
Proceedings of a Workshop
WORKSHOP OVERVIEW1
In recent years, significant progress has been made in the clinical development and use of various types of cancer immunotherapy, all of which rely on the immune system to fight cancer.2 The majority of new cancer drug applications submitted to the Food and Drug Administration (FDA) are for immunotherapies or combinations involving immunotherapies. One type of immunotherapy is an immune checkpoint inhibitor. Cells in the human body have proteins that regulate the immune system response to foreign invaders (e.g., cancer cells, microorganisms). However, cancer cells can coopt these “checkpoint” proteins and thwart the immune system’s ability to recognize and attack cancer cells. To help promote an immune response
___________________
1 The planning committee’s role was limited to planning the workshop, and the Proceedings of a Workshop was prepared by the workshop rapporteurs as a factual summary of what occurred at the workshop. Statements, recommendations, and opinions expressed are those of the individual presenters and participants, and are not necessarily endorsed or verified by the National Academies of Sciences, Engineering, and Medicine, and they should not be construed as reflecting any group consensus.
2 For more information on immunotherapy in cancer, please see the National Cancer Policy Forum workshop proceedings Policy Issues in the Clinical Development and Use of Immunotherapy for Cancer Treatment. http://www.nationalacademies.org/hmd/Reports/2016/policy-issues-in-clinical-development-immunotherapy-for-cancer-treatment.aspx (accessed June 27, 2019).
to cancer, researchers have developed immune checkpoint inhibitors that enable T-cells to recognize cancer cells as foreign and to prevent deactivation of an immune system response. FDA approved the first immune checkpoint inhibitor drug in 2011—an antibody that blocks the CTLA-4 receptor protein expressed on T-cells.3 Since then, immune checkpoint inhibitor drugs have also been developed to inhibit PD-1 (a regulatory receptor found on T-cells) and PD-L1 (a receptor ligand that binds with PD-1 and is found abundantly on cancer cells, as well as on some normal cells).4
Immune checkpoint inhibitors have changed the standard of care for multiple types of cancer, and this progress has generated a great deal of new drug development activity. However, immune checkpoint inhibitors are often not curative, and there are still many types of cancer for which they have not yet been effective. Samir Khleif, biomedical scholar and professor of oncology at the Georgetown University Lombardi Comprehensive Cancer Center, said it is commonly believed that the effectiveness of using immune checkpoint inhibitors as monotherapy5 for cancer treatment has plateaued. Consequently, there is strong interest in combining checkpoint inhibitors with other cancer therapies to improve therapeutic effectiveness and patient outcomes. To examine the challenges and opportunities to develop combination cancer therapies that include immune checkpoint inhibitors, the National Cancer Policy Forum held a workshop, Advancing Progress in the Development of Combination Cancer Therapies with Immune Checkpoint Inhibitors. This workshop, held July 16–17, 2018, in Washington, DC, convened stakeholders with a broad range of expertise, including cancer researchers, clinicians, patient advocates, and representatives from industry, academia, and government. Presentations and panel discussions examined the current treatment landscape and various aspects of combination therapy development with PD-1/PD-L1 immune checkpoint inhibitors, including
- Strategies to select combinations for testing;
- The role of biomarkers;
___________________
3 See https://news.bms.com/press-release/rd-news/fda-approves-yervoy-ipilimumab-treatment-patients-newly-diagnosed-or-previousl (accessed February 15, 2019).
4 Examples of PD-1 and PD-L1 drugs include pembrolizumab, nivolumab, cemiplimab, atezolizumab, avelumab, and durvalumab. See https://www.cancer.org/treatment/treatmentsand-side-effects/treatment-types/immunotherapy/immune-checkpoint-inhibitors.html (accessed February 15, 2019).
5 Monotherapy is the use of a single cancer treatment, whereas combination therapy involves two or more therapies or modalities (e.g., drugs, radiation therapy, or surgery) in a regimen.
- Strategies to improve clinical trial design;
- Regulatory challenges and opportunities; and
- The use of data sharing and real-world evidence.
Roger Dansey, chief medical officer at Seattle Genetics, said that this workshop provides an opportunity for reflection on the past and future trajectory of PD-1/PD-L1 drug development. He noted that the explosion of PD-1/PD-L1 development is due to the availability of new agents with novel mechanisms of action, broad potential applicability across many cancer types, industry momentum in driving PD-1/PD-L1 programs forward, and the need for more effective cancer therapies that improve patient outcomes. Given these factors, Dansey said that clinical trials for PD-1/PD-L1 drug development “have preceded and run somewhat ahead of the science.” Thus, this workshop could serve as the “perfect time to hit the reset [button] and think through [how] we can do this more intelligently, more thoughtfully, more rationally, and [more collaboratively],” said Dansey. Khleif echoed these sentiments, and emphasized that careful thought and planning are needed to advance progress.
This Proceedings of a Workshop highlights a number of suggestions from individual participants regarding potential ways to improve the development of combination therapies using PD-1/PD-L1 immune checkpoint inhibitors. These suggestions are discussed throughout the proceedings and are summarized in Box 1. Appendix A includes the workshop Statement of Task and Appendix B lists the workshop agenda. Workshop presentations and the webcast have been archived online.6
THE LANDSCAPE FOR IMMUNE CHECKPOINT INHIBITOR THERAPY
Several workshop speakers provided an overview of the current landscape of PD-1/PD-L1 development and use in clinical practice. Ramy Ibrahim, chief medical officer of the Parker Institute for Cancer Immunotherapy, said that six drugs targeting either PD-1 or PD-L1 have been approved by FDA to treat 14 types of cancer and for one histology-agnostic indication (Tang et al., 2018c). He added that a 2017 Cancer Research Institute analysis found 164 active agents against PD-1/PD-L1 in the
___________________
6 See http://nationalacademies.org/hmd/Activities/Disease/NCPF/2018-JUL-16.aspx (accessed June 27, 2019).
immunotherapy development pipeline (Tang et al., 2018b); in 2018, this number rose to 196 (Tang et al., 2018a). PD-1/PD-L1 drugs are just one component of the larger phenomenon of immunotherapy development, which includes immune modulator drugs, cancer vaccines, cell therapies, oncolytic viruses, and CD3-targeted bispecific antibodies. In just 1 year,
there was a 67 percent increase in the number of active immunotherapy agents in the development pipeline: from 2,031 in September 2017 to 3,394 in September 2018 (Tang et al., 2018a).
Ibrahim said the number of clinical trials evaluating combination therapies using PD-1/PD-L1 drugs is increasing rapidly (see Figure 1a). The
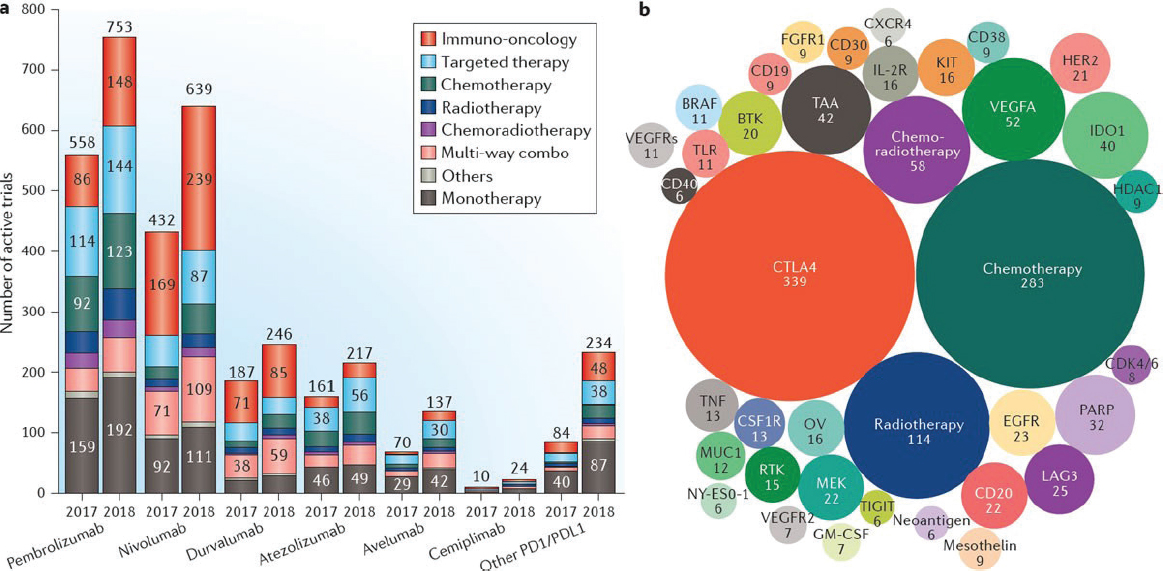
SOURCES: Ibrahim presentation, July 16, 2018; Tang et al., 2018c.
most common type of combination is a PD-1/PD-L1 drug with another immunotherapy drug, but PD-1/PD-L1 drugs are also being evaluated in combination with targeted therapies, chemotherapy, radiation therapy, radiation therapy plus chemotherapy, as well as other multi-way combination strategies. He noted that the largest share of clinical trials evaluating combination therapy with PD-1/PD-L1 drugs involves CTLA-4 inhibitors, but a growing number of clinical trials are assessing PD-1/PD-L1 drugs in combination with chemotherapy (see Figure 1b).
Ibrahim said there is strong scientific rationale for pursuing combination strategies using immune checkpoint inhibitors. However, conflicting clinical trial results with different combination strategies point to the challenges and complexities involved. In addition, preclinical findings may not be predictive of results in human trials. For example, in the context of prostate cancer, he said that preclinical evidence suggested radiation therapy plus immune checkpoint inhibitor therapy could provide a synergistic effect.7 Researchers had hypothesized that radiation-induced cell death would release tumor antigens that would enhance the antitumor activity of a CTLA-4 inhibitor in patients with metastatic, castration-resistant prostate cancer. “Unfortunately things are not that simple,” said Ibrahim, who noted there was no significant difference in overall survival among the patients receiving radiation therapy plus CTLA-4 immunotherapy versus those receiving radiation therapy plus placebo (Kwon et al., 2014).
However, Ibrahim said that when a PD-1 drug was given following chemotherapy plus radiation therapy for patients with unresectable stage III non-small cell lung cancer (NSCLC), researchers found significantly longer progression-free survival (Antonia et al., 2017). In addition, combining a PD-1 drug with chemotherapy as first line therapy for patients with advanced NSCLC resulted in significant improvements in overall response rate and progression-free survival, as well as a favorable trend toward overall survival (Borghaei et al., 2019).
The Impact of Immune Checkpoint Inhibitors on Cancer Treatment
A number of workshop participants described the striking effect that immune checkpoint inhibitor therapy has had on cancer care. Amy Abernethy, who at the time of the workshop was chief medical officer, chief
___________________
7 A synergistic effect is when two or more therapies combined have a greater effect than the sum of the effects when each therapy is given in isolation.
scientific officer, and senior vice president, oncology at Flatiron Health,8 illustrated the large uptake of PD-1/PD-LI therapy for patients with lung cancer over time (see Figure 2). In some cases, patients with advanced cancers and poor prognoses have achieved very durable responses and extended survival with PD-1/PD-L1 therapy. However, a number of workshop speakers noted that many patients do not benefit from immune checkpoint inhibitor therapy, and other patients benefit initially, but go on to develop resistance. Determining who is most likely to benefit from immune checkpoint inhibitor therapy has been an ongoing challenge, and multiple biomarkers to improve patient selection for therapy are in development or in use (see section on Biomarkers in Development or in Use for Immune Checkpoint Inhibitor Therapies).
Adverse events associated with immune checkpoint inhibitor therapy are also quite different from those experienced with other types of cancer therapy; similarly, dosing, therapeutic responses, and response timelines are markedly different from other cancer therapies (NASEM, 2016). With the rapid implementation of immune checkpoint inhibitor therapies into clinical practice, education is critically important, said Una Hopkins, administrative director of the cancer program at White Plains Hospital. Community-based oncologists, as well as other clinicians who care for patients with cancer, need to be aware of the potential adverse effects that patients might experience with immune checkpoint inhibitor therapies, as well as how to best intervene to mitigate these effects.
Roy Herbst, Ensign Professor of Medicine, chief of medical oncology, and director of the thoracic oncology research program at the Yale Cancer Center, said the advent of immune checkpoint inhibitors has ushered in a new era in lung cancer treatment (Herbst et al., 2018; Kazandjian et al., 2016). Herbst noted that three checkpoint inhibitor therapies—nivolumab, pembrolizumab, and atezolizumab—have been approved for patients with refractory9 NSCLC. In some cases, immune checkpoint inhibitor therapy has led to very durable treatment responses. For example, Herbst said one of the first patients with refractory NSCLC treated with nivolumab had a complete response to the therapy and is still healthy 8 years later. However,
___________________
8 In February 2019, Dr. Abernethy became Principal Deputy Commissioner of Food and Drugs at the Food and Drug Administration (FDA). The views expressed in this proceedings do not necessarily represent the official views or policies of FDA.
9 Refractory describes a disease that does not respond to treatment. See https://www.cancer.gov/publications/dictionaries/cancer-terms/def/refractory (accessed March 19, 2019).
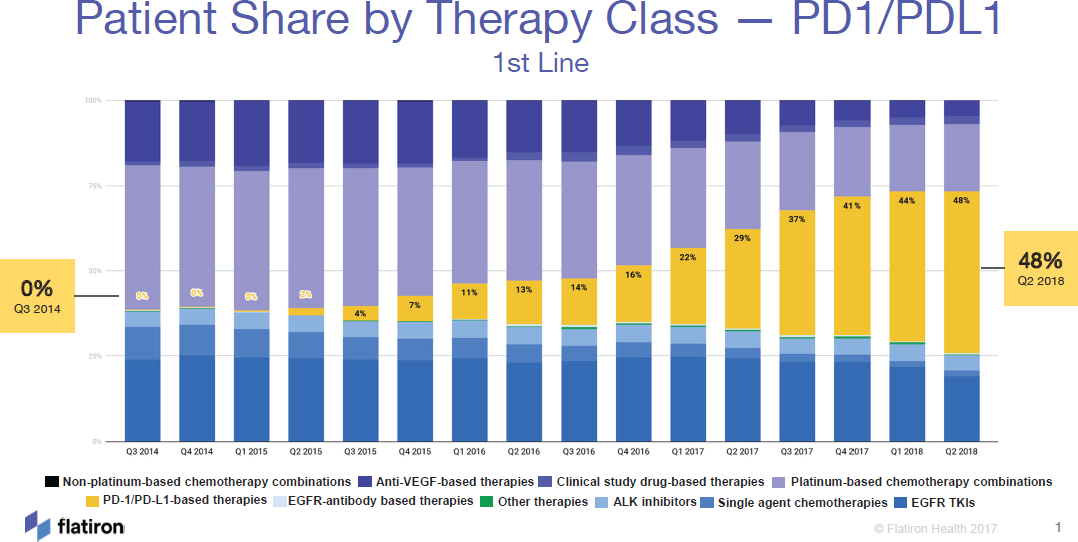
NOTE: ALK = anaplastic lymphoma kinase, EGFR = endothelial growth factor receptor, TKI = tyrosine kinase inhibitor, VEGF = vascular endothelial growth factor.
SOURCES: Abernethy presentation, July 17, 2018; Flatiron Health.
Herbst said this exceptional response to treatment is usually limited to only 10–15 percent of patients. He added that more than 50 percent of patients with lung cancer who receive immune checkpoint inhibitor therapy will eventually develop acquired resistance, illustrating that there is much room for improvement in patient outcomes.
Herbst described ongoing efforts to identify and validate biomarkers to better identify patients who will likely benefit from immune checkpoint inhibitor therapy. For example, he said high expression of PD-L1 in tumor cells (in at least 50 percent of cells) correlated with improved efficacy of pembrolizumab (Garon et al., 2015). However, more recently, researchers found that pembrolizumab improved overall survival compared with chemotherapy as first-line treatment for patients with advanced lung cancer whose PD-L1 expression level was greater than 1 percent (Lopes et al., 2018). Herbst stressed that additional work is needed to refine existing biomarkers and identify and validate new biomarkers.
Herbst added that investigators are evaluating combination therapy strategies in lung cancer, including combinations with targeted therapies, other checkpoint inhibitors, and chemotherapy. For example, he noted that the KEYNOTE 189 clinical trial showed that pembrolizumab combined with chemotherapy increased overall survival and progression-free survival in patients with metastatic NSCLC compared with chemotherapy plus placebo (Gandhi et al., 2018). He said this was a significant advance, but noted that two-thirds of patients receiving the combination still experienced disease progression at 1 year.
Herbst stressed that progress in improving patient outcomes will require additional work to better understand and appropriately deploy immune checkpoint inhibitor therapies. He noted that it has taken more than 20 years to develop effective strategies for targeted cancer drugs. “Now the same must be done for immuno-oncology, with even additional complexity,” he said, adding that immune checkpoint inhibitor therapies need to be evaluated in earlier stages of disease and in new types of cancers, and that investigators should leverage biomarkers to better determine mechanisms of sensitivity and resistance to better personalize immunotherapy.
Understanding Resistance to Immune Checkpoint Inhibitor Therapy
Several speakers discussed the challenge of resistance to immune checkpoint inhibitor therapies. Ibrahim said several strategies to overcome resistance to PD-1/PD-L1 are being tested, but results thus far have not
been very encouraging and there is a need to better understand the biology of resistance.
Herbst differentiated between primary and acquired resistance to immune checkpoint inhibitor therapies. Primary resistance occurs when there is no initial response to immune checkpoint inhibitor therapy; with acquired resistance, there is an initial response to therapy, but then a patient relapses or no longer experiences a response (Kim et al., 2018). He said a better understanding of the complex tumor microenvironment10 and the mechanisms underlying these types of resistance are key for improving combination therapy development. Elizabeth Jaffee, professor of oncology at Johns Hopkins University, added that the tumor microenvironment has multiple signaling pathways for suppressing T-cell trafficking and antitumor activity (Cui and Guo, 2016), but “we have yet to understand how these different signaling pathways interact to alter the balance between cancer development and anticancer immunity.”
Jaffee, Herbst, and Daniel Chen, vice president and global head for cancer immunotherapy development at Genentech/Roche, described three immune phenotypes (inflamed tumor, immune-excluded tumor, and immune desert tumor) as a way to understand why some tumors are more likely to be resistant or sensitive to immune checkpoint inhibitor therapies (Chen and Mellman, 2017; Herbst et al., 2014). An inflamed tumor has infiltration by several subtypes of immune cells, and is generally correlated with higher responses to PD-1/PD-L1 therapy. Non-inflamed (or “cold”) tumors—both immune-excluded and immune desert phenotypes—are more likely to have lower responses to checkpoint inhibitor therapies (Chen and Mellman, 2017).
Khleif further described mechanisms of immunotherapy resistance. He said that intrinsic tumor biology plays a role in primary resistance, and includes factors such as a lack of antigen presentation, and a T-cell deprived or immune-suppressive tumor microenvironment. For example, cancer cells can promote T-cell exclusion from the microenvironment through beta catenin signaling (Spranger et al., 2015).
Khleif said there are also treatment-specific mechanisms of resistance, such as low PD-L1 expression in some cancers or organs, or the presence of
___________________
10 The tumor microenvironment is composed of the normal cells, molecules, and blood vessels that surround a tumor cell. A tumor can alter its microenvironment, which can affect cancer growth and response to therapy. See https://www.cancer.gov/publications/dictionaries/cancer-terms/def/tumor-microenvironment (accessed April 9, 2019).
mutations. For example, one study found that mutations in the JAK-STAT pathway interfere with interferon-gamma receptor signaling and antigen presentation, contributing to acquired resistance (Zaretsky et al., 2016).
Khleif said that resistance may also occur due to incompatibility of combined immunotherapies (where immunotherapies do not work together in an additive or synergistic manner, and instead may negate each other’s effects) or immunotherapy-biologic incompatibility (where one component of an immunotherapy combination alters the tumor microenvironment such that the other component of the combination is unable to work as intended).
STRATEGIES FOR COMBINATIONS WITH PD-1/PD-L1 INHIBITORS
A number of workshop speakers described the rationale and strategies for testing immune checkpoint inhibitors in combination with other therapies. Jaffee said investigators are seeking to accomplish three primary goals by testing combination therapies:
- Enhancing efficacy of a single immune checkpoint inhibitor in an inflamed tumor;
- Increasing response to immunotherapy among patients with cold tumors; and
- Achieving higher response rates and more durable responses among patients who respond to immune checkpoint inhibitor therapies.
Chen noted that selecting potential combination strategies with immune checkpoint inhibitors is quite challenging because these drugs are broadly active, with complex biological effects on orthogonal signaling pathways. He added that a complex set of tumor, host, and environmental factors govern the strength and timing of anticancer immune responses (Chen and Mellman, 2013, 2017).
Khleif and Jaffee emphasized that investigators are exploring a variety of combination strategies to improve efficacy of immune checkpoint inhibitors and to overcome resistance, such as PD-1/PD-L1 therapies with other checkpoint inhibitors; targeted therapies; epigenetic agents; immune agonists; vaccines; chemotherapy; radiation or chemoradiation; and chimeric antigen receptor T-cell therapy. Chen added that combination strategies with immune checkpoint inhibitors could also be classified as combinations with the following:
- A standard-of-care regimen;
- An established in-class therapeutic;
- An investigational indication for an established agent; or
- A new molecular entity (new indication).
Katrin Rupalla, head of global regulatory strategy–oncology at Bristol-Myers Squibb, noted that combination strategies are more challenging if both agents have new mechanisms of action or are less-established agents in a specific indication.
Demonstration of Independent Action Versus Mechanism-Based Preclinical Data
Emmett Schmidt, lead of external collaborations at Merck Research Laboratories, described two approaches for selecting potential combinations in drug development: (1) selecting two therapies with demonstrated independent action versus (2) selecting combinations with mechanism-based preclinical data (Palmer and Sorger, 2017; Sharma et al., 2017).
Schmidt noted that historically, combination approaches in oncology have focused on combining agents with independent action. This approach is exemplified by clinical trials conducted by the Children’s Oncology Group, in which progressive improvements in the efficacy of multi-agent chemotherapy regimens have been achieved using trial designs with the standard of care plus an add-on chemotherapy agent. He said this approach has been very successful; the survival rate for childhood leukemia has risen dramatically from less than 10 percent in the 1960s to 90 percent today (Hunger and Mullighan, 2015).
Rupalla said that the checkpoint inhibitors nivolumab and ipilimumab—both approved as monotherapies—each have independent anticancer activity (Brahmer et al., 2010; Hamid and Carvajal, 2013; Topalian et al., 2012; Wang et al., 2014), and clinical data suggested that the combination would have greater activity and result in a better response among certain patient populations (Hammers et al., 2017; Larkin et al., 2015; Motzer et al., 2015). There are now three approvals for this combination: first line therapy for patients with unresectable or metastatic melanoma, first line therapy for patients with advanced renal cell carcinoma, and third line therapy for patients with metastatic colorectal cancer that has high microsatellite instability (MSI-H).
Khleif said preclinical models play an important role in identifying promising combination strategies using immune checkpoint inhibitor therapies, as well as identifying combinations that are unlikely to be effective. For example, investigators have conducted early-phase clinical trials of immune checkpoint inhibitors combined with an OX-40 T-cell receptor agonist (Aspeslagh et al., 2016), but he said data from a preclinical mouse model indicate that adding a PD-1 inhibitor to an OX-40 agonist and a tumor vaccine negates the effects of the OX-40 inhibitor/vaccine combination (Shrimali et al., 2017). “Science [should] guide us [on] how to move forward putting together combinations,” Khleif said.
Dan Theodorescu, director of the Samuel Oschin Comprehensive Cancer Center, added that functional genomics and preclinical models can also be leveraged to identify potential drugs to enhance the activity of immune checkpoint inhibitors (Tu et al., 2019). Using this approach, Theodorescu and colleagues identified a drug target, DDR2 (discoidin domain receptor 2) and evaluated potential FDA-approved drugs that target DDR2. Using preclinical mouse models representing five tumor histologies—bladder, breast, colon, sarcoma, and melanoma—Theodorescu and colleagues found that treatment with a DDR2 inhibitor (dasatinib) increased sensitivity to a PD-1 inhibitor compared with monotherapy (Tu et al., 2019). Theodorescu said these animal results are exciting, but he also cautioned that his mentor was fond of saying that we have been curing mice of cancer for decades, without equivalent progress in humans. “So when I see these [results] I remember those words, and I wonder then what can we do to make sure that we optimize these preclinical models so their [effectiveness] in actual patients is higher,” said Theodorescu. Schmidt agreed, and added that a major vulnerability of the mechanism-based approach to selecting potential combination strategies is the lack of representativeness of available preclinical models to reflect the diversity of human cancers and the immune environments of human tumors.
Schmidt said Merck has used both independent action and mechanism-based data to identify promising combination strategies with pembrolizumab. “Half of the trials have been motivated by combining things that have their own activity,” and the other half of clinical trials have been mechanism-driven, Schmidt said. He described the experience with a combination of pembrolizumab plus epacadostat, for which mechanism-based data suggested meaningful synergy, even though epacadostat had no independent activity as a monotherapy. A Phase I/II trial of the combination demonstrated promising antitumor activity in patients with advanced
melanoma (Hamid et al., 2017). However, the combination failed in a Phase III registration trial; in patients with unresectable or metastatic melanoma, the combination did not result in greater clinical benefit compared with pembrolizumab monotherapy (Long et al., 2018).
Chen noted that thus far, the biggest successes with combinations using immune checkpoint inhibitors have been with combinations of agents that have independent action. Chen said:
I do wonder whether [we should] rapidly prioritize and push forward those studies where we combine multiple active agents. And that doesn’t mean that you can’t have a combination where one drug doesn’t have single-agent activity, but perhaps that should be the minority of the studies moving forward.
Chris Boshoff, senior vice president and head of immuno-oncology at Pfizer Inc., agreed that combining two active agents is key, and he suggested that the next challenge is to better understand who benefits from which components of a particular combination. Boshoff said,
We can have two active agents, but when you combine them, there may be independence—meaning one population benefits from one, and another small population benefits from the other—and there are very few patients actually benefiting from both agents. . . . We don’t want to expose patients to two drugs if they can benefit from one.
Khleif voiced concerns about prioritizing combinations with independent activity:
I believe that if you have biologic data, [they are] what should guide us. The mere fact that one drug does not work on its own in immunotherapy, in my opinion, [isn’t a sufficient reason] not to combine it. . . . Some drugs might be very powerful in combination because the other drug is waiting for this change or this manipulation to happen to work better.
Jaffee agreed that science needs to drive the rationale for combinations, and added that improved knowledge of the following could aid in the development of optimal combination strategies:
- Inhibitory pathways that are co-expressed or upregulated in response to the PD-1/PD-L1 blockade;
- Primary and adaptive resistance to PD-1/PD-L1 blockade;
- The specific immune-suppressive populations within the tumor microenvironment; and
- Agonist signals that may enhance T-cell activation, prevent T-cell exhaustion,11 and induce immune memory.
Schmidt hypothesized that perhaps the best combination strategies may result when both independent action and mechanism-based data support the use of a combination (see Box 2). “I think where we’re going to achieve the greatest progress is probably where we use both of these sets of reasoning,” said Schmidt.
Sequencing Administration of Combination Strategies
Several speakers noted that sequencing of drug administration is an important consideration in the development of combination strategies. “The order of administration of checkpoint inhibitors makes a huge difference to the ultimate response rate and outcome,” said Adil Daud, professor of medicine and dermatology and director of the melanoma program at the University of California, San Francisco. For example, a Phase II trial found that patients with advanced melanoma who received nivolumab followed by ipilimumab had a better clinical response than patients who first received ipilimumab followed by nivolumab, although the former group also had a higher frequency of adverse events (Weber et al., 2016).
Herbst said more research should focus on sequencing strategies in combinations involving immune checkpoint inhibitors and chemotherapy. He said some patients might benefit from receiving immunotherapy followed by chemotherapy, and some of the Cooperative Groups are beginning to conduct these trials. Schmidt noted that sequencing had been critical to improvements in pediatric leukemia treatment.
Prioritization and Redundancy
A recurring theme at the workshop was the sheer quantity of research on immune checkpoint inhibitor therapies, and the implications for advancing progress in developing effective therapeutic approaches for patients with cancer. Herbst noted that cancer immunotherapy development has been described as a conundrum: Too many experimental drugs are being evaluated in too many clinical trials, and not enough patients are
___________________
11 T-cell exhaustion is a condition that can prevent optimal control of infections or tumors due to dysfunctional T-cell performance (Wherry, 2011).
available to participate in this research (Kolata, 2017). “I don’t think there are too many trials; there just aren’t enough of the right trials, and [enough] scientifically guided trials,” Herbst said.
The lack of comprehensive documentation of the landscape of immune checkpoint inhibitor clinical research and development is a significant challenge. “I think it would be a great resource if one of the foundations or groups put together lists of what is being studied and for what disease,” Herbst said. He noted that clinicaltrials.gov, for example, doesn’t often include all early--
phase studies, or information such as how many patients enrolled, the trial outcomes, and the biospecimens that were collected as part of the trial.
Schmidt added that “I don’t think that any of us would think that we should stop this pace.” He said the influx of activity is in direct response to unmet medical need among patients with cancer. Dansey noted that one of the concerns is whether we are getting maximum value out of the current approach. Herbst said a controlled effort is needed, with many trials, but also more effort to better define the role of biomarkers with combination therapies.
Dansey said the vast number of clinical trials with immune checkpoint inhibitors sounds daunting, but he noted that most of these trials are small, investigator-initiated signal-finding studies with 20 to 40 patients. Dansey said,
We can all understand why there is so much enthusiasm. There is a brand new class of agents that can really change the landscape . . . and constraining scientific discovery because the number of trials seems to be too high, from my perspective, seems like we are potentially getting concerned about a problem that actually may not really exist.
Ibrahim agreed with Dansey that innovation often stems from investigator-initiated studies, but described a number of challenges with these studies, including redundancy, their relevancy in moving the field forward, and the availability of the results. “In many cases, the same question is being asked by a different scientist [and] sometimes the data that we see from [these] studies cannot really inform next steps,” Ibrahim said. He added that there is a bias in publishing positive results, but the negative results could be just as informative.
Ibrahim compared the current drug development approach to the analogy of throwing spaghetti at the wall. He said there is an urgent need to learn from the existing data, and not add to the confusion with the deluge of ongoing immunotherapy research. While many people are focusing on developing the next combination therapy, Ibrahim noted that “there are very few who are spending time to try to understand why certain combinations did not work, or why are we seeing an early signal of activity, but then when you take it into a larger clinical trial, you don’t see the same effect.” Chen said he hopes the future of combination immunotherapy development will focus on more rational, science-based approaches. “I think [what] we’ve seen is a proliferation of a lot of studies, duplication of the same scientific hypothesis repeatedly. I’ve questioned how much we’ve actually learned, at least in the combination immunotherapy space, from throwing spaghetti against the wall.”
Louise Perkins, chief science officer of the Melanoma Research Alliance, cautioned that combination therapy strategies too often focus on convenience rather than on the underlying scientific rationale. Ibrahim agreed, and stressed that it is critical to prioritize research based on the strongest scientific hypotheses, rather than the most readily accessible combinations within a single development pipeline.
Workshop speakers also noted that the large number of clinical trials with very similar research questions may exhaust the number of potential patients enrolling in clinical trials. Ibrahim said that just because two agents combine well in terms of safety, that is not sufficient justification for testing the combination. Richard Pazdur, director of the FDA Oncology Center of Excellence, said FDA has encouraged industry sponsors to use a common control arm, due in part to the experience with clinical trials investigating immune checkpoint inhibitor combinations in renal cell cancer. Boshoff and others said that in the past 3 years, more than 2,000 patients with renal cell cancer have participated in different clinical trials, all with the same control arm therapy (sunitinib). Dansey questioned whether five different randomized trials with 2,000 patients were necessary in this case. “From an individual company perspective, it makes complete sense. From a broader patient resource perspective, actually it doesn’t make any sense at all,” Dansey said. Boshoff said that “hopefully in the future we can all work together and use this database of sunitinib outcomes in these Phase III studies as a potential synthetic arm.”
Steven Lemery, associate director of the FDA Division of Oncology Products 2, agreed there is a lot of redundancy. “I don’t know if it’s the best use of resources, [but] I can’t criticize any one company for developing a drug. This is the system we have today,” said Lemery.
Chen added that this is a remarkable era of immunotherapy development, where
we can talk about subtle differences between PD-1 and PD-L1 inhibitors . . . but the real challenge will be developing the next-generation molecule in this pathway. . . . Spending a ton of resources on developing 20 drugs that are very similar probably isn’t the best way to approach this, and we’re better off focusing on making a breakthrough.
Regulatory Considerations
Amy McKee, acting deputy director of the FDA Oncology Center of Excellence, provided an overview of FDA’s guidance on combination
therapy development. She provided key definitions relating to combination therapy development:
- Regimen: Two or more therapeutic products that are marketed separately, but are approved for use in combination based on one or more adequate and well-controlled trials.
- Cross-labeling: The inclusion of information in product labeling of two (or more) oncology products approved in a combination regimen for a specific indication.
More than a dozen applications are pending in the FDA Office of Hematology and Oncology for cross-labeling. She said these applications range from cross-labeling of two novel agents (both New Drug Applications [NDAs] and Biologics License Applications [BLAs]) to cross-labeling of two supplemental indications, and everything between those points.
Historically, FDA has required evidence for the contribution of effect for each agent in a regimen for which a label indication is sought. However, McKee said the ongoing consideration is the level of evidence that is acceptable for demonstration of the contributions for each component in the regimen. For companies developing novel-novel regimens, she noted that development and cross-labeling should follow the guidelines set forth in the FDA guidance on co-development.12
McKee said that when reviewing cross-labeling applications, FDA has to consider the current regulatory status of the product:
Is it approved or a new molecular entity? Is it approved in a specific disease or setting? What are all the available data that we have about a particular product, so that we can include it in the evaluation of a cross-labeling application? Is it a standard-of-care product in the community, even if it is not labeled as such? Is it new in that disease context, whereby we would probably require more information about that product? Is there a biologic rationale for why we should [potentially] cross-label across disease settings?
McKee provided several examples of recent cross-labeling approvals to help illustrate FDA’s thinking on acceptable levels of evidence for these decisions (see Box 3). She encouraged the drug development community to submit cross-labeling applications, noting that this could help address some of the redundancy in immune checkpoint inhibitor development. She
___________________
12 See https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm236669.pdf (accessed April 24, 2019).
added that supplemental BLAs and NDAs no longer require a user fee, so hopefully this will encourage greater use of cross-labeling as well.
Sean Khozin, associate director of the FDA Oncology Center of Excellence, discussed FDA efforts to improve the process for evaluating potential safety signals for Investigational New Drugs (INDs). Sponsors of an IND typically have to submit narrative safety reports describing certain adverse events within specified periods of time, Khozin noted. “It is a huge burden on sponsors and companies that have to collect this information, format it, and fax it or send it as a PDF file to FDA and other regulatory bodies,” Khozin said. He added that it is also a burden to FDA reviewers, who need to sift through these voluminous reports to deduce whether there are concerning safety signals related to the IND. He added that the number of safety reports has been increasing in recent years, but the majority of these reports are not informative for safety assessment (Jarow et al., 2016).
FDA’s INFORMED (Information Exchange and Data Transformation) program includes a project to facilitate quantitative review of IND safety data by digitizing the premarket safety report process (Khozin et al., 2016). These data can be quantitatively analyzed by FDA reviewers to identify safety signals, and Khozin described ongoing efforts using these data to generate algorithms to assess drug safety (Khozin et al., 2017). Khozin noted that an extension of this process has the potential to create a globally available, central electronic portal that would enable users to report and access adverse event data curated by a neutral third party (Levit et al., 2018).
BIOMARKERS IN DEVELOPMENT OR IN USE FOR IMMUNE CHECKPOINT INHIBITOR THERAPIES
Several workshop speakers discussed the role of biomarkers in advancing the development of combination therapies with immune checkpoint inhibitors. Daud said biomarkers are critical to help identify which patients will benefit from immune checkpoint inhibitor therapy and to rationally select combinations. David Rimm, professor of pathology and medicine at the Yale University School of Medicine, added that it is important to identify those patients for whom immune checkpoint inhibitors will not work because they also might be more susceptible to the toxicities of these therapies.
“It’s all about the biomarkers, and I think we really need to be thinking not just about the immune biomarkers associated with PD-1/PD-L1, but we need to be thinking about the other targeted pathways within the
tumor microenvironment, and [how we] can utilize biomarkers that measure those changes with the therapy, along with the PD-1 changes,” Jaffee said. She added that biomarkers could help to identify early responses to immune checkpoint inhibitor therapies and clarify mechanisms of action of contributing therapeutics, as well as identify the underlying mechanisms of action that may be responsible for immune-related toxicities. Biomarkers could also assess how the interaction of targeted combination pathways can be leveraged to optimize sequencing and dosing for combination therapies.
Types of Biomarker Tests
Several speakers discussed biomarker tests that are in development or in use for immune checkpoint inhibitor therapies, including
- Immunohistochemistry
- Genomics
- Expression signatures
- Multiplex fluorescence
- Circulating biomarkers
- Assessment of the microbiome
Lisa Butterfield, professor of medicine, surgery, immunology, and clinical and translational science at the University of Pittsburgh Hillman Cancer Center, said that biomarkers—for prediction, prognosis, and elucidation of mechanisms of action—are still largely exploratory, but validation, both analytical and clinical, is ongoing. Jaffee said investigators are leveraging rapidly developing technologies for biomarker discovery, including multiplex immunohistochemistry, mass cytometry, T-cell receptor sequencing, and immunoPET (positron emission tomography) imaging. She said these technologies are providing a better understanding of immune cell composition and functioning as well as the tumor microenvironment.
Immunohistochemistry Testing
Rimm said there are several immunohistochemistry (IHC)13 tests currently available to assess PD-L1 protein expression in a patient’s tumor,
___________________
13 Immunohistochemistry testing “use[s] antibodies to test for certain antigens (markers) in a sample of tissue. The antibodies are usually linked to an enzyme or a fluorescent dye.
which have been used as both as companion and complementary diagnostics.14 He said the availability of multiple tests for PD-L1 expression is a challenge because the tests are not identical, but they are used within the same clinical context. Comparisons have been undertaken to assess the performance and concordance of test results. For example, the Blueprint PD-L1 IHC Assay Comparison Project (Hirsch et al., 2017) found:
- Three of the four assays were closely aligned on tumor cell staining, but the fourth assay showed consistently fewer tumor cells stained.
- All of the assays demonstrated immune cell staining, but there was greater variability than with tumor cell staining.
- Despite similar analytical performance of PD-L1 expression for three assays, interchanging assays and cutoffs would lead to “misclassification” of PD-L1 status for some patients.
Rimm said that an analysis by the National Comprehensive Cancer Network (NCCN) also found that three of the four tests were essentially equivalent. The outlier test only identified 50 percent of the patients with positive results on the other three tests (Rimm et al., 2017). The analysis found high concordance among pathologists for PD-L1 expression on tumor cells, but not on immune cells.
Rimm said that the Blueprint 2 project was initiated to confirm results from the first Blueprint project and to assess the feasibility of harmonizing the clinical use of five independently developed, commercial PD-L1 immunohistochemistry tests (Tsao et al., 2018). This project involved 25 pathologists reading 81 cases, following a 1.5-day training course. This analysis confirmed the interchangeability of three of the tests, and also
___________________
When the antibodies bind to the antigen in the tissue sample, the enzyme or dye is activated, and the antigen can then be seen under a microscope. Immunohistochemistry is used to help diagnose diseases, such as cancer. It may also be used to help tell the difference between different types of cancer.” See https://www.cancer.gov/publications/dictionaries/cancer-terms/ def/immunohistochemistry (accessed February 27, 2019).
14 The FDA definition of a companion diagnostic is “a medical device, often an in vitro device, which provides information that is essential for the safe and effective use of a corresponding drug or biological product”; see https://www.fda.gov/medicaldevices/productsandmedicalprocedures/invitrodiagnostics/ucm407297.htm (accessed April 25, 2019). This is distinct from a complementary diagnostic test, which does not have a formal regulatory definition, but is described as a “test that aids in the benefit–risk decision making about the use of a therapeutic product” (Scheerens et al., 2017).
found concordance among pathologists similar to the NCCN analysis, but Rimm added that pathologists were unable to reliably classify immune cell PD-L1 protein expression.
Genomic Testing
Rimm said genomic testing of patients’ tumors can be divided into two types: (1) identifying specific genes in tumors that inactivate the immune system, such as mutations in the STK-11 gene (Skoulidis et al., 2018); or (2) assessing genes in a more global sense, such as assessment of the quantity of mutations in a tumor, or the tumor mutational burden (TMB) (Alexandrov et al., 2013).
Naiyer Rizvi, director of thoracic oncology at the Columbia University Medical Center, elaborated on TMB as a biomarker for immune checkpoint inhibitor therapy. Rizvi said patients who have cancers with a high TMB may be more likely to benefit from immunotherapy because higher numbers of tumor mutations can increase the presence of neoantigens,15 which can enhance an antitumor immune response (Rizvi et al., 2015). In support of this hypothesis, one study found a significant correlation between TMB and the objective response rates to immune checkpoint inhibitor therapy in an analysis of 27 tumor types (Yarchoan et al., 2017).
Another study found that lung cancer patients with a high TMB had a higher response rate in the nivolumab treatment group compared with the chemotherapy group, and progression-free survival was also longer. However, overall survival was similar between the treatment groups, regardless of TMB status (Carbone et al., 2017).
In the combination therapy setting, Rizvi said that TMB has also been predictive of response to therapy. Progression-free survival was significantly longer with first-line nivolumab plus ipilimumab compared with chemotherapy among patients with advanced NSCLC and a high TMB (Hellmann et al., 2018). He said this was an important result because it was the first trial to look prospectively at TMB as an endpoint in a Phase III setting.
Rimm noted that a major challenge of using TMB as a biomarker is the large number of assays currently in use and the lack of standardization
___________________
15 New antigens (or markers) of a cancer that cue a patient’s immune system to attack the cancer and eliminate it. See https://www.the-scientist.com/features/neoantigens-enable-personalized-cancer-immunotherapy-31743 (accessed April 15, 2019).
among them. “If you thought it was hard to standardize five assays for immunohistochemistry, we’re going to have real trouble standardizing the literally hundreds of TMB assays,” said Rimm. Recent studies have begun to compare TMB results derived from different methods for some types of cancer (e.g., Carbone et al., 2017). Rizvi noted that Friends of Cancer Research has convened a multi-stakeholder group—including representatives of test developers, drug development companies, FDA, the National Cancer Institute (NCI), and academia—to “review the current methods of TMB calculation and reporting and create a consensus solution on how best to standardize them” (Friends of Cancer Research, 2019). In the second phase of this project, the group will aim to create a universal reference standard using whole-exome sequencing and identify sources of variability after comparing TMB scores from targeted panels to the universal reference standard.
Rizvi said a related challenge is defining thresholds or cutpoints for determining which patients with “high” TMB will benefit from immune checkpoint inhibitor therapy. He added that thresholds may vary by tumor type, and the assessment of patient benefit may also need to be informed by PD-L1 expression, in addition to TMB status. However, in the context of combinations with PD-1/CTLA-4, Rizvi noted that PD-L1 expression may not need to be considered in combination with TMB because the benefit with combination PD-1/CTLA-4 therapy was irrespective of PD-L1 expression (Hellmann et al., 2018).
Rimm agreed that both variables may be helpful in identifying patients most likely to benefit, noting that biomarkers for TMB and PD-L1 expression identified distinct and independent population subgroups in a Phase II trial of nivolumab plus low-dose ipilimumab among patients with metastatic NSCLC (Ready et al., 2019). Another analysis also concluded that TMB and PD-L1 expression were independent variables (Rizvi et al., 2018).
Rizvi added that some of the emerging research suggests that individuals with both low PD-L1 expression and low TMB may not respond well to immune checkpoint inhibitor therapy. “So when we think about the clinical trials that we’re designing . . . I think you have to have TMB as part of your analysis to make sense of the data,” Rizvi said.
Expression Signatures
Benjamin Izar, instructor in medicine at Harvard Medical School, discussed the use of signatures derived from single-cell and bulk RNA
sequencing to predict response and resistance to immune checkpoint inhibitor therapy. He noted that bulk RNA sequencing is more commonly performed because many of the analytical approaches are well established, and data from a large number of patients can be obtained. A disadvantage of bulk RNA sequencing is that only one profile is obtained per patient, so it often does not reflect the heterogeneity of tumors. “Not only are we dealing with various cell types within a tumor, but also variability among those,” Izar noted.
Single-cell RNA sequencing enables the profiling of numerous cells, so that many measurements are obtained from a single patient sample. However, Izar noted that single-cell RNA sequencing is usually limited to relatively small cohorts because these studies are complex and resource intensive.
“The idea of this study was to combine and leverage the advantages of both of these [approaches] to try and make sense of what might be predictors of response or resistance to immunotherapies,” Izar said. He said a proof-of-concept study established the methods to generate usable single-cell transcriptome profiles and to develop data-clustering methods, as well as the analytical tools for understanding and robustly distinguishing cancer cells from non-malignant cells (Tirosh et al., 2016).
Single-cell RNA sequencing was conducted on 33 tumor samples from patients with melanoma, and investigators developed an algorithm that combined single-cell transcriptomes and bulk-RNA sequencing data from the Cancer Genome Atlas to identify genes that were over- or under-expressed in cancer cells and that were associated with T-cell exclusion. They also developed an algorithm to identify immune evasion, using single-cell RNA sequencing data from tumor samples of patients whose cancers were resistant to immune checkpoint inhibitor therapy. Even though these signatures were derived in different ways, Izar noted that they demonstrated overlap that was highly statistically significant, and termed this overlap “the resistance program.” Investigators validated the predictive value of this resistance program for progression-free survival and overall response in an independent cohort of patients with melanoma who received PD-1 inhibitor therapy (Izar et al., 2018; Jerby-Arnon et al., 2018).
Izar also reported on two other expression signatures that have been investigated for use with immune checkpoint inhibitor therapy. Ayers and colleagues (2017) analyzed mRNA expression in tumor samples of patients who received treatment with pembrolizumab. From this analysis, they developed an 18-gene expression signature of interferon-gamma respon-
sive genes, which correlated with clinical benefit. Validation studies are being performed in ongoing trials of pembrolizumab (Ayers et al., 2017). The second example was an evaluation of a T-cell effector gene expression signature in predicting clinical benefit of atezolizumab (a PD-L1 inhibitor) in patients with NSCLC in a Phase III trial (Kowanetz et al., 2017). Investigators found a gradient of improved progression-free survival benefit with atezolizumab therapy as the T-cell effector gene expression increased.
Multiplex Fluorescence
Multiplex fluorescence is a method that characterizes multiple proteins of a patient’s tumor and the tumor microenvironment using immunohistochemistry with fluorescence tagging. Rimm noted that multiplex fluorescence was used to demonstrate that patients who responded to immune checkpoint inhibitor therapy had preexisting tumor-infiltrating CD8 T-cells (Tumeh et al., 2014). He noted that his lab has also found that pretreatment infiltration of CD8 T-cells is highly predictive of response to immune checkpoint inhibitor therapy in patients with metastatic melanoma (Wong et al., 2019). Daud added that immune profiling demonstrated that the relative abundance of tumor-infiltrating CD8 T-cells predicts response to PD-1 inhibition (Daud et al., 2016).
Margaret Shipp, professor of medicine at Harvard Medical School, discussed the use of multiplex fluorescence in the context of her research in classical Hodgkin’s lymphoma (cHL). cHL is an unusual lymphoid malignancy; only a very small percentage of a tumor is composed of malignant cells (approximately 1–2 percent), while the remainder of the tumor is composed of extensive—but ineffective—inflammatory immune cell infiltrate. Chromosome 9p24.1 alterations are a defining feature of cHL, and this chromosome includes the PD-L1 and PD-L2 genes, as well as JAK2, a component of the JAK-STAT signaling pathway (Roemer et al., 2016). Shipp said this strongly suggested a genetic basis for reliance on PD-1 signaling as a mechanism of immune evasion in cHL, and this quickly prompted trials of nivolumab and of pembrolizumab to evaluate sensitivity to PD-1 blockade in cHL. In the CheckMate 205 clinical trial, patients with relapsed or refractory cHL whose tumors had higher levels of 9p24.1 alterations and higher PD-L1 expression were more likely to respond to nivolumab (Roemer et al., 2018).
“Because Hodgkin’s lymphoma is a disease where almost everybody has some level of PD-L1 expression, using an arbitrary cutpoint—like
1 percent PD-L1 expression used in some of the solid tumor trials—would not be meaningful in this setting. So, you really need to think about how you interpret biomarkers based on the biology of the disease,” said Shipp, noting that the objective response rate to nivolumab among patients with relapsed or refractory cHL was 69 percent (Armand et al., 2018). Shipp said
Hodgkin’s lymphoma has the highest response rate to PD-1 blockade thus far. We think there is an inherent genetic sensitivity for the PD-1 blockade, but clearly there are both responders to PD-1 blockade in Hodgkin’s lymphoma, and patients who either don’t respond or progress following initial responses, so it becomes very important to also understand biomarkers for response and resistance.
To characterize the tumor microenvironment of cHL and better understand the mechanisms of action of resistance to immune checkpoint inhibitor therapy, Shipp’s lab used multiplex immunofluorescence, digital image analysis, and mass cytometry (Cader et al., 2018; Carey et al., 2017). They found that the tumor microenvironment—including the presence of CD4 T-cells and PD-L1+ macrophages surrounding Hodgkin’s Reed-Sternberg cells—is likely to contribute to immune evasion.
Circulating Tumor Markers
Rimm briefly discussed circulating tumor markers, including circulating free DNA, as exploratory biomarkers for immune checkpoint inhibitor therapy. For example, researchers are assessing blood-based TMB as a predictive biomarker for atezolizumab (Velcheti et al., 2018). Jaffee added that these less invasive methods will provide the best opportunities for repetitive assessment and optimization of combination strategies. Theodorescu said one way to accelerate validation of biomarkers is to study circulating biomarkers before and after treatment in the neoadjuvant setting.16 Butterfield noted that it is important to collect both tumor tissue and blood biospecimens for biomarker discovery and validation: “I am personally of the opinion that eventually we will get back to the blood when we know exactly what we’re looking for. We will be able to find it there. It’s just, as an average of everything going on immunologically in the patient, it can be hard to find [blood-based biomarkers] at the outset,” said Butterfield.
___________________
16 Treatment given as a first step to shrink a tumor before the main treatment (usually surgery) is given. See https://www.cancer.gov/publications/dictionaries/cancer-terms/def/ neoadjuvant-therapy (accessed April 24, 2019).
The Microbiome
Christian Jobin, Gatorade Trust professor of medicine at the University of Florida, discussed whether the composition of an individual’s microbiome—the collection of microorganisms inhabiting a human host—can be used as a biomarker to distinguish patients who may be more likely to respond to immune checkpoint inhibitor therapies. Jobin said that an individual’s microbial composition is known to play roles in health in disease, including carcinogenesis and response to cancer therapies (Tsilimigras et al., 2017). He added that microbial genes can contribute to differences in drug metabolism that can alter the toxicity and efficacy of therapies.
He cautioned that research on the impact of the microbiome in cancer is nascent and primarily exploratory, but some exciting research studies have set the field in motion, including three recent papers in Science (Gopalakrishnan et al., 2018; Matson et al., 2018; Routy et al., 2018). These studies reported that patients can be stratified into responders and non-responders to immune checkpoint inhibitor therapy based on the composition of their intestinal microbiomes (Jobin, 2018). However, Jobin noted that there was no common signal among these studies. Gopalakrishnan and colleagues (2018) observed that the strongest predictors of response to immune checkpoint inhibitor therapy were bacterial diversity and abundance of Faecalibacterium and Bacteroidales bacteria in fecal samples. Matson and colleagues found that increased prevalence of eight microbial species, including Bifidobacterium longum, was associated with improved response to immune checkpoint inhibitor therapy, but two bacterial species were associated with non-responsiveness. Routy and colleagues found that increased prevalence of Akkermansia muciniphila was associated with improved response to immune checkpoint inhibitor therapy, and that exposure to antibiotics was associated with reduced responsiveness to therapy. Jobin said additional research is needed, including functional analyses with whole metagenome and RNA sequencing. Analyses of larger, multicenter patient cohorts could better characterize the influence of the microbiome on responsiveness to immune checkpoint inhibitor therapies.
Challenges with Biomarker Development
Biomarker Development for Single-Agent Versus Combination Therapy
Several speakers noted that biomarkers have been developed primarily for use with single-agent therapies. Chen said most of the work to develop
biomarkers for immunotherapy have focused on selecting patients likely to respond to PD-L1 and PD-1 inhibitors, but it is less clear whether these biomarkers will have clinical utility for combination therapy. “Many of these biomarkers—whether it’s microbiota, PD-L1 staining, [or] tumor mutation burden—may be good biomarkers for the benefit of cancer immunotherapy, but may not help us separate out which combination immunotherapy is best for a given patient,” he stressed. Rimm agreed, and added that “I don’t believe that we [understand] biomarkers for combinations yet, even though arguably that’s when we need them the most.” He added that biomarker research often follows clinical trials, and there is less incentive to incorporate biomarkers in the design of early-phase studies because “no one wants to eliminate patients who might respond.” Rimm suggested devising new strategies for incorporating biomarkers in Phase II and III studies.
Defining Thresholds for Biomarker Results
Ronald Kline, medical officer with the Center for Medicare & Medicaid Innovation at the Centers for Medicare & Medicaid Services, noted that biomarkers are rarely dichotomous variables; consequently, investigators need to define thresholds for positive or negative biomarker results. He said that decisions about where to set a threshold for a biomarker result have significant clinical and cost implications. Kline inquired, “How do you define, as scientists, what’s positive and what’s negative, and how do you create a framework that allows us to rationally look at different test results?” Lemery noted that this is particularly challenging if different thresholds are set for different tests that measure the same biomarker. Rimm noted that the decision to set a threshold for a biomarker test should be informed by calculating the predictive value and the area under the receiver operating characteristic curve for the test.17 Richard Schilsky, senior vice president and chief medical officer at the American Society of Clinical Oncology (ASCO), stressed the importance of lifecycle learning, and determining when there is sufficient evidence to confirm that a biomarker test identifies a population of patients likely to benefit, while also recognizing that every test is always going to be imperfect. “Once you’re in the market, you still have a lot to learn, because typically you have much broader use in much more heterogeneous populations,” he said.
___________________
17 See Mandrekar, 2010, for more information on area under the curve.
Biomarkers for Site-Agnostic Indications
Lemery discussed the use of biomarkers to identify molecular changes in a tumor that can be targeted by a therapy without consideration of the type of cancer a patient has. He said that the first FDA approval for a site-agnostic indication of a drug was for pembrolizumab in adult and pediatric patients who have any type of advanced cancer with MSI-H as a result of DNA mismatch repair deficiency.18
Lemery said that Le and colleagues (2015) first presented data on the clinical benefit of pembrolizumab for patients with MSI-H cancers in 2015. He noted that high microsatellite instability is found across many different tumor types, and is associated with increased TMB (Le et al., 2017). Lemery said the supplemental BLA for pembrolizumab in patients with MSI-H cancers was approved in 2017 based on objective response rates and durability of responses across 14 tumor types. Other considerations for the approval included a strong biological rationale and the unmet medical need of patients due to the lack of effective treatment options (Marcus et al., 2019). “Rather than requiring a separate development program in each different tumor type, which take years and years if not decades . . . we approved it for all the patients with [MSI-H cancers],” Lemery noted.
Schilsky raised concerns about the lack of a companion diagnostic for measuring MSI-H status because “there are a lot of ways of assessing MSI status—there are multiple platforms, there is more than one definition, and some of [the platforms] are more subjective than others” (e.g., immunohistochemistry versus polymerase chain reaction [PCR] versus next generation sequencing). In addition, there may be wide variation and uncertainty about the response rates and durations of response seen across patients with various tumor types, due to the small populations under study. He noted that the data for the MSI-H sBLA approval was based on 150 patients, of which 90 had colorectal cancers. “Certainly not every kind of cancer is represented in this dataset, and for most of the cancers that are represented, the number of patients with that cancer was very small, far smaller than you would include in even a Phase II study, to try to assess the efficacy of the treatment in that particular tumor type.” He added that a fundamental question for the development of site-agnostic biomarkers is “how much evidence is sufficient to actually conclude that a drug works
___________________
18 See https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm560040.htm (accessed April 19, 2019).
well across a variety of histologies, where the drug indication is defined by a common biomarker?”
Lemery responded that there are postapproval requirements, including the development of immunohistochemistry testing for assessing mismatch repair deficiency and nucleic-acid based testing for assessing MSI status, and that Merck has enrolled more than 400 patients with MSI-H cancers to further evaluate the response rate to pembrolizumab in different tumor types. Schilsky agreed that post-marketing data will be critical to further define the populations in which these drugs work and how well they work, noting that biomarker prevalence and predictive value may vary across tumor types. He added that biomarkers are often present at very low frequency in the population, so identifying patients with a particular biomarker can be challenging.
Stanton Gerson, director of the Case Comprehensive Cancer Center, inquired whether certain tissue-agnostic approvals will need to be reconsidered in the future, if new research clarifies individuals who are unlikely to respond to therapy and should not be exposed to the potential toxicities. Schilsky said that a mechanism to collect relevant data is needed, and that FDA should consider labeling changes based on accumulating knowledge about patient outcomes. “For example, we should have a national registry of pembrolizumab use in all MSI-high tumors, so that we actually know over time exactly how the drug is being used in practice,” he said.
Schilsky and Lemery also discussed the implications of site-agnostic therapy on clinical trial designs. Schilsky questioned whether it is even possible to conduct a randomized controlled trial to determine if a drug should be approved in a histologically agnostic indication, and what type of control arm would be appropriate in this context. Lemery said it may not be ethical or feasible to conduct a randomized controlled trial if investigators are assessing a very rare biomarker for therapy selection and the therapy has an unprecedented patient response, especially in the disease-refractory context.
Availability of Biospecimens for Biomarker Discovery and Development
Butterfield said variability in the quality of biospecimens available for study hinders the development of useful biomarkers for immune checkpoint inhibitor therapies—many biospecimens have not been collected, processed, and stored under standardized conditions, and are thus not useful for biomarker development. She pointed to an editorial from Nature Biotechnology stating (No sample left behind, 2015):
The reality is that most immune profiling efforts remain at a pilot scale. To truly galvanize clinically actionable insights, researchers will need to integrate datasets of sufficient diversity and statistical power. This will require greater attention to how samples are acquired and analyzed and community agreement on how [to] store, share, and interpret data.
George Weiner, director of the Holden Comprehensive Cancer Center at The University of Iowa, agreed that the availability of high-quality biospecimens is a major constraint to future research. “The opportunity to learn from [biospecimens] is being lost, because we’re not [collecting] them in a consistent and reliable way,” said Weiner. He added that the costs for storing biospecimens for future research purposes is an impediment. Butterfield added that the language in consent documents may also limit the availability of biospecimens for future research purposes.
Butterfield said that biospecimens should be banked to answer specific questions from a trial, but also “to answer the questions we think of while the trial is enrolling and the field has moved forward, [or] when the trial is complete, and the field has moved forward yet again. We are going to need more specimens very broadly banked to be able to answer all of those questions.”
Butterfield noted that the International Society for Biological Therapy of Cancer–Society for Immunotherapy of Cancer (SITC), FDA, and NCI developed recommendations for incorporating biomarker studies in clinical trials and standard operating procedures for biospecimen collection, processing, storage, and analysis to study immunologic questions (Butterfield et al., 2011). Since then, Butterfield noted that the field has rapidly progressed, and the SITC Immune Biomarkers Committee has organized a number of working groups to review state-of-the-art technologies and to identify opportunities to advance progress in immunotherapy research.19 Ibrahim added that the Partnership for Accelerating Cancer Therapies,20 a component of the Cancer Moonshot, is working to identify, develop, and validate immunotherapy biomarkers (Baker et al., 2018). He noted that the Parker Institute is also working on standardizing biospecimen collection processes in immunotherapy clinical trials.
Butterfield also suggested several opportunities to improve the measurement of immune biomarkers in clinical trials of combination therapies:
___________________
19 See https://www.sitcancer.org/research/biomarkers (accessed April 22, 2019).
20 See https://fnih.org/what-we-do/programs/partnership-for-accelerating-cancer-therapies (accessed April 22, 2019).
- Place a greater emphasis on tumor biopsies in the trial design;
- Incorporate assessments to determine whether the target molecules get to the tumor site and show activity;
- Assess previously identified candidate biomarkers in new settings;
- Use high-throughput technologies for hypothesis generation; and
- Explore innovative data analysis strategies.
CLINICAL TRIAL DESIGN STRATEGIES FOR COMBINATION IMMUNOTHERAPY
Many participants emphasized the importance of improving clinical trial design for testing combinations with immune checkpoint inhibitors. Workshop participants discussed:
- Ensuring that clinical trials answer questions that are important to patients (e.g., quality of life outcomes);
- Selecting appropriate endpoints for assessing response and patient outcomes; and
- Prioritizing trial designs that can enhance efficiency and answer multiple questions simultaneously, such as master protocols or shared controlled arms.
As discussed in the previous section, workshop speakers also highlighted the need to better integrate biomarkers in the design of trials in order to identify patients who are likely to benefit from combinations with immune checkpoint inhibitors, as well as those individuals who are unlikely to respond, so that they can avoid the toxicity of these therapies.
Patient-Centered Trial Outcomes
Linda House, president of the Cancer Support Community (CSC), urged researchers to examine the patient experience in clinical trials for combination therapies with immune checkpoint inhibitors. “We really haven’t talked about how our patients are living with their cancer and living with the treatments that we’re giving them,” House said. She said the CSC has been collaborating with FDA to identify opportunities to integrate patient experience outcomes in the design of clinical trials. “We have to collect these data because decisions are being made downstream that [affect] patients without the benefit of this [knowledge],” said House.
She noted that the CSC has a cancer experience registry, and in reviewing the latest data on patients with melanoma and lung cancer, she said their top five concerns about cancer and its treatment were maintaining physical activity, fatigue, the ability to eat and maintain nutrition, worry about the future, and the financial burden of treatment. She noted that clinical trials of new cancer treatments often do not provide data on these types of outcomes.
Perkins agreed that clinical trials should answer patient-centered questions, and added that patient advocates should be active participants in the drug development process. She cited the ASCO–Friends of Cancer Research effort to modernize patient eligibility criteria,21 and called for immunotherapy clinical trials to better reflect the real-world patient populations, including patients with melanoma who have brain metastases. Daud agreed with this suggestion and encouraged the clinical trials community to reduce the number of exclusion criteria so that a wider variety of patients can participate. Broad accessibility is needed to ensure that data are available to inform the treatment decisions of diverse patient populations.
Hopkins and Perkins advocated for the concept of a “clinical trial in a box,” which would enable community oncology care providers to quickly and efficiently enroll patients in a clinical trial. Hopkins also emphasized the importance of understanding the patient population served in a community hospital setting when deciding which trials to open. She noted that industry-sponsored trials are often not available in small community cancer centers because of the smaller numbers of potential patients, but she said developing a framework to quickly open a trial at a cancer center would be appealing to industry trial sponsors and would broaden patient access to cutting-edge clinical research.
Herbst agreed that more community cancer centers should be able to participate in clinical trials, particularly because most patients with cancer are treated in this setting. Herbst also suggested that clinical trials should be designed with the community center in mind and seek input from the leaders in community cancer care at the stage of trial design to ensure that the trials can be done outside of an academic center.
___________________
21 See https://www.asco.org/research-progress/clinical-trials/clinical-trial-eligibility-criteria (accessed April 23, 2019).
Endpoints for Clinical Trials
Ahmad Tarhini, professor and director of the melanoma and skin cancer program at the Cleveland Clinic Taussig Cancer Institute, said more consideration should be given to the types of endpoints used in clinical trials for immune checkpoint inhibitors. Jaffee agreed, and noted that the standard RECIST (Response Evaluation Criteria In Solid Tumors) criteria do not provide adequate assessment of immunotherapies (Wolchok et al., 2009). For example:
- Antitumor response for immunotherapies may take longer when compared to chemotherapy; and
- Clinical responses to immunotherapies can occur after conventional indications of progression are observed (pseudoprogression).
Jaffee noted that immune-modified response criteria have been developed to account for these differences (Hodi et al., 2018). For example, the immune-modified criteria acknowledge that durable, stable disease may actually reflect an antitumor immune response.
“With immunotherapeutics, we are still learning about tumor assessment endpoints and the effects on overall survival,” said Marc Theoret, associate director for immunotherapeutics with the FDA Oncology Center of Excellence. He added that more work is needed to clarify which endpoints can be used to assess clinical benefit or treatment effects of a combination therapy, and to help decide whether the combination should be evaluated at later stages of drug development.
Dosing, Sequencing Administration, and Treatment Duration for Combinations
Another challenge when designing clinical trials for combination therapies is determining the appropriate dose, sequencing of therapies, and treatment durations. Chen noted that with single-agent cancer therapies, determining optimal dosing and treatment duration is already quite challenging. With the added complexity of immunotherapy and multi-agent regimens, these decisions become even more complex. Theoret added that attributing adverse reactions for a combination therapy can also be difficult. Theoret and Tarhini noted that Phase I trials will help collect information on appropriate dosing schedules. Dansey also suggested that dose and
scheduling decisions for combination trials be informed by clinical trials conducted in community-based practices in order to include data from more heterogeneous populations.
Chen stressed that “we could spend 3 to 5 years doing Phase I trials if we started to test these critically important questions, and I think that is the crux of what we face for combination immunotherapy.” Jaffee suggested that more pharmacodynamic studies be conducted to help with these decisions.
Trial Designs to Promote Efficiency
Several speakers discussed opportunities to improve the efficiency of clinical trials assessing combinations with immune checkpoint inhibitors, including different types of master protocols and clinical trials with shared controlled arms.
Tarhini noted that the ever-expanding array of immune therapies in cancer creates a need for greater efficiency in clinical trials. “Our current classic model is a series of clinical trials testing one or two questions at a time in a single disease. This comes at a major cost in terms of time and, obviously, the financial cost,” said Tarhani. It can also limit the number of concurrent trials that can be conducted because multiple trials will be recruiting similar types of patients.
Master Protocols for Combination Trials
Tarhini provided an overview of the concept of master protocols for immunotherapy. He said there are several types of master protocols, as shown in Box 4, that can efficiently and effectively test multiple targeted/immunotherapy agents or therapeutic strategies in relatively small patient subpopulations (Redman and Allegra, 2015; Renfro and Sargent, 2017; Woodcock and LaVange, 2017). He noted that master protocols “could be a major methodologic innovation over the traditional approach of a series of clinical trials, where we may have an overall systematic approach to a disease with more efficient screening, [thereby] increasing the speed of drug development.”
Tarhini highlighted a number of considerations for designing master protocol trials, such as
- Eligibility criteria;
- Interim analyses and stopping rules;
- Statistical analysis plans;
- Inclusion of biomarkers (e.g., PD-L1, MSI-H, TMB, expression signatures);
- Whether trials are tumor agnostic or focus on a specific disease;
- Appropriate endpoints to assess patient outcomes; and
- Whether to use randomization or historical controls.
He also described several challenges. Master protocols require collaboration among competing pharmaceutical companies, which can be challenging to navigate, and these trials are also more logistically and operationally cumbersome, and require more resources to conduct. Identi-
fying meaningful short-term study endpoints that may enable the adaptive designs often used with master protocols can also be more complex with combinations involving immune checkpoint inhibitors.
Tarhini suggested that the NCI-sponsored National Clinical Trials Network (NCTN)22 should conduct more master protocol trials evaluating combination therapies using immune checkpoint inhibitors. Herbst noted that a public–private partnership involving NCTN and a number of organizations launched Lung-MAP (Lung Cancer Master Protocol), an umbrella
___________________
22 See https://www.cancer.gov/research/areas/clinical-trials/nctn (accessed April 24, 2019) and https://www.lung-map.org/about-lung-map (accessed April 24, 2019).
trial evaluating numerous drugs in patients with advanced NSCLC. Herbst added that this trial has been challenging to design because the landscape of lung cancer treatment has changed so quickly over the past 5 years. The trial is now focusing on testing immunotherapy combinations that might work in the refractory disease setting. He added that a major feature of this trial is the collection of biospecimens for correlative research.
Ibrahim added that the Parker Institute is developing the infrastructure to enable launch of collaborative master protocol trials using combination agents coming from different pharmaceutical companies or academic centers. “We feel that this can generate rich mechanistic and molecular data to inform how best to combine different agents,” said Ibrahim. Ibrahim noted that master protocols could also introduce some standardization across ongoing studies, as well as reduce redundancies, especially in regard to small, investigator-initiated studies.
Shared or External Control Arms
As noted earlier, many trials involving immune checkpoint inhibitors are using the same comparator treatment, which is creating redundancy. Pazdur advocated for reducing the number of patients allocated to the control arms of these trials by using a common control arm. McKee said that FDA approached several companies to suggest the use of a common control arm, assuring companies that the statistical evaluation would only involve comparisons of the company’s investigational drug to the common control arm, and not to the investigational agents from the other companies. However, she said there was no interest in pursuing this approach at the time, probably due to the fear that this could lead to comparisons among competing drugs in development. Dansey noted that another reason that companies may have resisted the proposal was because it can be very difficult to construct and negotiate this type of trial. Herbst agreed, and added that master protocol trials sometimes use common control arms, “but each of the arms has different eligibility criteria, so then you almost have to construct a separate control arm for each of the different drugs . . . it gets very hard.”
Dansey and Chen inquired whether FDA would be willing to consider the use of external control arms23 to reduce the number of patients who
___________________
23 According to FDA, an external control “consists of patients who are not part of the same randomized study as the group receiving the investigational agent.” See https://www.fda.gov/downloads/guidances/ucm073139.pdf (accessed April 26, 2019).
are allocated to control arm therapies. Chen suggested that control arms be reduced by bolstering them with real-world datasets. “Certainly from a patient perspective, [it would be beneficial] to have fewer patients on a control arm—especially when some of the control arms are chemotherapy [in the refractory lung cancer setting],” said Herbst. Dansey asked if there was a way to build “a simulation component into [trials]—either for multiple arms or for controls arms—to try and innovate the clinical trial process [and] make it more efficient?” Chen noted that advances in statistical modeling could help in this context: “What you need is a common set of modeling assumptions that then allows you to correct for factors that may not be the same across trials, and then you have a control arm dataset that really is balanced.”
McKee noted that the agency is open to considering novel statistical designs and real-world data, and suggested that companies discuss these ideas with FDA. Pazdur voiced some caution about using external control arms, especially if it reduces randomization. Pazdur said,
Randomization is a very important tool . . . the purpose of randomization [is] to balance the things you don’t know about. We may think we are smart about these diseases, but we don’t know about a lot of prognostic factors or other factors that may have an influence on overall survival or even time to progression.
However, Pazdur noted that the agency is very interested in working with the big data companies to assess whether it is possible to build in randomization using real-world data. “We would really like to attempt to preserve the concept of randomization. That doesn’t mean that you have to do it in the context of a clinical trial—it could be looked at in the context of real-world data. I think that is going to be a challenge for us . . . but that is where I see the future of drug development occurring,” Pazdur said. The FDA Oncology Center of Excellence is conducting Project: Switch,24 which is assessing:
- Whether well-matched external control arms, based on prior clinical trials, can be used to make inferences regarding the effect of a new drug; or
- Whether an external control arm could be used in place of a control arm in ongoing randomized controlled trials for patients with rare cancers, poor prognoses, and few treatment options.
___________________
24 See https://www.fda.gov/NewsEvents/Speeches/ucm629942.htm (accessed April 24, 2019).
Martin Murphy, chief executive officer of the CEO Roundtable on Cancer, noted that Project Data Sphere,25 an open-access data platform that facilitates clinical trials data sharing, is working to develop an external control arm based on historical clinical trials data for small cell lung cancer (SCLC) (Bertagnolli et al., 2017).26 He added that industry, academia, and regulatory partners are also collaborating on a project to study rare adverse events, such as myocarditis, in patients who have received immune checkpoint inhibitor therapies (Neilan et al., 2018).
REAL-WORLD DATA
Abernethy discussed how real-world data can advance the development of immune checkpoint inhibitor therapy. She cited FDA’s definitions of real-world data and real-world evidence27:
- Real-world data are the data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources, such as electronic health records, claims and billing activities, product and disease registries, patient-generated data including in home-use settings, and data gathered from other sources, such as mobile devices.
- Real-world evidence is the clinical evidence regarding the usage and potential benefits or risks of a medical product derived from analysis of real-world data. Real-world evidence can be generated by different study designs or analyses, such as randomized trials, including large simple trials and pragmatic trials, as well as observational studies (prospective and/or retrospective).
“Real-world data offer another kind of discovery. [They work] in concert with our traditional clinical trials and our traditional research mechanisms,” said Abernethy. For example, Flatiron Health’s real-world dataset illustrating the shift in the standard of care for lung cancer over time can inform the development of new clinical trials for immune checkpoint
___________________
25 See https://projectdatasphere.org/projectdatasphere/html/home (accessed May 1, 2019).
26 See https://www.biospace.com/article/releases/project-data-sphere-builds-on-five-years-of-success-by-expanding-open-access-data-platform-and-broadening-research-programs-that-accelerate-new-cancer-patient-therapies (accessed May 1, 2019).
27 See https://www.fda.gov/ScienceResearch/SpecialTopics/RealWorldEvidence/default.htm (accessed April 26, 2019).
inhibitor therapy: “If we are thinking about what our comparator arms are, the comparator is changing across this period,” said Abernethy.
Abernethy said real-world data can be analyzed to determine who uses immune checkpoint inhibitor therapies in clinical practice settings and how these drugs perform. A real-world cohort of patients with metastatic NSCLC who received nivolumab or pembrolizumab were older at treatment initiation and had a higher prevalence of smoking history compared with clinical trial cohorts (Khozin et al., 2018a). Researchers found that this real-world cohort had shorter overall survival compared with clinical trial results, and suggested that this difference may be due to the narrow eligibility criteria of these clinical trials (Khozin et al., 2018b). Khozin stressed that real-world data provide information about patients who are typically excluded from clinical trials, such as patients with organ dysfunction, brain metastases, HIV, or a history of autoimmune diseases.
Abernethy noted that the 21st Century Cures Act28 mandates the Secretary of the Department of Health and Human Services to establish a program evaluating the potential role of real-world evidence in supporting the approvals of new drug indications or in satisfying post-marketing requirements. In response, FDA has released a framework for the its real-world evidence program.29 “If we are going to use [real-world data] . . . in support of making decisions about how to take better care of patients, we better make sure that these are data that we can rely on and, importantly, that these data are adequately representative of the populations for whom we care,” Abernethy stressed.
Abernethy highlighted a checklist of features associated with high-quality real-world data, including the following (Miksad and Abernethy, 2018):
- Clinical depth: Data granularity to enable appropriate interpretation and contextualization of patient information.
- Completeness: Inclusion of both structured and unstructured information supports thorough understanding of patient clinical experience.
- Longitudinal follow-up: Ability to review treatment history and track patient outcomes over time.
___________________
28 Public Law 114-255, December 13, 2016. See https://www.govinfo.gov/content/pkg/PLAW-114publ255/pdf/PLAW-114publ255.pdf (accessed May 7, 2019).
29 See https://www.fda.gov/downloads/ScienceResearch/SpecialTopics/RealWorldEvidence/UCM627769.pdf (accessed April 26, 2019).
- Quality monitoring: Systematic processes implemented to ensure data accuracy and quality.
- Timeliness/recency: Timely monitoring of treatment patterns and trends in health care to derive relevant insights.
- Scalability: Efficient processing of information with data model that evolves with standard of care.
- Generalizability: Representativeness of the data cohorts to the broader patient population.
- Complete provenance: Robust traceability throughout the chain of evidence.
Collaboration and Real-World Data Sharing
Gaurav Singal, vice president of data strategy and product development at Foundation Medicine, noted that real-world data are emerging as key enablers in oncology research, with numerous efforts under way to leverage and link real-world datasets across academic medical centers, consortia, and industry. One example of an industry-driven collaboration is the Clinicogenomic Database, assembled by Foundation Medicine and Flatiron Health, to combine patients’ clinical data with their genomic sequencing data (Agarwala et al., 2018; Singal et al., 2019). Singal said that exploratory analyses of the Clinicogenomic Database found replication of previously described associations between clinical and genomic characteristics, driver mutations and response to targeted therapy, and TMB and response to immunotherapy among patients with advanced NSCLC (Singal et al., 2019). Singal noted that this was an important first step “in what will be an evolving effort to determine where exactly one can reliably use real-world data—collected, integrated, [and] linked in a somewhat . . . novel way.”
He said that a major challenge of this collaboration was developing a methodology to bring together deidentified patient datasets in a manner that preserved patient privacy by complying with the Privacy Rule promulgated under the Health Insurance Portability and Accountability Act of 1996.30 Flatiron Health and Foundation Medicine used a tokenizing
___________________
30 The HIPAA Privacy Rule “establishes national standards to protect individuals’ medical records and other personal health information and applies to health places, health care clearinghouses, and those health care providers that conduct certain health care transactions electronically.” See https://www.hhs.gov/hipaa/for-professionals/privacy/index.html (accessed March 29, 2019).
technology from a third-party clearinghouse to merge these datasets, and added that the companies are able to re-link the datasets on a quarterly basis using this technology (see Figure 3). “I share this because it wasn’t a trivial problem to solve . . . many of the sorts of opportunities we have as a community to share [really depends on] the methods,” Singal stressed.
Abernethy agreed, saying this “is going to be the most important part of data sharing.” She described a pilot project led by Friends of Cancer Research—involving six collaborating partners—with the aim of correlating real-world clinical endpoints with overall survival among patients with advanced NSCLC who received immune checkpoint inhibitor therapy.31 “It was quite remarkable that all of these organizations . . . agreed to work together because we all saw this as a common problem to solve together . . . how do we specify endpoints, how do we come up with a common data model, [and] how do we do this [kind of] work,” said Abernethy. As a group, the organizations developed a workplan for assessing the performance of real-world endpoints across multiple datasets, and then examined whether the various datasets could achieve a similar level of correlation and statistical significance using the common framework.
Abernethy described a number of challenges that need to be overcome to advance real-world data sharing. For example, she said ensuring that the data are accurate, complete, and uniform is difficult. “Getting the data cleaned [is] hard. This is an expensive endeavor, and often takes time, money, and will.” She stressed the importance of promoting common data standards and documenting data quality in order to aggregate large datasets and perform meaningful analyses. She said that a critical challenge is incomplete mortality data among real-world datasets. This threatens the validity of conclusions derived from real-world data, because lengthened survival could be attributed to missing mortality data, rather than a reflection of actual patient outcomes.
WRAP-UP
Dansey reflected that this workshop convened experts with diverse perspectives to consider solutions to ongoing challenges in the development of combination cancer therapies with immune checkpoint inhibitors. The state of the science is changing rapidly, and workshop speakers emphasized
___________________
31 See https://www.focr.org/sites/default/files/pdf/RWE_FINAL%207.6.18.pdf (accessed April 30, 2019).
NOTE: FH = Flatiron Health; FMI = Foundation Medicine; ID = identification; SFTP = Secure File Transfer Protocol.
SOURCES: Singal presentation, July 17, 2018; Singal et al., 2019.
how these therapies are transforming the landscape of cancer treatment, especially for patients with lung cancer, but there are still many challenges to overcome in developing effective combination therapies. Speakers discussed potential opportunities to streamline the deluge of drug development research in this area based on prioritization of rational, science-based combination strategies. He noted the importance of biomarker research—both in refining and standardizing current biomarkers, as well as in the identification, development, and validation of new biomarkers—to better select patients who might benefit from immune checkpoint inhibitor therapy and to prioritize combination strategies for evaluation in clinical trials. To conduct this research, he stressed the critical importance of collecting, processing, and storing biospecimens under standardized conditions. A number of speakers highlighted opportunities to improve clinical trials for evaluating combination strategies, such as prioritizing trials that are designed to answer questions that are important to patients; selecting appropriate endpoints; and using novel designs, such as master protocols, shared control arms, or external control arms. Discussion also focused on how real-world data could build on and improve the evidence base from clinical trials. Dansey added that it was very encouraging to hear that FDA is receptive to considering innovative approaches and has encouraged drug developers to consult with the agency to discuss these opportunities further. Speakers also highlighted the promise of collaborations among industry, academia, and regulatory partners to advance progress in the development of combination therapies with immune checkpoint inhibitors.
REFERENCES
Agarwala, V., S. Khozin, G. Singal, C. O. O’Connell, D. Kuk, G. Li, A. Gossai, V. Miller, and A. P. Abernethy. 2018. Real-world evidence in support of precision medicine: Clinicogenomic cancer data as a case study. 2018. Health Affairs 37(5):765–772.
Alexandrov, L. B., S. Nik-Zainal, D. C. Wedge, S. A. Aparicio, Jr., S. Behjati, A. V. Biankin, G. R. Bignell, N. Bolli, A. Borg, A.-L. Børresen-Dale, S. Boyault, B. Burkhardt, A. P. Butler, C. Caldas, H. R. Davies, C. Desmedt, R. Eils, J. E. Eyfjörd, J. A. Foekens, M. Greaves, F. Hosoda, B. Hutter, T. Ilicic, S. Imbeaud, M. Imielinski, N. Jäger, D. T. W. Jones, D. Jones, S. Knappskog, M. Kool, S. R. Lakhani, C. López-Otín, S. Martin, N. C. Munshi, H. Nakamura, P. A. Northcott, M. Pajic, E. Papaemmanuil, A. Paradiso, J. V. Pearson, X. S. Puente, K. Raine, M. Ramakrishna, A. L. Richardson, J. Richter, P. Rosenstiel, M. Schlesner, T. N. Schumacher, P. N. Span, J. W. Teague, Y. Totoki, A. N. J. Tutt, R. Valdés-Mas, M. M. van Buuren, L. van’t Veer, A. VincentSalomon, N. Waddell, L. R. Yates, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain,
J. Zucman-Rossi, P. Andrew Futreal, U. McDermott, P. Lichter, M. Meyerson, S. M. Grimmond, R. Siebert, E. Campo, T. Shibata, S. M. Pfister, P. J. Campbell, and M. R. Stratton. 2013. Signatures of mutational processes in human cancer. Nature 500:415.
Antonia, S. J., A. Villegas, D. Daniel, D. Vicente, S. Murakami, R. Hui, T. Yokoi, A. Chiappori, K. H. Lee, M. de Wit, B. C. Cho, M. Bourhaba, X. Quantin, T. Tokito, T. Mekhail, D. Planchard, Y.-C. Kim, C. S. Karapetis, S. Hiret, G. Ostoros, K. Kubota, J. E. Gray, L. Paz-Ares, J. de Castro Carpeño, C. Wadsworth, G. Melillo, H. Jiang, Y. Huang, P. A. Dennis, and M. Özgüroğlu. 2017. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. New England Journal of Medicine 377(20):1919–1929.
Armand, P., A. Engert, A. Younes, M. Fanale, A. Santoro, P. L. Zinzani, J. M. Timmerman, G. P. Collins, R. Ramchandren, J. B. Cohen, J. P. De Boer, J. Kuruvilla, K. J. Savage, M. Trneny, M. A. Shipp, K. Kato, A. Sumbul, B. Farsaci, and S. M. Ansell. 2018. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm Phase II Checkmate 205 trial. Journal of Clinical Oncology 36(14):1428–1439.
Aspeslagh, S., S. Postel-Vinay, S. Rusakiewicz, J. C. Soria, L. Zitvogel, and A. Marabelle. 2016. Rationale for anti-OX40 cancer immunotherapy. European Journal of Cancer 52:50–66.
Atkins, M. B., E. R. Plimack, I. Puzanov, M. N. Fishman, D. F. McDermott, D. C. Cho, U. Vaishampayan, S. George, T. E. Olencki, J. C. Tarazi, B. Rosbrook, K. C. Fernandez, M. Lechuga, and T. K. Choueiri. 2018. Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: A non-randomised, open-label, dose-finding, and dose-expansion Phase IB trial. The Lancet Oncology 19(3):405–415.
Ayers, M., J. Lunceford, M. Nebozhyn, E. Murphy, A. Loboda, D. R. Kaufman, A. Albright, J. D. Cheng, S. P. Kang, V. Shankaran, S. A. Piha-Paul, J. Yearley, T. Y. Seiwert, A. Ribas, and T. K. McClanahan. 2017. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. The Journal of Clinical Investigation 127(8):2930–2940.
Baker, R. G., A. X. Hoos, S. J. Adam, D. Wholley, J. H. Doroshow, D. R. Lowy, L. A. Tabak, and F. S. Collins. 2018. The partnership for accelerating cancer therapies. The Cancer Journal 24(3):111–114.
Barker, A. D., C. C. Sigman, G. J. Kelloff, N. M. Hylton, D. A. Berry, and L. J. Esserman. 2009. I-SPY 2: An adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clinical Pharmacology & Therapeutics 86(1):97–100.
Berry, S. M., J. T. Connor, and R. J. Lewis. 2015. The platform trial: An efficient strategy for evaluating multiple treatments. JAMA 313(16):1619–1620.
Bertagnolli, M. M., O. Sartor, B. A. Chabner, M. L. Rothenberg, S. Khozin, C. Hugh-Jones, D. M. Reese, and M. J. Murphy. 2017. Advantages of a truly open-access data-sharing model. New England Journal of Medicine 376(12):1178–1181.
Borghaei, H., C. J. Langer, S. Gadgeel, V. A. Papadimitrakopoulou, A. Patnaik, S. F. Powell, R. D. Gentzler, R. G. Martins, J. P. Stevenson, S. I. Jalal, A. Panwalkar, J. C. Yang, M. Gubens, L. V. Sequist, M. M. Awad, J. Fiore, S. Saraf, S. M. Keller, and L. Gandhi. 2019. 24-month overall survival from KEYNOTE-021 cohort G: Pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous non-small cell lung cancer. Journal of Thoracic Oncology 14(1):124–129.
Brahmer, J. R., C. G. Drake, I. Wollner, J. D. Powderly, J. Picus, W. H. Sharfman, E. Stankevich, A. Pons, T. M. Salay, T. L. McMiller, M. M. Gilson, C. Wang, M. Selby, J. M. Taube, R. Anders, L. Chen, A. J. Korman, D. M. Pardoll, I. Lowy, and S. L. Topalian. 2010. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. Journal of Clinical Oncology 28(19):3167–3175.
Butterfield, L. H., A. K. Palucka, C. M. Britten, M. V. Dhodapkar, L. Hakansson, S. Janetzki, Y. Kawakami, T. O. Kleen, P. P. Lee, C. Maccalli, H. T. Maecker, V. C. Maino, M. Maio, A. Malyguine, G. Masucci, G. Pawelec, D. M. Potter, L. Rivoltini, L. G. Salazar, D. J. Schendel, C. L. Slingluff, Jr., W. Song, D. F. Stroncek, H. Tahara, M. Thurin, G. Trinchieri, S. H. van Der Burg, T. L. Whiteside, J. M. Wigginton, F. Marincola, S. Khleif, B. A. Fox, and M. L. Disis. 2011. Recommendations from the iSBTc-SITC/FDA/NCI workshop on immunotherapy biomarkers. Clinical Cancer Research 17(10):3064–3076.
Cader, F. Z., R. C. J. Schackmann, X. Hu, K. Wienand, R. Redd, B. Chapuy, J. Ouyang, N. Paul, E. Gjini, M. Lipschitz, P. Armand, D. Wu, J. R. Fromm, D. Neuberg, X. S. Liu, S. J. Rodig, and M. A. Shipp. 2018. Mass cytometry of Hodgkin lymphoma reveals a CD4+ regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 132(8):825–836.
Carbone, D. P., M. Reck, L. Paz-Ares, B. Creelan, L. Horn, M. Steins, E. Felip, M. M. van den Heuvel, T.-E. Ciuleanu, F. Badin, N. Ready, T. J. N. Hiltermann, S. Nair, R. Juergens, S. Peters, E. Minenza, J. M. Wrangle, D. Rodriguez-Abreu, H. Borghaei, G. R. Blumenschein, L. C. Villaruz, L. Havel, J. Krejci, J. Corral Jaime, H. Chang, W. J. Geese, P. Bhagavatheeswaran, A. C. Chen, and M. A. Socinski. 2017. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. New England Journal of Medicine 376(25):2415–2426.
Carey, C. D., D. Gusenleitner, M. Lipschitz, M. G. M. Roemer, E. C. Stack, E. Gjini, X. Hu, R. Redd, G. J. Freeman, D. Neuberg, F. S. Hodi, X. S. Liu, M. A. Shipp, and S. J. Rodig. 2017. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 130(22):2420–2430.
Chen, D. S., and I. Mellman. 2013. Oncology meets immunology: The cancer-immunity cycle. Immunity 39(1):1–10.
Chen, D. S., and I. Mellman. 2017. Elements of cancer immunity and the cancer-immune set point. Nature 541(7637):321–330.
Cui, Y., and G. Guo. 2016. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. International Journal of Molecular Sciences 17(11):1942.
Daud, A. I., K. Loo, M. L. Pauli, R. Sanchez-Rodriguez, P. M. Sandoval, K. Taravati, K. Tsai, A. Nosrati, L. Nardo, M. D. Alvarado, A. P. Algazi, M. H. Pampaloni, I. V. Lobach, J. Hwang, R. H. Pierce, I. K. Gratz, M. F. Krummel, and M. D. Rosenblum. 2016. Tumor immune profiling predicts response to anti–PD-1 therapy in human melanoma. The Journal of Clinical Investigation 126(9):3447–3452.
Friends of Cancer Research. 2019. Tumor mutational burden. https://www.focr.org/tmb (accessed April 15, 2019).
Gandhi, L., D. Rodríguez-Abreu, S. Gadgeel, E. Esteban, E. Felip, F. De Angelis, M. Domine, P. Clingan, M. J. Hochmair, S. F. Powell, S. Y.-S. Cheng, H. G. Bischoff, N. Peled, F. Grossi, R. R. Jennens, M. Reck, R. Hui, E. B. Garon, M. Boyer, B. Rubio-Viqueira, S. Novello, T. Kurata, J. E. Gray, J. Vida, Z. Wei, J. Yang, H. Raftopoulos, M. C. Pietanza, and M. C. Garassino. 2018. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. New England Journal of Medicine 378(22):2078–2092.
Garon, E. B., N. A. Rizvi, R. Hui, N. Leighl, A. S. Balmanoukian, J. P. Eder, A. Patnaik, C. Aggarwal, M. Gubens, L. Horn, E. Carcereny, M.-J. Ahn, E. Felip, J.-S. Lee, M. D. Hellmann, O. Hamid, J. W. Goldman, J.-C. Soria, M. Dolled-Filhart, R. Z. Rutledge, J. Zhang, J. K. Lunceford, R. Rangwala, G. M. Lubiniecki, C. Roach, K. Emancipator, and L. Gandhi. 2015. Pembrolizumab for the treatment of non–small-cell lung cancer. New England Journal of Medicine 372(21):2018–2028.
Gopalakrishnan, V., C. N. Spencer, L. Nezi, A. Reuben, M. C. Andrews, T. V. Karpinets, P. A. Prieto, D. Vicente, K. Hoffman, S. C. Wei, A. P. Cogdill, L. Zhao, C. W. Hudgens, D. S. Hutchinson, T. Manzo, M. Petaccia de Macedo, T. Cotechini, T. Kumar, W. S. Chen, S. M. Reddy, R. Szczepaniak Sloane, J. Galloway-Pena, H. Jiang, P. L. Chen, E. J. Shpall, K. Rezvani, A. M. Alousi, R. F. Chemaly, S. Shelburne, L. M. Vence, P. C. Okhuysen, V. B. Jensen, A. G. Swennes, F. McAllister, E. Marcelo Riquelme Sanchez, Y. Zhang, E. Le Chatelier, L. Zitvogel, N. Pons, J. L. Austin-Breneman, L. E. Haydu, E. M. Burton, J. M. Gardner, E. Sirmans, J. Hu, A. J. Lazar, T. Tsujikawa, A. Diab, H. Tawbi, I. C. Glitza, W. J. Hwu, S. P. Patel, S. E. Woodman, R. N. Amaria, M. A. Davies, J. E. Gershenwald, P. Hwu, J. E. Lee, J. Zhang, L. M. Coussens, Z. A. Cooper, P. A. Futreal, C. R. Daniel, N. J. Ajami, J. F. Petrosino, M. T. Tetzlaff, P. Sharma, J. P. Allison, R. R. Jenq, and J. A. Wargo. 2018. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359(6371):97–103.
Hamid, O., and R. D. Carvajal. 2013. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opinion on Biological Therapy 13(6):847–861.
Hamid, O., T. F. Gajewski, A. E. Frankel, T. M. Baure, A. J. Olszanski, J. J. Luke, A. S. Balmanoukian, E. V. Schmidt, B. Sharkey, J. Maleski, M. J. Jones, and T. C. Gangadhar. 2017. 1214O Epacadostat plus pembrolizumab in patients with advanced melanoma: Phase 1 and 2 efficacy and safety results from ECHO-202/KEYNOTE-037. Annals of Oncology 28(Suppl 5):v428–v448.
Hammers, H. J., E. R. Plimack, J. R. Infante, B. I. Rini, D. F. McDermott, L. D. Lewis, M. H. Voss, P. Sharma, S. K. Pal, A. R. A. Razak, C. Kollmannsberger, D. Y. C. Heng, J. Spratlin, M. B. McHenry, and A. Amin. 2017. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: The CheckMate 016 study. Journal of Clinical Oncology 35(34):3851–3858.
Hellmann, M. D., T.-E. Ciuleanu, A. Pluzanski, J. S. Lee, G. A. Otterson, C. Audigier-Valette, E. Minenza, H. Linardou, S. Burgers, P. Salman, H. Borghaei, S. S. Ramalingam, J. Brahmer, M. Reck, K. J. O’Byrne, W. J. Geese, G. Green, H. Chang, J. Szustakowski, P. Bhagavatheeswaran, D. Healey, Y. Fu, F. Nathan, and L. Paz-Ares. 2018. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. New England Journal of Medicine 378(22):2093–2104.
Herbst, R. S., J. C. Soria, M. Kowanetz, G. D. Fine, O. Hamid, M. S. Gordon, J. A. Sosman, D. F. McDermott, J. D. Powderly, S. N. Gettinger, H. E. Kohrt, L. Horn, D. P. Lawrence, S. Rost, M. Leabman, Y. Xiao, A. Mokatrin, H. Koeppen, P. S. Hegde, I. Mellman, D. S. Chen, and F. S. Hodi. 2014. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515(7528):563–567.
Herbst, R. S., D. Morgensztern, and C. Boshoff. 2018. The biology and management of non-small cell lung cancer. Nature 553(7689):446–454.
Hirsch, F. R., A. McElhinny, D. Stanforth, J. Ranger-Moore, M. Jansson, K. Kulangara, W. Richardson, P. Towne, D. Hanks, B. Vennapusa, A. Mistry, R. Kalamegham, S. Averbuch, J. Novotny, E. Rubin, K. Emancipator, I. McCaffery, J. A. Williams, J. Walker, J. Longshore, M. S. Tsao, and K. M. Kerr. 2017. PD-L1 immunohistochemistry assays for lung cancer: Results from phase 1 of the blueprint PD-L1 IHC assay comparison project. Journal of Thoracic Oncology 12(2):208–222.
Hodi, F. S., M. Ballinger, B. Lyons, J. C. Soria, M. Nishino, J. Tabernero, T. Powles, D. Smith, A. Hoos, C. McKenna, U. Beyer, I. Rhee, G. Fine, N. Winslow, D. S. Chen, and J. D. Wolchok. 2018. Immune-modified response evaluation criteria in solid tumors (imRECIST): Refining guidelines to assess the clinical benefit of cancer immunotherapy. Journal of Clinical Oncology 36(9):850–858.
Huang, Y., J. Yuan, E. Righi, W. S. Kamoun, M. Ancukiewicz, J. Nezivar, M. Santosuosso, J. D. Martin, M. R. Martin, F. Vianello, P. Leblanc, L. L. Munn, P. Huang, D. G. Duda, D. Fukumura, R. K. Jain, and M. C. Poznansky. 2012. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proceedings of the National Academy of Sciences 109(43):17561–17566.
Hunger, S. P., and C. G. Mullighan. 2015. Acute lymphoblastic leukemia in children. New England Journal of Medicine 373(16):1541–1552.
Hyman, D. M., I. Puzanov, V. Subbiah, J. E. Faris, I. Chau, J.-Y. Blay, J. Wolf, N. S. Raje, E. L. Diamond, A. Hollebecque, R. Gervais, M. E. Elez-Fernandez, A. Italiano, R.-D. Hofheinz, M. Hidalgo, E. Chan, M. Schuler, S. F. Lasserre, M. Makrutzki, F. Sirzen, M. L. Veronese, J. Tabernero, and J. Baselga. 2015. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. New England Journal of Medicine 373(8):726–736.
Izar, B., L. Jerby-Arnon, A. Rotem, P. Shah, D. Liu, G. Zhang, B. Schilling, O. Rozenblatt-Rosen, G. Marie Boland, F. Hodi, K. Flaherty, E. Allen, B. Johnson, D. Schadendorf, C. Yoon, L. Garraway, and A. Regev. 2018. Single-cell RNA-sequencing and -imaging of melanoma ecosystems reveals sources of resistance to immune checkpoint blockade. Journal of Clinical Oncology 2018 ASCO Annual Meeting 36(Suppl, Abstr 3074).
Jarow, J. P., S. Casak, M. Chuk, L. A. Ehrlich, and S. Khozin. 2016. The majority of expedited Investigational New Drug safety reports are uninformative. Clinical Cancer Research 22(9):2111–2113.
Jerby-Arnon, L., P. Shah, M. S. Cuoco, C. Rodman, M. J. Su, J. C. Melms, R. Leeson, A. Kanodia, S. Mei, J. R. Lin, S. Wang, B. Rabasha, D. Liu, G. Zhang, C. Margolais, O. Ashenberg, P. A. Ott, E. I. Buchbinder, R. Haq, F. S. Hodi, G. M. Boland, R. J. Sullivan, D. T. Frederick, B. Miao, T. Moll, K. T. Flaherty, M. Herlyn, R. W. Jenkins, R. Thummalapalli, M. S. Kowalczyk, I. Canadas, B. Schilling, A. N. R. Cartwright, A. M. Luoma, S. Malu, P. Hwu, C. Bernatchez, M. A. Forget,
D. A. Barbie, A. K. Shalek, I. Tirosh, P. K. Sorger, K. Wucherpfennig, E. M. Van Allen, D. Schadendorf, B. E. Johnson, A. Rotem, O. Rozenblatt-Rosen, L. A. Garraway, C. H. Yoon, B. Izar, and A. Regev. 2018. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175(4):984–997.
Jobin, C. 2018. Precision medicine using microbiota. Science 359(6371):32–34.
Kazandjian, D., D. L. Suzman, G. Blumenthal, S. Mushti, K. He, M. Libeg, P. Keegan, and R. Pazdur. 2016. FDA approval summary: Nivolumab for the treatment of metastatic non–small-cell lung cancer with progression on or after platinum-based chemotherapy. Oncologist 21(5):634–642.
Khozin, S., M. Chuk, T. Kim, G. Kim, R. Pazdur, S. De, and S. Sahoo. 2016. Regulatory watch: Evaluating the potential for digital submission of expedited premarket safety reports to the FDA. Nature Reviews Drug Discovery 15(10):670–671.
Khozin, S., G. Kim, and R. Pazdur. 2017. Regulatory watch: From big data to smart data: FDA’s informed initiative. Nature Reviews Drug Discovery 16(5):306.
Khozin, S., A. P. Abernethy, N. C. Nussbaum, J. Zhi, M. D. Curtis, M. Tucker, S. E. Lee, D. E. Light, A. Gossai, R. A. Sorg, A. Z. Torres, P. Patel, G. M. Blumenthal, and R. Pazdur. 2018a. Characteristics of real-world metastatic non-small cell lung cancer patients treated with nivolumab and pembrolizumab during the year following approval. Oncologist 23(3):328–336.
Khozin, S., K. R. Carson, J. Zhi, M. Tucker, S. E. Lee, D. E. Light, M. D. Curtis, M. Bralic, I. Kaganman, A. Gossai, P. Hofmeister, A. Z. Torres, R. A. Miksad, G. M. Blumenthal, R. Pazdur, and A. P. Abernethy. 2018b. Real-world outcomes of patients with metastatic non-small cell lung cancer treated with programmed cell death protein 1 inhibitors in the year following U.S. regulatory approval. The Oncologist (Epub ahead of print).
Kim, T. K., R. S. Herbst, and L. Chen. 2018. Defining and understanding adaptive resistance in cancer immunotherapy. Trends in Immunology 39(8):624–631.
Kolata, G. 2017. A cancer conundrum: Too many drug trials, not enough patients. The New York Times. https://www.nytimes.com/2017/08/12/health/cancer-drug-trialsencounter-a-problem-too-few-patients.html (accessed April 12, 2019).
Kowanetz, M., W. Zou, M. McCleland, D. R. Gandara, S. Gadgeel, A. Rittmeyer, F. Barlesi, K. Park, D. Shames, H. Koeppen, M. Ballinger, A. Sandler, and P. Hegde. 2017. MA 05.09 pre-existing immunity measured by Teff gene expression in tumor tissue is associated with atezolizumab efficacy in NSCLC. Journal of Thoracic Oncology 12(11):S1817–S1818.
Kwon, E. D., C. G. Drake, H. I. Scher, K. Fizazi, A. Bossi, A. J. van den Eertwegh, M. Krainer, N. Houede, R. Santos, H. Mahammedi, S. Ng, M. Maio, F. A. Franke, S. Sundar, N. Agarwal, A. M. Bergman, T. E. Ciuleanu, E. Korbenfeld, L. Sengelov, S. Hansen, C. Logothetis, T. M. Beer, M. B. McHenry, P. Gagnier, D. Liu, and W. R. Gerritsen. 2014. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. The Lancet Oncology 15(7):700–712.
Larkin, J., V. Chiarion-Sileni, R. Gonzalez, J. J. Grob, C. L. Cowey, C. D. Lao, D. Schadendorf, R. Dummer, M. Smylie, P. Rutkowski, P. F. Ferrucci, A. Hill, J. Wagstaff, M. S. Carlino, J. B. Haanen, M. Maio, I. Marquez-Rodas, G. A. McArthur, P. A. Ascierto, G. V. Long, M. K. Callahan, M. A. Postow, K. Grossmann, M. Sznol,
B. Dreno, L. Bastholt, A. Yang, L. M. Rollin, C. Horak, F. S. Hodi, and J. D. Wolchok. 2015. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. New England Journal of Medicine 373(1):23–34.
Le, D. T., J. N. Uram, H. Wang, B. R. Bartlett, H. Kemberling, A. D. Eyring, A. D. Skora, B. S. Luber, N. S. Azad, D. Laheru, B. Biedrzycki, R. C. Donehower, A. Zaheer, G. A. Fisher, T. S. Crocenzi, J. J. Lee, S. M. Duffy, R. M. Goldberg, A. de la Chapelle, M. Koshiji, F. Bhaijee, T. Huebner, R. H. Hruban, L. D. Wood, N. Cuka, D. M. Pardoll, N. Papadopoulos, K. W. Kinzler, S. Zhou, T. C. Cornish, J. M. Taube, R. A. Anders, J. R. Eshleman, B. Vogelstein, and L. A. Diaz. 2015. PD-1 blockade in tumors with mismatch-repair deficiency. New England Journal of Medicine 372(26):2509–2520.
Le, D. T., J. N. Durham, K. N. Smith, H. Wang, B. R. Bartlett, L. K. Aulakh, S. Lu, H. Kemberling, C. Wilt, B. S. Luber, F. Wong, N. S. Azad, A. A. Rucki, D. Laheru, R. Donehower, A. Zaheer, G. A. Fisher, T. S. Crocenzi, J. J. Lee, T. F. Greten, A. G. Duffy, K. K. Ciombor, A. D. Eyring, B. H. Lam, A. Joe, S. P. Kang, M. Holdhoff, L. Danilova, L. Cope, C. Meyer, S. Zhou, R. M. Goldberg, D. K. Armstrong, K. M. Bever, A. N. Fader, J. Taube, F. Housseau, D. Spetzler, N. Xiao, D. M. Pardoll, N. Papadopoulos, K. W. Kinzler, J. R. Eshleman, B. Vogelstein, R. A. Anders, and L. A. Diaz. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349):409–413.
Levit, L. A., R. P. Perez, D. C. Smith, R. L. Schilsky, D. F. Hayes, and J. M. Vose. 2018. Streamlining adverse events reporting in oncology: An American Society of Clinical Oncology research statement. Journal of Clinical Oncology 36(6):617–623.
Long, G. V., R. Dummer, O. Hamid, T. Gajewski, C. Caglevic, S. Dalle, A. Arance, M. S. Carlino, J. J. Grob, T. M. Kim, L. V. Demidov, C. Robert, J. Larkin, J. Anderson, J. E. Maleski, M. M. Jones, S. J. Diede, and T. C. Mitchell. 2018. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. Journal of Clinical Oncology 2018 ASCO Annual Meeting 36(Suppl, Abstr 108).
Lopes, G., Y.-L. Wu, I. Kudaba, D. Kowalski, B. C. Cho, G. Castro, V. Srimuninnimit, I. Bondarenko, K. Kubota, G. M. Lubiniecki, J. Zhang, D. A. Kush, and T. Mok. 2018. Pembrolizumab versus platinum-based chemotherapy as first-lone therapy for advanced metastatic NSCLC with a PD-L1 tumor proportion score ≥ 1%: Open-label, phase 3 KEYNOTE-042 study. 2018 ASCO Annual Meeting Abstract LBA4.
Mandrekar, J. N. 2010. Receiver operating characteristic curve in diagnostic test assessment. Journal of Thoracic Oncology 5(9):1315–1316.
Marcus, L., S. J. Lemery, P. Keegan, and R. Pazdur. 2019. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clinical Cancer Research (Epub ahead of print).
Matson, V., J. Fessler, R. Bao, T. Chongsuwat, Y. Zha, M.-L. Alegre, J. J. Luke, and T. F. Gajewski. 2018. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 359(6371):104–108.
Miksad, R. A., and A. P. Abernethy. 2018. Harnessing the power of real-world evidence (RWE): A checklist to ensure regulatory-grade data quality. Clinical Pharmacology & Therapeutics 103(2):202–205.
Mitchell, T. C., O. Hamid, D. C. Smith, T. M. Bauer, J. S. Wasser, A. J. Olszanski, J. J. Luke, A. S. Balmanoukian, E. V. Schmidt, Y. Zhao, X. Gong, J. Maleski, L. Leopold, and T. F. Gajewski. 2018. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). Journal of Clinical Oncology (Epub ahead of print).
Motzer, R. J., B. Escudier, D. F. McDermott, S. George, H. J. Hammers, S. Srinivas, S. S. Tykodi, J. A. Sosman, G. Procopio, E. R. Plimack, D. Castellano, T. K. Choueiri, H. Gurney, F. Donskov, P. Bono, J. Wagstaff, T. C. Gauler, T. Ueda, Y. Tomita, F. A. Schutz, C. Kollmannsberger, J. Larkin, A. Ravaud, J. S. Simon, L.-A. Xu, I. M. Waxman, and P. Sharma. 2015. Nivolumab versus everolimus in advanced renal-cell carcinoma. New England Journal of Medicine 373(19):1803–1813.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2016. Policy issues in the clinical development and use of immunotherapy for cancer treatment: Proceedings of a workshop. Washington, DC: The National Academies Press.
Neilan, T. G., M. L. Rothenberg, L Amiri-Kordestani, R. J. Sullivan, R. M. Steingart, W. Gregory, S. Hariharan, T. A. Hammad TA, J, Lindenfeld, M. J. Murphy, J, J. Moslehi, and the Checkpoint Inhibitor Safety Working Group. 2018. Myocarditis associated with immune checkpoint inhibitors: An expert consensus on data gaps and a call to action. The Oncologist. 23(8):874–878.
No sample left behind. 2015. Nature Biotechnology 33:1010.
Palmer, A. C., and P. K. Sorger. 2017. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 17(7):1678–1691.
Ready, N., M. D. Hellmann, M. M. Awad, G. A. Otterson, M. Gutierrez, J. F. Gainor, H. Borghaei, J. Jolivet, L. Horn, M. Mates, J. Brahmer, I. Rabinowitz, P. S. Reddy, J. Chesney, J. Orcutt, D. R. Spigel, M. Reck, K. J. O’Byrne, L. Paz-Ares, W. Hu, K. Zerba, X. Li, B. Lestini, W. J. Geese, J. D. Szustakowski, G. Green, H. Chang, and S. S. Ramalingam. 2019. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): Outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. Journal of Clinical Oncology (Epub ahead of print).
Redman, M. W., and C. J. Allegra. 2015. The master protocol concept. Seminars in Oncology 42(5):724–730.
Renfro, L. A., and D. J. Sargent. 2017. Statistical controversies in clinical research: Basket trials, umbrella trials, and other master protocols: A review and examples. Annals of Oncology 28(1):34–43.
Rimm, D. L., G. Han, J. M. Taube, E. S. Yi, J. A. Bridge, D. B. Flieder, R. Homer, W. W. West, H. Wu, A. C. Roden, J. Fujimoto, H. Yu, R. Anders, A. Kowalewski, C. Rivard, J. Rehman, C. Batenchuk, V. Burns, F. R. Hirsch, and Wistuba, II. 2017. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non-small cell lung cancer. JAMA Oncology 3(8):1051–1058.
Rizvi, N. A., M. D. Hellmann, A. Snyder, P. Kvistborg, V. Makarov, J. J. Havel, W. Lee, J. Yuan, P. Wong, T. S. Ho, M. L. Miller, N. Rekhtman, A. L. Moreira, F. Ibrahim, C. Bruggeman, B. Gasmi, R. Zappasodi, Y. Maeda, C. Sander, E. B. Garon, T. Merghoub, J. D. Wolchok, T. N. Schumacher, and T. A. Chan. 2015. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–128.
Rizvi, H., F. Sanchez-Vega, K. La, W. Chatila, P. Jonsson, D. Halpenny, A. Plodkowski, N. Long, J. L. Sauter, N. Rekhtman, T. Hollmann, K. A. Schalper, J. F. Gainor, R. Shen, A. Ni, K. C. Arbour, T. Merghoub, J. Wolchok, A. Snyder, J. E. Chaft, M. G. Kris, C. M. Rudin, N. D. Socci, M. F. Berger, B. S. Taylor, A. Zehir, D. B. Solit, M. E. Arcila, M. Ladanyi, G. J. Riely, N. Schultz, and M. D. Hellmann. 2018. Molecular determinants of response to anti–programmed cell death (PD)-1 and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. Journal of Clinical Oncology 36(7):633–641.
Roemer, M. G., R. H. Advani, A. H. Ligon, Y. Natkunam, R. A. Redd, H. Homer, C. F. Connelly, H. H. Sun, S. E. Daadi, G. J. Freeman, P. Armand, B. Chapuy, D. de Jong, R. T. Hoppe, D. S. Neuberg, S. J. Rodig, and M. A. Shipp. 2016. PDL1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. Journal of Clinical Oncology 34(23):2690–2697.
Roemer, M. G. M., R. A. Redd, F. Z. Cader, C. J. Pak, S. Abdelrahman, J. Ouyang, S. Sasse, A. Younes, M. Fanale, A. Santoro, P. L. Zinzani, J. Timmerman, G. P. Collins, R. Ramchandren, J. B. Cohen, J. P. D. Boer, J. Kuruvilla, K. J. Savage, M. Trneny, S. Ansell, K. Kato, B. Farsaci, A. Sumbul, P. Armand, D. S. Neuberg, G. S. Pinkus, A. H. Ligon, S. J. Rodig, and M. A. Shipp. 2018. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. Journal of Clinical Oncology 36(10):942–950.
Routy, B., E. Le Chatelier, L. Derosa, C. P. M. Duong, M. T. Alou, R. Daillère, A. Fluckiger, M. Messaoudene, C. Rauber, M. P. Roberti, M. Fidelle, C. Flament, V. Poirier-Colame, P. Opolon, C. Klein, K. Iribarren, L. Mondragón, N. Jacquelot, B. Qu, G. Ferrere, C. Clémenson, L. Mezquita, J. R. Masip, C. Naltet, S. Brosseau, C. Kaderbhai, C. Richard, H. Rizvi, F. Levenez, N. Galleron, B. Quinquis, N. Pons, B. Ryffel, V. Minard-Colin, P. Gonin, J.-C. Soria, E. Deutsch, Y. Loriot, F. Ghiringhelli, G. Zalcman, F. Goldwasser, B. Escudier, M. D. Hellmann, A. Eggermont, D. Raoult, L. Albiges, G. Kroemer, and L. Zitvogel. 2018. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 359(6371):91–97.
Scheerens, H., A. Malong, K. Bassett, Z. Boyd, V. Gupta, J. Harris, C. Mesick, S. Simnett, H. Stevens, H. Gilbert, P. Risser, R. Kalamegham, J. Jordan, J. Engel, S. Chen, L. Essioux, and J. A. Williams. 2017. Current status of companion and complementary diagnostics: Strategic considerations for development and launch. Clinical and Translational Science 10(2):84–92.
Sharma, P., S. Hu-Lieskovan, J. A. Wargo, and A. Ribas. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168(4):707–723.
Shrimali, R. K., S. Ahmad, V. Verma, P. Zeng, S. Ananth, P. Gaur, R. M. Gittelman, E. Yusko, C. Sanders, H. Robins, S. A. Hammond, J. E. Janik, M. Mkrtichyan, S. Gupta, and S. N. Khleif. 2017. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunology Research 5(9):755–766.
Singal, G., P. G. Miller, V. Agarwala, G. Li, G. Kaushik, D. Backenroth, A. Gossai, G. M. Frampton, A. Z. Torres, E. M. Lehnert, D. Bourque, C. O’Connell, B. Bowser, T. Caron, E. Baydur, K. Seidl-Rathkopf, I. Ivanov, G. Alpha-Cobb, A. Guria,
J. He, S. Frank, A. C. Nunnally, M. Bailey, A. Jaskiw, D. Feuchtbaum, N. Nussbaum, A. P. Abernethy, and V. A. Miller. 2019. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. Journal of the American Medical Association 321(14):1391–1399.
Skoulidis, F., M. E. Goldberg, D. M. Greenawalt, M. D. Hellmann, M. M. Awad, J. F. Gainor, A. B. Schrock, R. J. Hartmaier, S. E. Trabucco, L. Gay, S. M. Ali, J. A. Elvin, G. Singal, J. S. Ross, D. Fabrizio, P. M. Szabo, H. Chang, A. Sasson, S. Srinivasan, S. Kirov, J. Szustakowski, P. Vitazka, R. Edwards, J. A. Bufill, N. Sharma, S. I. Ou, N. Peled, D. R. Spigel, H. Rizvi, E. J. Aguilar, B. W. Carter, J. Erasmus, D. F. Halpenny, A. J. Plodkowski, N. M. Long, M. Nishino, W. L. Denning, A. Galan-Cobo, H. Hamdi, T. Hirz, P. Tong, J. Wang, J. Rodriguez-Canales, P. A. Villalobos, E. R. Parra, N. Kalhor, L. M. Sholl, J. L. Sauter, A. A. Jungbluth, M. Mino-Kenudson, R. Azimi, Y. Y. Elamin, J. Zhang, G. C. Leonardi, F. Jiang, K. K. Wong, J. J. Lee, V. A. Papadimitrakopoulou, Wistuba, II, V. A. Miller, G. M. Frampton, J. D. Wolchok, A. T. Shaw, P. A. Janne, P. J. Stephens, C. M. Rudin, W. J. Geese, L. A. Albacker, and J. V. Heymach. 2018. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRASmutant lung adenocarcinoma. Cancer Discovery 8(7):822–835.
Socinski, M. A., R. M. Jotte, F. Cappuzzo, F. Orlandi, D. Stroyakovskiy, N. Nogami, D. Rodriguez-Abreu, D. Moro-Sibilot, C. A. Thomas, F. Barlesi, G. Finley, C. Kelsch, A. Lee, S. Coleman, Y. Deng, Y. Shen, M. Kowanetz, A. Lopez-Chavez, A. Sandler, M. Reck, and IMpower150 Study Group. 2018. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. New England Journal of Medicine 378(24):2288–2301.
Spranger, S., R. Bao, and T. F. Gajewski. 2015. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523(7559):231–235.
Stein, S., M. J. Pishvaian, M. S. Lee, K. H. Lee, S. Hernandez, A. Kwan, B. Liu, W. Grossman, K. Iizuka, and B. Ryoo. 2018. Safety and clinical activity of 1L atezolizumab + bevacizumab in a phase Ib study in hepatocellular carcinoma (HCC). Journal of Clinical Oncology 36(15 Suppl):4074.
Suzuki, H., H. Onishi, J. Wada, A. Yamasaki, H. Tanaka, K. Nakano, T. Morisaki, and M. Katano. 2010. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. European Journal of Immunology 40(1):197–203.
Tang, J., L. Pearce, J. O’Donnell-Tormey, and V. M. Hubbard-Lucey. 2018a. Trends in the global immuno-oncology landscape. Nature Reviews Drug Discovery 17:783.
Tang, J., A. Shalabi, and V. M. Hubbard-Lucey. 2018b. Comprehensive analysis of the clinical immuno-oncology landscape. Annals of Oncology 29(1):84–91.
Tang, J., J. X. Yu, V. M. Hubbard-Lucey, S. T. Neftelinov, J. P. Hodge, and Y. Lin. 2018c. The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nature Reviews Drug Discovery 17:854.
Tian, L., A. Goldstein, H. Wang, H. Ching Lo, I. Sun Kim, T. Welte, K. Sheng, L. E. Dobrolecki, X. Zhang, N. Putluri, T. L. Phung, S. A. Mani, F. Stossi, A. Sreekumar, M. A. Mancini, W. K. Decker, C. Zong, M. T. Lewis, and X. H. F. Zhang. 2017. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544:250.
Tirosh, I., B. Izar, S. M. Prakadan, M. H. Wadsworth, 2nd, D. Treacy, J. J. Trombetta, A. Rotem, C. Rodman, C. Lian, G. Murphy, M. Fallahi-Sichani, K. Dutton-Regester, J. R. Lin, O. Cohen, P. Shah, D. Lu, A. S. Genshaft, T. K. Hughes, C. G. Ziegler, S. W. Kazer, A. Gaillard, K. E. Kolb, A. C. Villani, C. M. Johannessen, A. Y. Andreev, E. M. Van Allen, M. Bertagnolli, P. K. Sorger, R. J. Sullivan, K. T. Flaherty, D. T. Frederick, J. Jane-Valbuena, C. H. Yoon, O. Rozenblatt-Rosen, A. K. Shalek, A. Regev, and L. A. Garraway. 2016. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352(6282):189–196.
Topalian, S. L., F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton, and M. Sznol. 2012. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. New England Journal of Medicine 366(26):2443–2454.
Tsao, M. S., K. M. Kerr, M. Kockx, M. B. Beasley, A. C. Borczuk, J. Botling, L. Bubendorf, L. Chirieac, G. Chen, T. Y. Chou, J. H. Chung, S. Dacic, S. Lantuejoul, M. Mino-Kenudson, A. L. Moreira, A. G. Nicholson, M. Noguchi, G. Pelosi, C. Poleri, P. A. Russell, J. Sauter, E. Thunnissen, I. Wistuba, H. Yu, M. W. Wynes, M. Pintilie, Y. Yatabe, and F. R. Hirsch. 2018. PD-L1 immunohistochemistry comparability study in real-life clinical samples: Results of blueprint phase 2 project. Journal of Thoracic Oncology 13(9):1302–1311.
Tsilimigras, M. C., A. Fodor, and C. Jobin. 2017. Carcinogenesis and therapeutics: The microbiota perspective. Nature Microbiology 2:17008.
Tu, M. M., F. Y. F. Lee, R. T. Jones, A. K. Kimball, E. Saravia, R. F. Graziano, B. Coleman, K. Menard, J. Yan, E. Michaud, H. Chang, H. A. Abdel-Hafiz, A. I. Rozhok, J. E. Duex, N. Agarwal, A. Chauca-Diaz, L. K. Johnson, T. L. Ng, J. C. Cambier, E. T. Clambey, J. C. Costello, A. J. Korman, and D. Theodorescu. 2019. Targeting DDR2 enhances tumor response to anti–PD-1 immunotherapy. Science Advances 5(2):eaav2437.
Tumeh, P. C., C. L. Harview, J. H. Yearley, I. P. Shintaku, E. J. Taylor, L. Robert, B. Chmielowski, M. Spasic, G. Henry, V. Ciobanu, A. N. West, M. Carmona, C. Kivork, E. Seja, G. Cherry, A. J. Gutierrez, T. R. Grogan, C. Mateus, G. Tomasic, J. A. Glaspy, R. O. Emerson, H. Robins, R. H. Pierce, D. A. Elashoff, C. Robert, and A. Ribas. 2014. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515(7528):568–571.
Velcheti, V., E. S. Kim, T. Mekhail, C. Dakhil, P. Stella, X. Shen, S. Hu, S. Paul, D. Shames, C. Yun, S. Phan, and M. Socinsk. 2018. Prospective clinical evaluation of blood-based tumor mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC): Interim B-F1RST results. Paper presented at 2018 ASCO Annual Meeting: Chicago, IL.
Vergote, I., M. Teneriello, M. A. Powell, D. S. Miller, A. A. Garcia, O. N. Mikheeva, T. Pinter, M. Bidzinski, C. L. Cebotaru, J. Fan, M. Ren, N. Meneses, Y. Funahashi, T. Kadowaki, J. P. O’Brien, and R. T. Penson. 2013. A phase II trial of lenvatinib in patients with advanced or recurrent endometrial cancer: Angiopoietin-2 as a predictive marker for clinical outcomes. Journal of Clinical Oncology 31(15 Suppl):Abstract 5520.
Wang, C., K. B. Thudium, M. Han, X. T. Wang, H. Huang, D. Feingersh, C. Garcia, Y. Wu, M. Kuhne, M. Srinivasan, S. Singh, S. Wong, N. Garner, H. Leblanc, R. T. Bunch, D. Blanset, M. J. Selby, and A. J. Korman. 2014. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunology Research 2(9):846–856.
Weber, J. S., G. Gibney, R. J. Sullivan, J. A. Sosman, C. L. Slingluff, D. P. Lawrence, T. F. Logan, L. M. Schuchter, S. Nair, L. Fecher, E. I. Buchbinder, E. Berghorn, M. Ruisi, G. Kong, J. Jiang, C. Horak, and F. S. Hodi. 2016. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. The Lancet Oncology 17(7):943–955.
Wherry, E. J. 2011. T cell exhaustion. Nature Immunology 12(6):492–499.
Wolchok, J. D., A. Hoos, S. O’Day, J. S. Weber, O. Hamid, C. Lebbe, M. Maio, M. Binder, O. Bohnsack, G. Nichol, R. Humphrey, and F. S. Hodi. 2009. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clinical Cancer Research 15(23):7412–7420.
Wong, P. F., W. Wei, J. W. Smithy, B. Acs, M. I. Toki, K. R. Blenman, D. Zelterman, H. M. Kluger, and D. L. Rimm. 2019. Multiplex quantitative analysis of tumor-infiltrating lymphocytes and immunotherapy outcome in metastatic melanoma. Clinical Cancer Research 25(8):2442–2449.
Woodcock, J., and L. M. LaVange. 2017. Master protocols to study multiple therapies, multiple diseases, or both. New England Journal of Medicine 377(1):62–70.
Yarchoan, M., A. Hopkins, and E. M. Jaffee. 2017. Tumor mutational burden and response rate to PD-1 inhibition. New England Journal of Medicine 377(25):2500–2501.
Yasuda, S., M. Sho, I. Yamato, H. Yoshiji, K. Wakatsuki, S. Nishiwada, H. Yagita, and Y. Nakajima. 2013. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clinical and Experimental Immunology 172(3):500–506.
Zaretsky, J. M., A. Garcia-Diaz, D. S. Shin, H. Escuin-Ordinas, W. Hugo, S. Hu-Lieskovan, D. Y. Torrejon, G. Abril-Rodriguez, S. Sandoval, L. Barthly, J. Saco, B. Homet Moreno, R. Mezzadra, B. Chmielowski, K. Ruchalski, I. P. Shintaku, P. J. Sanchez, C. Puig-Saus, G. Cherry, E. Seja, X. Kong, J. Pang, B. Berent-Maoz, B. Comin-Anduix, T. G. Graeber, P. C. Tumeh, T. N. Schumacher, R. S. Lo, and A. Ribas. 2016. Mutations associated with acquired resistance to PD-1 blockade in melanoma. New England Journal of Medicine 375(9):819–829.
Zheng, J., A. Chang, J. Larkin, R. J. Motzer, M. Amantea, C. Bello, D. Pavlov, M. Geraldes, M. Martignoni, A. Di Pietro, and G. Andrews. 2017. Potential impact of avelumab+axitinib (A+AX) on tumor size (TS) compared with historical data of sunitinib (S) as evaluated by a modeling and simulation (MS) approach. Paper presented at ESMO 2017 Congress: Madrid, Spain.




























































