketing the tests without their having yet been cleared. The company successfully responded to that warning letter in 2014 following a cascade of class action litigation inspired by the letter. According to Roller, the complaints that were filed in those lawsuits (all of which were or are being settled, she noted) were similar to consumer protection issues
raised by FTC, such as misrepresentation to the consumer (e.g., the marketing of the product implied to consumers that it had scientific support and FDA approval) and inadequate data security practices (e.g., consumers were unaware that third parties would have access to their genetic information).
Summary
In closing, Roller recapped issues that she believes merit further consideration as nutrigenomics moves forward, such as the adequacy of
- existing CMS and FDA frameworks for regulating genetic tests, including LDTs that are used in DTC products and services;
- existing FDA regulatory framework to accommodate nutrigenomics claims for foods without triggering “drug” status;
- existing claim substantiation guidance with respect to claims promoting genetic tests and foods in the context of nutrigenomics, such as what “competent and reliable scientific evidence” should mean in this context; and
- data security and disclosure practices concerning the personal genetic information that is being collected.
DISCUSSION
Following Roller’s presentation, she, Janssens, and Schork participated in an open discussion with the audience, summarized here.
Nutrition in the Disruptive Frontier: Innovation Versus Regulation
An audience member commented on how the “disruptive frontier” being described at the workshop really challenges the way nutrition is understood. As examples, she mentioned Steven Zeisel’s discussion of medical food as a potential category for classification in the context of nutrigenomics (Zeisel’s presentation is summarized in Chapter 4) and Schork’s discussion of N-of-1 studies and real-world evidence as new ways to measure the impact of nutritional interventions. She asked the speakers to reflect on the balance between maintaining a long-term outlook and continuing to foster this type of innovation so that nutrigenomics products can reach the people who really need them, as well as the need for oversight to control these developments from food safety and communication perspectives.
Roller responded, “I think that right now is about as favorable an environment for bringing those questions to the FDA as we have had in a number of years.” In addition to the 23andMe clearance showing “some creativity,” she mentioned a health claim that had been cleared earlier in
2017 concerning a food allergen, commenting on how the claim addressed the kind of health condition for which previous health claims had not been cleared. She cautioned, however, that while the environment is favorable, it is important to be very particular when requesting a response from FDA.
Schork added that relatively little time had elapsed since the 21st Century Cures Act was introduced, so the fact that there have not yet been many examples of such requests may simply reflect the fact that investigators have not had enough time to develop new study designs or ways to collect data in the real world. He agreed with Roller that currently, FDA is willing and eager to listen to the scientific community, more so than it has been in the past. As another example of a new development, he mentioned that it is now possible to register digital therapeutics (e.g., apps on smart-phones) with FDA as health technologies, so that if approved, they can be prescribed by physicians instead of pills. “So there’s a lot of really forward thinking that goes on,” he said, adding that “of course the data is going to have to pass muster, but there’s a lot of receptivity for novel ideas.”
The Flexibility of the FDA Regulatory System
An audience member observed that currently, risk is structured by “put[ting] things very neatly into boxes with boundaries.” For example, there is a definition for food, a definition for dietary supplements, and so on, and FDA is structured that way as well; that is, devices and biologics and drugs are each considered separately. Yet, the audience member pointed out that the discussion at the workshop had revolved around a systems approach whereby testing “that” leads to “this” and to “this.” “But we don’t have a regulatory system that puts those puzzle pieces together,” she said, or an agency that is structured to deal with a holistic approach. She asked how, moving forward, the systems-level, or holistic, nature of nutrigenomics will be managed from a regulatory perspective when the regulatory system is not structured to handle such an approach.
Roller replied that, although FDA has customarily interpreted its statutes in particular ways, in fact this simply reflects a general principle of administrative law. That is, she explained, when an agency interprets its own statute, the courts will defer to the agency. So if the agency has been interpreting a statute in a particular way but decides that the world is changing and a different approach is needed, it can change its approach—it simply needs to justify the change. Roller again emphasized the flexibility and authority FDA has to be creative.
The audience member also commented on the fact that there is no established format for labeling medical foods, in contrast with dietary supplements and conventional foods. The term “medical food” does not appear on foods that are intended to be medical foods, she elaborated,
while many foods in the marketplace that do not meet the definition of medical food are labeled as such. Thus, she said, consumers cannot tell by looking at the package whether a food is a medical food (as defined by FDA). Roller replied that only one kind of food is required to have a label indicating what it is, and that is dietary supplements. For all the other food categories, she explained, what is important is that the product meet the requirements for that kind of food. She reiterated that FDA has flexibility, in this case with its approach to medical foods. In her opinion, something not being well defined can actually be a “good thing.”
Transparency of 23andMe
Ahmed El-Sohemy commented on Janssens’s approval of 23andMe’s handling of its DTC genetic testing. Janssens clarified that she has, in fact, criticized 23andMe many times in the past and that there are still things about the company she does not like, mainly in relation to its presentation of health risks. But she does like its transparency and how clearly it informs consumers that their risks will need to be updated as new material becomes available, and that their current risks are based only on “what the science at this moment knows about your genes.” She recalled that her own thrombosis risk “decreased” from 24 percent to 9 percent when the company refined incidence rates to be sex-specific.
The Ethics of Sharing Information with Patients
An audience member commented on the many medical centers in the United States that ask their patients to provide DNA so they can conduct whole-exome sequencing and then bank those data. He asked whether it was ethical not to give that information back to the patients. To him, it appears that information is being collected about many SNPs, or alleles, that could be of value to people’s health but is not being shared.
“That is a difficult question,” Janssens replied. After clarifying that only data are collected and that data do not become information until they are interpreted by an algorithm or specialist, she expressed uncertainty about whether it was ethical not to share that information. She posed a different question: Is it ethical to give something to the patient when the patient does not know what to do with it? “I think in health care,” she said, “doctors should give answers to questions, and not just answers, but that’s a personal opinion.”
Roller added that, based on her observations and in the context of litigation at the federal level, it appears that genetic information is being treated in much that same way as social security numbers. In other words, she clarified, disclose all the material facts and ensure that consumers know
what they are getting. But for her, the question is, Is it the same kind of information, or is it something more significant? Right now, she observed, there is no legal framework for dealing with these kinds of ethical issues. She added that in the complaints she had mentioned during her presentation, people have mentioned property rights. “It’s not settled right now,” she said, “but I do think it’s an issue that does deserve further consideration.”
Janssens added that the American College of Medical Geneticists has developed a list of variants that it calls “actionable mutations.” These are variants, she clarified, that need to be returned when people undergo sequencing, such as the BRCA mutation for breast cancer.
Is Nutrigenomics Premature or Is It Ready for Prime Time?: The Level of Evidence Needed
In response to points made by Janssens during her presentation, El-Sohemy pointed out, first, that the Academy of Nutrition and Dietetics had formally withdrawn its position on nutrigenomics and was not just updating it, as Janssens had mentioned; and, second, that some letters to the editor have asserted that the Pavlidis et al. (2015) study, which Janssens had discussed, should be retracted. Additionally, El-Sohemy pointed out that his company, Nutrigenomix, has been asked by practitioners to provide APOE testing to predict response to blood lipids, but that the company decided not to include that gene in its panel because of the potential for unintended consequences (i.e., due to its association with a risk for Alzheimer’s disease). He then asked Janssens what kind of evidence she would need to see to agree that nutrigenomics should be available and used.
Janssens replied that in her opinion, proper evidence does not stop with demonstrating a robust gene–diet outcome association. That is the start, she said, “but you need to show that changing a diet really compensates that genetic disadvantage.” So, for example, if a certain genotype has been shown to be associated with higher blood pressure, she believes it needs to be demonstrated that by changing diet, blood pressure in individuals with that genotype can be lowered to a level that is similar to that associated with other genotypes. Whether developing this evidence requires a randomized controlled trial or an observational study, she suggested starting with the latter; then, when sufficient observational evidence exists, see if a randomized controlled trial is still needed. She explained that she holds this view because the field is changing too rapidly to rely on randomized controlled trials. She emphasized that in her opinion, what is not needed is more evidence on clinical outcomes, and thus she discouraged the type of long study needed to show whether a change in diet affects clinical outcome. “If you can show that the intermediate factor changes, for me, that’s enough,” she said.






















