
Central nervous system (CNS) therapies historically have been restricted to small molecules because large molecules do not get into the brain very well, said David Bredt, global head of neuroscience discovery at Johnson & Johnson Pharmaceutical Group. The field has tried to develop monoclonal antibodies for neurological diseases, but thus far has been unsuccessful, he said. Gene therapy offers an alternate way of selecting CNS targets, but as noted by Jeffrey Kordower in Chapter 2, delivery of gene-targeted therapies to their targets has proved challenging. New technologies that incorporate novel capsid proteins and/or carefully selected promoters are in development to ensure that therapies are expressed in the cells of interest in the CNS, said Bredt.
When choosing the appropriate modality, several issues need to be considered, said Beverly Davidson, director of the Raymond G. Perlman Center for Cellular and Molecular Therapeutics at the Children’s Hospital of Philadelphia and professor of pathology and laboratory medicine at the Perelman School of Medicine, University of Pennsylvania. Whether targeting RNA or DNA, the delivery approach must be chosen with safety in mind, said Davidson. For example, the safety of chronic expression versus redosing or regulated expression is an important consideration when targeting RNA. When targeting DNA and RNA, the delivery modality should match the biodistribution goal of the therapy, or redosing may need to be considered. Whether targeting the genome or RNA with sustained delivery, Davidson noted the importance of considering immune responses. Genome editing raises additional safety considerations, including consideration of what happens at the editing site.
For gene addition approaches, Davidson noted the importance of fitting the genetics to the therapy being developed, that is, determining whether the
optimal target is the gene itself or a modifier of the gene. Other considerations include the region or cell type to be targeted, and for loss-of-function disorders, whether cross-correction—the ability of non-transduced cells to take up genetically modified gene products through receptor binding—can be exploited. She added that the tolerability of gene products also needs to be addressed, with expression fine-tuned if a gene product is not well tolerated.
DELIVERY OF GENE-TARGETED THERAPIES TO THE CENTRAL NERVOUS SYSTEM
Sarah DeVos, project leader at Denali Therapeutics, described three different modalities for delivery of gene-targeting therapeutics to the CNS:
- Naked gene therapy, where the genetic material is delivered without a viral capsid to modulate where the gene is expressed.
- Peripheral adeno-associated virus (AAV) gene therapy, in which the vector delivers its cargo to a peripheral organ such as the liver, where the gene is expressed and protein generated and could be secreted.
- Central AAV gene therapy, where the vector delivers the gene via direct injection into the brain or intravenously if the vector is capable of crossing the blood–brain barrier.
Blood–brain barrier penetration is a hurdle for both naked gene therapy and peripheral approaches, said DeVos. However, she has shown that both the naked gene therapy approach with antisense oligonucleotides and the central approach with AAV have potential for repressing the expression of the microtubule-associated protein tau gene, more commonly referred to simply as tau, one of the two hallmark pathogenic proteins in Alzheimer’s disease and several other forms of dementia known as tauopathies.
Gene-targeted therapies and immunotherapies also have the potential to elicit an immune response from the person being treated, which can reduce the efficacy of a treatment, particularly if redosing is needed. Ronald Crystal, professor and chair of the department of genetic medicine at the Weill Medical College of Cornell University, said more work and better models are needed to determine whether immunosuppression is necessary, when to immunosuppress, and with what drugs.
ASO Therapies
DeVos demonstrated in mouse models expressing mutant human tau that repressing tau expression with antisense oligonucleotides (ASOs) reversed
tau pathology (DeVos et al., 2017). One advantage of this ASO approach, she said, is that because it is not permanent, its safety can be assessed even if the full biological function of a protein is not fully understood.
Anastasia Khvorova, professor in the RNA Therapeutics Institute at the University of Massachusetts Medical School, said that for oligonucleotide therapeutics, safe delivery to the CNS can also be achieved by defining the chemical and structural architecture of the oligonucleotide to have the desired pharmacokinetic properties, that is, absorption, distribution, metabolism, and excretion. Once this structural backbone has been defined, the oligonucleotide can be easily reprogrammed with different sequences to silence genes on demand.
This design approach has been used to generate three types of oligonucleotide therapeutics that are currently in clinical development, said Khvorova (Khvorova and Watts, 2017) (see Figure 3-1). The simplest type, called steric-blocking oligonucleotides, simply bind to mRNA to block translation or alter slicing. Spinraza®, the only approved oligonucleotide therapy in the CNS, is an example of a steric-blocking antisense product. The second class of antisense compounds described by Khvorova require interaction with the enzyme ribonuclease H to cleave the targeted RNA and downregulate gene activity. RNA interference (RNAi), the most complex type of oligonucleotide therapeutic, interacts with other cellular proteins to form the RNA-induced silencing complex and silence gene expression.1 Over the past two decades, Khvorova said the chemistry has evolved substantially to enhance the pharmacokinetic and pharmacodynamic properties of small interfering RNAs (siRNAs), increase their potency and duration of effect, and chemically stabilize the oligonucleotides without interfering with their protein machinery interactions.
In August 2018, patisiran became the first RNAi-based therapeutic to receive Food and Drug Administration (FDA) approval.2 Although the drug treats a peripheral rather than CNS disease (polyneuropathy), Akshay Vaishnaw, president of research and development at Alnylam Pharmaceuticals, Inc., used it as a case study to exemplify what has been learned about RNAi in the clinic. The lessons learned from the development of patisiran can be applied to the development of RNAi for CNS diseases, he said. The Phase 3 studies of patisiran showed first that RNAi can be used to suppress a target for long periods of time, and that this translates into neurological benefit in patients, said Vaishnaw. The drug not only halted progres-
___________________
1 For an overview of RNA interference (RNAi), see https://www.umassmed.edu/rti/biology/how-rnai-works (accessed August 9, 2019).
2 FDA News Release, August 10, 2018: FDA approves first-of-its-kind targeted RNA-based therapy to treat a rare disease. See https://www.fda.gov/news-events/press-announcements/fda-approves-first-its-kind-targeted-rna-based-therapy-treat-rare-disease (accessed June 6, 2019).
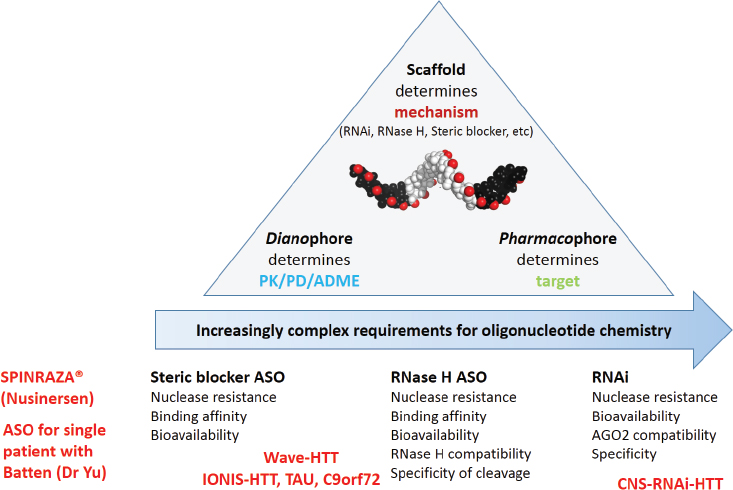
NOTES: ADME = absorption, distribution, metabolism, and excretion; ASO = antisense oligonucleotide; PD = pharmacodynamics; PK = pharmacokinetics; RNAi = ribonucleic acid interference; RNase H = ribonuclease H.
SOURCE: Presented by Anastasia Khvorova, April 23, 2019.
sion of disease, but was also associated with improvement in a composite neuropathy score that assesses symptoms across several sensory and motor domains. He noted that the drug was also well tolerated with an acceptable safety profile.
Because oligonucleotides do not cross the blood–brain barrier in healthy adults, they need to be delivered directly to the cerebral spinal fluid either by intrathecal or intracerebroventricular (ICV) injection or by implantation of an Ommaya reservoir, said Khvorova. Her lab has developed RNAi oligonucleotides that get robustly distributed throughout the brain after a single ICV injection. In a Huntington’s disease (HD) mouse model, ICV injection of a siRNA directed against the mutant huntingtin gene (HTT) demonstrated significant protein silencing for up to 6 months, said Khvorova. Yet, while silencing huntingtin protein expression in a mouse model is exciting, the human brain is much larger and more diverse. Because of this, her lab has been working with two larger animal models: sheep and non-human
primates. In both models, they have shown that a single ICV injection of siRNA results in widespread delivery throughout the cortex and distribution to deep brain structures. Most importantly, a single ICV injection of siRNA efficiently silenced huntingtin protein expression in the cortex and deep brain structures of non-human primates, said Khvorova.
Vaishnaw and colleagues are developing another approach, which they call a conjugate-based approach, to target siRNAs to the CNS. To target the liver in the development of patisiran, they used N-acetylgalactosamine as the ligand, conjugated to an siRNA that targets mutant forms of the transthyretin gene (Adams et al., 2018). For delivery to the CNS, they have identified other novel ligands conjugated to siRNA targeting SOD1 (superoxide dismutase 1, a gene implicated in familial amyotrophic lateral sclerosis) or b-catenin (used as a control). In rat models, they demonstrated dose-dependent protracted and reversible silencing of these genes following a single or multiple dose delivered intrathecally, said Vaishnaw. He added that while siRNAs are known to be cleared rapidly from the CNS, the conjugated siRNAs have far superior uptake by neuronal tissues relative to unconjugated siRNAs or to ASOs. They have also tested this approach in non-human primates with similar results, including uptake in all key cell types of the CNS (neurons, microglia, and astrocytes).
Viral Delivery of Gene Therapy
Two FDA-approved gene therapy products, Luxturna and Zolgensma, use a viral vector—AAV—to deliver therapeutic genes to their targets, and while many other viruses have been used for gene therapy applications, AAV remains the most widely used (Lundstrom, 2018). The first approval of an AAV gene therapy product—Glybera—in 2012 opened the floodgates of funding for development of gene therapies and has enabled the field to flourish, said Robert Kotin, scientific founder of Generation Bio and adjunct professor at the University of Massachusetts Medical School. Currently several different serotypes of AAV are being used as vectors in gene therapy clinical trials, said Kotin. All are derived from non-pathogenic dependoparvoviruses, which have proven to be safe and effective in numerous clinical trials. They vary in terms of tissue specificity, transduction efficiency, and antigenicity (Rayaprolu et al., 2013).
According to R. Jude Samulski, co-founder of the AAV gene therapy company AskBio and former director of the University of North Carolina Gene Therapy Center, AAV gene therapies are defined by four characteristics: (1) the capsid, which determines what cells to target; (2) the transgene, which can be altered and optimized for better translation; (3) the promoter, which regulates gene transcription; and (4) the mode of production. Petra Kaufmann added that what goes into the engineering and design of viral
vectors is largely empirical. It requires finding a promoter that provides a certain tropism and the appropriate level of expression, she said.
Challenges associated with AAV therapeutics include the limited capsid capacity and preexisting and cross-reacting neutralizing antibodies, which limit dosing to a single administration, said Samulski. Switching the formulation of a therapy by switching serotypes or genetically modifying the capsid may provide superior vectors. For example, using directed evolution, which is discussed further in Chapter 6, vectors have been developed that selectively cross the seizure-compromised blood–brain barrier and transduce cells in the CNS (Gray et al., 2010). This approach could be used to deliver gene therapy for seizure disorders, said Samulski.
In their efforts to develop therapies that would repress production of tau, DeVos and colleagues are developing a longer lasting central gene therapy approach by packaging into an AAV capsid a zinc finger protein fused to a transcription factor that recognizes and represses tau. She said they hope to couple this with an AAV variant called AAV-PHP.B. PHP.B is a capsid protein that more efficiently transduces neurons after intravenous injection in rodents (Challis et al., 2019; Deverman et al., 2016). Recent developments on engineered capsids are described in Chapter 6.
To determine the biodistribution of an AAV vector in vivo, Crystal and colleagues have developed a non-invasive technique by covalently labeling the AAV capsid with a positron emitter that can be detected using positron emission tomography. In non-human primates, they have shown that intravenously administered vectors home primarily to the liver, but also to the bone marrow. Even following direct injection into the brain, some of the vectors travel to the bone marrow and liver, said Crystal. In animals that were hyperimmunized (to mimic a person with preexisting immunity), they showed that when the vector was readministered several months later, nearly all of the vector homed to the spleen. This kind of information could be important clinically because it can help assess whether immunosuppression should be used before, during, or after administration of gene therapy, said Crystal.
IDENTIFYING THERAPEUTIC TARGETS
Gene-targeted therapies for CNS disorders discussed thus far have focused primarily on monogenic disorders; however, there has also been progress in developing gene-targeted approaches for more common complex polygenic disorders such as Parkinson’s disease (PD). Dopaminergic therapies currently available for PD address motor symptoms of disease, but do not impact disease progression, said Asa Abeliovich, founder and chief executive officer of Prevail Therapeutics. However, PD is not just a motor disease. Non-motor symptoms, which often become
dominant and lead to the most serious morbidity, are not the result of dopaminergic neuron loss in the mid-brain, said Abeliovich. To have meaningful disease modification requires thinking about PD as a brain-wide disease, he said.
For more than 100 years it has been known that intracellular protein inclusions called Lewy bodies are the main pathological finding in PD, which suggested that lysosome dysfunction may be a core mechanism underlying the disease, said Abeliovich. Genetic studies since then have identified many PD-associated genes that play a direct role in lysosome function and trafficking, he said (Abeliovich and Gitler, 2016). One of these is the glucocerebrosidase gene, GBA1. GBA1 mutations cause the lysosomal storage disorder Gaucher disease and are the most common genetic risk factors for PD (Sidransky et al., 2009), affecting all aspects of PD including severity and age of presentation, progression, and risk of progression to dementia, said Abeliovich. For example, mutations in GBA1 increase the likelihood that a patient will progress to cognitive dysfunction and dementia. He added that there is a range of severity associated with the more than 300 known GBA mutations and a gene dosage effect such that homozygotes have more aggressive cognitive decline than heterozygotes (Liu et al., 2016a). Even mutations in just one copy of GBA1 are associated with an increased risk of PD, said Abeliovich.
Several different gene therapy approaches have been developed for Gaucher disease, said Abeliovich. A lentiviral vector used to transduce hematopoietic stem cells with the GBA gene was shown to prevent Gaucher disease progression in mouse models (Dahl et al., 2015). Abeliovich said that while this vector is not delivered efficiently to the brain, the study showed that through cross-correction, transduction of only about 10 percent of the cells drove sufficient GBA expression to correct symptoms. AAV vectors delivering GBA1 to the CNS have also been shown to lower α-synuclein levels in synuclein transgenic mice (Sardi et al., 2013), and intravenous injection of AAV-PHP.B-GBA1 prevented formation of α-synuclein inclusions in PD mouse models (Morabito et al., 2017).
Davidson noted that the capacity for cross-correction is not universal. When designing gene therapies intended to take advantage of cross-correction, one must first determine whether the gene products are well secreted and well tolerated. Beautiful cross-correction has been achieved for several different lysosomal storage diseases, she said, but some gene products need to be reengineered for better secretion. If the gene products are well secreted, they may have deleterious effects. For example, in the Davidson laboratory, an attempt in mouse models to advance gene therapy for progranulin deficiency by cross-correction with AAV9 showed that expressing progranulin elicited massive T cell infiltration and disappearance of the hippocampus (Amado et al., 2019). This raises the question
of whether overexpressing other proteins in other diseases could have untoward effects and whether this problem could be dealt with by turning protein expression off or expressing the protein in a pulsatile manner, said Davidson. For secreted proteins, these questions need to be answered in a stepwise fashion, she said.
Most of the therapies discussed this far are intended to treat loss-of-function disorders. For gain-of-function disorders, other challenges must be considered, said Davidson. She focused her comments on microsatellite expansion disorders in which short sequences of nucleotides are repeated up to 1,000 times in coding or non-coding regions of the genome (Gao and Richter, 2017). More than 40 disorders are caused by microsatellite expansions, including HD, spinocerebellar ataxia, Fragile X disease, and many others, said Davidson.
Targeting the DNA in these disorders by editing out the toxic gain-of-function gene presumably could represent a one-and-done approach. She and her colleagues are developing CRISPR/Cas3 gene editing approaches to treat HD. To develop this approach, they needed to consider how, where, and when to intervene and what part of the gene to target. Given that HD progresses over 15 to 20 years with massive degeneration of the basal ganglia and cortical structures, Davidson and colleagues concluded that the treatment should be initiated very early in the disease course, while there were still cells left to treat. Selecting which part of the gene to target was informed by earlier research by Gillian Bates and colleagues, who showed that the toxic form of the HTT protein results from aberrant splicing of exon 1 in mutant HTT (Sathasivam et al., 2013). More recently, Laura Ranum and colleagues showed that repeat expansions can be translated in both the sense and antisense directions, and that the resulting proteins (called repeat-associated non-ATG, or RAN proteins) are very toxic (Banez-Coronel et al., 2015). These findings suggested to Davidson that the entire length of the mutant expanded allele should be deleted.
An investigator in her laboratory, Alex Monteys, answered the question of how to intervene, said Davidson. He devised a CRISPR strategy that took advantage of single nucleotide polymorphisms linked to the mutant allele, but not present on the normal allele so that he could selectively edit out only the HTT gene that contained the mutation, leaving the non-mutated HTT gene intact to carry out its normal functions (Monteys et al., 2017). This enabled him to dramatically reduce the levels of mutant HTT mRNA and protein levels in injected areas of the brain in HD mouse
___________________
3 Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) genes provide an efficient, accurate, rapid, and inexpensive method for genome editing. To learn more about CRISPR-Cas, see https://www.broadinstitute.org/files/news/pdfs/PIIS0092867415017055.pdf (accessed June 3, 2019).
models. Davidson’s lab has also demonstrated the ability to silence the locus using a novel approach called CRISPR interference.
To advance these approaches, Davidson said more efficient methods are needed to improve delivery of the therapies to larger brains (e.g., with lipid nanoparticles) or alternatively, to improve the safety of virally delivered editing machinery.










