
1
Introduction
Every single day that you get up out of bed, you’re fighting a battle, and when you take that first breath … it’s a breath of pain. You assess mentally. Okay, where are all the places that are hurting right now … and you actually have to take several deep breaths to push the circulation through your body.
—Tosin O. (Open Session Panelist)
Sickle cell disease (SCD) refers to a group of inherited red blood cell (RBC) disorders resulting from a mutation in hemoglobin, which impedes regular blood flow and leads to painful vaso-occlusive episodes and other severe complications (CDC, 2017b). Present at birth, SCD causes lifelong acute and chronic complications throughout the whole body. This debilitating, multi-system condition affects approximately 100,000 people in the United States and millions globally1 (ASH, 2016; Mulumba and Wilson, 2015). Childhood mortality due to SCD has declined in the United States due to medical advances and preventive services, but despite this progress life expectancy and quality of life for people living with SCD are lower than for those without the disease (Lubeck et al., 2019; Piel et al., 2017), necessitating action to improve health and outcomes and reduce this disparity.
___________________
1 This text has changed since the prepublication release of this report to more accurately reflect the estimates of prevalence of sickle cell disease identified in the literature.
SCOPE OF WORK
Charge to the Committee
The Office of Minority Health at the Office of the Assistant Secretary for Health at the U.S. Department of Health and Human Services (HHS) requested that the National Academies of Sciences, Engineering, and Medicine (the National Academies) convene a committee to develop a strategic plan and blueprint to address SCD in the United States. The Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action was established in response to this request. As part of its work, the committee was asked to develop a framework that provides guidance on the best approaches to addressing pertinent issues in SCD such as health care disparities, stigma, race and biases, access to care, workforce development, transitions in care, innovations needed, curative treatments, and the role of patient advocacy and community engagement. Box 1-1 shows the committee’s Statement of Task. In addressing the Statement of Task, the committee defined its scope as addressing challenges of SCD and sickle cell trait (SCT)2 in the United States and creating an action plan for prolonging healthy lives through the delivery of high-quality and equitable care to individuals with SCD. The committee found it necessary to acknowledge the global burden of the disease in low- and middle-income countries in order to display the full context of its impact; this was especially important for areas where research and findings from other countries were essential to fill in knowledge gaps in the United States. The committee also addressed ethical concerns pertaining to areas such as screening, treatment, and research.
STUDY PROCESS AND INFORMATION GATHERING
This section presents the process that the committee used to identify and evaluate the scientific literature related to SCD and the Statement of Task, committee areas of expertise, how the literature search was conducted, and the evaluation criteria used to screen and categorize literature for the chapters.
Committee Expertise and Meetings
The National Academies appointed a team of 14 multidisciplinary experts to the Committee on Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; their expertise spanned epidemiology,
___________________
2 SCT is not a disease but refers to when an individual inherits one sickle gene from one parent and a normal gene from another parent. SCT carriers live normal lives and do not experience symptoms associated with SCD.
hemoglobinopathies, pediatrics, hematology, oncology, emergency medicine, psychology, care management and delivery, pain management, health disparities, health economics, health policy, ethics, treatment of diseases associated with SCD, research, and workforce development.
The committee convened five times; it held public information-gathering sessions at each of those meetings and invited panelists to present on specific topics of interest to the committee. Full descriptions of the open session panels are included in Appendix A. The first meeting included the
presentation of the charge to the committee by the Assistant Secretary for Health and Office of Minority Health. Between the in-person meetings, committee members held deliberative sessions to review the literature, discuss the evidence base, and write the report.
Literature Search
The committee conducted a comprehensive literature search through the National Academies Research Center. The search encompassed a wide array of terms related to SCD and its complications, as detailed in Appendix B. The search was restricted to 1990–2019 except for the search terms: bias, stigma, discrimination, and racism, which had no date bounds to capture the experiences of minority populations in the health care system. The committee performed additional targeted searches, as needed, to supplement the landscape literature review and expanded their search to related patient populations when information on certain topics could not be found for SCD. Because there have been relatively few large, rigorous studies conducted on SCD and SCT, the committee decided not to employ specific criteria for including/excluding literature that was evaluated as part of this report, thus allowing for smaller, observational, and qualitative studies to be included in the committee’s analysis. The committee also examined abstracts of conference presentations to allow for inclusion of emerging research on SCD and SCT.
Conceptual Approaches for Information Gathering
Life-Span Approach
SCD causes various challenges at different stages in life, so the committee conceptualized the tasks for this report by assessing the needs of people living with SCD over the life span. For example, dactylitis occurs mainly in children, whereas heart failure and chronic leg ulcers typically affect adults. Age compounds the complications and impact of SCD on the affected individual’s body (Oyedeji et al., 2019; Quinn et al., 2010; Sandhu and Cohen, 2015; Swanson et al., 2011). Children may also experience strokes, which can negatively affect cognitive function, impede their performance in school, and explain long-term cognitive function limitations in adulthood, which can manifest as poor pain coping or non-adherence to prescribed treatments (Crosby et al., 2015; Gold et al., 2008; Greenham et al., 2016; Swanson et al., 2011). These long-term cognitive limitations could be a contributing factor to the high rate of unemployment in the SCD population (Swanson et al., 2011). Taking a life-span approach provides the
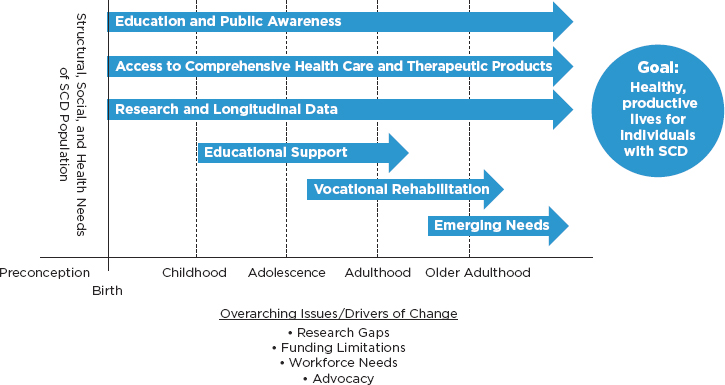
NOTE: SCD = sickle cell disease.
opportunity to identify specific areas for continued research and policy to improve care for individuals as they transition from pediatric to adult care and into old age. Figure 1-1 provides a graphical representation of some of the categories of interventions that the committee identified as crucial at different stages of life.
Person-Centric SCD Care Approach
Considering that more people with SCD are living into adulthood, it is important to understand how to manage this chronic disease across the life span, with collaborative input from physicians, those living with SCD and their families, and relevant community stakeholders. The committee decided that effective management of SCD needs to happen in the context of a team-based comprehensive care model that places the patient and his or her needs at the center. Consequently, the committee referred to a previous National Academies report titled Epilepsy Across the Spectrum (IOM, 2012) that provides a detailed description of a model used to provide care for epilepsy, which in turn was based on the so-called Chronic Care Model (see Figure 1-2). Developed by Edward Wagner and his colleagues, this model emphasizes the need for transformation in the health care delivery system to proactively keep individuals with chronic diseases healthy. Both
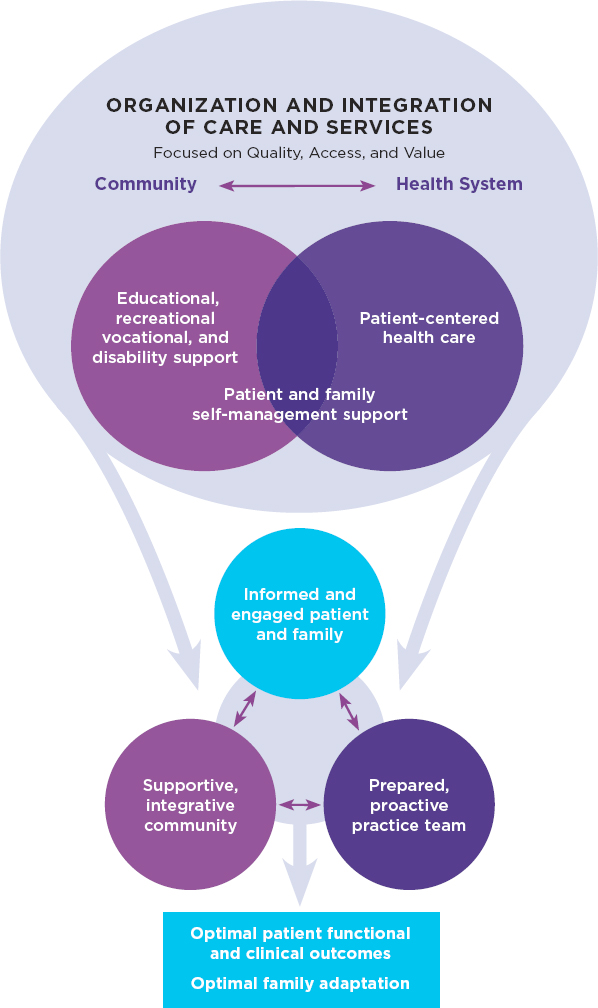
NOTE: SCD = sickle cell disease.
SOURCES: IOM, 2012; originally adapted from Wagner, 1998. Republished with permission of American College of Physicians - Journals from Chronic Disease Management: What Will It Take to Improve Care for Chronic Illness?, E. H. Wagner, volume 1, 1998; permission conveyed through Copyright Clearance Center, Inc.
the health system and the community have key roles to play in equipping individuals with chronic conditions such as SCD to self-manage their conditions in pursuit of optimal outcomes (IOM, 2012; Wagner, 1998; Wagner et al., 2001, 2005). Similar care models for a variety of chronic diseases have been implemented, such as for cystic fibrosis (CF), which is discussed in Chapter 5, while others are being piloted by the Centers for Medicare & Medicaid Services and can be adapted for the SCD population.
OVERVIEW OF SCD AND SCT
SCD is a group of genetic blood disorders. A point mutation in the gene that codes for beta globin, one of the two types of amino acid chains that compose the adult hemoglobin, leads to an abnormal hemoglobin named hemoglobin S (HbS). HbS polymerizes under low oxygen states, forming elongated structures within the RBCs that deform their shape from a biconcave, donut-shaped disc to a sickle or crescent shape, from which the name of the disease derives (Booth et al., 2010; CDC, 2017b; Malowany and Butany, 2012; Mentzer and Wang, 1980; NHLBI, n.d.; Telen et al., 2019). James Herrick first discovered and named SCD in Chicago in 1910; he described the sickle RBCs he was examining as “peculiar, elongated and sickle-shaped red blood corpuscles” (ASH, 2008; Herrick, 1910, p. 517). In 1949 Linus Pauling postulated that SCD was caused by the presence of an abnormal hemoglobin molecule (ASH, 2008), representing the first identification of a molecular disease (Eaton, 2003).
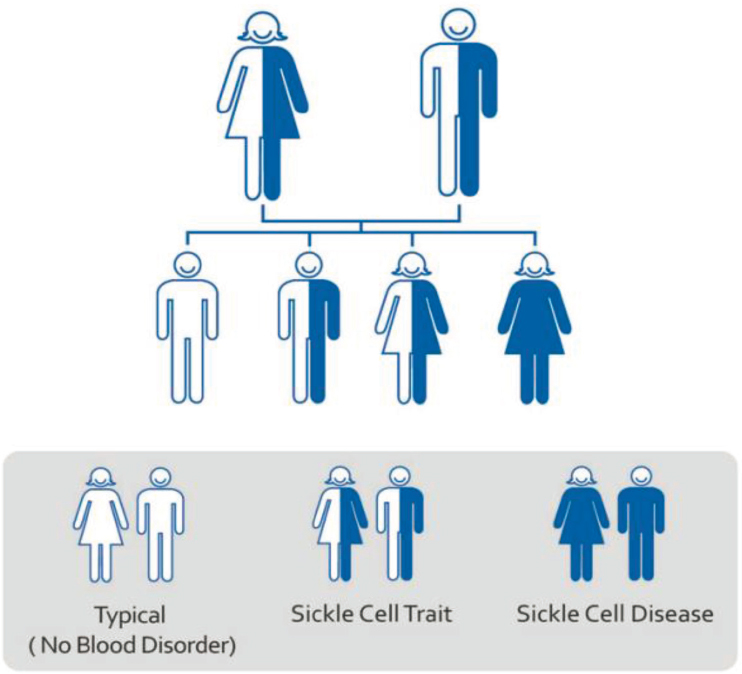
The inheritance of a single copy of the mutation leads to SCT (heterozygous carrier), a mostly benign condition, while the inheritance of two copies of the mutation (HbSS) or one copy of the HbS mutation in combination with a copy of certain other hemoglobin mutations leads to SCD (see Figure 1-3). Thus, SCD is an autosomal recessive disease, because only the individual that inherits two mutated beta globin chains is affected. Table 1-1 describes SCT and the common SCD genotypes and provides the appropriate nomenclature.
SCD collectively denotes a set of syndromes that, if untreated, can be highly morbid and deadly (Dampier, 2019; Habara and Steinberg, 2016; NIH, 2019; Quinn, 2016; Williams and Weatherall, 2012).
The homozygous inheritance of HbS, also known as sickle cell anemia (SCA), is the most prevalent and severe form of the disease and also the most researched (Dampier, 2019; Habara and Steinberg, 2016). SCD also occurs as compound heterozygotes for HbS and other hemoglobin variants, including HbC, HbE, HbD, and HbO/Arab or beta thalassemia mutations (Habara and Steinberg, 2016; Serjeant, 2013; Williams and Weatherall, 2012). Hemoglobin Sb0 –thalassemia (HbSb0) is clinically similar to HbSS and sometimes jointly referred to as SCA (NHLBI, 2014). HbSC has
SOURCE: CDC, 2017b.
moderate clinical severity, and HbSb+ is generally a milder genotype, although all individuals with SCD, regardless of the genotype, are at risk of severe complications (NIH, 2019; Quinn, 2016).
If both parents have SCT, each child will have a 50 percent chance of inheriting SCT and a 25 percent chance of inheriting SCD, while there is a 25 percent chance that the child will inherit neither SCT nor SCD and have non-mutated hemoglobin.
The origins of the sickle gene have been traced to sub-Saharan Africa (with earlier theories hypothesizing an independent, additional origin of the gene elsewhere). HbS confers partial protection from Plasmodium falciparum malaria, a major infectious killer in the tropics; hence, the mutation provides a survival advantage to individuals with SCT and has been conserved throughout evolution (Luzzatto, 2012; Williams and Weatherall, 2012). The trans-Atlantic slave trade and, later, global migration patterns
TABLE 1-1 Common SCD Genotypes, Nomenclature, and Mutational Products
| Genotype | Common Diagnostic Term | Types of Beta Globin Gene Mutation Product |
|---|---|---|
| Homozygous SS | Hemoglobin SS disease; sickle cell anemia | Two hemoglobin S genes (HbSS) |
| Compound Heterozygous SC | Hemoglobin SC disease | One hemoglobin S gene and one hemoglobin C gene (HbSC) |
| Compound Heterozygous SD | Hemoglobin SD disease | One hemoglobin S gene and one hemoglobin D gene (HbSD) |
| Compound Heterozygous SE | Hemoglobin SE disease | One hemoglobin S gene and one hemoglobin E gene (HbSE) |
| Compound Heterozygous SO | Hemoglobin SO disease | One hemoglobin S gene and one hemoglobin O gene (HbSO) |
| Compound Heterozygous S Beta Thalassemia Zero | S/B0 thalassemia Hemoglobin S/B0 thalassemia | One hemoglobin S gene and one hemoglobin beta thalassemia zero gene (HbSb0-thalassemia) |
| Compound Heterozygous S Beta Thalassemia Plus | S/B plus thalassemia Hemoglobin S/B+ thalassemia | One hemoglobin S gene and one hemoglobin beta thalassemia plus gene (HbSb+-thalassemia) |
| Sickle Cell Trait (Not a form of sickle cell disease) | Sickle Trait Hemoglobin AS | One hemoglobin A gene and one hemoglobin S gene (HbAS) |
contributed to the spread of the disease across the world (Dampier, 2019; Piel et al., 2010; Schroeder et al., 1990; Solovieff et al., 2011; Williams and Weatherall, 2012).
The HbS gene can now be found among every ethnic group, with the highest prevalence seen in individuals from sub-Saharan Africa and India and their descendants across the world; other areas of relatively high prevalence are the Middle East and Mediterranean basin (CDC, 2017a; Williams and Weatherall, 2012).
Epidemiology of SCD and SCT in the United States
An estimated 100,000 Americans are affected by SCD, and approximately 1 million to 3 million individuals in the United States are carriers of SCT, including approximately 8 to 10 percent of African Americans (ASH, n.d.; Hassell, 2010). Each year, 1,800 to 2,000 infants are born with SCD, including every 1 out of 365 African American births and 1 out of 16,300 Hispanic American births (CDC, 2018). An analysis of 2010 data from state newborn screening (NBS) programs found that the incidence of SCT in participating states was 15.5 cases per 1,000 newborns overall, including
73.1 cases per 1,000 African American newborns; 6.9 cases per 1,000 Hispanic newborns; 3.0 cases per 1,000 Caucasian American newborns; and 2.2 cases per 1,000 Asian, Native Hawaiian, or other Pacific Islander newborns (Ojodu et al., 2014). The total number of babies born with SCT in 2010 was estimated to be greater than 60,000 (CDC, 2018). Although SCD is relatively rare in the United States, there are millions of affected individuals across the globe.
The population living with SCD is concentrated along Southern and Eastern states in the United States (see Figure 1-4). This distribution has implications for access to care and state programming and policies. The map in Figure 1-4, which is based on a publication from almost 10 years ago, uses data estimated from NBS and therefore does not account for individuals with SCD born outside of those states or before universal NBS was implemented.
The mortality rate from SCD has historically been high, with most people not living past childhood due to a lack of access to proper care and a lack of treatment. However, in recent decades the mortality rate for children with SCD has been steadily decreasing, which can be attributed to preventative services, such as pneumococcal vaccines and the use of prophylactic penicillin to prevent sepsis. The Centers for Disease Control and Prevention
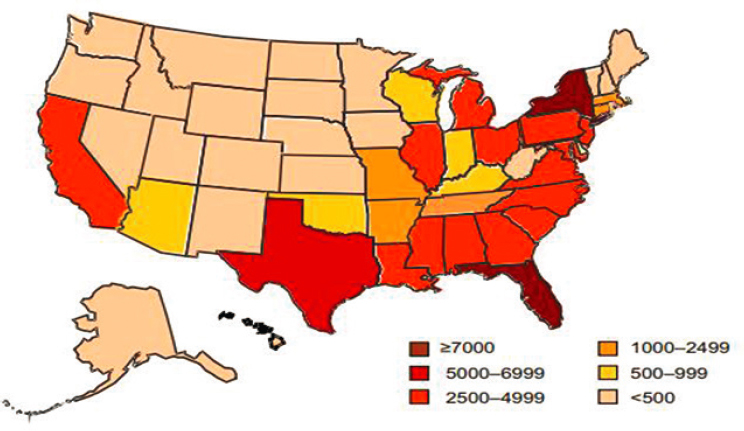
SOURCE: Reprinted from the American Journal of Preventive Medicine, 38 (4 Suppl), K. L. Hassell, Population Estimates of Sickle Cell Disease in the U.S., S512–S521, Copyright (2010), with permission from Elsevier.
reported that SCD-related deaths for African American children aged 4 and younger decreased by 42 percent between 1999 and 2002 (CDC, 2017a). Despite medical advances, however, individuals living with SCD continue to experience barriers to access care and knowledgeable providers, and their average life expectancy remains 20–30 years lower than that of the average American (Lubeck et al., 2019; Piel et al., 2017). A recent simulation modeling study showed that projected quality-adjusted life expectancy for individuals with SCD was 33 years, compared with 67 years for the non-SCD cohort (Lubeck et al., 2019).
Diagnosis of SCD
SCD can be easily diagnosed by blood testing. The classical blood test is hemoglobin electrophoresis, which identifies and measures different types of hemoglobin, including HbS, in the blood (Mentzer and Wang, 1980). However, there are numerous other tests that are more suitable in specific situations or in determined subpopulations. For instance, most NBS programs employ high-performance liquid chromatography or isoelectric focusing to identify children with SCD (Naik and Haywood, 2015). Typically, a positive result by NBS is followed by a confirmatory test by the same or different method.
Prenatal diagnosis through chorionic villus sampling of fetal DNA and amniocentesis are also available (Mentzer and Wang, 1980; Yenilmez and Tuli, 2016). These tests may pose some risks and raise ethical and social concerns, as discussed in Chapter 3. Point-of-care diagnostic strategies for SCD and SCT are being developed and may be particularly advantageous in settings with limited resources (McGann and Hoppe, 2017; Steele et al., 2019).
Clinical Complications and Comorbidities
RBCs harboring high levels of HbS have a shorter life span (approximately 20 days versus approxinately 120 days for normal RBCs), which results in hemolysis, the premature destruction of RBCs. Hemolysis in turn results in anemia when the production of RBCs cannot compensate for their premature destruction (NHLBI, n.d.). In recent decades, thanks to in vitro studies and mouse models of SCD, multiple mechanisms of disease have been elucidated. Because of hemolysis and other changes to the integrity of RBCs, SCD results in a chronic inflammatory state that affects multiple other cell types. RBCs, white clood cells, platelets, and endothelial cells become hyperadhesive, causing them to stick to each other and to the walls of the blood vessels, thereby impeding blood flow to the organs and causing ischemia (lack of oxygen) and infarction (death of tissues). Chronic and
acute ischemia and the rapid restoration of blood flow lead to a cascade of downstream effects that predispose individuals with SCD to numerous complications (Sundd et al., 2019; Telen et al., 2019).
The severe, acute pain episodes that result from the sickling of RBCs and vaso-occlusion are known as the most common complication of SCD; however, SCD is also a complex multi-system disease, characterized by other acute and chronic clinical complications, which are discussed in Chapter 4.
Causes of Death for SCD Patients
Individuals in the United States with SCD have a life expectancy that is 30 years less than their same-ethnicity peers (Platt et al., 1994). While childhood mortality rates have declined to the point that 98 percent of children now survive to at least age 18, one study found that adult (> 19 years) mortality rates increased at a rate of 1 percent per year during the study period (1979–2005) (Lanzkron et al., 2013). Early death occurs in all SCD genotypes, including the compound heterozygous sickle cell syndromes, particularly during the delicate transition period from pediatric to adult care (Blinder et al., 2013).
Advances in the science, treatment, and management of SCD have led to improvements in survival, with most children living to at least 18 years of age (Hulihan et al., 2017). Despite this progress, morbidity and mortality remain high, especially among adults. A proportion of SCD deaths can be attributed to acute chest syndrome, stroke, pulmonary hypertension, and infection (Fitzhugh et al., 2005). However, a large proportion of deaths are sudden and undefined. A study of 306 autopsies conducted on patients with SCD found that almost 41 percent had experienced sudden and unexpected deaths (Manci et al., 2003). This incidence is slightly higher than that recorded in another study of 141 adults treated by a single physician at one institution. That study, which also used autopsy reports to determine the cause of death, classified approximately 24 percent of those deaths as sudden. The leading causes of death were determined to be pulmonary hypertension, renal failure, sepsis, thromboembolism, and cirrhosis (Darbari et al., 2015). In a recent study of 486 individuals identified from the California SCD Data Collection Program who died at a median age of 45 years, most were in the hospital (63 percent) and emergency department (15 percent), signaling that they may have been receiving care for an acute event that became life threatening (Johnston et al., 2020).
Health-Related Burden
Data from the Global Burden of Disease project suggest that sickle cell disorders in the United States alone are annually responsible for 744 deaths,
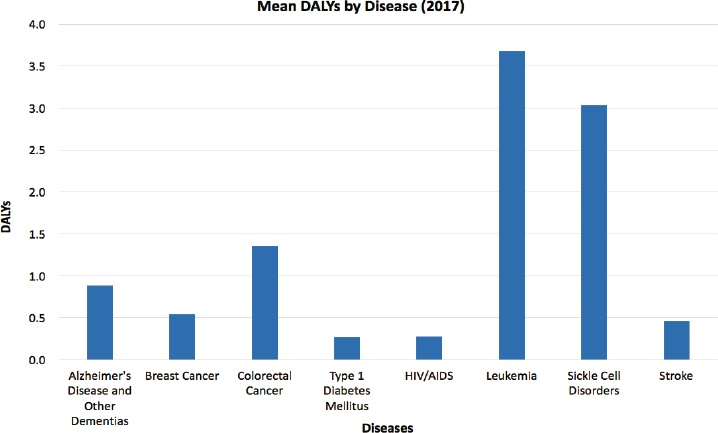
NOTE: DALY = disability-adjusted life-year.
SOURCE: Global Health Data Exchange, 2019.
29,284 years of life lost, and 3,984 disability-adjusted life-years (DALYs) lost (Coles and Mensah, 2017). In terms of DALYs, the burden of SCD on individual patients exceeds that of numerous other severe illnesses, including Alzheimer’s disease, breast cancer, colorectal cancer, type 1 diabetes mellitus, HIV/AIDS, leukemia,3 and stroke (Global Health Data Exchange, 2019) (see Figure 1-5). DALYs measure the potential years of life lost due to premature death and the years of healthy life lost due to disease or disability (WHO, 2006a). Another way of thinking of DALYs is as the additional expected years of life and healthy years of life that a patient would have enjoyed if he or she had never been diagnosed with the disease in question.
Available data suggest that SCD imposes a significant mortality and morbidity burden, but the quality and completeness of these data could be improved. Paulukonis et al. (2016) report SCD deaths using SCD surveillance data and find that about half of them are not captured by government mortality databases. It is unclear whether SCD patients who are invisible to government statistics die at higher or lower rates than their peers. This uncertainty suggests the value of expanding existing SCD surveillance and
___________________
3 This category includes acute lymphoid leukemia, acute myeloid leukemia, chronic lymphoid leukemia, and chronic myeloid leukemia.
patient registries to capture more complete data on health-related burden and other key metrics of patient well-being.
United States Versus Global Burden
In 2006 the World Health Organization declared SCD a global health issue and challenged countries to identify solutions to aid individuals living with the disease (WHO, 2006b). As mentioned before, available estimates indicate that 100,000 Americans currently have SCD. Globally, about 300,000 people are born with SCD each year (ASH, 2016; Thien and Thien, 2016). There are approximately 100,000,000 people worldwide who carry the SCT gene (ASH, n.d.). In some African countries, approximately 10–40 percent of the population carry the gene (ASH, 2016).
In developing countries, the burden of SCD is high; 90 percent of children do not live into adulthood (Sickle Cell Disease Coalition, n.d.). A systematic review conducted by Wastnedge et al. (2018) identified 67 studies (from literature published from 1980 through 2017) on incidence and mortality data in children under age 5. Africa experiences the highest SCD birth prevalence and mortality rate. The birth prevalence in Africa was 1,125 per 100,000 live births, compared with 43 per 100,000 live births in Europe (Wastnedge et al., 2018). Mortality data for SCD in children are limited; only 15 of the identified studies in the systematic review contained mortality data. Wastnedge et al. (2018) reported a pooled estimate of mortality per 100 years of child observation of 7.30 for Africa, 0.11 for Europe, and 1.06 for the United States (Wastnedge et al., 2018). The burden of SCD has been decreasing in the United States, especially in children; from 1999 through 2002 the mortality rate decreased by 68 percent for 0- to 3-year-olds, 39 percent for 4- to 9-year-olds, and 24 percent for 10- to 14-year-olds (CDC, 2017a). The child survival rate has increased to 94 percent in the United States. Even with this progress, the U.S. survival rate still lags behind the rate in Britain, which has a 99 percent survival rate for SCD, with an estimated median age of survival of 67 years (DeBaun et al., 2019; Gardner et al., 2016).
The economic costs associated with SCD also present a significant burden. In the United States, from 1989 through 1993 the 75,000 hospitalizations per year among people with SCD were estimated to cost $475 million annually (Ashley-Koch et al., 2000). SCD-related costs in the United States have now risen to $2 billion per year (CDC Foundation, 2019). In developing countries, information on the economic burden is limited. Proper care management for SCD in the United States and internationally needs to be addressed in an effort to decrease mortality rates and medical expenditures.
THE SICKLE CELL PATIENT AND THE HEALTH CARE SYSTEM
SCD has long been considered a childhood disease because survival to adulthood was uncommon due to the high rates of fatal infections in early childhood. Efforts to understand and address the disease have thus been focused on the pediatric population, which has resulted in an improved survival into young adulthood; for example, between 1999 and 2002 mortality rates decreased by 42 percent among African American children under the age of 4, thanks in part to the introduction of a vaccine against invasive pneumococcal disease (CDC, 2017a). However, mortality and morbidity rates increase dramatically as individuals transition into young adulthood and adulthood, likely because there is no standardized system to appropriately transition children into adult care. Other likely factors contributing to this increase are individuals’ lack of necessary knowledge and skills to make effective decisions about their care as they get older and their anxiety over receiving care from an unfamiliar provider (described in Chapter 6). Finally, health care personnel issues and resources play a role, as SCDexpert-led, multidisciplinary health care teams focusing on comprehensive care appear to be more prevalent and accessible in pediatrics than in adult care (Treadwell et al., 2011).
The health care needs of individuals living with SCD have been neglected by the U.S. and global health care systems, causing them and their families to suffer (Bahr and Song, 2015). Many of the complications that afflict individuals with SCD, particularly pain, are invisible. Pain is only diagnosed by self-reports, and in SCD there are few to no external indicators of the pain experience. Nevertheless, the pain can be excruciatingly severe and requires treatment with strong analgesics. Individuals with SCD often face discrimination by health care providers who do not see visible signs to corroborate the reports of pain and, with the frequent recurrence of crisis, tend to characterize the repeat acute care visits by individuals with SCD, a majority of whom are African American, as “drug-seeking” behavior (Jenerette and Brewer, 2010).
The SCD community has developed a significant lack of trust in the health care system due to the nearly universal stigma and lack of belief in its reports of pain, a lack of trust that has been further reinforced by historical events, such as the Tuskegee experiment, in which researchers deliberately withheld treatment from African Americans with syphilis in order to track the progression of the disease (CDC, 2015). This pervasive disengagement of an entire disease population from health care and research is partly responsible for the absence of new treatments to help improve care (Braveman and Gottlieb, 2014).
The neglect of SCD by the health care system extends to research into the natural history of the condition and the utility of different interventions
as well as to a lack of investigation into new treatments. There have been significant disparities in research funding for SCD compared with similar rare genetic disorders of childhood, as discussed for CF later in this chapter.
The decrease in survival rates for SCD following transition into adult care is fueled by the lack of evidence-based clinical practice guidelines across the life span, particularly for aging adults who continue to experience accelerated mortality. This has led to the current situation in which there is a pressing need to address treatment options for SCD, to improve the delivery of care and ensure optimal access to high-quality care with treatments to prevent subsequent morbidity and mortality, and to develop curative therapies. There is also an imperative to establish an informed workforce to apply these interventions.
POLICY MAKING AND FUNDING FOR SCD
Legislative Activity
Since the discovery of SCD, relatively few federal legislations with direct or indirect impact on SCD care, research, and funding have been enacted (see Figure 1-6).
The first landmark legislation for SCD was the National Sickle Cell Anemia Control Act of 1972 (Public Law 92-294), which authorized funding for screening and counseling programs, research programs for health care professionals, and medical training on SCD treatment and prevention. It also authorized the creation of comprehensive sickle cell research and treatment centers (Manley, 1984) and education clinics under the National Institutes of Health (NIH).
The Rare Diseases Act of 2002 (Public Law 107-280) was created to amend the Public Health Service Act; it authorized the creation of an Office of Rare Diseases under NIH. The purpose of this act was to provide a national research agenda, to offer educational opportunities for researchers, and to increase the development of diagnostics and treatments for those with rare diseases such as SCD (Public Law 107-280).
In 2004 the American Jobs Creation Act of 2004 (Public Law 108-357) was signed into law, amending Title XIX (Medicaid) of the Social Security Act. Section 712 of the act authorized primary and secondary medical services and treatment for individuals with SCD as medical assistance under the Medicaid program. It also directed the administrator of the Health Resources and Services Administration (HRSA) to conduct a demonstration program to develop and establish systemic mechanisms, including a national coordinating center, to improve the prevention and treatment of SCD. This act established the Sickle Cell Disease Treatment Demonstration Program, with the purpose of funding regional coordinating centers that
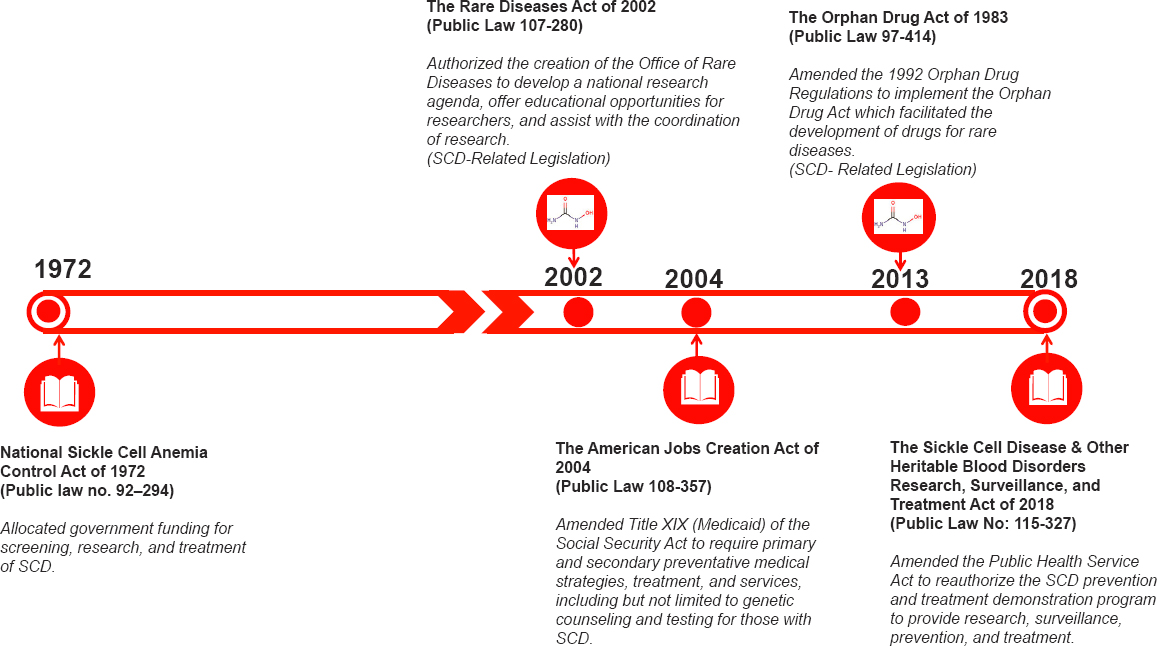
NOTE: SCD = sickle cell disease.
would work to form networks that would support processes to improve and treat SCD (Public Law 108-357).
In 2013 the Orphan Drug Act of 1983 (Public Law 97-414) was amended to address and encourage the development of drugs for people with rare diseases. This amendment provided clarity concerning the regulatory language used and suggested improvements for the drug designation process; drugs such as hydroxyurea that were previously only available to adults were made available to children. The amendment also paved the way for the development of Endari, the second drug approved for SCD by the U.S. Food and Drug Administration (FDA). In late 2019 FDA approved two additional drugs for SCD: Adakveo (crizanlizumab) and Oxbryta (voxelotor) (Global Blood Therapeutics, 2019; Novartis, 2019). Figure 1-7 provides a timeline of all FDA approvals of drugs for treating SCD.
The most recent legislative action for SCD took place on December 18, 2018, with the signing into law of the Sickle Cell Disease and Other Heritable Blood Disorders Research, Surveillance, Prevention, and Treatment Act of 2018 (Public Law 115-327). That act amended part A of Title XI of the Public Health Service Act to reauthorize HHS to support data collection on SCD and promote public health activities on heritable blood disorders. The legislation seeks to improve SCD treatment, research, monitoring, and prevention. As of the development of this report, the section of the legislation related to data collection on certain blood disorders had not yet been funded by Congress.
Funding
Funding for SCD has historically been low and has decreased over the years. For example, appropriations for the National Sickle Cell Anemia Control Act of 1972 (Public Law 92-294) authorized $85 million, and the American Jobs Creation Act of 2004 (Public Law 108-357) authorized $50 million over 5 years, for medical services and treatment for individuals with SCD.
It is also well documented that SCD receives less federal and private funding than other conditions. Some attribute this difference to the history of discrimination against the racial and ethnic minority population most affected by SCD (Haywood et al., 2014). Figure 1-8 compares NIH funding between 2008 and 2016 for SCD with its funding for CF, which mostly affects white Americans and affects fewer people (approximately 30,000). Average annual NIH funding per affected individual for CF was almost four times more than the average annual funding per affected individual for SCD during the period under review (Farooq and Strouse, 2018). The funding difference was even more stark for funds from private foundations ($342 million for CF compared with $6.4 million for SCD between 2008 and 2012) (Farooq and Strouse, 2018) (see Figure 1-9).
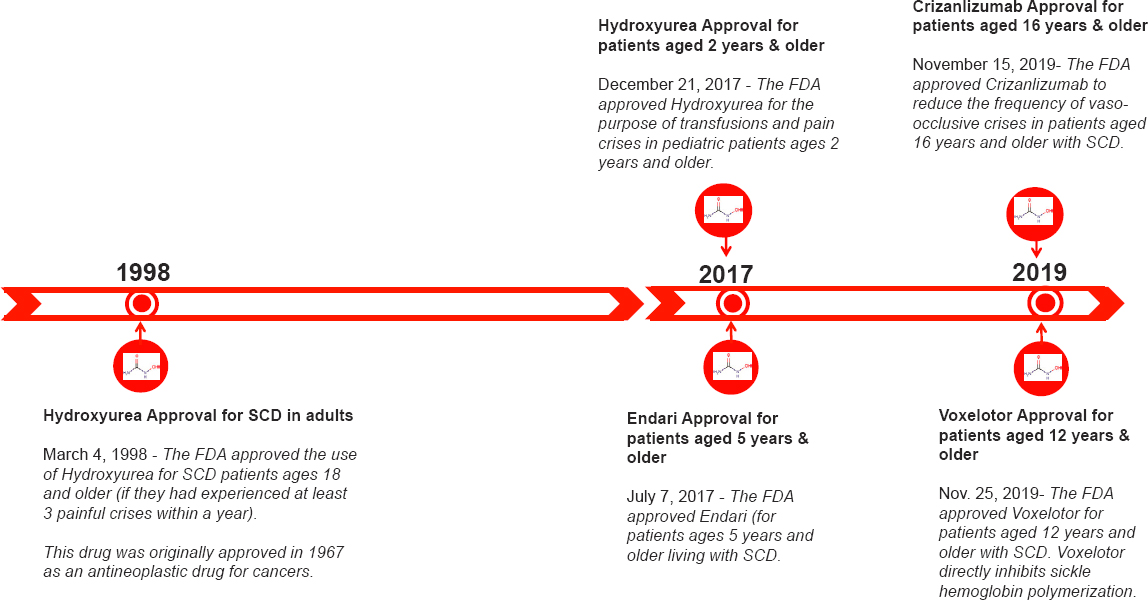
NOTE: FDA = U.S. Food and Drug Administration; SCD = sickle cell disease.
SOURCE: Data from ClinicalTrials.gov.
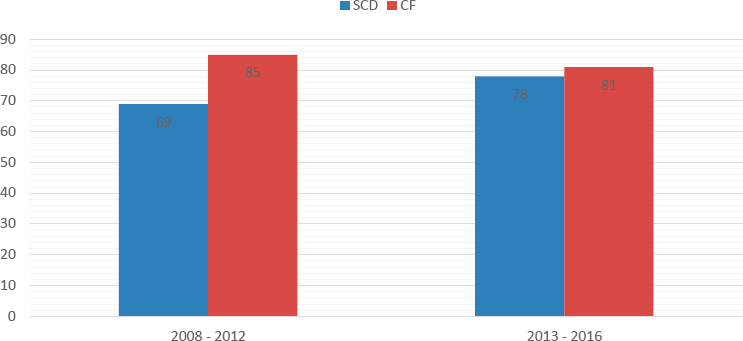
NOTE: CF = cystic fibrosis; SCD = sickle cell disease.
SOURCE: Adapted from Farooq and Strouse, 2018.
KEY SCD ACTORS
There are several actors actively engaged in SCD research, treatment, and advocacy. This section provides a brief description of such actors, including the federal and state governments, health care providers, payers, health professional associations, and industry as well as other stakeholders from the scientific community, advocacy groups, and patients and families (see Table 1-2).
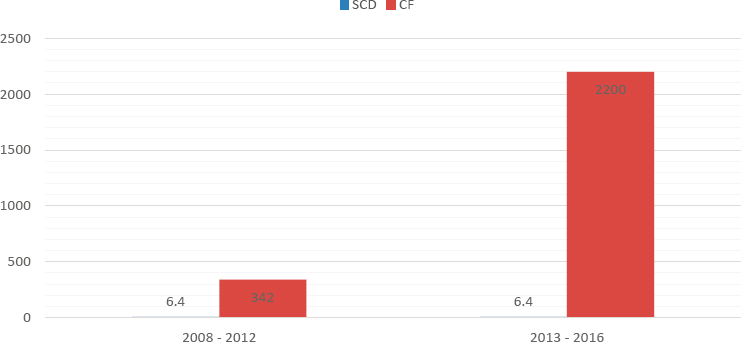
NOTE: CF = cystic fibrosis; SCD = sickle cell disease.
SOURCE: Adapted from Farooq and Strouse, 2018.
TABLE 1-2 Description of SCD Key Actors
| Stakeholder | Description |
|---|---|
| Federal Government |
Federal agencies’ work in sickle cell disease (SCD) includes
|
| Health Care Providers | Health care systems, including multi-hospital systems and stand-alone community clinics, have programs that provide services to individuals with SCD. Large academic health systems, which are located primarily in urban areas, may run specialized sickle cell centers or clinics. A wide variety of health care providers, including doctors, nurses, and allied health professionals are also key to care delivery (see Chapters 5 and 6). |
| Payers | Public and private payers, together with individuals with SCD and their families, bear the substantial costs associated with SCD care (see Chapter 2). |
| Health Professional Associations | Professional associations and organizations, specifically for medical specialties that often treat SCD patients such as the American Society of Hematology and the American College of Emergency Physicians, have efforts to promote awareness of SCD and to provide ongoing education of members and other activities to improve care for individuals with SCD (see Chapter 6). |
| Industry | Several pharmaceutical and biopharmaceutical companies are actively developing therapeutic products for SCD (see Figure 1-7 for approved SCD therapies and Chapter 7 and Appendix I for therapeutic products in development). Some of these companies also partner with stakeholders for advocacy and fund services for individuals with SCD and their families. |
TABLE 1-2 Continued
| Stakeholder | Description |
|---|---|
| Research Community | Beyond federal agencies, other entities fund or conduct SCD research. A couple of examples are the Patient-Centered Outcomes Research Institute, which has funded a robust portfolio of SCD projects (PCORI, n.d.) and the California Institute for Regenerative Medicine, which was created by the state of California to fund research on stem cell–based therapies for multiple diseases, including SCD (CIRM, n.d.). Researchers at several large academic medical centers are also actively conducting research and enrolling patients in clinical trials. |
| State Agencies | Multiple state agencies, including departments of health, education, and social work, among others, run programs that benefit individuals with SCD. The availability and types of such programs vary by state. Some states also have detailed state-level SCD action plans even though the status of the implementation of these programs is unclear, per publicly available information. |
| Patient Advocacy Groups and Community-Based Organizations | Patient advocacy groups and community-based organizations may function at the federal, state, local, or even community level and may take on various structures, such as virtual organizations or being based at a health care institution. The roles of these organizations also vary and are discussed in Chapter 8. |
| Patients and Families | Individuals living with SCD and their families are integral to improving SCD care because of their lived experience with the disease. They have a role to play in SCD research, health care, and advocacy. |
ORGANIZATION OF THE REPORT
This report contains 9 chapters and 14 appendixes. Chapter 2 presents background information on the societal, individual, and environmental factors that affect SCD patients; the impact of SCD on mental health; the economic burden on people living with SCD and their families; and the distribution of both private and public insurance for patients. Chapter 3 provides a detailed description of screening, registries, and surveillance. This chapter examines the importance of communication for screening results, the use of that data, the ethical implications of screening, and the limitations and strengths of using surveillance, screening, or registries. Chapter 4 summarizes management approaches for SCD care as it pertains to disease-modifying agents, therapies, the treatment of complications, mental health, non-pharmacologic therapies, psychosocial support, and disease self-management education. This chapter describes evidence-based strategies, the effectiveness of current therapeutics approaches, and the opportunities for maximizing the use of current resources; it also
summarizes the available evidence on SCT. Chapter 5 reviews the organization and delivery of SCD care. This chapter delves into how and where people with SCD should receive care and what types of care they should receive; the issues with the transition from pediatric to adult care; what comprehensive SCD care encompasses; and the geographic, financial, and socioeconomic barriers to care. Chapter 6 addresses the current state of quality of SCD care and the workforce needs to deliver high-quality care. This chapter provides details on the transition of care, the indicators for high-quality care, how to engage health care professionals, the attitudes of health care providers, and the opportunities for training programs for health professionals. Chapter 7 centers on innovative and curative therapies. This chapter highlights the perspectives of those with SCD, current therapies, the process for clinical trials, reimbursement policies and lifetime costs, and the reform of health care delivery. Chapter 8 focuses on the landscape of patient organizations advocating for and providing services to the SCD population, community engagement, and the importance of education and awareness for people with SCD and the public. Chapter 9 presents the strategic plan and blueprint for action created by the committee to address SCD. The recommended actions detailed in the blueprint are linked to the conclusions and recommendations in the chapters.
The committee’s conclusions and recommendations are presented at the end of each chapter. The references used follow each chapter. Appendix A contains the agendas of the open meetings and a list of presentation topics. Appendix B describes the literature review strategy with the terms used by the National Academies Research Center. Appendix C provides the committee and staff biographies. Appendix D details the results of a brief survey that the Association of Public Health Laboratories conducted on behalf of the committee. Appendixes E, F, and G give brief descriptions of the California SCD Data Collection Program, the 24-hour sickle cell program at Grady Memorial Hospital, and the Emory Adult Cystic Fibrosis Program Protocol, respectively. Appendix H provides information on SCD programs funded by HRSA. Appendix I lists select treatments that are currently under development. Appendix J provides information on other models for training hematologists. Appendix K provides a list of some of the community-based organizations and patient advocacy groups in the United States. Appendix L is a summary of the committee’s strategic plan and blueprint for action. Appendix M is a guide of the chapters in this report that discuss SCT. Finally, Appendix N is a glossary of key terms used in the field of SCD and research and referenced in this report.
REFERENCES
ASH (American Society of Hematology). 2008. Milestones in sickle cell disease. https://www.hematology.org/About/History/50-Years/1533.aspx (accessed June 5, 2019).
ASH. 2016. State of sickle cell disease. http://www.scdcoalition.org/pdfs/ASH%20State%20of%20Sickle%20Cell%20Disease%202016%20Report.pdf (accessed June 5, 2019).
ASH. n.d. Sickle cell trait. https://www.hematology.org/Patients/Anemia/Sickle-Cell-Trait.aspx (accessed June 5, 2019).
Ashley-Koch, A., Q. Yang, and R. Olney. 2000. Sickle hemoglobin (Hb S) allele and sickle cell disease: A huge review. American Journal of Epidemiology 151(9):839–845.
Bahr, N. C., and J. Song. 2015. The effect of structural violence on patients with sickle cell disease. Journal of Health Care for the Poor and Underserved 26(3):648–661.
Blinder, M. A., F. Vekeman, M. Sasane, A. Trahey, C. Paley, and M. S. Duh. 2013. Age-related treatment patterns in sickle cell disease patients and the associated sickle cell complications and healthcare costs. Pediatric Blood & Cancer 60(5):828–835.
Booth, C., B. Inusa, and S. K. Obaro. 2010. Infection in sickle cell disease: A review. International Journal of Infectious Disease 14(1):e2–e12.
Braveman, P., and L. Gottlieb. 2014. The social determinants of health: It’s time to consider the causes of the causes. Public Health Reports 129(Suppl 2):19–31.
CDC (Centers for Disease Control and Prevention). 2015. U.S. Public Health Service syphilis study at Tuskegee: The Tuskegee timeline. https://www.cdc.gov/tuskegee/timeline.htm (accessed August 1, 2019).
CDC. 2017a. Data & statistics on sickle cell disease. https://www.cdc.gov/ncbddd/sicklecell/data.html (accessed June 5, 2019).
CDC. 2017b. What is sickle cell disease? https://www.cdc.gov/ncbddd/sicklecell/facts.html (accessed June 5, 2019).
CDC. 2018. Incidence of sickle cell trait in the U.S. https://www.cdc.gov/ncbddd/sicklecell/features/keyfinding-trait.html (accessed June 5, 2019).
CDC Foundation. 2019. Improving the lives of people with sickle cell disease. https://www.cdcfoundation.org/sites/default/files/files/2019CDCFSickleCell.pdf (accessed June 5, 2019).
CIRM (California Institute for Regenerative Medicine). n.d. Stem cell gene therapy for sickle cell anemia. https://www.cirm.ca.gov/our-progress/video/stem-cell-gene-therapy-sickle-cell-anemia-donald-kohn (accessed June 5, 2019).
Coles, E., and G. A. Mensah. 2017. The burden of heart, lung, and blood diseases in the United States, 1990 to 2016: Perspectives from the National Heart, Lung, and Blood Institute. Global Heart 12(4):349–358.
Crosby, L. E., N. E. Joffe, M. K. Irwin, H. Strong, J. Peugh, L. Shook, K. A. Kalinyak, and M. J. Mitchell. 2015. School performance and disease interference in adolescents with sickle cell disease. Physical Disabilities: Education and Related Services 34(1):14–30.
Dampier, C. 2019. New and emerging treatments for vaso-occlusive pain in sickle cell disease. Expert Review of Hematology 12(10):857–872.
Darbari, D. S., J. Kwagyan, S. Rana, V. R. Gordeuk, and O. Castro. 2015. 47 causes of death in adult sickle cell disease patients at Howard University. Journal of Investigative Medicine 53(2):S395.
DeBaun, M., D. Ghafuri, M. Rodeghier, P. Maitra, S. Chaturvedi, A. Kassim, and K. Ataga. 2019. Decreased median survival of adults with sickle cell disease after adjusting for left truncation bias: A pooled analysis. Blood 133(6):615–617.
Eaton, W. A. 2003. Linus Pauling and sickle cell disease. Biophysical Chemistry 100(1–3):109–116.
Farooq, F., and J. Strouse. 2018. Disparities in foundation and federal support and development of new therapeutics for sickle cell disease and cystic fibrosis. Blood 132(Suppl 1):4687.
Fitzhugh, C., N. Lauder, J. Jonassaint, F. R. Gilliam, M. J. Telen, and L. M. De Castro. 2005. Morbidity and associated sudden death in sickle cell disease. Blood 106(11):2348.
Gardner, K., A. Douiri, E. Drasar, M. Allman, A. Mwirigi, M. Awogbade, and S. Thein. 2016. Survival in adults with sickle cell disease in a high-income setting. Blood 128(10):1436–1438.
Global Blood Therapeutics. 2019. FDA approves Oxbryta™ (voxelotor), the first medicine specifically targeting the root cause of sickle cell disease. https://ir.gbt.com/news-releases/news-release-details/fda-approves-oxbrytatm-voxelotor-first-medicine-specifically (accessed February 19, 2020).
Global Health Data Exchange. 2019. Secondary global health data exchange—GBD results tool 2019. http://ghdx.healthdata.org/gbd-results-tool (accessed June 10, 2019).
Gold, J. I., C. B. Johnson, M. J. Treadwell, N. Hans, and E. Vichinsky. 2008. Detection and assessment of stroke in patients with sickle cell disease: Neuropsychological functioning and magnetic resonance imaging. Pediatric Hematology and Oncology 25(5):409–421.
Greenham, M., A. Gordon, V. Anderson, and M. T. Mackay. 2016. Outcome in childhood stroke. Stroke 47(4):1159–1164.
Habara, A., and M. H. Steinberg. 2016. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease. Experimental Biology and Medicine (Maywood) 241(7):689–696.
Hassell, K. 2010. Population estimates of sickle cell disease in the U.S. American Journal of Preventive Medicine 38(4):S512–S521.
Haywood, C., Jr., M. Diener-West, J. Strouse, C. P. Carroll, S. Bediako, S. Lanzkron, J. Haythornthwaite, G. Onojobi, and M. C. Beach. 2014. Perceived discrimination in health care is associated with a greater burden of pain in sickle cell disease. Journal of Pain and Symptom Management 48(5):934–943.
Herrick, J. 1910. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. JAMA (formerly Archives of Internal Medicine) 6(5):517–521.
Hulihan, M., K. L. Hassell, J. L. Raphael, K. Smith-Whitley, and P. Thorpe. 2017. CDC grand rounds: Improving the lives of persons with sickle cell disease. Morbidity and Mortality Weekly Report 66(46):1269–1271.
IOM (Institute of Medicine). 2012. Epilepsy across the spectrum: Promoting health and understanding. Washington, DC: The National Academies Press.
Jenerette, C. M., and C. Brewer. 2010. Health-related stigma in young adults with sickle cell disease. Journal of the National Medical Association 102(11):1050–1055.
Johnston, E. E., O. O. Adesina, E. Alvarez, H. Amato, S. Paulukonis, A. Nichols, L. J. Chamberlain, and S. Bhatia. 2020. Acute care utilization at end of life in sickle cell disease: Highlighting the need for a palliative approach. Journal of Palliative Medicine 23(1):24–32.
Lanzkron, S., C. P. Carroll, and C. Haywood, Jr. 2013. Mortality rates and age at death from sickle cell disease: U.S., 1979–2005. Public Health Reports 128(2):110–116.
Lubeck, D., I. Agodoa, N. Bhakta, M. Danese, K. Pappu, R. Howard, M. Gleeson, M. Halperin, and S. Lanzkron. 2019. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Network Open 2(11):e1915374.
Luzzatto, L. 2012. Sickle cell anaemia and malaria. Mediterranean Journal of Hematology and Infectious Diseases 4(1):e2012065.
Malowany, J., and J. Butany. 2012. Pathology of sickle cell disease. Seminars in Diagnostic Pathology 29(1):49–55.
Manci, E. A., D. E. Culberson, Y. M. Yang, T. M. Gardner, R. Powell, J. Haynes, Jr., A. K. Shah, V. N. Mankad, and Investigators of the Cooperative Study of Sickle Cell Disease. 2003. Causes of death in sickle cell disease: An autopsy study. British Journal of Haematology 123(2):359–365.
Manley, A. F. 1984. Legislation and funding for sickle cell services, 1972–1982. The American Journal of Pediatric Hematology/Oncology 6(1):67–71.
McGann, P. T., and C. Hoppe. 2017. The pressing need for point-of-care diagnostics for sickle cell disease: A review of current and future technologies. Blood Cells Molecules and Diseases 67:104–113.
Mentzer, W., and W. Wang. 1980. Sickle-cell disease: Pathophysiology and diagnosis. Pediatric Annals 9(8):10–22.
Mulumba, L. L., and L. Wilson. 2015. Sickle cell disease among children in Africa: An integrative literature review and global recommendations. International Journal of Africa Nursing Sciences 3:56–64.
Naik, R. P., and C. Haywood, Jr. 2015. Sickle cell trait diagnosis: Clinical and social implications. Hematology 2015:160–167.
NHLBI (National Heart, Lung, and Blood Institute). 2014. Evidence-based management of sickle cell disease: Expert panel report, 2014. Bethesda, MD: National Heart, Lung, and Blood Institute.
NHLBI. n.d. Hemolytic anemia. https://www.nhlbi.nih.gov/health-topics/hemolytic-anemia (accessed January 9, 2020).
NIH (National Institutes of Health). 2019. Your guide to understanding genetic conditions: Sickle cell disease. https://ghr.nlm.nih.gov/condition/sickle-cell-disease#inheritance (accessed June 5, 2019).
Novartis. 2019. New novartis medicine Adakveo® (crizanlizumab) approved by FDA to reduce frequency of pain crises in individuals living with sickle cell disease. https://www.novartis.com/news/media-releases/new-novartis-medicine-adakveo-crizanlizumab-approved-fda-reduce-frequency-pain-crises-individuals-living-sickle-cell-disease (accessed August 1, 2019).
Ojodu, J., M. M. Hulihan, S. N. Pope, and A. M. Grant. 2014. Incidence of sickle cell trait—United States, 2010. Morbidity and Mortality Weekly Report 63(49):1155–1158.
Oyedeji, C., J. J. Strouse, R. D. Crawford, M. E. Garrett, A. E. Ashley-Koch, and M. J. Telen. 2019. A multi-institutional comparison of younger and older adults with sickle cell disease. American Journal of Hematology 94(4):e115–e117.
Paulukonis, S. T., J. R. Eckman, A. B. Snyder, W. Hagar, L. B. Feuchtbaum, M. Zhou, A. M. Grant, and M. M. Hulihan. 2016. Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Reports 131(2):367–375.
PCORI (Patient-Centered Outcomes Research Institute). n.d. Explore our portfolio of funded projects. https://www.pcori.org/research-results?keywords=sickle+cell#search-results (accessed August 1, 2019).
Piel, F. B., A. P. Patil, R. E. Howes, O. A. Nyangiri, P. W. Gething, T. N. Williams, D. J. Weatherall, and S. I. Hay. 2010. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nature Communications 1(1):104.
Piel, F. B., M. H. Steinberg, and D. C. Rees. 2017. Sickle cell disease. New England Journal of Medicine 377(3):305.
Platt, O. S., D. J. Brambilla, W. F. Rosse, P. F. Milner, O. Castro, M. H. Steinberg, and P. P. Klug. 1994. Mortality in sickle cell disease. Life expectancy and risk factors for early death. New England Journal of Medicine 330(23):1639–1644.
Quinn, C. T. 2016. Minireview: Clinical severity in sickle cell disease: The challenges of definition and prognostication. Experimental Biology and Medicine (Maywood) 241(7):679–688.
Quinn, C. T., Z. R. Rogers, T. L. McCavit, and G. R. Buchanan. 2010. Improved survival of children and adolescents with sickle cell disease. Blood 115(17):3447–3452.
Sandhu, M. K., and A. Cohen. 2015. Aging in sickle cell disease: Co-morbidities and new issues in management. Hemoglobin 39(4):221–224.
Schroeder, W., E. Munger, and D. Powars. 1990. Sickle cell anaemia, genetic variations, and the slave trade to the United States. The Journal of African History 31(2):163–180.
Serjeant, G. R. 2013. The natural history of sickle cell disease. Cold Spring Harbor Perspectives in Medicine 3(10):a011783.
Sickle Cell Disease Coalition. n.d. Spread the word. http://www.scdcoalition.org/get-involved/spread-the-word.html (accessed June 5, 2019).
Solovieff, N., S. W. Hartley, C. T. Baldwin, E. S. Klings, M. T. Gladwin, J. G. Taylor, G. J. Kato, L. A. Farrer, M. H. Steinberg, and P. Sebastiani. 2011. Ancestry of African Americans with sickle cell disease. Blood Cells, Molecules, and Diseases 47(1):41–45.
Steele, C., A. Sinski, J. Asibey, M. D. Hardy-Dessources, G. Elana, C. Brennan, I. Odame, C. Hoppe, M. Geisberg, E. Serrao, and C. T. Quinn. 2019. Point-of-care screening for sickle cell disease in low-resource settings: A multi-center evaluation of HemoTypeSC, a novel rapid test. American Journal of Hematology 94(1):39–45. https://doi.org/10.1002/ajh.25305.
Sundd, P., M. Gladwin, and E. Novelli. 2019. Pathophysiology of sickle cell disease. Annual Review of Pathology: Mechanisms of Disease 14:263–292.
Swanson, M. E., S. D. Grosse, and R. Kulkarni. 2011. Disability among individuals with sickle cell disease: Literature review from a public health perspective. American Journal of Preventive Medicine 41(6):S390–S397.
Telen, M. J., P. Malik, and G. M. Vercellotti. 2019. Therapeutic strategies for sickle cell disease: Towards a multi-agent approach. Nature Reviews Drug Discovery 18(2):139–158.
Thein, M. S., and S. L. Thein. 2016. World Sickle Cell Day 2016: A time for appraisal. Indian Journal of Medical Research 143(6):678–681.
Treadwell, M., J. Telfair, R. W. Gibson, S. Johnson, and I. Osunkwo. 2011. Transition from pediatric to adult care in sickle cell disease: Establishing evidence-based practice and directions for research. American Journal of Hematology 86(1):116–120.
Wagner, E. H. 1998. Chronic disease management: What will it take to improve care for chronic illness? Effective Clinical Practice 1(1):2–4.
Wagner, E. H., B. T. Austin, C. Davis, M. Hindmarsh, J. Schaefer, and A. Bonomi. 2001. Improving chronic illness care: Translating evidence into action. Health Affairs 20(6):64–78.
Wagner, E. H., S. M. Bennett, B. T. Austin, S. M. Greene, J. K. Schaefer, and M. Vonkorff. 2005. Finding common ground: Patient-centeredness and evidence-based chronic illness care. Journal of Alternative and Complementary Medicine 11(Suppl 1):S7–S15.
Wastnedge, E., D. Waters, S. Patel, K. Morrison, M. Goh, D. Adeloye, and I. Rudan. 2018. The global burden of sickle cell disease in children under five years of age: A systematic review and meta-analysis. Journal of Global Health 8(2):021103.
WHO (World Health Organization). 2006a. Years of life lost (percentage of total). https://www.who.int/whosis/whostat2006YearsOfLifeLost.pdf (accessed June 5, 2019).
WHO. 2006b. Fifty-Ninth World Health Assembly: Resolutions and decisions, annexes: WHA59/2006/REC/1. Geneva, Switzerland: World Health Organization.
Williams, T. N., and D. J. Weatherall. 2012. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harbor Perspectives in Medicine 2(9):a011692.
Yenilmez, E. D., and A. Tuli. 2016. New perspectives in prenatal diagnosis of sickle cell anemia. In B. Inusa, ed. Sickle cell disease: Pain and common chronic complications. London: IntechOpen.
This page intentionally left blank.




























