
Summary1
There are approximately 100,000 people living with sickle cell disease (SCD) in the United States and millions more globally. Sickle cell trait (SCT) is even more prevalent and occurs in 1–3 million Americans and 8–10 percent of African Americans in the United States. Current estimates indicate that about 300,000 people are born with SCD each year worldwide and that more than 100 million people across the globe live with SCT. The sickle gene is found in every ethnic group, not just among those of African descent.
Since its discovery in 1910 by James Herrick, SCD has received relatively little attention and few resources from the scientific, clinical, and public health communities compared with other genetic disorders, such as cystic fibrosis (CF). Until December 2018 there was only one drug approved by the U.S. Food and Drug Administration (FDA) for the condition. A contributing factor to this lack of awareness and resources is that the affected population, which is primarily composed of racial and ethnic minorities, contends with persistent discrimination in the health care system and racism in society at large. As described by Keith Wailoo, a medical historian who has extensively studied the history of SCD, “Sickle cell disease is a microcosm of how issues of race, ethnicity, and identity come into conflict with issues of health care.” Thus, individuals with SCD have suffered from a lag in the development of treatments and cures as well as an often strained relationship with health care providers and limited resources for advocacy efforts.
___________________
1 Citations and references for all facts and figures mentioned in the Summary are included in the subsequent chapters of this report.
To accelerate progress for those living with SCD, the Office of Minority Health at the Office of the Assistant Secretary for Health (OASH) at the U.S. Department of Health and Human Services (HHS) asked the Health and Medicine Division of the National Academies of Sciences, Engineering, and Medicine (the National Academies) to develop a strategic plan and blueprint to address SCD in the United States. (The full charge to the committee [Statement of Task] is provided in Box S-1.) This report is the answer to that request.
SCD and SCT status are currently identified at birth through universal newborn screening (NBS) in all 50 states, the District of Columbia, and U.S. territories. NBS has been highly successful at ensuring early access to much needed care, such as prophylactic penicillin for young children to avoid sepsis, which has saved countless children’s lives. Despite the effectiveness of NBS, there are wide variations in states’ short- and long-term follow-up practices regarding screening results (see Chapter 3). Most states also track individuals only if they remain within the same state, thus missing those who move out of the state. Additionally, while NBS identifies newborns who have SCT, there are currently no standardized practices for short- and long-term follow-up for carriers. This has important implications because of emerging evidence that SCT status might be a risk factor for certain clinical complications and because it is important for reproductive decision making. NBS also misses a large proportion of the SCD and SCT population who was born outside of the United States or before universal NBS was implemented in the country.
The genetic mutation responsible for SCD causes an individual’s red blood cells to distort into a C or sickle shape, reducing their ability to transport oxygen throughout the body. These sickled red blood cells break down rapidly, become very sticky, and develop a propensity to clump together, which causes them to become stuck and cause damage within blood vessels. The result is reduced blood flow to distal organs, which leads to physical symptoms of incapacitating pain, tissue and organ damage, and early death.
Pain is the hallmark of SCD, and individuals with SCD experience both acute and chronic pain. SCD pain is complex because it can be influenced by the disease pathophysiology as well as by psychological and social factors. The disease can also affect every organ in the body, as discussed in Chapter 4. While death rarely occurs among children with SCD in the United States, with 98 percent surviving to 18 years of age, SCD has a persistently high mortality rate in adults, and end-organ damage is the major driver of this mortality. Fatigue and emotional distress, such as anxiety and depression, become more prevalent with age and, along with chronic pain, pose a high burden and cause significant disability, which is under-recognized. In addition, childhood mortality for SCD remains very high in resource-poor countries.
Care delivery for SCD is inadequate. This stems in part from the fact that for most of the 20th century SCD was considered primarily a childhood disease because most affected individuals did not survive into adulthood. High childhood mortality rates for SCD led to an increased emphasis on improving the infrastructure for pediatric care in the United States and conducting research to prevent early death from infections.
The emphasis on early interventions for SCD led to seminal studies that provided the evidence base for clinical guidelines for the prevention
and management of pediatric complications, such as the implementation of guidelines for prophylaxis against pneumococcal sepsis and stroke. Unfortunately, over the years there has not been a parallel development of guidelines and infrastructure for care delivery to adults living with SCD. Persistent gaps in the understanding of the natural history of SCD, predictors and biomarkers of morbidity and mortality, and the pattern of emergence of organ damage and other sources of disability persist, thus limiting optimal care delivery, particularly for adults (see Chapter 4).
Individuals who are transitioning from pediatric to adult SCD care are at a particularly high risk for morbidity and mortality because the robust care delivery systems available to pediatric patients are not replaced by matching or adequate resources for the adult patients (see Chapters 5 and 6). The lack of dedicated facilities and personnel caring for adult patients is compounded by the rising complexity of the disease, the emergence of comorbidities, and the vulnerability of individuals through the challenging period of adolescence and young adulthood. Young adults with SCD report being unprepared to engage optimally with and navigate the adult health care system independently after years of support in the pediatric care system. Areas of particular unmet need center on the domains of independence, self-care, vocation, and insurance coverage. When these factors are coupled with the aforementioned stigma, racism, and discrimination within the health care setting and society more broadly, individuals living with SCD often find themselves facing a solitary battle. This battle includes having to advocate for themselves in the health care and public sectors, at work, in schools, and sometimes even in their families.
Finally, even when interventions are well established, the delivery of appropriate and comprehensive care is uneven (see Chapter 6). Some of this may reflect payment mechanisms that do not support coordinated care. The majority of those with SCD are publicly insured, and covered services may vary across states. Furthermore, health care providers may not be well versed in accessing appropriate and available enabling services,2 or such enabling services may not exist. There is a lack of awareness among providers, especially those who do not regularly encounter patients with SCD, of the available clinical practice guidelines for evidence-based SCD care, which leads to inconsistent or substandard care. Members of a health care team may also be unaware of the implicit biases that influence their interactions with and medical decision making for those individuals living with SCD (see Chapters 2 and 6).
___________________
2 Enabling services are defined as patient services that are intended to improve access to health care and create better health outcomes. Some examples include health education, case management, and transportation.
Improving the organization of care delivery can help ensure that individuals have access to appropriate services and can also enhance the quality of the care that is delivered, in turn improving the overall quality of life (QOL) for individuals living with SCD while increasing their access to new therapies. Two therapeutic products have been recently approved by FDA (voxelotor and crizanlizumab), and researchers are actively pursuing improved curative options by broadening access to stem cell transplant and developing gene therapy protocols (see Chapter 7). In addition to increasing access to new interventions, service delivery should be restructured by creating multidisciplinary care teams that can support delivery of whole-person care necessary to improve physical and social functioning and QOL for individuals with SCD.
The restructuring of care delivery would also provide a platform for patient advocacy groups and community-based organizations (CBOs) that have played an integral role in driving policy and much needed programming for the SCD population. These groups provide education about SCD and SCT, genetic support, psychosocial support, camps, care coordination, case referral, and transition assistance, among other services. However, it should be noted that patient advocacy groups and CBOs are under-resourced and are thereby limited in their ability to serve their constituents. Furthermore, there are no guidelines or unified infrastructure for the operation of these organizations and advocacy groups. Despite their key role for the SCD population, they also have traditionally not been recognized as part of the SCD care delivery system (see Chapter 8).
One challenge that has stifled progress in SCD is the lack of funding. To successfully garner attention and resources for those affected by SCD and SCT, there is a need for public education and awareness about the disease and the burden it places on individuals and the health care system. The contribution of racism, discrimination, mistrust of the health care system, socioeconomic disadvantage, and inadequate services across many sectors experienced by ethnic minorities in the United States cannot be overemphasized. However, the problems of access to high-quality health and social services are not exclusive to SCD, and the strategies developed to address them in other conditions can serve as a guide. CF and hemophilia, for instance, which are both rare and inheritable diseases, have well-organized and well-funded health care delivery systems despite there being smaller numbers of affected individuals (approximately 30,000 and 20,000, respectively, in the United States). Amyotrophic lateral sclerosis attracted widespread public attention and efforts fueled by social media, which resulted in dramatically increased funding. Other rare diseases have also benefited from an infusion of federal and private funding. These diseases set valuable precedents for addressing the needs of the SCD population.
SCOPE OF WORK
Charge to the Committee
The Office of Minority Health at OASH at HHS commissioned the National Academies’ Health and Medicine Division to develop a strategic plan and blueprint to address SCD in the United States. A committee was formed to direct the study titled Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. The charge to the committee (Statement of Task) is shown in Box S-1.
The Committee’s Approach
To accomplish its task, the committee focused the report to address the most salient issues surrounding SCD and SCT in the United States. Thus, most of the literature reviewed for the report originates from the United States. However, because the majority of the SCD population resides outside of the United States, seminal evidence and developments from other countries were included in the report where necessary. The bulk of the report focuses on the needs of the SCD population because this is the area of greatest need.
Conceptual Framework
Life-span approach
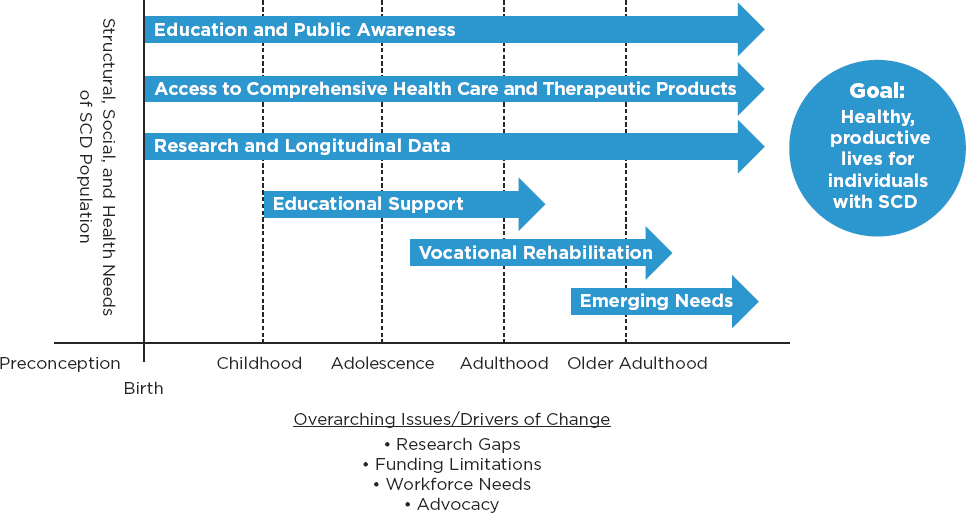
In its assessment, the committee considered the needs and specific challenges at different stages in life. While most SCD complications are common to both children and adults, specific ones may be more prevalent at different ages. For example, dactylitis (painful swelling of the hands and feet), stroke, and enlarged spleen are all common in children, whereas retinopathy, pulmonary hypertension, heart failure, chronic leg ulcers, and cognitive burdens are more prevalent in the adult population.
The non-health-related needs of younger and older individuals with SCD may also vary. For instance, children who experience overt or silent strokes as a result of SCD develop cognitive impairment that may warrant additional educational support in order to increase the chances for academic success. With age, these cognitive challenges coupled with the additional burden of chronic organ damage may necessitate vocational rehabilitation support, specific workplace accommodations, or support to facilitate changes in vocation. Finally, the life-span approach is appropriate because existing resources for children and adults with SCD vary. High-quality care is currently better established for children, although there is room for improvement; quality indicators for adults with SCD are significantly underdeveloped and clinical guidelines for high-quality care lack a strong evidence base. As individuals with SCD survive into adulthood, information is needed about the appropriate service needs, and this need may require longitudinal
NOTE: SCD = sickle cell disease.
data systems that will inform evolving care and service needs over time. The committee conceptualized the life-span approach in Figure S-1 in order to represent the need for targeted interventions at different life stages.
It is also critical to include a focus on QOL factors and on understanding how QOL may change over a lifetime; this is a common approach in the study of other chronic conditions, such as cancer and cardiovascular diseases.
Person-centric SCD care
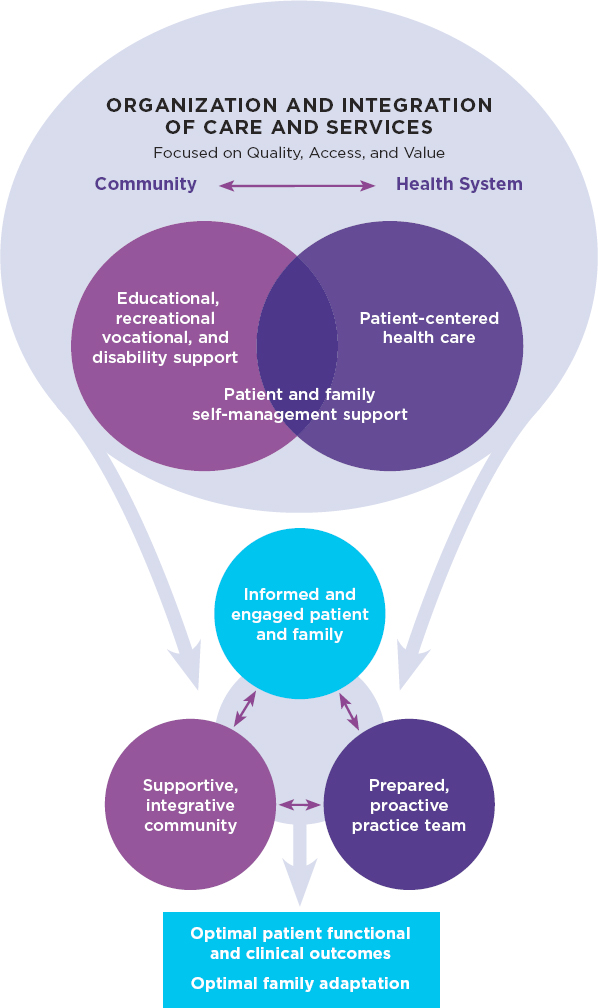
SCD is a genetic lifelong condition; as the survival rate continues to improve for children, it must be managed as a chronic disease, which requires an ongoing person-centric, collaborative approach to care management. Building on previous National Academies work on epilepsy, another debilitating medical condition diagnosed in childhood, the SCD committee based its recommendations at least in part on the epilepsy model of care (see Figure S-2), which is built on Wagner’s Chronic Care Model. Wagner’s model emphasizes the foundational partnership between the care team and an activated and empowered patient as being crucial for care delivery. The model also underscores the importance of family members and community service providers in the care delivery system. This is especially relevant for the SCD population, which relies heavily on services provided by advocacy groups and CBOs.
The main focus for any model of care should be the individuals with SCD and their families, rather than the health care system. Bearing in mind
NOTE: SCD = sickle cell disease.
SOURCES: IOM, 2012; originally adapted from Wagner, 1998. Republished with permission of American College of Physicians - Journals from Chronic Disease Management: What Will It Take to Improve Care for Chronic Illness?, E. H. Wagner, volume 1, 1998; permission conveyed through Copyright Clearance Center, Inc.
the individual’s preferences, needs, and values, systemic efforts are required to facilitate access to comprehensive care and empower individuals to self-manage and remove barriers to treatment. Health care and social services are designed to benefit patients and improve their health outcomes, QOL, and ability to be productive. The engaged, supported, and empowered patients are then able to work collaboratively with the care delivery team and community resources to effectively manage their care.
There is also a need to establish acceptable minimum standards for the delivery of the complex care that individuals living with SCD require across the life span. Several centers of excellence in SCD have made efforts to establish learning collaboratives that define these care delivery standards, leveraging quality improvement measures to ensure that every patient has access to standardized care at all times, with ongoing data monitoring to track the processes’ effectiveness. For example, the SCD Emergency Department Learning Collaborative recently supported quality improvement interventions across three sites to improve time to first analgesia for acute SCD pain, and the Hemoglobinopathy Learning Collaborative has focused on strategies that result in more coordinated and appropriate care in order to achieve fewer complications, acute care visits, and hospitalizations; enhanced QOL; and more compassionate and respectful treatment from the health care system. Expanding the reach of such collaboratives will help spread nationally agreed-upon standards of care to other sites, with ongoing efforts to improve the consistency and ultimately the quality of care for individuals living with SCD. (See Chapter 6 for more information.)
RECOMMENDATIONS
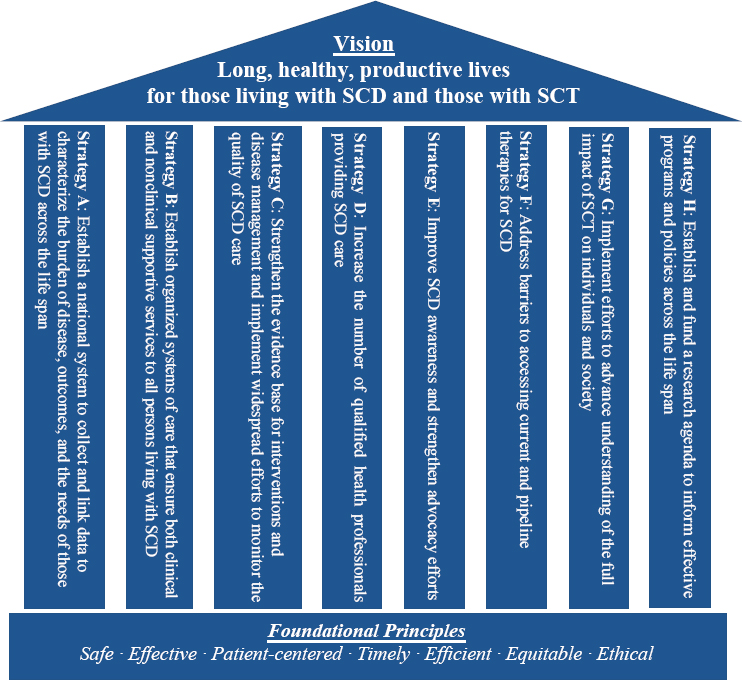
Against the contextual backdrop described in the preceding sections, the committee developed a strategic plan and blueprint for SCD action and identified strategies and specific actions (or recommendations) for improving care and outcomes. The vision for the strategic plan is to ensure “long, healthy, productive lives for those living with SCD and those with SCT.” The committee found that the core message of the Institute of Medicine report Crossing the Quality Chasm: A New Health System for the 21st Century still holds true today for the SCD population, which has not benefited from medical science advances as much as the general population or even at the same rate as those living with other rare and heritable diseases, such as CF and hemophilia. There is insufficient up-to-date information about the SCD population to appropriately inform programming and policies that address the population’s specific needs. Finally, evidence-based interventions (preventative, acute, and post-acute services) that apply to the general population are not always available to individuals living with SCD.
The committee determined that, at minimum, the strategic plan and blueprint should ensure that the SCD population receives the same high-quality care that every American is entitled to. The committee based the foundational principles for action on the six aims for the health care system identified in the Crossing the Quality Chasm report, namely that health care be safe, effective, patient-centered, timely, efficient, and equitable. According to the authors of the report, “a health care system that achieves major gains in these six areas would be far better at meeting patient needs.” The committee felt that, due to the history of marginalization and racism experienced by the majority of the affected population, it was important to add a seventh principle: that health care be ethical.
The strategic plan (see Figure S-3) is made up of a strategic vision, eight overarching strategies or “pillars” that support the vision, and foundational
NOTE: SCD = sickle cell disease; SCT = sickle cell trait.
principles, which undergird the strategic plan. The strategies take into account the multifaceted needs of the SCD and SCT population and the equally multidimensional interventions required to meet these needs. The strategies are equally important and need to be approached with the same amount of urgency.
The committee has also proposed a blueprint for implementing the strategic plan. The blueprint offers recommended actions for each of the strategies in the strategic plan. These action steps are the recommendations identified in the report’s various chapters after a thoughtful review of the available evidence. The action steps or recommendations are enumerated with the chapter that contains the supporting evidence and listed in order of implementation timeframe. The committee offers timeframes for accomplishing each of the recommendations. The timeframes take into account the complexity of the activity, the level of resources needed to accomplish the task, and the existence of current programs that can serve as vehicles for advancing action. Activities are also prioritized by actions that need to occur sequentially.
In order to make meaningful and sustained progress on the strategic plan, OASH at HHS should appoint an oversight body with members from across HHS agencies to oversee the roll-out of the strategic plan and blueprint. The appointment of the oversight body should be immediate, and the current HHS Sickle Cell Disease Workgroup, which has representation from 11 HHS agencies, would be one option for such an interagency group.
Finally, to ensure continued progress, the oversight body should conduct regular assessments of the implementation of the strategic plan, with the first evaluation occurring no more than 5 years after the release of this report.
Chapter 9 includes a complete description of the components of the strategic plan and blueprint.
___________________
3 This text was revised since the prepublication release of this report to correct American Association of Family Practitioners to American Academy of Family Physicians.
___________________
4 This text was revised since the prepublication of the report to include the timeline for implementation of this strategy. The prepublication version of the report listed the timeline as “Ongoing.”


















