
6
Delivering High-Quality Sickle Cell Disease Care with a Prepared Workforce
There needs to be a concise way of capturing that information and understanding what the implications are of different treatments and different delivery systems on different people, capture that somehow, and be able to feed that back to the clinician and the patient that are in the midst of trying to make a really important treatment decision.
—Sara van G. (Open Session Panelist)
High-quality care for individuals living with sickle cell disease (SCD) should be evidence-based and accompanied by clear, measurable metrics that assess quality and improve performance. Care should be delivered by a well-trained workforce that is willing and able to provide the necessary services. Chapter 4 discussed the myriad acute and chronic complications that individuals living with SCD experience, and Chapter 5 detailed the comprehensive health care and health-related services that individuals living with SCD and sickle cell trait (SCT) need for optimal health outcomes. This chapter examines the state of evidence associated with clinical practice guidelines for managing the care of children and adults with SCD and the current state of health system performance assessment in delivering those services. This chapter also discusses strategies for addressing the obstacles to developing a cadre of health professionals who are prepared to deliver high-quality care. As discussed in Chapter 5, the committee recommends the use of a multidisciplinary team of providers to address the complex care needs of individuals living with SCD.
Some health care providers may be uncomfortable with providing SCD care because of a lack of knowledge and understanding about the clinical condition and the affected population. Clinical practice guidelines are an effective way of standardizing care and informing health care providers (especially non-experts) of the appropriate services that individuals living with SCD need. Commensurate endorsed quality metrics allow individual providers and systems to measure how well they adhere to available guidelines in providing such care and the consistency of this application to “every patient, every time.”
GUIDELINES FOR HIGH-QUALITY SCD CARE
Introduction
The discussion in this section is guided by the quality framework from two prior National Academies of Sciences, Engineering, and Medicine (the National Academies) publications, Crossing the Quality Chasm: A New Health System for the 21st Century (IOM, 2001) and Crossing the Global Quality Chasm: Improving Health Care Worldwide (NASEM, 2018). As noted in Figure 6-1, achieving quality requires ongoing attention by the health care system to provide care that is safe, effective, accessible/timely, efficient, equitable, and person-centered (NASEM, 2018).
Health care organizations and clinicians assess how well they are achieving these quality aims by assessing performance on metrics indicative of high-quality care. To foster the delivery of high-quality SCD care, health care providers need information and tools that synthesize available knowledge into clinical practice, and clinicians, organizations, and payers
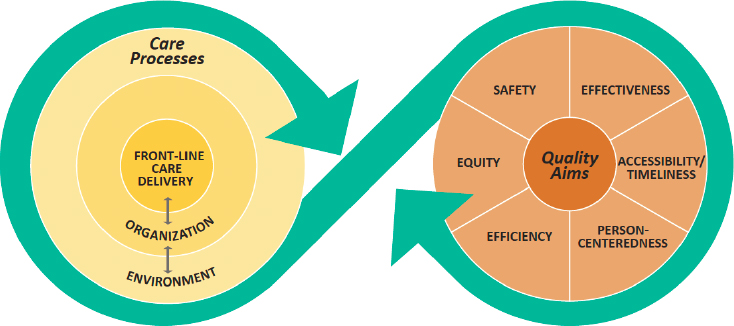
SOURCE: NASEM, 2018.
need to be able to measure, reward, and identify opportunities for improving quality. Quality measures, performance indicators, and clinical practice guidelines are all relevant tools. Where there is a strong evidence base, defined as well-conducted randomized controlled trials and robust data to support performance tracking, these evidence-based SCD services can be defined as quality measures.
Quality measures are tools that help us measure or quantify health care processes, outcomes, patient perceptions, and organizational structure and/or systems that are associated with the ability to provide high-quality health care and/or that relate to one or more quality goals for health care. (CMS, 2020)
There are currently two measures for SCD care that have been endorsed by the National Quality Forum (NQF) (discussed in this chapter). Quality or performance indicators are “standardized evidence-based measures of health care quality that can be used … to measure and track clinical performance and outcomes” (AHRQ, n.d.). Quality indicators are used to benchmark actual performance against recommended practices; these are collected for reporting purposes and may be tied to payment. When the evidence base is insufficiently robust or still being gathered for services that the expert consensus sees as beneficial, clinical practice guidelines can be developed to guide care delivery. “Clinical practice guidelines are statements that include recommendations intended to optimize patient care that are informed by a systematic review of evidence [and expert opinion] and an assessment of the benefits and harms of alternative care options” (IOM, 2011a, p. 15). These guidelines help to standardize care by translating the existing research into recommendations for clinicians on the most beneficial services for different patient populations.
The structural aspects of health system design, such as the availability of a trained workforce and the use of data collection systems, are critical to adopting, measuring, and implementing quality measures and performance indicators.
Development of SCD-Specific Clinical Guidelines
As early as 1972 there were efforts to provide treatment guidelines for SCD, which were led by the National Heart, Lung, and Blood Institute (NHLBI) and a group of funded investigators who were aligned with the establishment of the SCD comprehensive care centers (Prabhakar et al., 2010; Smith et al., 2006). In 1984 NHLBI published the first set of national guidelines for SCD management, which were subsequently updated in 1989, 1995, 1999, and 2002 (NHLBI, 2002). In 2002 the American Academy of Pediatrics’ (AAP’s) Section on Hematology/Oncology and its Committee on Genetics published guidelines specific to SCD management for children with SCD (AAP, 2002).
In 2009 NHLBI convened an expert panel to develop guidelines for SCD management, which included health care professionals in areas such as pediatric and adult hematology, family medicine, and evidence-based medicine (Yawn et al., 2014). The expert panel, along with an independent methodology committee, reviewed the literature, rated the quality of evidence, and evaluated the strength of the recommendations. The quality of evidence was assessed using a modified Grading of Recommendations Assessment, Development and Evaluation (GRADE) framework (Balshem et al., 2011). The NHLBI expert panel’s process adapts the GRADE process and rates the quality of recommendations as “strong,” “moderate,” or “weak.” The panel therefore added the “moderate” category to the regular GRADE framework of “strong” or “weak” recommendations to capture research that is from either low-quality randomized controlled trials or large, well-conducted observational studies. The expert panel also made consensus recommendations based on evidence-based practice guidelines from other entities, such as the U.S. Preventive Services Task Force (USPSTF) and the Advisory Committee on Immunization Practices (Yawn et al., 2014).
Before finalizing its recommendations, the expert panel sought input from a number of associations with expertise in SCD, including AAP, the American Society of Hematology (ASH), and the American Society of Pediatric Hematology/Oncology (ASPHO). The resulting guidelines, Evidence-Based Management of Sickle Cell Disease, were published in 2014 (Yawn et al., 2014). Highlights of the NHLBI clinical guidelines covering health maintenance, the management of acute and chronic complications associated with SCD, and the use of hydroxyurea (HU) and transfusion therapy is included in Box 6-1.
Only a few of the strongly recommended NHLBI guidelines are supported by high-quality evidence. The majority of the recommended health care services had moderate or low evidence, indicating a huge gap in the evidence base for SCD interventions. Despite these gaps, the expert panel indicated that moderate-strength recommendations can be used to develop protocols to guide care delivery (Yawn et al., 2014). Recommended services with low-quality evidence represent areas where some variation in care may be acceptable because the services may be appropriate for only a subset of the SCD population. There is a need to generate research to fill in the evidence gaps for strongly recommended services where the supporting evidence base was moderate or weak or for services with moderate or weak recommendations.
Despite the NHLBI guidelines being a considerable contribution to improving the quality of care delivered, experts have noted that they have shortcomings, which offer valuable lessons for developing the next round of clinical guidelines for SCD. Citing recommendations from the Institute of Medicine (IOM) report Clinical Practice Guidelines We Can Trust, which offers criteria by which guidelines should be assessed, experts note that the NHLBI panel did not include patient representation, thus missing
the opportunity to solicit patient perspective and preferences. Additionally, the perspective of health professional associations representing clinical specialties that have expertise in the management of some of the clinical complications associated with SCD (e.g., chronic kidney disease [CKD], pulmonary hypertension, obstructive lung disease, and stroke) was not solicited in the development of the guidelines (DeBaun, 2014; Thompson, 2014). Also, there are prevalent SCD complications that are notably missing from the NHLBI guidelines, such as asthma, screening for and management of pulmonary hypertension, and hematopoietic stem cell transplant (DeBaun, 2014; Thompson, 2014).
In addition to the 2014 NHLBI guidelines, ASH initiated an effort in 2016 to develop guidelines for screening, diagnosis, and management for five SCD-related areas: transfusion support, cardiopulmonary and kidney disease, cerebrovascular disease, pain, and stem cell transplantation. The Mayo Clinic Evidence-Based Practice Research Center led the systematic review of evidence for the ASH work. At the time of the development of this report, ASH had released guidelines for the screening, diagnosis, and management of SCD-related cardiopulmonary and renal complications (Liem et al., 2019) and transfusion support (Chou et al., 2020). Final guidelines are in development from ASH for cerebrovascular disease, transplantation, and the management of acute and chronic pain (ASH, n.d.).
National Quality Forum–Endorsed SCD-Specific Measures
NQF was created in 1999 in response to the report of the President’s Advisory Commission on Consumer Protection and Quality in the Healthcare Industry (NQF, n.d.c). The commission concluded that an organization like NQF should be created to improve the measurement and reporting of quality health care indicators. An NQF endorsement of quality measures signifies a rigorous review of the science and evidence base supporting the measure and input from key stakeholders, including patients and families, to develop consensus about measures that warrant a “best in class” designation (NQF, n.d.a).
NQF-endorsed measures are used widely at the federal, state, and local levels for payment and reporting. Currently, NQF has endorsed two measures related to SCD for measuring high-quality care for children (NQF, 2018):
- NQF measure #2797, “Transcranial Doppler Ultrasonography Screening among Children with Sickle Cell Anemia: The percentage of children ages 2 through 15 years old with sickle cell anemia (Hemoglobin SS) who received at least one transcranial Doppler (TCD) screening within a year.”
- NQF measure #3166, “Antibiotic Prophylaxis Among Children with Sickle Cell Anemia: The percentage of children ages 3 months to 5 years old with sickle cell anemia (SCA, hemoglobin [Hb] SS) who were dispensed appropriate antibiotic prophylaxis for at least 300 days within the measurement year.”
The evidence on adherence to NQF-endorsed measures in SCD care is not robust. The following section discusses the available information on adherence to these measures.
Transcranial Doppler Screening
TCD screening is a significant advance in identifying and preventing stroke in children and adolescents with SCD. However, studies have consistently demonstrated that fewer than half of all eligible patients have received proper TCD screening. Raphael et al. (2008) found that the average yearly TCD screening rate for eligible pediatric patients (207 in the evaluation) was 45 percent. Eckrich et al. (2013) found that among a cohort of 338 children with SCD who were publicly insured, the cumulative incidence rates of annual TCD screening increased from 2.5 percent in 1997 to 68.3 percent in 2008. While screening increased significantly over the study period, 31 percent of children did not receive TCD screening over the entire study period. Reeves et al. (2016) conducted a retrospective cross-sectional study using Medicaid claims data from 2005 to 2010 and found that among 4,775 children and adolescents (2–16 years old), TCD screening rates increased over the 6-year study period from 22 percent to 44 percent (p < 0.001). The authors also found that screening rates varied substantially across states and that the receipt of well-child visits was associated with higher odds of a TCD screening. In a retrospective chart review of 195 children ages 2–16 years who were eligible for TCD screening, Hussain et al. (2015) found that only 41 percent had achieved the standard of care. Bundy et al. (2016) conducted a retrospective cohort study of children aged 2–5 years and found that only 25 percent of the children had received one or more TCD screenings during the 14-month study period. The children who were most likely to receive a TCD (42 percent) were those with two or more hematologist visits.
A lack of knowledge about TCD guidelines has been identified as a barrier to TCD screening among some physicians. Reeves et al. (2015) conducted a survey of primary care, neurology, and hematology physicians to explore the factors that influence physician adherence to TCD screening guidelines for children with SCD. They found variation in the degree to which physicians felt well informed about screening guidelines. Of the 276 survey respondents, more primary care providers (PCPs) reported
not feeling well informed (66 percent) than neurologists (25 percent) and hematologists (6 percent). Bollinger et al. (2011) found that a lack of knowledge in caregivers may also be a barrier that prevents children with SCD from receiving annual TCD screening.
Penicillin Prophylaxis
Infants and young children with SCD are susceptible to bacteremia and meningitis due to Streptococcus pneumonia; penicillin prophylaxis decreases episodes of pneumococcal bacteremia. Kanter et al. (2017) evaluated commercial and Medicaid claims data for children with SCD and found that more than 80 percent of insured children aged 1–5 received a prescription for penicillin prophylaxis. However, other studies suggest that a prescription does not guarantee receipt of or optimal adherence to the medication, so the rates of adherence are much lower. For example, Beverung et al. (2014) conducted a retrospective cohort study using Wisconsin Medicaid claims data and found that only 18 percent of eligible children 5 years or older were adherent, as defined by a medication possession ratio1 of greater than 80 percent (18.18%, 95% confidence interval [CI] 11.31–25.05). Bundy et al. (2016) conducted a retrospective cohort study (using Maryland Medicaid claims data) of 266 children aged 2–5 years and found that 30 percent had consistently filled prophylactic antibiotic prescriptions. Having more than two hematologist visits or generalist visits that were not for well-child care was associated with more consistent antibiotic prophylaxis. Finally, Reeves et al. (2018) evaluated Medicaid claims data from six states for children aged 3 months to 5 years with SCA (2005–2012) and found that only 18 percent received at least 300 days of antibiotics. Furthermore, well-child visits were found to be associated with increased odds of receiving at least 300 days of antibiotics (odds ratio [OR] 1.08, 95% CI 1.02–1.13).
These findings indicate that there is a need to promote adherence to NQF-endorsed measures. Some strategies for promoting uptake of measures are discussed later in the chapter.
Recommendations Adapted from Other Stakeholder Groups
Several other associations for health professionals have developed evidence-based recommendations for managing care for adults and children that are relevant for the SCD population. Organizations whose recommendations appear in the 2014 NHLBI guidelines include USPSTF, the Advisory Committee on Immunization Practices, the World Health Organization, the
___________________
1 The medication possession ratio is calculated as follows: (sum of days supplied) / [(number of days from first encounter) – (number of days hospitalized)] (Beverung et al., 2014).
Centers for Disease Control and Prevention (CDC), and the American Pain Society. The consensus-adapted recommendations for SCD care pertain primarily to beneficial preventive services and include immunizations, screening for hepatitis C virus (HCV) and retinopathy, contraception use, reproductive counseling, and opioid use during pregnancy, as shown in Table 6-1. The NHLBI guidelines also included adapted consensus guidelines for chronic pain management. As with other recommended services, the National Academies SCD committee’s literature review identified very little data on adherence to recommended vaccinations and preventive services in the SCD population. The next section briefly reviews the available information.
Immunizations
Pneumococcal vaccination
Individuals with SCD are at extremely high risk of fatal pneumococcal infection and therefore are routinely immunized with pneumococcal conjugate vaccine (PCV) and pneumococcal polysaccharide vaccine (PPSV, sometimes referred to as PPV). Neunert et al. (2016) examined retrospective medical record and claims data to identify eligible children with SCA aged 24–36 months between January 1, 2004, and December 31, 2008; they found that of 125 children, 73.6 percent received PPV as recommended. Similarly, Beverung et al. (2014) found that 77 percent of eligible children received PCV7,2 and 50 percent of children less than 18 years of age received PPSV23 at least once in a 4-year period, whereas 38 percent of adults over 18 years of age had received PPSV23 at least once in a 4-year period. Ter-Minassian et al. (2019) reviewed the medical records of all persons with SCD seen at Kaiser Permanente Mid-Atlantic States (KPMAS) and the Adult Sickle Cell Disease Program at Johns Hopkins Hospital (JHH) from January 1, 2014, to December 31, 2015, to assess quality of care. Among 146 KPMAS patients, 85 percent had documentation of ever receiving PCV13 or PPSV23, but only 52 percent had received both. Among 308 JHH patients, 87 percent had documentation of receipt of either PCV13 or PPSV23, but only 30 percent had both.
Influenza vaccine
Influenza vaccination is recommended in the United States for everyone 6 months and older and may be even more important for people with SCD, for whom infections can be more serious and associated with additional SCD-related complications. There are several studies evaluating the rates of influenza vaccination for persons with SCD.
___________________
2 PCV7 was the first pneumococcal conjugate vaccine licensed by the U.S. Food and Drug Administration. The pneumococcal conjugate vaccine recommended currently is PCV13, which offers protection against an increased number of types of pneumococcal infection. For more information, see https://www.cdc.gov/vaccines/vpd/pneumo/public/index.html (accessed December 23, 2019).
Beverung et al. (2014) found that only 30.3 percent of children and 11.6 percent of adults were considered adherent3 over a 5-year period. The percent of non-adherence (zero vaccinations per influenza season) among children was 17.5 percent and 38.2 percent in adults (Beverung et al., 2014). Bundy et al. (2016) conducted a retrospective cohort study of children aged 2–5 years and found that 41 percent received at least one influenza immunization during the study period. Children with two or more hematologist visits were most likely to be immunized (62 percent versus 35 percent for children without a hematologist visit).
Ter-Minassian et al. (2019) reviewed the medical records of persons with SCD seen at KPMAS and JHH for adherence to seasonal influenza vaccination. They found documentation of influenza vaccination in 75 percent of KPMAS participants and 51 percent of JHH participants.
Meningococcal vaccination
Ter-Minassian et al. (2019) also reviewed the medical records of persons with SCD seen at KPMAS and JHH to assess meningococcal vaccination rates. Vaccination rates were low prior to 2016, with only 24 percent of KPMAS and 17 percent of JHH patients having had documentation of meningococcal vaccination.
The little evidence available on adherence to immunization guidelines suggests that there is room to promote adherence to recommended immunizations for individuals with SCD. At least one of the consensus-adapted recommendations—influenza vaccination—is NQF endorsed, meaning that it can be included in reporting and payment programs to promote accountability and adherence. The National Academies SCD committee was able to identify only limited evidence on adherence to the other strongly recommended services for SCD.
PATIENT-CENTERED DIMENSIONS OF HIGH-QUALITY CARE
The available guidelines for management of SCD focus primarily on managing the disease and associated complications and thus miss other important dimensions of high-quality care that pertain to the impact of the disease on an individual’s health-related quality of life (HRQOL) or their experience with the health care system, especially in care transitions for children from pediatric care into adult care.
___________________
3 “Adherence rate was calculated by dividing the number of influenza vaccines received by the number of eligible influenza seasons.… Individuals were considered adherent if their vaccination rate was greater or equal to 0.80 per influenza season.” Other categories of adherence were moderate (vaccination rate of 0.05–0.79 per season) and low (vaccination rate of 0.01–0.49 per season) (Beverung et al., 2014).
Health-Related Quality of Life
HRQOL refers to the effects of health, illness, and treatment on an individual’s quality of life (QOL) (Ferrans et al., 2005; Wilson and Cleary, 1995). Seminal work on HRQOL proposed measuring patient outcomes in five areas: biological function, symptoms, functional status, general health perceptions, and overall QOL (Ferrans et al., 2005; Wilson and Cleary, 1995).
Tools for measuring HRQOL have been adapted over time to assess the health effects of specific illnesses or conditions. In 2002 stakeholders participating in NHLBI gatherings that were focused on the treatment needs of individuals living with SCD identified the need to document patient-reported outcomes (PROs) (Treadwell et al., 2014). In response to this need, efforts began to develop SCD-specific tools as part of an NHLBI-sponsored Adult Sickle Cell Quality of Life Measurement Information System (ASCQ-Me) project (Treadwell et al., 2014). ASCQ-Me was developed as a set of self-administered items that assess the impact of SCD on adult functioning (Keller et al., 2017; Treadwell et al., 2014). The health domains assessed by the ASCQ-Me include emotional impact, pain impact, sleep impact, social functioning impact, stiffness impact, pain episodes, and an SCD medical history checklist (Keller et al., 2017; Treadwell et al., 2014).4 In a recent systematic literature review of PRO instruments used in SCD, the ASCQ-Me was considered to have strong content validity and internal reliability and good construct validity with respect to psychometric properties (Sarri et al., 2018). The tool was recently validated in a UK population (Cooper et al., 2019).
In addition, to assess the experiences of adults living with SCD in accessing care and the quality of care received, the ASCQ-Me Quality of Care (ASCQ-Me QOC) tool was developed. The tool has four domains focusing on access, provider communication, emergency department (ED) care, and ED pain treatment. Results based on the ASCQ-Me QOC indicate that adults with SCD report deficiencies in ED care related to race and their disease condition (Evensen et al., 2016).
It should be noted that in the design of the ASCQ-Me QOC a number of domain questions (access and provider communication) are very similar to or identical to those found in the Consumer Assessment of Healthcare Providers and Systems (CAHPS)5 surveys (Evensen et al., 2016). These surveys were designed to understand patient experiences with health care. There are a number of surveys; some surveys ask about a patient’s experience with
___________________
4 For more information on the tool, see www.HealthMeasures.net/ASCQ-Me (accessed January 6, 2020).
5 For more information on the CAHPS surveys, see https://www.ahrq.gov/cahps/aboutcahps/cahps-program/index.html (accessed January 6, 2020).
providers (clinician and groups), while others ask about experiences with care delivered in facilities (e.g., adult and child hospitals). Supplements to these surveys have been developed, such as one that provides questions specific to children with chronic conditions. While a CAHPS survey specific to SCD has not been developed (e.g., parallel to the CAHPS Cancer Care Survey6), items from the various surveys could be helpful in understanding the patient experience of care for individuals with SCD.
Efforts to develop a tool to assess the HRQOL of children living with SCD resulted in the development of the Pediatric QOL™—Sickle Cell Disease Module (PedsQL™ SCD Module). The module is a self- or parent proxy-administered survey that assesses HRQOL in children aged 2–18 years (Panepinto et al., 2013). The PedsQL™ SCD Module comprises nine scales: (1) pain and hurt, (2) pain impact, (3) pain management and control, (4) worry I, (5) worry II, (6) emotions, (7) treatment, (8) communication I, and (9) communication II (Panepinto et al., 2013). In the systematic review of PRO instruments, the PedsQL™ SCD Module was found to have strong internal reliability, but its other psychometric properties were unclear (Sarri et al., 2018). The SCD module complements other non-SCD-specific PRO instruments such as the PedsQL™ 4.0 Generic Core Scales (Varni et al., 2001) and the PedsQL™ Multidimensional Fatigue Scale (Panepinto et al., 2014), which assess other aspects of HRQOL in children.
A number of other tools specific to the SCD population have been developed with a focus on pain and self-efficacy. With respect to pain assessment, Zempsky et al. (2013) developed the Sickle Cell Disease Pain Burden Interview–Youth for use in youth and young adults aged 7–21 years. The brief tool, which consists of seven items, is administered by interview and inquires about days of pain and the impact of pain in the past month. Questions assess the impact of pain on mood, functional ability, and QOL. The tool has strong internal reliability and good content validity, construct validity, and test-retest reliability (Sarri et al., 2018).
According to Edwards et al. (2001), disease self-efficacy refers to an individual’s beliefs about his or her ability to achieve a desired health outcome. With respect to SCD and self-efficacy, Clay and Telfair (2007) and Edwards et al. (2000) used a nine-item scale focusing on the ability of adults and adolescents living with SCD to engage in daily functional activities. They found that high levels of self-efficacy were related to fewer physical, psychological, and total SCD symptoms (Clay and Telfair, 2007; Edwards et al., 2000). The instrument was assessed as having strong internal reliability in the recent systematic review (Sarri et al., 2018).
___________________
6 For information on the CAHPS Cancer Care Survey, see https://www.ahrq.gov/cahps/surveysguidance/cancer/index.html (accessed January 6, 2020).
The above-cited SCD-specific tools provide an opportunity to develop a better understanding of the burden that SCD imposes on the QOL of individuals living with SCD and their experiences with providers and health systems. HRQOL outcomes also point to areas of clinical practice that may need improvement, such as deficiencies in how patients are assessed and treated for pain in EDs as well as non-clinical interventions and strategies to better support individuals in developing self-efficacy, carrying out activities of daily living, and social functioning.
Patient Engagement in Care Through Shared Decision Making
Shared decision making (SDM) is defined as “a process of communication in which clinicians and patients work together to make informed health care decisions that align with what matters most to patients and their individual concerns, preferences, goals, and values” (NQF, n.d.b).7 NQF issued a national call to action to ensure that SDM in clinical practice is a standard of care for all patients (NQF, n.d.b). SDM in SCD care is critical but evolving.
Decisions about disease-modifying therapies for SCD often require patient adherence in order to be optimally effective (e.g., HU) or involve significant risks (e.g., bone marrow transplant, CRISPR gene editing). These decisions are complex and preference sensitive, and patients/families should be informed and involved in making them. This requires an effective partnership between patients and families and clinicians based on trust and clear communication.
Results from a study of physicians’ perceptions of patient decisional needs and physicians’ approaches to decision making showed that physicians tended to use two approaches to treatment-related decision making (Bakshi et al., 2017). One approach was characterized by the physician advocating for a specific treatment plan with the objective of convincing the patient to accept that plan. The second approach was characterized by the physician emphasizing the need to discuss all treatment approaches. Bakshi et al. (2017) found that which approach a physician used was influenced by a number of factors, including the characteristics of the patient, disease severity, the nature of the therapies, institutional frameworks, and other decision characteristics. Another study that directly observed dialogue about initiation of HU between patients and clinicians found that clinicians did not uniformly present the risks of HU and that patient concerns about
___________________
7 For more information on NQF shared decision making, see http://www.qualityforum.org/National_Quality_Partners_Shared_Decision_Making_Action_Team_.aspx (accessed January 6, 2020).
HU were not always raised and discussed (Lee et al., 2018). A recent qualitative study of patients’ perspectives on the process of deciding whether to take HU and physician communication found that providers who involved patients in SDM empowered those patients to start HU treatment (Jabour et al., 2019). However, patients who perceived that their providers were not attentive to their concerns reported having disengaged from HU treatment (Jabour et al., 2019).
Researchers are exploring ways to develop decision aids to assist in SDM. Crosby et al. (2015, 2019) developed an HU decision aid that increased HU knowledge and decreased decisional conflict. Other decision aids for transplant and non-transplant treatments exist for SCD (Sullivan et al., 2018). For example, Sickle Options is a website that provides information on treatment options, risks, and benefits and relates them to personal values.8 A Cochrane review of 105 studies on decision aids for a variety of clinical decisions found that when the aids are used, people improve their knowledge of options, feel better informed, and have a better understanding of what matters most to them (high-quality evidence), and they probably have more accurate expectations of the benefits and harms of options and participate more in decision making (moderate-quality evidence) (Stacey et al., 2017). Given the paucity of research in this area specific to SCD, there are ample research opportunities to examine SDM and the effectiveness of decision aids in improving patient knowledge and decision making in SCD care.
Pain Management
Pain management is a source of frustration for both patients and providers and an impetus for discrimination, confusion, and dissatisfaction; as noted by Anie et al. (2012, p. 1), “sickle cell pain assessment is a crucial and difficult task.” There are some best practices for pain control, but these have not been disseminated widely or implemented outside of institutions serving high numbers of patients with SCD. ASH is creating guidelines for treating SCD-related pain, but the recommendations are still in draft form (ASH, 2019).
There are a number of reasons that pain management for individuals living with SCD is difficult. For example, a number of researchers and health care professionals have noted the difficulty in treating the pain episodes of individuals living with SCD because of the subjectivity associated with the experience of pain (Okwerekwu and Skirvin, 2018). Many providers
___________________
8 For more information on Sickle Options, see http://sickleoptions.org/en_US (accessed January 6, 2020).
underestimate the severity of these pain episodes in spite of the fact that “pain management should be based on patient-reported severity,” according to the 2014 NHLBI sickle cell management guidelines (Yawn and John-Sowah, 2015).
There is also a great deal of variability in the intensity, duration, and frequency of pain episodes, as discussed in Chapter 4 (Geller and O’Connor, 2008). Given the challenges with defining, measuring, and treating pain episodes, it is not surprising that there is little in the quality improvement literature about effective pain management. Added to this is that clinician attitudes and race- and disease-based discrimination significantly affect the implementation of evidence-based guidelines (e.g., administering pain medication in the ED within 30 minutes) and clinical outcomes for individuals living with SCD.
Pain management for individuals living with SCD must be seen in the context of the current opioid crisis. Sinha et al. (2019), for example, collected qualitative data from 15 interview subjects about their perceptions of pain management from 2017 to 2018, after the 2016 CDC guidelines for chronic pain management were published. Interviewees reported that their opioid prescriptions were managed more closely by their health care providers and that their opioid use was monitored more closely than previously. This stigmatization of opioid use in managing SCD pain in light of the opioid crisis has negatively affected the care that these individuals are receiving (Sinha et al., 2019).
The National Academies has published several reports that can be used with this current report to create an effective system of care for people living with SCD and SCT, including Crossing the Quality Chasm (IOM, 2001), Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care (IOM, 2003), Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research (IOM, 2011b), and The Role of Nonpharmacological Approaches to Pain Management (NASEM, 2019), and Telfer and Kaya (2017) suggest an integrated approach to treating sickle cell pain in the absence of a standard protocol.
Transition from Pediatric to Adult Care
As described in Chapter 5, adolescents and young adults who transfer from pediatric to adult care encounter many challenges that affect their care. Individuals may lack the knowledge (e.g., SCD complication history) and skills (e.g., decision making, communication) needed to take an active role in their care and successfully navigate the adult health care system (Jordan et al., 2013). Compounding the transition to adult health care are the many other life transitions that young adults experience (e.g., emotional—establishing new friendships; social—living independently;
academic—graduating from high school/college/vocational program) (de Montalembert and Guitton, 2014). Despite the complexity of the transition to adult care, the field has identified several indicators of a successful transition for young adults living with SCD.
When I was in peds, I got amazing care. I was really encouraged to know … what worked for me. And then when I transition[ed] to adult, it was totally different. I started being accused of drug seeking just because I knew what dose of Dilaudid I needed in the ER.
—Teonna W. (Open Session Panelist)
Indicators of a Successful Transition
National organizations have developed position statements, guidance, and white papers defining a high-quality transition process for youth with chronic conditions and individuals with SCD. One available source of guidance, for example, is Got Transition™, discussed in Chapter 5. NHLBI provides guidance on adolescent health care and transitions in its report The Management of Sickle Cell Disease. That guidance, developed by a multidisciplinary group of experts, highlights transition readiness based on developmental age, transition discussions before transfer, the introduction of adult providers in the pediatric setting, and a focus on the coordination of care (NHLBI, 2002). In 2015, ASPHO and the Association of Pediatric Hematology/Oncology Nurses developed a policy statement for the transition of individuals living with SCD from pediatric to adult health, which identifies the following elements as essential: transition planning, multidisciplinary transition team, transfer process, and confirmation of transfer. In 2017 an expert panel from the Sickle Cell Adult Provider network used a modified Delphi process to identify nine quality indicators for transition, including five that were identified by Oyeku and Faro (2017) in their review of SCD quality improvement indicators for transition from pediatric to adult care: (1) communication between pediatric and adult providers, (2) time to first visit at the adult provider, (3) patient self-efficacy, (4) QOL, and (5) trust with the adult provider (Oyeku and Faro, 2017; Sobota et al., 2017).
The efforts mentioned above to provide guidance and indicators for the transition from adolescent to adult care share common elements, suggesting agreement across the field and a strong foundation for the development of SCD transition quality indicators. Despite this agreement, transition metrics are not routinely collected and tracked across programs, which has resulted in variations in the quality of care in the United States and other countries (Treadwell et al., 2018). The next section briefly reviews some
additional outcomes and process indicators identified through the work of various organizations that can be measured and tracked to improve the quality of transitions.
Insurance coverage and access to adult providers
Young adults with SCD may have limited access to health insurance (de Montalembert and Guitton, 2014). They may lose their health insurance coverage as they move from their parents’ plan to their own. Enrolling in and maintaining health insurance, particularly public health insurance, can be a challenging and time-consuming process, as discussed in Chapter 2. This lack of or delay in obtaining insurance coverage is a significant obstacle in accessing care and may seriously affect quality and continuity of care as young adults may avoid regular care and become more reliant on urgent or emergency care.
Transition education and readiness assessment
There is a need to assess the quality of transition planning and preparation, as individuals with SCD are generally unprepared for transition. The implementation of the transition core elements appears to be challenging. Okumura et al. (2008) found that 89 percent of providers in the survey supported a systematic transition process, but Telfair et al. (2004a,b) found that only 67 percent reported integrating transition core elements in their practice.
It has been shown that young adults may have a poor understanding of their medical history (Williams et al., 2015). DeBaun and Telfair (2012) identified educational milestones for the transition period to improve the transition process. Milestones include the adolescent having knowledge about his or her SCD phenotype, having the ability to articulate the most important components of his or her medical history, being able to manage pain according to a pain plan, and being aware of preventive measures for SCD complication, to name a few (DeBaun and Telfair, 2012). Additional research is needed to describe the interplay between cognition and disease knowledge in pediatric SCD (Hood et al., 2019). Young adults are able to identify specific health topics and barriers (e.g., mistrust, maintaining health insurance, employment difficulties, and managing stress) to be addressed in an individualized transition program (Bemrich-Stolz et al., 2015). Despite high interest in learning about transition (Williams et al., 2015), young adults may not attend transition-specific activities (Sivaguru et al., 2015). Lebensburger et al. (2012) supports starting the transition process early to better meet the needs of the patients from the beginning.
Routine assessment of transition readiness is variable across the United States; only certain SCD clinics and centers have integrated this into usual care. Several tools for measuring transition readiness exist, including the Transition Readiness Assessment Questionnaire and the Transition Intervention Program—Readiness for Transition assessment (Treadwell et al., 2016).
Communication/coordination of adult and pediatric providers
Inadequate communication between adult and pediatric providers may cause poor continuity of care (Treadwell et al., 2011), which sets the stage for recurrent hospitalizations, mistrust, and poorer health outcomes (Hunt and Sharma, 2010). Interventions targeting care coordination have led to improvements in the transfer of care and in overall quality. Hankins et al. (2012) piloted a transition program that helped adolescents identify an adult medical home. Data indicated that the program was feasible and showed promise for improving the transfer process. A patient-centered medical home may be another approach to improving coordination of transition care. In this model the pediatric provider conducts regular monitoring, documents complications and treatments, and communicates with an adult provider during and immediately after transition (Kelly et al., 2002).
Time to first visit with adult provider
Centers with well-established SCD transition programs use a registry to track eligibility, participation, and transfer completion. For example, the Duke University Medical Center conducted a retrospective chart review of individuals with SCD aged 18–23 years to identify care gaps (e.g., time to transfer), successful transition rates (completed appointment at an adult clinic 1 year post-transition), and associated factors (Hill et al., 2014). The data showed that only one-third of patients transitioned successfully and that a care gap exists for 18- to 20-year-olds (Hill et al., 2014). This study supports the importance of tracking individuals with SCD to improve the quality of care pre- and post-transition.
Quality of life
In the transition period, QOL indicators such as Pediatric QOL and Adult Sickle Cell QOL can help identify and prioritize health problems, facilitate discussions between the adolescent and providers, and identify areas needing greater medical, emotional, or social support.
Patient self-efficacy
Self-efficacy (the individual’s confidence in his or her ability to manage SCD on a daily basis) affects transition readiness (Treadwell et al., 2016). A 2015 review and 2016 multisite study found that youth with higher self-efficacy were more ready for transition (Molter and Abrahamson, 2015; Treadwell et al., 2016). Another 2015 study found that adults living with SCD reported improvements in autonomy and self-efficacy after transition (Bemrich-Stolz et al., 2015). Consequently, many transition programs target increasing patient self-management skills as a way of increasing self-efficacy. Moreover, 50 percent of SCD clinics responding to a survey about transition services and needs reported that they had a written list of desirable self-management skills (Sobota et al., 2011).
To better prepare young adults with SCD for the transfer to adult care, there is a need to help them develop increased confidence in their life skills.
In an effort to overcome neurocognitive or health literacy challenges that may interfere with attaining self-management skills (e.g., medication adherence), some programs have begun to integrate technology (e.g., videos, apps) into their care. Additional research is needed to understand essential components for increasing self-management skills and the most effective modalities (e.g., groups, in person, online, mobile apps) (Matthie et al., 2015).
PROMOTING UPTAKE OF RECOMMENDATIONS FOR SCD CARE
There are a number of beneficial and effective services recommended for SCD care; two of these recommended services are endorsed by NQF as quality measures. Several other specialty organizations (ASH, ASPHO, American College of Emergency Physicians (ACEP), AAP, American Academy of Family Physicians, American College of Physicians) and non-specialty organizations and governmental entities (CDC, USPSTF, NHLBI) have also developed consensus guidelines/recommendations to support quality metrics for the general population and SCD that should be applied consistently to the SCD population. In 2011 a panel of experts recommended 41 indicators for managing SCD care for children, including a subset of eight indicators9 that they believe will result in considerable improvement on QOL and health outcomes for children with SCD (Wang et al., 2011). The 41 indicators covered 18 topic areas, including some of the areas identified throughout this report as having opportunity for improvement, such as genetic counseling, transition to adult care, hematopoietic stem cell transplant, and comprehensive planning (Wang et al., 2011). These prior efforts can inform future attempts to expand indicators for high-quality SCD care. Internationally, standards of SCD care developed by the National Health Service and the Sickle Cell Society in the United Kingdom are valuable resources for SCD care management in the United States (Public Health England, 2019; Sickle Cell Society, 2018).
Clinical guidelines and metrics that are supported by evidence are, however, not widely applied because there is a lack of systematic efforts to foster the development of evidence-based learnings from iterative quality improvement in the SCD management. As a result, the quality and outcomes for SCD lag behind those of other rare, inheritable, and chronic diseases, such as cystic fibrosis (CF), hemophilia, and juvenile idiopathic arthritis. There is a need to implement and measure these underused guidelines and metrics.
The two NQF-endorsed measures can be immediately measured for public reporting and tied to payment. For 2019 the Centers for Medicare &
___________________
9 The eight indicators cover the following topics: timely assessment and treatment of pain and fever, comprehensive planning, penicillin prophylaxis, transfusion, and the transition to adult care (Wang et al., 2011).
Medicaid Services (CMS) announced that it would be maintaining the current core set of pediatric quality measures that states use for reporting the performance of Medicaid programs (CMS, 2018). Freed (2019) said CMS had “missed a historic opportunity to definitively address a national shame, the poor state of care provided to children with sickle cell disease,” because the Pediatric Measure Application Partnership (P-MAP) had recommended certain changes, such as to include the two NQF-endorsed measures in the core measure set. P-MAP is a multi-stakeholder panel that guides the U.S. Department of Health and Human Services (HHS) in selecting performance measurements for federal health programs.
QUALITY INDICATORS FOR SCT AND GENETIC COUNSELING FOR SCD AND SCT
Although SCT may be considered clinically benign, it affects nearly 300 million individuals worldwide (Grant et al., 2011). As stated in Chapter 3, all U.S. newborn screening (NBS) programs are state-based, so there are no requirements for them to report data to a national source. There are also no federal standards or consensus definitions for reporting or governing NBS data (Therrell et al., 2015). In 2009, CDC hosted a meeting on SCT and invited key stakeholders to engage in discussions to identify gaps in public health, health care delivery, epidemiologic research,10 and community-based outreach and to develop an agenda for the future for SCT. The meeting deliberations culminated in recommendations for community awareness and education, screening practices, ethical and legal issues, prevention of negative health outcomes, and epidemiological and clinical research (Grant et al., 2011). The National Academies SCD committee was unable to find any data on adherence to these guidelines other than efforts referenced in Chapters 3 and 5 and found no studies specifically examining quality.
Many individuals with SCT are identified through state NBS programs. Notification of NBS results varies widely by state. No best practices have yet been established for notifying families of results that improve a family’s ability to inform their child about his or her SCT and educate him or her about the implications. Programs that inform families of NBS results in person should be explored (Salm et al., 2012).
There are no guidelines or best practices for genetic counseling for individuals with SCT during adolescence or young adulthood, yet this is typically the time when family planning and decisions about contraception occur. When providing genetic counseling to women about their risk for having a child with SCD, it is important that the correct test is used
___________________
10 For more information on epidemiological research, see https://www.sciencedirect.com/topics/medicine-and-dentistry/cohort-effect (accessed August 3, 2020).
to assess hemoglobinopathy trait status. Hemoglobin electrophoresis and other hemoglobin quantitative separation methods are best (Naik and Haywood, 2015). Solubility tests, such as Sickledex and Sickle Cell Screen, are misleading. These tests do not distinguish SCD from SCT, nor do they detect the presence of other hemoglobin variants that may place a couple at risk for having a child with SCD, and false negatives and false positives are common. These tests should not be used for genetic counseling. Protocols for screening children and adults for SCD or SCT should use hemoglobin electrophoresis or other reliable hemoglobin separation methods, and organizations such as the National Collegiate Athletic Association and others should adopt this screening approach (Tubman and Field, 2015). Other tests such as a complete blood count may be necessary to detect beta thalassemia carriers, who also need genetic counseling. Quality indicators should be developed and tracked to ensure that appropriate tests are used, particularly for pregnant women (ACOG, 2017). In addition, individuals should be screened once in their lifetime, with confirmatory testing when appropriate. Repeated testing for a genetic state is not cost effective. Methods to store information about SCT and other hemoglobinopathy traits should be part of the health record, and NBS results should be stored and available to individuals when requested.
The National Library of Medicine developed a Newborn Screening Coding and Terminology Guide to standardize NBS test results (NLM, 2018). Another effort to standardize definition and data collection is the Hemoglobinopathy Uniform Medical Language Ontology (HUMLO) Project (NIH, 2008). The goal of HUMLO was to create an ontology for researchers and federal programs. Similarly, the PhenX Project sought to identify consensus measures for genetics that could be used in large-scale genomic studies (Hamilton et al., 2011).
There are no consensus guidelines on when or how to screen (e.g., methods, specific tests) individuals living with SCT for clinical complications (see Chapter 4). This is a notable gap in the literature, particularly because there is strong evidence for some clinical complications (e.g., CKD) (Naik et al., 2018b).
An NQF-like endorsement mechanism is needed for all indicators of high-quality SCD services across the life span, similar to what has been done for asthma, children from low-income families, and other groups. This will require developing measure sets, similar to the ones in Figure 6-2, that incorporate core metrics relevant for all adults and children in addition to core metrics specifically relevant for individuals with SCD and SCT.
Developing and accrediting comprehensive care centers will facilitate adherence to quality indicators and existing and future quality measures, because these centers’ accreditation and performance can be tied to providing recommended services for SCD.
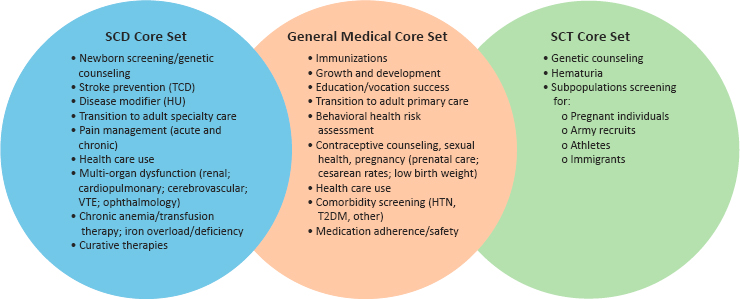
NOTE: HTN = hypertension; HU = hydroxyurea; T2DM = type 2 diabetes mellitus; TCD = transcranial Doppler screening; VTE = venous thromboembolism.
SUMMARY
The evidence supporting the guidelines for SCD care is highly variable, relying on expert consensus but highlighting research gaps. The evidence for a comprehensive, systematic approach to quality care delivery for SCD is lacking, and the science of quality in SCD care appears to lag behind other diseases in several areas:
- The majority of SCD quality indicators are supported with poor-quality evidence, often with consensus expert recommendation as the highest level of evidence available.
- There are multiple sets of quality indicators, but none are comprehensive or universally accepted or applied.
- There is a lack of data streams to measure adherence to quality indicators, requirements for centers to report their adherence to quality metrics, incentives to promote reporting, and public transparency of reports when available.
- There is neither funding nor a priority established for quality metrics in SCD, even in the face of poor performance of clinical systems. Furthermore, there has been no formal, systematic quality improvement of training programs.
There is no metric to support compassionate care in the health care system, particularly in light of the outright mistreatment of individuals with SCD presenting with pain—the most notable feature of SCD outside of death. Continued inaction over the past 18 years and a lack of required certifications has set back quality in SCD care, perpetuated mistrust of the health care system, and delayed the development of life-saving treatments
and standards of care. In the absence of robust evidence from randomized controlled trials, the quality of health care can be improved by leveraging learning collaboratives across health systems to foster the consistent application of available evidence-based guidelines and to obtain data on the effectiveness of consensus guidelines in improving outcomes. The disparities in applying available guideline-based care, the lack of care coordination within the delivery system and across the life span, and other structural deficiencies all contribute to poor outcomes in the SCD population. Defining and measuring high-quality care is a key step to improving outcomes.
THE AVAILABILITY OF A TRAINED AND PREPARED WORKFORCE
The committee believes that achieving the goal of delivering high-quality SCD health care to all will rest in part on the availability of an active, highly trained workforce. This section will describe the challenges associated with accessing such a workforce.
Multidisciplinary Teams
Treatment for people with SCD is best managed by a multidisciplinary team of professionals delivering comprehensive care. Traditionally, hematologists coordinate the management of care and liaise with other specialties, as they generally have the most familiarity with the multiple SCD complications and presentations (Grosse et al., 2009). In addition to medical providers, the members of the comprehensive care team should include behavioral health providers (e.g., psychologists, neuropsychologists, and psychiatrists), social workers, dietitians, physiotherapists, massage therapists, community health workers (CHWs), care coordinators, and school liaison officers. Furthermore, strong partnerships with community health services and voluntary agencies enhance the likelihood of the multidisciplinary team’s success (Okpala et al., 2002). Comprehensive care has several beneficial effects, such as improved QOL, reduced ED visits, and shorter hospital admissions (Okpala et al., 2002; Vichinsky, 1991).
The importance of social services and psychological support should not be underestimated. Services that may be needed include psychological treatment (e.g., mindfulness techniques and cognitive behavioral therapy), neuropsychological testing, general information and advice, disability assistance, transportation, financial allowances, practical help in the home or school, and assistance with care transition. However, access to these services is often institution dependent and further reduced in adult care. Such services often require specialized clinical experience and technical expertise and are harder to access outside of comprehensive care centers.
Although SCD is more common than CF in the United States, only a minority of individuals with SCD are seen at specialized centers (Smith et al., 2006). In stark contrast, there are more than 115 comprehensive, multidisciplinary CF care centers in the United States that are accredited and funded by the Cystic Fibrosis Foundation (CFF). CFF also maintains a national registry of CF patients who attend accredited centers (CFF, 2019). Using expert panels made up of physicians and scientists, it published consensus guidelines for best practices in CF care (e.g., guidelines for the management of infants with CF) (CFF et al., 2009; Conway et al., 2014). CFF also supports quality improvement activities to improve medical and care process at CF centers (Godfrey and Oliver, 2014; Grosse et al., 2009). A comprehensive network of care that can surveil health outcomes, develop best practices, and monitor the quality of care for SCD does not exist.
Strategies recommended in the IOM report Retooling for an Aging America: Building the Healthcare Workforce (IOM, 2008) to increase health workers for the geriatric population may be applicable to SCD. These strategies include the following:
- Congress should allocate money in the budget to monitor the workforce.
- Hospitals should encourage resident training across all settings.
- All licensure, renewal, and maintenance of certification activities should measure competence in areas relevant to the topic.
- States and the federal government should increase minimum training standards in this topic.
- Public and private payers should include financial incentives to increase the number of topic specialists in all professions.
- Payers should promote and reward models of care shown to be effective.
- Federal agencies should promote advances in technology to increase the efficiency and safety of caregiving.
- Public, private, and community organizations should provide funding and ensure that adequate training opportunities are available in the community for informal caregivers.
Table 6-1 presents the essential care team members (core team) required to provide high-quality care for individuals with SCD, the specific roles for care team members, the level of training/experience necessary, and potential barriers to an available and well-prepared workforce. The committee employed a variety of methods in determining team members. First, the committee identified specialists and clinical services historically involved in SCD care. Next, the committee identified providers and specialists essential for addressing the routine, acute, and subacute care needs of individuals
| SCD Team Member or Service | Guideline for Provider Training | Team Member Role | Workforce Barrier | |
|---|---|---|---|---|
| First Level Core Team Members | Hematologist/physician with expertise in SCD | Expertise in the evidence-based management of individuals with SCD | Provide core treatment for individuals with SCD; coordinate overall management and liaise with other specialties. | There is a workforce shortage because older hematologists are retiring and fewer new physicians are entering the field. |
| Emergency medicine | Knowledge of evidence-based management of individuals with SCD | Recognize the acute manifestations of SCD, provide early pain management and resuscitation, and quickly determine patient disposition. | Negative provider attitudes toward individuals with SCD are a barrier to the delivery of guideline-adherent pain care. | |
| PCP | Knowledge of evidence-based management of individuals with SCD | Recognize manifestations of SCD and implement clinical advances in treatment in clinical practice; assist with patient education, medication/transfusion monitoring, and disease complication screening follow-up. | There are not enough PCPs with the comprehensive knowledge and expertise to care for individuals with SCD. There is a reluctance to devote time and effort to support a rarely seen population. |
|
| Nurse practitioner/physician assistants | Knowledge of evidence-based management of individuals with SCD | Assist with patient education, medication/transfusion monitoring, and disease complication screening follow-up. | There is no formal training with the SCD population. | |
| Nurse care coordinator and nurses with SCD expertise | Knowledge of evidence-based management of individuals with SCD | Conduct initial assessment in clinics, assist with appointment scheduling, address patient-specific insurance issues, administer infusions in cooperation with the pharmacist, and perform exchange blood transfusions. | There is no formal training with the SCD population (negative provider biases and attitudes can influence care). Nurse care coordinators are only available at larger institutions. |
| Blood bank and transfusion medicine services | Knowledge of evidence-based management of individuals with SCD | Provide testing for extended RBC antigens and RBC alloimmunization. Exchange transfusion services. |
Individuals with SCD commonly become alloimmunized to RBC antigens—a contributing factor is that the donor base is predominantly Caucasian. | |
| Psychologist/psychiatrist | Comfortable discussing evidence-based management of individuals with SCD | Provide psychological support for mental issues, such as depression, anxiety, pain management, grief counseling, transition to adult care, and pharmacological treatment. | Knowledge gaps about SCD and negative provider attitudes contribute to poor communication. | |
| Neuropsychologist | Comfortable discussing evidence-based management of individuals with SCD | Assess for cognitive challenges, as individuals with SCD are at high risk of neuropsychological problems (i.e., cognitive, emotional, social, or behavioral). | Social, cultural, linguistic/communication, and financial issues interfere with making and keeping appointments for traditionally scheduled neuropsychological evaluations. | |
| Social worker | Comfortable discussing evidence-based management of individuals with SCD | Assess the specific needs to ensure that services are provided to meet the identified needs; address social determinants of health and barriers to care. | Available options for social services are often inadequate for the specific needs of individuals with SCD. |
continued
TABLE 6-1 Continued
| SCD Team Member or Service | Guideline for Provider Training | Team Member Role | Workforce Barrier | |
|---|---|---|---|---|
| Second Level Core Team Members | Pulmonologist | Knowledge of evidence-based management of individuals with SCD | Assess and provide treatment recommendations for pulmonary complications, which include ACS, asthma, lower airway obstruction, and airway hyper-responsiveness/bronchodilator response. | There is limited formal training with the SCD population. |
| Neurologist | Knowledge of evidence-based management of individuals with SCD | Assess and provide treatment recommendations for neurologic complications, which include stroke, silent cerebral infarct, moyamoya syndrome, posterior reversible encephalopathy syndrome, cerebral fat embolism, and cerebral venous sinus thrombosis. | There is limited formal training with the SCD population. | |
| Dentist/dental hygienist | Knowledge of evidence-based management of individuals with SCD | Assess and provide treatment for dental complications, which include caries or cavities, tooth hypomineralization, orofacial pain, neuropathy, facial swelling, pallor of oral mucosa, malocclusions, infections, pulpal necrosis, cortical erosions, medullary hyperplasia, and abnormal trabecular spacing. | There is limited formal training with the SCD population. | |
| Ophthalmologist | Knowledge of evidence-based management of individuals with SCD | Screen and provide treatment recommendations on vision complications. Every part of the eye can be affected by microvascular occlusions in SCD. The major cause of vision loss is proliferative sickle cell retinopathy. Screening before age 10 is recommended. | There is limited formal training with the SCD population and a lack of evidence regarding the optimal management of sickle retinopathy. |
| Nephrologist | Knowledge of evidence-based management of individuals with SCD | Assess and provide treatment for renal manifestations, which include hyperfiltration, hypertrophy, impaired urinary concentration, microalbuminuria, macroalbuminuria, hematuria, acute and chronic kidney injury, and end-stage renal disease. | SCD presents unique challenges in the marked heterogeneity of renal involvement. Current therapeutic approaches are largely adopted from other kidney diseases. | |
| Obstetrician/gynecologist | Knowledge of evidence-based management of individuals with SCD | Perform SCD screening and adult patient care through prenatal screening, folic acid supplementation, and pregnancy management. | Many OB/GYNs who care for individuals with SCD are not consistent with the College Practice Guidelines on the screening of certain target groups and folic acid supplementation. | |
| Pharmacist | Knowledge of evidence-based management of individuals with SCD | Provide pharmacologic management of SCD pain, which may involve the use of non-opioid medications, opioids, and adjuvants; HU monitoring; patient education and health maintenance counseling. | The focus of training is oncology rather than benign hematology. | |
| Genetic counselor | Knowledge of evidence-based management of individuals with SCD | Provide premarital and prenatal counseling and testing. | This role requires additional sensitivity when working with a majority black population. | |
| Transition coordinator | Knowledge of evidence-based management of individuals with SCD | Evaluate patients for transfer readiness; prepare for transition and undertake an ongoing discussion about the transition process when patients reach their early teens; coordinate transfer to adult providers. | This will often be a part-time position shared with nursing or social work and only available at larger institutions. | |
| Community health worker | Basic knowledge of evidence-based management of individuals with SCD | Assist with medication adherence, transitions of care, patient education, and education/vocation assistance. | There are no national standards for training, which is designed by the organizations that employ CHWs. |
continued
TABLE 6-1 Continued
| SCD Team Member or Service | Guideline for Provider Training | Team Member Role | Workforce Barrier | |
|---|---|---|---|---|
| Ancillary Team Members | Patient and family advisory group(s) | Basic knowledge of evidence-based management of individuals with SCD | Identify and drive projects important to individuals with SCD. Engage in decisions that affect quality of care. Promote patient- and family-centered care. Advise and advocate for children and their families. | This is only available at larger institutions. |
| Data quality analyst | Knowledge of evidence-based management of individuals with SCD | Develop and implement databases, identify trends, and interpret data from SCD clinical research. | There is no formal training with the SCD population. This position is only available at larger institutions conducting clinical research. | |
| Palliative care/integrative medicine | Basic knowledge of evidence-based management of individuals with SCD | Provide therapies, including non-pharmacological approaches to improve QOL. Integrative medicine may also include herbal remedies, diet and exercise interventions, acupuncture, acupressure, and aromatherapy. | Palliative care model has limited use with SCD population. There is no formal training with the SCD population. This position is only available at larger institutions. | |
| Physical therapist | Basic knowledge of evidence-based management of individuals with SCD | Provide therapies to reduce stress and pain and improve range of motion, which can include aerobic and breathing exercises, massages, or transcutaneous electrical nerve stimulation to disrupt pain sensors and relax muscles. | There is no formal training with the SCD population. |
| Education/vocational rehabilitation services | Basic knowledge of evidence-based management of individuals with SCD | Assist with obtaining special education services for youth (school intervention); provide counseling and assistance related to educational attainment, vocational counseling, job training opportunities, and academic and employment accommodations. | There is no formal training with the SCD population. This position is only available at larger institutions. | |
| Financial advisor/counselor | Basic knowledge of evidence-based management of individuals with SCD | Assist with completing insurance applications and paying medical bills. | There is no formal training with the SCD population. This position is only available at larger institutions. | |
| Pastoral care | Basic knowledge of evidence-based management of individuals with SCD | Provide emotional and spiritual support; explore spiritual questions and concerns. | There is no formal training with the SCD population. This position is only available at larger institutions. |
NOTE: ACS = acute chest syndrome; CHW = community health worker; HU = hydroxyurea; OB/GYNs = obstetricians and gynecologists; PCP = primary care provider; QOL = quality of life; RBC = red blood cell; SCD = sickle cell disease.
SOURCES: Informed by Agrawal et al., 2019; CFF, n.d.; Farooq and Testai, 2019; McIntosh, 2016; Mulimani et al., 2016; Okpala et al., 2002; Scott, 2016; Simon et al., 2016; Wilkie et al., 2010; and National Academies SCD committee judgment.
with SCD, as discussed in Chapter 4. Finally, the committee considered multidisciplinary care team models from other chronic, inheritable diseases, such as CF (CFF, n.d.).
The SCD care team members are classified as core or supplementary based on their level of involvement in managing the clinical and psychosocial complications of SCD (see Chapter 4) or in decreasing disparities in care (e.g., CHWs). The committee also considered disciplines currently providing care that are perceived as beneficial by clinicians or patients (e.g., financial counselors). Some members of the core team, such as hematologists and ED physicians, currently treat children and adults with SCD in the United States. However, the committee determined that PCPs, nurses, and other advanced practice practitioners, with the right level of preparation, could manage the care of patients with SCD as needed. Second-level core team members assist with managing SCD complications but are designated as level 2 because they may not serve the entire SCD population and instead focus on individuals with a specific complication or need (e.g., a pulmonologist may work with patients with pulmonary hypertension, but not all patients with SCD will have pulmonary complications). Ancillary team members may provide services on an ad hoc basis or may provide enabling services but influence the way that care is delivered or managed by addressing key social contributors (determinants) of health.
TRAINING THE NEXT GENERATION OF SCD CARE PROVIDERS
There have been some efforts by national organizations and health care systems to recruit and retain the SCD workforce of the future. For example, ASH offers a training series for hematologists and internists to guide them in starting an adult SCD clinic. However, additional programming and resources are needed to foster the development of a well-trained multidisciplinary SCD workforce. In addition, a lack of diversity among health care providers may contribute to disparities in health care service. Having providers who are similar to the patients in important dimensions, such as race, ethnicity, and language, promotes effective communication and can improve patient–provider relationships. Diversity among faculty, staff, and trainees enhances creative problem solving and fosters robust decision making, innovation, and productivity both in the health care setting and the workplace in general (Alsan et al., 2018; Cohen et al., 2002; Forbes Insights, 2011; Hunt et al., 2015). Thus, to identify the best and brightest, national organizations and the health care system should implement specific recruitment and retention strategies that address identified gaps in training and professional development; this is essential for achieving and maintaining high-quality care. The next section summarizes current efforts
in workforce development and identifies gaps and opportunities for training in hematology, emergency medicine, primary care, and non-hematologic disciplines.
Hematology11
The growing need for greater clinical and research training in benign hematology has long been recognized. The number of physicians entering the field is decreasing, and many are not engaged in research (Hoots et al., 2015; Soffer and Hoots, 2018). Minorities are also under-represented in hematology/medical oncology, with only about 6 percent of individuals identifying as black/African American and about 8 percent as Hispanic (Santhosh and Babik, 2020). Further exacerbating the problem, hematology/medical oncology trainees receive little early clinical exposure to nonmalignant hematology (Marshall et al., 2018).
Curriculums in combined programs of hematology/medical oncology have been hypothesized to contribute to a shortage of researchers in benign hematology (Naik et al., 2018a). Of the 134 fellowship programs in the United States, 132 are combined double-board programs for hematology/medical oncology; only two institutions currently offer hematology-only programs (Naik et al., 2018a). Further reducing opportunities for training, the Accreditation Council for Graduate Medical Education no longer mandates the number of months required for nonmalignant hematology instruction in fellowships. Traditionally, one-third of a fellow’s time used to be spent in nonmalignant hematology training (Wallace et al., 2015). These difficulties are mirrored outside of the United States. For example, in UK medical schools, rotations are offered in cardiology, surgery, and psychiatry, but there is no dedicated rotation in hematology. Instead, exposure to hematology is largely tested as part of a syllabus focused on pathology (Mandan et al., 2016).
Intellectual curiosity and stimulation are significant determining factors for medical students and internal medicine residents when choosing a focus (Marshall et al., 2018). Hematology covers a wide breadth of topics that relate to both malignant and nonmalignant diseases, which makes hematologists important in advancing the research and treatment of many diseases. Third-year fellows have described benign hematology as more “complicated” and “overwhelming” than malignant hematology or medical oncology (Bernstein et al., 2017). Currently, there is little early clinical exposure to benign hematology that highlights its intellectual excitement (Marshall et al., 2018). Less than 6 percent of graduates plan to practice primarily in nonmalignant hematology (Todd et al., 2004). Importantly,
___________________
11 See Appendix J for additional training models for hematologists.
even at programs where exposure to benign hematology is mandatory, such as the University of Pennsylvania, only around one out of seven or eight fellows is training in benign hematology (Loren, 2009).
Additionally, there are generally fewer available mentors than in other specialties. A lack of or ineffective mentorship has been observed for potential hematology fellows, which can contribute to difficulty in hiring and retaining junior faculty, disillusionment with academia, and reduced grant funding (Straus et al., 2013). Fellows in hematology/oncology training programs have reported that mentorship opportunities tend to happen “randomly,” rather than individuals being able to actively seek out a relationship. Additionally, there are generally more available mentors in oncology, which can play a decisive role in the career decision process (Wallace et al., 2016). Without effective mentorship, it is difficult to generate excitement and inspire the same enthusiasm, and there are fewer networking opportunities to develop other productive relationships (Sambunjak et al., 2010).
Medical students and fellows have a misconception that there are fewer jobs available in hematology than in oncology. Instead, projections indicate a shortage in both hematology and oncology. Overall the demand for hematology/oncology services is projected to grow 40 percent, whereas the supply may grow only 25 percent (Yang et al., 2014). In 2013, a survey of practice-based hematology was sent to 36 percent of ASH’s membership (5,081 physicians) (ASH, 2015). One-quarter of respondents indicated that they were considering retirement in the next 5 years (ASH, 2015; Tejaswini, 2015). Compounding the problem, many of these physicians specialize in benign hematology, and there is no pool of new fellows to replenish them. Patient loads are already so high that several cancer centers are searching for hematologists to join their faculty (Hoots et al., 2015; Sharma et al., 2019).
Starting salaries for hematology providers tend to be lower than other hematology and oncology specialties; pediatric hematologists/oncologists are among the 20 specialties with the lowest annual earnings (approximately $223,000) according to the Doximity 2019 physician compensation report, which includes responses from 90,000 U.S. physicians (Doximity, 2019). Compounding this comparatively low earning rate, according to a survey of 236 hematology–oncology fellows training at 56 participating cancer centers, 37 of them graduated with more than $100,000 in debt (Horn et al., 2011). Higher debt levels have been shown to increase stress and influence the likelihood of considering income potential when choosing a specialty (Rohlfing et al., 2014). For those seeking grant funding in hematological research, the outlook is no better. A National Institutes of Health (NIH) physician–scientist workgroup found that the number of investigators submitting and receiving NIH grants in the field declined by
about 70 percent over the past 13 years (Hoots et al., 2015). Because of these challenges, basic science advances in hematology await translation into clinical practice, and the care of nonmalignant hematology patients is suffering (Sharma et al., 2019; Soffer and Hoots, 2018).
The Division of Blood Diseases and Resources of NHLBI collaborated with ASH to convene a series of two overlapping working groups in 2012 and 2013 to identify potential strategies to address the declining clinical and research workforce in hematology (Hoots et al., 2015). They identified the following strategies:
- Early identification of future young physician–scholars as high school students and undergraduates and provision of summer internships and travel awards to conferences, particularly targeting students from diverse backgrounds. Introduction to hematologic practice and discovery should also start early through the Internet, television (e.g., documentaries), and science-based programming.
- During medical school, the introduction to hematology course could be moved to the first rather than second year of instruction to increase interest. Demonstrations for medical students in the classroom and hospital and clinic settings can illustrate the wide range of hematologic practice and the high impact of hematologic discoveries. If possible, it would also be beneficial for medical students to attend a comprehensive sickle cell clinic either on site or at an affiliated institution. Supplemental training could include rotation in the blood bank or attendance at a local blood center education program.
- Partnerships with the pharmaceutical industry may provide teaching and learning opportunities through translational and clinical trials.
- Grant funding mechanisms are needed for early career investigators. NHLBI and the National Institute of Diabetes and Digestive and Kidney Diseases have a short-term pilot program that funds hematologic laboratory programs for up to 6 months of mentored experiences for pre- and post-doctoral trainees. The ASH Alternative Training Pathway Grant also funds innovative training experiences that combine hematology with another field, such as pharmacology, or in combined pediatric/adult hematology.
Easing the financial burden from student loans and offering financial incentives for research on and treatment of the SCD population is another way to attract health care professionals to specialize in SCD care. The current federal loan repayment programs (LRPs) offered through NIH and the Health Resources and Services Administration (HRSA) can be further
enhanced to attract more providers to the SCD workforce. The National Health Service Corps (NHSC) LRP
offers primary care medical, dental, and mental and behavioral health care providers the opportunity to have their student loans repaid, while earning a competitive salary, in exchange for providing health care in urban, rural, or tribal communities with limited access to care. (HHS, 2019)
The current program offers $30,000–$50,000 to eligible applicants, depending on the health professional shortage area (HPSA) they work in, for 2 years of full-time service; applicants also have a half-time service option. Similarly, medical and dental students and other allied health providers seeking to enter primary care careers may be eligible for NHSC scholarships if they commit to providing service in a designated HPSA (HHS, 2020a). These opportunities need to be disseminated to students and health professionals who may be interested in providing care to the SCD population but who have concerns regarding student loan debt. Recently in response to the opioid epidemic, Congress directed HRSA to establish the NHSC Substance Use Disorder Workforce LRP to attract a workforce to provide the comprehensive care needed to effectively treat substance use disorders (HHS, 2020b). The committee believes that due to the dire need to develop and sustain the SCD workforce, establishing an LRP specifically for the SCD workforce could help to achieve this goal.
NIH also offers intramural and extramural LRPs of up to $50,000 annually for researchers in return for a commitment to conduct research relevant to the mission of NIH (NIH Division of Loan Repayment, n.d.). These programs are, however, limited (eight LRPs total) and competitive, which means that SCD clinicians and researchers are competing with those working with much larger disease populations. Considering the multitude of research gaps identified for SCD and SCT, NIH is encouraged to expand these LRPs, including designating SCD as a health disparity to incentivize early career SCD researchers.
Emergency Medicine
Individuals with SCD often require care in the ED, which may sometimes act as a safety net for those who have no other option or no access to a medical home (Yusuf et al., 2010). Emergency medicine care providers are often suspicious of SCD patients who have frequent ED visits (Shapiro et al., 1997) and may view patients with high ED use as addicts and label them “drug-seeking.” These inaccurate beliefs can lead to some individuals with SCD avoiding the ED for fear of being perceived as malingering or opioid dependent. Negative provider attitudes can affect redosing of pain
medication (Glassberg et al., 2013) and result in longer wait times at the ED (Haywood et al., 2013) (see Chapter 5).
Glassberg (2017), an emergency medicine physician at the Mount Sinai Comprehensive Sickle Cell Program at the Icahn School of Medicine at Mount Sinai, has developed four tenets for improving ED care for patients with SCD (see Figure 6-3): reducing negative provider attitudes, reducing the time to first dose of analgesic medication, improving ED pain care beyond the first dose, and improving ED patient safety.
Supplemental education about SCD disease-related processes for emergency providers has proved successful. Examples include a workshop led by six SCD experts to improve provider knowledge of common acute and chronic physiologic complications, pain pathophysiology, and best practices for analgesic management in the ED setting. The workshop assessed pre- and post-workshop knowledge and had its greatest impact on shifting
NOTE: ED = emergency department; FMECA = failure mode, effects, and criticality analysis; IV = intravenous; NHLBI = National Heart, Lung, and Blood Institute; PCA = patient-controlled analgesia; PDSA = plan-do-study-act; QI = quality improvement; SCD = sickle cell disease.
SOURCE: Glassberg, 2017. Republished with permission of the American Society of Hematology; permission conveyed through Copyright Clearance Center, Inc.
the perception of ED providers regarding addiction in the SCD population (Tanabe et al., 2013). Similarly, a short video intervention that included an adult hematologist discussing challenges about seeking treatment for pain with SCD patients reduced negative provider attitudes (Haywood et al., 2011). More recently, another short online video intervention was delivered by e-mail to emergency providers; it included both provider and patient perspectives regarding the challenges in managing in ED care for patients with SCD pain and resulted in improved provider attitudes about SCD (Singh et al., 2016).
Other models to improve care in the ED include a multidisciplinary SCD committee, such as the program at Jefferson University Hospitals (JUH). Aside from internists, who provide both inpatient and outpatient care, this multidisciplinary team includes an outpatient sickle cell nurse, a sickle cell social worker, a psychiatrist, specialized sickle cell hematologists, and nurse practitioners (JUH, n.d.). The program focuses on long-term care for patients with SCD. Patients can attend monthly self-support group meetings coordinated by a JUH social worker who teaches techniques to reduce stress and teaches the patients about their disease and its complications. The program also offers a 24/7 observation unit (JUH, n.d.).
Improving ED Management for Individuals with SCD is another innovative program that warrants replication on a larger scale. The program has advanced the treatment of SCD-related pain in the ED, leading to more rapid pain relief. The multidisciplinary team of physicians, nurses, social workers, and individuals living with SCD works to increase awareness and decrease negative provider attitudes about SCD patients. The team has also created a user-friendly treatment algorithm to help guide ED treatment (CCNC Sickle Cell Task Force, n.d.), a webpage devoted the care of patients with SCD in the ED that is an educational resource for providers (Duke University School of Medicine, 2012), and an SCD toolbox app that offers providers access to the latest evidence-based guidelines and to an SCD specialist (Improving Sickle Cell Care in North Carolina and Community Care of North Carolina, n.d.). In addition, the program holds an annual educational conference aimed at improving knowledge and open communication among providers, patients, and families.
The Emergency Department of Sickle Cell Care Coalition (EDSC3) is a national forum whose goal is to improve the emergency care of patients with SCD in the United States. ACEP created the coalition in collaboration with multiple public, private, and professional partners. EDSC3 primarily focuses on disseminating research findings to local and national stakeholders, supporting the education of ED providers and patients regarding the appropriate management of SCD-related pain and other complications, supporting advocacy and community outreach efforts, and supporting the development of appropriate metrics to improve the emergency care of
patients with SCD. EDSC3 is an excellent resource for training-related issues for emergency medicine providers working with SCD patients in the ED; it also includes links to podcasts with SCD experts and a blog supporting conversations regarding equity, fact sheets, training modules, pain consortium materials, and protocols (EDSC3, n.d.).
Primary Care
High-quality primary care treatment can limit preventable and costly interactions with the health care system. Currently, however, there are insufficient PCPs with the comprehensive knowledge and expertise to care for people with SCD (Mainous et al., 2015), and PCPs’ greatest challenge is adequate pain management (Utuama et al., 2015). Parents of children with SCD feel that current care from PCPs is inadequate (Raphael et al., 2013). Additionally, when children with SCD experience more health care barriers (i.e., experience of marginalization), it is more challenging to receive high-quality care that is accessible, comprehensive, and well coordinated (Jacob et al., 2016).
Project Extension for Community Healthcare Outcomes (ECHO) is a rapidly growing national network of telementoring hubs for training and knowledge sharing, with the goal of improving capacity and access to specialty care for rural and underserved populations. SCD ECHO hubs are regionally based and managed by a designated regional coordinating center. Their virtual learning teleECHO™ programs are open to community providers, PCPs, and SCD stakeholders in the United States and across the world. HRSA funds and manages this program, which is a relatively low-cost option that can mentor and train PCPs, offer feedback on difficult cases, and share knowledge and expertise through monthly didactics and clinical case presentations. The program is a proven way to increase knowledgeable PCPs (Shook et al., 2016). In 2020, HRSA, in collaboration with the Office of Minority Health at HHS, launched a 6-month pilot of a national SCD ECHO, the Sickle Cell Disease Training and Mentoring Program (HHS, n.d.). The program, which is targeted at PCPs, offers a telehealth series taught by a hematologist covering topics such as pain management, HU, and preventive services (HHS, n.d.). Regional hubs have been successful in increasing PCP knowledge about SCD (Shook et al., 2016); the committee recommends an evaluation of the program at the end of the pilot to determine its effectiveness and opportunities for scaling.
“The Department of Health and Human Services has given grants to nine facilities to develop programs to improve services for patients with SCD” (Butterfield, 2013). For the Johns Hopkins University School of Medicine, this grant led to the development of the Improving Health Outcomes and Medical Education for Sickle Cell Disease (iHOMES) Network.
The iHOMES team found that 40 percent of community physicians surveyed were uncomfortable with their ability to provide ambulatory care or manage comorbidities for SCD patients. More than half felt uncomfortable managing SCD pain and medications. The aims of the network are twofold (Butterfield, 2013). First, it has expanded physician training by having internal medicine residents at JHH provide primary care to SCD patients. Second, it offers community providers a number of support mechanisms (JHM, n.d.).
Non-Hematologic Medical Providers
The complex nature of SCD creates care needs that require medical and allied health expertise from providers other than hematologists, emergency medicine providers, and PCPs (Okpala et al., 2002). Other members of the SCD medical treatment team include, but are not limited to, nurses and nurse practitioners, physician assistants, blood bank and transfusion specialists, neurologists, radiologists, dentists, ophthalmologists, obstetrician/gynecologists, genetic counselors, integrative medicine specialists, nephrologists, orthopedists, pulmonologists, psychologists, and social workers (see Table 6-1). Training related to SCD-related complications varies widely, with most providers receiving little to no specific training (Hanik et al., 2014). There are currently no uniform practices or standardized protocols, which can make it challenging for providers to administer care. Provider racial or cultural biases also affect clinical care and treatment decisions (Bulgin et al., 2018) (see Chapter 2).
Knowledge gaps about SCD and the best standard of care contribute to poor communication between patients and SCD specialists (Azonobi et al., 2014) and to poor patient satisfaction and outcomes. The most difficult hurdle is that most providers serve very few SCD patients (Mainous et al., 2015). Because interactions are so infrequent, providers may be somewhat reluctant to devote time and effort to support a rarely seen population. Furthermore, the unpredictable and often persistent nature of the pain and other complications associated with SCD pose a difficult challenge for providers (Utuama et al., 2015). There are no known objective measures that indicate the presence or severity of this pain, which introduces a great deal of clinical uncertainty into the patient–provider relationship (Matthias et al., 2010) (see Chapter 5).
It may be challenging for some providers to improve patient outcomes because they do not have a sufficient volume of cases. In the quality literature, higher volumes generally correlate with improved outcomes. The exact reasons are not known, but they are suspected to follow from the opportunity to develop increased provider skill, multidisciplinary teams, and access to specialized resources. Other fields have overcome this barrier using
distance learning and consultation and telementoring (e.g., TeleECHO for HCV).
In addition to the challenge of prevailing health care professionals’ workforce shortages in the United States, which is not specific to the SCD population, most of the care team members identified in Table 6-1 have limited or no formal training in the SCD population and so would need to be prepared to effectively deliver care. Yet, there are systemic challenges (e.g., reimbursement models) that also contribute to the workforce challenges. There are a few models that offer strategies for preparing disease-specific specialists, including the HIV specialist model, conferred through the American Academy of HIV Medicine (AAHIVM).
The American Academy of HIV Medicine’s HIV Specialist Model
AAHIVM convened a group of individuals who had been offering HIV care in order to establish standards for delivering high-quality care. Several types of health care providers, including physicians, nurse practitioners, physician assistants, and pharmacists, can receive a professional credential (e.g., HIV specialist, expert, or pharmacist) if they actively care for more than 25 HIV patients (AAHIVM, 2020). The goals of the credentialing process are to (1) improve the quality of HIV care, (2) broaden patient access to quality care, and (3) expand the number of HIV-specialized medical care providers (AAHIVM, 2020; Sweet, n.d.). AAHIVM offers a core curriculum and training program that provides basic knowledge on HIV for new providers and information about clinical advances and complex topics for experienced providers (AAHIVM, 2020; Sweet, n.d.). This credentialing model could be applied to health professionals who are interested in treating the SCD population.
CONCLUSIONS AND RECOMMENDATIONS
Despite national efforts to improve quality in SCD care, that quality remains poor for most individuals in the United States. National quality metrics have not been developed, resulting in significant variations. Some SCD programs track and perform well on consensus-based quality indicators (e.g., transition readiness assessment); however, data suggest that the overall performance on the few evidence-based quality metrics identified (e.g., penicillin, stroke prevention) is poor. This overall inadequate state of care hinders the advancement of comprehensive, multidisciplinary, effective, data-driven programs for individuals living with SCD and SCT. Pain management is also an area where there is a lack of quality measures.
Routine preventive care is necessary for both pediatric and adult patients with SCD, and some routine indicators, such as developmental
assessment in pediatrics, need to be performed more frequently due to SCD-related complications. Quality indicators for SCD acute, subacute, and chronic care have been identified in the literature but are not endorsed or tracked by any national entity. For example, NHLBI guidelines recommend offering HU at 9 months of age, yet uptake ranges from 14 to 28 percent for clinical samples. Similarly, TCD is known to detect the risk of stroke, but patients receive it about 23–45 percent of the time. Barriers to the implementation of evidence-based or consensus guidelines need to be quantified, defined, measured, and tracked to improve the system of care.
Adolescents and young adults who transfer from pediatric to adult care encounter many challenges that affect their quality of care. Several national organizations and quality initiatives have developed transition metrics. Data indicate that although these individuals may be participating in programs to prepare them for transition, a high number do not successfully transfer to an adult provider. These youth also tend to use the ED more than other individuals with SCD, which is another indicator that the quality of care during this time may be inadequate. New SCD transition programs and initiatives using quality improvement methods to improve the transition process hold promise for decreasing disparities in care experienced during this time.
To develop a body of endorsed relevant quality metrics that can be adequately disseminated and tracked to measure their impact, there needs to be an NQF-like entity that formulates, tracks, and transparently reports on the adherence of systems to these metrics for SCD, similar to what has been done for asthma, children from low-income families, and other groups. There should also be a parallel organization that monitors performance and provides guidance on mitigating poor performance, with an appropriate accountability that could be tied to payment/reimbursement.
Access to providers and the availability of a trained, prepared workforce are key issues that have not been adequately addressed in SCD care. The lack of adult hematologists and other providers further contributes to variations in care. Some individuals receive treatment from multidisciplinary teams with experience managing SCD, while others have providers with limited awareness of SCD and its many complications. There are limited educational and training opportunities for providers and trainees who wish to specialize in SCD care. Along with the lack of diversity in the workforce, there is a need for competency training in the sociocultural factors in the patient experience of care in order to improve relationships between patients and health care teams. This chapter identifies the team members needed to provide high-quality care for all SCD patients, the team members needed to care for individuals who experience specific complications (e.g., pulmonary hypertension, stroke), and training gaps and opportunities. Individuals with SCD may also experience sociodemographic challenges that
can affect their receipt of high-quality care. Ancillary team members, such as financial counselors, can help to mitigate sociodemographic barriers or increase resilience factors (e.g., pastoral care), thereby improving access to and delivery of care. Provider certification models (e.g., HIV expert) and hub-and-spoke models, such as Project ECHO, can help to build and maintain a well-prepared SCD workforce.
SCT is even less well understood, and little is known about the relevant quality of care. There is also a need to develop national reporting guidelines. Instituting the best practices of SCT notification and genetic counseling would vastly improve the care provided and allow for the examination of clinical outcomes.
Conclusion 6-1: Many of the approaches to SCD management lack a good evidence base, particularly to support current treatment strategies. The lack of evidence-based guidelines results in inconsistent treatment approaches across centers and providers, reduced access to care, and poor outcomes. Strategies are needed to enhance the evidence base to support the treatment of SCD.
Conclusion 6-2: The evidence for a comprehensive, systematic approach to the delivery of high-quality care for SCD is lacking, and the science of quality in SCD care lags behind other diseases in several areas: SCD clinical guidelines are supported with poor quality evidence; there are multiple sets of guidelines, but none are universally accepted or cover the services needed for comprehensive care; adherence to clinical guidelines and quality indicators is not measured, reported, required, incentivized, or included in public transparency reports; and there is evidence of a lack of funding and priority for the development of quality metrics in SCD. There is ample evidence from other disease areas that can be leveraged for SCD care while research is generated to fill in the gaps in the evidence base.
Conclusion 6-3: There is a need for provider- and patient-focused strategies to increase the uptake of guidelines and metrics for SCD that have a strong evidence base.
Conclusion 6-4: Optimal treatment for people with SCD is best managed by a multidisciplinary team of professionals delivering comprehensive high-quality care. Traditionally, hematologists have coordinated the management of care and liaised with other specialties. Comprehensive, high-quality care delivery for SCD requires the availability and preparedness of a multidisciplinary
team of providers—behavioral health providers (e.g., psychologists, neuropsychologists, and psychiatrists), social workers, dietitians, physiotherapists, massage therapists, community health workers, care coordinators, and school liaison officers. Substantial barriers, such as an aging workforce, workforce shortages, negative provider attitudes, and a lack of training, have contributed to a limited workforce for SCD care.
___________________
12 This text was revised since the prepublication release of this report to correct American Association of Family Practitioners to American Academy of Family Physicians.
REFERENCES
AAHIVM (American Academy of HIV Medicine). 2020. American Academy of HIV Medicine credentialing handbook. Washington, DC: American Academy of HIV Medicine.
AAP (American Academy of Pediatrics). 2002. Health supervision for children with sickle cell disease. Pediatrics 109(3):526.
ACOG (American College of Obstetricians and Gynecologists). 2017. Carrier screening for genetic conditions. Committee opinion no. 691. Obstetrics and Gynecology 129:e41–e55.
Agrawal, S., W. B. Burton, D. Manwani, D. Rastogi, and A. De. 2019. A physicians survey assessing management of pulmonary airway involvement in sickle cell disease. Pediatric Pulmonology 54(7):993–1001.
AHRQ (Agency for Healthcare Research and Quality). n.d. AHRQ quality indicators. https://www.qualityindicators.ahrq.gov (accessed April 9, 2020).
Alsan, M., O. Garrick, and G. C. Graziani. 2018. Does diversity matter for health? Experimental evidence from Oakland. NBER Working Paper No. 24787. https://www.nber.org/papers/w24787.pdf (accessed July 1, 2020).
Anie, K. A., H. Grocott, L. White, M. Dzingina, G. Rogers, and G. Cho. 2012. Patient self-assessment of hospital pain, mood and health-related quality of life in adults with sickle cell disease. BMJ Open 2(4):1–6.
ASH (American Society of Hematology). 2015. Adapting to changes in practice-based hematology. ASH Clinical News, May 15. https://www.ashclinicalnews.org/spotlight/adapting-to-changes-in-practice-based-hematology (accessed April 12, 2020).
ASH. 2019. ASH draft recommendations on sickle cell disease-related pain. https://www.hematology.org/Clinicians/Guidelines-Quality/Documents/9558.aspx (accessed February 28, 2020).
ASH. n.d. ASH clinical practice guidelines on sickle cell disease. https://www.hematology.org/SCDguidelines (accessed February 28, 2020).
Azonobi, I. C., B. L. Anderson, V. R. Byams, A. M. Grant, and J. Schulkin. 2014. Obstetrician-gynecologists’ knowledge of sickle cell disease screening and management. BMC Pregnancy and Childbirth 14(1):356.
Bakshi, N., C. B. Sinha, D. Ross, K. Khemani, G. Loewenstein, and L. Krishnamurti. 2017. Proponent or collaborative: Physician perspectives and approaches to disease modifying therapies in sickle cell disease. PLOS ONE 12(7):e0178413.
Balshem, H., M. Helfand, H. J. Schunemann, A. D. Oxman, R. Kunz, J. Brozek, G. E. Vist, Y. Falck-Ytter, J. Meerpohl, S. Norris, and G. H. Guyatt. 2011. Grade guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 64(4):401–406.
Bemrich-Stolz, C. J., J. H. Halanych, T. H. Howard, L. M. Hilliard, and J. D. Lebensburger. 2015. Exploring adult care experiences and barriers to transition in adult patients with sickle cell disease. International Journal of Hematology and Therapy 1(1):PMC4756764.
Bernstein, E., N. A. Podoltsev, and A. Lee. 2017. Teaching hematology to fellows: A qualitative study. Blood 130(Suppl 1):5641.
Beverung, L. M., D. Brousseau, R. G. Hoffmann, K. Yan, and J. A. Panepinto. 2014. Ambulatory quality indicators to prevent infection in sickle cell disease. American Journal of Hematology 89(3):256–260.
Bollinger, L. M., K. G. Nire, M. M. Rhodes, D. J. Chisolm, and S. H. O’Brien. 2011. Caregivers’ perspectives on barriers to transcranial Doppler screening in children with sickle-cell disease. Pediatric Blood & Cancer 56(1):99–102.
Bulgin, D., P. Tanabe, and C. Jenerette. 2018. Stigma of sickle cell disease: A systematic review. Issues in Mental Health Nursing 39(8):675–686.
Bundy, D. G., J. Muschelli, G. D. Clemens, J. J. Strouse, R. E. Thompson, J. F. Casella, and M. R. Miller. 2016. Preventive care delivery to young children with sickle cell disease. Journal of Pediatric Hematology and Oncology 38(4):294–300.
Butterfield, S. 2013. “Sic”ing primary care physicians on sickle cell disease. https://acpinternist.org/archives/2013/04/sickle-cell.htm (accessed December 15, 2019).
CCNC (Community Care of North Carolina) Sickle Cell Task Force. n.d. Emergency department vaso-occlusive crisis management: Adults and children. https://sickleemergency.duke.edu/sites/default/files/ccnc-voc-protocol.pdf (accessed December 15, 2019).
CFF (Cystic Fibrosis Foundation). 2019. 2018 patient registry: Annual data report. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-RegistryAnnual-Data-Report.pdf (accessed March 6, 2019).
CFF. n.d. Your CF care team. https://www.cff.org/Care/Your-CF-Care-Team (accessed December 15, 2019).
CFF, D. Borowitz, K. A. Robinson, M. Rosenfeld, S. D. Davis, K. A. Sabadosa, S. L. Spear, S. H. Michel, R. B. Parad, T. B. White, P. M. Farrell, B. C. Marshall, and F. J. Accurso. 2009. Cystic Fibrosis Foundation evidence-based guidelines for management of infants with cystic fibrosis. Journal of Pediatrics 155(6 Suppl):S73–S93.
Chou, S. T., M. Alsawas, R. M. Fasano, J. J. Field, J. E. Hendrickson, J. Howard, M. Kameka, J. L. Kwiatkowski, F. Pirenne, P. A. Shi, S. R. Stowell, S. L. Thein, C. M. Westhoff, T. E. Wong, and E. A. Akl. 2020. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Advances 4(2):327–355.
Clay, O. J., and J. Telfair. 2007. Evaluation of a disease-specific self-efficacy instrument in adolescents with sickle cell disease and its relationship to adjustment. Child Neuropsychology 13(2):188–203.
CMS (Centers for Medicare & Medicaid Services). 2018. 2019 updates to the child and adult core health care quality measurement sets. https://www.medicaid.gov/sites/default/files/federal-policy-guidance/downloads/cib112018.pdf (accessed March 5, 2020).
CMS. 2020. Quality measures. https://www.cms.gov/Medicare/Quality-Initiatives-PatientAssessment-Instruments/QualityMeasures/index.html (accessed February 27, 2020).
Cohen, J. J., B. A. Gabriel, and C. Terrell. 2002. The case for diversity in the health care workforce. Health Affairs 21(5):90–102.
Conway, S., I. M. Balfour-Lynn, K. De Rijcke, P. Drevinek, J. Foweraker, T. Havermans, H. Heijerman, L. Lannefors, A. Lindblad, M. Macek, S. Madge, M. Moran, L. Morrison, A. Morton, J. Noordhoek, D. Sands, A. Vertommen, and D. Peckham. 2014. European Cystic Fibrosis Society standards of care: Framework for the cystic fibrosis centre. Journal of Cystic Fibrosis 13:S3–S22.
Cooper, O., H. McBain, S. Tangayi, P. Telfer, D. Tsitsikas, A. Yardumian, and K. Mulligan. 2019. Psychometric analysis of the Adult Sickle Cell Quality of Life Measurement Information System (ACSQ-ME) in a UK population. Health and Quality of Life Outcomes 17(1):74.
Crosby, L. E., L. M. Shook, R. E. Ware, and W. B. Brinkman. 2015. Shared decision making for hydroxyurea treatment initiation in children with sickle cell anemia. Pediatric Blood & Cancer 62(2):184–185.
Crosby, L. E., A. Walton, L. M. Shook, R. E. Ware, M. Treadwell, K. L. Saving, M. Britto, J. Peugh, E. McTate, S. Oyeku, C. Nwankwo, and W. B. Brinkman. 2019. Development of a hydroxyurea decision aid for parents of children with sickle cell anemia. Journal of Pediatrics Hematology and Oncology 41(1):56–63.
de Montalembert, M., and C. Guitton. 2014. Transition from paediatric to adult care for patients with sickle cell disease. British Journal of Haematology 164(5):630–635.
DeBaun, M. R. 2014. The challenge of creating an evidence-based guideline for sickle cell disease. JAMA 312(10):1004–1005.
DeBaun, M. R., and J. Telfair. 2012. Transition and sickle cell disease. Pediatrics 130(5):926–935.
Doximity. 2019. 2019 physician compensation report: Third annual study. https://s3.amazonaws.com/s3.doximity.com/press/doximity_third_annual_physician_compensation_report_round4.pdf (accessed March 6, 2020).
Duke University School of Medicine. 2012. Emergency department sickle cell disease: Crisis management and beyond. https://sickleemergency.duke.edu (accessed December 15, 2019).
Eckrich, M. J., W. C. Wang, E. Yang, P. G. Arbogast, A. Morrow, J. A. Dudley, W. A. Ray, and W. O. Cooper. 2013. Adherence to transcranial Doppler screening guidelines among children with sickle cell disease. Pediatric Blood & Cancer 60(2):270–274.
EDSC3 (Emergency Department Sickle Cell Care Coalition). n.d. About the EDSC3. https://www.acep.org/by-medical-focus/hematology/sickle-cell (accessed November 19, 2019).
Edwards, R., J. Telfair, H. Cecil, and J. Lenoci. 2000. Reliability and validity of a self-efficacy instrument specific to sickle cell disease. Behaviour Research and Therapy 38(9):951–963.
Edwards, R., J. Telfair, H. Cecil, and J. Lenoci. 2001. Self-efficacy as a predictor of adult adjustment to sickle cell disease: One-year outcomes. Psychosomatic Medicine 63(5):850–858.
Evensen, C. T., M. J. Treadwell, S. Keller, R. Levine, K. L. Hassell, E. M. Werner, and W. R. Smith. 2016. Quality of care in sickle cell disease: Cross-sectional study and development of a measure for adults reporting on ambulatory and emergency department care. Medicine (Baltimore) 95(35):e4528.
Farooq, S., and F. D. Testai. 2019. Neurologic complications of sickle cell disease. Current Neurology and Neuroscience Reports 19(4):17.
Ferrans, C. E., J. J. Zerwic, J. E. Wilbur, and J. L. Larson. 2005. Conceptual model of health-related quality of life. Journal of Nursing Scholarship 37(4):336–342.
Forbes Insights. 2011. Global diversity and inclusion: Fostering innovation through a diverse workforce. New York: Forbes.
Freed, G. L. 2019. A missed opportunity to address a national shame: The case of sickle cell disease in the United States. JAMA Pediatric 173(8):715–716.
Geller, A. K., and M. K. O’Connor. 2008. The sickle cell crisis: A dilemma in pain relief. Mayo Clinic Proceedings 83(3):320–323.
Glassberg, J. A. 2017. Improving emergency department-based care of sickle cell pain. Hematology: American Society of Hematology—Education Program 2017(1):412–417.
Glassberg, J. A., P. Tanabe, A. Chow, K. Harper, C. Haywood Jr., M. R. DeBaun, and L. D. Richardson. 2013. Emergency provider analgesic practices and attitudes toward patients with sickle cell disease. Annals of Emergency Medicine 62(4):293–302.
Godfrey, M. M., and B. J. Oliver. 2014. Accelerating the rate of improvement in cystic fibrosis care: Contributions and insights of the Learning and Leadership Collaborative. BMJ Quality & Safety 23(Suppl 1):i23–i32.
Grant, A. M., C. S. Parker, L. B. Jordan, M. M. Hulihan, M. S. Creary, M. A. Lloyd-Puryear, J. C. Goldsmith, and H. K. Atrash. 2011. Public health implications of sickle cell trait: A report of the CDC meeting. American Journal of Preventive Medicine 41(6 Suppl 4):S435–S439.
Grosse, S. D., M. S. Schechter, R. Kulkarni, M. A. Lloyd-Puryear, B. Strickland, and E. Trevathan. 2009. Models of comprehensive multidisciplinary care for individuals in the United States with genetic disorders. Pediatrics 123(1):407–412.
Hamilton, C. M., L. C. Strader, J. G. Pratt, D. Maiese, T. Hendershot, R. K. Kwok, J. A. Hammond, W. Huggins, D. Jackman, H. Pan, D. S. Nettles, T. H. Beaty, L. A. Farrer, P. Kraft, M. L. Marazita, J. M. Ordovas, C. N. Pato, M. R. Spitz, D. Wagener, M. Williams, H. A. Junkins, W. R. Harlan, E. M. Ramos, and J. Haines. 2011. The PhenX toolkit: Get the most from your measures. American Journal of Epidemiology 174(3):253–260.
Hanik, M., K. M. Sackett, and L. L. Hartman. 2014. An educational module to improve healthcare staffs’ attitudes toward sickle cell disease patients. Journal for Nurses in Professional Development 30(5):231–236.
Hankins, J. S., R. Osarogiagbon, P. Adams-Graves, L. McHugh, V. Steele, M. P. Smeltzer, and S. M. Anderson. 2012. A transition pilot program for adolescents with sickle cell disease. Journal of Pediatric Health Care 26(6):e45–e49.
Haywood, C., S. Lanzkron, M. T. Hughes, R. Brown, M. Massa, N. Ratanawongsa, and M. C. Beach. 2011. A video-intervention to improve clinician attitudes toward patients with sickle cell disease: The results of a randomized experiment. Journal of General Internal Medicine 26(5):518–523.
Haywood, Jr., C., P. Tanabe, R. Naik, M. C. Beach, and S. Lanzkron. 2013. The impact of race and disease on sickle cell patient wait times in the emergency department. American Journal of Emergency Medicine 31(4):651–656.
HHS (U.S. Department of Health and Human Services). 2019. National Health Service Corps loan repayment program. https://nhsc.hrsa.gov/sites/default/files/NHSC/loan-repayment/nhsc-lrp-fact-sheet.pdf (accessed February 3, 2020).
HHS. 2020a. National Health Service Corps scholarship program: School year 2020–2021 application & program guidance. https://nhsc.hrsa.gov/sites/default/files/NHSC/scholarships/nhsc-scholarship-application-program-guidance.pdf (accessed March 6, 2020).
HHS. 2020b. National Health Service Corps substance use disorder loan repayment program. https://nhsc.hrsa.gov/sites/default/files/NHSC/loan-repayment/sud-lrp-application-guidance.pdf (accessed March 6, 2020).
HHS. n.d. STAMP: Sickle Cell Disease Training and Mentoring Program. https://www.minorityhealth.hhs.gov/sicklecell/#stamp (accessed February 3, 2020).
Hill, S., G. Maslow, L. Walker, S. Johnson, and N. Shah. 2014. Growing pains—Determination of transfer and transition from pediatrics to adult outpatient clinics for patients with sickle cell disease (SCD). Blood 124(21):3518.
Hood, A. M., A. A. King, M. E. Fields, A. L. Ford, K. P. Guilliams, M. L. Hulbert, J. M. Lee, and D. A. White. 2019. Higher executive abilities following a blood transfusion in children and young adults with sickle cell disease. Pediatric Blood & Cancer 66(10):e27899.
Hoots, W. K., J. L. Abkowitz, B. S. Coller, and D. M. DiMichele. 2015. Planning for the future workforce in hematology research. Blood 125(18):2745–2752.
Horn, L., E. Koehler, J. Gilbert, and D. H. Johnson. 2011. Factors associated with the career choices of hematology and medical oncology fellows trained at academic institutions in the United States. Journal of Clinical Oncology 29(29):3932–3938.
Hunt, S. E., and N. Sharma. 2010. Transition from pediatric to adult care for patients with sickle cell disease. JAMA 304(4):408–409; author reply 409.
Hunt, V., D. Layton, and S. Prince. 2015. Why diversity matters. https://www.mckinsey.com/business-functions/organization/our-insights/why-diversity-matters (accessed October 28, 2019).
Hussain, S., F. Nichols, L. Bowman, H. Xu, and C. Neunert. 2015. Implementation of transcranial Doppler ultrasonography screening and primary stroke prevention in urban and rural sickle cell disease populations. Pediatric Blood & Cancer 62(2):219–223.
Improving Sickle Cell Care in North Carolina and Community Care of North Carolina. n.d. SCD toolbox: A sickle cell disease toolbox to improve care. https://www.acep.org/globalassets/uploads/uploaded-files/acep/by-medical-focus/hematology---edsc3/scd-toolbox-flier_v2-final.pdf (accessed January 9, 2020).
IOM (Institute of Medicine). 2001. Crossing the quality chasm: A new health system for the 21st century. Washington, DC: National Academy Press.
IOM. 2003. Unequal treatment: Confronting racial and ethnic disparities in health care. Washington, DC: The National Academies Press.
IOM. 2008. Retooling for an aging America: Building the health care workforce. Washington, DC: The National Academies Press.
IOM. 2011a. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press.
IOM. 2011b. Relieving pain in America: A blueprint for transforming prevention, care, education, and research. Washington, DC: The National Academies Press.
Jabour, S. M., S. Beachy, S. Coburn, S. Lanzkron, and M. N. Eakin. 2019. The role of patient–physician communication on the use of hydroxyurea in adult patients with sickle cell disease. Journal of Racial and Ethnic Health Disparities 6(6):1233–1243.
Jacob, E., C. Childress, and J. D. Nathanson. 2016. Barriers to care and quality of primary care services in children with sickle cell disease. Journal of Advanced Nursing 72(6):1417–1429.
JHM (Johns Hopkins Medicine). n.d. Johns Hopkins sickle cell disease ECHO. https://www.hopkinsmedicine.org/Medicine/sickle/providers/index.html?_ga=2.65953070.1018478254.1561329500-1735271705.1561329500 (accessed January 9, October 28, 2020).
Jordan, L., P. Swerdlow, and T. D. Coates. 2013. Systematic review of transition from adolescent to adult care in patients with sickle cell disease. Journal of Pediatric Hematology and Oncology 35(3):165–169.
JUH (Jefferson University Hospitals). n.d. Comprehensive sickle cell program. https://hospitals.jefferson.edu/departments-and-services/comprehensive-sickle-cell-program.html (accessed 2019).
Kanter, J., C. Dampier, I. Agodoa, R. Howard, S. Wade, V. Noxon, and S. K. Ballas. 2017. Quality of care in United States children with sickle cell anemia. Blood 130(Suppl 1):2098.
Keller, S., M. Yang, C. Evensen, T. Cowans, and the American Institutes for Research. 2017. Adult sickle cell quality of life measurement information system: ASCQ-Me user’s manual. http://www.healthmeasures.net/images/ASQMe/ASCQ-Me_Scoring_Manual.pdf (accessed February 27, 2020).
Kelly, A. M., B. Kratz, M. Bielski, and P. M. Rinehart. 2002. Implementing transitions for youth with complex chronic conditions using the medical home model. Pediatrics 110(Suppl 3):1322–1327.
Lebensburger, J. D., C. J. Bemrich-Stolz, and T. H. Howard. 2012. Barriers in transition from pediatrics to adult medicine in sickle cell anemia. Journal of Blood Medicine 3:105–112.
Lee, J., W. Callon, J. C. Haywood, S. M. Lanzkron, P. Gulbrandsen, and M. C. Beach. 2018. What does shared decision making look like in natural settings? A mixed methods study of patient–provider conversations. Communication & Medicine 14(3):217–228.
Liem, R. I., S. Lanzkron, T. D. Coates, L. DeCastro, A. A. Desai, K. I. Ataga, R. T. Cohen, J. Haynes, Jr., I. Osunkwo, J. D. Lebensburger, J. P. Lash, T. Wun, M. Verhovsek, E. Ontala, R. Blaylark, F. Alahdab, A. Katabi, and R. A. Mustafa. 2019. American Society of Hematology 2019 guidelines for sickle cell disease: Cardiopulmonary and kidney disease. Blood Advances 3(23):3867–3897.
Loren, A. 2009. Hematology and Goliath: Ensuring the future of benign hematology in a world of combined hematology–oncology fellowships. Hematologist 16(4):2.
Mainous, A. G., R. J. Tanner, C. A. Harle, R. Baker, N. K. Shokar, and M. M. Hulihan. 2015. Attitudes toward management of sickle cell disease and its complications: A national survey of academic family physicians. Anemia 2015:853835.
Mandan, J., H. S. Sidhu, and A. Mahmood. 2016. Should a clinical rotation in hematology be mandatory for undergraduate medical students? Advances in Medical Education and Practice 7:519–521.
Marshall, A. L., S. Jenkins, J. Mikhael, and S. D. Gitlin. 2018. Determinants of hematology–oncology trainees’ postfellowship career pathways with a focus on nonmalignant hematology. Blood Advances 2(4):361–369.
Matthias, M. S., A. L. Parpart, K. A. Nyland, M. A. Huffman, D. L. Stubbs, C. Sargent, and M. J. Bair. 2010. The patient–provider relationship in chronic pain care: Providers’ perspectives. Pain Medicine 11(11):1688–1697.
Matthie, N., C. Jenerette, and S. McMillan. 2015. Role of self-care in sickle cell disease. Pain Management Nursing 16(3):257–266.
McIntosh, I. D. 2016. Health human resources guidelines: Minimum staffing standards and role descriptions for Canadian cystic fibrosis healthcare teams. Canadian Respiratory Journal 2016:PMC4904508.
Molter, B. L., and K. Abrahamson. 2015. Self-efficacy, transition, and patient outcomes in the sickle cell disease population. Pain Management Nursing 16(3):418–424.
Mulimani, P., S. K. Ballas, A. B. L. Abas, and L. Karanth. 2016. Treatment of dental complications in sickle cell disease. Cochrane Database of Systematic Reviews 2016(12):CD011633.
Naik, R. P., and C. Haywood, Jr. 2015. Sickle cell trait diagnosis: Clinical and social implications. Hematology: American Society of Hematology—Education Program 2015:160–167.
Naik, R. P., K. Marrone, S. Merrill, R. Donehower, and R. Brodsky. 2018a. Single-board hematology fellowship track: A 10-year institutional experience. Blood 131(4):462–464.
Naik, R. P., K. Smith-Whitley, K. L. Hassell, N. I. Umeh, M. de Montalembert, P. Sahota, C. Haywood, Jr., J. Jenkins, M. A. Lloyd-Puryear, C. H. Joiner, V. L. Bonham, and G. J. Kato. 2018b. Clinical outcomes associated with sickle cell trait: A systematic review. Annals of Internal Medicine 169(9):619–627.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2018. Crossing the global quality chasm: Improving health care worldwide. Washington, DC: The National Academies Press.
NASEM. 2019. The role of nonpharmacological approaches to pain management: Proceedings of a workshop. Washington, DC: The National Academies Press.
Neunert, C. E., R. W. Gibson, P. A. Lane, P. Verma-Bhatnagar, V. Barry, M. Zhou, and A. Snyder. 2016. Determining adherence to quality indicators in sickle cell anemia using multiple data sources. American Journal of Preventive Medicine 51(1 Suppl 1):S24–S30.
NHLBI (National Heart, Lung, and Blood Institute). 2002. The management of sickle cell disease. https://www.nhlbi.nih.gov/files/docs/guidelines/sc_mngt.pdf (accessed September 22, 2019).
NIH (National Institutes of Health). 2008. Research portfolio online reporting tools (RePORT). https://report.nih.gov/crs/View.aspx?Id=713 (accessed September 22, 2019).
NIH Division of Loan Repayment. n.d. Eligibility and programs. https://www.lrp.nih.gov/eligibility-programs (accessed February 3, 2020).
NLM (National Library of Medicine). 2018. About the newborn screening coding and terminology guide. https://newbornscreeningcodes.nlm.nih.gov/nb/sc/about (accessed March 5, 2020).
NQF (National Quality Forum). 2018. Strengthening the core set of healthcare quality measures for children enrolled in Medicaid and CHIP, 2018: Final report. Washington, DC: U.S. Department of Health and Human Services.
NQF. n.d.a. What NQF endorsement means. https://www.qualityforum.org/Measuring_Performance/ABCs/What_NQF_Endorsement_Means.aspx (accessed February 27, 2020).
NQF. n.d.b. National Quality Partners™ shared decision making action team. http://www.qualityforum.org/National_Quality_Partners_Shared_Decision_Making_Action_Team_.aspx (accessed February 28, 2020).
NQF. n.d.c. NQF’s history. http://www.qualityforum.org/about_nqf/history (accessed March 12, 2020).
Okpala, I., V. Thomas, N. Westerdale, T. Jegede, K. Raj, S. Daley, H. Costello-Binger, J. Mullen, C. Rochester-Peart, S. Helps, E. Tulloch, M. Akpala, M. Dick, S. Bewley, M. Davies, and I. Abbs. 2002. The comprehensiveness care of sickle cell disease. European Journal of Haematology 68(3):157–162.
Okumura, M. J., M. Heisler, M. M. Davis, M. D. Cabana, S. Demonner, and E. A. Kerr. 2008. Comfort of general internists and general pediatricians in providing care for young adults with chronic illnesses of childhood. Journal of General Internal Medicine 23(10):1621–1627.
Okwerekwu, I., and J. A. Skirvin. 2018. Sickle cell disease pain management. U.S. Pharmacists 43(3):12–18.
Oyeku, S. O., and E. Z. Faro. 2017. Rigorous and practical quality indicators in sickle cell disease care. Hematology 2017(1):418–422.
Panepinto, J. A., S. Torres, C. B. Bendo, T. L. McCavit, B. Dinu, S. Sherman-Bien, C. Bemrich-Stolz, and J. W. Varni. 2013. PedsQL™ sickle cell disease module: Feasibility, reliability, and validity. Pediatric Blood & Cancer 60(8):1338–1344.
Panepinto, J. A., S. Torres, C. B. Bendo, T. L. McCavit, B. Dinu, S. Sherman-Bien, C. Bemrich-Stolz, and J. W. Varni. 2014. PedsQL™ multidimensional fatigue scale in sickle cell disease: Feasibility, reliability, and validity. Pediatric Blood & Cancer 61(1):171–177.
Prabhakar, H., C. Haywood, Jr., and R. Molokie. 2010. Sickle cell disease in the United States: Looking back and forward at 100 years of progress in management and survival. American Journal of Hematology 85(5):346–353.
Public Health England. 2019. Sickle cell and thalassaemia screening programme: Standards. https://www.gov.uk/government/publications/sickle-cell-and-thalassaemia-screening-programme-standards (accessed March 5, 2020).
Raphael, J. L., P. B. Shetty, H. Liu, D. H. Mahoney, and B. U. Mueller. 2008. A critical assessment of transcranial Doppler screening rates in a large pediatric sickle cell center: Opportunities to improve healthcare quality. Pediatric Blood & Cancer 51(5):647–651.
Raphael, J. L., T. L. Rattler, M. A. Kowalkowski, B. U. Mueller, T. P. Giordano, and D. C. Brousseau. 2013. Associations of care in a medical home and health care utilization among children with sickle cell disease. Journal of the National Medical Association 105(2):157–165.
Reeves, S. L., H. J. Fullerton, K. J. Dombkowski, M. L. Boulton, T. M. Braun, and L. D. Lisabeth. 2015. Physician attitude, awareness, and knowledge regarding guidelines for transcranial Doppler screening in sickle cell disease. Clinical Pediatrics 54(4):336–345.
Reeves, S. L., B. Madden, G. L. Freed, and K. J. Dombkowski. 2016. Transcranial Doppler screening among children and adolescents with sickle cell anemia. JAMA Pediatrics 170(6):550–556.
Reeves, S. L., A. C. Tribble, B. Madden, G. L. Freed, and K. J. Dombkowski. 2018. Antibiotic prophylaxis for children with sickle cell anemia. Pediatrics 141(3):e20172182.
Rohlfing, J., R. Navarro, O. Z. Maniya, B. D. Hughes, and D. K. Rogalsky. 2014. Medical student debt and major life choices other than specialty. Medical Education Online 19(1):25603.
Salm, N., E. Yetter, and A. Tluczek. 2012. Informing parents about positive newborn screen results: Parents’ recommendations. Journal of Child Health Care 16(4):367–381.
Sambunjak, D., S. E. Straus, and A. Marusic. 2010. A systematic review of qualitative research on the meaning and characteristics of mentoring in academic medicine. Journal of General Internal Medicine 25(1):72–78.
Santhosh, L., and J. M. Babik. 2020. Trends in racial and ethnic diversity in internal medicine subspecialty fellowships from 2006 to 2018. JAMA Network Open 3(2):e1920482.
Sarri, G., M. Bhor, S. Abogunrin, C. Farmer, S. Nandal, R. Halloway, and D. A. Revicki. 2018. Systematic literature review and assessment of patient-reported outcome instruments in sickle cell disease. Health and Quality of Life Outcomes 16(1):99.
Scott, A. W. 2016. Ophthalmic manifestations of sickle cell disease. Southern Medical Journal 109(9):542–548.
Shapiro, B. S., L. J. Benjamin, R. Payne, and G. Heidrich. 1997. Sickle cell-related pain: Perceptions of medical practitioners. Journal of Pain and Symptom Management 14(3):168–174.
Sharma, D., N. Wallace, E. A. Levinsohn, A. L. Marshall, K. Kayoumi, J. Madero, M. Homer, R. Reynolds, J. Hafler, N. A. Podoltsev, and A. I. Lee. 2019. Trends and factors affecting the U.S. adult hematology workforce: A mixed methods study. Blood Advances 3(22):3550–3561.
Shook, L. M., C. B. Farrell, K. A. Kalinyak, S. C. Nelson, B. M. Hardesty, A. G. Rampersad, K. L. Saving, W. J. Whitten-Shurney, J. A. Panepinto, R. E. Ware, and L. Crosby. 2016. Translating sickle cell guidelines into practice for primary care providers with Project ECHO. Medical Education Online 21(1):33616.
Sickle Cell Society. 2018. Standards for the clinical care of adults with sickle cell disease in the UK. https://www.sicklecellsociety.org/wp-content/uploads/2018/05/Standards-for-the-Clinical-Care-of-Adults-with-Sickle-Cell-in-the-UK-2018.pdf (accessed March 5, 2020).
Simon, E., B. Long, and A. Koyfman. 2016. Emergency medicine management of sickle cell disease complications: An evidence-based update. Journal of Emergency Medicine 51(4):370–381.
Singh, A. P., C. Haywood, Jr., M. C. Beach, M. Guidera, S. Lanzkron, D. Valenzuela-Araujo, R. E. Rothman, and A. F. Dugas. 2016. Improving emergency providers’ attitudes toward sickle cell patients in pain. Journal of Pain and Symptom Management 51(3):628–632.
Sinha, C. B., N. Bakshi, D. Ross, and L. Krishnamurti. 2019. Management of chronic pain in adults living with sickle cell disease in the era of the opioid epidemic: A qualitative study. JAMA Network Open 2(5):e194410.
Sivaguru, H., S. M. Kemp, R. Crowley, G. Hann, D. A. Yardumian, M. Roberts-Harewood, and O. Wilkey. 2015. An evaluation of the transition to adult care for young patients with sickle cell disease. Archives of Disease in Childhood 100(Suppl 3):A168–A169.
Smith, L. A., S. O. Oyeku, C. Homer, and B. Zuckerman. 2006. Sickle cell disease: A question of equity and quality. Pediatrics 117(5):1763–1770.
Sobota, A., E. J. Neufeld, P. Sprinz, and M. M. Heeney. 2011. Transition from pediatric to adult care for sickle cell disease: Results of a survey of pediatric providers. American Journal of Hematology and Oncology 86(6):512–515.
Sobota, A. E., N. Shah, and J. W. Mack. 2017. Development of quality indicators for transition from pediatric to adult care in sickle cell disease: A modified Delphi survey of adult providers. Pediatric Blood Cancer 64(6).
Soffer, E., and W. K. Hoots. 2018. Challenges facing the benign hematology physician–scientist workforce: Identifying issues of recruitment and retention. Blood Advances 2(3):308.
Stacey, D., F. Légaré, K. Lewis, M. J. Barry, C. L. Bennett, K. B. Eden, M. Holmes-Rovner, H. Llewellyn-Thomas, A. Lyddiatt, R. Thomson, and L. Trevena. 2017. Decision aids for people facing health treatment or screening decisions. Cochrane Database of Systematic Reviews 2017(4):CD001431.
Straus, S. E., M. O. Johnson, C. Marquez, and M. D. Feldman. 2013. Characteristics of successful and failed mentoring relationships: A qualitative study across two academic health centers. Academic Medicine 88(1):82–89.
Sullivan, K. M., M. Horwitz, I. Osunkwo, N. Shah, and J. J. Strouse. 2018. Shared decision-making in hematopoietic stem cell transplantation for sickle cell disease. Biology of Blood and Marrow Transplantation 24(5):883–884.
Sweet, D. n.d. Value of credentialing. https://aahivm.org/value-of-credentialing (accessed March 6, 2020).
Tanabe, P., A. Stevenson, L. DeCastro, L. Drawhorn, S. Lanzkron, R. E. Molokie, and N. Artz. 2013. Evaluation of a train-the-trainer workshop on sickle cell disease for ED providers. Journal of Emergency Nursing 39(6):539–546.
Tejaswini, D. 2015. Post-fellowship career decision-making in a changing hematology practice landscape. Hematologist 12(3):14–15.
Telfair, J., L. R. Alexander, P. S. Loosier, P. L. Alleman-Velez, and J. Simmons. 2004a. Providers’ perspectives and beliefs regarding transition to adult care for adolescents with sickle cell disease. Journal of Health Care for the Poor and Underserved 15(3):443–461.
Telfair, J., J. E. Ehiri, P. S. Loosier, and M. L. Baskin. 2004b. Transition to adult care for adolescents with sickle cell disease: Results of a national survey. International Journal of Adolescent Medicine and Health 16(1):47–64.
Telfer, P., and B. Kaya. 2017. Optimizing the care model for an uncomplicated acute pain episode in sickle cell disease. Hematology: American Society of Hematology—Education Program 2017(1):525–533.
Ter-Minassian, M., S. Lanzkron, A. Derus, E. Brown, and M. A. Horberg. 2019. Quality metrics and health care utilization for adult patients with sickle cell disease. Journal of the National Medical Association 111(1):54–61.
Therrell, Jr., B. L., M. A. Lloyd-Puryear, J. R. Eckman, and M. Y. Mann. 2015. Newborn screening for sickle cell diseases in the United States: A review of data spanning 2 decades. Seminars in Perinatology 39(3):238–251.
Thompson, A. 2014. Is the NIH expert report on sickle cell disease a “clinical practice guideline we can trust?” The Hematologist 11(6):14.
Todd, R. F., S. D. Gitlin, and L. J. Burns. 2004. Subspeciality training in hematology and oncology, 2003: Results of a survey of training program directors conducted by the American Society of Hematology. Blood 103(12):4383–4388.
Treadwell, M., J. Telfair, R. W. Gibson, S. Johnson, and I. Osunkwo. 2011. Transition from pediatric to adult care in sickle cell disease: Establishing evidence-based practice and directions for research. American Journal of Hematology 86(1):116–120.
Treadwell, M. J., K. Hassell, R. Levine, and S. Keller. 2014. Adult Sickle Cell Quality-of-Life Measurement Information System (ASCQ-ME): Conceptual model based on review of the literature and formative research. Clinical Journal of Pain 30(10):902–914.
Treadwell, M., S. Johnson, I. Sisler, M. Bitsko, G. Gildengorin, R. Medina, F. Barreda, K. Major, J. Telfair, and W. R. Smith. 2016. Self-efficacy and readiness for transition from pediatric to adult care in sickle cell disease. International Journal of Adolescent Medicine and Health 28(4):381–388.
Treadwell, M., M. DeBaun, and J. Tirnauer. 2018. Transition from pediatric to adult care: Sickle cell disease. https://www.uptodate.com/contents/transition-from-pediatric-to-adult-care-sickle-cell-disease (accessed April 12, 2020).
Tubman, V. N., and J. J. Field. 2015. Sickle solubility test to screen for sickle cell trait: What’s the harm? Hematology: American Society of Hematology—Education Program 2015:433–435.
Utuama, O., K. Carter-Wicker, J. Herbert, R. Gibson, A. Kutlar, A. Logan, M. M. Kabongo, and T. Adamkiewicz. 2015. Sickle cell disease: Challenges and comfort in providing care by family physicians: Blood 126(23):5570.
Varni, J. W., M. Seid, and P. S. Kurtin. 2001. PedsQL™ 4.0: Reliability and validity of the Pediatric Quality of Life Inventory™ version 4.0 generic core scales in healthy and patient populations. Medical Care 39(8):800–812.
Vichinsky, E. P. 1991. Comprehensive care in sickle cell disease: Its impact on morbidity and mortality. Seminars in Hematology 28(3):220–226.
Wallace, P. J., N. T. Connell, and J. L. Abkowitz. 2015. The role of hematologists in a changing United States health care system. Blood 125(16):2467–2470.
Wallace, N. H., J. P. Hafler, M. E. Hurwitz, N. A. Podoltsev, J. Lacy, and A. I. Lee. 2016. Factors influencing hematology career choice in hematology and oncology fellows at a major academic institution. Blood 128(22):3538.
Wang, C. J., P. L. Kavanagh, A. A. Little, J. B. Holliman, and P. G. Sprinz. 2011. Quality-of-care indicators for children with sickle cell disease. Pediatrics 128(3):484–493.
Wilkie, D. J., B. Johnson, A. K. Mack, R. Labotka, and R. E. Molokie. 2010. Sickle cell disease: An opportunity for palliative care across the life span. Nursing Clinics 45(3):375–397.
Williams, C. P., C. H. Smith, K. Osborn, C. J. Bemrich-Stolz, L. M. Hilliard, T. H. Howard, and J. D. Lebensburger. 2015. Patient-centered approach to designing sickle cell transition education. Journal of Pediatric Hematology and Oncology 37(1):43–47.
Wilson, I. B., and P. D. Cleary. 1995. Linking clinical variables with health-related quality of life: A conceptual model of patient outcomes. JAMA 273(1):59–65.
Yang, W., J. H. Williams, P. F. Hogan, S. S. Bruinooge, G. I. Rodriguez, M. P. Kosty, D. F. Bajorin, A. Hanley, A. Muchow, N. McMillan, and M. Goldstein. 2014. Projected supply of and demand for oncologists and radiation oncologists through 2025: An aging, better-insured population will result in shortage. Journal of Oncology Practice 10(1):39–45.
Yawn, B. P., and J. John-Sowah. 2015. Management of sickle cell disease: Recommendations from the 2014 expert panel report. American Family Physician 92(12):1069–1076.
Yawn, B. P., G. R. Buchanan, A. N. Afenyi-Annan, S. K. Ballas, K. L. Hassell, A. H. James, L. Jordan, S. M. Lanzkron, R. Lottenberg, W. J. Savage, P. J. Tanabe, R. E. Ware, M. H. Murad, J. C. Goldsmith, E. Ortiz, R. Fulwood, A. Horton, and J. John-Sowah. 2014. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 312(10):1033–1048.
Yusuf, H. R., H. K. Atrash, S. D. Grosse, C. S. Parker, and A. M. Grant. 2010. Emergency department visits made by patients with sickle cell disease: A descriptive study, 1999–2007. American Journal of Preventive Medicine 38(4 Suppl):S536–S541.
Zempsky, W. T., E. A. O’Hara, J. P. Santanelli, T. M. Palermo, T. New, K. Smith-Whitley, and J. F. Casella. 2013. Validation of the Sickle Cell Disease Pain Burden Interview–Youth. Journal of Pain 14(9):975–982.






















































