
5
Harnessing Quantum Dynamics in the Time and Frequency Domains
When we watch a movie, we follow a plot and learn how a sequence of events unfolds in time. We also learn which interactions are important in driving events forward toward the end of the story. One of the fundamental goals of atomic, molecular, and optical (AMO) science is to assemble molecular movies of how electrons, atoms, and molecules interact with each other in real time, using ultrashort pulses of light or particles to take a series of “snapshots” that follow the dynamics. This will allow us to understand and subsequently control diverse processes such as how chemical reactions take place from the earliest stage to the end products; how the photoprotection mechanism in our DNA works to limit damage from ultraviolet radiation; or how a laser pulse can switch a current on and off in an insulator more than a trillion times per second. These dynamical processes take place on ultrafast time scales and are often exceedingly difficult to both measure and understand, since they generally involve strong correlations between different constituents, as well as transfer of energy among different degrees of freedom. The observation and control of such coupled dynamics thus require advanced investigative tools both experimental and theoretical.
For direct time domain access, the unprecedented development of ultrafast light sources as described in Chapter 2 has revolutionized the capabilities of AMO science to make molecular movies. This is illustrated in Figure 5.1, which shows how different types of dynamics take place on different characteristic time scales and are associated with different characteristic energies. For electrons, the natural time scale is tens to hundreds of attoseconds (10−17 to 10−15 s), and this can routinely be accessed by attosecond pulses from high harmonic generation (HHG)-based,
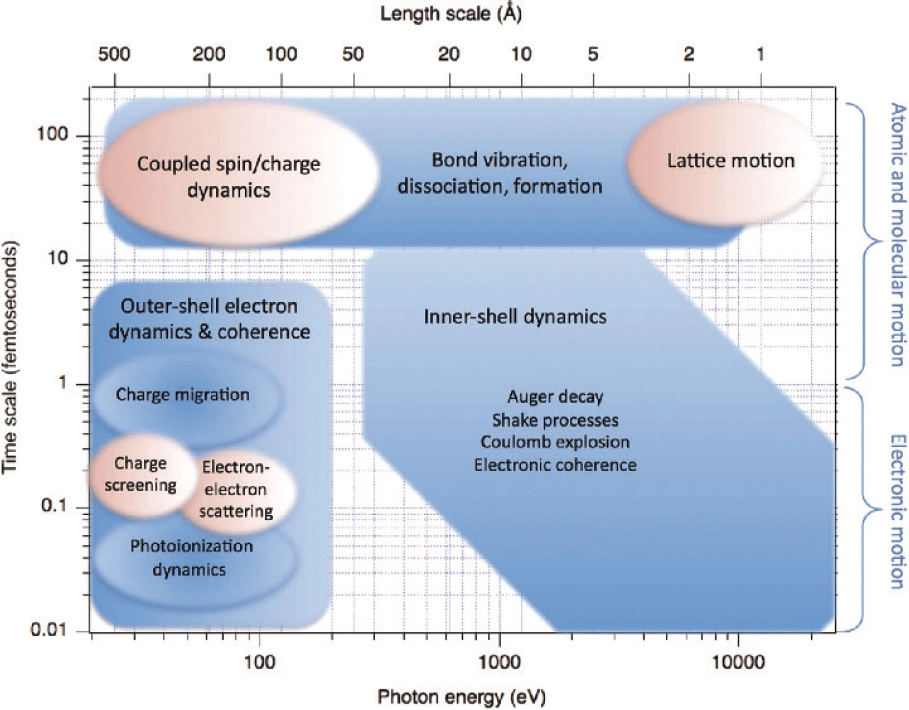
table-top sources of extreme ultraviolet (XUV) and soft X-ray radiation. The dynamics of the heavier nuclei takes place on time scales of tens to hundreds of femtoseconds (10−14 to 10−12 s). The femtosecond pulses of hard X-ray radiation currently available from accelerator-based X-ray free-electron laser (XFEL) sources, with attosecond capabilities planned for the future, can access inner-shell electrons and probe the ensuing dynamics of the nuclei (termed molecular dynamics).
Because inner-shell transitions have energies that are characteristic of the local environment, these X-ray pulses allow scientists to focus on individual atoms even when embedded inside complex molecules. Another advantage of hard X-ray radiation is its very short wavelength, which is comparable to chemically relevant lengths scales, determined by bond-lengths of a few angstroms, and therefore allows for high spatial resolution when taking direct pictures of molecular structure via scattering of light. Ultrafast pulses of electrons have the same advantage, and recent developments have also allowed the making of molecular movies in which such electron pulses are taking the pictures. This chapter begins with a discussion on the making of molecular movies, starting with the attosecond electron dynamics and continuing with the femtosecond molecular dynamics.
Dynamical processes can also be accessed in the frequency domain, as evident from decades of knowledge gained from spectroscopy and collision physics. Although collisions are inherently time-dependent, collision physics often deals with phenomena at a fixed energy, and efforts by theory and experiment are devoted to understanding energy-dependent reaction rates. Such rates are frequently needed to model state-to-state collisional processes in gaseous environments, especially those with observable resonance phenomena. Collision theory, which connects with the observables of atomic and molecular spectroscopy, frequently involves solving the time-independent Schrödinger equation provided no time-dependent observation is carried out. Such theoretical descriptions are of interest when overall reaction rates for various processes are needed—for example, to describe how a gas cools in a terrestrial or atmospheric environment, or the chain of reactions that produces desired chemical reaction products. Even though the underlying phenomena may involve auto-ionization and other rapid electron transfer phenomena that occur in the femtosecond or attosecond time frame, such processes can still be treated within the framework of time-independent quantum calculations of the collisional dynamics. Collision experiments that measure such phenomena are highly sophisticated, and theory has made tremendous progress in its capability to describe increasingly complex reactions involving four or five atoms, but it is a challenge to push quantum theory capabilities to the point where it can handle systems with multiple complex reactants. This chapter also discusses dynamics accessed in the frequency domain, through collision physics theory and experiment, and through frequency comb spectroscopy.
Last, the committee discusses the novel physics that can be done with the extreme light sources of today. Chapter 2 described the extreme intensities that can be reached both in the optical regime, at pentawatt (PW) laser facilities, and in the X-ray regime, at XFEL facilities. The penultimate section of Chapter 5, “Novel Physics with Extreme Light,” outlines some of the exciting science both within and beyond AMO that can be explored using such light sources. This chapter ends with a list of findings and recommendations.
CHALLENGES AND OPPORTUNITIES
Direct experimental access to out-of-equilibrium dynamics and coupling between charges, spins, and atoms, together with their interpretation using advanced theoretical models, represents a grand challenge for both scientific and technological applications. Here, the committee outlines a number of exciting challenges and opportunities for the coming decade:
- Harnessing of coherent electron dynamics: Attosecond technology is enabling the measurement and control of electron dynamics in atomic, molecular, and condensed-phase systems. An exemplar is the concept of light-wave electronics in which a current in a semiconductor can be controlled with the electric field from a strong laser pulse, with potential for impact on the speed of electronic devices.
- Making of molecular movies, from electron to structural dynamics: The goal is tracking and controlling the flow of energy from electronic excitation through structural and chemical changes, spanning subfemtosecond to nanosecond time scales. This includes disentangling strong correlations between electrons, nuclei, and spins, with implications spanning the range from fundamental questions about energy transfer, to biological processes such as light harvesting, photoprotection, and to new molecular devices. In particular, recent advances in ultrafast X-ray sources, spectroscopy, and diffraction methods have us on the cusp of producing molecular movies starring individual molecules.
- Complex reaction dynamics and collision physics: The improvement of theoretical and experimental techniques that can predict reaction rates and unravel complex reaction dynamics of increasingly complex systems of atoms, molecules, ions, and photons is needed for deepening our understanding of chemical transformation processes and plasma environments.
- Extreme physics with extreme light sources: Light sources with extreme intensities, both in the infrared/optical regime and the X-ray regime, will allow for unprecedented studies of matter in extreme conditions with potential for numerous applications. X-ray strong-field physics, laser-assisted pair creation at the Schwinger limit, or laser-driven electron acceleration raise both fundamental questions and have significant potential for impact beyond AMO.
ATTOSECOND SCIENCE: THE TIME SCALE OF THE ELECTRON
As discussed above, electrons in matter have very fast dynamics—on the time scale of tens to hundreds of attoseconds. Enormous progress in attosecond science and technology over the past decade means that researchers now routinely produce attosecond pulses and study attosecond processes using table-top laser facilities.
But what do we mean when we talk about “electron dynamics,” since quantum mechanics teaches us that electrons should be described by their probability densities, which are distributed in space around the heavier nuclei in atoms and molecules? Electron dynamics in general means changes in the electron distribution following excitation. In the case of ionization, removal of an electron from an atom or molecule leads to a charge imbalance and therefore a rearrangement of the other electrons. Some examples of electron dynamics that can be measured and timed are photoemission (the absorption of a photon that leads to the release of an electron—how long does this take and which factors determine it?), ultrafast decay processes (for example, the filling of a hole after removing an electron from an inner shell), and charge migration (the coherent oscillatory motion of the electron cloud in a molecule resulting from the localized removal of an electron from a molecule). Some of these processes, and how to measure them, are described in more details below.
Fundamental Questions on the Attosecond Time Scale
The photoelectric effect, which was first explained by Einstein more than 100 years ago, describes the emission of an electron following the absorption of a photon whose energy exceeds the system’s binding energy. Starting in 2010, a series of experimental and theoretical works have explored the dynamics of the photoelectric effect. In the 2010 experiment, an attosecond XUV pulse freed electrons from two different orbitals in a neon atom (s and p orbitals) and their emission times were compared. The measurement showed a difference in the electron emission time from the two states ranging from 10-100 attoseconds. Continued progress in ultrafast laser development and metrology has allowed the accurate measurement of photoemission times in a range of systems, from atoms to semiconductors, as illustrated in Box 5.1. In the interaction between the ion core and the departing electron, the emission time can be interpreted in terms of the scattering phase imparted as the electron makes its way out from the ion core; this provides a concept of time in the quantum mechanical description of this process.
In a related set of experiments, the photoelectron emission resulting from strong-field ionization has been accurately timed using the so-called attoclock. In these measurements, the attosecond timing is provided by the rotating polarization of a near-circularly polarized laser field that tunnel-ionizes the atom. The polarization clock makes a full rotation every optical laser cycle, namely 2,700 as, and the electron will thus be emitted in different directions at different times during the cycle. In this way, attosecond dynamics are probed without the necessity of making an attosecond pulse.
Theory has played a crucial role in understanding the photoemission measurements. Fully quantum mechanical calculations of the dynamics of multiple
correlated electrons are possible only for small atomic systems, but these have allowed researchers to use measurements in helium as an absolute timing reference. Concepts gleaned from electron-ion scattering, well-known from collision physics, have been essential in the interpretation of these experiments, which are really measurements of the scattering phase, in terms of timing. Large-scale calculations of surface- and bulk-electron structure and dynamics have been necessary to understand emission times and laser interactions from condensed-phase materials.
Another fundamental ultrafast process is the decay that takes place in atoms, molecules, and solids. Molecules and most atoms have a range of core excited states that are inherently unstable. These have very short lifetimes, on the time scale of a few femtoseconds. Holes created in inner shells, for example, are often filled on this time scale by the decay of an electron in a higher-lying shell by emitting a photon (fluorescence) or another electron (Auger). This decay process can even happen between nearby atoms in a molecule or cluster so that an electron from one atom decays to a core hole in a neighboring atom. This process, called interatomic Coulombic decay, has recently attracted a lot of attention as it has been realized that it happens in a wide range of system and is important in some photobiological processes. As another example, coherent excitations from the valence band to the conduction band in insulators are thought to last only a few femtoseconds before they decohere by coupling to the crystal lattice degrees of freedom. These types of decay process can now be accessed in experiments combining ultrafast strong-field and attosecond methods.
Finally, charge migration is an important example of ultrafast coherent electron motion that occurs in a molecule following the rapid removal of an electron from either a core- or a valence shell to create a localized hole. This creates a charge imbalance in the molecule and can lead to the hole migrating across the molecule and back, on the time scale of one to a few femtoseconds, as has been reported by several groups for different molecules. Charge migration is strongly influenced by electron correlations, and is a precursor to more permanent charge- and structural rearrangements that happen over longer time scales. Calculations have suggested that there is in fact a universal attosecond response of many different molecules to a localized ionization event. Charge migration is an excellent exemplar for the earliest stages of a molecular movie, both in terms of the electron dynamics, which starts out coherent, and in terms of how the coherence is lost due to electron-nuclear coupling. Charge migration thus provides an exciting challenge for both experiment and theory in ultrafast AMO science.
Intense Laser-Matter Interactions: A Gateway to Attosecond Science
In the first decade of the new millennium, the field of attosecond science was synonymous with the generation of attosecond light pulses in atomic gases via high harmonic generation. This is because the underlying physics of this generation
process, based on an intense laser field (above 1014 W/cm2) interacting with an atom or molecule, is inherently attosecond in nature. However, the attosecond light pulses are only one possible outcome of this strong-field interaction, and as illustrated above, attosecond science now drives a range of applications ranging from fundamental questions in quantum mechanics to filming molecular movies.
The physics of intense laser-matter interaction is well established by a plethora of experiments and theoretical analyses, which culminated in a widely accepted intuitive picture, the semiclassical recollision model. In this model, an atom or molecule subjected to an intense linearly polarized laser field is viewed in three steps, as illustrated in Figure 5.2. In the first step, an electron wave packet (EWP) is promoted to the continuum by tunnel ionization at some phase of the field. The wave packet then evolves under the combined influence of the field and atomic potential (step 2) until it either escapes or recollides with the parent ion after approximately one-half of an optical cycle (step 3). As illustrated in the figure, the recollision of the EWP with the parent ion leads to dipole emission, yielding high-energy photons (the high harmonics); the production of high-energy electrons; and sometimes multiple electron ionization. Most importantly, the initial tunnel ionization process, whose rate is exponential with the laser field, sets a time scale for the EWP that is inherently subcycle and therefore at the attosecond level. It is this EWP timing that is imprinted on all subsequent recollision processes.

The three-step recollision model illustrates how the essence of strong-field attosecond science is the control of the EWP, which conveys additional ultrafast information via different observables. In high harmonic spectroscopy (HHS), as described in more detail below, the high harmonic light emerging from a molecule carries information about the molecular structure and dynamics in its spectral amplitude and phase. Figure 5.2 also illustrates another method for extracting molecular dynamics, termed laser-induced electron diffraction (LIED). This is the elastically scattered EWP—marked in the bottom left of the figure. The momentum distribution of the elastically scattered EWP process conveys information on the molecular structure, and LIED measurement provide a novel means for spatial-temporal imaging of molecular dynamics.
The recollision model has also been very influential in strong-field and ultrafast theory, through calculations based on the single-active-electron approximation, both at the ab initio and phenomenological level. In the past decade in particular, calculations based on the three-step recollision model, including quantitative calculations of the ionization probability, core and continuum dynamics, and complex rescattering cross-sections, have been used to interpret a number of experiments employing HHS and LIED to study atomic and molecular structure and dynamics.
From a fundamental perspective, harnessing the ultrafast and semiclassical nature of the EWP has been a major research thrust. AMO scientists have recognized that the scaling-laws of strong-field physics mean that experiments benefit from longer wavelength fundamental fields relative to the near infrared—for instance, leading to higher-energy photons and electrons while using lower intensity. The semiclassical three-step behavior described above has been firmly established using different laser platforms ranging from mid-infrared (1-5 microns) to terahertz generation. This strategy not only enables more applications but also has resulted in the discovery of new strong-field phenomena such as those described below.
Strong-Field Attosecond Physics in Solids and Nanostructures
The insights of the recollision model are applicable not only to describing gas-phase processes, but also to the physics associated with a crystalline material interacting with an intense long-wavelength laser field. For example, nonlinear absorption across the bandgap in semiconductors and insulators generates emission through electron-hole recombination. Solids also provide additional processes absent in gas-phase systems. For example, a nonlinear current induced in a single band via Bloch oscillations generates harmonics. Studies have shown that both the recollision and the Bloch oscillation processes have a subcycle response (attoseconds) to the driving field. The interplay between these different mechanisms results in rich physics, and currently a variety of long- and short-range order in materials, including bulk and nanostructures, are under investigation. This is
a very active area of research since it has the potential of providing a novel probe of band-structure dynamics, and perhaps the development of an on-chip vacuum ultraviolet (VUV) light source.
Nanostructures interacting with intense, few-cycle pulses also exhibit recollision model behavior, such as producing high-energy electrons and high-harmonic photons on very fast time scales as described above, with the physics of the dynamics again richer than in atoms. Exposing a metal nanotip to an intense field produces an electron energy distribution quite analogous to the well-known phenomenon of above-threshold ionization in atoms. Depending on the carrier envelope phase, electrons are emitted either from a single sub-500-attosecond burst or from two; the latter case leads to spectral interference. The interpretation is consistent with the coherent elastic scattering of a field-driven EWP. Unlike an atom or molecule, however, the nanotip produces a plasmonic enhancement of the local field, which decays rapidly from the tip. Thus, these experiments require only an unamplified ultrafast laser oscillator, since the intensities needed are orders of magnitude lower than the atomic case. Harnessing field-driven EWPs may provide new time-resolved methods for surface science. Low-energy electron diffraction, with electrons originating from and probing the surface yielding time scales of ~100 as, might come into reach.
Attosecond Metrology: Methods for Clocking Electron Dynamics
The uncertainty principle says that one cannot simultaneously determine the precise energy and the precise timing of a physical process. As a result, an ultrashort attosecond pulse will in general excite a range of processes with different characteristic energies. To be able to resolve individual processes, attosecond metrology often relies on pump-and-probe experiments in which an ultrafast process is initiated with a pump pulse, and then probed by a second pulse that arrives some time later. The time resolution in the experiment is then determined by the precise control of the delay between the two pulses (which can be at the attosecond level), and the energy resolution is determined by the method of detection. By measuring how the absorption of an attosecond X-ray pulse changes as a function of this time delay (termed Attosecond Transient Absorption Spectroscopy, or ATAS), researchers have for example measured few-femtosecond lifetimes of states created by ionization or excitation, and specifically have observed the correlated motion of two quasi-bound electrons in helium, which “beats” with a periodicity of about 1 fs.
The standard picture of absorption is that of a quantum of energy being lost from the light field and stored in the material. However, in the time domain picture, absorption can be interpreted as the destructive interference between the electromagnetic field of the input light and the light resulting from a coherent oscillation created in the material. This means that the absorption process can be interrupted and controlled by a probe infrared laser field. In the spectral domain, this manifests
as a change of the basic shape of the absorption peak in the X-ray spectrum, and has wide-ranging consequences for potential control of the temporal and spatial properties of X-ray pulses. Absorption along one direction can, for example, be refashioned as emission along a different direction, leading to the modulation of X-ray light by an optical field.
In HHS, the time resolution is provided by the time-frequency ordering of the returning EWP in the semiclassical recollision model: the electrons that give rise to the higher photon energies return later than the low-energy ones. This means that different frequencies in the harmonic spectrum carry information about different recollision times, at the sub-laser-cycle (and thus attosecond) time scale, and the resulting chirp of the emitted harmonic radiation has been dubbed the attochirp. HHS has been used in a wide range of applications, measuring atomic and molecular structure and dynamics, both at the electronic (charge migration) and nuclear (dissociation) level, and more recently to characterize both structural and dynamical features in solids.
There are also several attosecond clocking methods based on measuring photoelectrons, in particular measuring their spectral yield as a function of a time delay that is known with attosecond precision. This is the basis for attosecond streaking and RABBITT (both discussed in Chapter 2) and the attoclock. The timing of the photoelectric effect (see Box 5.1) was performed using the attosecond streaking method. Lastly, the ability to control and measure the polarization properties of high-order harmonics has exciting implications for the characterization of spin, magnetic, and chiral properties and dynamics of gas- and condensed-phase samples. This polarization control, which has been demonstrated and explored in detail in this past decade, is achieved by generating harmonics with counter-rotating two-color laser fields, leading to harmonic radiation with any choice of linear, elliptical, or circular polarization. The chiral properties of a sample can then be probed either through the imprint of the sample on the harmonic properties (similar to HHS), or by examining the response of the sample subjected to polarized XUV or X-ray light.
Challenges for Theory
Calculating attosecond electron dynamics from first principles in molecular systems, like biomolecules, is one of the premier computational challenges today and will remain so for the foreseeable future. The fundamental difficulty is the rapid scaling of the computational effort with the number of active electrons. This is particularly true because most attosecond dynamics involves ionization, which necessitates being able to describe multielectron continuum as well as bound states. Progress in the past decade has allowed a number of fully ab initio studies of correlated electron dynamics in helium and other two-electron systems, treating strong-field double-ionization, two-XUV-photon ionization, and questions
of photoionization timing as discussed above. Efforts are underway to gather a number of these highly validated theoretical approaches in a common Atomic and Molecular Physics Gateway, which will include access to software suites and documentation for other research groups. Calculations in larger systems can in principle be attacked by including multiple, time-dependent configuration interactions. This has been done for small systems using configurations including single and double excitations, and it is an active area of research to extend such approaches to more complex and/or nonlinear systems. An alternative, approximate treatment using time-dependent density functional theory (TDDFT) has the attractive feature that it can be scaled to large systems. TDDFT has been shown to be accurate enough for single ionization and charge migration studies, but also raises open questions about how to calculate certain observables from the density, as well as its reliability to describe processes with correlations beyond the mean-field level.
The next-level problem in calculating coherent electron dynamics will be to include the coupling to the inevitable nuclear dynamics and the ensuing loss of electronic coherence. For molecules, only hydrogen-like systems with two nuclei and two electrons have been treated fully ab initio, for example for studies of electron localization upon strong-field ionization, or the influence on nuclear dynamics on transient absorption spectra. Calculations of ultrafast electron dynamics in condensed-phase systems have been undertaken by a number of groups in the past few years, as experimental interest has flourished as described above. Most of these have been done in periodic, crystalline systems in which many-body interactions can be managably treated, for instance using the semi-conductor Bloch equations. However, also for these systems, the loss of electronic coherence via for instance coupling to the nuclear degrees of freedom has generally been treated phenomenologically. Much work still needs to be done, for both molecular and condensed-phase systems.
The Future of Attosecond Science
Attosecond science is beginning to impact our understanding in condensed-phase systems, with implications for both XUV source development and ultrafast metrology. For example, progress on the implementation and understanding of HHG in solids suggests that compact and efficiently engineered sources of ultrafast XUV light are feasible. The use of pump-probe methods to determine attosecond photoemission times from metals, and attosecond lifetimes of conduction band electrons in semiconductors, means that attosecond metrology is capable of probing the fastest dynamics even in such highly correlated systems.
The concept of lightwave electronics is particularly exciting. The increase in speed of modern electronics is a consequence of shrinking electronic devices. However, the sizes are rapidly approaching the ultimate nanoscale limit; thus, the clock rate (processing speed) has been limited to a few gigahertz for more than
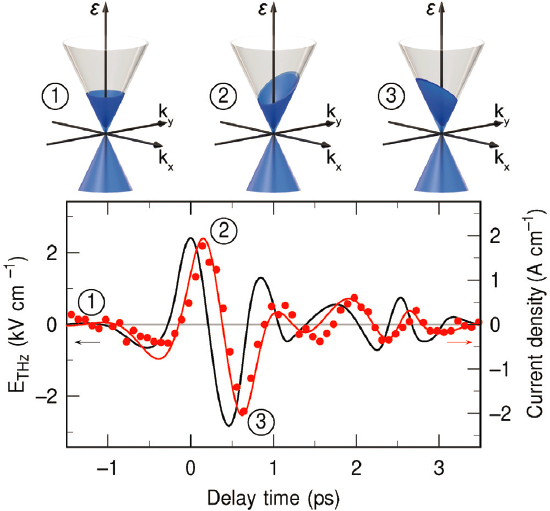
a decade. Recent experiments have demonstrated lightwave control of electronic currents in insulators, in which a strong laser field induces and modulates a time-dependent electric current in the material (see Figure 5.3). These laser-induced currents are generally reversible and nearly dissipation-free. Given that strong laser fields can be produced with optical periods close to the petahertz level, this suggests that lightwave-driven currents in transistors could result in speed-up of many orders of magnitude in the near future.
Another exciting prospects for attosecond science is the impending availability of intense attosecond pulses in the soft and hard X-ray regime from XFELs, which would enable the initiation of electron dynamics (e.g., charge migration) from inner valence or core electrons that are in general highly localized on specific atoms within a molecule. In combination with pump-and-probe capabilities, such pulses would thus allow for both spatial and temporal resolution of attosecond electron dynamics.
THE MOLECULAR TIME SCALE: FEMTO- TO PICOSECONDS
At the heart of any chemical reaction is explosive transformation: old bonds in a molecule are broken and new ones formed. In photochemistry, rapid ionization of a system leads to rapid electron motion, which leads to atomic displacements, which leads to chemical changes. Light harvesting in chromophores, the photoprotection mechanism in our DNA, and more generally optically driven molecular devices are all examples of processes in which an initial electronic excitation is coupled to nuclear motion that leads to chemical or structural changes. In the previous section, the committee discussed that the initial electron motion takes places on sub-to-few femtosecond time scales and often involves electron-electron correlation. This section focuses on primarily the subsequent molecular dynamics that happens over tens to thousands of femtoseconds and involves both electron-nuclear and nuclear-nuclear correlations.
Molecular Dynamics and the Concept of Femtosecond Molecular Movies
The nuclear dynamics discussed above includes both motion, such as vibrations, rotations, and structural/configurational changes of the molecule, and changes in the charge-, spin-, or oxidation state. Electrons always play a role in the nuclear dynamics, since the molecule will always seek to be in the configuration with the lowest electronic energy. The interaction between the electronic and nuclear degrees of freedom can often be thought of in the Born-Oppenheimer approximation, in which the electron and nuclear dynamics are treated separately (based on the different time scales for their dynamics) so that the electronic density simply creates the potential surfaces on which the nuclei move. However, there are also many interesting instances in which the electronic and nuclear dynamics is much more directly coupled, so that the Born-Oppenheimer approximation breaks down. This is often seen in the form of conical intersections (CIs)—where different molecular geometries give rise to electronically excited states with the same energies. At such points, an electronic excitation in one nuclear configuration can therefore result in population transfer to a different nuclear configuration, thereby driving structural changes in the molecule. These CIs, spanning the space between the physics of coherent electron dynamics and the chemistry of transformation, have been topics of intense interest in the ultrafast AMO community, and have been documented both in naturally occurring transformations as well as controlled by ultrafast light pulses.
Ultrafast pulses of light or electrons are ideal tools to see the nuclear motion directly in the time domain, as first demonstrated by one of the the founders of “femtochemistry,” Ahmed Zewail, for which he was awarded the 1999 Nobel Prize in Chemistry. An example of such a femtosecond molecular movie is illustrated in Box 5.2. The individual pictures in the movie can consist of any measurement
that is sensitive to the instantaneous electronic or structural configuration of the molecule, and it can be direct in the form of actual images such as scattering patterns, or indirect in the form of absorption or emission spectra, or the detection of ions or photoelectrons. Several of these approaches are discussed in more detail in the subsections below.
The access to and control of ultrafast light pulses, ranging from the optical through the hard X-ray spectral range, continues to drive immense progress in time-resolving fundamental processes in molecules and beyond. An exemplar for advances in ultrafast technology in the past decade, as well as an illustration of coupled electron-nuclear dynamics driving a configurational change, is the photo-induced ring-opening reaction in 1,3-cyclohexadiene. This well-known photobiological reaction plays a role in vitamin D synthesis, and involves an opening of the initial ring-shaped molecule to the final chain-shaped 1,3,5-hexatriene molecule, which takes place on the 100 fs time scale. The ring-opening reaction involves a reshuffling of single and double electronic bonds, and proceeds through a conical intersection. Progress in ultrafast metrology has allowed this reaction to be probed directly in the time domain, in three different types of experiments in the past decade: (1) hard X-ray diffraction as illustrated in Box 5.2; (2) soft X-ray absorption spectroscopy; and (3) ultrafast electron diffraction, both of which are discussed in the sections below.
The Ubiquity of Ultrafast X-Rays
The unprecedented development of ultrafast XUV and X-ray sources in the past decade, both XFEL sources and XUV table-top sources based on HHG as described in Chapter 2, has revolutionized the capabilities of AMO science to probe ultrafast dynamics. The short duration and high photon energies of these modern X-ray sources enable scientists to focus on individual atoms, even when embedded in complex molecules, and to view electronic and nuclear motion on their intrinsic time and space scales.
Soft and hard X-ray pulses generally probe different aspects of the electronic and nuclear dynamics, and different types of experiments can therefore elucidate the connection between the initial electronic dynamics and the subsequent molecular dynamics in a molecule that has been excited by an optical or X-ray pump pulse. As also discussed in the section “Attosecond Science: The Time Scale of the Electron,” soft X-ray and XUV pulses address the structure and dynamics of valence electrons with their moderate binding energies, whereas hard X-ray pulses with their very short wavelengths can be used to resolve the location of nuclei directly through scattering images, as was illustrated in Box 5.2.
These experiments also benefit from the extension of well-known X-ray spectroscopy techniques into the femtosecond domain, and from an unprecedented
degree of control over the initial preparation of the sample, the relative timing of the pump and probe pulses, and the extraction of information from the detection step. In sample preparation, in particular, the ability to align and orient gas-phase molecules using lasers means that the molecular dynamics can often be explored in three dimensions, as the ion and photoelectron yield can be measured as a function of the relative orientation of the molecule and the polarization of the light field. Several methods allow for detection of all or several dynamics constituents in coincidence, vastly increasing the amount of information gained about how the dynamics proceeded. Among these are velocity map imaging and Cold Target Recoil Ion Momentum Spectroscopy (COLTRIMS).
X-ray absorption and photoemission spectroscopy can also be used to infer molecular dynamics via the measurement of characteristic electronic transitions, especially between core- and valence-level states. This is because the electronic excitations energies are characteristic of the nuclear configuration, including its charge-, oxidation, and spin-state. This is the basis for time-resolved X-ray absorption spectroscopy “movies,” an example of which is illustrated in Figure 5.4 for the dissociation of tetrafluoromethane. In this molecule, the increase in the
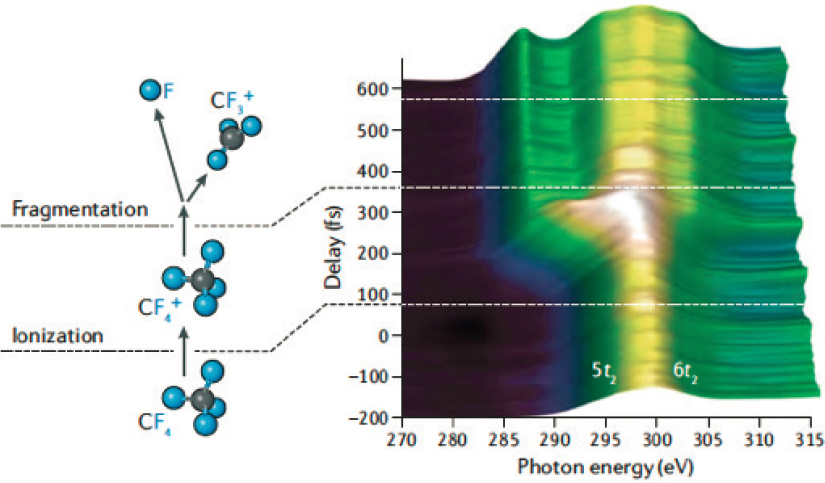
internuclear separation between the F atom and the remaining CF3+ group gives rise to a characteristic new peak in the core-level absorption spectrum.
Time-resolved soft X-ray absorption spectroscopy was also used to investigate the ring-opening of cyclohexadiene. By following the evolution of characteristic core-to-valence transitions with delay, AMO scientists were able to directly reveal the intermediate (transient) electronic state that the ring-opening reaction proceeds through, the so-called pericyclic minimum. This represents information complementary to the purely structural information about the location of the nuclei during the reaction, helping to shed light on the coupling of the electronic and nuclear degrees of freedom.
The characterization of electrons or ionized fragments resulting after X-ray ionization or excitation of deeply bound states can also elucidate the interaction between the electronic and molecular dynamics. Electronic kinetic energies, for example, whether from direct photoelectrons or Auger electrons, are sensitive both to particular bond lengths (as also discussed above) near the atomic site from which they originated, and to the electronic states involved in the photochemical reaction. In a recent example, researchers were able to solve a long-standing controversy involving the photoprotection mechanism of the DNA-compound thymine. By combining Auger electron and soft X-ray absorption spectroscopy, and via comparisons with theory, they found that after UV photoexcitation, the molecule relaxes into the protected (less chemically reactive) state via a conical intersection in less than 100 fs.
Hard X-ray radiation is ideal for taking direct pictures of molecular structure through scattering images, because their short wavelengths, comparable to common bond lengths, permit very high spatial resolution. Hard X-ray radiation was used in the direct imaging of the ring-opening reaction illustrated in Box 5.2. This type of imaging through diffraction, using ultrafast pulses of light or electrons, is discussed in the following section.
Molecular Imaging Through Ultrafast Diffraction
A multitude of diffraction techniques exist to take “pictures” of molecular samples during a dynamical process, and more broadly to image the structure of noncrystalline samples at the atomic level. The scattering pattern on the detector is not in general a straightforward image of the sample (like that formed by a camera) but has to be interpreted using sophisticated Fourier-transform-based algorithms. Coherent diffractive imaging (CDI), for example, takes advantage of the high spatial coherence of the laser-like ultrafast X-ray pulses from HHG and XFEL sources to retrieve both amplitude and phase information from the diffraction pattern, leading to increased sensitivity and contrast. For imaging of gas-phase ensembles, the phase can often be retrieved directly from the Fourier transform of
the scattered data. For more complex (e.g., crystalline) samples, the phase retrieval is based on hundreds or thousands of iterations of an algorithm that goes back and forth between the measured diffraction pattern on the detector and the real-space image of the sample. In each iteration, more constraints can be applied since the high quality of the X-ray beam allows for oversampling of the spatial structure of the sample. By controlling the polarization of the X-ray light, or in combination with its X-ray absorption spectroscopy, CDI enables mapping of the elemental, chemical, and magnetic properties of complex matter at the nanoscale.
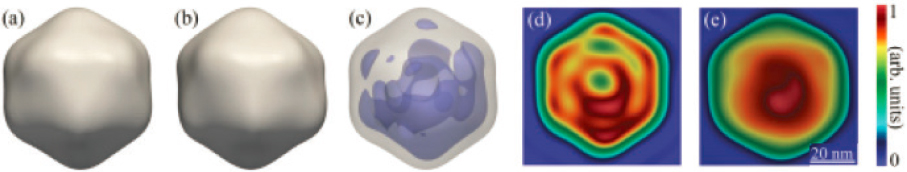
Single-particle imaging (SPI) has long been a dream of several communities, both as an element of making molecular movies, but more generally for imaging in structural biology and materials science. Inroads are now being made toward this goal, thanks to the advent of the very intense and short pulses of X-ray light available at XFEL facilities. Imaging a single biomolecule presents a number of technical and computational challenges. First, the scattering from a single molecule is very weak, and even when using intense X-ray sources, the scattering pattern from many different individual molecules, generally with different orientations, must be analyzed using CDI techniques discussed above, and added together to give three-dimensional (3D) information. Second, the intense X-ray light causes the molecules to explode. However, if the pulse is short enough, then the scattering pattern can be formed before the damage occurs. A global SPI initiative, launched in 2014 and drawing on significant multidisciplinary effort from research groups around the world, has been devoted to resolving current and future challenges in SPI. In 2015, researchers presented the first 3D measurements of a single biological sample, the large 750-nm-diameter mimivirus, with a spatial resolution of 125 nm. More recently, several smaller viruses have been imaged with spatial resolutions below 10 nm, as illustrated in Figure 5.5.
Ultrafast electron diffraction (UED) is a complement to ultrafast X-ray diffraction; it benefits from larger electron scattering cross-sections. Advances in the production and stability of femtosecond pulses of high-energy electrons have allowed a number of studies of dynamics using an optical pump and a UED probe. Recent examples include mapping the ultrafast photo-induced dissociation dynamics of CF3I molecules in the gas phase via a conical intersection that allows the transfer of the initial electronic excitation energy to the nuclear degrees of freedom responsible for the dissociation. In another recent study, the very high spatial resolution provided by the electron beam allowed researchers to follow the evolution of individual carbon-carbon bonds during the ring-opening reaction in cyclohexadiene (also studied in the X-ray scattering experiment in Box 5.2), yielding not only a detailed movie of the ring opening itself but also evidence that the chain-shaped molecule after opening bends back and forth between two different configurations.
In LIED, the ultrafast electron “pulse” is generated by a strong laser field as the rescattering EWP discussed in the context of the three-step model for intense laser-matter interactions above (see Figure 5.2). This self-probing of a system set in motion by strong-field ionization has proven to have high resolution in both space and time, with recent measurements of the ultrafast dissociation of acetylene, for example, or the laser-induced structural deformation of the C60 cage.
Using Light to Control Reaction Paths
Light can be used not only to initiate and probe a dynamical process but also to control how the process unfolds. This is generally referred to as quantum control. The past decade has continued to see extensive developments. There are several different approaches to quantum control including ones that exploit optical lattices in reduced dimensionality, and ones that exploit excitation to and from, and dynamics on, different potential energy surfaces, designing system-specific pump-probe sequences. Some methods have their own names, like optimal control and coherent control, respectively.
The optimal control approach relies on the ability to control a range of parameters influencing the shape of an ultrafast light pulse that interacts with the system of interest so that a desired outcome can be achieved, often using a feedback loop and some number of iterations. Early experiments used precise shaping of the driving laser pulse to control ionization, dissociation, or emission properties in small quantum systems, as well as reaction pathways in chemical reactions, for example, via light-induced conical intersections. For current and future studies, the continued progress in machine learning and big-data analysis enables the control of a much larger range of laser and environmental parameters, potentially making outcomes more robust and repeatable.
The coherent control approach involves building creative control scenarios by harnessing the underlying quantum mechanics—for example, exploiting interferences between different pathways to the same final product state to control, for example, product branching ratios, photoassociation, enantiomeric selectivity, state-to-state control, and spin-orbit entanglement in atom pairs. Other recent applications include biexciton control in quantum dots, pulse train control, and producing chains of entangled ion-atom pairs. It also has opened a view into foundational issues such as examinations of the role of “nontrivial” quantum effects (such as interference, nonlocality, and entanglement) in control scenarios, considerations of classical limits on control, and recognition of the role of in-principle distinguishable pathways in diminishing control. Important challenges remain in the area of understanding and controlling decoherence.
Quantum Calculations of Molecular Dynamics
Predicting the nuclear motion and structural dynamics for hundreds of femtoseconds after some initial excitation requires the description of both electronic processes, such as excitation and charge transfer, and nuclear dynamics facilitated by transitions between electronic states. Even without taking into account electronic coherence, this is a grand-challenge-level computational problem, and major efforts are dedicated to its solution.
One such theory approach, termed ab initio nonadiabatic molecular dynamics, uses methods from quantum chemistry to solve the time-independent electronic Schrödinger equation to obtain electronic structure, simultaneously with quantum descriptions of the nuclear motion and transitions between electronic states. Recent progress has allowed a number of high-level comparisons to experimental results, showing very good quantitative agreement with theoretical predictions—for example, for both of the UED experiments described above, on the ring-opening reaction and the dissociation of CF3I. A key bottleneck for going forward is the molecular size and degree of correlations that can be modeled with such approaches, due to both scaling with system size issues and the vastly different time scales that need to be computed for the electronic and nuclear motions. Improvements in the treatment of quantum effects in the nuclear dynamics and the solution of the electronic structure problem for large molecules are both critical to further progress. In order to even qualitatively model conical intersections, where multiple electronic states are degenerate, multireference electronic structure methods are needed. At the same time, dynamic electron correlation effects can be important in changing the location and energetics of conical intersections. Thus, both static and dynamic electron correlation effects must be included. In order to simulate and understand charge and energy transport in large-scale molecular assemblies (relevant in application areas such as artificial photosynthesis and batteries), it is
therefore critical to improve methods for incorporating static and dynamic electron correlation simultaneously with nuclear motion.
New conceptual approaches to these problems are critical, but it is also important that they be adapted to and implemented on fast and efficient computer architectures. The emergence of graphical processing units (GPUs) as computational engines for physics and chemistry, has highlighted the importance of “stream processing”—that is, extreme data parallelism—as well as exploitation of heterogeneous architectures such as combinations of CPUs and GPUs. Some of the algorithms for electronic structure theory and molecular dynamics have been recast in a form suitable for efficient execution on these architectures, which will almost certainly play a key role in the path to exascale computing. Much more effort along these lines is needed, especially in a manner that creates building blocks that are easily reused and retooled so that they remain relevant when new conceptual approaches appear.
The Role of Quantum Chemistry in Spectroscopy and Dynamics
The calculation of molecular energy spectra, for both ground-state and excited-state molecules, is the basis for comparison with and predictions for a wide range of experiments in high-precision or time-resolved spectroscopy. A number of large-scale packages for quantum chemistry calculations exist, and are continuously being developed and applied. The need to treat molecules with a large number of quantum constituents, and with highly correlated electrons, drives developments in quantum chemistry, including both improvements in precision and in the scaling of resources with the number of electrons. Recent notable developments include advances in various forms of quantum Monte Carlo (QMC) as well as the development of methods in density-matrix renormalization group (DMRG) better able to deal with quantum chemistry. While QMC methods have been considered the gold standard for accuracy for several decades, they have until recently been computationally costly. In the past decade, advances in the initial trial wave functions used in QMC have allowed both improvements in precision and computational scaling for a range of moderately sized systems. Similarly, DMRG methods have been gaining attention in the quantum chemistry community recently, also for their potential to both increase precision and computational efficiency. A recent example of the latter includes studies of long chain molecules for which the computational time scales linearly with chain length.
The Future of Time-Resolved Molecular Dynamics
Modern ultrafast X-ray sources provide shorter pulses, a wider selection of photon energies, and higher brilliance than ever before, and will in general enable better resolved dynamics in both space and time. For example, the increased brightness that will soon be available at the upgraded LCLS facility (LCLS-II)
makes it very likely that the next decade will see time-resolved dynamics at the single-particle level. Similarly, the impending availability of X-ray pump, X-ray probe capabilities, using intense pulses for which the photon energy and the relative delay can be controlled, will enable nonlinear spectroscopy studies that involve multiphoton transitions. Nonlinear spectroscopy—for example, Coherent Anti-Stokes Raman Scattering—using a carefully timed series of optical pulses, has long been a favorite tool for studying electron and molecular dynamics, and its extension to the X-ray regime is an active area of research. Its extension to the X-ray regime would allow making “complete” molecular movies spanning both spatially resolved, attosecond, core, and valence electron dynamics, as well as couplings to femtosecond nuclear dynamics. For instance, one could track the ultrafast passage through a conical intersection and its impact on electronic coherence. Additionally, ultrafast AMO science will have far-reaching consequences for applications to photobiology. Figure 5.6 shows an example of the photo-induced dynamics in the retinal-binding protein bacteriorhodopsin. Retinal is a light-sensitive molecule
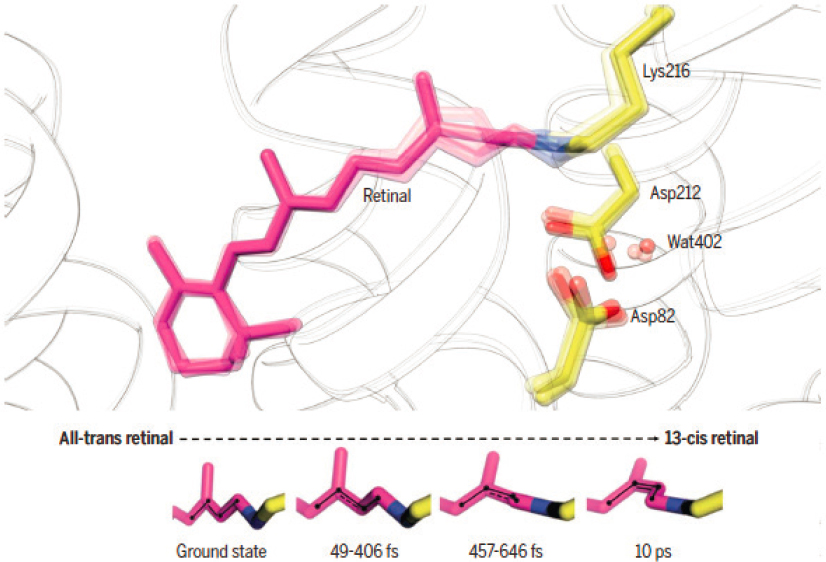
that is used biologically in a range of light-harvesting and light-energy transfer processes, and its structural rearrangement (isomerization) takes place in tens to hundreds of femtoseconds, making it among the fastest known in photobiology. Researchers captured its transformation in real time using time-resolved ultrafast X-ray scattering.
Last, optical pump and X-ray probe studies will enable a wide range of studies of new phases of matter, including quantum materials, induced by coherent light-matter couplings. Recent exciting examples include the demonstration of light-induced superconductivity, and control of topological phases via the polarization of the excitation light source. A theoretical prediction for the ability to switch a material between a topological insulator and a conducting semi-metal using femtosecond laser sources could potentially be experimentally validated using diffraction of hard X-ray pulses from soon-to-be-available XFEL sources.
FREQUENCY-DOMAIN APPROACHES TO DYNAMICS: COLLISIONS AND CORRELATIONS
The sections above have discussed how the increased temporal resolution provided by femtosecond laser sources enables the study of ultrafast dynamics directly in the time domain. Likewise, the unprecedented spectral resolution provided by phase-controlled continuous wave lasers enables the study of ever finer energy structures of matter. For example, it is now possible to investigate optical transitions with a resolution approaching 1 part in 1016. Many new scientific thrusts have emerged through this quest, such as tests of fundamental physics, the development of sensors of increasing sensitivity, and the search for new physics beyond the standard model.
For much of the past half-century, efforts by theory and experiment were mostly devoted to understanding energy-dependent reaction rates, in contrast to the recent emphasis on time-dependent processes that has been enabled by the advent of ultrafast lasers and detection techniques. Complementary to time-resolved experiments, the energy-resolved results from collision studies is what is frequently needed to model state-to-state collisional processes in gaseous environments, especially those with observable resonance phenomena. Advances in both time- and frequency-domain approaches are still needed, in order to achieve deep understanding and accurate predictive power of quantum mechanical processes, especially at low collisional energies or temperatures. In particular, significant fundamental progress is needed to extend current theoretical capabilities to quantitatively treat and unravel collision events involving four or more atoms or small molecules.
It should also be stressed that the possibilities are not restricted to “time-only” resolution or “frequency-only” resolution. In some cases, it is desirable to obtain both time and frequency resolution of dynamical processes simultaneously, and
this is possible within the usual constraints imposed by the frequency-time uncertainty principle.
Frequency Combs—A New Frontier in Broadband and High-Precision Spectroscopy
Optical frequency combs offer enormous potential owing to the simultaneous availability of vast spectral coverage and high spectral resolution, and they open a new frontier for coherent spectroscopy and broad-bandwidth, high-resolution quantum control of molecular dynamics. As discussed in Chapter 2, these combs emerge when phase stabilization is applied to a periodic train of femtosecond mode-locked laser pulses, yielding control over both the repetition frequency and the optical carrier with respect to the pulse envelope. The broad spectral coverage of the resulting comb provides phase control of optical frequency markers across intervals of many hundreds of terahertz. Scientists can thus measure atoms and molecules by combining high-sensitivity, precise frequency control, broad spectral coverage, and high resolution in a single experimental platform.
Recent applications of cavity-enhanced direct frequency comb spectroscopy include sensitive and multiplexed trace-molecule detection for various species, as well as precise quantum control of atomic transitions via coherent pulse accumulations. More advanced spectroscopic and quantum control capabilities are being created by developing frequency comb sources in the deep ultraviolet and mid-infrared spectral regions. These distant spectral regions can in fact be coherently connected, thus in principle allowing the simultaneous study and control of vibrational and electronic molecular dynamics. The use of multiple frequency combs in pump-probe-type experiments has enabled increased resolution in both the temporal and spectral domain, and has for example yielded femtosecond time resolution of finely resolved optical properties in solids.
Real-time Chemical Kinetics
A key recent success of frequency comb spectroscopy is its application to precise determination of real-time kinetics of important chemical reactions. The study of chemical reactions requires a comprehensive characterization of the dynamics of reactants, intermediates, and products. Because of their short-lived nature, reaction intermediates present the highest challenges. A new experimental capability has emerged and was recently applied to an important chemical reaction, OH+CO, under ambient conditions. This reaction has served as a benchmark system for kinetics and dynamics studies of complex-forming bimolecular reactions for the past four decades because of its importance in atmospheric and combustion chemistry (see Figure 5.7). With multiple elementary chemical reactions that produce two
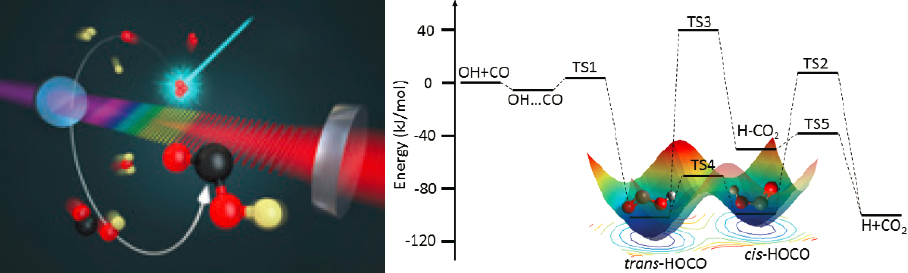
transient intermediates (trans-HOCO and cis-HOCO) and products (H+CO2) on a multidimensional potential energy surface, this reaction has been challenging to understand. Mid-infrared frequency comb spectroscopy allowed the first direct observation of trans-DOCO and cis-DOCO intermediates from OD+CO at thermal reaction conditions. (OD is a deuterated version of OH.) Together with measurements of D+CO2 products, the experiment has allowed the full determination of rate coefficients for isomerization and branching ratios for all channels of this important multistep reaction.
Studying the Complex Structure of Large Molecules
In Chapter 3, the exciting prospect of studying non-equilibrium dynamics of strongly interacting quantum systems is highlighted. Large and complex molecules present such an opportunity where a frequency-domain approach can reveal the complex structure that connects directly to the dynamics governing intramolecular energy flow between different degrees of freedom. This provides another important application opportunity for comb spectroscopy. Spectroscopic identification of larger room temperature molecules is nearly impossible because of spectral congestion. Cooling molecules to low temperatures drastically simplifies molecular spectra by enhancing the population of lower ro-vibrational states. The use of comb spectroscopy then opens the exploration of the complex energy-level structure of polyatomic molecules by providing a simultaneous map of the many relevant infrared transitions. In the case of C60, the first quantum-state resolved spectroscopy was demonstrated in 2019. The observed transitions between individual ro-vibrational states reveal fundamental details of the quantum mechanical structure of C60, including its remarkable icosahedral symmetry and nuclear-spin statistics. The possibilities for understanding and controlling complex quantum systems have thus been greatly advanced.
Studying Dynamics Through Collision Physics
Many of the challenges involved in advancing our understanding of collisional phenomena boil down to the theoretical treatment of correlations, which arise in a variety of different contexts. In quantum chemistry, “correlations” are sometimes narrowly defined as electron-electron correlations that go beyond a Hartree-Fock independent electron model. But in fact, correlations are broader than this, as electron motion is correlated with nuclear motion in processes such as dissociative attachment or Penning ionization. At the experimental level, probing correlations is challenging because this normally requires multiparticle detection in coincidence, which leads to low count rates. Increasingly complex reactive processes can now be studied experimentally, through the enhancements over the years of technologies such as COLTRIMS and the so-called reaction microscope.
Penning Ionization
A prominent example of a fundamental study involving correlation between electronic and nuclear motions in a small molecule, at the interface between chemical physics and AMO physics, is the challenging process of Penning ionization. This is a complex dynamical process in which an excited atom collides with a neutral atom or molecule, and leads to ejection of an electron, producing a positively charged atomic or molecular ion. Penning ionization has been observed and studied theoretically for decades, but until recently, with comparatively limited experimental evidence for the quantum mechanical nature of this process. In a remarkable experimental advance, it has now become possible to study this process in a low-temperature collision, below 1 degree Kelvin for excited helium atoms colliding with molecular hydrogen. In this regime, quantum mechanical resonances have been observed for the first time for such a complicated rearrangement collision.
Reactive Processes Involving Physics: Topological Physics
Examples of excellent progress in the past decade include far better understanding of reactive processes that involve topological physics, which include physics related to conical intersections. In ultracold AMO physics context, as in condensed-matter physics, the word “topological” is often used to refer to many-particle phases of matter, but even systems with a few electrons and atoms involved exhibit topological features in their quantum mechanical behavior, especially in the context of reactive collisions. For instance, Jahn-Teller and Renner-Teller systems in molecular physics inherently involve correlated electron and nuclear degrees of freedom, and in some cases, Rydberg state interactions as well. Now calculations of the dissociative attachment process, such as electrons colliding with an ammonia molecule (NH3) to produce an atomic hydrogen negative ion (H−), can be carried
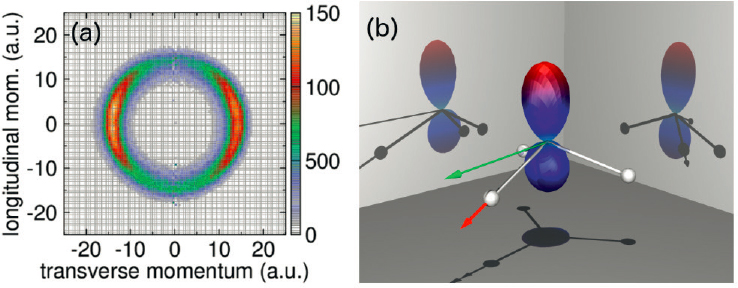
out and tested in great detail by comparison with experiment. This exemplifies many advances of both theory and experiment to be able to measure specialized properties such as the angle of escape of the H− ion by using COLTRIMS and related techniques (see Figure 5.8). (See also the Chapter 3 section on Topological Matter with Cold Atoms for the role of topology in ultracold science contexts.)
Broad Impact of Collision Dynamics Studies
While collision physics has tremendous intellectual challenges remaining—for example, to achieve a deeper understanding of reactions involving polyatomic molecules—there are additional strong practical motivations for advancing theory and experiment in this arena. The following examples showcase some of those subjects in need of better understanding of collision physics, especially reaction rates in a variety of environments:
- Impact on astrophysics: Extensive experimental and theoretical headway has emerged from studies of electron collisions with a variety of hydrogen-rich molecules that are very important in astrophysical environments—molecular ions like HeH+, H2+, H3+, CH3+, NH4+, to name just a few. There is now an opportunity to build on this progress by tackling systems that would have been unimaginably complicated to consider treating theoretically, just one or two decades ago.
One impressive development in experimental studies of low-energy small-molecule reactive collisions involving molecular ions, electrons, and
-
neutral atoms or molecules has been the emergence of cryogenic storage rings (CSRs). The poster child for this technology is the CSR that recently became operational in Heidelberg, which is capable of cooling molecular target ions down to around 10 degrees above absolute zero, and is thus able for the first time to accurately mimic conditions in many astrophysical environments such as interstellar clouds.
Likewise, ion-atom collisions leading to charge-exchange processes emitting X rays that are detected by space-based telescopes can yield information on both the projectile and the target. Highly energetic events such as in the solar wind or supernova explosions lead to highly charged ions propagating through space. When these ions encounter neutral atoms, they can readily capture one or more electrons from the atom. The preferential route is via velocity matching, and so the electrons are likely to be captured to an excited state, which then decays by X-ray emission. The observation of the resulting X rays tells us their source ion and also the target atom. Accordingly, X-ray space-based telescopes are observing the emission of such charge-exchange processes and yielding information on the ingredients of their origin. The charge-exchange process is rather challenging to calculate due to its two-center (ion and atom) nature. A quantitative description of charge-exchange processes is possible for relatively simple ions and atoms, but can also be exceeding complicated.
- Plasma modeling of fusion plasmas such as those in the International Thermonuclear Experimental Reactor (ITER): The goal of the large-scale ITER project is to create abundant energy via nuclear fusion with the fuel being derived from seawater. While the building stage is now in full swing, there are still many uncertainties in energy balances inside the fusion plasma. As diverters will be made from tungsten, such impurities in the plasma will lead to radiative losses. There will also be other partially ionized impurities in the plasma that will also lead to radiative losses. Quantum collision theory is necessary to make accurate estimates of all of the radiative losses and free electron production due to ionizing collisions.
- Ion scattering on soft tissue to determine the stopping power of ions in the body for the purpose of ion therapy treatment of cancer: In the past decade or so, a new cancer treatment modality has been developed known as hadron therapy. The idea is an extension of proton therapy, namely to bombard tumors with even heavier projectiles such as carbon ions, and utilize the Bragg peak to destroy the tumor without affecting the healthy cells surrounding it. Many children have had their brain cancers completely cured with hadron therapy without the typical severe side-effects associated with conventional X-ray radiation therapy. The key aspect is to accurately calculate the stopping power of heavy ions in soft tissue, with liquid water
- being a reasonable starting point. As the therapy is extremely expensive, the goal is to broaden its applicability to organs that may be moving due to the breathing of the patient during the therapy application. The determination of accurate stopping powers for ions is vital to ensure that the Bragg peak occurs at the tumor, and so away from healthy tissue.
- Positronium scattering on antiprotons to form antihydrogen: Further experimental progress toward understanding antimatter and questions about possible CPT violation, as is addressed in Chapter 6, requires greater numbers of antihydrogen atoms to be created. One technique for antihydrogen creation is guided by calculations of positronium (Ps) interactions with antiprotons. It has been shown that a rapid increase in antihydrogen production can be obtained via laser-excitation of Ps to utilize excited states, but the scaling is not as simple as classically expected. Generally, quantum collision theory will continue to provide guidance on the various mechanisms for producing antihydrogen.
The Future of Frequency Domain Studies of Dynamics and Correlations
Many challenges remain in the field of collision physics, to treat more complex open-shell atoms, ions, and molecules and their reactivity, with or without external fields present. These are challenging problems that require a community of theoretical and experimental experts and capabilities. These involve quantal, semiclassical, or in some cases classical treatments of atomic or molecular energy levels or potential-energy surfaces, and of collisional dynamics such as electron-atom or electron-molecule scattering, ion-atom collisions, and atomic or molecular photoionization. It is imperative that the AMO community continue to invest in these capabilities, which impact so many other different fields in addition to having fundamental interest and importance in their own right.
Also in the frequency domain, with the revolutionary impact on precision metrology and ultrafast science brought by the recent development of optical frequency combs in the visible and mid-infrared spectral regions, we can naturally expect to extend this coherent spectral coverage to the extreme ultraviolet (XUV) ends of the spectrum. Ultrashort, ultraintense laser pulses at a high repetition rate can be frequency converted from visible to the VUV and XUV regions through high harmonic generation inside a femtosecond enhancement cavity. When the optical coherence from pulse to pulse is maintained, a frequency comb in the XUV spectral region can be generated, providing exceptional phase coherence for high-resolution spectroscopy, precision measurement, and quantum control. This is one of the best examples highlighting the advantage of joint control in both the time and frequency domains for optical fields.
NOVEL PHYSICS WITH EXTREME LIGHT
As described in Chapter 2, the past decade has seen the appearance of a number of extreme light sources. XFEL facilities around the world have welcomed their first user experiments, including in the United States, Europe, and Asia. And Petawatt laser facilities continue to expand the limits for the highest intensity possible, with the European ELI project expected to reach intensities of 1023 W/cm2 in the near future. This chapter ends with a brief discussion of the exciting physics that can be done using such light sources, spanning dynamics in AMO and beyond.
Extreme Physics with XFEL Laser Light
XFEL sources provide coherent laser pulses of X-ray light with record-setting intensities and pulse durations. A number of XFEL experiments in this first decade have been performed on AMO systems, addressing fundamental questions and characterizing the capabilities of the light source. The sections above have described the capabilities enabled by the short duration of these pulses for dynamical studies. Other types of studies are enabled by the extreme intensity of these light sources.
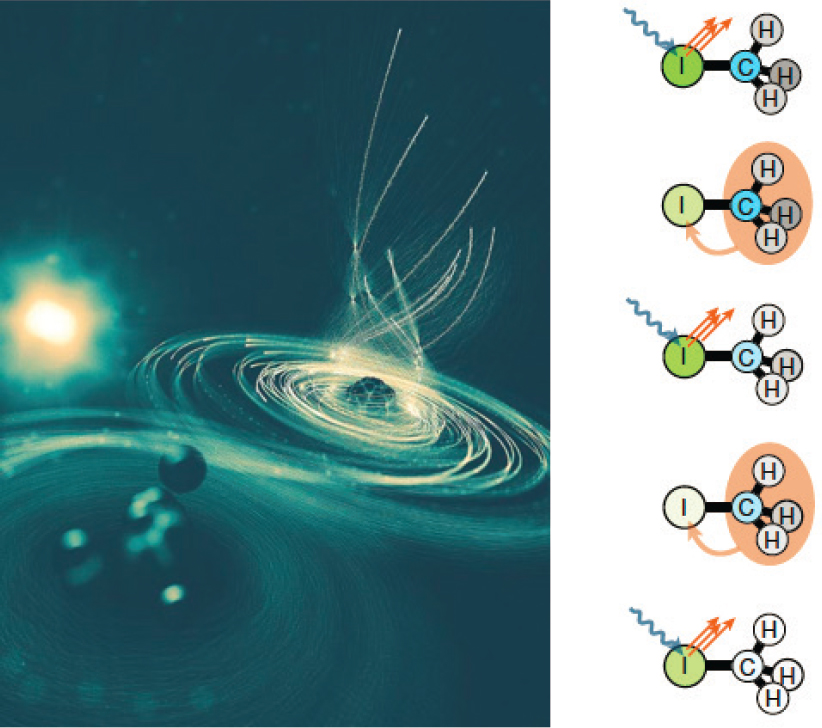
One example of such studies is extreme ionization. The number of X-ray photons in a ~100 fs pulse from the Linac Coherent Light Source (LCLS) is so large compared to previous third-generation light sources that it is possible to strip the majority of electrons from small atomic or molecular systems. Early LCLS experiments on such extreme ionization of rare gas atoms (leading to Ne10+, Ar12+, Xe36+) demonstrated that sequential, multi-step single-photon absorption by far dominates over direct multiphoton processes, even at the high intensities. In addition, the stripping away of electrons radically differed from intense optical pulses. In the latter case, valence electrons are removed sequentially, while intense X rays strip electrons from the inside out. In fact, the X-ray ionization is so rapid that it competes on the natural Auger time scale. More recently, researchers have shown that extreme ionization of small molecules is different from that of isolated atoms, and it can lead to the formation of a so-called molecular black hole (see Figure 5.9). This is because in a molecule, a charge imbalance created by X-ray ionization on one end of the molecule can be mitigated by electrons donated from the other end of the molecule. This charge transfer process is fast enough that it takes place even during the ultrashort (~100 fs) LCLS pulse, and means that more electrons can be removed from the molecule than from the group of individual atoms constituting the molecule. The interpretations of these experiments have been aided by large-scale calculations developed to describe the extremely fast ionization and rearrangement dynamics taking place during the X-ray pulse. These calculations combine quantum chemistry calculations of electronic structure with the coupled solutions of millions of equations describing the coupling between electronic states via transitions and decay.
The extreme intensity of these novel sources also enables nonlinear optics in the X-ray regime. It is notoriously difficult to induce nonlinear optical processes in the X-ray spectral region, and only recently have low-order nonlinear optical processes such as second harmonic generation been observed experimentally. This is because nonlinear optical processes rely on a nonlinear response in some material, which is much weaker in the X-ray region than in the visible region. This means that the high X-ray intensities provided by XFELs provide an opportunity for studying X-ray nonlinear optics. Recent experiments have demonstrated two-photon atomic ionization of core electrons and nonlinear Compton scattering of X-ray photons in a solid target. Surprisingly, in the latter case, the researchers found that they could not explain the final photon energy of the scattered photon with the standard, semiclassical descriptions of Compton scattering. This finding illustrates how little is known about how matter interacts with intense X-ray fields in particular, and with extreme light fields more generally, and points toward the need for future studies.
Reaching Beyond AMO Science Using Petawatt Laser Sources
The recent report on opportunities in intense ultrafast laser science1 details the current and future scientific opportunities in this field, so here the committee gives only a brief overview. The applications of super-intense laser light have a broad reach with the potential to impact astrophysics, nuclear physics, materials science, and medical applications among others. For example, intense-field generated high-density plasmas containing relativistic particles, and their interaction with the electromagnetic field, are of interest in astrophysics for laboratory-scale investigations of the physics of exotic objects such as gamma-ray bursts, giant planets, and pulsar winds. Likewise, secondary sources of high-energy photons, protons, electrons, or neutrons that can be generated at very high laser intensity can be of interest to both nuclear physics and provide table-top rather than accelerator-size environments for experiments. These secondary sources have also shown a lot of promise for medical applications in both imaging and radiotherapy treatment, and for national security.
Future of Applications of Extreme Light Sources
The field of short-wavelength nonlinear optics is only just opening up. With next-generation HHG sources of intense XUV light, and current-and-next generation XFEL sources of intense soft and hard X-ray light, we will be learning much more about the nonlinear response of atoms, molecules, and solids to intense short-wavelength radiation. The semiclassical model for the interaction of matter
___________________
1 National Academies of Sciences, Engineering, and Medicine, 2018, Opportunities in Intense Ultrafast Lasers: Reaching for the Brightest Light, The National Academies Press, Washington, DC.
with intense infrared laser light has been hugely influential in guiding experimental and conceptual progress in ultrafast and strong-field AMO science. It is an open question as to where the strong-field limit lies in the X-ray regime and in what way it will shape the development of new experiments and concepts. While it is unlikely that one-electron tunnel ionization would ever be dominant, a strong-field response in the X-ray regime could for instance involve a collective, many-electron response that would necessitate thinking beyond single-active-electron physics.
Another extreme-light setting for a “semiclassical” or recollision model is that of ultrarelativistic electrons—for example, created by ultra-intense, next-generation PW lasers—and their parent ions. The so-called Schwinger limit (above 1029 W/cm2) is when the vacuum becomes unstable to electron-positron pair production, and the “classical” trajectories of charged particles will be completely disrupted even by single-photon emission. In analogy with strong-field atomic physics, coherent recollisions will become important as the electron kinetic energy greatly exceeds its rest mass. As discussed in Chapter 2, the Schwinger limit is not reachable with present-day lasers, but may be within reach in the next decade, potentially via collision of focused petawatt laser beams with ultrarelativistic electrons.
FINDINGS AND RECOMMENDATIONS
In this chapter, the committee discussed the study of dynamics spanning the attosecond to the microsecond time scale, and how to access it in both the time domain and the frequency domain. The committee has discussed current and future progress in the making of time-domain molecular movies, spanning attosecond, coherent electron dynamics, through femtosecond molecular dynamics and beyond. It has been shown how improved resolution in the time domain, provided by modern ultrafast light and electron sources, has enabled fundamental investigations of time delays in photoionization, charge migration in molecules, and dynamics near conical intersections that are foundational not just to AMO physics, but also to materials science, chemistry, and photobiology. The unprecedented intensity available from XFELs has finally put time-resolved imaging of individual molecules within reach and has opened new fields of multiphoton and nonlinear X-ray physics, where the behavior of matter under extreme conditions can be explored. The committee also discussed how improved sensitivity in the frequency domain, both that provided by modern frequency-comb light sources, amurind by advances in collision physics, allows complementary studies of dynamical processes.
Finding: This is a unique time in ultrafast science due to the ubiquity and controllability of ultrafast light sources spanning the terahertz through the hard X-ray regime. The development and application of these sources have driven much of the progress described in this chapter.
Finding: Control of ultrafast electron dynamics in molecular and condensed-phase systems has significant potential for impact well beyond AMO science, including at the technological and industrial levels. Likewise, continued development of molecular movies will drive advances at the fundamental level, and promises societal benefits through improved understanding of photo-driven biological processes.
Finding: Continued progress on these challenges will require the combined expertise of multiple principal investigators (PIs) and mid-scale infrastructure, either because they involve advanced facilities with many different elements, or because they are inherently multidisciplinary in nature, covering AMO, condensed-matter physics, chemistry, laser technology, and large-scale computation. Funding mechanisms similar to the Physics Frontiers Centers or Multidisciplinary University Research Initiatives, in which multiple PIs from experiment and theory work toward a common goal, are crucial.
Key Recommendation: U.S. federal agencies should invest in a broad range of science that takes advantage of ultrafast X-ray light source facilities, while maintaining a strong single principal investigator funding model. This includes the establishment of open user facilities in mid-scale university-hosted settings.
Finding: There has been widespread use of data typically gathered in collision physics, which are needed for applications and analysis in astrophysics, plasma physics, and nuclear medicine among others, as well as a decline in university-supported collision physics groups.
Recommendation: National laboratories and NASA should secure the continuation of collision physics and spectroscopy expertise in their research portfolios.



































