
Successful development of gene-targeted therapies for central nervous system (CNS) disorders over the past few years—particularly the approval of the antisense oligonucleotide (ASO) Spinraza® in 2016 for spinal muscular atrophy (SMA); Luxturna™ in 2017 for a rare form of inherited retinal dystrophy; and most recently in 2019, the gene replacement therapy Zolgensma® for the treatment of SMA1—were built on a long history of gene therapy products that were tested in clinical trials, but never made it to the clinic, said Lamya Shihabuddin. Lessons from both successful and unsuccessful trials are equally informative, said Shihabuddin.
APPROVED GENE-TARGETED THERAPIES FOR MONOGENIC CENTRAL NERVOUS SYSTEM DISORDERS
Single-gene or monogenic disorders, although each are relatively rare, offer perhaps the most straightforward approach for gene-targeted therapies. Indeed, said Biogen’s Chris Henderson, the gene-targeted CNS therapies that have been successful so far have all targeted monogenic disorders in children.
___________________
1 Zolgensma® was approved about 1 month after the workshop.
Starting with familial forms of disease makes sense because it allows investigators to work out how to design a modality to achieve the large effect sizes needed. The next step, he said, will be to find better targets for sporadic forms of disease and to move from treating childhood-onset to adult-onset disorders.
Nusinersen: An Antisense Oligonucleotide Treatment for Spinal Muscular Atrophy
In December 2016, the ASO medication nusinersen (Spinraza®) became the first Food and Drug Administration (FDA)-approved drug to treat SMA (Paton, 2017), a rare autosomal-recessive neuromuscular disorder and the leading genetic cause of infant mortality, caused by mutations or deletions in the survival motor neuron 1 (SMN1) gene, which encodes the SMN protein (Farrar and Kiernan, 2015). Nusinersen was approved by the European Medicines Agency (EMA) in June 2017.
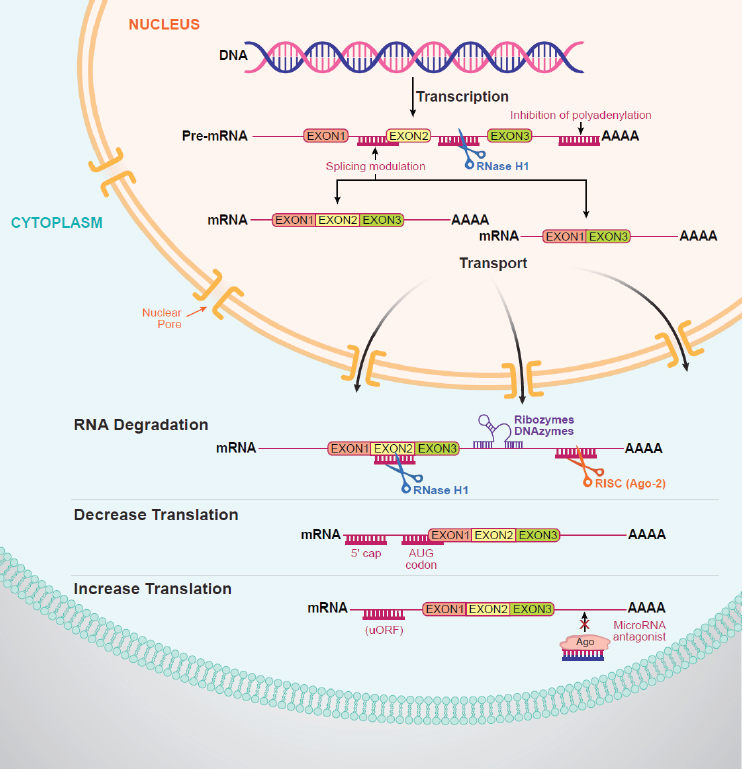
C. Frank Bennett, senior vice president of research at Ionis Pharmaceuticals, used the approval of nusinersen as an example of the potential of ASOs to treat genetically based CNS disorders. Once synthetic ASOs bind to RNA, they can evoke different mechanisms to modulate its function, including degradation of the RNA, modulating splicing, or decreasing or increasing translation of a particular protein (see Figure 2-1). There are currently six approved antisense drugs on the market, said Bennett.
Mutation or deletion of SMN1 causes a deficiency in production of the SMN protein, which results in motor neuron dysfunction. Late in human evolution, a duplication of SMN1 resulted in a second gene called SMN2, which differs from SMN1 by just 5 to 11 nucleotides, said Bennett. However, the SMN2 gene has a change in one nucleotide within exon 7, which results in alternative splicing of the exon, with approximately 80 percent of transcripts skipping exon 7, which produces a truncated protein that is rapidly degraded. The remaining 20 percent of transcripts contain exon 7 and produce full-length, fully functional SMN protein.
SMA presents in a variable manner, said Bennett, ranging from type 1 infantile onset, which is associated with a very short life expectancy, to type 3 later onset, associated with a near-normal life expectancy. Disease severity correlates with the number of copies of the SMN2 gene. Thus, most children with SMA type 1 have two copies of SMN2, begin to show symptoms before 6 months of age, and are never able to sit. Children with SMA type 2 usually have three copies of SMN2, begin to show symptoms after 6 months of age, are able to sit and stand but not walk, and have a shortened life expectancy. Children with SMA type 3 usually have three or four copies of SMN2, begin to show symptoms after 6 months of age, and have a near-normal life expectancy; although they may learn to walk, muscle weakness
SOURCE: Presented by C. Frank Bennett, April 23, 2019.
and skeletal deformities eventually lead to a non-ambulatory state for most of these children (Finkel et al., 2014; Rudnik-Schoneborn et al., 2009).
Nusinersen targets the SMN2 pre-mRNA, binding to a sequence in the SMN2 pre-mRNA, which modulates splicing and leads to production of full-length SMN2 mRNA and SMN2 protein (Hua et al., 2010). Preclinical studies established proof of mechanism and biology, determined the pharmacokinetic and pharmacodynamic relationship, optimized delivery methods, and demonstrated a lack of toxicity, said Bennett (Paton, 2017).
To design a clinical program, Bennett and colleagues (including partners at Biogen) relied on natural history studies published by the Pediatric Neuromuscular Clinical Research Network for SMA (Finkel et al., 2014; Kaufmann et al., 2012). These studies allowed them to demonstrate, for example, in an open-label Phase 2 study in infants with SMA type 1 that the survival benefit in patients treated with nusinersen differed markedly from the natural history of the disease and that benefits were greatest when infants were treated as early in the disease as possible, even before symptom onset (Finkel et al., 2016). Regulators were concerned, however, that these differences between study participants and natural history controls might reflect improved care of SMA patients or that healthier patients were being preselected for the study, he said. Thus, the sponsors designed a very large sham-controlled study. FDA acknowledges that an improvement in motor function scores instead of survival benefit might be acceptable, said Bennett. Fortunately, he said, the trial design included a prespecified interim analysis. This analysis showed that nusinersen improved both survival and motor function; these observations resulted in early termination of the trial (Finkel et al., 2017) and eventual approval by both FDA and EMA.
Voretigene Neparvovec: Gene Therapy for an Ultrarare Genetic Eye Disease
Kathleen Reape, chief medical officer at Spark Therapeutics, focused her presentation on clinical and regulatory challenges associated with the development of voretigene neparvovec (Luxturna®), a gene therapy product approved by FDA in 2017 and EMA in 2018 for the treatment of an ultrarare inherited retinal disease caused by biallelic mutations in the RPE65 gene. RPE65 encodes an enzyme necessary for production of a vitamin A derivative in photoreceptors (Bennett et al., 2012). The absence of a functional enzyme leads to the buildup of toxic precursors, the death of photoreceptors, and progressive loss of vision. Voretigene supplies a copy of the gene encoding the enzyme and restores the visual cycle, said Reape.
One of the early decisions the developers made was to base the indication on a molecular diagnosis rather than clinical symptoms, because even though the hallmark feature of the disorder is nyctalopia, or night blindness, only individuals specifically with biallelic RPE65 mutations will benefit from the therapy, said Reape. Requiring a molecular diagnosis led to certain hurdles in terms of participant recruitment and enrollment for clinical trials given the limited availability of genotyping. This continues to be a challenge for the commercial product, she said.
Development of voretigene benefited from the availability of a naturally occurring large animal model of the condition—the Briard dog—and extensive preclinical work by Jean Bennett and colleagues at the University of Pennsylvania (Bennett et al., 2012; Bennicelli et al., 2008). The dogs
enabled definition of the dose range for the first clinical trial and provided insight into the expected magnitude and onset of the treatment response, said Reape. Other challenges to overcome in the development of voretigene included balancing the potential benefit of the treatment with the risk of surgical delivery via subretinal injection in children, the selection of primary and secondary endpoints, and the choice of a control group.
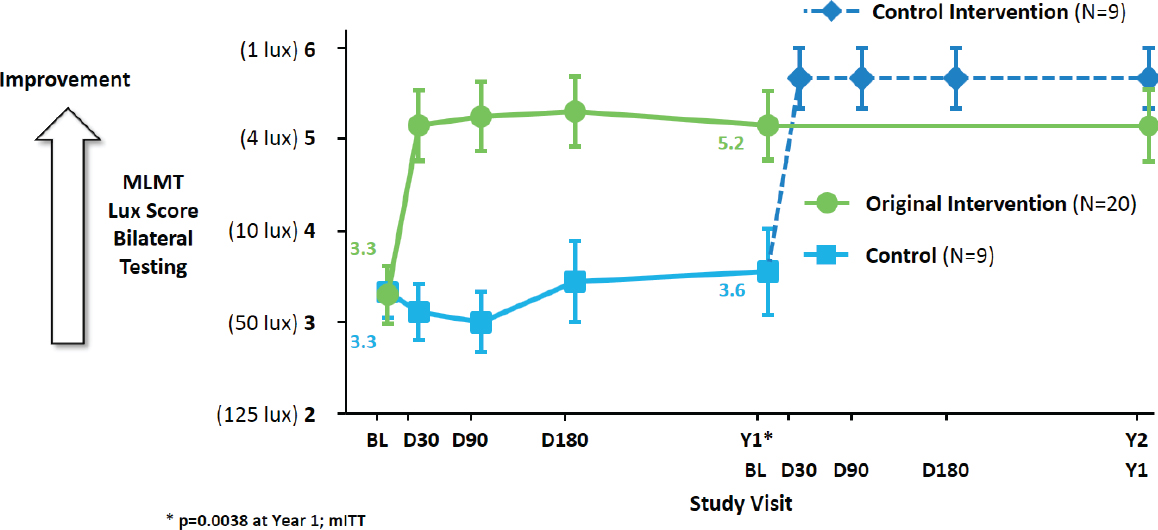
Individuals with biallelic RPE65 mutations progressively lose the ability to detect light, resulting in impaired navigation and other vision-dependent activities (Chung et al., 2018). Because existing functional vision tests were found to inadequately capture the effect of illumination on speed and accuracy of navigation, investigators developed and validated a novel endpoint—the multiluminance mobility test (MLMT)—which tests functional vision and incorporates components of visual field, visual acuity, and light sensitivity, said Reape (Chung et al., 2018). The MLMT tests the performance of participants navigating a course with obstacles and arrows to follow at seven different light levels with a minimum number of errors in a prespecified amount of time. The primary endpoint was the mean change in lowest passing light level. Reape said the investigators created 12 different standardized courses that were presented in a randomized fashion to minimize learning effects. They used an independent reading center with two masked adjudicators using a detailed grading protocol with clearly defined parameters for what constituted errors, passes, and fails. Secondary endpoints included full-field light sensitivity threshold testing, monocular MLMT performance, and visual acuity, as well as exploratory endpoints assessing visual fields, she said, adding that participants or parents also completed a visual function questionnaire to assess activities of daily living relevant to vision loss (Russell et al., 2017a). Reape noted the importance of including both subjective and objective endpoints for these types of studies to ensure that any change measured is clinically meaningful.
Choosing a control group for this trial was complicated, said Reape. Often for ultrarare diseases, natural history data can be used; however, such data did not exist for this condition. Using the contralateral eye was also considered, but raised concerns that in developing children, leaving one eye untreated could cause other problems or that the rate of degeneration may not be symmetrical in the two eyes, leading to incorrect conclusions. In addition, in the real-world setting, each eye would be dosed separately within a relatively short period of time. Reape said the final decision was to use a two-to-one randomization, have the untreated control group observed for 1 year, and then offer control subjects the opportunity to cross over and receive this treatment. Because the trial included young children, the company rejected the idea of using a sham surgical procedure; thus, the trial was an open-label (unmasked) study. Results from the original untreated control group after crossing over and receiving treatment confirmed that the
magnitude and onset of the treatment effect was similar among participants (see Figure 2-2).
In these trials of rare conditions that enroll such small numbers of participants, capturing a meaningful incidence of adverse events is difficult, said Reape. She also noted that the labeling reflects the difficulty of separating out adverse events caused by the product versus the administration procedure, vitrectomy, and subretinal injection, adding that when there is a risk associated with the administration procedure, it is especially important to identify the population that is likely to benefit.
AVXS-101: Clinical Phase Gene Replacement Therapy for SMA
Another gene-targeted treatment for SMA was described by Petra Kaufmann, vice president, research and development translational medicine at AveXis.2 AVXS-101 (Zolgensma®) is a gene replacement therapy product designed to treat the root cause of SMA by targeting the mutation in SMN1, said Kaufmann. AVXS-101 delivers the gene via a vector constructed from the adeno-associated virus 9 (AAV9), which is able to cross the blood–brain barrier and reach motor neurons in the spinal cord. Inside the AAV9 capsid shell, a double-stranded piece of DNA has been created from the relevant parts of the transgene controlled by a promoter that enables sustained production of the missing SMN1 protein and flanked by inverted terminal repeats, which accelerate transcription of the transgene and production of a full-length functioning protein (Powell et al., 2015) (see Figure 2-3).
Kaufmann cited preclinical studies in a mouse model of SMN that showed a dose-dependent increase in survival when treated with an AAV9-SMN construct (Foust et al., 2010), and another study in newborn-to-3-year-old cynomolgus macaques demonstrating that AAV9 injected into the cerebrospinal fluid crosses the blood–brain barrier and delivers its transgene to motor neurons, resulting in restricted expression of the gene in the central nervous system (Bevan et al., 2011). Another study in a pig model of SMA showed that presymptomatic treatment with AAV9-SMN prevented the development of SMA symptoms (Duque et al., 2015).
These preclinical studies led to a Phase 1 clinical trial in 15 infants with SMA1, with 3 patients receiving a low dose and 12 receiving the intended higher dose (Mendell et al., 2017). The primary outcome was safety; a secondary efficacy outcome was survival free of permanent ventilation by Bilevel Positive Airway Pressure for more than 16 hours per day, and an exploratory outcome was a decline on the Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders motor function scale (Finkel et al., 2014).
___________________
2 This therapy was approved by FDA in May 2019, approximately 1 month after the workshop.
SOURCES: Presented by Kathleen Reape, April 23, 2019; Russell et al., 2017a,b.
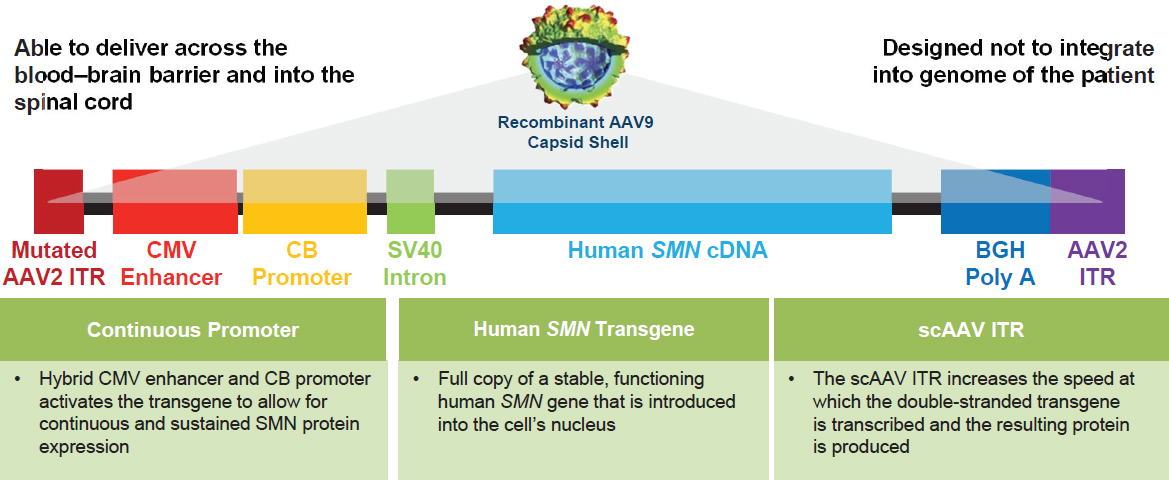
NOTES: AAV2 = adeno-associated virus serotype 2; AAV9 = AAV serotype 9; BGH Poly A = bovine growth hormone polyadenylation; CB = chicken b-actin; cDNA = complementary DNA; CMV = cytomegalovirus; ITR = inverted terminal repeat; scAAV = self-complementary AAV; SMA = spinal muscular atrophy; SMN = survival motor neuron; SV = simian virus.
SOURCE: Presented by Petra Kaufmann, April 23, 2019.
Natural history data were used as a control. Kaufmann noted the difficulty of conducting natural history studies in patients with rare and serious diseases such as SMA. “It’s a real gift of altruism on the part of families to participate over often more than a year in a study when there is no treatment,” she said. She credited not only the families, but also the SMA Foundation for funding the Pediatric Neuromuscular Clinical Research Network study, and the National Institute of Neurological Disorders and Stroke for funding the National Network for Excellence in Neuroscience Clinical Trials SMA Infant Biomarker Study, which enrolled and for 24 months followed 26 infants with genetically confirmed SMA and 27 age, sex, and birthweight-matched healthy infants. Among the SMA cohort, 12 died and 2 received invasive ventilatory support (Kolb et al., 2017).
Kaufmann briefly described the large effect size observed in the Phase 1 study: 11 of the 12 patients who got the proposed dose could sit briefly, and some even walked. At the long-term follow-up, they had not lost milestones, and some had even gained. The demonstrated safety as well as transformative improvements on efficacy endpoints led to a multicenter Phase 3 trial. Kaufmann said that interim data from this Phase 3 study demonstrated improved survival and motor function improvement. On May 24, 2019, FDA approved Zolgensma® as the first gene therapy to treat children younger than age 2 with SMA (FDA, 2019).
Kaufmann noted that the clinical studies also showed that younger patients, some of them presymptomatic, did better than older patients. Even those less than 6 weeks old with two or three copies of the mutated SMN2 gene, that is, those predicted to have a severe form of SMA, did well, suggesting that the greatest benefit is in patients treated early. The lesson learned from this observation, she said, is the importance of early diagnosis so that patients can be treated as early as possible.
One patient in the Phase 3 study made good progress in terms of motor function but, sadly, died from SMA. Kaufmann described how the parents provided an altruistic gift by consenting to an autopsy, which provided a unique opportunity to investigate biodistribution of the vector. This case study showed that, similar to what has been seen in animal studies, the vector was broadly distributed in all tissues evaluated, including all regions of the spinal cord. SMN expression was also demonstrated in motor neurons of this patient, she said.
UNSUCCESSFUL GENE THERAPY TRIALS: LESSONS LEARNED
In efforts to identify an effective treatment for Parkinson’s disease (PD), Jeffrey Kordower, the Alla V. and Solomon Jesmer Professor of Aging and Neurological Sciences at the Rush University Medical Center, has conducted several gene therapy and cell replacement trials, all of which have failed.
Although disappointing, Kordower said these trials have also provided valuable lessons relevant to ongoing and future gene-targeting trials.
In the late 1990s, Kordower and colleagues used intracerebroventricular injections of glial-derived neurotrophic factor (GDNF) to treat a man with PD (Kordower et al., 1999). The treatment not only failed to improve the man’s parkinsonian symptoms, but also was associated with substantial side effects. Kordower said the trial was not supported by preclinical data and never should have been performed. Poor distribution of GDNF in the brain indicated that a better delivery method was needed. Indeed, Kordower said that most gene therapy trials fail because of delivery problems.
Next, they tested a gene therapy approach using GDNF delivered by a lentiviral vector to monkeys that had been given injections of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a neurotoxin that induces degeneration of nigrostriatal neurons and motor deficits similar to those seen in patients with PD (Kordower et al., 2000). While these animals improved far more than control animals, and a subsequent Phase 1 human trial showed adequate safety and tolerability, a Phase 2 trial failed. Kordower said the reason is that progression of PD results from a combination of brain pathologies (Buchman et al., 2019). Reversing nigrostriatal dysfunction alone is not sufficient, he said.
A subsequent trial of a different neurotrophic factor called neurturin also failed, said Kordower. Inadequate delivery of the agent to the brain again was the culprit, although this time the reason was that in order to avoid a long surgical procedure, the investigators injected too low of a dose of the protein to the brain. There was also another problem. As shown originally by Patrick Aebischer, Christophe Lo Bianco, and colleagues in 2004, GDNF has no beneficial effect in a synucleinopathy model of PD (Lo Bianco et al., 2004), said Kordower. He and his colleagues showed in a recent autopsy study that even patients with prodromal PD have nerve fibers filled with α-synuclein (Chu et al., 2018). There was no way this approach was going to work, said Kordower.
Kordower said the failures of GDNF trials illustrate the multiple points where development of gene-targeted therapies can go wrong. First, he cautioned that preclinical data can be misleading. Because animals do not develop PD in nature, the models used in preclinical studies may not accurately reflect the disease process in humans. Preclinical studies, he said, should be designed not to be successful, but to inform clinical trials so that those trials will be successful. He argued that many failed clinical trials are the result of translational scientists moving forward without a clear understanding of the human disease or the patient population they are trying to treat. Finally, he stressed the importance of interpreting data with rigor and honesty. Adhering to these guidelines will save time and money while not compromising the safety of trial participants, he said.
This page intentionally left blank.












