
5
Microbial Dimension to Human Development and Well-Being
The first day of the workshop concluded with a plenary presentation that delved into how microbes are not only associated with various noncommunicable diseases (NCDs) but also how they are integral to normal human physiological functions. Rob Knight, founding director of the Center for Microbiome Innovation, University of California, San Diego, explored how a holistic, complex view of the relationship between microbes and people can change the way health is approached. He began his presentation by explaining that microbes outnumber humans, even within our own bodies. The typical human body consists of 30 trillion human cells and 39 trillion microbial cells—by that measure, humans are only 43 percent “human” (Sender et al., 2016). The microbial dimension is even more astounding when looking at genetics instead of cells. The efforts of the Human Microbiome Project (HMP) have revealed that each human has around 20,000 human genes,1 a measly comparison to the 2 to 20 million microbial genes in human bodies (Knight et al., 2017; NIH, 2019). By this measure, Knight stated that “we are actually 1 percent human at best” (Turnbaugh et al., 2007).
INFLUENCE OF THE MICROBIOME
Multiple systems in the body are affected by the human microbiome (Blacher et al., 2017). Microbes in the gut represent about 2 kilograms of
___________________
1 The HMP was established by the National Institutes of Health to generate resources that allow the characterization of the human microbiome and analysis of its role in human health and disease. For more information on the HMP, see https://hmpdacc.org (accessed August 16, 2019).
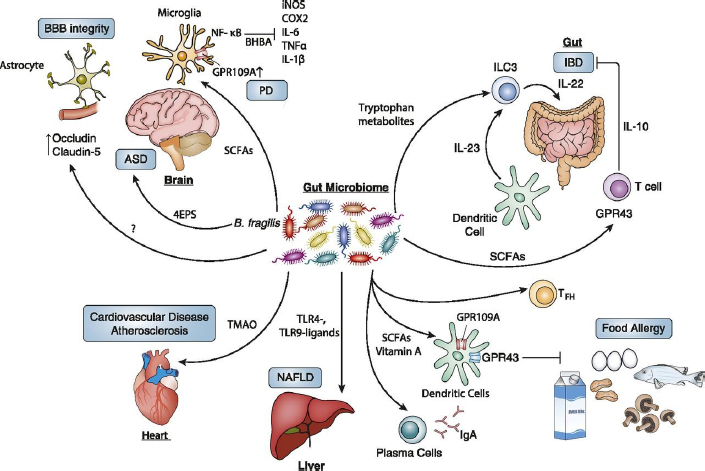
NOTE: ASD = autistic spectrum disorder; BBB = blood–brain barrier; IBD = inflammatory bowel disease; NAFLD = nonalcoholic fatty liver disease; PD = Parkinson’s disease; SCFA = short chain fatty acid; TMAO = trimethylamine-N-oxide.
SOURCES: Knight presentation, June 11, 2019; Blacher et al., 2017.
the typical person’s biomass (Qin et al., 2010; Forbes et al., 2016). These gut microbes can affect the body in obvious ways, such as inflammatory bowel disease, as well as more surprising ways, such as food allergies, liver disease, and cardiovascular disease (Blacher et al., 2017) (see Figure 5-1). Detailed mechanistic accounts also demonstrate how microbes can have a huge effect on the human brain in diseases like Parkinson’s disease, multiple sclerosis, and autism (Turnbaugh et al., 2007; Kirby and Ochoa-Repáraz, 2018; Sharon et al., 2019). Knight noted that research based only on the human genome—such as genomewide association studies or systems biology—excludes 99 percent of the genes in the human body. Because these microbial genes encode the vast majority both of unique biochemical functions and of antigens, this type of work ignores the genes that can be changed and modified throughout humans’ lifetimes to promote health and rather focuses instead on the 1 percent that are fixed when humans are conceived.
GLOBAL MICROBIAL BIODIVERSITY CRISIS
Despite the significant number of microbial genes in humans, however, humans are losing them (Smits et al., 2017). Studies of populations of people
with preindustrial lifestyles that are more characteristic of the past million years—rather than the past 100 years—have revealed major groups of bacteria (an entire phylum) that are not found in people living postindustrial lifestyles today (Clemente et al., 2015; Jha et al., 2018). To illustrate the potential consequences of dwindling global microbial diversity, Knight drew the analogy of turning a complex and diverse rain forest into a cityscape in which only the rats and pigeons survive. In the 1960s, conservationist and marine biologist Rachel Carson’s work documented how DDT and other pesticides wiped out birds and other components of large-scale ecosystems (Carson, 1962). More recently, other scientists documented similar effects when humans eliminate microbes early in life and thus affect the inner human ecosystem, which may be growing into a biodiversity crisis (Blaser, 2014).
Knight explained that throughout the twentieth century, single-pathogen diseases such as measles and tuberculosis (TB) fell to advances in medicine and public health, while the rates of multiple sclerosis, Crohn’s disease, type 1 diabetes, and asthma have skyrocketed. As presented in a review article in 2002, none of those chronic diseases were known to be linked to the microbiome (Bach, 2002). Today, all four of them—and dozens more—are known to be associated with the microbiome in humans (Pascal et al., 2017; Frati et al., 2018; Kirby and Ochoa-Repáraz, 2018; Zheng et al., 2018). Analogs of those diseases can be caused and cured in animal models through microbiome manipulation (Zheng et al., 2018). This raises questions about the unintended consequences of attempts to eradicate single pathogens such as TB and measles on the rest of the microbiome, he added.
COMPLEXITY OF THE HUMAN MICROBIOME
To better understand the complexity of the human microbiome, the HMP was launched by the National Institutes of Health, with a follow-up project that looked at specific diseases and recently published the results (Proctor et al., 2019). Samples were collected from about 250 healthy people, from up to 18 sites on the body at up to three time points, for a total of 4.5 trillion bases of DNA—about 1,500 human genome equivalents. Knight explained that this provided an unprecedented amount of DNA sequence data about the human microbiome (Caporaso et al., 2010; Human Microbiome Project et al., 2012). The abundance of DNA sequence data posed challenges for researchers, he noted. Similarly, the sheer volume of microbial data that can be gathered from the gut of a single individual can make it impractical for use diagnostically or therapeutically.
Knight’s lab is trying to help make these complex multivariate profiles useful for comparison between people and within a single person over time to address the massive degree of complexity in the human microbiome.
When processed with Qiime, a platform developed by Knight’s lab over the past 10 years, HMP data can be mapped using principal coordinates, which reduces the dimensionality of microbiome data to summarize the microbial community compositional differences in two or three dimensional scatterplots (Goodrich et al., 2014). Each point on the map represents a microbiome, and the distance between points represents how composition-ally different the samples are from one another (see Figure 5-2). The maps reveal that different parts of the body are like different continents, with the mouth, skin, vaginal, and fecal communities quite distinct from one another. Knight added that although the microbiomes included on the map come from healthy people, many factors can affect the microbiome, including age, gender, the time of day the sample was collected, and what the person ate the day before.
To build this evidence base, the Earth Microbiome Project,2 which aims to characterize global microbial taxonomic and functional diversity on Earth, crowdsourced tens of thousands of samples from scientists around the world (Gilbert et al., 2014). Knight noted that environmental microbiomes are just as different from each other as the mouth and gut microbiomes of one individual. The mouth can be thought of as a coral reef, complex mineralized structures covered with biofilms. The mouth is as far from the

SOURCES: Knight presentation, June 11, 2019; Knight, 2018.
___________________
2 For more information on the Earth Microbiome Project, see http://www.earthmicrobiome.org (accessed August 13, 2019).
gut in terms of its microbiome ecology as the water in a coral reef is from the dirt in a prairie, he explained. Knight and his colleagues did not expect that a few feet along the length of a human body could make as much difference in terms of microbes as thousands of miles across the Earth’s surface in completely different environments (Thompson et al., 2017).
To demonstrate the relevance of a person’s position on this microbial map, Knight described work carried out on Clostridium difficile (C. difficile), a hospital-acquired infection that is rapidly increasing in incidence in the United States. Microbiome mapping illustrates that stool samples from patients with C. difficile have microbiomes that are profoundly altered compared to healthy stool microbiomes (Schubert et al., 2014). A study provided fecal transplants to patients with C. difficile from donors with samples in the healthy microbiome region (Kelly et al., 2016). The microbiomes of patients who received stool transplants quickly moved from an unhealthy to a healthy state within days. These patients had to fail antibiotic therapy for 2 years to qualify for the trial and within a short time after the transplant, they were producing firm stool for the first time in years. The changes in these C. difficile profiles are complex and multidimensional, but they can be simplified by using a microbiome map that plots the changes on a single axis from sick to healthy along one dimension.
Origins of Human Microbial Complexity
Microbiome maps can also be used to trace the origins of a human body’s complex microbial communities. The way a person is born has a huge effect on those microbial origins. Babies delivered vaginally have microbial communities similar to their mother’s vaginal microbe communities, but the starting point is completely different for babies delivered via cesarean section, who have communities throughout their bodies that are all similar to their mother’s specific skin communities (Dominguez-Bello et al., 2010). Additionally, antibiotics, birth mode, and diet affect the microbiome in early life (Bokulich et al., 2016). He explained that it is possible to predict much about children at age 10—from their weight to their cognitive performance—from their microbiome a few days after birth. In particular, breast milk seems to reverse the adverse effects of the combination of antibiotics and cesarean sections on the microbiome and phenotype (Knight, 2018). The University of California, San Diego, has a new breast milk research center, the Mother–Milk–Infant Center of Research Excellence, which is studying components of breast milk that could be added to formula for families that cannot breastfeed.
Knight presented a series of images that demonstrated the development of an infant’s fecal biome community over time. The fecal biome of a vaginally delivered infant starts out in the vaginal area of the microbiome map, and
over the next 2 years it moves to the fecal adult state at varying speeds (Koenig et al., 2011). From one week to the next, children are microbially more different from themselves 1 week prior than the difference between any two healthy adults in the HMP, Knight illustrated. If a child receives antibiotics for an ear infection, for example, it causes a massive regression of the microbiome, unwinding months of normal development (Yassour et al., 2016). He noted that the microbiome of some children will recover rapidly within a couple of weeks and continue to move toward the healthy adult fecal status. However, he said many adults and children are less resilient to antibiotics in terms of microbiome and phenotype. A major challenge is to determine why some people are less resilient and then develop a way to predict who is at risk.
These microbial development effects are so strong that they can be picked up cross-sectionally across populations at a specific point in time, Knight noted. A comparison of the adult state of the microbiome over the first 3 years of life in rural Malawians, Amerindians from the Amazonas of Venezuela, and U.S. populations from metropolitan areas found that the approach to the adult state is generally complete by age 3 (Yatsunenko et al., 2012). However, the final states of the microbiomes were completely different in the non-Western populations compared to the Western populations, with different infectious and noninfectious disease profiles. Knight noted that the HMP and other large-scale projects are focusing on Western adults; thus mapping the microbiome of children and non-Westerners depicts completely different configurations. This underscores the importance of building microbial growth curves for children in diverse populations, which can be used to measure the effects of breastfeeding, antibiotics, and delivery mode of maternal drugs. It could also be used to predict the effect of falling off the microbiome “growth curve,” he added.
RESEARCH DIRECTIONS TO EXPLORE MICROBIOME COMPLEXITY
Knight described some of the research directions that could help provide a more realistic understanding of this complex relationship of microbes to infectious diseases, NCDs, and normal physiological functioning. The war against antimicrobial resistance is failing, and new approaches are urgently needed, he cautioned. Although much work has been devoted to combating individual species or entire taxonomic groups of microbes, much less work has gone into strengthening the host, which has great potential. It is well established that one way to strengthen the host is through diet, but the details of how to strengthen the host against infection and chronic disease through diet are yet unknown.
Knight remarked that there have been many attempts to supplement iron in childhood anemic populations but, in general, those attempts release
iron-limited pathogens that cause diarrheal diseases and other problems (Lonnerdal, 2017). Research using individual studies have discovered that Salmonella enterica serovar Typhimurium, an enteric bacterial infection transmitted through contaminated food or water, can be treated by a combination of copper, L-arginine, vitamin C, and linalool (Haque, 2012; Ghosh et al., 2019). However, he said more global approaches are needed to rapidly profile large numbers of pathogens, micronutrients, and dietary ingredients and assess their effects. This applies to viruses as well as pathogens, he added. A mouse model has demonstrated a reduction in mortality among mice exposed to influenza that were supplemented with the molecule desaminotyrosine, which is naturally produced by gut bacteria from flavonoids in the diet (Steed et al., 2017). Treating the exposed mice with antibiotics led to around 90 percent mortality, while giving them antibiotics plus desaminotyrosine brought mortality down to about 30 percent (Steed et al., 2017). Furthermore, the study found that certain microbes (Clostridium orbidscindens) had to be present to make the compound from the dietary ingredient. This is an example of why diet, microbiology, and immunity have to be integrated to comprehend the full picture, he said.
To contribute more robust research and an integrated picture of diet, microbiology, and immunity, Knight has cofounded the Global FoodOmics Project, which is running thousands of food samples through a mass spectrometer to find out what is in food products at the point of sale or at the point of consumption.3 These data are cross-referenced with the American Gut Project, which has collected more than 20,000 stool samples contributed by citizen scientists, as well as performing meta-analyses with their other clinical projects.4 He presented a picture of a molecular network from an early iteration of the project, which reveals the complete biotransformation of food in the human gut. In new mouse models, researchers have been 3D scanning conventionally raised, pathogen-free, and germ-free mice, then dicing them to sequence and running the mass spectrometer on each piece. They are then able to reconstruct full 3D models with any specific bacterium or molecule highlighted in the structure in the mouse. This technique has been used to demonstrate how soyasaponin accumulates in a germ-free mouse but not in a specific-pathogen-free mouse.5 Conversely, soyasapogenol accumulates in the specific-pathogen-free mouse that has the necessary bacteria,6 but it does not accumulate in the germ-free mouse. Both soyasapogenol and soyasaponin have been shown to modify immune
___________________
3 For more information on Global FoodOmics, see https://sites.google.com/site/globalfoodomics/project-description (accessed August 12, 2019).
4 For more information on the American Gut Project, see http://humanfoodproject.com/americangut (accessed August 12, 2019).
5 Soyasaponin is a phytochemical found in vegetables, beans, and herbs.
6 Soyasapogenol is created by cleaving the bond in soyasaponin.
response (Guang et al., 2014). The complete transformation of bioassay profiles and other metabolic details can also be observed in the mice through this work.
Knight called for a coordinated global effort to build a more comprehensive repository of information about the microbiomes of people with much more diverse demographic, geographical, and health-related characteristics. This effort should be catalyzed by the potential strides to be made against the sheer number of conditions that have now been linked to the gut microbiome, such as inflammatory bowel disease, irritable bowel syndrome, food allergies, asthma, cardiovascular disease, diabetes, multiple sclerosis, autism, and Parkinson’s disease, he said. For instance, large amounts of money have been channeled into researching the genetics of human obesity, yet he noted that the accuracy in predicting obesity from human genes is significantly lower (57 percent) than predictions using microbial genes (90 percent). This holds for a large number of traits. Studies of microbial contribution versus host genetic contributions to phenotypes, and their microbial-genetic associations, show that microbial contribution dominates for traits such as lactose consumption and waist circumference, while traits such as height are determined by genetics (Rothschild et al., 2018). Because the microbial associations are so much stronger, Knight noted, traits can be associated with microbes with 10-fold fewer test subjects than are required for genetic associations. He also mentioned that dogs and their owners can be matched with a fair degree of precision by their shared microbes (Coelho et al., 2018). He added that people who are overweight tend to have overweight dogs and cats, suggesting that obesity may be transmissible microbially. While the environment and geography have a strong influence on human gut microbiota variations, he cautioned using generalized microbiota-based diagnostic models among various locations (He et al., 2018).
To assess causality rather than association, Knight and his collaborators are using a technology called gnotobiotic mice, which are mice raised in a bubble with no microbes of their own. To establish causality, these mice are then colonized with microbes that are thought to affect their phenotype. When gnotobiotic mice were colonized with microbes from an obese human, they became obese; when colonized with microbes from a lean human, the mice remained lean (Ridaura et al., 2013). Similar work has been done on inflammatory bowel disease, colon cancer, Parkinson’s disease, multiple sclerosis, and most recently for autism (Sharon et al., 2019). It has also been done for many other traits including response to drugs and response to diet, particularly for components like emulsifiers and artificial sweeteners that are associated with highly individualized responses (Faith et al., 2011; Smits et al., 2016; Zimmermann et al., 2019). He explained that the approach can also be used to evaluate susceptibility to a wide range of bacterial, viral, and fungal pathogens that can be transmitted from humans
to mice on an individual level. This establishes that microbial genes can be causally implicated—at least to the extent to which the animal models are a good model of those diseases.
In his concluding remarks, Knight described his vision of future tools to track the microbiome (see Box 5-1) and outlined a set of research needs, including the following:
- More work integrating food, drugs, microbiology, immunology in studies with multiple data layers and large enough study populations to get adequate statistical power
- More comparative studies of microbiomes in populations where burdens of infectious and chronic disease differ
- More prospective longitudinal studies that integrate microbiology and immunology, infection, and chronic disease
- More translational work relating animal studies to humans
- More longitudinal studies with defined perturbations using already-approved compounds, especially in early life
- Better molecular and data science tools
DISCUSSION
Following Knight’s presentation, workshop participants had the opportunity to ask questions. The discussion covered topics on specific factors that shape the human microbiome, as well as the public health implications of microbiome research.
Factors Shaping the Human Microbiome
Srinath Reddy, president of the Public Health Foundation of India, asked about the nature of epigenetic changes in the microbial genes and long-term effects beyond changing the character of the colony, as well as whether environmental exposures are more likely to lead to epigenetic changes in the huge pool of genes in the microbiome. Knight replied that little is known about the epigenetics of the bacteria, but there is evidence of the epigenetic process of DNA methylation in the bacterial genomes themselves. Many bacterially produced metabolites, such as the histone inhibitor butyrate, have effects on the host epigenome. He said that recent publications have reported strong links between a person’s microbes, host methylation, and histone modification markers. Knight explained that progress is being hampered, however, because the laboratories working on the microbiome are different than the ones that study epigenetics. Both types of laboratories require complex and expensive technology, making it challenging to fund projects at a level at which both of those techniques can be combined. He added that systems-level understanding is also being informed by research into metabolite profiles that lead to epigenetic changes and by profiling different tissues around the body—changes in the microbiomes in mice cause observable gene expression changes in their brains, for example.
Mary Wilson, clinical professor of epidemiology and biostatistics at the University of California, San Francisco, asked about research on the antibacterial activity of nonantibiotic pharmaceuticals, some of which are used to treat chronic conditions. Knight replied that a large number of pharmaceuticals (including metformin, antipsychotic drugs, and selective serotonin reuptake inhibitors) are not traditionally considered to be antibiotics, yet they have large effects on individual gut microbes (Maier et al., 2018; Spaniogiannopoulos and Turnbaugh, 2018). There is a dearth
of knowledge, however, because only a few dozen compounds have been tested, and they have only been tested against a limited range of microbiomes from Western adults, Knight added. It is not yet understood how those pharmaceuticals interact with the environment, or have downstream effects, in the microbiomes of fish, seawater, or soil. The effect of microplastics on the environment is a related issue, he added. Evidence is emerging that microbes attach easily to fibrils and that spatial structuring substantially modifies their activity in the gut (Tropini et al., 2017). It is likely that the same is true for microplastics and other nonnutritive substances that are commonly ingested, he said.
Kevin Olival, senior research scientist at EcoHealth Alliance, asked about the role of viruses, particularly bacterial phages, in shaping human microbial communities. Knight replied that the prospect of understanding phage bacterial dynamics is exciting. However, it will require large sample sizes and a dense time series, which are currently prohibited by the cost of shotgun metagenomics,7 though the cost is likely to decrease and enable such studies in coming years. He noted that phage therapy has been remarkably clinically effective as a last-ditch measure for patients who had exhausted all other treatment options. Learning how to use phage to intentionally modify a microbiome has great potential, he said, given that total synthesis of a phage with a specific sequence is already possible. Phage engineering will need to be integrated with the microbiome research to carry out this type of work.
Relatedly, Dennis Carroll, director of the Emerging Threats Division at the U.S. Agency for International Development, asked Knight to comment on viruses and fungi in the microbiome. Knight explained that the microbiomes of Westerners tend to have relatively few genera of fungi, mostly Candida, Saccharomyces, and a few others, but people in Asian countries tend to have a much more diverse repertoire of fungi and microbial eukaryotes. The hope was that shotgun metagenomics would revolutionize knowledge about everything except bacteria, which are the only components that can be picked up efficiently and with decent taxonomic resolution by most other methodologies. However, a key limitation has been the lack of reference databases, particularly for viruses and fungi. Shotgun metagenomics provides many short fragments of DNA, which need to be matched up using reference databases. It would cost about $4 million to sequence every type strain of bacteria, he added. This is beyond the budget of most individual laboratories and falls between the mandates of different agencies that could
___________________
7 Shotgun metagenomics is an environmental sequencing method that allows researchers to sample all genes in all organisms present in a given sample. The method aims to enable researchers to evaluate bacterial diversity and function and to detect the abundance of microbes in different types of environments.
potentially fund it. He emphasized that sequencing all of the type strains and as many fungi from human and animal stool isolates as possible would be an immensely valuable resource that would greatly increase the efficiency of all metagenomic studies.
Paul Miller, chief scientific officer at Artizan Biosciences, noted that much interest centers on the interplay between the gut microbiota, the gut epithelial, and the immune system, yet most of measurements and comparisons use stool samples. He asked about the use of biopsies and other sources of data that are closer to the epithelial side than the end product. Knight said that in general, biomarkers obtainable from stool are good but not always as good as biomarkers obtained from biopsy specimens. Stool is much more useful for biomarker studies than for mechanistic studies, which benefit from tissue sampling. He added that stool is more practical for daily sampling in longitudinal studies, because variation in the microbiome itself—even on a daily time scale—allows for predicting future-state immunologically linked phenotypes such as inflammatory bowel disease.
Public Health Implications of Microbiome Research
Jay Varma, senior advisor at the Africa Centres for Disease Control and Prevention, asked if microbiome research corroborates the current “super foods” trend and claims about how diet can boost a person’s immunity. Knight replied that there is much emerging work on the intersection of microbiome research with public health advice about diet. The available evidence supports certain public health recommendations. For example, eating more fiber is beneficial for the production of butyrate in the gut by the microbes that ferment fiber (Baxter et al., 2019).8 Evidence about artificial sweeteners and emulsifiers demonstrates how they are bad for the gut microbiome (Harpaz et al., 2018), which is consistent with policy recommendations. On the horizon are personalized dietary recommendations that have the potential for large effects, he said. Part of the reason why supermarket tabloid diets are broadly ineffective is that they tend to be based on a single person, rather than larger sample sizes. The aim with dietary recommendations is to discover a relatively small number of types of microbes in the gut that can inform public health recommendations by developing microbe-detection tests coupled with discrete evidence-based recommendations based on that test. He said that currently available data are not poised to change any public health recommendations at present, but the individual effect sizes of these findings are large. Some people are negatively affected by artificial
___________________
8 Butyrate is a short-chain fatty acid that supports health through a number of mechanisms, such as immune system regulation (Baxter et al., 2019).
sweeteners or red meat, while others are not; this depends on the details of bacteria in the person’s gut.
Finally, referring back to Knight’s presentation that noted humans carry 1 percent of human genes, Rima Khabbaz, director of the National Center for Emerging and Zoonotic Infectious at Diseases at the U.S. Centers for Disease Control and Prevention, asked about studies that incorporate hosts to inform the development of risk assessments and personal recommendations. Knight said that an increasing number of studies are incorporating both hosts and microbial genomes, showing that microbiome associations with the phenotype were much greater than with the host. Personalized recommendations are still on the horizon, he said. Individual differences are known to be large, but there is no test available to evaluate a sample and integrate it into the data frame. To make this type of data useful and actionable at the individual level, he said, will require input from federal agencies and others to develop standards for integrating individual samples into consistent reference ranges, as well as work to standardize databases across specific geographical locations.
This page intentionally left blank.














