
CHAPTER SIX
Carbon Mineralization of CO2
Carbon mineralization is an emerging approach to remove carbon dioxide (CO2) from the air and/or store it in the form of carbonate minerals such as calcite or magnesite. Mineralization occurs naturally during weathering of silicate materials (e.g., olivine, serpentine, and wollastonite) and rocks rich in Ca and Mg, particularly peridotite, which composes Earth’s upper mantle and basaltic lava formed by partial melting of the upper mantle. The glossary provides short definitions for these and other geological terms.
This chapter reviews the kinetics of carbon mineralization and the major pathways for capturing and storing CO2, discusses the amount and cost of CO2 that could potentially be stored, and lays out a research agenda. The carbon mineralization methods described in this chapter include those that store enriched CO2 in carbonate minerals as well as those that both remove CO2 from air and store it in carbonate minerals. The methods reviewed here emulate and accelerate spontaneous, natural processes, making use of the abundant chemical potential energy that is available where rocks from Earth’s deep interior are emplaced at and near the surface, where they are far from equilibrium with the atmosphere and hydrosphere. Because they utilize this naturally available chemical energy, these methods may offer a low cost means to mitigate greenhouse gas emissions. And because the CO2 is locked into solid carbonate minerals, storage has a strong potential to be permanent and nontoxic.
INTRODUCTION
Storing CO2 via reaction with common silicate rocks and minerals has been considered for almost 30 years (Lackner et al., 1995; Seifritz, 1990). More recently, the possibility of carbon mineralization using alkaline industrial waste products such as steel slag has been added to the palette of options (e.g., Gadikota et al., 2015; Huijgen et al., 2007; Pan et al., 2012; Sanna et al., 2014). Below the Committee summarizes carbon mineralization reactions and methods for capturing and storing CO2 in carbon minerals.
Carbon Mineralization
In general, the extent of mineral carbonation depends on (1) available CO2 dissolved in solution, (2) available alkalinity in solution, and (3) chemical conditions that foster
available alkalinity via mineral dissolution and carbonate precipitation. Although low pH accelerates mineral dissolution (e.g., Pokrovsky et al., 2004) and higher pH favors carbonate precipitation (e.g., Harrison et al., 2013), experiments have demonstrated that silicate dissolution and carbonate precipitation can be combined in a single step (Chizmeshya et al., 2007; Gadikota et al., 2015; O’Connor et al., 2005).
A key source of alkalinity for Mt to Gt storage of CO2 via carbon mineralization is Mg-rich, Ca-bearing, highly reactive rocks from Earth’s deep interior, including mantle peridotite, basaltic lava, and ultramafic intrusions. Such rocks are rich in olivine and pyroxene minerals. The mineral wollastonite (CaSiO3) reacts more rapidly than olivine and pyroxene, but it is not found in large quantities and has a limited geographical distribution.
In carbon mineralization, CO2 reacts with minerals rich in Ca and Mg to form carbonates, such as calcite (CaCO3), magnesite (MgCO3), and dolomite (CaMg(CO3)2), and often quartz (SiO2). Some idealized reactions are as follows:
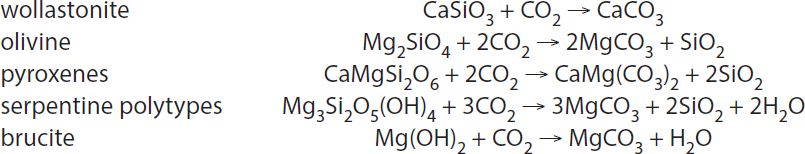
Wollastonite, olivine, and brucite react relatively rapidly, as do fibrous serpentines with high surface area to volume ratios, such as asbestiform chrysotile. Ca and Mg in alkaline industrial wastes react at about the same rate as wollastonite. A more extensive discussion of kinetic data for carbon mineralization appears below (see section “Carbon Mineralization Kinetics”).
These reactions are spontaneous and exothermic; carbonate minerals are the “ground state” for CO2 in near-surface rock systems such as Mg-Ca-C-O-H and Mg-Ca-Si-C-O-H (Figure 6.1).
Methods for Capturing and Storing CO2
Carbon mineralization methods are either aimed at storing CO2 in carbonate minerals (referred to as solid storage) or both removing CO2 from air and storing it in carbonate minerals (referred to as combined mineral capture and storage). Solid storage can be accomplished in three ways:
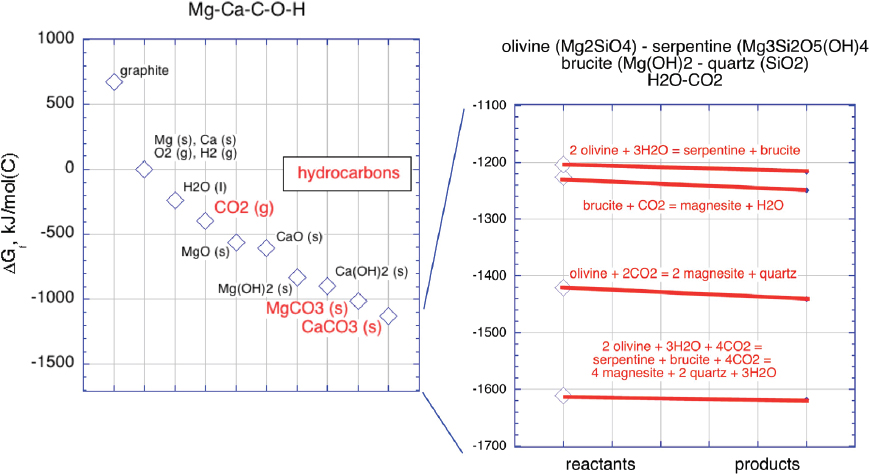
NOTE: The rectangle labeled “hydrocarbons” encompasses the range of Gibbs free energies of formation for common hydrocarbon species (e.g., methane, ethane, and butane) per mole of carbon.
- ex situ carbon mineralization—solid reactants are transported to a site of CO2 capture, then reacted with fluid or gas rich in CO2,
- surficial carbon mineralization—CO2-bearing fluid or gas is reacted with mine tailings, alkaline industrial wastes, or sedimentary formations rich in reactive rock fragments, all with a high proportion of reactive surface area, and
- in situ carbon mineralization—CO2-bearing fluids are circulated through suitable rock formations at depth.
Combined mineral capture and storage may be accomplished through surficial or in situ carbon mineralization processes discussed above, using natural surface waters rather than fluids enriched in CO2. These methods most closely emulate natural processes of CO2 uptake. In contrast, direct air capture systems require additional technologies and energy input for CO2 storage.
Laboratory research on ex situ carbon mineralization has been relatively extensive, but studies to date have used a wide range of reagants and conditions, making it difficult
to compare results. Research on surficial carbonation of mine tailings and other fine-grained solid reactants is at an intermediate scale of readiness, ripe for larger scale field experiments. In situ carbon mineralization CO2 storage in basaltic rocks has been studied in two medium-scale experiments and warrants further exploration at the scale of Mt/y CO2. Natural examples of extensive in situ mineral capture and storage in ultramafic rocks, rapid reaction rates in laboratory studies, and the potential for permanent storage of hundreds of trillions of tons of CO2 warrant continued basic research into this little-studied, high-risk, high-reward opportunity.
Mineral capture and storage using mine tailings can be implemented at a low cost but has a limited capacity. Strikingly, mining and grinding rock for the purpose of mineral capture and storage potentially has about the same cost as direct air capture systems, within uncertainties for both. In situ mineral capture and storage potentially has a lower cost than surficial methods, and a giant storage capacity, but involves uncertain feedbacks between permeability, reactive surface area, and reaction rate, and so remains a topic of basic, theoretical research.
Pumping is a substantial cost for mineral capture involving natural, CaOH-rich alkaline water in peridotite aquifers or water in CO2 exchange equilibrium with air, due to low concentrations of dissolved carbon species. In such cases, mineralization requires co-location with geothermal power plants and/or significant thermal gradients to drive convection. Surficial mineral capture would generate many km3 of rock products per Gt of CO2 per year, posing potential transport and storage problems. Furthermore, the size, injectivity, permeability, geomechanics, and microstructure of key subsurface reservoirs for in situ mineral capture and storage remain almost entirely unexplored.
CARBON MINERALIZATION KINETICS
Surprisingly, despite a plethora of recent reviews of various aspects of ex situ mineral carbonation (Gadikota et al., 2015; Pan et al., 2012; Power et al., 2013a, 2013c; Sanna et al., 2014), there have been few comparisons of carbon mineralization rates, as a function of temperature, partial pressure of CO2 [P(CO2)], and other key variables. One reason for this is the ongoing pursuit of parallel lines of inquiry, involving (1) a single reaction step, combining CO2-rich fluid with solid reactants, (2) a two-stage process, involving dissolution of solid reactants—typically at low pH—followed by introduction of CO2 and precipitation of carbonate minerals—typically at high pH, and (3) pretreatment of solid reactants, for example by rapid heating. Nevertheless, in the two-stage process, it is generally considered that mineral dissolution is the rate-limiting step, so it is possible to compare mineral dissolution rates (as an upper bound on the combined
rate of two-stage processes) and full carbon mineralization rates, for example using grain size data from experiments to calculate geometric surface area (median or mean grain diameter squared x pi), and presenting rates in mass fraction per second for 50 to 70 micron grains (Kelemen et al., 2011; Matter and Kelemen, 2009).
Here, we update this comparison, incorporating new data and using units more familiar to many geoscientists (mol/(m2s)). Figure 6.2 provides some of this information. Where necessary and possible, we use geometric surface area to calculate rates because more detailed surface area measurements are not available for many of the data sets we wish to compare. Huge variability in measured rates—with consistency among different experiments on the same set of fluid and mineral reactants—suggests that the uncertainty in surface area is of relatively minor importance in this type of comparison. Because of time constraints, there is no comparable review in this report of rates as a function of temperature, P(CO2) and other factors for alkaline, industrial waste materials.
After experiments indicated that wollastonite (CaSiO3) was among the fastest reacting silicates (e.g., O’Connor et al., 2005), some groups have focused on carbonation of this mineral. Indeed, it may be a reasonable analogue for carbonation of calcium silicates in industrial wastes, such as steel slag for example. However, the U.S. Geological Survey (USGS) website for wollastonite (U.S. Geological Survey, 2018d) indicates that less than 1M tons of natural wollastonite per year were produced in 2016 and 2017, from global reserves estimated to be approximately 100M tons. Economic wollastonite deposits are restricted almost entirely to narrow “skarns,” formed by reaction between granitic magmas and limestone or marble host rocks. The USGS reserve estimate for wollastonite includes data on skarns that are profitable to mine at $200-350/t of wollastonite (depending on purity and crystal shape) plus income due to sale of byproducts. When fully carbonated, a ton of wollastonite can store 0.33 tons of CO2, so these costs correspond to $600-1,000/tCO2. Even if these are the only costs of CO2 storage using wollastonite, these costs are more than 10 times higher than the cost of storing CO2 in subsurface pore space ($10-20/t). This is not surprising. Typical costs for hard rock mining in large deposits are approximately $10/t of rock. For smaller mines, the cost is higher. If a deposit contains less than 10 percent wollastonite, the extraction cost will be more than $100/t. Thus, looking beyond skarns, marbles with minor amounts of wollastonite formed by prograde metamorphism are not a practical source of wollastonite for carbon mineralization.
Because olivine, (Mg,Fe)2SiO4, reacts with CO2-bearing fluids almost as rapidly as wollastonite, it is the most important mineral constituent of the upper mantle. Olivine remains a major constituent of partially hydrated (serpentinized) peridotite massifs
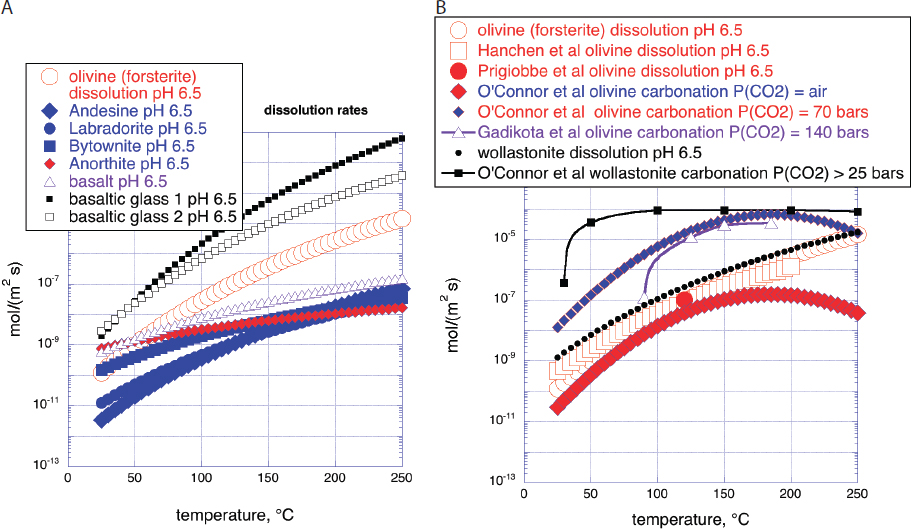
NOTES: All panels have the same horizontal and vertical axis ranges, a linear temperature range of 0 to 250°C, and a logarithmic range of rates from 10-13 to 10-2 mol/(m2s). Red open circles in all four panels are dissolution rates of Mg-endmember olivine (forsterite) at pH 6.5 based on fit to compiled data by Palandri and Kharaka, 2004. Panel A: Rates of mineral carbonation and dissolution: “Hanchen et al olivine dissolution pH 6.5” and “Prigiobbe et al olivine dissolution pH 6.5”: steady state dissolution of San Carlos olivine (91% forsterite) in CO2-bearing aqueous fluid at pH 6.5 (Hanchen et al., 2006, 2008; Prigiobbe et al., 2009). “O’Connor et al olivine carbonation”: Fit to data of O’Connor et al., 2005 by Kelemen and Matter, 2008 for carbonation of olivine (91% forsterite) at different partial pressures of CO2, in 1M NaCl, 0.64M NaHCO3 aqueous fluid in 1 hour at pH ~ 6.5. “Gadikota et al olivine carbonation”: Full carbonation of olivine at different partial pressures of CO2, in 0.64M NaHCO3 aqueous fluid, with and without NaCl over 3 hours at pH ~ 6.5 (Gadikota et al., 2014). “Wollastonite dissolution pH 6.5”, calculated from Palandri and Kharaka, 2004. “O’Connor et al wollastonite carbonation”: Full carbonation of wollastonite at different partial pressures of CO2, in 1M NaCl, 0.64M NaHCO3 aqueous fluid (O’Connor et al., 2005). Panel B: Rates of mineral carbonation at elevated P(CO2) in aqueous fluid with 1M NaCl and 0.64M NaHCO3 for 3 hours (Gadikota et al., 2018), allowing direct comparison of carbon mineralization rates for olivine (Figure 6.2A), plagioclase feldspar (labradorite, with a molar Ca/(Ca+Na) ratio of 0.54), a rock composed of plagioclase feldspar, olivine, and iron oxides (anorthosite, with a molar Ca/(Ca+Na) ratio in plagioclase of 0.98 and a molar Mg/(Mg+Fe) ratio in olivine of 0.66) and a basaltic lava (molar Ca/(Ca+Na) 0.60, molar Mg/(Mg+Fe) 0.48). NOTE: Carbon mineralization rates for basalt, olivine and shale in “damp”, H2O-bearing, supercritical CO2 are approximately the same as carbonation rates for the same materials in aqueous fluid with the same P(CO2) (Loring et al., 2011, 2013; Schaef et al., 2011, 2013).
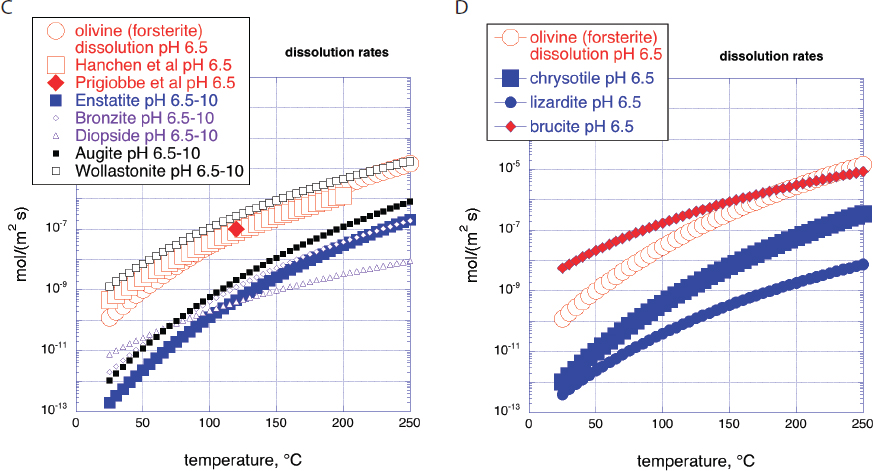
exposed at Earth’s surface, and much research has focused on this mineral. Olivine in the mantle and in ultramafic intrusions tends to contain 88 to 92 mole percent of the Mg-endmember forsterite (Mg2SiO4), so that simplified carbonation reactions such as those below (see “Carbon Mineralization Kinetics”) often omit discussion of the Fe-endmember fayalite.
Engineered “pH swing” methods (e.g., Park and Fan, 2004) were thought to be essential for olivine carbonation because of the perceptions that (a) aqueous fluids with high P(CO2) have intrinsically low pH, too low for extensive carbonate precipitation, and (b) olivine dissolves slowly at neutral to high pH. Breakthrough experiments by O’Connor et al., 2005 showed that using NaHCO3 to buffer pH (to ~ 6.5 at experimental conditions) yielded rapid olivine carbonation. As concentrations of NaHCO3 before and after experiments were approximately the same in these and subsequent, similar experiments (Chizmeshya et al., 2007; Gadikota et al., 2014), it is apparent that NaHCO3
is not a net source of carbon to form carbonate minerals, but instead acts as a kind of catalyst. Dissolved NaHCO3 buffers pH in a range where both olivine dissolution and carbonate precipitation are relatively rapid. In turn, it is thought that carbonate precipitation provides a continuous sink for dissolved Mg and maintains a high olivine dissolution rate in experiments (Gadikota et al., 2014), even at low water/rock ratios. In natural systems, and proposed, engineered, in situ carbon mineralization involving subsurface reaction of CO2-bearing fluids with olivine-rich rocks, use of NaHCO3 would probably not be required to achieve the same effect, because reaction path modeling has repeatedly shown that low pH, CO2-rich fluids reacting with olivine-rich (ultramafic) rocks are rapidly buffered to high pH (e.g., Bruni et al., 2002; Paukert et al., 2012) in a spontaneous pH swing.
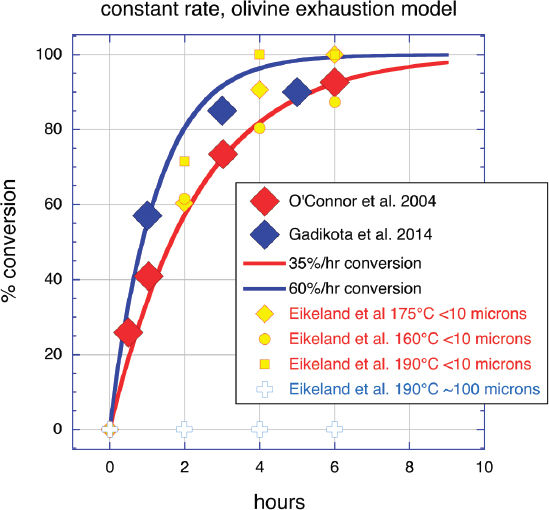
Another important characteristic of olivine carbonation experiments has been the lack of observed passivation. Although an SiO2-rich layer on the surface of partially dissolved olivine has been observed (Bearat et al., 2006; Chizmeshya et al., 2007), and might cause slowing of reaction rates over time, this is not borne out in the results of time-series experiments. Figure 6.3 shows our new plot of time series experiments by O’Connor et al., 2005, Gadikota et al., 2014, and Eikeland et al., 2015. Although the data sets show slightly different rates, each can be fit with a constant rate of olivine carbonation, continuing until olivine is greater than 90 percent consumed, illustrating that passivation does not significantly reduce olivine carbonation rates, at least for grain sizes less than approximately 100 microns. We tentatively ascribe this lack of passivation during olivine carbonation to the process of reaction-driven cracking, and/or to various related processes such as the formation of etch pits on reacting olivine surfaces as discussed later in this chapter. Another factor may be buffering of pH—by addition of NaHCO3 in experiments, by natural water/rock reactions in peridotite—which has been observed to suppress the precipitation of iron oxides, which may form passivating layers in lower pH conditions (Gadikota et al., 2014).
This lack of passivation during olivine carbonation does not appear to be common for other mineral and rock reactants. An ongoing problem in comparing carbon mineralization rates is the lack of kinetic experiments with different solid reactants with the same fluid composition. Gadikota et al., 2018 performed a series of experiments to begin to address this problem (Figure 6.2B). These data provide quantitative support for the view that, in NaHCO3-rich aqueous solutions, olivine carbonation is faster than carbonation of plagioclase feldspar, mafic intrusions (anorthosite, 55 wt% plagioclase feldspar, 35% olivine, 5% pyroxene, 3% Fe-oxides), and basaltic lavas in general (~ 0 to 20% olivine, 0 to 40% pyroxene, ~ 60% plagioclase feldspar). Importantly, carbonation rates for all reactants except olivine decreased with time, presumably due to passivation. This may indicate that passivation will be a problem in engineered systems—ex
situ or in situ—requiring extensive carbonation of Mg and Ca in basaltic reactants with abundant plagioclase feldspar and pyroxene minerals, and relatively little olivine.
The data in Figure 6.2C illustrate the basis for the widely held view that olivine-rich ultramafic rocks such as mantle peridotite dissolve faster than basalt and the plutonic equivalent of basalt, gabbro. Basalt and gabbro are rich in alumino-silicate minerals such as plagioclase feldspar with molar Ca/(Ca+Na) less than 80 percent (most commonly, labradorite, with molar Ca/(Ca+Na) from 50 to 70%), low solubilities in aqueous fluids, and slow intracrystalline diffusion. However, the data also illustrate that amorphous basaltic glass is an important exception to the rule that peridotite reacts faster than basaltic lava. Amorphous glass forms when portions of basaltic lava flows cool faster than the time required for nucleation and growth of crystals. Basaltic lava formations with abundant glassy horizons could provide exceptionally good reactants for carbon mineralization. We are not aware of regional compilations of glass abundance and distribution
in basaltic lavas, but because glass forms where basalt cools very quickly, it is likely that glass is most abundant in lavas that erupted in submarine settings (particularly hyaloclastite breccias composed of quenched fragments of flow surfaces), and in cinder cones and other formations formed by explosive eruption of small lava droplets into air.
In addition to igneous rocks such as peridotite and basalt, and their constituent minerals, some alteration products of peridotite have received considerable attention as possible reactants for carbon mineralization. In the context of the broad range of rates in Figure 6.3, dissolution rate data are sufficiently similar to results of more detailed study with a variety of different fluid reactants and experimental conditions that we can succinctly summarize relative rates using the dissolution data alone, in Figure 6.2D. Serpentine minerals dissolve much more slowly than olivine, consistent with slow carbonation of another serpentine polymorph, antigorite, under conditions favoring rapid olivine carbonation. By contrast, at low temperature brucite dissolves much faster than olivine, consistent with many studies indicating that brucite (and some fibrous, asbestiform chyrsotile) undergoes rapid carbonation during exposure of ultramafic mine tailings to air.
Figure 6.4 compares rates of carbon mineralization at elevated P(CO2) but at ambient surface temperatures, which are useful for evaluating proposed methods for direct capture of CO2 from air, and for proposed storage methods such as “sparging” CO2-rich gas through mine tailings and industrial waste heaps (Assima et al., 2013a, 2014c; Harrison et al., 2013).
Finally, recognition that mine tailings rich in serpentine are abundant, and that carbonation of asbestiform chrysotile provides the benefit of mitigating an environmental hazard, has led to remarkable number of studies of carbonation of amorphous (dehydroxylated) products of rapid heat-treatment of serpentine, as a proposed feedstock for ex situ carbon mineralization using captured CO2 from a hydrocarbon-fueled power plant (Balucan et al., 2011; Balucan and Dlugogorski, 2013; Dlugogorski and Balucan, 2014; Fedoročková et al., 2012; Ghoorah et al., 2014; Larachi et al., 2010, 2012; Li et al., 2009; Maroto-Valer et al., 2005; Mckelvy et al., 2004; O’Connor et al., 2005; Sanna et al., 2014) or using flue gas without CO2 capture (Hariharan et al., 2014, 2016; Hariharan and Mazzotti, 2017; Pasquier et al., 2014; Werner et al., 2013, 2014). In the studies listed in the previous sentence, heat treatment is considered a relatively minor step, in terms of cost and CO2 production, if it is accomplished using waste heat from a power plant, which is also the site of carbon capture and ex situ mineral carbonation. Waste heat may not be sufficient if a temperature as high as 600°C is required (e.g., Liu and Gadikota, 2018). If so, the cost of heat treatment could become considerable. Use of heat treatment to increase the rate of serpentine carbonation is less relevant to methods for storage of CO2 captured from air. Moreover, costs of CO2 storage via ex
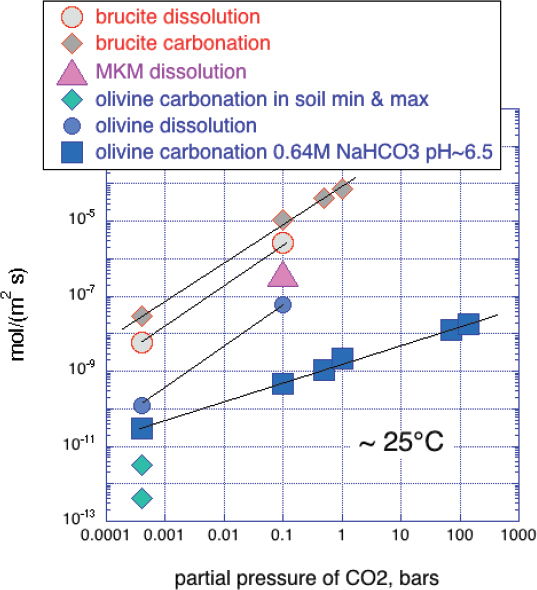
NOTES: Brucite, MKM (Mt. Keith Mine, brucite-rich, serpentine mine tailings) and olivine dissolution rates at 0.1 bar P(CO2) (Greg Dipple, personal communication, 2018). Steady state dissolution rates for brucite and olivine at 0.0004 bars P(CO2) from Palandri and Kharaka, 2004. Olivine carbonation in soil: Renforth et al., 2015. Brucite carbonation: Harrison et al., 2013. Olivine carbonation 0.64M NaHCO3 pH ~ 6.5 calculated with Kelemen and Matter, 2008 fit to data of O’Connor et al., 2005 for reaction extent in 1 hour.
situ carbon mineralization using heat-treated serpentine and CO2-rich fluids remain higher than injection of CO2 into subsurface pore space (Table 6.1).
Conclusions
Geologically abundant olivine, in mantle peridotites and some ultramafic intrusions and lava flows, together with the mineral brucite produced by hydrous alteration of olivine, provide the best reactants for in situ carbon mineralization. Wollastonite reacts faster than olivine, but it is not geologically abundant (for more information, see the section on carbon mineralization kinetics earlier in this chapter). Minerals such as typical plagioclase feldspar with molar Ca/(Ca+Na) less than 80 percent—major constituents of basaltic lithologies—react more slowly than olivine and undergo passivation at a relatively early extent of conversion. However, basaltic formations are more
TABLE 6.1 Solid Storage of CO2 via Carbon Mineralization at Elevated P(CO2) and/or Temperature
| Method | Weight fraction CO2/hr | t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction in 1 y at rate in table | Gt/y CO2 | $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Ex situ carbon mineralization | ||||||||||
| Peridotite and olivine | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2), with or without pH swing process and heat treatment | 0.1-0.3 | Some fraction of 0.2 Gt ultramafic tailings per year | 0.1-0.5 | 0.02-0.1 | 50-100 | Recent comprehensive reviews by Bodenan et al., 2014; Chizmeshya et al., 2007; Gadikota and Park, 2015; Gadikota et al., 2014; Gerdemann et al., 2007; Giannoulakis et al., 2014; Khoo et al., 2011; O’Connor et al., 2005; Pan et al., 2012; Power et al., 2013a, 2013c; Sanna et al., 2013, 2014 | 2, 3 | ||
| Fits to data by Kelemen and Matter, 2008; Kelemen et al., 2011; Matter and Kelemen, 2009 | ||||||||||
| Brucite | Sparging of elevated P(CO2), ambient temperature gas through brucite powder + H2O | 0.03-0.3 | Some fraction of 0.2 | 0.03-0.1 | 0.01-0.02 | ~ 10–20? | Harrison et al., 2013 | 2 | ||
| Brucite | Manufacture of brucite from mine tailings via reaction at elevated temperature with NaOH-H2O solution, plus sparging with CO2-rich gas | 0.3? | Some fraction of 0.2 | 0.03-0.1 | 0.01-0.02 | 200-600 | Madeddu et al., 2015 | 2 | ||
| Serpentine | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2), with or without pH swing process and heat treatment | Much slower than olivine | Some fraction of 0.2 | 0.03-0.1 | 0.01-0.02 | 200-600 | Bodenan et al., 2014; Gerdemann et al., 2007; Huijgen et al., 2007; Khoo et al., 2011; O’Connor et al., 2005; Sanna et al., 2013, 2014 | 2, 4 |
| Method | Weight fraction CO2/hr | t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction in 1 y at rate in table | Gt/y CO2 | $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Serpentine | Elevated temperature and pressure reaction with flue gas or water saturated in flue gas, and/or with prior heat treatment | Still slower than olivine, passivation problems | Some fraction of 0.2 | 0.03-0.1 | 0.01-0.02 | 200-600 | Hariharan et al., 2013; Hariharan and Mazzotti, 2017; Mazzotti personal communication, 2017; Sanna et al., 2013, 2014; Werner et al., 2011, 2013, 2014 | 2, 4 | ||
| Wollastonite | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) | 0.2-0.6 | 0.00055 | 0.1 | 0.3 | 0.0002 | 80-160 | Gerdemann et al., 2007; Giannoulakis et al., 2014; Huijgen et al., 2007; O’Connor et al., 2005; U.S. Geological Survey, 2016 | 1 | |
| Steel and blast furnace slag | Elevated temperature and pressure reaction with purified CO2 or water saturated at | Faster than wollastonite | 0.17-0.50 | 0.01-0.20 | 0.002-0.1 | 75-100 | Gomes et al., 2016; Huijgen et al., 2007; Renforth et al., 2011; Sanna et al., 2013, 2014 | 5 |
| high P(CO2), with or without pH swing process and heat treatment | 5 | |||||||||
| Cement waste | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.42-2.1 | 0.016-0.25 | 0.001-0.3 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Gomes et al., 2016; Renforth et al., 2011; Sanna et al., 2013, 2014 | 5 | ||
| Construction and demolition waste | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 1.4-5.8 | 0.08-0.11 | 0.1-0.6 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Renforth et al., 2011 | 5 |
| Method | Weight fraction CO2/hr | t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction in 1 y at rate in table | Gt/y CO2 | $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Other municipal solid waste | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 1.3 | 0.016-0.25 | 0.004-0.16 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Hoornweg and Bhada-Tata, 2012; Sanna et al., 2013, 2014 | 5 | ||
| Coal ash in general | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.4-0.6 | 0.022-0.29 | 0.0002-0.09 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Gomes et al., 2016; Sanna et al., 2013, 2014 | 5 |
| Lignite ash | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.03-0.06 | 0.03-0.10 | 0.0009-0.006 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Renforth et al., 2011; Sanna et al., 2013, 2014 | 5 | ||
| Anthracite ash | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.02-0.05 | 0.01-0.10 | 0.0002-0.005 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Renforth et al., 2011 | 5 |
| Method | Weight fraction CO2/hr | t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction in 1 y at rate in table | Gt/y CO2 | $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Bituminous ash | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.15-0.28 | 0.003-0.020 | 0.0004-0.006 | Probably steel slag cost divided by ratio of weight fraction CO2 for steel slag to this commodity | Renforth et al., 2011 | 5 | ||
| Red mud, residue of Al2O3 extraction from bauxite | Elevated temperature and pressure reaction with purified CO2 or water saturated at high P(CO2) ± various pretreatment steps | Faster than wollastonite | 0.12 | 0.04-0.07 | 0.00001-0.006 | ~150 | Gomes et al., 2016; International Aluminium Institute, 2018; Sanna et al., 2014 | 5 |
| Other alkaline wastes, mostly produced at rates less than 0.01 Gt per year | See comprehensive list in Sanna et al., 2014 | |||||||||
| Ultramafic mine tailings at ambient temperature | ||||||||||
| Ultramafic mine tailings | Sparging CO2-rich gas through mine tailings | 0.03-0.30 | 1E8-1E9 | 0.200 | 0.03-0.10 | 0.006-0.02 | 10-30 | Assima et al., 2013a; Harrison et al., 2013; U.S. Geological Survey, 2018a, b, c | ||
| In situ mineral carbonation | ||||||||||
| Peridotite | Drill to depth where temp > 90°C, inject CO2-rich fluid at P(CO2) > 60 bars | 0.1-0.3 | 3E8-1E9 | 1-100 | 1E5-1E8 | 0.1-0.6 | 0.1-60 | 10-30 | Kelemen and Matter, 2008; Kelemen et al., 2011, 2016; Matter and Kelemen, 2009 | 6 |
| Method | Weight fraction CO2/hr | t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction in 1 y at rate in table | Gt/y CO2 | $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| Basaltic lava, specific CarbFix and Wallula sites | Drill to depth where temp > 25°C, inject CO2-rich fluid at P(CO2) > 60 bars | >0.0003 at CarbFix, ~ 0.001 at Wallula | >10,000 | 0.00003 | 0.01-0.25 | 3E7-8E6 | 10-30 | Aradóttir personal communication 2017; Gislason and Oelkers, 2014; Matter et al., 2016; Sigfusson et al., 2015; Xiong et al., 2018 | 6 | |
| Basaltic lava, global onland flood basalts | Drill to depth where temp > 25°C, inject CO2-rich fluid at P(CO2) > 60 bars | 1-100 | 1E5-1E6 | 0.01-0.25 | 0.01-25 | 10-30 | McGrail et al., 2017a for Columbia River Basalt and Deccan Trapps, plus our extrapolation to include Siberian Trapps, Karoo Basalts, Afar Volcanic Province, and Parana Basalts | 7 |
| Basaltic lava, global, submarine, suitable sites with sedimentary cap rocks | Drill to depth where temp > 25°C, inject CO2-rich fluid at P(CO2) > 60 bars | 1-100 | 8E4-4E5 | 0.01-0.25 | 0.01-25 | 200-400 | Goldberg and Slagle, 2009; Goldberg personal communication, 2017 | 7 |
NOTES:
1. USGS estimates for production and global resource.
2. Production and CO2 capacities assuming that peridotite, olivine, brucite, and/or serpentine are derived from serpentinized, ultramafic mine tailings. If ultramafic rock were mined and milled for the purpose of CO2 storage, then the storage reservoir becomes 100s of millions to 100s of billions of tons of rock, multiplied by the CO2 weight fraction achieved by a specific storage process, and the additional cost is $10/t divided by the CO2 weight fraction captured.
3. Little or no evidence for olivine passivation with reaction progress; almost 100% carbonation obtained within a few hours.
4. Reduces asbestos content of mine tailings.
5. Commonly reduces environmental hazards of alkaline waste products.
6. Ignores possible negative feedbacks, destruction of permeability, and armoring of reactive surface areas via carbonate crystallization, much more significant for peridotite with initial porosity ~1% than for basaltic lava with initial porosity > 10%.
7. Assumes CO2 injection into 10% pore space in submarine lava flows, eventually followed by carbon mineralization.
abundant than ultramafic rocks, and are present in huge volumes relatively close to some population centers. For in situ carbon mineralization, where it may be possible to access vast amounts of rock via injection into high-porosity, high-permeability lava flows, the local rate and total extent of reaction may not be limiting factors.
In altered ultramafic rocks, the minor mineral brucite (0 to 10% by weight), together with asbestiform chrysotile serpentine, provides the best reactant for combined mineral capture and solid storage at low-temperature, near-surface conditions, for example in mine tailings. For solid storage alone, sparging CO2-rich gas through mine tailings and perhaps alkaline industrial wastes could yield up to million-fold accelerations in carbon mineralization rates, compared to uptake of CO2 from air in unmodified tailings.
Because the Ca-bearing component in many alkaline industrial waste products is similar in composition to the rapidly carbonating mineral wollastonite (Figure 6.2A), but less fully crystalline and thus potentially faster to dissolve and react, alkaline wastes are likely to react somewhat faster than the minerals whose kinetic data have been summarized here. However, relatively low Ca and Mg concentrations in some industrial wastes limit the CO2 storage capacity per ton of solid reactant (Table 6.1).
Finally, where basaltic lava formations contain horizons rich in amorphous glass, these react more rapidly than olivine, and provide excellent targets for in situ mineral carbonation. Basaltic glass can also provide exceptionally good material for ex situ mineral carbonation.
EX SITU CARBON MINERALIZATION
An influential review by Mazzotti et al., 2005 concluded that known methods for ex situ carbon mineralization, coupled with CO2 capture from flue gas and other point sources, were too expensive for implementation at scale. For example, they estimated that such processes would approximately double the cost of electricity from a coal-fired power plant. Recent review papers (Gadikota and Park, 2015; Giannoulakis et al., 2014; Huijgen et al., 2007; Khoo et al., 2011; Sanna et al., 2014) confirm that CO2 storage via ex situ carbon mineralization remains significantly more expensive than storage of supercritical CO2 in deep sedimentary formations. (A generic estimate of energy requirements appears in Appendix E.) Moreover, scaling up ex situ carbon mineralization poses the problem of transporting and storing gigatons of carbonated solid material, which potentially involves large transport costs and unknown environmental impacts.
Table 6.1 lists possible solid reactants proposed for ex situ carbon mineralization. These reactants include minerals, rocks, and industrial wastes that could be combined with high-CO2 fluids in reactors at elevated temperature and pressure. The mineral olivine, and rocks containing tens of percent olivine, such as mantle peridotite, ultramafic intrusions, and basaltic lavas, are widely available and react rapidly, and therefore are the most commonly considered solid reactants for carbon mineralization.
Many industrial processes produce alkaline waste materials, such as steel slag, construction and demolition wastes, and cement kiln dust, that are rich in metal cations and have relatively low SiO2 and Al2O3 contents. These materials react readily with CO2 to form carbonate minerals at rates comparable to, or slightly faster than, the fastest reacting natural silicate minerals (i.e., wollastonite and olivine). Peak rates for wollastonite carbonation in alkaline industrial waste are achieved at approximately 100°C (Figure 6.2A), lower than the optimal temperature for olivine carbonation (~185°C), leading to a possible cost savings (Greeshma Gadikota, personal communication, 2017). Cumulative annual production rates of these materials, excluding mine tailings (discussed below), are estimated to have a total CO2 uptake capacity of 0.5 to 1.0 Gt/y (e.g., Renforth et al., 2011; Sanna et al., 2014).
The cost of carbon mineralization is highly dependent on the path and materials used. Nonetheless, a consistent theme is that the cost for proposed CO2 storage via ex situ mineralization is about 10 times higher than the cost of storage of injected CO2 in subsurface pore space beneath an impermeable cap rock, even when the long-term cost of monitoring potential leaks is included for storage in pore space. For this reason, recent reviews (e.g., Gadikota et al., 2015; Huijgen et al., 2007; Pan et al., 2012; Sanna et al., 2014) focus on producing value-added materials that can be sold, to offset some or all of the cost of the CO2 capture and mineralization process. In particular, the focus falls on building materials, because these are used at a global rate exceeding 10 Gt/y and involve storage for decades to centuries. Gadikota et al. (2015) write, “Replacing 10% of building materials with minerals carbonated with anthropogenic CO2 [could] reduce CO2 emissions by 1.6 Gt/year.” One specific idea is to use a significant fraction of carbonated material, produced by ex situ reaction of CO2 with minerals or industrial waste, as aggregate in concrete. Given that the carbonation process adds cost, compared to other sources of aggregate for concrete, research focuses on the possibility of producing aggregate that has added, desirable properties in the final concrete product (e.g., Gadikota et al., 2015). Another idea is to use the reaction products in the supply chain for conversion of CO2 to methane (CH4) and more complex hydrocarbons.
Carbon capture, utilization, and storage is the subject of a separate National Academies of Sciences, Engineering, and Medicine study.1
ENHANCED CARBONATION OF ULTRAMAFIC MINE TAILINGS AND SEDIMENTS
Mine tailings provide “low-hanging fruit” as rock reactants for CO2 removal from air and for CO2 storage via carbon mineralization, because of their high surface area to volume ratios compared to subsurface geological formations, and because they are quarried and mined for other reasons. Fresh and, more commonly, partially serpentinized ultramafic rocks from Earth’s mantle are mined for Cr and Ni. Ultramafic intrusions, with Mg to Si ratios approaching 2 and abundant olivine, are mined for platinum-group elements, Cr, and diamonds. Ultramafic lava flows (komatiites) and tectonically exposed and weathered mantle peridotites are an important source of Cr and Ni. Mafic intrusions comparable in composition to basaltic lavas are mined for platinum group elements, Cr and Ni.
Uptake capacity for complete olivine and brucite carbonation in these materials is significant: 0.62 tons of CO2 per ton of olivine reactant, 0.76 tons of CO2 per ton of brucite reactant, and approximately 0.4 to 0.5 tons of CO2 per ton of pyroxene and serpentine. Based on our informal assessment of the volume of existing mines, we estimate that the total mass of existing ultramafic tailings is less than 10 billion tons, of which an unknown proportion has already been carbonated due to natural weathering processes. Annual production of new ultramafic mine tailings is about 200 million tons (Power et al., 2013c). Dipple and co-workers (Harrison et al., 2013; Power et al., 2011, 2013c; Wilson et al., 2014) emphasize that at surface conditions (~10-30°C, P(CO2) ~ 0.004 atm) most minerals in ultramafic and mafic rocks are relatively slow to react, compared to brucite and some asbestiform chrysotile. Thus, they focus on rapid carbonation of labile magnesium contained in the latter two minerals. Typically, labile Mg comprises approximate 3 wt% of partially to fully serpentinized, ultramafic mine tailings, with an approximate maximum 10 wt% (Greg Dipple, personal communication, 2017). Carbonation of this Mg (~24 gm/mol) in newly produced mine tailings would consume less than 36 million tons of CO2 per year.
Because this storage capacity is small, the question arises whether one could mine ultramafic rock for the purpose of creating fine-grained rock reactants for CO2 mineral capture from air and storage. Quarrying, crushing, and grinding of mine tailings
___________________
1 See Developing a Research Agenda for Utilization of Gaseous Carbon Waste Streams, http://nas-sites.org/dels/studies/gcwu/ (accessed January 28, 2019).
cost approximately $10/t (InfoMine, 2018). Additional costs per ton to speed carbon mineralization (e.g., spreading tailings in thin sheets, stirring them, providing access to air flow) might be negligible. Carbonating 1 to 10 wt% labile Mg, consuming approximately 2 to 18 wt% CO2, would correspond to $55-500/tCO2. This cost is comparable to estimates for direct air capture systems (Chapter 5) plus subsequent storage in deep sedimentary formations (Chapter 7), within the uncertainties of all values. On the one hand, creating tailings solely to consume labile Mg for rapid CO2 capture from air and solid storage would produce 5-50 Gt of tailings (2-17 km3) per Gt of CO2 captured. To put these volumes in context, this corresponds to a layer 3-30 microns thick over the 510 million km2 area of the oceans, 0.1-1.2 millimeters thick over the 14 million km2 of arable land worldwide, or 10-100 m thick over Washington, D.C. (177 km2), per Gt of CO2 captured from air and stored. Depending on location and societal preferences, transporting and storing or disposing of these tailings could be problematic.
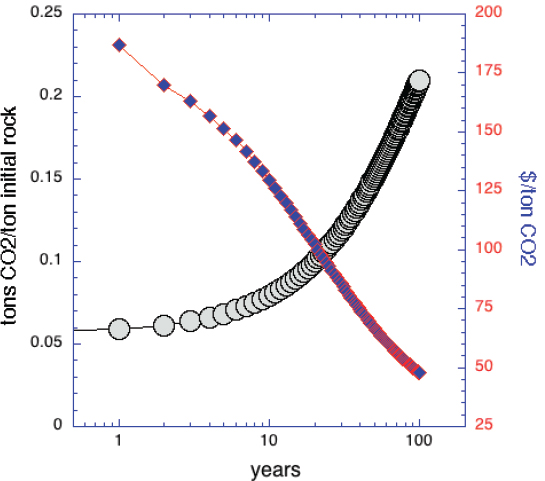
In addition to rapid carbonation of labile Mg, some olivine and a small amount of non-asbestos serpentine in ultramafic tailings will also undergo carbonation over decades of weathering. To calculate how much, we use the data in Figure 6.4, the assumption of constant reaction rates (e.g., Figure 6.3), a brucite conversion rate (mass fraction per second) of 3 10-8/s, and olivine and serpentine conversion rates that are 102 and 104 times smaller, respectively. This calculation yields the estimate that carbon mineralization of typical mine tailings with 3 wt% labile Mg (7.2 wt% brucite, 40 wt% olivine, 52.8 wt% serpentine) could capture and store about 21 wt% CO2 in 100 years (Figure 6.5). If the cost of carbonation is not much greater than the initial cost of quarrying and grinding (~ $10/ton of rock), this would cost $48/tCO2—somewhat less than the lowest estimated costs of direct air capture systems—and produce about 1.7 km3 of tailings per Gt of CO2 captured and stored (a 10 m layer over Washington, D.C.). However, extensive areas of thinly layered tailings would have to be maintained for many decades. It might be possible to reach a similar goal by dispersing ultramafic tailings on coastlines or onto shallow seafloor (Hartmann et al., 2013; Köhler et al., 2010, 2013; Montserrat et al., 2017; Rigopoulos et al., 2018; Schuiling and Krijgsman, 2006), but environmental impacts and barriers to social acceptance are uncertain.
Rates of ultramafic mineral carbonation at ambient surface temperatures increase with elevated CO2 pressure (Figure 6.4), suggesting that sparging CO2-rich gas or fluids through tailings piles could accelerate CO2 uptake in mine tailings for solid storage. Given the cost estimates in the previous two paragraphs, this approach is likely to be more expensive than storage of supercritical CO2 in sedimentary formations (about $10-20/t; see Chapter 7), but could provide a local solution where, for example, mining companies or geothermal power plants wish to store CO2 emitted on site and/or add value via carbon offsets.
NOTES: The initial rock reactant contains 7.2 wt% brucite, 40 wt% olivine, and 52.8 wt% serpentine, with constant reaction rates (mass fraction per second) of 3 10-8, 3 10-10, and 3 10-12, respectively. Passivation effects, and the cost of methods to ameliorate them, are not included here. Formation of crusts within a tailings pile (e.g. Wilson et al., 2014, Figure 3) and carbonate coatings on grains (e.g., Wilson et al., 2014, Figure 2) can cause passivation. Some of this could be overcome by stirring the tailings and similar low-cost methods.
Grinding ultramafic or mafic basaltic rock reactants to smaller sizes than typical of mine tailings, and spreading them in agricultural soil, forest soil, or along beaches has been suggested as a means for CO2 removal from air (e.g., Schuiling and Krijgsman, 2006). This idea has recently been experimentally evaluated and reviewed in several papers (Beerling et al., 2018; Edwards et al., 2017; Hartmann et al., 2013; Kantola et al., 2017; Köhler et al., 2010, 2013; Meysman and Montserrat, 2017; Montserrat et al., 2017; Moosdorf et al., 2014; Renforth et al., 2015; Renforth and Henderson, 2017; Rigopoulos et al., 2018; Taylor et al., 2016, 2017; ten Berge et al., 2012). Most of these studies use olivine as the mineral reactant, because brucite and asbestiform chrysotile are not abundant in most candidate rock formations, asbestos is a serious health hazard, and most other serpentine minerals are slow to react at surface conditions (see “Carbon Mineralization Kinetics”). Some studies also consider basaltic lava as a reactant.
Carbon mineralization for different rates and grain sizes is illustrated in Figure 6.6. Carbon uptake in soils occurs through the interaction of air with dissolved alkalinity, which in some cases is known to release Ca at rates of 10-15 to 10-16 mol/(cm2 s)
(Renforth et al., 2015). The reaction of seawater with olivine ground to about 1 micron has comparable rates (Köhler et al., 2013; Montserrat et al., 2017; Rigopoulos et al., 2018). Using the approach of Renforth (2012), an energy cost of $0.05-0.30/kWh, and $10/t for mining and crushing olivine to tailings size, we calculate that this method costs approximately $25-105/t.
Similarly, Renforth et al. (2009, 2015) have shown that carbonate precipitation in soil at brownfield sites modified with demolition rubble is 3 times higher than the value of average C content in urban areas, corresponding to a storage potential of 30 ± 10-2 kg CO2/m2. This increase in carbonate precipitation is attributed to the leaching of alkalinity from olivine and/or Ca-rich building materials that are pulverized during demolition to increase the reactive surface area and mix within the top layer of soil.
Microbial processes may enhance weathering rates in soil, because microbial degradation of organic matter generates chelating agents and organic and inorganic acids. These microbial processes also accelerate mineral carbonation by increasing the local CO2 partial pressure to 10-100 times atmospheric concentration (Power et al., 2009, 2013a).
These studies suggest that incorporating finely ground olivine in agricultural soils or broadcasting it into the surface ocean is sufficiently fast and inexpensive to be competitive with direct air capture systems. A concern is that minor constituents in olivine, such as Ni and Cr, could accumulate in soil or water over time. Oxidized Ni and Cr compounds in water and food constitute significant health hazards at low concentrations.
IN SITU CARBON MINERALIZATION
In situ storage—via circulation of CO2-rich fluids (CO2-rich water or H2O-bearing supercritical CO2) in appropriate formations to form subsurface carbonate minerals—addresses many of the problems of ex situ solid storage but remains a largely speculative alternative. Most investigations of in situ solid storage, and in situ capture and storage, focus on mafic and ultramafic rock formations, particularly large provinces of basaltic lava and large massifs of mantle peridotite, because of their high abundance, widespread geographical distribution, and rapid carbon mineralization rates.
Kinetic studies (see “Carbon Mineralization Kinetics”) suggest that in situ carbon mineralization in ultramafic rocks (with high molar Mg to Si ratios) and in glass-rich basaltic lava flows can potentially consume tens of weight percent of injected CO2 within a few years. Experimental studies of reaction kinetics are consistent with the results
of pilot experiments on carbon mineralization in mafic and ultramafic formations, as summarized in the following sections.
It is important to add that a study estimating carbon mineralization rates in a specific sandstone in a depleted oil reservoir (Benson et al., 2005) led to the widespread impression that in situ carbon mineralization is so slow that it would not become a significant factor for large-scale storage of CO2 for thousands of years after injection. However, this result is not applicable to carbon mineralization in basaltic lavas and ultramafic rocks (Figure 6.7; Benson, personal communication, 2017).
NOTES: For a given extent of dissolution (X), the initial particle diameter (D0; m) can be related to dissolution rate (Wr; mol m-2 s-1) through a shrinking core model
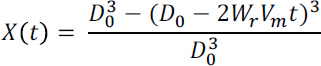 where Vm is the molar volume of the material (m3 mol-1 assumed here to be 40 cm3 mol-1, although the molar volume of natural primary silicate minerals ranges from this up to >100 cm3 mol-1), and t is the dissolution time (s). Grinding produces a particle size range. Here we use a gamma distribution previously coupled to a shrinking core model (Gbor and Jia, 2004)
where Vm is the molar volume of the material (m3 mol-1 assumed here to be 40 cm3 mol-1, although the molar volume of natural primary silicate minerals ranges from this up to >100 cm3 mol-1), and t is the dissolution time (s). Grinding produces a particle size range. Here we use a gamma distribution previously coupled to a shrinking core model (Gbor and Jia, 2004)
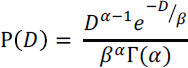 where α and β are empirically-derived coefficients that describe the variability of particle size. A maximum mean particle size was simulated with a typical particle size distribution from comminution to achieve 90% dissolution. Dissolution rate ranges are shown for brucite (Mg(OH)2), chrysotile serpentine (Mg3Si2O5(OH)4), minerals in the plagioclase feldspar solid solution [Na endmember albite (Alb - NaAlSi3O8), Ca endmember anorthite (Ano - CaAl2Si2O8), oligoclase (Olg – 10-30% Ano), andesine (30-50% Ano), labradorite (Lab – 50-70% Ano), bytownite (Byt – 70-90% Ano)], wollastonite (CaSiO3), and Mg-rich olivine (Mg2SiO4), derived using rate data from Palandri and Kharaka (2004) for all minerals except chrysotile serpentine (Thom et al., 2013), assuming pH from 3 to 7, 25°C (blue bars) and 180°C (orange bars), and negligible effects of mineral solution saturation. Rates determined from mineral dissolution experiments are shown for (a) basalt added to artificial soil (~10°C; Manning et al., 2013), (b) basalt/dunite/hazburgite added to seawater (25°C, Rigopoulos et al., 2018), (c) Mg-rich olivine added to soil (25°C, ten Berge et al., 2012), (d) Mg-rich olivine added to seawater (25°C Montserrat et al., 2017), and (e) Mg-rich olivine added to soil (19°C, Renforth et al., 2015). Figure and calculations courtesy of Phil Renforth, personal communication, 2018.
where α and β are empirically-derived coefficients that describe the variability of particle size. A maximum mean particle size was simulated with a typical particle size distribution from comminution to achieve 90% dissolution. Dissolution rate ranges are shown for brucite (Mg(OH)2), chrysotile serpentine (Mg3Si2O5(OH)4), minerals in the plagioclase feldspar solid solution [Na endmember albite (Alb - NaAlSi3O8), Ca endmember anorthite (Ano - CaAl2Si2O8), oligoclase (Olg – 10-30% Ano), andesine (30-50% Ano), labradorite (Lab – 50-70% Ano), bytownite (Byt – 70-90% Ano)], wollastonite (CaSiO3), and Mg-rich olivine (Mg2SiO4), derived using rate data from Palandri and Kharaka (2004) for all minerals except chrysotile serpentine (Thom et al., 2013), assuming pH from 3 to 7, 25°C (blue bars) and 180°C (orange bars), and negligible effects of mineral solution saturation. Rates determined from mineral dissolution experiments are shown for (a) basalt added to artificial soil (~10°C; Manning et al., 2013), (b) basalt/dunite/hazburgite added to seawater (25°C, Rigopoulos et al., 2018), (c) Mg-rich olivine added to soil (25°C, ten Berge et al., 2012), (d) Mg-rich olivine added to seawater (25°C Montserrat et al., 2017), and (e) Mg-rich olivine added to soil (19°C, Renforth et al., 2015). Figure and calculations courtesy of Phil Renforth, personal communication, 2018.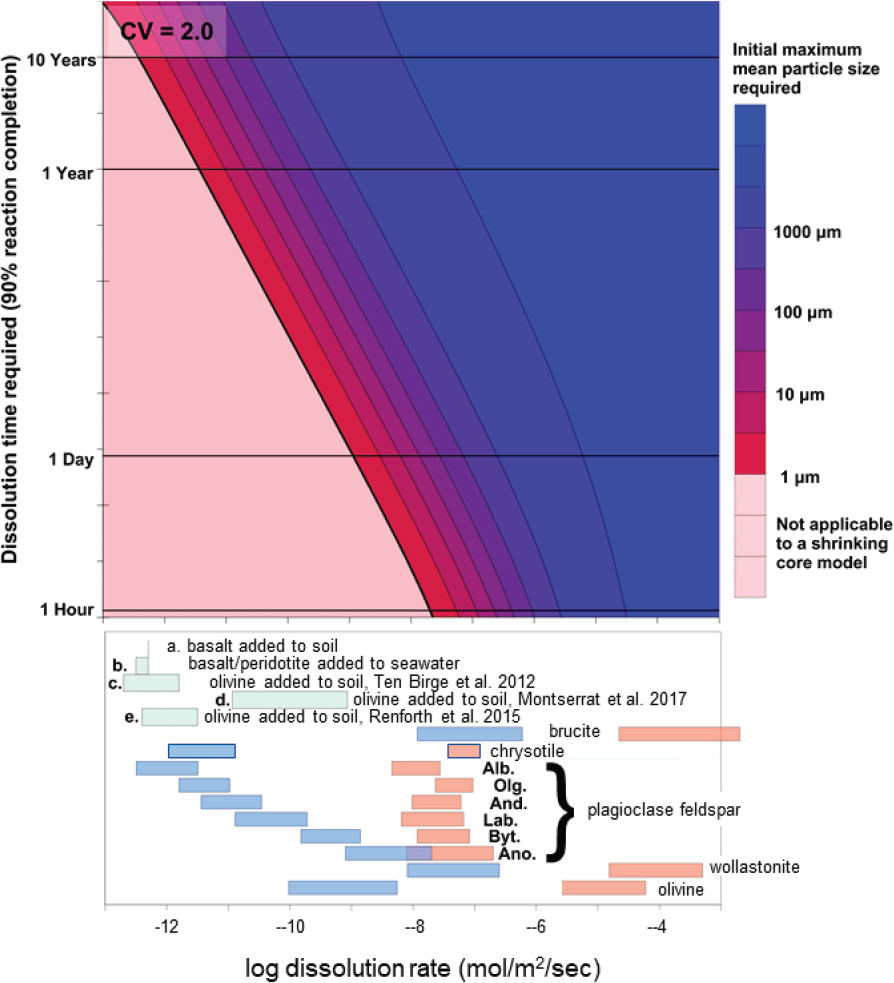
In situ Carbon Mineralization in Basalt
The committee’s November 2017 workshop included two presentations on in situ carbon mineralization experiments in basalt: the ongoing CarbFix experiment in Iceland and the recently completed Wallula Project in Washington State. Both experiments involved extensive characterization of the composition, structure, and hydrology in thick sequences of basaltic lavas, followed by injection of CO2-rich fluids to investigate storage in pore space and as solid carbonate minerals. The following information is derived from these presentations and from published reviews of CarbFix (e.g., Aradóttir et al., 2011; Gislason et al., 2010; Gunnarsson et al., 2018; Matter et al., 2011, 2016; Snæbjörnsdóttir et al., 2017) and Wallula (e.g., McGrail et al., 2014, 2017a, 2017b).
The Wallula project injected 977 tons of water-saturated, supercritical CO2 at a depth of 828 to 886 meters. Side cores from the main borehole wall revealed the presence of abundant, newly formed carbonate minerals precipitated by reaction of the basalt with injected CO2, consistent with the composition of water in the borehole. Extensive surface studies and borehole observations for several years revealed no leakage of CO2 from the highly permeable horizon into which it was injected. Via experiments on drill core from the same basalt formation as the host for the Wallula injection, Xiong
NOTES: Secondary trapping mechanisms vary widely depending on geology, structure, and hydrology. Lefthand panel, typical sedimentary reservoir, after Krevor et al., 2015. Right hand panel, peridotite reservoir, approximated using data from Figure 6.5.
et al. (2018) inferred a CO2 mineralization rate of 0.04 wt%/y (mass fraction of ~ 10^(-11)/s) for this site, consistent with other laboratory experiments on basalt and its constituent minerals, discussed above, and the observed rate of carbon mineralization at CarbFix, estimated below. Over the time span of several years to decades, passivation of reactive surfaces may decrease the mineralization rate. For this and other reasons, it is not known how much of the injected CO2 formed carbonate minerals, and how much still remains within fluid in pore space at the Wallula site.
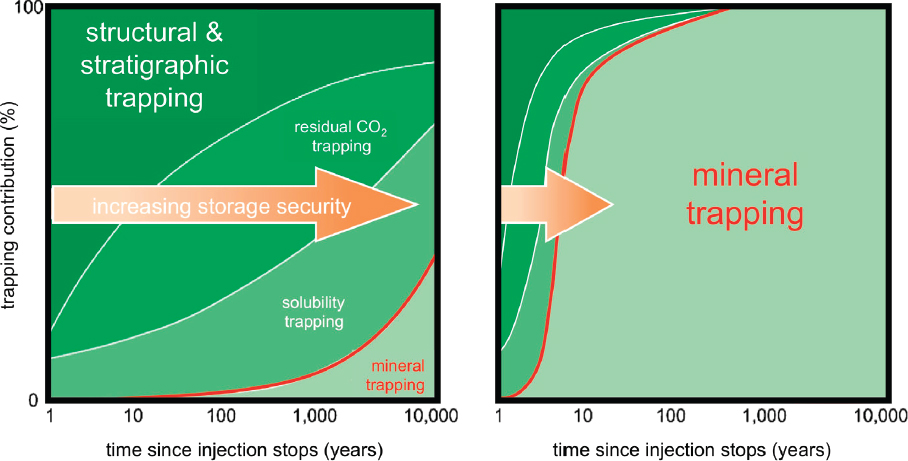
The CarbFix experiment is being conducted by the geothermal power company Reykjavik Energy, together with a consortium of research scientists. In addition to investigating proposed methods for CO2 storage, this project has the goal of storing CO2 and hydrogen sulfide co-produced with geothermal fluid at a specific power plant. Phase I of CarbFix injected about 200 tCO2 into highly permeable, fractured basalts at a depth of 500 m (ambient temperature ~ 20-50°C, porosity ~ 10%). At this depth, CO2 solubility in water is not high, and CO2 rich fluids are not supercritical. As a result, the project employs the novel technique of separately injecting H2O and CO2 with proportions adjusted to ensure complete solubility of CO2 into aqueous fluid at the target depth (Figure 6.8). This technique is known as solution trapping.
CarbFix also used a novel tracer technique, injecting SF6 and other conserved tracers together with labeled 14C-rich CO2. Because of considerable anisotropy in the groundwater flow path at the test site, production and monitoring wells can be confidently placed downstream from the injection well. The tracers allow researchers to see the injected fluid pulse arrive at the production wells (Figure 6.9). Notably, after an initial small pulse in 14C at the production well, the carbon concentration and the proportion of 14C in carbon returned to near-ambient levels, indicating nearly complete loss of carbon along the approximate 100 m flow path from injection to production well. It is inferred that this carbon is lost via reaction of the injected fluid with host basalt to form carbonate minerals.
CarbFix Phase II is continuing with the methodology of Phase I, but with a deeper and wider range of injection depths (~1,500 meters at higher temperature) and a large increase in CO2 flux. Cumulatively, more than 20,000 tons of CO2 have been injected, with tracer results continuing to indicate nearly complete loss of carbon along an approximate 2,000 m flow path. Phase II is close to the target scale for routine operation, accommodating most of Reykjavik Energy’s CO2 emissions at a specific geothermal power plant, and also disposing of hydrogen sulfide produced at the power plant.
A rough calculation of the volume of rock infused with injected CO2-rich aqueous fluid can be made if we assume that most of the flow is confined to a vertical interval of 10-100 m, over a width of 100 m over the flow path of 100-2,000 m in Phase I
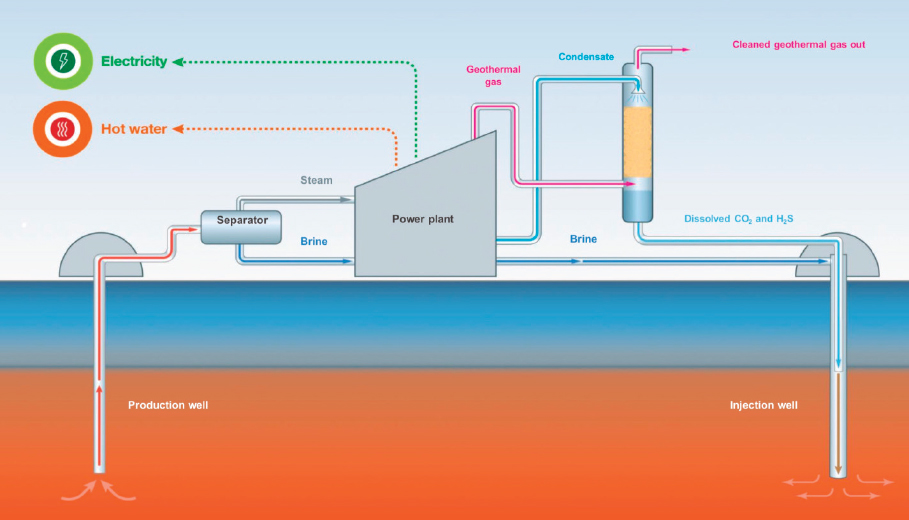
NOTES: CO2 and SO2 are important constituents of the geothermal gas that are separated from other gas constituents and co-injected with geothermal brine effluent from the power plant. CO2 + SO2 are injected as a separate gas phase, until they are mixed with aqueous brine at a depth in proportions such that all gases dissolve in water.
SOURCE: Aradóttir, personal communication (2017).
and Phase II. (The actual volume probably widens from injection well to production well, but for this calculation we hold the width constant.) Given this assumption, the infused rock volume is about 105 to 2 107 m3, with a density of about 2.8 t/m3, yielding ≤ 56 Mt of rock in the Phase II injection volume. For Phase II, then, the mass fraction of CO2 within this volume is approximately 3.6 10-4 (~0.7 wt% carbonate minerals) and the CO2 uptake rate to date, in mass fraction per second, has been approximately 10-11/s over 3 years. This figure corresponds to approximately 5 10-9 mol/m2/s if the rock reactants can be approximated as cubes or spheres 1 mm in size. This rate is similar to those calculated using experimental data on plagioclase feldspar dissolution (Figure 6.2C) and experimental rates for full basalt carbonation (Figure 6.2B). It is possible that the initial grain size of the most reactive minerals is less than 1 mm and/or that the surface area per grain is larger than it is for cubes or spheres. If so, the rate of CO2 consumption in the CarbFix experiment may be limited by CO2 supply, rather than the local reaction rate, and could be substantially higher if CO2 were injected more rapidly.
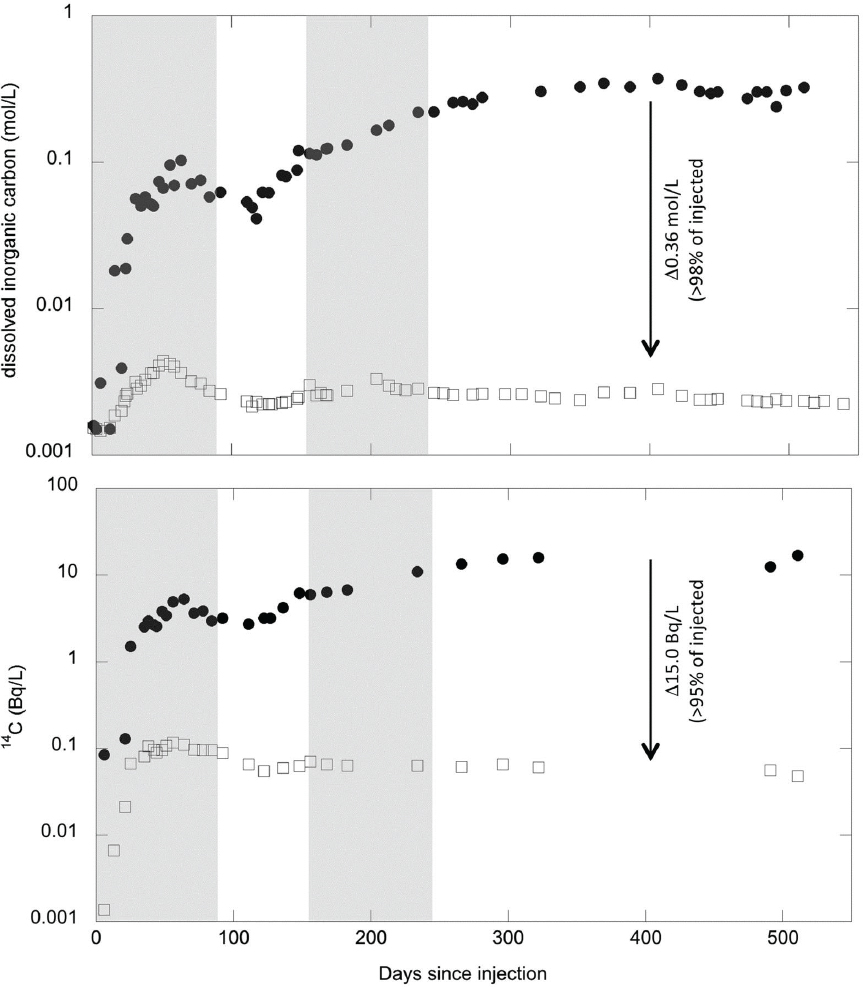
NOTE: The observed deficit in carbon concentration and 14C are consistent with loss of consumption of almost all injected CO2 along the flow path to form solid carbonate minerals.
SOURCE: Matter et al., 2016.
On the horizon, four concerns emerge. The first is to determine which minerals are reacting. A large variety of alteration minerals are present in Icelandic and Columbia River basalts, including zeolites, clays, and other fine-grained materials that may react rapidly. Often, such alteration phases comprise just a few percent of basaltic lavas. If alteration minerals are significant reactants in carbon mineralization, it will be important to determine their abundance to predict the capacity of the basalt reservoir at the current reaction rates. Alternatively, perhaps igneous, rock-forming minerals are the main solid reactants in carbon mineralization. This would be good news, because rock-forming minerals are probably more abundant than alteration minerals in the injection horizon.
A second concern is the possibility of passivation of reactive mineral surfaces over time, either by incongruent mineral dissolution leaving SiO2-rich residual crusts, or by precipitation of newly formed, solid reaction products that armor the surface of solid reactants. As noted above, passivation has been observed in experiments on full carbonation of basalt and plagioclase feldspar (e.g., Gadikota et al., 2018). Basalt formations may be more susceptible to armoring of reactive surfaces with new reaction products than peridotite formations.
A third concern is clogging of pore space. To date there has been no observed reduction of permeability in the CarbFix storage reservoir (Aradóttir, personal communication, 2017). And if the small proportion of carbonate produced is widely distributed over a large fraction of the reservoir volume, one would not expect to see an appreciable change. For highly localized precipitation fronts, higher injection rates, and/or injection over longer times, permeability changes could become appreciable. However, clogging of pore space in basaltic formations, which have initial porosity of approximately 10 percent, is likely to be less important than clogging in peridotite, which have approximately 1 percent fracture-dominated porosity.
A fourth concern—or interesting variable—is the spatial distribution of carbonate mineral precipitation vs dissolution. After solution trapping at depth, the injected fluid at CarbFix has a low pH (~ 3) and is undersaturated in carbonate minerals. At low fluid to rock ratios, far from the injection site, pH will increase due to reaction with host basalt and newly formed carbonate minerals along the flow path. There must be an annulus around the injection site where existing carbonate minerals dissolve in low pH fluid and new carbonate will not precipitate. As the fluid to rock ratio increases around the injection well over time, this low pH annulus is likely to grow, with a widening volume of dissolution of newly formed carbonate pushing the zone of new carbonate precipitation outward, as predicted via modeling by Aradóttir et al., 2012.
The topics discussed in the previous four paragraphs can be examined to some extent
and by forward models (e.g., Snæbjörnsdóttir et al., 2017). In addition, the actual evolution of dissolution and crystallization fronts depends on many variables whose values are poorly known. Models will need to be calibrated and validated using extensive observations. Fortunately, CarbFix Phase II is predicted to continue for several more years, and Reykjavik Energy hopes to make CO2 (+ hydrogen sulfide) injection a standard part of its operating procedure. Note that CarbFix was not envisioned as part of a process to remove CO2 from air, but instead as a means to store emissions from a geothermal power plant. However, recent addition of a ClimeWorks direct air capture unit at the CarbFix site serves as a concrete reminder that direct air capture systems, together with in situ carbon mineralization in basaltic lavas, could be a potent combination to achieve negative emissions.
The CarbFix methodology can be seen as a two-stage storage technique, first trapping CO2 dissolved in water at depth, and then converting the dissolved CO2 to solid carbonate minerals over the time groundwater flows over 2,000 m. If necessary, carbon-depleted water could be produced downstream and recycled, reducing overall water consumption. Because of the first step (solution-trapping), there is no requirement for an impermeable caprock to avoid CO2 leakage. With an estimated cost of $30/t (Aradóttir, personal communication, 2017) for nth-of-a-kind methods of this type, this process costs approximately $10-20/tCO2 more than the estimated cost of injection into subsurface pore space and could be a locally preferred option in some regions. This prospect opens vast expanses of volcanic provinces, on land and in submarine, near-shore environments, as potential storage reservoirs (Figure 6.10).
Looking more generally at the global potential for in situ carbon mineralization in basalt formations, Tables 6.1 and 6.2 indicate that both cost and tonnage are favorable for this approach, which could potentially accommodate tens of gigatons of CO2 per year for decades. It is important to realize that basaltic lava formations—at depths of approximately less than 1,000 m, with moderately high porosity (~ 10%), and with an impermeable cap rock—can be considered appropriate reservoirs for storage of supercritical CO2 in subsurface pore space. This approach has the added benefit that at least some of the stored CO2 will be converted to carbonate within a few years or decades, reducing the risk of leakage and the need for monitoring as time goes by. Such hybrid methods, combining storage in pore space and relatively rapid carbon mineralization, may represent the best mix for CO2 storage in regions with appropriate geology, as emphasized in a series of papers on onshore and near-shore marine basalt formations by Goldberg and co-workers (Goldberg and Slagle, 2009; Goldberg et al., 2008, 2010).
There has been little focus on in situ mineral capture and storage of CO2 in basalt formations. This is due to relatively slow laboratory rates of carbon mineralization in
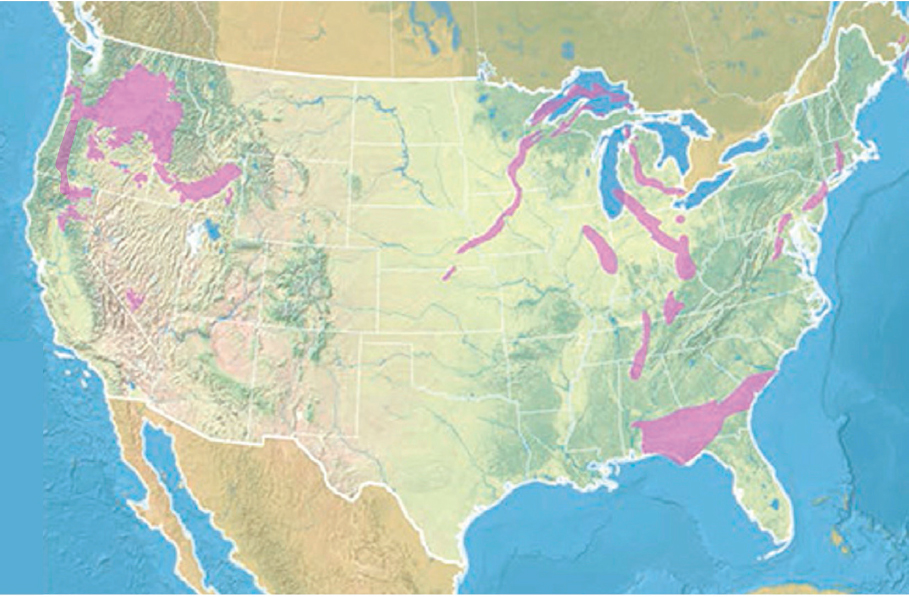
NOTES: In addition to the resources depicted here, there are large, onland and near-shore basalt provinces on all continents and many ocean islands. Table 6.1 lists the potential size of the basalt reservoir at 105 to 106 Gt of rock, with a potential to contain up to 25% CO2, based on estimates from McGrail et al. (2017a) for the capacity of the Columbia River and Deccan Trapps flood basalts, extended to include other large basalt provinces onland (Parana, Siberia, North Atlantic) and near shore (Kerguelen, Iceland). We are not aware of more detailed, comprehensive reviews of the global CO2 storage capacity of onland basalts. Goldberg and Slagle (2009) estimate the global carbon storage capacity of seafloor basalts. Modified from https://www.netl.doe.gov/coal/carbon-storage/faqs/carbon-storage-faqs#types (accessed January 28, 2019).
basalt and minerals common in basalt (particularly plagioclase feldspar) compared to ultramafic rocks and their common minerals (e.g., olivine, asbestiform chrysotile, and brucite; Figures 6.2 and 6.3). However, the potential for rapid carbon mineralization in basaltic glass (Figure 6.2C) should not be overlooked.
In Situ Carbon Mineralization in Peridotite and Other Ultramafic Rock Formations
The classic paper of Barnes and O’Neil (1969) demonstrated that mantle peridotite near Earth’s surface undergoes low temperature hydration (serpentinization) and carbonation at appreciable rates. The best-studied natural example of this process is the
TABLE 6.2 Capture of CO2 from Air and Surface Waters Plus Solid Storage, via Carbon Mineralization at Ambient P(CO2) and Temperature
| Location | Method | Grain size and crack spacing microns | Natural rate t/y CO2 | Rate weight fraction CO2/y | Rate t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction at rate in table | Cost $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Natural processes | ||||||||||||
| Mine tailings rich in brucite and fine chrysotile | Mt. Keith Mine, AU | Natural percolation | 20-200 | 4.0E+04 | 3.6E-03 | 9.0E+06 | 0.011 | 0.03-0.1 | 0 | Wilson et al., 2014 | 1, 2 | |
| Mine tailings rich in brucite and fine chrysotile | Diavik Mine, CA | Natural percolation | 20-200 | 3.0E+04 | 4.0E-05 | 1.6E+04 | 0.002 | 0.03-0.1 | 0 | Wilson et al., 2011 | 1, 2, 3 | |
| Serpentinite mine tailings | Black Lake Mine, CA | Natural percolation | 20-200 | 6.0E+03 | 5.0E-05 | 2.0E+04 | 0.120 | 0.03-0.1 | 0 | Pronost et al., 2012 | 1, 2 | |
| Global ultramafic mine tailings | Global | Natural percolation | 20-200 | 0.200 | <10 | 0.03-0.1 | 0 | Dipple and Kelemen, personal communication, 2017 | ||||
| Fractured peridotite aquifers | Oman, depth <3 km | Natural groundwate circulation | 1–1E6 r | 3.3E-07 | 1.0E+03 | 50,000 | 0.60 | 0 | Kelemen and Matter, 2008 | 4, 5, 6 | ||
| Location | Method | Grain size and crack spacing microns | Natural rate t/y CO2 | Rate weight fraction CO2/y | Rate t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction at rate in table | Cost $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Global fractured peridotite aquifers | On land, depth below surface <3 km | Natural groundwater circulation | 2–1E6 | 3.3E-07 | 1.0E+03 | 1E5-1E6 | 0.60 | 0 | Kelemen et al., 2011 | 4, 5, 6 | ||
| Global fractured peridotite aquifers | Seafloor, depth below seafloor <3 km | Natural groundwater circulation | 3–1E6 | 3.3E-07 | 1.0E+03 | ~1E8 | 0.60 | 0 | Kelemen et al., 2011 | 4, 5, 6 | ||
| Soils contaminated with Ca-rich building waste | Newcastle, UK | Natural percolation | 1-100 | 4.6E-03 | 9.2E+06 | ~ 0.015 | 0 | Manning and Renforth, 2013; Renforth et al., 2009; Washbourne et al., 2015 | 3, 7 | |||
| Enhanced processes | ||||||||||||
| Nearly pure Mg(OH)2 from brucite mine | Lab experiment | Air sparged through brucite + H2O | 2-40 | 9.9E-05 | 2.5E+05 | 0.200 | <10 | 0.03-0.1 | 10-30 | Harrison et al., 2013 | 1 | |
| Bioleaching and microbial carbonate precipitation | Proposed | Bioengineered | 3.6E-3–3.6E-2 | 0.200 | <10 | 0.03-0.1 | 10-30 | Power et al., 2010, 2011, 2013a | 7 | |||
| Thinner distribution, stirring of ultramafic mine tailings | Generic | Mechanical | 3.6E-3–3.6E-2 | 0.200 | <10 | 0.03-0.1 | Assima et al., 2013a; Harrison et al., 2013; Power et al., 2013c; Wilson et al., 2014 | 7 | ||||
| Ground peridotite or basalt on soils, beaches | Lab experiment | Grinding and broadcast | 0.01-1.0 | 5E-11–5E-10 | 0.025-0.25 | 28,000 | 0.05? | 25-115 | Hartmann et al., 2013; Köhler et al., 2013; Montserrat et al., 2017; Renforth, 2012; Renforth et al., 2015; Rigopoulos et al., 2018 | 3, 8-11 |
| Location | Method | Grain size and crack spacing microns | Natural rate t/y CO2 | Rate weight fraction CO2/y | Rate t CO2/km3/y | CO2 reservoir production Gt rock/y | CO2 reservoir Gt rock | Max CO2 weight fraction at rate in table | Cost $/t CO2 | References | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Produce alkaline water from peridotite aquifers | Generic | Drill, pump if necessary | NA | ~10? | Kelemen et al., 2016 | 5, 12, 13 | ||||||
| Circulate water through high permeability peridotite aquifers via thermal convection | Generic | Drill to depth where temp ~90°C, flow and recharge driven by hydrothermal convection, permeability 10-12 m2 | 1–1E6 | 1.5E-4–1.5E-3 | 4.5E5–4.5E6 | 1E5-1E8 | 0.60 | 30-60 | Kelemen et al., 2011, 2016 | 5, 14 | ||
| Circulate water through low permeability peridotite aquifers via pumping | Too costly | Drill to depth where temp ~ 90°C, flow and recharge driven by pumping, permeability 10–14 m2 | 1–1E6 | 1.5E-4–1.5E-3 | 4.5E5–4.5E6 | 1E5-1E8 | 0.60 | 3,000-6,000 | Kelemen et al., 2016 | 5, 11, 14 |
| Circulate water through peridotite aquifers combined with geothermal power generation | Generic | Reduce cost of geothermal power generation via carbon offsets | 1–1E6 | 1.5E-4–1.5E-3 | 4.5E5–4.5E6 | 0.60 | <0? | Kelemen et al., 2011, 2016 | 5, 14 | |||
| Mine ultramafic rock to create tailings for carbon removal | Generic | Mining and grinding | 20–200 | 1E5–1E6 | 0.03–0.1 | 100–300 | Dipple and Kelemen, personal communication, 2017 | 15 | ||||
| Artificial mix of organic waste and quarry fines | Lab experiment | Natural percolation | 2–3400 | 8.81E-04 | 1.76E+06 | 2E4–5E4 | ~ 0.015 | 50–100? | Manning and Renforth, 2013; Renforth et al., 2009; Washbourne et al., 2015 | 3, 8, 9 |
NOTES
1. Assumed density of tailings 2.5 t/m3.
2. Impacts may be confined to mine tailings sites, assumed density of tailings 2.5 t/m3.
3. Assumed depth of carbonation in tailings and soil 1 m.
4. Assumed density of partially serpentinized natural peridotite 3 t/m3.
5. No known geochemical contamination; water from peridotite-hosted aquifers is within EPA safe drinking water limits.
6. Rates of natural CO2 uptake tabulated here may be confined to upper 15 m, but reservoir size tabulated here is upper 3 km.
7. Maximum of 10× acceleration compared to fastest known natural rate.
8. Assumed density of soil 2 t/m3.
9. Total mass is global area of arable soil to depth in (3).
10. Ni and Cr may accumulate in soils over time.
11. $10 to mine and grind to tailings size, 300 to 350 kWh/ton CO2 to mill from tailings to 1 micron, assumed cost of energy $0.05 to 0.30/kWh.
12. If drilled at existing spring sites, pumping should be minimal, cost is simply cost of typical water well.
13. Once calcite is precipitated and water reaches normal pH, discharge to local drainages.
14. Crystallization of carbonate minerals in subsurface could destroy permeability and armor reactive surfaces, or alternatively could maintain or enhance permeability and reactive surface area via reaction-driven cracking (Jamtveit et al., 2008; Kelemen and Hirth, 2012; MacDonald and Fyfe, 1985; Zhu et al., 2016).
15. $10 per ton of rock for mining and grinding to typical tailings size.
alteration of mantle peridotite in the Samail ophiolite (e.g., Neal and Stanger, 1985), a block of oceanic crust and mantle peridotite thrust onto the Arabian continental margin from 96 to 70 million years ago, and now exposed by faulting and erosion in northern Oman and the eastern United Arab Emirates. Present-day carbon mineralization forms carbonate veins in fractures, within partially serpentinized peridotites at ambient, near-surface temperature and pressure, and large travertine terraces of chemically deposited calcite (CaCO3) on the surface. Ongoing rates of carbonation, constrained by 14C geochronology and other data, are about 1 gm CO2/m3/y (1,000 t/km3/y) in a weathering horizon that may have an average depth of approximately 15 m (Kelemen and Matter, 2008; Kelemen et al., 2011; Mervine et al., 2014; Streit et al., 2012). Carbonates in the rock matrix and in veins filling fractures constitute about 1 percent of peridotite in outcrop, on average. In the past, fully carbonated peridotites, known as listvenites, in which all Mg and Ca have combined with CO2 to form carbonate minerals, and SiO2 is present as quartz, formed in Oman at somewhat higher temperature, approximately 100°C, in the presence of fluids with high P(CO2) (de Obeso et al., 2017; Falk and Kelemen, 2015; Godard et al., 2017; Kelemen et al., 2017; Manning et al., 2017), similar to conditions for proposed in situ CO2 storage.
Research on engineered in situ carbon mineralization in peridotite is motivated by five key factors. First, the mineral olivine is the most abundant mineral in Earth’s upper mantle and remains abundant in most partially altered peridotites that have been exposed at and near the surface. At conditions accessible in the upper few km of Earth’s crust (temperatures from 50 to 300°C, and/or elevated P(CO2)), olivine reacts with CO2 in fluids to form solid carbonate minerals faster than any other common, rock-forming silicate mineral (see “Carbon Mineralization Kinetics”). In altered peridotites at low temperatures, the mineral brucite also reacts rapidly with CO2 to form solid carbonates. Olivine and brucite undergo rapid carbonation because their solubility in aqueous fluids is moderately high, and intra-mineral diffusion is relatively fast, compared to rock-forming alumino-silicate minerals such as plagioclase feldspars, which are abundant in basalt.
Second, high solubilities and rapid reaction rates for carbon mineralization in olivine and other minerals in mantle peridotite are due, in part, to the fact that they are far from CO2, H2O, and O2 exchange equilibrium with air and surface waters. Where mantle peridotite massifs are exhumed, they constitute an immense reservoir of chemical potential energy that drives rapid, spontaneous reaction and can be converted into both heat (Kelemen and Matter, 2008) and work (Kelemen and Hirth, 2012). Engineered systems that utilize this potential energy could, in principle, be among the least expensive routes to combined CO2 capture from air and solid storage. Proposed methods for engineered carbon mineralization in peridotite involve emulating natural
systems to harness this chemical potential to minimize external energy inputs and costs.
Third, as reviewed by Kelemen et al. (2011) and Kelemen and Manning (2015), carbonate veins are abundant in outcrops of mantle peridotite (Figure 6.11). Peridotite outcrops worldwide are also hosts to alkaline springs, rich in dissolved CaOH-, with little dissolved Mg and C (e.g., Barnes and O’Neil, 1969, 1971; Barnes et al., 1967, 1978; Clark and Fontes, 1990; Falk et al., 2016; Kelley et al., 2001; Launay and Fontes, 1985; Mervine et al., 2014; Neal and Stanger, 1985). Alkaline spring waters are interpreted as products of precipitation of Mg-carbonate minerals during reaction of groundwater with peridotite, together with dissolution of Ca-bearing silicates in peridotite (e.g., pyroxenes and plagioclase). At the surface, alkaline spring water combines directly with CO2 from air to form calcium carbonate (CaCO3) in locally extensive travertine terraces (Figure 6.12). 14C data indicate that most of these large travertine deposits form in less than 20,000 years (Kelemen and Matter, 2008; Kelemen et al., 2011; Mervine et al., 2014; Streit et al., 2012).

SOURCES: Two lefthand images from Kelemen, personal communication, 2017. Top right: Kelemen and Matter, 2008. Bottom right: Falk and Kelemen, 2015.
Fourth, fully carbonated peridotites (listvenites), in which all Mg has combined with CO2 to form carbonate minerals, while Si remains in pure SiO2 minerals (quartz, chalcedony, and/or opal), are exposed in Oman (Falk and Kelemen, 2015; Lacinska et al., 2014; Nasir et al., 2007; Stanger, 1985; Wilde et al., 2002) and elsewhere around the world (e.g., Akbulut et al., 2006; Beinlich et al., 2012, 2014; Boschi et al., 2009; Garcia del Real et al., 2016; Halls and Zhao, 1995; Hansen et al., 2005; Quesnel et al., 2013, 2016; Tominaga et al., 2017; Ulrich et al., 2014). Figure 6.13 illustrates listvenites in Oman. Many of these record replacement at ~ 100°C, within the temperature range where carbon mineralization rates are high. The presence of listvenites reveals that there are natural pathways to complete reaction under such temperature conditions, despite potential negative feedbacks discussed in the “Feedbacks between reaction and fluid flow during in situ carbon mineralization” section.
Fifth, when fully carbonated, initially olivine-rich peridotite (dunite) can incorporate 40 wt% CO2—a 60 percent increase in solid mass, relative to the initial mass of olivine. For example, iron-free olivine (Mg2SiO4, ~ 41 gm/mol) + 2 CO2 (2 × 44 gm/mol) forms two moles of magnesium carbonate (MgCO3) plus one mole of quartz (SiO2). This, combined with the abundance of peridotite within 3 km of Earth’s surface, yields storage reservoirs capable in principle of holding 105 to 108 Gt CO2 in solid form (Kelemen et al., 2011, 2016; Tables 6.1 and 6.2).

SOURCE: Images from Kelemen and Matter, 2008.

NOTE: Inset is a back-scattered electron image of listvenite, in which dark grey areas are magnetite, light grey areas are quartz, and bright grey and white areas are Cr-rich oxide minerals.
SOURCE: Images from Falk and Kelemen, 2015.
The combination of these five factors has sustained basic research on natural and engineered in situ carbon mineralization in peridotite massifs for a decade. However, there have been no field-scale investigations of engineered, in situ carbonation of peridotite. This may be due to several factors, including the lower initial porosity and permeability of peridotite relative to basalt, the lower abundance of peridotite outcrops, and their location far from population centers and point-source CO2 emitters. As a result of this lack of experience, assessment of the potential pros and cons of in situ mineral carbonation in peridotite remains highly speculative, a topic for basic research and initial engineering assessment.
Feedbacks between reaction and fluid flow during in situ carbon mineralization
In natural and engineered systems, rapid and extensive carbon mineralization could be inhibited by negative feedback processes. Loss of CO2 by reaction along the initial stages of a reactive fluid pathway could limit supply of CO2 to rocks more distal from an injection well. Filling of pore space with reaction products may reduce permeability and armor reactive surfaces, forming a solid diffusive boundary layer between fluid and solid reactants. These negative feedbacks may commonly cause peridotite carbonation (and hydration and oxidation) to be self-limiting, preserving lithologies in outcrop that are far from equilibrium with surface conditions.
The reactions outlined in the Introduction all involve large increases in the solid volume, via addition of CO2 (± H2O, ± O2) from the fluid into the solid phases coupled with the low density of solid products relative to solid reactants. If large volumes of other components were dissolved from the rock volume, then perhaps the net change in solid volume change would be small. However, we infer from nearly constant ratios of major cations (Mg/Si, Mg/Fe) in fully carbonated peridotites, compared to CO2-free peridotite reactants that there has been very little dissolution and transport of material out of the rock system (Kelemen et al., 2017), consistent with decades of prior studies (e.g., Coleman and Keith, 1971; Malvoisin, 2015). Near-surface peridotites have fracture-dominated porosity of approximately 1 volume percent. Small increases in the solid volume and precipitation of minerals in pore space, in such a limited porosity network, could have large, negative impacts on permeability, potentially limiting carbon mineralization in peridotite reacting with CO2-rich fluids.2
Nevertheless, natural alkaline springs, formed as a result of subsurface peridotite carbonation and hydration, persist for 10s of thousands to 100s of thousands of years (Früh-Green et al., 2003; Kelemen and Matter, 2008; Kelemen et al., 2011; Ludwig et al., 2006; 2011; Mervine et al., 2014, 2015), indicating that the underlying reactive flow network does not clog or exhaust reactive surface area on this timescale. In addition, observations summarized in the first few paragraphs of this section indicate that complete carbonation, in which all Mg- and Ca-cations combine with CO2, does occur.
A positive feedback mechanism that may explain the persistence of geologically rapid peridotite carbonation, extending over long times and continuing to 100 percent completion, is reaction-driven cracking in which volume expansion due to
___________________
2 The negative feedbacks outlined above are most likely during subsurface carbon mineralization in peridotite using CO2-rich fluid reactants for CO2 storage. In contrast, clogging of porosity may be less important for slow uptake of CO2 from circulating surface waters, in methods for CO2 removal from air and solid storage.
carbonation causes large differential stresses, which in turn cause fractures, maintaining or enhancing permeability and reactive surface area (Figure 6.14; Jamtveit et al., 2008; 2009; Kelemen and Hirth, 2012; MacDonald and Fyfe, 1985; O’Hanley, 1992; Rudge et al., 2010; Ulven et al., 2014a, 2014b; Zhu et al., 2016). In experiments on olivine powder, as noted above, the lack of passivation (Figure 6.3) may be a very small-scale result of the reaction-driven cracking process, in which observed layers of amorphous SiO2 on olivine surfaces (Béarat et al., 2006; Chizmeshya et al., 2007) fracture and spall off dissolving grains, or are “pushed” from the surface by precipitating reaction products. Available chemical potential energy to drive reaction-driven
NOTES: Blue rock reacts with black fluid to form white solid products, with volume increase and associated stress concentrations forming red cracks. In turn, cracks provide rapid fluid access deeper into the rock. This discrete element model provides clear simulations of fracture formation at the grain scale, but does not incorporate Darcy flow within the porous fracture network.
cracking in peridotite undergoing carbonation or hydration is large, more than sufficient to fracture rocks (Kelemen and Hirth, 2012). The overall concept seems simple enough, and this process has been observed in peridotite carbonation experiments (Zhu et al., 2016).
Nevertheless, in other experimental tests of olivine carbonation and hydration, volume change and fractures were not observed (van Noort et al., 2017), and permeability dropped with increasing reaction progress (Andreani et al., 2009; Godard et al., 2013; Hövelmann et al., 2012). The reasons for this are not yet clear. It is becoming apparent that micro- and nano-scale properties of fluid-rock systems, such as fluid-mineral surface energy, and related characteristics such as sorptivity and disjoining pressure (Evans et al., 2018; Lambart et al., 2018; Zheng et al., 2018) may play a significant role in locating the crucial bifurcation between self-limiting negative feedbacks (clogging) and accelerating, positive feedbacks (cracking) (Figures 6.15 and 6.16). In addition, viscous and/or frictional dissipation of stress, for example in weak reaction products such as gypsum (Skarbek et al., 2018) and brucite (Moore and Lockner, 2004, 2007; Morrow et al., 2000; Zheng et al., 2018), may also reduce crystallization pressure before stresses due to volume expansion become high enough to generate new fractures.
Other processes—such as selective, local dissolution and precipitation processes (Lisabeth et al., 2017; Peuble et al., 2018) and/or crack propagation from etch pits along dislocation boundaries and other defects in olivine crystals (Daval et al., 2011; Grozeva et al., 2017; Klein et al., 2015; Lisabeth et al., 2017; Malvoisin et al., 2017; McCollom et al., 2016; Plümper et al., 2012; Rouméjon and Cannat, 2014; Velbel, 2009)—may also play a role in sustaining permeability and fluid flow. Perhaps complete carbonation in natural systems is relatively slow, and thus cannot be engineered on a human time scale, as suggested by van Noort et al. (2017). However, in the competition between (1) volume expansion and stress accumulation and (2) processes that relax elastic stresses such as viscous flow or frictional sliding along existing fractures, it seems likely that reaction-driven cracking happens when the rates of reaction and volume change are maximized.
In summary, despite the initial, apparent simplicity of the feedbacks in peridotite carbonation, understanding them and developing predictive hypotheses validated by experimental and field observations is an increasingly complex and interesting research field. And again, continued research on this topic is justified by geologic observations of fully carbonated peridotite. If natural systems can do it, it is likely to be possible to design engineered systems that emulate this process. Moreover, understanding of the feedbacks that lead to reaction-driven cracking could be applied to geothermal power generation, in situ mining for, for example, uranium, and extraction of oil and gas

SOURCE: Evans et al., 2018.
SOURCE: Evans et al., 2018.
from tight reservoirs, as well as to CO2 capture and storage. In other applications, it is desirable to prevent reaction-driven cracking, for example in well-bore cement and in caprocks above storage reservoirs for supercritical CO2 fluid. Ongoing research seeks to outline a “phase diagram” delineating the conditions for reaction-driven cracking, and the surrounding parameter space dominated by clogging and armoring of reactive surfaces.
Solid storage of CO2 via in situ carbon mineralization in peridotite
Kinetic data summarized in the “Carbon Mineralization Kinetics” section yield empirical predictions of olivine carbonation rates (mass fraction olivine ≤ 75 microns) such as:
| G = 1.15 10-5 (P(CO2) bars)1/2 exp[0.000334(T°C-185)2] | (Kelemen and Matter, 2008; further validated in Gadikota et al., 2014) |
Rather than guess the effective grain size (and/or fracture spacing) in natural peridotite aquifers, we use observed rates in the near-surface weathering horizon as a calibration point, calculate relative rate enhancements using this expression, and derive a scaled rate from the product of the observed rate and the relative rate enhancement. This approach indicates that olivine-rich peridotite could, in principle, consume more than 1 Gt CO2/km3 peridotite/y, at temperatures greater than approximately 150°C, and CO2 partial pressures greater than approximately 60 bars (Kelemen and Matter, 2008). Even at somewhat lower temperatures, approximates 100°C, carbon mineralization in peridotite reacting with high P(CO2) fluids could achieve rates of 300 Mt CO2/ km3/y. For drilling costs of $3M to $6M, and compression costs of $10/t of injected fluid, over 10 years this process could lead to solid storage3 of 3 Gt CO2 at $10-20/t ton. Figure 6.17 illustrates how solid storage of 3 Mt/y CO2 might be achieved at this cost, by injecting CO2-rich fluid into a rock volume of 107 m3 (10-2 km3) surrounding a single 3 km deep borehole.
CO2 removal from air and solid storage via in situ carbon mineralization in peridotite
CO2 removal from air, in addition to solid storage, may be achieved using engineered methods that closely emulate natural carbon mineralization in peridotite. During
___________________
3 Although the term “storage” might imply accumulation for future use, the committee uses this term interchangeably with the term “sequestration” in accordance with the literature reviewed.
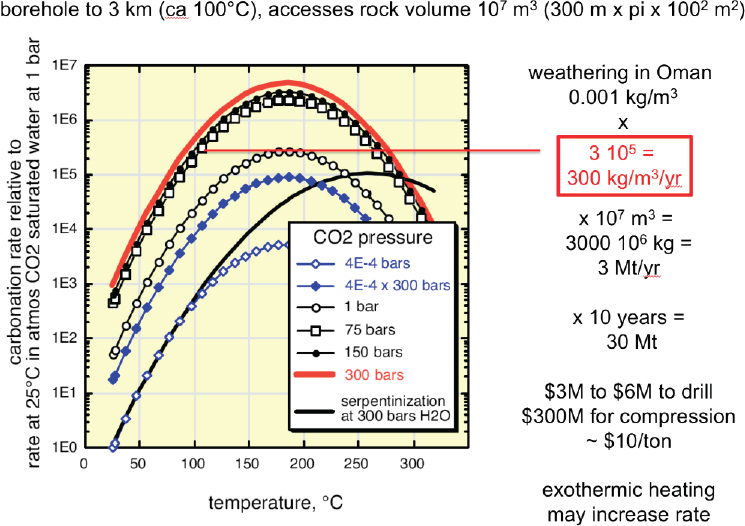
NOTES: Values of rate enhancement relative to the rate at 25°C in air, due to elevated temperature and P(CO2), are from Kelemen and Matter, 2008, fit to data of O’Connor et al., 2005, consistent with experimental data of Gadikota et al., 2014. Rate of peridotite carbonation due to weathering in Oman is from Kelemen and Matter, 2008 and Kelemen et al., 2011. Drilling costs from, e.g., Augustine et al., 2006, Bloomfield and Laney, 2005, and Shevenell, 2012. CO2 compression costs from, e.g., McCollum and Ogden, 2006 and Rubin et al., 2015.
weathering, shallow groundwater, equilibrated with atmospheric CO2, reacts with peridotite in the subsurface, in a system closed to CO2 exchange with the atmosphere. This quickly reduces dissolved carbon concentrations to zero, via precipitation of Mg- and Ca-carbonate minerals in veins (Figure 6.11). Along the reaction path, pH rises to 11.5 or more, and the concentration of dissolved Ca2- increases to approximately 400 ppm. When these alkaline waters reach the surface, they combine with atmospheric CO2 to form calcite, CaCO3, in some places creating extensive travertine deposits (Figure 6.12; e.g., Barnes and O’Neil, 1969; Barnes et al., 1978; Clark and Fontes, 1990; Kelley et al., 2001; Launay and Fontes, 1985; Neal and Stanger, 1985).
Engineered CO2 removal from air could begin with production of C-depleted alkaline water from existing aquifers in peridotite, to form travertine at the surface. The amount of CO2 that could be captured via this simple and relatively inexpensive method is uncertain, because the size, permeability, productivity and recharge rates of
alkaline, peridotite-hosted aquifers are unknown. This represents an obvious, relatively low-cost research opportunity.
In parallel, alkaline aquifers could be replenished via enhanced circulation of surface water through subsurface peridotite formations. For this method, it is important to consider the cost of pumping fluid. Because of the dilute concentration of carbon-species in water equilibrated with air at 1 bar (~100 ppm CO2), every penny spent pumping water corresponds to approximately $100/tCO2. Thus, it may be best to rely on thermal convection to drive fluid circulation. In this context, the temperature contrast between surface water and the target aquifer, and the permeability of the subsurface peridotite aquifer, are critical variables. Where thermal convection is sufficient to drive circulation (DT > 50°C, permeability > 5 10-13 m2), drilling expenses similar to those for geothermal wells, amortized over a 20-year lifespan, yield highly approximate, estimated costs of less than $100/tCO2 (Kelemen et al., 2016).
In lower permeability formations (permeability < 5 10-14 m2) added costs due to the energy requirements for pumping fluid rise above $1,000/t. Such high costs would render CO2 capture via circulation of surface water impractical. An exception could be co-located geothermal power generation and CO2 capture. Large geothermal power plants commonly employ pumps at the surface and within boreholes, optimizing the flow rate to generate maximum electricity at a minimum pumping rate. In this mix, carbon offsets for CO2 capture could increase revenues directly, and/or allow additional pump pressure, more rapid fluid flow rates, and additional electrical generation.
Individual injection wells probably can capture a maximum of approximately 1,000 tCO2/y via carbon mineralization without significant pumping. Capturing gigatons of CO2 is possible, simply via drilling more wells. The number of wells required to capture 1 Gt/y CO2 in this way is approximately equal to the number of operating oil and gas wells in the United States. However, there are no obvious economies of scale that would reduce the cost of capture per ton.
Some key uncertainties require medium-scale field testing. In particular, the evolution of reaction progress and permeability during flow of fluid in a crystalline rock aquifer containing hierarchical fracture networks is difficult to predict. While modeling efforts are advisable, there really is no substitute for experiments at a scale that is tens to hundreds of times larger than the spacing of key fracture sets.
As is evident from Tables 6.1 and 6.2, the potential reservoir for CO2 capture and storage in peridotite formations is enormous, with a capacity greater than 105 Gt CO2. Both rates and costs of CO2 capture from subsurface circulation of water saturated in air are potentially competitive with other methods to achieve CO2 removal from
air, and as a consequence proposed methods warrant intensified, basic research to delineate the conditions for cracking rather than clogging, and to determine a variety of important physical properties of fractured, partially serpentinized, subsurface peridotite. Additionally, field tests should shed light on the potential for contamination of local water supplies,4 induced earthquakes, and other negative impacts.
If pathways can be found to positive feedback regimes—in which carbon mineralization causes small-scale fractures that maintain or enhance permeability and reactive surface area—then the cost of in situ carbon mineralization in peridotite could be relatively low (e.g., Figure 6.17), similar to estimated costs for nth-of-a-kind basalt carbonation processes similar to CarbFix, and approximately the same as for injection of CO2 into pore space. Where peridotite is abundant, then, this could be the optimal storage method for CO2. And finally, as for direct air capture via in situ carbon mineralization driven by circulation of surface water through peridotite, in situ CO2 storage via injection of CO2-rich fluids into peridotite could be combined with geothermal power generation.
In the lower 48 United States, potential sites for in situ CO2 storage in peridotite are abundant near both coasts (Krevor et al., 2009). In order to realize enhanced reaction rates at elevated temperature (Figure 6.18), areas of high heat flow are preferred, and these are localized in the western states. One of the largest peridotite massifs in North America, the Trinity peridotite in northern California, dips beneath the Cascade volcanic front (Fuis et al., 1987), an area of high heat flow with elevated temperatures at shallow depth (Bonner et al., 2003; Ingebritsen and Mariner, 2010), ideal for both geothermal power production and carbon mineralization in peridotite. Similarly, smaller bodies of peridotite flank the geothermal area in the Geysers region of northern California (Sadowski et al., 2016), near the Calpine power plant, which is the largest geothermal power plant in the world.
IMPACT POTENTIAL
Geologic storage of CO2 in ultramafic, mafic, and sedimentary formations has the potential to accommodate 10s of billions of tons of CO2 per year, and ultimately store 1000s of trillions of tons. This chapter has focused on geologic storage in ultramafic formations, and Chapter 7 discusses storage in sedimentary formations. Together with increasing the carbon content of soils and the oceans, geological storage is one of a
___________________
4 Preliminary data indicate concentrations of dissolved metals such as Ni and Cr, which are abundant in peridotite, are very low in peridotite-hosted aquifers, well within EPA safe drinking water limits (Amelia Vankeuren, personal communication, 2016).
NOTES: In addition to these U.S. resources, peridotite in blocks of oceanic crust and mantle thrust onland (ophiolites) is found on all continents, with a total peridotite mass between 1014 and 1015 tons within 3 km of the surface. The largest is the “Samail ophiolite,” in the Sultanate of Oman and the United Arab Emirates. It contains approximately 5 × 1013 tons of peridotite within 3 km of the surface (Kelemen and Matter, 2008). Other individual ophiolites including peridotite bodies with a similar scale are in New Caledonia, Papua New Guinea, and Albania. The gigantic Bushveld igneous layered intrusion in South Africa contains more than 1013 tons of peridotite and is very close to a district of intensive coal mining and coal-fired power generation, supplying electricity to most of the country. The similar Stillwater intrusion in Montana contains almost 1012 tons of peridotite. Continental mantle peridotite massifs (exclusive of ophiolites), including the Beni Boussera massif in Morocco and the Ronda massif in southeastern Spain, have a combined mass of about 1012 tons.
SOURCE: Krevor et al., 2009.
few options that could accommodate CO2 removed from air at this (required) scale. Processes that involve billions of tons of fluid and rock inevitably have a substantial impact. It remains to be seen whether society is willing to tolerate these impacts in order to mitigate past and continuing greenhouse gas emissions.
Although greenhouse gas mitigation is the primary target, carbon mineralization has secondary benefits. In particular, the development of engineered methods to control feedbacks between chemical and physical processes could have other important applications, such as extraction of oil and gas from tight reservoirs, in situ solution mining (e.g., for uranium), and production of geothermal power. In addition, in some cases ex situ carbon mineralization acts to mitigate environmental hazards. For
example, fibrous asbestos poses a significant health hazard where it is present at the surface, as in mine tailings. In this context, carbonation of chrysotile asbestos reduces the hazard. Similarly, carbonation of some kinds of alkaline industrial wastes can significantly ameliorate the risk of chemical contamination. Moreover, there is a small but robust market for magnesium carbonate, MgCO3, which is sold for $100-1,000/t depending on purity.
All of the potential negative impacts of carbon storage in sedimentary formations—particularly contamination of water resources and induced earthquakes (see Chapter 7)—may also apply to carbon mineralization projects. Preliminary data suggest that the risk of water contamination risk is small. For example, Ni and Cr concentrations in natural carbon mineralization systems are orders of magnitude lower than U.S. Environmental Protection Agency (EPA) limits for safe drinking water (Amelia VanKeuren, personal communication, 2017). Whether this observation holds true for engineered systems will need to be tested. Earthquakes may be triggered because in situ subsurface carbon mineralization increases the solid volume, increasing stresses that could cause fracture. Theoretical considerations and observations of the extent of naturally formed carbonate veins suggest that most reaction-driven fracture events are likely to be small, magnitude 1 or less. As a general rule, the size of fracture events should be limited by the low yield strength of fractured rocks within a few km of Earth’s surface, which is the likely target depth for in situ carbon mineralization. However, larger scale deformation associated with Mt/y-scale CO2 injections could possibly lead to larger-scale failures and will need to be modeled and monitored.
SUMMARY: COST AND CAPACITY OF CARBON MINERALIZATION METHODS
This chapter outlined several proposed methods for engineered acceleration of natural carbon mineralization processes, to achieve either solid storage of CO2, or combined mineral capture from air and storage. Here we summarize the data in Tables 6.1 and 6.2 to provide a concise evaluation of the cost and capacity for various proposed methods, as compared to direct air capture systems and/or storage of supercritical CO2 fluid in pore space.
Most ex situ methods for solid storage via carbon mineralization—using fluids enriched in CO2 compared to air and water in CO2 exchange equilibrium with air—are significantly more expensive than storage of supercritical CO2 fluid in subsurface pore space. However, in some cases carbon mineralization acts to mitigate environmental hazards, such as asbestos in ultramafic mine tailings, and toxins in alkaline
industrial wastes, which may add value. Research on utilization of reaction products, for example for building materials, or in the supply chain for conversion of CO2 to CH4 and more complex hydrocarbons, is ongoing. Carbon capture, utilization, and storage is the subject of a separate National Academies study on “Developing a Research Agenda for Utilization of Gaseous Carbon Waste Streams.”5
Surficial and in situ methods of solid storage, using fluids enriched in CO2, may be cost competitive with storage in pore space (Figure 6.19). They may also have some potential advantages because storage is in permanent, inert carbonate minerals with little risk of groundwater contamination, and may be a regionally appropriate technology.
Combined mineral capture from air and solid storage, via surficial processes using existing ultramafic mine tailings, could be a relatively inexpensive and straightforward technology, but has a limited storage capacity. Current, highly approximate cost estimates suggest that mining, crushing, and perhaps additional milling of appropriate lithologies—with a focus on mantle peridotite—for the purpose of mineral capture from air and storage via enhanced weathering may be cost-competitive with direct air capture systems, within the uncertainties for cost estimates for each type of process (Figure 6.20).
In situ mineral capture and storage is potentially cost-competitive with direct air capture systems, and offers a gigantic storage potential, if negative feedbacks such as clogging can be avoided, and positive feedbacks such as reaction-driven cracking can be harnessed.
All of these avenues for mitigating CO2 emissions via carbon mineralization warrant continued, accelerated research programs, including laboratory experiments, numerical modeling, investigation of social and regulatory factors, and pilot projects in the United States.
RECOMMENDED RESEARCH AGENDA
Carbon Mineralization Kinetics
Comparison of rates for different mineral, rock, and synthetic solid reactants is hampered by a lack of consistency in experimental methods. Comparative studies of carbonation rates for different materials, at the same temperature, pressure, P(CO2), grain size, surface area, and fluid composition, remain a high priority that is achievable
___________________
5 See http://nas-sites.org/dels/studies/gcwu (accessed January 28, 2019).
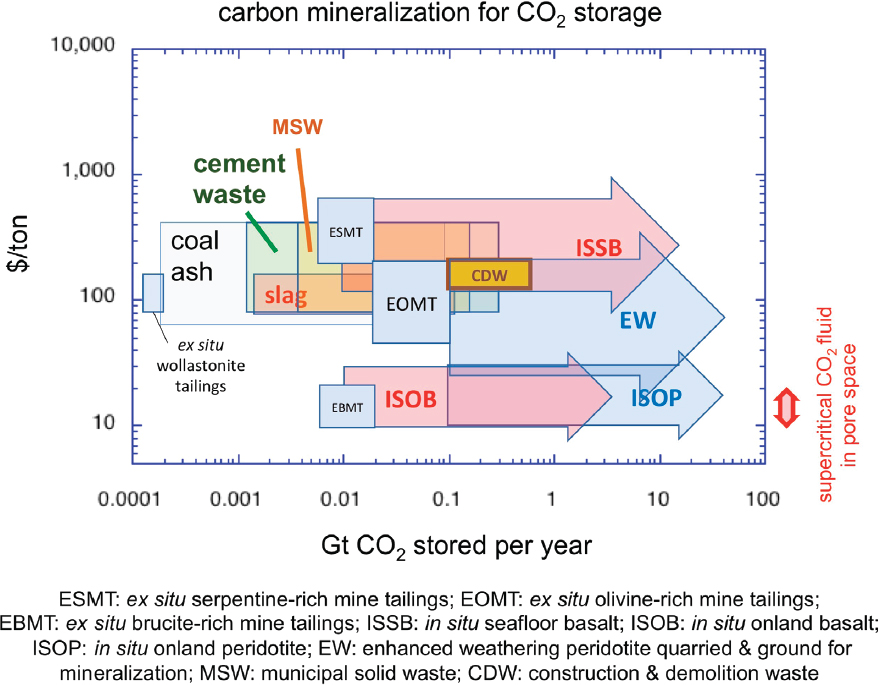
NOTE: Costs should be compared to the cost of storage of supercritical CO2 in subsurface pore space, ~$10-20/tCO2 (Chapter 7).
at low cost. Our compilation is missing key data for carbon mineralization rates in alkaline industrial wastes. A compilation of experimental data for these materials, allowing comparison using units such as mol/(m2 s), or mass fraction per second at a common grain size, is a worthy research goal in itself, and would likely reveal a need for additional, comparative experimental studies.
Ex Situ Carbon Mineralization
Laboratory studies of ex situ carbon mineralization processes for CO2 storage have been extensive and are ongoing. Due to the relatively high cost of ex situ mineralization compared to storage of supercritical CO2 in pore space (Table 6.1), the focus on ex situ methods in the past decade has been on producing economically valuable commodities as well as storing captured CO2. Carbon capture, utilization and storage
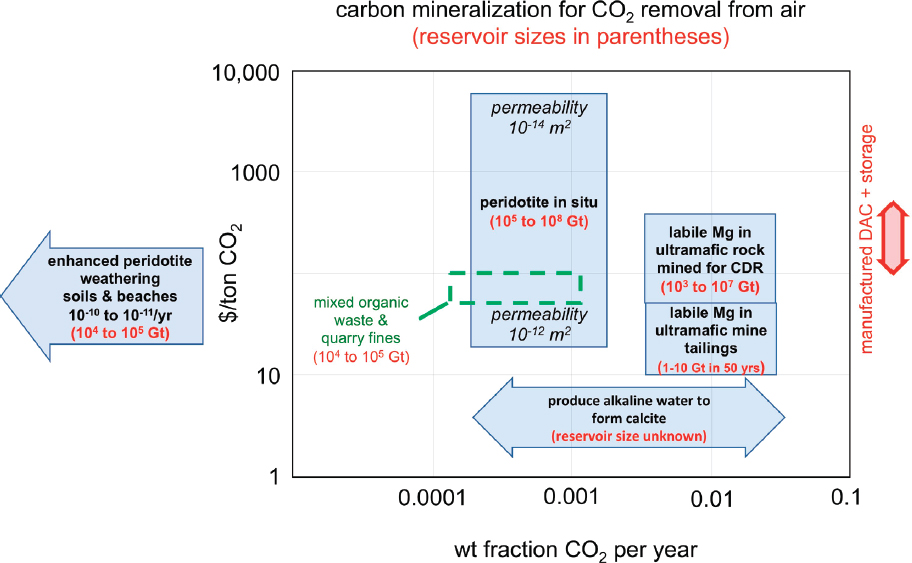
NOTE: The size of potential storage reservoirs is indicated in parentheses for each proposed method. Costs are compared to the cost of direct air capture systems, ~$100-600/t CO2 (Chapter 5).
is the subject of a separate National Academies study on “Developing a Research Agenda for Utilization of Gaseous Carbon Waste Streams.”6 Creation of carbon-added, value-added aggregate for concrete is one area of active work, as is air-to-fuels via conversion of captured CO2 to hydrocarbons. Rock reactants, particularly mantle peridotite, could potentially be used to achieve this conversion (e.g., McCollom et al., 2010), although direct air capture technologies may be a less expensive source of CO2. Research in these directions is partly supported by the Department of Energy (DOE), and there seems to be substantial grant and venture-capital support.
___________________
6 See http://nassites.org/dels/studies/gcwu (accessed January 28, 2019).
Surficial Carbon Mineralization in Mine Tailings, Industrial Wastes, and Sediments
Based on extensive research by a few groups focused on this topic, surficial carbon mineralization for solid storage or for mineral capture and storage is ready for kiloton to megaton per year field experiments, together with extensive field inventories and laboratory characterization of the reactivity of various potential solid reactants. Field experiments on mine tailings and industrial wastes may be particularly good opportunities for university-industry and/or government-industry partnerships, as illustrated by a recently initiated collaboration involving the diamond-producer DeBeers and several academic research groups (Mervine et al., 2017). Significant limitations are the relatively small mass of accumulated and annually produced of mine-tailings, and the relatively limited CO2 storage capacity of industrial wastes with low concentrations of Mg and Ca. More effort needs to be focused on potential effects of geochemical contamination, because in many cases implementation is likely to be close to surface water and groundwater resources. In the United States, carbonation of ultramafic mine tailings could be combined with efforts to reduce asbestos hazard at these same sites. Carbonation of industrial wastes to mitigate hazards is of potential interest to EPA and DOE.
A basic research area with high potential, but whose effectiveness and cost are currently impossible to assess, is microbial acceleration of carbon mineralization in mine tailings (e.g., Power et al., 2013a). This should be funded as laboratory-based investigation by several research groups.
Another research priority concerns optimization of various pretreatment strategies, including heat pretreatment, grinding/comminution processes, and new technological approaches such as microwave treatment, to increase the rapidly reacting labile Mg content of tailings and/or to reduce process costs per ton of CO2. Such approaches are currently used to enhance metal extraction but could also be followed to enhance carbon mineralization. For example, grinding to separate small sulfide or metal oxide minerals for liberation and flotation may exceed the amount of grinding needed to access labile Mg. Thus, it may be possible to reduce the cost and energy intensity of quarrying and grinding rock for carbon mineralization alone. Moreover, where the goals are both metal extraction and carbon mineralization, parts of ore deposits with high labile Mg concentrations could be mined, even if these include rock volumes with a relatively low ore grade. Industry-academic partnerships, with research funding from the National Science Foundation (NSF), seem best suited to address such optimization issues.
Progress on verification protocols and regulatory and pricing frameworks is another priority. Because of recent, damaging failures of tailings dams in the United States and Canada, the current regulatory and social environment discourages innovation in the design of tailings storage facilities. Creating a safe research space to explore innovation could ultimately lead to safer and more efficient operations. Funding could come from current and prospective industry partners.
Some research groups have begun investigating hybrids of ex situ methods (bringing solid reactants to the site of CO2 capture) and in situ methods (transporting captured CO2 to large rock reservoirs). Work has focused on local modifications to ultramafic mine tailings, particularly mine tailings from highly altered serpentinites that include abundant brucite (and asbestiform chrysotile) (Alt et al., 2007; Assima et al., 2012, 2013a, b, 2014a, b, c, d; Bea et al., 2012; Gadikota et al., 2014; Hansen et al., 2005; Harrison et al., 2013, 2015, 2016; Larachi et al., 2010, 2012; McCutcheon et al., 2014, 2015; Mervine et al., 2017; Power et al., 2007, 2009, 2010, 2011, 2013a, b, c, 2016; Pronost et al., 2011, 2012; Sarvaramini et al., 2014; Thom et al., 2013; Wilson et al., 2006, 2009a, b, 2010, 2011, 2014). Suggested modifications are as simple as stirring tailings, depositing them in thinner layers, and sprinkling water on them to transport atmospheric CO2 deeper into the pile for direct air capture. Other modifications have been more innovative, such as introducing CO2-rich gas or fluid into slurry pipes that transport tailings. Two recent studies demonstrate the potential for up to a million-fold acceleration of carbon mineralization rates by sparging CO2-rich gas through mine tailings (Assima et al., 2013a; Harrison et al., 2013). Their experiments achieved rates at ambient surface temperatures that approach or exceed the highest laboratory rates for carbon mineralization at elevated temperature and pressure, at least for CO2 uptake of 3 to 10 wt%. This is a promising avenue of research, which should be pursued in both laboratory experiments and field-scale pilot studies at several mine sites. Similar techniques might also be applied to alkaline industrial waste heaps.
Carbon mineralization in crushed ultramafic materials may be viable in peridotite-rich alluvial gravels, which have high surface area to volume ratios and are present along some tectonic plate boundaries. A study of one ancient deposit found that it was already extensively carbonated (Beinlich et al., 2010). However, other basins with large volumes of peridotite gravel should be investigated to determine the extent to which they offer potential reactants for engineered carbon mineralization. For example, peridotite-rich sediments derived from mechanical weathering of the Samail ophiolite in Oman and the United Arab Emirates are present in km-thick formations beneath the Batinah coastal plain (e.g., Al Lazki et al., 2002) and in the extensive Barzaman Formation south and west of the ophiolite (Lacinska et al., 2014; Radies et al., 2004; Styles
et al., 2006). While some of the ultramafic clasts in these rocks are extensively altered, subsurface exploration for less altered, more reactive potential CO2 reservoirs in these settings is warranted. In the United States, this exploration might be best undertaken by the USGS.
In Situ Carbon Mineralization in Basaltic Lavas
The Wallula and CarbFix projects have established the viability of combined CO2 storage in pore space and carbon mineralization in basaltic lavas. In particular, the strategy used at CarbFix, combining solution trapping of CO2 dissolved in water with rapid carbon mineralization, demonstrates that this combination need not consume huge volumes of water, as many feared, and opens the opportunity for extensive CO2 storage in relatively accessible, easily characterized, near-surface basalt formations.
The logical next step is megaton per year experiments in the United States. To date, the emphasis has been on the large volume Columbia River and East Coast Triassic Flood Basalt Provinces (Figure 6.10). In addition, the volcanic Cascade volcanic arc, especially in Oregon and northernmost California, may offer reservoirs with high storage potential. Potential basalt reservoirs will have layers that are rich in highly reactive, amorphous volcanic glass, have high permeability, and have relatively high temperatures at shallow depth. Ideally, these layers will be bounded by low permeability barriers, with little faulting or deformation. However, an impermeable caprock to prevent leakage is not required for the solution trapping strategy used at CarbFix. This strategy might be appropriate for shallow, glassy lava flows that are more reactive than most flood basalt formations. In moving from small- to medium-scale pilot experiments, research groups will have to focus on potential chemical contamination of nearby aquifers and surface waters, and on the risk of induced earthquakes.
This extensive scoping—focused on the reactivity and capacity of reservoir rocks and the availability of impermeable cap rocks—should be funded by DOE and USGS, potentially in collaboration with state governments and/or industry. Medium-scale pilot projects are likely to cost 10s of millions of dollars per year, with substantial funding coming from DOE (Appendix F provides a schematic budget for such a project). States that have implemented carbon taxes or offer other incentives for carbon management may be willing partners, together with industries seeking offsets in these states. Once sites for pilot experiments are chosen, it will be important to implement the lessons of CarbFix in designing tracer studies. Experiments should focus on optimization of one- and two-phase injection strategies, particularly for shallow reservoirs where solution
trapping is necessary. Longer duration, higher flux experiments should examine evolution of subsurface reaction fronts, determine the nature of local carbon mineralization reactions (i.e., what minerals are reacting, at what grain sizes, and at what rates?), and especially feedbacks affecting permeability and reactive surface area. Recommended laboratory experiments and numerical modeling studies to address these feedbacks for in situ carbon mineralization in basaltic lavas are essentially identical to those for in situ mineralization in ultramafic rocks (see the next section, “In situ Carbon Mineralization in Ultramafic Rocks”).
To date, there has been little interest in combined mineral capture and storage in basalt reservoirs, probably, because there are few examples of extensive, natural carbon mineralization in basalt at low- temperature, near-surface conditions, and because laboratory studies have found slower carbon mineralization rates in basalts compared to ultramafic rocks such as mantle peridotites. (An exception is glassy basaltic lavas.) As a result, pilot studies in most basalt formations will be focused on injection of CO2-rich fluids to achieve a combination of storage in pore space and solid storage. Because injection of CO2 is subject to federal regulations governing Class VI wells, extensive site characterization, monitoring, and post-injection site closure operations will be required.7
Experimental carbon mineralization rates and capacities for different combinations of basalt types and fluid compositions vary widely. Quantifying the controls on basalt carbonation rates is another high priority, comparatively low-cost opportunity for laboratory research, which should be largely supported by DOE.
In Situ Carbon Mineralization in Ultramafic Rocks
In situ carbon mineralization in ultramafic rocks, mainly in large massifs of tectonically exposed mantle peridotite, represents a high-risk, high-reward opportunity, requiring extensive basic research to evaluate its practicality, combined with one or more small-scale field experiments over the next decade.
Ultramafic rock tends to have low porosity and extensive fracturing. These characteristics create the potential for important feedbacks between carbon mineralization and permeability. Consequently, extensive studies of chemo-mechanical processes are required to determine the conditions favoring reaction-driven cracking and other positive feedbacks, as well as the conditions leading to clogging of pore space and passivation via armoring of reactive surfaces. A crucial research goal is to create a
___________________
7 See https://www.epa.gov/uic/class-vi-guidance-documents (accessed January 28, 2019).
“phase diagram” delineating the conditions favoring positive and negative feedbacks for carbon mineralization in ultramafic rocks.
If reaction-driven cracking were well understood, it would be possible to engineer conditions that generate ramified fracture networks at the grain scale. Such approaches could be valuable for a variety of technologies, including CO2 capture and storage, geothermal power generation, in situ mining, and extraction of oil and gas from low permeability reservoirs. Similarly, avoiding reaction-driven cracking is important for ensuring the long-term integrity of impermeable cap rocks and well cement in boreholes for subsurface CO2 and hydrocarbon reservoirs.
Increasingly, researchers are realizing that nano-scale material characteristics (mineral fluid surface energy, sorptivity, disjoining pressure) may play a key role in controlling the bifurcation between clogging and cracking. Because these characteristics vary in unpredictable ways from one material to another, research is moving away from simple analog systems (hydration or carbonation of CaO, MgO, and CaSO4) toward experiments involving the most geologically relevant rock formations (e.g., peridotite and basalt for CO2 capture and storage, shale for oil and gas, and sandstone for uranium). A growing community is working on these topics, inspired in part by the problems and promise of in situ mineral carbonation, and the dialog among different research groups around the world promises to be productive. U.S. participation and leadership in this emerging basic science field deserves continued, focused support from NSF and DOE.
The above topics are ideal for laboratory and numerical modeling approaches and will be particularly fruitful where the two are combined and constrained using field observations of natural systems. Both NSF and DOE fund basic research on feedbacks during reactive fluid transport. Expanded support from NSF and DOE would enable the integration of laboratory, modeling, and field approaches.
In parallel with laboratory research and numerical modeling, we recommend funding for two or three small- to medium-scale (~1 to 100 kt/y) field experiments. Such experiments would investigate the viability of in situ carbon mineralization during (a) mineral capture and storage of CO2 via circulation of surface water through peridotite and (b) solid storage of CO2 via circulation of CO2-rich fluids through peridotite. They could also be used to optimize engineered methods to achieve rapid carbon mineralization and minimize negative feedbacks.
We envision multistep pilot projects on in situ carbon mineralization in ultramafic rocks, with gradually increasing cost, ambition, and risk and funded by DOE, USGS, and/or state sources, ideally in combination with industry partners. Such projects will first require scoping efforts—characterizing physical properties and the suitability of
rock formations for possible pilot projects and assessing the long-term potential for significant CO2 storage reservoirs. Scoping will also include a significant component of policy research, public outreach, and investigation of political and social factors in nearby communities and regions.
Assuming that geologically and socially appropriate sites can be identified, a next step would be to produce water from existing, alkaline, carbon-depleted aquifers for direct uptake of CO2 from air to form carbonate minerals in travertine deposits on the surface, and dissolved bicarbonate in surface water. The size, permeability, productivity, and physical and chemical recharge rates of such aquifers are unknown and could readily be evaluated for several sites at a relatively low cost. Experiments could then progress to injecting recycled water into peridotite aquifers, with continued characterization of injectivity, permeability, volume, and subsurface rates of subsurface CO2 mineralization. Pending a successful outcome of this step (CO2 removal from circulating fluids, no sustained decrease in permeability), experiments could progress to investigating deeper circulation into hotter rock formations, with faster carbon mineralization rates. A final set of experiments could include injecting fluids with high CO2 concentration to evaluate proposed storage of CO2 captured elsewhere. The steps involving production and re-injection of groundwater depend on state and local regulations but might be relatively straightforward to implement. Because injection of fluids enriched in CO2 is subject to federal regulations governing Class VI wells, the final set of experiments would require significantly more site characterization, monitoring, and post-injection site closure operations.
The likely cost of such a phased experiment, carried to completion, is estimated as $10-20M/y, based on the costs of CarbFix Phase I (Aradóttir, personal communication, 2017) and a notional budget for a large-scale experiment on CO2 storage in basalt (Appendix F, Table F.3). Because carbon mineralization rates in peridotite are optimal at ~185°C, there is potential synergy between carbon mineralization and geothermal power generation. The best region to explore this synergy in the United States is northern California, where carbon management incentives and an active geothermal industry might facilitate combined DOE, state, and industry participation.
Improved Public Dissemination and Impact Assessment
Beyond the research to advance understanding of basic scientific processes and develop pilot-scale tests of carbon mineralization technologies for CO2 removal and storage, the committee identified three other areas in need of additional research. Included in the research agenda is the call for the development of a database for
carbon mineralization to ensure that the results of the research activities are disseminated broadly to the research community. Further, research is also needed to examine the social and environmental impacts of an expanded extraction industry to meet the needs of scaling up these NETs. Finally, the committee believes research is needed to assess the environmental impacts of minerals additions to terrestrial, coastal, and marine environments. This research would be shared with the research agenda for terrestrial carbon removal and sequestration (Chapter 3).
Cost of the Research Agenda
There is far less experience and data on even kiloton-per-year storage of CO2 via carbon mineralization, let alone megaton- and gigaton-per-year processes, compared to experience and data on storage of supercritical CO2 in deep sedimentary formations. This is true largely because the economic incentives and technical expertise for injecting supercritical CO2 for enhanced oil recovery are not matched by opportunities associated with carbon mineralization. Significant decadal progress on this research agenda requires a mix of basic and applied research aimed toward the same overall outcome (Table 6.3).
Implementation of the Research Agenda
While the research topics enumerated above appeal to research scientists, it can be difficult to obtain federal grant support for such studies, in part because the research falls between the perceived domains of the NSF Directorate for Geosciences, where many of the research topics may be viewed as “too applied,” and DOE, where the same topics may be viewed as “too theoretical.” Some DOE programs bridge the gap between basic and applied science, but funding flows primarily to the National Laboratories. An example is DOE’s Crosscutting Subsurface Technology and Engineering Research, Development, & Demonstration (SubTER) initiative,8 which covers National Laboratory investigations on hydraulic fracture and induced earthquakes related to oil and gas extraction. Developing a similar grants program at DOE would enable univ ersity research on cracking versus clogging during reactive transport of fluids in fractured porous media, chemo-mechanical feedback during carbon mineralization, geothermal energy production, and in situ solution mining.
___________________
8 See https://www.energy.gov/subsurface-science-technology-engineering-and-rd-crosscut-subter (accessed January 28, 2019).
TABLE 6.3 Carbon Mineralization Research Agenda Budget
| Typical Academic Grants (Approximate annual amount $500K) | National Lab and USGS Projects (Approximate annual amount $1.5M) | Cost/y | Years | Total | Barriers the research will address, or new frontiera | |
|---|---|---|---|---|---|---|
| Basic Research | ||||||
| Kinetics | 5 | 2 | $5.5M | 10 | $55M | STU: experiments at common T, P(CO2), fluid composition to establish quantitative framework and enable comparisons and optimization. |
| Rock mechanics | 6 | 2 | $6M | 10 | $60M | Frontier: exploration of positive and negative feedbacks between reaction and fluid flow, for in situ carbon mineralization, in situ mining, geothermal power generation, extraction of oil and gas from tight reservoirs, ensuring integrity of reservoir caprock and wellbore cement. |
| Numerical modeling | 6 | 2 | $6M | 10 | $60M | As above |
| Field studies | 4 | 2 | $5M | 10 | $50M | As above |
| Scoping for pilot sites | 0 | 5 | $7.5M | 5 | $37.5M | STU, OENV, G |
| Development of a resource database for carbon mineralization | 4 | 0 | $2M | 5 | $10M | STU, PBSU |
| Examining the social and environmental impact of an expanded extraction industry for the purpose of carbon removal | 10 | 0 | $5M | 10 | $50M | C, OENV, PBSU |
| Reactive mineral additions to soils | 3 | 0 | $3M | 10 | $30M | Frontier: Assess weathering rates of reactive minerals (i.e., olivine) added to agricultural soils and effects on agricultural productivity, soil carbon, nutrient use, water use, and albedo. Determine the carbon storage limits that can be achieved with mineral addition. |
| Studying the environmental impact of mineral addition to terrestrial, coastal and marine environments | 8 | 4 | $10M | 10 | $100M | STU, C, OENV, G, PBSU |
| Number of academic/national lab/industrial partnerships | Total cost/project/year | |||||
| Pilot Studies | ||||||
| Ex situb | 2 | $250K | $500K | 10 | $5M | C, EN, OENV, G, M&V, PBSU |
| Mine tailings, alkaline wastes | 4 | $250K | $1M | 10 | $10M | STU, C, EN, OENV, G, M&V, PBSU |
| Soils, beaches | 3 | $1M | $3M | 10 | $30M | Frontier: Challenging field-scale characterization of slow rates over large areas with poor controls on natural inputs and outputs |
| In situ basaltc | 1 | $10M | $10M | 10 | $100M | STU,C, EN, OENV, G, M&V, PBSU |
| in situ peridotitec | 1 | $10M | $10M | 10 | $100M | Frontier: No prior experiments on field scale, initial low porosity requires stimulation and/or practical knowledge of positive feedbacks such as reaction-driven cracking. |
a STU = scientific and technical understanding, C = cost, EN = energy use, OENV = other environmental concerns, G = governance, M&V = monitoring and verification, PBSU = practical barriers to scale-up, Frontier = long term research.
b Lab kinetics above, relevant to all categories, but leave larger-scale ex situ studies to the carbon capture, utilization, and storage community.
c Constant annual cost listed here, phased project with increasing annual expenses for successive phases described in text and Appendix F, Table F.3.
Research topics spanning NSF and DOE domains could be facilitated by establishing a formal NSF-DOE partnership, focused on funding research on greenhouse gas mitigation (not restricted to CO2 removal from air). Such a partnership could resemble several successful initiatives within NSF’s Directorate for Geosciences, such as the RIDGE, MARGINS, and GeoPRISMS Programs, in which the Earth Sciences and Marine Sciences Divisions cooperated to fund multidisciplinary research “across the shoreline.” Examples of interagency cooperation of this kind include volcano monitoring and investigations of dark matter.








































































