
3
Introduction to the Study of Generational Effects
The transmission of health effects to his or her children, grandchildren, and subsequent generations resulting from a veteran’s exposures is referred to as generational inheritance. A basic knowledge of both genetics1 and a newer field—epigenetics2—is necessary to appreciate the potential biological mechanisms underlying heritable effects across generations. The potential impact of deployment exposures on veterans and their descendants must be considered in the broader context of a veteran’s genome and epigenome as well as of exposures to other stressors (biological, chemical, psychological, radiological, or physiological) before, during, and after deployment that can also have genetic and epigenetic effects. This chapter describes how environmental exposures can affect the genome and epigenome, explains how those changes can be transmitted to children and subsequent generations, and provides a brief background on the evolving field of generational health effects. The committee cautions, however, that not all adverse effects on reproduction or fetal development are the result of genetic or epigenetic mechanisms: during gestation, on the developing embryo and fetus.
A more detailed description of the concepts of genetics and epigenetics as they apply to deployment-related exposures is presented below. Both genetic and epigenetic mechanisms have emerged as possible mediators of exposure-related impacts on the health of veterans and subsequent generations. As such, the committee’s review of the literature included studies on genetic and epigenetic effects in its assessment of deployment exposures, and these studies are described in chapters 4 through 7. The committee recognizes that the principles of genetics and epigenetics can be complicated areas of study and that the information summarized in this chapter is at times highly technical and contains terminology that may not be familiar to all readers. A glossary of the key terms used in this report is presented in Appendix B.
___________________
1Genetics is the study of DNA and the genes it encodes, sometimes referred to as the genome when referencing the totality of the DNA (and all the genes) of an organism.
2Epigenetics is the study of processes that cause heritable changes in gene expression without changing the DNA sequence. The epigenome is comprised of chemical modifications made to DNA and the histone proteins that make up chromatin. “Heritable” in this context can refer to inheritance not only between parents and offspring (generational or meiotic inheritance of epigenetic alterations) but also between parent and daughter cells (mitotic inheritance).
HERITABLE TARGETS FOR DEPLOYMENT-RELATED EXPOSURES
According to current knowledge, the main biological targets for deployment-related exposures that could potentially mediate heritable effects across generations are the genome (and ribonucleic acid [RNA] linked to the genome), mitochondrial DNA, and the epigenome.
The Genome
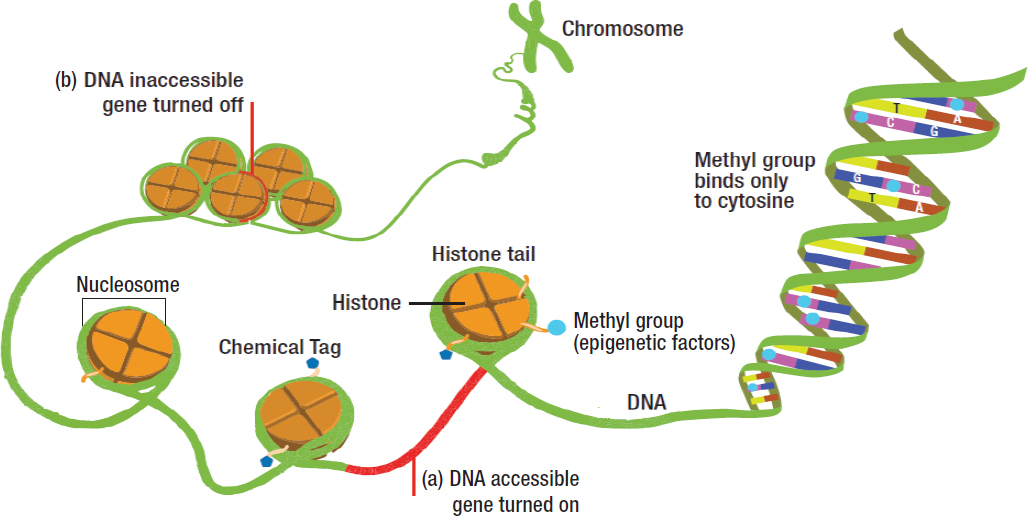
The human genome encodes the genetic information necessary for human development in DNA molecules, and, with a few exceptions, all cells in the body contain an exact copy of this genome. Except for identical twins and triplets, everyone has a unique genome, which remains relatively stable throughout a lifetime. Human cells contain both nuclear and mitochondrial DNA, which together carry the instructions (referred to as the genetic “code”) for making all the proteins within a human cell. Nuclear DNA is organized into chromosomes, which are made up of chromatin, a term used to describe DNA packaged with its associated proteins (see Figure 3-1). It has been estimated that the nuclear genome encodes in total about 20,000 genes, which through a number of different mechanisms give rise to the more than 6 million types of proteins needed by cells to function (Ponomarenko et al., 2016).
The human genome has 23 pairs of chromosomes (46 total chromosomes). Every individual inherits one set of 23 chromosomes from its mother and another 23 from its father. The sperm and the egg (the mature male and female germ cells, also called gametes) have only one set of 23 chromosomes each. These germ cells are thus different from other cells in the body, which are called somatic cells. Each copy of a gene is called an allele, and we inherit two alleles of every gene, one from each parent. The
SOURCE: NHGRI, 2016.
exceptions are the sex chromosomes, X and Y, where genes contained on the male Y chromosome are only inherited from the father. When the sperm and the egg (oocyte) unite, the new cell that is formed, called a zygote, combines the chromosomes from each parent. As a result of this combination, the newly formed zygote carries characteristics inherited from each parent, with the exception of mitochondrial DNA (mtDNA), which is inherited solely from the mother. The unique features of mitochondria and their DNA are discussed further below.
The building blocks of DNA are the bases that make up the genome: adenine, thymine, cytosine, and guanine (A, T, C, G). Errors in copying genes and gene rearrangements can occur during the course of cell replication, and individual DNA bases can mutate, either spontaneously or after exposure to DNA-damaging agents. Throughout the course of hundreds of thousands of years, mutations have accumulated in the human genome that have given rise to the phenotypic variations now seen among humans.
Mutations in DNA can occur for many reasons, including environmental exposures such as ultraviolet light and chemical carcinogens. However, if these exposures are limited to somatic cells and no mutations occur in the sperm or the oocyte, the mutations will not be passed to future generations. While some mutations are benign, others can be lethal or change the DNA in ways that give rise to or alter susceptibility to disease. The latter types of mutations can cause birth defects and diseases such as cystic fibrosis, leukemias, hemochromatosis, and sickle cell anemia. Some DNA mutations may even be beneficial and protective against diseases, such as by conferring resistance to malaria or to viral infections (Williams, 2016). Because some mutations are only harmful when present in both alleles, carriers of harmful mutations on only one allele can lead healthy lives but may pass the mutated allele to their offspring. In the latter case, if both parents are healthy carriers of one copy of the same disease-causing mutation, the offspring has a 25% chance of inheriting two mutation-carrying alleles and becoming affected. Mutations may also be acquired during the formation of the gametes or of the zygote, resulting in new mutations that the offspring genome carries but that are absent in either parent.
While genetics focuses on DNA and mutations as mechanisms of inheritance and disease, the field of epigenetics focuses on mechanisms that can cause diseases that are not dependent upon DNA mutations. Like DNA mutations, some epigenetic alterations may be transmitted across cell divisions (i.e., from mother to daughter cells) and generations (i.e., from parent to offspring). The modifications associated with epigenetic inheritance can affect both the DNA itself, as happens with DNA methylation, and also the histone proteins, which together with DNA constitute a nucleosome, the structural unit of chromatin (see Figure 3-1).
The transmission of heritable traits to future generations in response to environmental exposures of the parent or parents aligns with the so-called Barker hypothesis, also known as the developmental origins of health and disease (DOHaD) (Skogen and Overland, 2012). DOHaD hypothesizes that early life events can influence health and disease susceptibility for the entire life of an individual. Thus, in addition to DNA mutations and small variations in DNA sequence within populations (polymorphisms), environmental exposures and life experiences may also affect an individual’s susceptibility to disease. For example, both famine-influenced and obesity-influenced nutrition in utero has been associated with disease in adulthood, including type-2 diabetes and cardiovascular mortality (Langley-Evans, 2014). The diversity of the human genome may contribute to differences in susceptibility to environmental and occupational exposures. The immediate effects of those differences may be an accelerated or diminished response to harmful exposures, such as the hostile environment of a war zone. Only when environmental and lifestyle factors as well as genetic and epigenetic variation are appropriately considered will it be possible to fully understand the impact of deployment-related exposures on generational health.
The Mitochondrial Genome
In addition to changes in the nuclear genome and epigenome discussed above, environmentally driven changes in mtDNA represent another potential mechanism for environmental inheritance. Mitochondria are the energy powerhouses of cells, and they possess a genome that is distinct from that of the nucleus. The mitochondrial genome encodes 37 genes that give rise to 13 proteins crucial to cellular bioenergetic functions. A unique feature of mtDNA is that it is transmitted to subsequent generations exclusively through the maternal lineage. When mtDNA is damaged by mutations, maternally inherited disorders, termed mitochondrial encephalomyopathies, may occur. Because there are multiple copies of mtDNA, mitochondrial diseases arise when a significant proportion of affected mitochondria carry a mutation that can then be passed to the offspring. Mitochondria are extremely sensitive to environmental sources of oxidative stress because, unlike nuclear DNA, mtDNA lacks histones and DNA repair mechanisms (Kucej et al., 2008).
The Epigenome
By definition, epigenetic modifications influence the chromatin structure and gene expression without altering the underlying sequence of DNA (Barbara and Abdilla, 2017). The epigenome refers to the entire collection of epigenetic modifications of chromatin, which are sometimes called epigenetic marks. The main epigenetic marks are the methylation of DNA and histone modifications (Murr, 2010; Robertson, 2005). Small RNAs that interact directly with DNA to modulate its function also function as epigenetic modulators and play a role in epigenetic inheritance. A large number of proteins make up the epigenetic machinery that adds, removes, or acts on epigenetic marks. This machinery modifies chromatin in ways that change genome structure and function—for example, modifying how DNA is condensed into chromosomes—and determines when genes are expressed or silenced (Atlasi and Stunnenberg, 2017; Youn, 2017). The inheritance of epigenetic changes is a mechanism (in addition to DNA mutation) by which traits can be passed from parent to children—in this case, without changes in the sequence of DNA. Additional details on the epigenome are provided below.
Unlike the genome, which is mostly static and unique to each individual, the epigenome is dynamic and changes during the course of development, across the passage of time, and from one cell type to another. In fact, the way that multiple cell types and diverse tissues in the human body originate from a single cell is accounted for almost entirely by differences in the epigenome. While each person has only one genome, each cell type of the body has its own epigenome, which can change over time in response to the internal and external environment. Thus, epigenomes are considerably more malleable than genomes and as a result are inherently more vulnerable to environmental exposures. The timing of exposure is also important (described below), and development is a time of rapid changes in the epigenome, as the zygote rapidly transitions from the sperm and egg epigenomes to the new epigenomic programming that will direct the development of all cells and tissues of the body. Thus, the epigenome of developing germ cells in the embryo and fetus is quite susceptible to environmental exposures (Fraser and Lin, 2016; Xu and Xie, 2018).
Some epigenetic modifications to DNA and histones as well as modifiers such as noncoding RNAs (ncRNAs) can interact and reinforce or negate each other to influence patterns of gene expression. This means that an exposure affecting one type of epigenetic factor could also influence (reinforce or negate) another type of epigenetic factor. For example, when somatic cells are exposed to benz(a)pyrene, the initial effect is an inhibition of repressive histone marks at selective sites in the genome, along with the promotion of activating histone marks at other sites; these initial epigenetic alterations in turn affect DNA
methylation (i.e., they inhibit DNA methylation), with the overall result being an increased expression of affected genes (Teneng et al., 2011). Thus, while an exposure may initially lead to an effect on one type of epigenetic factor (e.g., histone modification), generational effects caused by that exposure could be mediated by a different type of epigenetic factor, such as DNA methylation.
The epigenetic machinery—the enzymes that “read, write, and erase” epigenetic marks—controls DNA methylation, histone marks, ncRNAs, and chromatin-remodeling proteins and is essential for the proper installation and maintenance of epigenetic functions. There are several pathways by which environmental exposures can affect the epigenome, resulting in effects that persist long after the exposure ceases—or even across generations (Walker, 2016). For example, toxicants such as endocrine disruptors can activate signaling pathways within the cell that change the activity of the epigenetic machinery and alter patterns of epigenetic marks. This reprogramming of the epigenome can alter gene expression in the progeny of exposed cells or individuals (Uzumcu et al., 2012). Other variations in the epigenome may result either from DNA mutations that directly affect the genes encoding components of the epigenetic machinery and alter epigenetic control or else from a modulation of regulatory control that compromises the functionality of the epigenetic machinery (Ramos et al., 2018). The environment may also influence the availability of cofactors needed for normal epigenetic processes to occur, such as the iron or oxygen needed for demethylase “erasers” or dietary folate that provides methyl groups for both DNA and histone methylation (Crider et al., 2012; Walker, 2016). In addition to epigenetic “readers, writers, and erasers,” many other proteins can associate with chromatin, including those that change chromatin structure to influence gene expression. The activity of these chromatin remodelers can also be influenced by environmental exposures (Bariar et al., 2013; Biswas and Rao, 2018). While these proteins may not themselves be considered part of the epigenome, they can influence epigenetic marks and thus change the epigenome in ways that are heritable. Examples of these types of proteins would be the high-mobility group proteins and the SWI/SNF ATPases that modulate chromatin structure (Hepp et al., 2014).
Epigenetic patterns vary from one cell type to another, and virtually all tissues have mixed cell populations. Therefore, when a sample of mixed cell types is analyzed, care must be taken to determine that the changes in both DNA and histone methyl marks are due to changes in the epigenome and not simply to changes in the relative cell populations in the sample. This cell heterogeneity may complicate the interpretation of the effects of exposures. For example, Jaffe and Irizarry (2014) reported cell composition changes across age in whole blood and demonstrated that cellular composition can explain much of the observed variability in the measured DNA methylation. This implies that if an exposure such as PM2.5 changes the cell composition of blood—for example, because of an immune response without any changes in methylation patterns themselves—changes in the methylation profile of blood can be observed even when no methylation changes have occurred in any cell. Future studies will need to eliminate or account for this type of confounder. This limitation applies to both human and animal studies that use tissue-based sampling. Methods to address this issue with single-cell approaches are being developed (Angermueller et al., 2016; Karemaker and Vermeulen, 2018).
DNA Methylation
DNA methylation is of interest because of the numerous examples of diseases caused by an increase in methylation (that is, hypermethylation) associated with the suppression of gene expression as well as by a loss of methylation (that is, hypomethylation) associated with increases in gene expression. Because genes are expressed differently in each cell type, gene-specific methylation patterns are one way to specify a cellular response. DNA methylation can be modified (both increased and decreased) in response to the environment. Altered DNA methylation patterns can then be copied and passed on to
daughter cells during cell division, allowing the transmission of altered methylation patterns to daughter cells, which is termed mitotic inheritance. Modified DNA methylation caused by an environmental exposure can also occur in germ cells and be passed on to future generations. This is termed meiotic inheritance and can lead to generational effects.
Depending on the timing of the exposure during development, DNA methylation changes can profoundly affect germ cells (Ly et al., 2015; Wu et al., 2015). DNA methylation is also involved in genomic imprinting, an important process that occurs during gametogenesis and early embryogenesis when gene expression patterns are established in a parent-of-origin-dependent manner, directing genes to be expressed solely from DNA inherited from the mother or from the father (McEwen and Ferguson-Smith, 2010). Imprinted genes are a classic example of genes that are marked in a sex-specific manner in the male or female germ line, often by DNA methylation, such that only one of the parental genes will be expressed in the offspring (with the gene from the other parent being methylated and therefore silenced). The methylated imprinted gene in the sperm or egg is thus inherited by the offspring and represents an example of how epigenetic changes in germ cells can be “remembered” and passed on to the next generation. The methylation silencing of the maternal or paternal gene in genomic imprinting is critical for normal development, as dysregulation of this process can result in a number of disorders, including Angelman, Prader-Willi, and Beckwith-Weidemann syndromes (Butler, 2009). Experimental studies show that toxicants’ effects on the germ cell’s epigenome can result in long-lasting marks on the chromatin which remain through cell divisions and across generations and which can affect health (Wei et al., 2015).
DNA methylation can occur at approximately 28 million cytosine-adjacent-to-guanine (CpG) sites in the human genome. While other DNA bases can also be methylated, CpG methylation is by far the most prevalent and best understood mechanism for how DNA methylation regulates gene expression; however, non-CpG methylation and hydroxymethylation are emerging marks of interest (Jang et al., 2017). The precise location of DNA methylation at CpG sites provides context for how methylation affects gene expression. Both global (genome-wide) changes and site-specific effects that are more nuanced and only affect small regions of the genome (e.g., specific genes) can be influenced by the environment.
Histones and Other Chromatin-Associated Proteins
In the cell nucleus, DNA wraps around eight histones (two each of the core histones 2a, 2b, 3, and 4) to form the nucleosome and thus chromatin. Another histone (histone 1) links proteins between nucleosomes (see Figure 3-1). Histones are subject to several types of chemical modifications, including methylation, acetylation, sumoylation, and phosphorylation, and they can participate in both the activation and the repression of gene expression. Importantly, as is the case with DNA methylation patterns, patterns of epigenetic histone marks, specifically methyl marks, can be both mitotically and meiotically inherited. Thus, exposures that alter histone methyl marks could potentially cause generational health effects. Indeed, alterations in histone marks exhibit a much greater dynamic range in terms of their effects on gene expression than changes in DNA methylation (Anonymous, 2015). At the cellular level, alterations in histone methylation can result in changes in gene expression that can be both quantitative (i.e., high versus low levels of transcription) as well as qualitative (i.e., is a gene expressed or not), whereas alterations in DNA methylation primarily result in qualitative changes in gene expression—for example, gene silencing caused by increased promoter methylation.
Noncoding RNAs
There are at least 48 different noncoding classes of RNAs in addition to the protein-coding messenger RNAs (mRNAs) in the human genome. These ncRNAs vary in size from 20–30 nucleotides in length for the small ncRNAs to longer than 200 nucleotides for the long ncRNAs. They participate in a wide array of cellular processes, from protein synthesis to epigenetic regulation and inheritance. Although examples can be found throughout the plant and animal kingdoms, this report focuses on using rodent models to inform and contrast with what is known about humans. For example, X-inactive specific transcript, a long ncRNA, is a major effector of X-chromosome inactivation. Various small ncRNAs, such as microRNAs (miRNAs) which act as scaffolds for histone and DNA methyltransferases, also participate in the epigenetic regulation of the genome in many species (Holoch and Moazed, 2015), along with transfer RNA fragments and Piwi-interacting RNAs which limit the action of transposable elements to protect the integrity of the genome (Julinano et al., 2011; Schorn et al., 2017).
Given their important functions in human germ cells, miRNAs are particularly relevant to the study of generational inheritance. While acting directly at the epigenetic level, miRNAs can also act as post-transcriptional regulators of gene expression in mammals (Virant-Klun et al., 2016). Through complementary base pairing, the miRNA can specifically destabilize and silence mRNA and modulate gene expression without altering the DNA sequence. For example, miRNAs, such as mir-181c, affect DNA methylation at fertilization by regulating DNA methyltransferase, which affects embryo pluripotency (Xu et al., 2013).
In males, epigenetic inheritance is thought to be mediated, in part, by a wide spectrum of sperm RNAs that are delivered at fertilization. The sperm RNAs are likely to participate in early embryonic development until the point of zygotic genome activation (Sendler et al., 2013; Yuan et al., 2016). From a mechanistic standpoint, sperm RNAs may have immediate effects in the zygote: extending calcium oscillations in the embryo to signal fertilization, early cell-fate decisions, genome restructuring through interaction with repetitive elements, or direct modification of the genome (Jodar et al., 2017). Similarly, various types of short and long noncoding RNAs are present in oocytes (Svoboda, 2017). Evidence is emerging that oocyte-derived miRNAs can influence gene expression in early embryos (Eckersley-Maslin et al., 2018).
Paternal exposures to toxicants can affect sperm RNA, and this may explain the patterns of paternal inheritance. Mouse models have shown that sperm RNAs can be affected by psychological and physiological factors, with the altered RNAs being passed to offspring (Yeshurun and Hannan, 2018). In the context of transgenerational inheritance, mouse sperm RNAs appear to have the capacity to be integrated into an offspring’s genome, although at a very low rate (Spadafora, 2016). The influence of paternal diet and age on mouse sperm RNAs is well documented. For example, paternal obesity can lead to an increased risk of metabolic disease in the offspring, irrespective of maternal body weight (Sharma and Rando, 2017). This parallels changes in patterns of DNA methylation and levels of various sperm RNAs. In contrast to these recent studies in males, little is known at this point regarding the role of female, small ncRNAs in the intergenerational inheritance of effects (Clarke and Vieux, 2015).
PARADIGMS OF INHERITANCE
This report uses two terms to describe paradigms of generational inheritance: “intergenerational” and “transgenerational.” Whether the effects of parental exposures on offspring are considered “inter” or “trans” generational is determined by the timing of the exposure relative to conception and by whether the exposure has a direct or an indirect effect on the health of descendants (see Figure 3-2).
In the upper panel in Figure 3-2, men and nonpregnant women (i.e., the F0 generation) may be exposed to harmful agents, but the exposure ends before conception, and subsequent generations are not
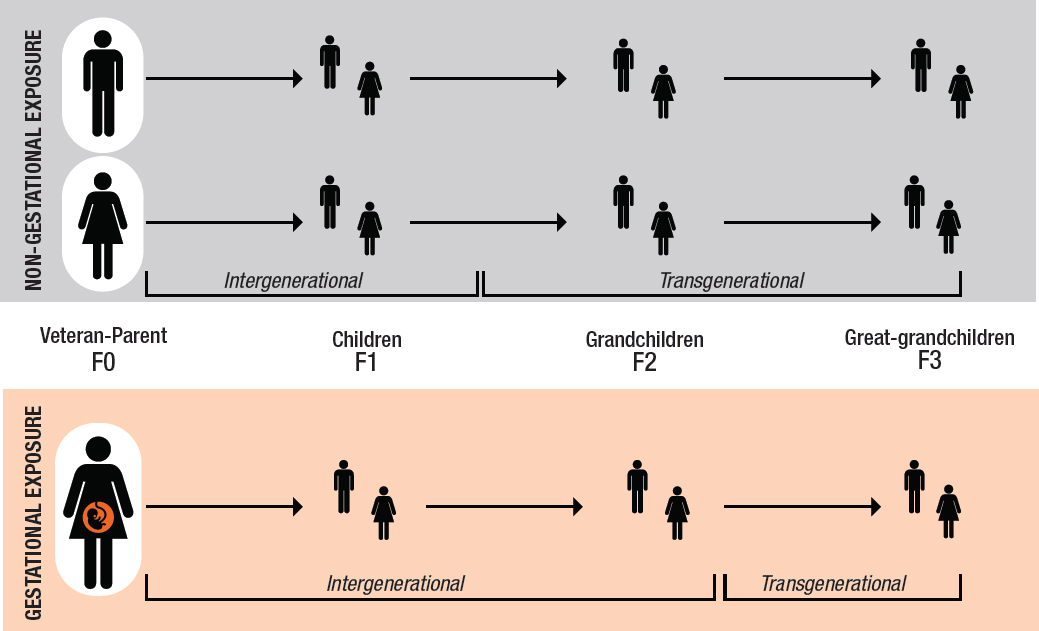
directly exposed. However, if a male or female veteran’s germ cells are exposed to a toxicant, his or her children, the F1 generation, may be considered to have been exposed as well via the exposed gametes giving rise to what is called an intergenerational effect. The observed effect in the F1 generation may be due to genetic or epigenetic changes or both. The effects that are then passed on to the children of the veteran’s children—that is, the grandchildren, the F2 generation, who were not exposed to the toxicant directly or via the germ cells—are called transgenerational effects.
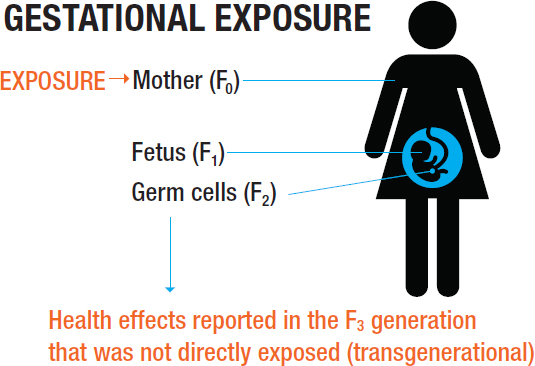
When a pregnant woman is exposed to a toxicant (the lower panel in Figure 3-2), three generations are directly exposed: the mother (F0), her fetus (the F1 generation), and the germ cells of the developing fetus. Because fetal germ cells give rise to the F2 generation, that generation is also considered to have been exposed. In Figure 3-3, effects of the maternal exposure observed in the children and grandchildren (F1 and F2 generations) constitute intergenerational effects, while effects observed in the great-grandchildren—that is, the F3 generation, the first generation to not have been exposed directly or via germ cells—are considered to be transgenerational effects.
These differences in direct and indirect exposure to a toxicant as well as the timing of that exposure represent important variables to be considered in evaluating and interpreting the results of studies designed to examine reproductive, developmental, and intergenerational and transgenerational effects. These variables are discussed in the following sections and in subsequent chapters.
SOURCE: NTP, 2018.
TIMING OF EXPOSURES AND OUTCOMES
Exposures that lead to reproductive effects in men or women can occur at any time. Parental exposures that may affect the development of an embryo, a fetus, or a child can occur before conception (during the development or maturation of germ cells or gametes) (see Figure 3-4) or during pregnancy (see Figure 3-5). The timing of exposure is particularly important to understanding whether genetic or epigenetic effects can be inherited. Mechanistically, the timing of exposure and of effects on genetic and epigenetic processes is important in the regulation of normal germ cell development (including the regulation of gene expression) and in the establishment of epigenetic marks that are passed onto the offspring at the time of fertilization.
One consideration with respect to evaluating exposure–disease associations in an intergenerational or transgenerational context is that certain exposures may have differing effects depending on when they occur prior to or across an individual’s lifecourse. In fact, exposure during the preconception and prenatal time periods may be particularly important to consider, as these may be particularly vulnerable critical windows of growth and development with later life effects on chronic disease risk in the offspring. This concept of windows of vulnerability has led to the DOHaD hypothesis discussed earlier in the chapter. A critical period is defined as a specific window of time during which an exposure can affect development resulting in a subsequent risk of disease. In this hypothesis, the exposure only has an effect during a finite time period; for example, studies of vaginal epithelial changes in women born to mothers who used diethylstilbestrol during pregnancy indicate that unless diethylstilbestrol exposure was begun before the 18th week of gestation, the chance of vaginal epithelial changes in the female children was very small (Glaze, 1984). Thus, an exposure can have an irreversible effect on disease risk in the offspring and future generations later in life. In addition to critical periods, sensitive periods also have to be considered. A sensitive period is similar to a critical period in that exposure during this period of possibly rapid biological change can affect disease risk, but the impact of the exposure on disease risk can be altered outside of this time period (Kuh and Ben-Shlomo, 2004). Thus, effects caused by exposure during a sensitive period may be reversed or exacerbated by later life exposures.
These concepts come from the framework of lifecourse epidemiology. The World Health Organization (2000) has embraced a lifecourse epidemiology approach using conceptual models to evaluate exposure–disease questions; four models are discussed here. These different models should be considered when designing studies, as well as the appropriate statistical methods that could involve effect modification, and carefully considering confounding and other forms of potential bias.
One proposed model is the critical period model, which occurs when an exposure has an immutable effect on later life disease risk through alterations to the structure or function of organs, tissues, or body systems. For example, prenatal exposure to trichloroethylene has been associated with cardiac defects in children, and such defects may lead to heart problems later in the child’s life (see Chapter 7 for a discussion of the developmental effects of trichloroethylene).
The second model is the critical period model with later effect modifiers. For this, an exposure has an effect on later life disease risk for the same reasons mentioned above, but later life exposures can alter the impact of the exposure. For example, a veteran’s exposure to certain deployment toxicants might alter the metabolism of his or her children, and the metabolic alternations might affect the future development of metabolic diseases such as obesity and type 2 diabetes. However, a healthy diet and physical activity in childhood and early adulthood could act as effect modifiers to decrease the risk of developing these conditions in later life. In essence, these two lifecourse models are equivalent to the respective critical and sensitive periods discussed earlier.
SOURCE: Adapted from Trounson et al., 2013, with permission.
A third model is that of cumulative risk, where exposures are independent and uncorrelated. In the model, the accumulation of not just one, but multiple exposures could impact later life disease risk. For example, if a service member is exposed to a chemical both on the base and at a single time during a patrol, the two exposures are not correlated, but the exposures could interact synergistically or antagonistically to affect the risk of subsequent disease.
Finally, the fourth model is where exposures accumulate but are also correlated. In this case, the correlated exposures result in clustering or chains and pathways of risk. An example of this with respect to generational health effects is the establishment of a “warrior caste” where children and grandchildren of military parents serve in the military as well (Schafer, 2017). In these military families, exposures may be correlated or clustered resulting in disease risk accumulation that might affect multiple generations.
Toxic Insult to Sperm or Eggs Prior to Conception
Animal models and epidemiological studies have demonstrated a significant role of sex in the response of parental germ cells to toxicants. Because the timing of germ cell production and maturation is vastly different for men and for women (refer to Figure 3-4 for details), environmental exposures will produce different effects depending on whether the mother or the father was exposed and the timing of the exposure.
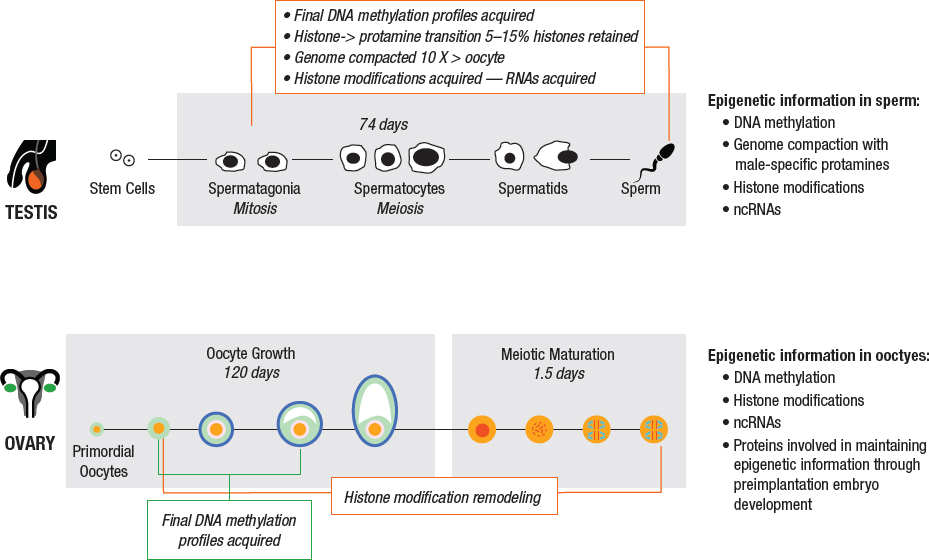
The primary reproductive organs are the testes in males and the ovaries in females, which are responsible for producing sperm and oocytes, respectively, and also hormones. In men, testosterone stimulation of the testis causes germ cells to develop from spermatogonia to spermatozoa, a process called spermatogenesis. Spermatogenesis takes about 74 days, with an additional 10 days or so for these cells to further mature as they pass through the epididymis (Amann et al., 2008; Heller et al., 1969) (Figure 3-4, top panel). The sperm cell epigenome undergoes a number of modifications during spermatogenesis, including changes in DNA methylation patterns and histone modifications, as part of an extensive chromatin remodeling process that occurs during male germ cell development.
At birth, females are thought to have about 1 million primordial germ cells (PGCs; oocytes). In response to hormonal stimulation beginning at puberty, individual oocytes within the ovary grow and differentiate within the follicles over a period of about 4 months prior to ovulation (Figure 3-4, bottom panel). Key epigenetic marks, such as DNA methylation and epigenetic marks on imprinted genes, are acquired during this growth phase, and exposure of the oocyte to toxicants during this time might adversely affect the viability of the oocyte, fetus, or child.
Male-Mediated Effects on Offspring Due to Fathers’ Exposures
For men, the exposures of most concern are those that permanently affect the male germ line stem cells since these cells continue to produce the precursors of sperm throughout a man’s life. Although small-scale studies (Robbins et al., 1997) found no genetic effects following a complete cycle of spermatogenesis subsequent to receiving chemotherapy for cancer, in larger studies, both genetic (e.g., Tempest et al., 2008) and epigenetic (e.g., Shnorhavorian et al., 2017) effects in sperm have been found to persist for years after treatment ended. The larger studies suggest that chemotherapy-induced abnormalities in male germ line stem cells have the potential to affect the children of these men.
After the stem cell phase, epigenetic changes can be incorporated into sperm in two main ways: at the time of the repackaging of DNA and through RNAs included with the sperm in semen. Key epigenetic modifications normally occur in male germ cells during the repackaging of chromatin. This genome compaction using protamines makes sperm chromatin unique. Protamine compaction reduces the genome size and, functionally, renders the sperm genome transcriptionally and translationally inert by condensing DNA to an almost crystalline form. This process is brought about as DNA exchanges its chromatin through a final wave of methylation, acetylation, and exchange of packaging proteins. At each stage of the compaction process, the genome and epigenome are susceptible to exposure-related modifications.
During epididymal transit, RNAs are added to the sperm (Jodar et al., 2017; Johnson et al., 2015). RNAs reach the epididymal fluid from multiple sites throughout the body (Cossetti et al., 2014) and then reside within the sperm (Johnson et al., 2015). The incorporation of RNAs with sperm from throughout the body provides a direct mechanism to epigenetically communicate exposures to gametes. For example, impacts on expression of sperm miRNAs have been identified in association with smoking (Marczylo et al., 2012), but it has not been established that those miRNAs are actually delivered by the sperm to the zygote. An understanding of the mechanisms by which epigenetic changes are incorporated into sperm is beginning to emerge, but much research is still needed to establish how these epigenetic changes are retained or recoded for the next and subsequent generations.
Female-Mediated Effects on Offspring Due to Mothers’ Exposures
Unlike men, women are born with their full complement of germ cells (i.e., oocytes). Thus, exposures of women that affect oocytes that enter the reproductive cycle could result in abnormal reproductive
outcomes and effects in the next generation. Studying the effects of toxic exposures on oocytes is challenging because of the small numbers of available cells and the difficulty in obtaining oocytes, even in animal models. There are several examples of intergenerational effects on the health of offspring following the exposure of females to high-fat or low-protein diets or to diabetic states (Clarke and Vieux, 2015). Several defects were identified in oocytes that could underlie the health effects of these maternal exposures in the offspring, including mitochondrial defects, lipid abnormalities, meiotic spindle abnormalities, and DNA methylation defects in imprinted genes. Other relevant studies of female-mediated effects via altered epigenetic marks or gene expression in the offspring have examined the effects of alcohol “binges” (VandeVoort et al., 2015), high dietary sugar intake (Chaffin et al., 2014), and maternal zinc deficiency (Tian and Diaz, 2012, 2013).
Toxic Insult to the Mother and Fetus
Exposure of a pregnant mother to a toxicant might adversely affect her developing embryo or fetus. Although the detection of a pregnancy in a deployed female service member is reason for her evacuation from a war zone, toxic exposures during the earliest period of gestation and before a pregnancy can pose a threat to the developing fetus.
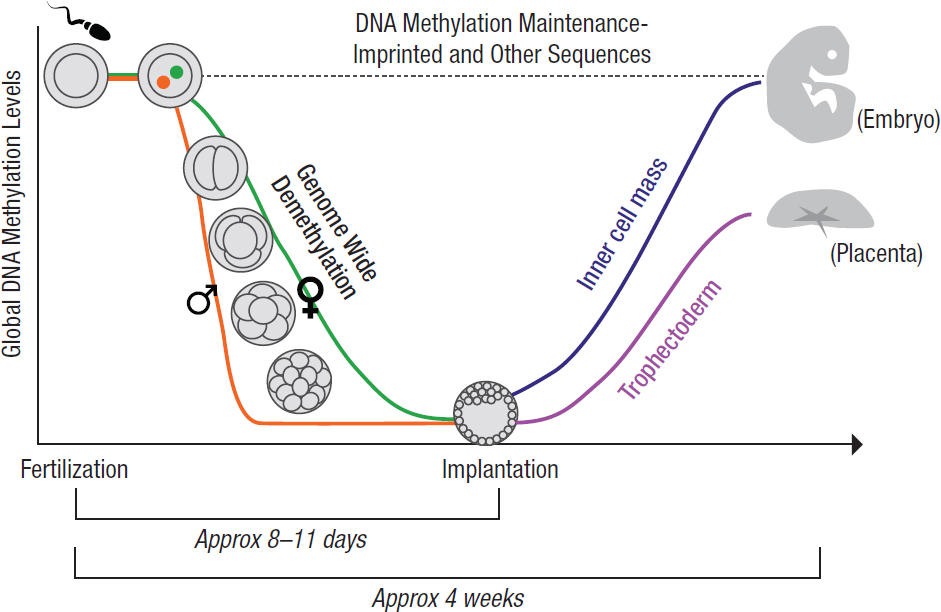
During the first week of gestation, the zygote develops into the multicellular blastocyst, which implants into the uterus and goes on to develop into an embryo and portions of the placenta (see Figure 3-5). As noted earlier, an important phase of epigenetic reprogramming occurs at this time of development (i.e., between fertilization and implantation at about 1 week of pregnancy) during which most, but not all, of the epigenetic marks in the blastocyst cells are erased. This time is the beginning of embryonic stem cell differentiation, the process that ultimately decides the fate of individual cells (Messerschmidt et al., 2014). At this early blastocyst stage, various chemical, genetic, and cellular interactions determine if these earliest stem cells differentiate into the inner cellular mass (ICM) or into the trophectoderm. The trophectoderm is the outer covering of cells which eventually forms the placental interface between mother and fetus. The trophectoderm is necessary for successful embryonic development, as it transfers nutrients and oxygen from the mother to the developing fetus. The ICM is the bundle of cells that will eventually form the fetus itself. Stem cells in the ICM differentiate into organ systems, although some stem cells will remain to participate in tissue renewal and repair. Epigenetic patterns in both DNA and histone methylation are then reacquired in a cell- and tissue-specific manner as the embryo and placenta continue to develop (Developmental Biology Interactive, n.d.).
The reprogramming of epigenetic information in the first week of life has been shown in studies in mice to be particularly susceptible to the external environment (McGowan and Roth, 2015; Sunde et al., 2016). To emphasize the complexity of the challenge of understanding intergenerational effects, it should be noted that for epigenetic marks to have an effect on future generations they need to be malleable enough to be affected by a subtle environmental effect but, at the same time, stable enough to survive the reprogramming that occurs during the formation of the early embryo and the germ line and during development.
The window of exposure during pregnancy relative to the developmental stage of the embryo or fetus is critically important. The period of organogenesis that occurs within the first 3 months of the pregnancy follows a tightly orchestrated series of biochemical events that give rise to the different organ systems. Different organs develop at different times as a function of cell–cell and cell–matrix interactions which define the survival of the developing embryo (Chen et al., 2009; Hill, 2018). Therefore, exposures during this critical window can affect different organs depending not only on the nature of the exposure itself but also on the timing of the exposure. It is important to note that the ways in which certain organ
systems, such as the nervous system and the lung, mature can extend the window of susceptibility (Rice and Barone, 2000; Shi et al., 2007).
Toxic Insult to Fetal Somatic Cells
In utero exposures of fetal somatic (non-germ-cell) tissues can cause birth defects or may affect the health of the child after birth or later in life, depending on the organ affected and the timing of the exposure. Post-fertilization growth and developmental trajectories occur at different rates for male and female offspring, and studies have shown that epigenetic changes reflect this by taking place differently in male and female offspring (Faulk and Dolinoy, 2011; Kanherkar et al., 2014). Furthermore, the placenta produces signals that are specific to the sex of the fetus, so exposures during pregnancy that affect the placenta can produce birth outcomes that are specific to fetal sex. Therefore, a maternal exposure can have very different impacts on pregnancy and fetal development for girls versus for boys.
Toxic Insult to the Germ Cells of the Fetus
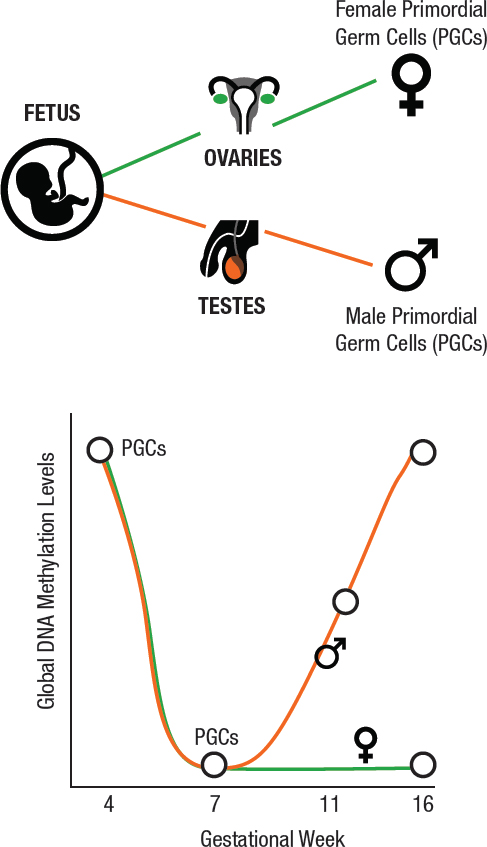
A new area of concern is the potential generational effects associated with the exposure of fetal germ cells to toxicants during the mother’s pregnancy. Both animal and human studies suggest that adverse health effects in the grandchildren can result from a woman’s exposures while pregnant (i.e., an intergenerational effect) (Lane et al., 2014). In contrast to the effects on fetal somatic cells, exposures of the developing fetal reproductive organs can affect the future reproductive function of a child or may be passed on to the next generation. Fetal germ cells undergo a dramatic epigenetic reprogramming of DNA methylation and histone modifications during the first 16 weeks of pregnancy (see Figure 3-6). After the PGCs in the ovary of a female fetus enter their final mitotic phase, they quickly arrest until puberty (see Figure 3-6). At puberty, the oocytes gain epigenetic marks as they mature (see Figure 3-4). In comparison, PGCs in the testes of a male fetus gain epigenetic marks before birth (see Figure 3-6) and undergo meiosis at puberty (see Figure 3-4). That is, epigenetic marks are gained in male germ cells in the fetal period, whereas epigenetic marks are gained in female germ cells as adults. These relationships are important because the genome/epigenome is most vulnerable to environmental exposures when it is undergoing mitosis.
Timing of Outcomes
The consequences of deployment on the health of a veteran’s descendants (generational effects) can be viewed within the framework of the DOHaD hypothesis that early life experiences can program the pathologies of old age (Xin et al., 2015). Epigenetic modifications are thought to be largely responsible for DOHaD “reprogramming,” whereby the developing epigenome is “reprogrammed” by environmental exposures in a way that persists for the life of the individual and can result in adverse health effects in adulthood. Prenatal insults, such as malnutrition and stress, can lead to an increase in common metabolic and cardiovascular diseases in adulthood (Barker, 2004). Similarly, in some cases adverse childhood outcomes, such as cancer and neurodevelopmental effects, are thought to be related to prenatal exposures to toxicants, such as, respectively, benzene and organophosphate pesticides. Other exposures, such as to endocrine-disrupting chemicals which mimic the biological actions of endogenous hormones, have been found to have later life effects on adult metabolic function in mice (La Merrill et al., 2014) and also behavioral and neurological outcomes in both humans and animals (Gore et al., 2015). Other adult diseases thought to originate from developmental exposures include impaired reproductive capability
in animals (Gore, 2008; Ho et al., 2017) and mental illness in humans and animals (Guintivano and Kaminsky, 2016).
Early life development and pubertal onset are also maturation periods which can trigger the manifestation of latent effects of developmental reprogramming and can be significant factors in disease presentation. For instance, neurodevelopmental disorders such as autism, attention deficit and hyperactivity disorder, and early-onset schizophrenia have a profound sex bias, with an increased incidence of presentation in males, supporting the concept that vulnerabilities and resiliencies to preconception or prenatal insults may be unique to the sex of the fetus (Kern et al., 2017; McCarthy, 2016; Waddell and McCarthy, 2012; Werling, 2016).
EPIDEMIOLOGIC GENERATIONAL AND EPIGENETIC STUDIES
The paucity of information about exposures associated with generational effects, combined with the current lack of understanding about male and female germ cell reprogramming, make it difficult to determine whether there is an increased risk of health effects among children or grandchildren. It is clear from the few epidemiological studies in which risk factors for generational health outcomes have been examined that much less is known about paternal exposures than about maternal exposures, as most of these studies have focused on maternal experiences during pregnancy. However, some epidemiological studies examining men exposed to the Swedish famines or to the Dutch “Hunger Winter” have attempted to identify periods of development where germ cells may be more susceptible to environmental stresses (Vansant et al., 2016).
In epidemiologic studies the interpretation of epigenetic patterns is complicated by disease status. Many studies use a case-control study design, where study participants are recruited based on disease status (i.e., cases versus controls), and samples are collected after disease development among the cases. In this scenario, any change in an epigenetic pattern among cases may have arisen after the disease onset in response to differences in treatments, diets, exercise, and metabolism. Therefore, it is difficult to distinguish between pre-disease epigenetic changes and those that are the result of such post-disease factors. Therefore, a better study design to assess the effects of an exposure on epigenetic changes and resulting disease would examine epigenetic patterns in tissue samples collected before disease onset. This design would ensure that epigenetic changes preceded the onset of the disease and were not a downstream consequence of the disease or treatment.
It is also important in epigenetic research to assess appropriate tissue samples. The most accessible human biospecimen for studying DNA methylation or histone biomarkers is blood, most commonly white blood cells, but these may not be the best target for studying the disease of interest. For example, the N-methyl-D-aspartate (NMDA) receptor is found in glutamatergic neurons in the brain and plays a key role in schizophrenia. The DNA methylation pattern of the NMDA receptor gene in white blood cells is different than the methylation pattern in the brain (Walton et al., 2016); therefore, white blood cells are poor surrogates for studying epigenetic changes affecting NMDA receptor function in schizophrenia. This highlights the need for appropriate tissue samples, but research may be hindered by the relative lack of access to human brain and other tissue.
Recent studies on the effects of parental exposures on genetic and epigenetic inheritance leading to generational health effects are intriguing and provide preliminary evidence of the potential for research in this area. Epigenetics provides a new mechanism that may help explain generational effects, and while the study of epigenetics has received much attention in the press, the field is still in its infancy. New and rapidly developing “omics”3 technologies promise to elucidate biomarkers that may be used to follow exposure-related effects across generations and help establish what, if any, effects may be occurring in children and grandchildren. Omic technologies will be crucial for unraveling the mechanisms underlying such generational health effects.
ANIMAL MODELS
There are several advantages to using animal models for the study of generational effects. Most animal models have much shorter generation times (days to months rather than decades); have many physiological and molecular properties in common with humans; and, in comparison with human studies,
___________________
3Omics refers to the study of genomic, proteomic, metabolomic, and associated information to study biological function and response.
are relatively inexpensive. In addition, they can facilitate the study of the molecular basis for how chemical exposures cause generational effects. This is particularly true for studies in which the cells with the affected epigenome, including germ cells, might be hard to access or study in humans. For example, some cell types and tissues such as neurons or lung tissue are rarely available from humans. Animals—mice, in particular—are also useful for inter- and transgenerational studies because they reach sexual maturity quickly and have a short gestational age and reproductive cycle, which permits researchers to observe three or more generations within a year. An important caveat is that it is important to pay attention to differences between species in such factors as metabolism, susceptible developmental stages, and lifespans when considering the applicability of findings in animal models to humans.
In animal studies, transgenerational outcomes following maternal exposures have been examined more extensively than paternal modes of transmission. One important limitation is that there are few data on maternal exposures that occur only prior to pregnancy, as might be experienced by female service members exposed to toxicants or stressors prior to conceiving. In contrast, paternal studies (including those focused on stress) suggest that exposures experienced prior to mating can affect future offspring via germ cell reprogramming. For instance, recent animal models of paternal stress have shown a wide window of germ cell vulnerability across spermatogenesis and maturational periods. Recent paternal studies have examined the effects of social defeat, chronic stress, dietary challenges, endocrine disrupters, and conditioned fear on offspring (Chan et al., 2018). In the case of social defeat stress, the offspring from defeat-subjected fathers showed changes in behavioral and physiological stress reactivity (Dietz et al., 2011). Similarly, in a mouse model of conditioned fear stress, males exposed to a shock paired with a specific odor passed on this conditioning effect to their first- and second-generation offspring (Dias and Ressler, 2014). Studies in male mice exposed to chronic stress prior to mating also showed a significant epigenetic reprogramming of their germ cells. This reprogramming resulted in long-term changes in offspring brain development and in their stress reactivity throughout their lives (Rodgers et al., 2015).
SUMMARY
This chapter provides an overview of the genetic and epigenetic mechanisms that have emerged as possible mediators of exposure-related impacts on the health of veterans’ children and grandchildren. For genetic mechanisms, a new environmentally mediated mutation could arise in a germ cell (sperm or oocyte), and this mutation could then be passed on to descendants. For epigenetic changes, the mechanisms are less clear. There is a growing body of research suggesting that health effects in descendants are not mediated by germ cell mutations and are therefore not directed by genetic mutations. These effects likely arise by environmentally mediated changes in the epigenome. The primary candidate mechanisms that have been implicated in the generational inheritance of health effects include changes in DNA methylation and histone modifications that regulate transcription and also changes in noncoding RNAs that regulate both transcription and translation. These mechanisms are a major focus of the field of epigenetics.
REFERENCES
Amann, R.P. 2008. The cycle of the seminiferous epithelium in humans: A need to revisit? Journal of Andrology 29(5):469–487.
Angermueller, C., S.J. Clark, H.J. Lee, I.C. Macaulay, M.J. Teng, T.X. Hu, F. Krueger, S. Smallwood, C.P. Ponting, T. Voet, G. Kelsey, O. Stegle, and W. Reik. 2016. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nature Methods 13(3):229–232.
Anonymous. 2015. 2. Relationship between different epigenomic marks: DNA accessibility and methylation, histone marks, and RNA. Nature 14310. doi:10.1038/nature14310.
Atlasi, Y., and H.G. Stunnenberg. 2017. The interplay of epigenetic marks during stem cell differentiation and development. Nature Reviews Genetics 18:643–658.
Barbara, M. A., and Y. Abdilla. 2017. An Introduction to Epigenetics. Neonatal Network 36(3):124–128.
Bariar, B., C.G. Vestal, and C. Richardson. 2013. Long-term effects of chromatin remodeling and DNA damage in stem cells induced by environmental and dietary agents. Journal of Environmental Pathology, Toxicology and Oncology : Official Organ of the International Society for Environmental Toxicology and Cancer 32(4):307–327.
Barker, D.J.P. 2004. Developmental origins of adult health and disease. Journal of Epidemiology and Community Health 58:114–115.
Biswas, S., and C.M. Rao. 2018. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. European Journal of Pharmacology 17:S0014-2999(18)30466-7. doi: 10.1016/j.ejphar.2018.08.021 [Epub ahead of print].
Butler, M.G. 2009. Genomic imprinting disorders in humans: A mini-review. Journal of Assisted Reproduction and Genetics 26(9–10):477–486.
Chaffin, C.L., K.E. Latham, N.R. Mtango, U. Midic, and C.A. VandeVoort. 2014. Dietary sugar in healthy female primates perturbs oocyte maturation and in vitro preimplantation embryo development. Endocrinology 155(7):2688–2695.
Chan, J.C., B.M. Nugent, and T.L. Bale. 2018. Parental advisory: Maternal and paternal stress can impact offspring neurodevelopment. Biological Psychiatry 83(10):886–894.
Chen, S., W. Fitzgerald, J. Zimmerberg, H.K. Kleinman, and L. Margolis. 2009. Cell–cell and cell–extracellular matrix interactions regulate embryonic stem cell differentiation. Stem Cells 25(3):553–561.
Clarke, H.J., and K.F. Vieux. 2015. Epigenetic inheritance through the female germ-line: The known, the unknown, and the possible. Seminars in Cell and Developmental Biology 43:106–116.
Cossetti, C., L. Lugini, L. Astrologo, I. Saggio, S. Fais, and C. Spadafora. 2014. Soma-to-germline transmission of RNA in mice xenografted with human tumour cells: Possible transport by exosomes. PLOS ONE 9(7):e101629.
Crider, K.S., T.P. Yang, R.J. Berry, and L.B. Bailey. 2012. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Advances in Nutrition 3(1):21–38.
Developmental Biology Interactive. n.d. Embryonic stem cell differentiation and trophectoderm development in primates. http://www.devbio.biology.gatech.edu/vertebrate-development/mammals-2/primates-embryonic-stem-cell-differentiation-andtrophectoderm-development/ (accessed July 11, 2018).
Dias, B.G., and K.J. Ressler. 2014. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nature Neuroscience 17(1):89–96.
Dietz, D.M., Q. Laplant, E.L. Watts, G.E. Hodes, S.J. Russo, J. Feng, R.S. Oosting, V. Vialou, and E.J. Nestler. 2011. Paternal transmission of stress-induced pathologies. Biological Psychiatry 70(5):408–414.
Eckersley-Maslin, M. A., C. Alda-Catalinas, and W. Reik. 2018. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nature Reviews Molecular Cell Biology 19(7):436–450.
Faulk, C., and D.C. Dolinoy. 2011. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics 6(7):791–797.
Fraser, R., and C.J. Lin. 2016. Epigenetic reprogramming of the zygote in mice and men: on your marks, get set, go! Reproduction 152(6):R211–R222.
Glaze, G.M. 1984. Diethylstilbestrol exposure in utero: Review of literature. Journal of the American Osteopathic Association 83(6):435–438.
Gore, A.C. 2008. Developmental programming and endocrine disruptor effects on reproductive neuroendocrine systems. Frontiers in Neuroendocrinology 29(3):358–374.
Gore, A.C., V.A. Chappell, S.E. Fenton, J.A. Flaws, A. Nadal, G.S. Prins, J. Toppari, and R.T. Zoeller. 2015. The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocrine Reviews 36(6):593–602.
Guintivano, J., and Z. A. Kaminsky. 2016. Role of epigenetic factors in the development of mental illness throughout life. Neuroscience Research 102:56–66.
Heller, C.G., G.V. Heller, and M.J. Rowley. 1969. Human spermatogenesis: An estimate of the duration of each cell association and of each cell type. In C. Gual and F.J.G. Ebling (eds.). Progress in Endocrinology (Proceedings of the Third International Congress of Endocrinology, Mexico). Amsterdam: Excerpta Medica. Pp. 1012–1018.
Hepp, M.I., V. Alarcon, A. Dutta, J.L. Workman, and J.L. Gutiérrez. 2014. Nucleosome remodeling by the SWI/SNF complex is enhanced by yeast high mobility group box (HMGB) proteins. Biochimica et Biophysica Acta 1839(9):764–772.
Hill, M.A. 2018. Embryology: Timeline human development. August 7. https://embryology.med.unsw.edu.au/embryology/index.php/Timeline_human_development (accessed August 8, 2018).
Ho, S.M., A. Cheong, M. Adgent, J. Veevers, A. Suen, N.C. Tam, Y.K. Leung, W.N. Jefferson, and C.J. Williams. 2017. Environmental factors, epigenetics, and developmental origin of reproductive disorders. Reproductive Toxicology 68:85–104.
Holoch, D., and D. Moazed. 2015. RNA-mediated epigenetic regulation of gene expression. Nature Reviews Genetics 16(2): 71–84.
Jaffe, A.E., and R.A. Irizarry. 2014. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biology 15(R31):1–9.
Jang, H.S., W.J. Shin, J.E. Lee, and J.T. Do. 2017. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8(6):148.
Jodar, M., A. Soler-Ventura, and R. Oliva. 2017. Semen proteomics and male infertility. Journal of Proteomics 162:125–134.
Johnson, G.D., P. Mackie, M. Jodar, S. Moskovtsev, and S.A. Krawetz. 2015. Chromatin and extracellular vesicle associated sperm RNAs. Nucleic Acids Research 43(14):6847–6859.
Juliano, C., J. Wang, and H. Lin. 2011. Uniting germline and stem cells: The function of Piwi proteins and the piRNA pathway in diverse organisms. Annual Review Genetics 45:447–469.
Kanherkar, R.R., N. Bhatia-Dey, E. Makarev, and A.B. Csoka. 2014. Cellular reprogramming for understanding and treating human disease. Frontiers in Cell and Developmental Biology 2:67.
Karemaker, I.D., and M. Vermeulen. 2018. Single-cell DNA methylation profiling: Technologies and biological applications. Trends in Biotechnology. https://doi.org/10.1016/j.tibtech.2018.04.002.
Kern, J.K., D.A. Geier, K.G. Homme, P.G. King, G. Bjørklund, S. Chirumbolo, and M.R. Geier. 2017. Developmental neurotoxicants and the vulnerable male brain: A systematic review of suspected neurotoxicants that disproportionally affect males. Acta Neurobiologiae Experimentalis Journal 77(4):269–296.
Kucej, M., B. Kucejova, R. Subramanian, X.J. Chen, and R.A. Butow. 2008. Mitochondrial nucleoids undergo remodeling in response to metabolic cues. Journal of Cell Science 121(11):1861–1868.
Kuh, D., and Y. Ben-Shlomo. 2004. A life course approach to chronic disease epidemiology, 2nd Ed. New York: Oxford University Press.
La Merrill, M., E. Karey, E. Moshier, C. Lindtner, M.R. La Frano, J.W. Newman, and C. Buettner. 2014. Perinatal exposure of mice to the pesticide DDT impairs energy expenditure and metabolism in adult female offspring. PLOS ONE 9(7):1–10.
Lane, M., R.L. Robker, and S.A. Robertson. 2014. Parenting from before conception. Science 345(6198):756–760.
Langley-Evans, S.C. 2014. Nutrition in early life and the programming of adult disease: A review. Journal of Human Nutrition and Dietetics 28(Suppl 1):1–14.
Ly, L., D. Chan, and J. M. Trasler. 2015. Developmental windows of susceptibility for epigenetic inheritance through the male germline. Seminars in Cell and Developmental Biology 43:96–105.
Marczylo, E.L., A.A. Amoako, J.C. Konje, T.W. Gant, and T.H. Marczylo. 2012. Smoking induces differential miRNA expression in human spermatozoa: A potential transgenerational epigenetic concern? Epigenetics 7(5):432–439.
McCarthy, M.M. 2016. Sex differences in the developing brain as a source of inherent risk. Dialogues in Clinical Neuroscience 18(4):361–372.
McEwen, K.R., and A.C. Ferguson-Smith. 2010. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics & Chromatin 3:2.
McGowan, P.O., and T.L. Roth. 2015. Epigenetic pathways through which experiences become linked with biology. Development and Psychopathology 27(2):637–648.
Messerschmidt, D.M., B.B Knowles, and D. Solter. 2014. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes & Development. 28(8):812–828.
Murr, R. 2010. Interplay between different epigenetic modifications and mechanisms. Advances in Genetics 70:101–141.
NHGRI (National Human Genome Research Institute). 2016. Epigenomics fact sheet. https://www.genome.gov/27532724/epigenomics-fact-sheet/ (accessed July 23, 2018).
NTP (National Toxicology Program). 2018. State of the Science Evaluation for Transgenerational Inheritance of Health Effects. https://ntp.niehs.nih.gov/pubhealth/hat/selected/trans/index.html (accessed August 8, 2018).
Ponomarenko, E.A., E.V. Poverennaya, E.V. Ilgisonis, M.A. Pyatnitskiy, A.T. Kopylov, V.G. Zgoda, A.V. Lisitsa, and Alexander I. Archakov. 2016. The size of the human proteome: The width and depth. International Journal of Analytical Chemistry 2016:1–6. doi: 10.1155/2016/7436849.
Ramos, K.S., and P. Bojang. 2018. Long interspersed nuclear element (LINE-1/L1). In Comprehensive Toxicology, 3rd ed. Edited by C.A. McQueen. Waltham, MA: Elsevier. Pp. 626–643.
Rice, D., and S. Barone. 2000. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environmental Health Perspectives 108(Suppl 3):511–533.
Robbins, W.A., M. L. Meistrich, D. Moore, F.B. Hagemeister, H.U. Weier, M.J. Cassel, G. Wilson, B. Eskenazi, and A.J. Wyrobek. 1997. Chemotherapy induces transient sex chromosomal and autosomal aneuploidy in human sperm. Nature Genetics 16:74–78.
Robertson, K.D. 2005. DNA methylation and human disease. Nature Reviews Genetics 6(8):597–610.
Rodgers, A.B., C.P. Morgan, N.A. Leu, and T.L. Bale. 2015. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proceedings of the National Academy of Sciences 112(44):13699–13704.
Schorn, A.J., M.J. Gutbrod, C. LeBlanc, and R. Martienssen. 2017. LTR-retrotransposon control by tRNA-derived small RNAs. Cell 170(1):61–71.
Sendler, E., G.D. Johnson, S. Mao, R.J. Goodrich, M.P. Diamond, R. Hauser, and S.A. Krawetz. 2013. Stability, delivery and functions of human sperm RNAs at fertilization. Nucleic Acids Research 41(7): 4104–4117.
Sharma, U., and O.J. Rando. 2017. Metabolic inputs into the epigenome. Cell Metabolism 25(3):544–558.
Shi, W., S. Bellusci, and D. Warburton. 2007. Lung development and adult lung diseases. Chest 132(2):651–656.
Shnorhavorian, M., S.M. Schwartz, B. Stansfeld, I. Sadler-Riggleman, D. Beck, and M.K. Skinner. 2017. Differential DNA methylation regions in adult human sperm following adolescent chemotherapy: Potential for epigenetic inheritance. PLOS ONE 12(2):1–18.
Skogen, J. C. and S. Overland. 2012. The fetal origins of adult disease: A narrative review of the epidemiological literature. Journal of the Royal Society of Medicine Short Report 3(8):59.
Spadafora, C. 2016. Soma to germline inheritance of extrachromosomal genetic information via a line-1 reverse transcriptasebased mechanism. Bioessays 38(8):726–733.
Sunde, A., D. Brison, J. Dumoulin, J. Harper, K. Lundin, M.C. Magli, E. Van den Abbeel, and A. Veiga. 2016. Time to take human embryo culture seriously. Human Reproduction 31(10):2174–2182.
Svoboda, P. 2017. Long and small noncoding RNAs during oocyte-to-embryo transition in mammals. Biochemical Society Transactions 45(5):1117–1124.
Tempest, H.G., E. Ko, P. Chan, B. Robaire, A. Rademaker, and R.H. Martin. 2008. Sperm aneuploidy frequencies analysed before and after chemotherapy in testicular cancer and Hodgkin’s lymphoma patients. Human Reproduction 23(2):251–258.
Teneng, I., D.E. Montoya-Durango, J.L. Quertermous, M.E. Lacy, and K.S. Ramos. 2011. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 6(3):355–367.
Tian, X., and F.J. Diaz. 2012. Zinc depletion causes multiple defects in ovarian function during the periovulatory period in mice. Endocrinology 153(2):873–886.
Tian, X., and F.J. Diaz. 2013. Acute dietary zinc deficiency before conception compromises oocyte epigenetic programming and disrupts embryonic development. Developmental Biology 376(1):51–61.
Trounson, A., R. Gosden, and U. Eichenlaub-Ritter (Eds.). 2013. Biology and pathology of the oocyte, role in fertility, medicine and nuclear reprograming, 2nd Ed. Cambridge University Press.
Uzumcu, M., A.M. Zama, and E. Oruc. 2012. Epigenetic mechanisms in the actions of endocrine-disrupting chemicals: Gonadal effects and role in female reproduction. Reproduction in Domestic Animals = Zuchthygiene 47(0 4):338–347.
VandeVoort, C.A., K.N. Grimsrud, U. Midic, N. Mtango, and K.E. Latham. 2015. Transgenerational effects of binge drinking in a primate model: Implications for human health. Fertility and Sterility 103(2):560–569.
Vansant, G., S. Wallace, and L. Godderis. 2016. Effect of maternal and paternal nutrition on DNA methylation in the offspring: A systematic review of human and animal studies. Advances in Obesity, Weight Management & Control 4(4):00093.
Virant-Klun, I., A. Ståhlberg, M. Kubista, and T. Skutella. 2016. MicroRNAs: From female fertility, germ cells, and stem cells to cancer in humans. Stem Cells International 2016:3984937.
Waddell, J.M., and M. McCarthy. 2012. Sexual differentiation of the brain and ADHD: What is a sex difference in prevalence telling us? Current Topics in Behavioral Neurosciences 9:341–360.
Walker, C.L. 2016. Minireview: Epigenomic plasticity and vulnerability to EDC exposures. Molecular Endocrinology 30(8):848–855.
Walton, E., J. Hass, J. Liu, J.L. Roffman, R. Bernardoni, V. Roessner, M. Kirsch, G. Schackert, V. Calhoun, and S. Ehrlich. 2016. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophrenia Bulletin 42(2):406–414.
Wei, Y., H. Schatten, and Q.Y. Sun. 2015. Environmental epigenetic inheritance through gametes and implications for human reproduction. Human Reproduction Update 21(2):194–208.
Werling, D.M. 2016. The role of sex-differential biology in risk for autism spectrum disorder. Biology of Sex Differences 7(58):1–18.
WHO (World Health Organization). 2000. The implications for training of embracing a life course approach to health. International Longevity Centre–UK. WHO/NMH/HPS/00.2.
Williams, S.C.P. 2016. News feature: Genetic mutations you want. Proceedings of the National Academy of Sciences 113(10):2554–2557.
Wu, H., R. Hauser, S.A. Krawetz, and J.R. Pilsner. 2015. Environmental susceptibility of the sperm epigenome during windows of male germ cell development. Current Environmental Health Reports 2(4):356–366.
Xin, F., M. Susiarjo, and M.S. Bartolomei. 2015. Multigenerational and transgenerational effects of endocrine disrupting chemicals: A role for altered epigenetic regulation? Seminars in Cell & Developmental Biology 43:66–75.
Xu, Q., and W. Xie. 2018. Epigenome in early mammalian development: inheritance, reprogramming and establishment. Trends in Cell Biology 28(3):237–253.
Xu, Z., J. Jiang, C. Xu, Y. Wang, L. Sun, X. Guo, and H. Liu. 2013. MicroRNA-181 regulates CARM1 and histone aginine methylation to promote differentiation of human embryonic stem cells. PLOS ONE 8(1):e53146.
Yeshurun, S., and A.J. Hannan. 2018. Transgenerational epigenetic influences of paternal environmental exposures on brain function and predisposition to psychiatric disorders. Molecular Psychiatry March 8. doi: 10.1038/s41380-018-0039-z.
Youn, H.K. 2017. Methylation and demethylation of DNA and histones in chromatin: The most complicated epigenetic marker. Experimental & Molecular Medicine 49:e321.
Yuan, S., A. Schuster, C. Tang, T. Yu, N. Ortogero, J. Bao, H. Zheng, and W.Yan. 2016. Sperm-borne miRNAs and endosiRNAs are important for fertilization and preimplantation embryonic development. Development (Cambridge, England) 143(4):635–647.
This page intentionally left blank.






















