
2
The Job of Medicines Regulators in Today’s World
Ensuring the health of the public through safe and effective medicines is arguably the highest priority of a medicines regulator. Carrying out this function within an increasingly complex system of production and manufacturing, alongside rapidly evolving technology and innovations in medicines, creates a whole new set of challenges and opportunities for regulators. Collaborating with others facing similar issues can be viewed as one way of managing the increasing workload faced by virtually all regulatory authorities (RAs). The key is to understand how cooperation might build coalitions by leveraging finite resources to maximize the assets of each RA. Before delving into various aspects of cooperation for the regulation of medicines, it is important to understand some of the core functions of RAs and the environment in which they now work, one that has seen unprecedented changes due primarily to advances in technology and applications stemming from the Human Genome Project (Lesko and Woodcock, 2004).
CORE FUNCTIONS OF REGULATORS AND REGULATORY AUTHORITIES
According to the World Health Organization’s (WHO’s) 2002 multicountry study on effective drug regulation (Ratanawijitrasin and Wondemagegnehu, 2002), the basis for regulation of medicines resides in a country’s drug laws. It is up to the regulators to implement those laws—which are influenced by new developments and societal needs—by overseeing industry in the use of standards and guidelines. Key functions of regulators include “licensing, inspection of manufacturing facilities
and distribution channels, product assessment and registration, adverse drug reaction monitoring, quality control, control of drug promotion and advertising, and control of clinical drug trials” (Ratanawijitrasin and Wondemagegnehu, 2002, p. 2). At times, all of these functions are carried out under a single agency, while in other instances the responsibility falls on multiple agencies, possibly at differing levels of government, which can create fragmentation and decrease the effectiveness of regulation. Some RAs are given functions, such as drug manufacturing and procurement, that would be viewed as conflicts of interest with the agency’s mission of drug regulation. This situation speaks to the issue of financing. Most RAs are financed through a mix of government support and user fees, while a small number of RAs are entirely dependent on user fees. The financial structure of an agency, according to the WHO report, should not influence regulatory decisions; however, RAs with inadequate staff, poor working conditions, or poor financing may be subject to compromise (Babigumira et al., 2018).
REGULATORS WORKING TOGETHER
Most, if not all, well- and moderately well-resourced RAs have shifted to a risk-based approach to regulation. For each of them, this includes allocating regulatory resources more proportionately so that, for example, manufacturers of medicines with greater risk are inspected more frequently than those that present a much lower risk. The approach also includes working together with other RAs. Those with similar structures, functions, and national characteristics might find a more natural fit for working together, depending on the collaborative activity undertaken. Such activities can be divided into pre- and post-marketing efforts (Babigumira et al., 2018). Pre-marketing activities, as outlined in the U.S. Agency for International Development’s 2018 Risk-Based Resource Allocation Framework (Babigumira et al., 2018), include licensing of premises and persons, inspection, evaluation and registration, and quality control testing. Quality control testing can also occur post-marketing along with product quality monitoring and surveillance, safety/pharmacovigilance, and enforcement of pharmaceutical laws and regulations.
Good Regulatory Practices for Cooperation
The Organisation for Economic Co-operation and Development (OECD) describes good regulatory outcomes as “almost always a cooperative effort: by the regulator and other regulators, the regulated, and often the broader community” (OECD, 2014, p. 5). OECD notes further that governance arrangements can foster cooperative efforts and outlines areas
of good governance for regulators that could lay a foundation for strong cooperation and collaboration. Six of these areas are:
- role clarity—functions are clear and without conflicts;
- preventing undue influence and maintaining trust—objective, impartial, consistent decision making;
- accountability and transparency—accountable to government and the public;
- engagement—enhance and maintain public and stakeholder confidence;
- funding—protect independence but be transparent to heighten confidence in decisions; and
- performance evaluation—to understand impacts of own actions and drive improvements.
WHO is developing guidelines for good regulatory practices for RAs specific to medicines. While this and another WHO effort on good reliance practices remain works in progress, there does appear to be potential overlap between the two concepts whereby good regulatory practice involves reliance. WHO’s draft of the guidelines for good regulatory practice states that, “regulations should include sufficient administrative flexibility to allow for participation in international cooperation frameworks, such as for information-sharing, convergence, harmonization, work-sharing, reliance and recognition” (WHO, 2016b, p. 10).
RECOGNITION AND RELIANCE
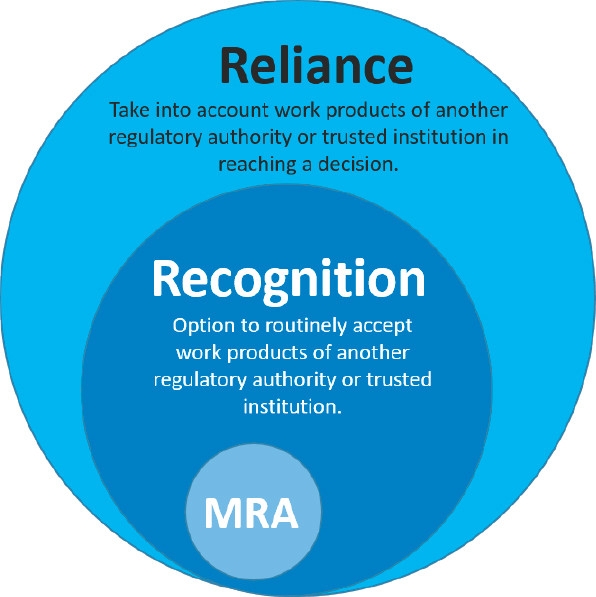
As part of its benchmarking tool, WHO (2018b) defines recognition as the routine acceptance of another RA’s—or other trusted institution’s—regulatory decisions based on the work products of that other agency. In contrast, reliance is defined as being more flexible, giving an RA in one jurisdiction the option of taking into account work products (e.g., inspection reports, scientific assessment reports, joint assessment reports produced together with another RA) of another authority or trusted institution in reaching its own decision. These definitions are presented in Box 2-1. A decision to routinely accept or to take into account the work of another regulator is the choice of the reliant RA and might be based on a variety of factors including the context in which a decision is being made, access to resources, and whether there is sufficient trust and confidence in the RA being relied on for information. But regardless of whether an RA uses recognition or reliance, in the end it is the responsibly of the RA to protect the health of its people and thus make the best regulatory decisions for its country. In this regard (illustrated in Figure 2-1), recognition can be viewed
as a subset, or a more stringent form of reliance, with the mutual recognition agreement (MRA) as the most stringent form of all.
Recognition and Reliance Among Well- and Lower-Resourced Regulatory Authorities
The way in which an RA views recognition and reliance may have more to do with how well-resourced that agency is in relation to another RA. For example, equally well-resourced RAs would typically take a horizontal perspective in sharing information rather than the more unidirectional approach that would likely result from lower-resourced RAs relying on the work of well-resourced or moderately well-resourced RAs. These concepts are explained in greater detail below.
Recognition and Reliance Among Well-Resourced Regulatory Authorities
In the case of recognition and reliance between well-resourced RAs, it is necessary for technical regulatory requirements as well as procedures in inspection/review of medicines manufacturers to be fairly close, if not identical. If a good manufacturing practice (GMP) itself is different between
SOURCES: Adapted from a figure created by Dr. Petra Doerr, Swissmedic, and presented by Emer Cooke, World Health Organization (WHO, 2019i).
country A and country B, joint inspections, regardless of how many times they may be conducted, cannot facilitate reliance on the inspection results. Because RAs involved in horizontal recognition and reliance arrangements are of similar or comparable capacities, the collaboration is usually mutual and “trust” here means that one agency has concluded that the other does as good a job as its own reviewers or inspectors.
Recognition and Reliance Among Lower-Resourced Regulatory Authorities
A lower-resourced RA can base its decisions on the work products of well-resourced, trusted RAs. The extent to which the reliance occurs can vary from critically examining inspection reports or reviews and potentially reaching different conclusions to accepting other agencies’ decisions without further examination. This type of recognition and reliance would not typically be considered mutual, and in such cases, regulations need
not be completely harmonized between the relying and the relied-on RAs. The “trust” here is from lower-resourced to well-resourced or moderately well-resourced RAs. Trust in these instances can be formed without such critical examination of the capabilities of the RA being relied on as is necessary for horizontal recognition and reliance, and can be naturally reputation-based because of the resource constraint involved. Such recognition and reliance apply to regulatory oversight during the pre- and post-approval phases. An example is the Caribbean Regulatory System (CRS), where numerous small states make up the Caribbean Community. Each state lacks the resources and capacity necessary to fully conduct medicines regulation on its own so the CRS uses a regional reliance mechanism—relying on trusted RAs within the Pan American Health Organization, the European Union (EU), and WHO prequalification—to help inform its decisions about which medicines to recommend for regional market authorization (PAHO, 2019b).
Trust as a Foundational Element
What underlies all recognition and reliance arrangements is trust. Medicines RAs cannot rely if they do not trust the provenance and findings of another RA. This means there are necessary early steps in building trust that, over time, can grow stronger through using common international standards (harmonization) and report formats, and through working together. Figure 2-2 illustrates trust in the regulatory sense and how it forms the backbone of any reliance process. Prior to executing formal reliance arrangements, RAs will likely need to pilot an established process of joint inspections/working together and even auditing of each other’s work. All of these efforts build confidence in the systems and procedures of the respective RAs. In contrast, other forms of reliance exist that take a more unidirectional approach to the medicines approval process by fully embracing the results of trusted RAs.
COOPERATION AND COLLABORATION TOOLS OF REGULATORY AUTHORITIES
Different tools are available for recognition and reliance across the product lifecycle. These are collectively referred to in this report as “arrangements.” Arrangements can be formal agreements put in place, for example, through Memoranda of Understanding (MOUs), confidentiality commitments, and MRAs; or they can be informal through the establishment of ad hoc committees, working parties, and other, more topic-specific working groups (EMA, 2019g). WHO’s draft guidelines for good regulatory practice note that “less formal practices include sharing of information, scientific

NOTE: COI = conflict of interest; PIC/S = Pharmaceutical Inspection Co-operation Scheme; RA = regulatory authority; WHO = World Health Organization.
collaboration, common risk assessment, joint reviews, and development of standards” (WHO, 2016b, p. 27). Some formal collaborative activities include what might be considered less formal interactions through regularly scheduled calls and meetings that build trust between regulators as they move toward greater confidence levels for reliance.
In many cases, pilot activities can be set up under the formal mechanism of confidential arrangements (also known as confidentiality commitments) between RAs. The arrangements differ, but generally speaking, they allow regulators to exchange confidential information that is not in the public domain. Pilots can be useful tools for building trust and confidence between regulators from different jurisdictions. One example is the International Generic Drug Regulators Pilot that began in 2012 as a collaborative arrangement among RAs from Australia, Canada, Chinese Taipei, the EU, and Switzerland (EMA, 2015). It was modeled on the EU system, which is a network of 50 RAs from 31 countries (28 Member States, Iceland, Liechtenstein, and Norway) that rely on each other through application of the mutual recognition principle and common regulations and procedures. Another example of a work-sharing arrangement (described in Box 2-2) is the Australia-Canada-Singapore-Switzerland (ACSS) Consortium. It started in 2007 as a pilot to maximize international cooperation and is now exploring opportunities for information and work sharing in such areas as:
- generic medicines registration,
- assessment reports for new prescription medicines,
- information sharing and investigations into post-market medicine safety,
- alignment of information technology systems for information sharing, and
- development of technical guidelines (TGA, 2019b).
MOUs are another tool frequently used by RAs to coordinate activities and share information. They are nonbinding agreements that can vary greatly in their scope depending on their purpose and the parties involved. Often, MOUs allow RAs a more formal route to clarifying the roles and responsibilities of the parties. A Memorandum of Cooperation (MOC) is similar to an MOU in that it is not legally binding. This tool has been used by Japan to set guidelines for cooperation between parties. Unlike MOUs and MOCs, recognition agreements can be legally binding.
Mutual Recognition Agreements
While reliance arrangements promote the sharing of information and facilitate fewer redundant inspections and other regulatory activities, MRAs allow agencies not just to rely on each other’s work products but also to recognize them as equal to the work of their own reviewers or inspectors. For the EU, MRAs allow for reliance on the GMP inspection systems of other RAs, information sharing based on inspections and quality, and the waiving of batch testing of those products imported into EU territories. It should be noted that while safeguarding the public’s health is intrinsic to MRAs, they are in the end “trade agreements that aim to facilitate market access” (EMA, 2019f). As such, they tend to be laborious to develop and negotiate and are labor-intensive to maintain; however, the required preparation and maintenance work is often deemed worthwhile as it prevents duplication of effort, makes precious resources available, and expedites approval time and market release, all of which are of great public health benefit. As is shown in later chapters of this report, these benefits are also achieved through the use of reliance arrangements that do not pose many of the challenges associated with MRAs. See Appendix B for a list of the MRAs that are a primary focus of this report.
Unilateral and Multilateral Recognition
Unilateral recognition allows one RA’s decisions to be informed partially or fully by the work of another. Some Latin American countries take such an approach. One example is Mexico’s RA, Comisión Federal para
la Protección contra Riesgos Sanitarios (COFEPRIS), which in 2012 established a unilateral agreement with the EU to use work carried out at the European Medicines Agency (EMA) during its single marketing authorization process (WHO, 2016a). COFEPRIS set up other similar unilateral processes with the U.S. Food and Drug Administration (FDA), Health Canada, Australia’s Therapeutic Goods Administration (TGA), and Swissmedic to expedite the approval of new medicines and their entry onto the Mexican market (Patel et al., 2019). It appears that other Latin American countries (i.e., Argentina, Dominican Republic, Ecuador, El Salvador, Paraguay, Peru, and Uruguay) may also use unilateral approaches based on information from what the countries deem to be trusted sources (Sravani et al., 2017). With this arrangement, those countries that are relied on are placed in a position whereby approval decisions in their jurisdiction have relevance and impacts beyond their own country or region.
The European Economic Area (EEA), composed of Iceland, Liechtenstein, and Norway, has MRAs with Australia, Canada, New Zealand, and Switzerland on GMP (and additionally, on good laboratory practice in the MRA between EEA and the European Free Trade Association/Switzerland) that are modeled on the EU MRAs. Another example is the mutual recognition arrangements of the Association of Southeast Asian Nations (ASEAN) Member States. In this case, all ASEAN member states are “obliged to recognize and accept the [GMP] inspection reports and certificates issued by ASEAN Inspection Services without duplicating GMP inspection in each other’s territory” (HSA, n.d.). COFEPRIS also has multilateral agreements with a number of regional and international organizations, with different scopes for different conformity assessment bodies (La Entidad Mexicana de la Acreditación, 2019).
Cooperation Through Harmonization
Broader efforts among countries for cooperation on aspects of the regulation of medicines have taken place under the auspices of regulatory harmonization (APEC, 2019; ICH, 2019; IPRP, 2019; PAHO, 2019a). Regulatory reliance may be facilitated by having RAs converge more than diverge on such matters as procedures and common formats for reports and forms, as appropriate, and as supported by international organizations such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, and by bringing regulators together for various collaborative and cooperative efforts (see Box 2-3 for a summary of harmonization, convergence, and reliance efforts through international organizations).
Strengthening Regulatory Systems for Medicines Through Transparency
RAs in low- and middle-income countries face multiple challenges that vary by country. According to Roth and colleagues (2018), these challenges tend to include access to medicines—in part because of limited commercial returns for industry—as well as access to evidence-based data, both pre- and post-market. In some instances, donor organizations, the pharmaceutical industry, and public–private partnerships have attempted to improve the supply of medicines in developing countries, but despite these efforts, a lag time persists. Typically, it takes 4 to 7 years between initial approval by one RA (generally in a developed country) and final approval (in the developing country) (Ahonkhai et al., 2016). Numerous reasons were suggested for the delay, including “failure to leverage or rely on the findings from reviews already performed by competent regulatory authorities, disparate requirements for product approval by the countries, and lengthy timelines by manufacturers to respond to regulatory queries” (Ahonkhai et al., 2016, p. 1).
Some efforts are under way to build regulatory capacity for accelerated registration through WHO prequalification (WHO, 2019c) and the work of the International Coalition of Medicines Regulatory Authorities in the area of GMP (ICMRA, 2017). As capacity is built within some RAs to better utilize information from trusted authorities, fuller access to information, reports, and data will likely become more valuable. In an effort to promote
greater openness and transparency, WHO, EMA, and FDA have sought to make some information publicly available (EMA, 2019b; FDA, 2015b; WHO, 2019d). These reports exclude confidential proprietary information, and in the case of WHO, are posted only on its website with the manufacturer’s approval. Similarly, openness between well-resourced and lower-resourced RAs would almost certainly require input from industry.
This page intentionally left blank.














