
Data privacy is a hot issue right now, according to Kristen Rosati, a partner at Coppersmith Brockelman, PLC. In the research community, there is broad support for open data and increased data accessibility, she said, with the acknowledgment that individual privacy must be protected. Concerns about the extent to which data sharing poses privacy risks were fueled recently by a study from scientists at the Imperial College London and the Université Catholique de Louvain in Belgium, and covered widely in the popular press, including The New York Times, demonstrating that nearly all Americans could be correctly re-identified in any “anonymized” dataset using only 15 demographic variables, such as zip code, date of birth, gender, etc. (Rocher et al., 2019).
Rosati described a complicated web of laws that exist in Europe, the United States, and individual states regulating privacy and security in research. The complexity arises, she said, because of different laws from different sources that impact different institutions and different types of information. In the European Union (EU), the General Data Protection Regulation (GDPR) applies to organizations within the European Economic Area (EEA)—including EU member states plus Iceland, Liechtenstein, and Norway as well as organizations outside EEA that offer goods or services to data subjects within EEA or monitor the behavior of data subjects within EEA. The regulations govern any data that directly or indirectly identifies a living individual, and adds special protections to what it deems sensitive data such as genetic, biometric, health, and demographic data. Importantly, said Rosati, because GDPR applies restrictions to the transfer of personal data, it impacts even researchers with organizations not directly required to comply with GDPR, for example, organizations that want to collaborate with organizations in Europe.
In the United States, multiple regulations add complexity to the web of laws, according to Rosati. The Health Insurance Portability and Accountability Act (HIPAA) regulates covered entities such as health care providers “and their business associates” and applies to “protected health information”; the Common Rule regulates federally funded research; the federal substance use disorder treatment regulations govern substance abuse information in a much more protective way than HIPAA does; the Food and Drug Administration (FDA) oversees clinical trials, which regulate a subset of data collected in support of drug approvals; and NIH policies and regulations provide for certificates of confidentiality. Layered on top of those regulations are state laws such as the California Consumer Protection Act and state health information confidentiality laws and state licensure requirements, said Rosati.
Each of these regulations define which data are protected somewhat differently as well as what constitutes de-identified information (under HIPAA) or non-identifiable information (under the Common Rule). More-
over, these regulations are in constant flux, said Rosati. For example, she said the Office for Human Research Protection and other agencies that enforce the Common Rule are expected to issue guidance soon on whether whole-genome analysis or other types of genetic information should be treated as identifiable information even if it is not paired with data elements that directly identify individuals. This could widen the difference in how the Common Rule and HIPAA interpret the identifiability of data, which will likely cause substantial confusion about the sharing of genomic information, said Rosati.
Rosati added that the patient privacy advocacy community is very active and has a voice in Congress about putting restrictions around the use of de-identified data without individual consent. This would be a disaster for research, she said, noting that the research community needs similar advocates to explain to the public and Congress the importance of data sharing and open access to data to advance good science. A better public policy would be to prohibit re-identification of individuals from de-identified datasets. Moreover, while security of patient information is important, Magali Haas, chief executive officer (CEO) and president of Cohen Veterans Bioscience, noted that patients are providing informed consent for a purpose, including for research. Haas and Clare Mackay, professor of imaging neuroscience at the University of Oxford, said that laws, regulations, and data access policies governing research should take participants’ perspectives into account.
CURRENT PROMISING PRACTICES TO PROTECT PRIVACY
One of the inspirations for this workshop was an experience described by Jonathan Cohen, Robert Bendheim and Lynn Thoman Professor of Neuroscience at Princeton University. His group was developing methods for putting real-time image analysis of fMRI data on the cloud and wanted to test it in a clinical trial. This type of analysis is helpful, and in some instances necessary to do in the cloud because the computing requirements are so high and may be out of reach of clinical facilities with access to relevant patient populations. Because Princeton does not have a medical school, his team partnered with William Hanson at the Perelman School of Medicine at the University of Pennsylvania (Penn Med). When they were ready to start the project, however, administrators at Penn Med were concerned that there were no standards to ensure these data could be shared in a way that protected the privacy of patients. Hanson said there were also issues with Penn Med’s Information Services department and Institutional Review Board (IRB) that had to be overcome to enable moving forward with the project. He explained that health systems such as Penn Med are extremely risk averse and under constant threat of data breaches by hackers
trying to fish for patient data in their systems. As the chief medical information officer at Penn Med, Hanson and his team worked to put together principles for access, use, and disclosure, starting with the principle that data sharing is in the interest of patient treatment. Patients who choose not to have their data shared must opt out.
Hanson said they are now working on a framework to guide all efforts around data access, use, and disclosure of patient information. These principles fall into six domains: lawful basis, institutional mission and values, trustworthiness and accountability, risk mitigation, strong security controls, and documentation (see Figure 3-1). They plan to apply these principles to specific use cases in order to operationalize the Penn Med position and convene a data governance committee to oversee the process. Hanson said that rather than viewing these protections as obstacles, he believes they facilitate research.
Many other models have also been developed to protect the privacy of patients. Mackay described three projects in the United Kingdom that have navigated the question of privacy protection in different ways:
- The UK Biobank, possibly the largest cohort study in the world with 500,000 individuals, multiple data types, and multiple institutions, but a single principal investigator and single IRB.1
- The Wellcome Center for Integrative Neuroimaging (WIN), a study taking place at a single institution with multiple study types, principal investigators, and IRBs.2
- Dementias Platform UK (DPUK), a substudy of the Field platform (which also includes Health Data Research UK, or HDRUK) that is taking place at multiple institutions with multiple study types, IRBs, consent, and principal investigators.3
The message Mackay delivered to the workshop is that one size does not fit all. Each project has a different governance model and different infrastructure designed to fit the types of data gathered, the study participants, and the users of the data.
The UK Biobank, for example, is open by design, said Mackay. Participants consent upon enrollment to having their data shared, not only imaging data, but also a range of sensitive health and personal information. Data access is governed through a data access committee; researchers apply
___________________
1 For more information, see https://www.ukbiobank.ac.uk (accessed November 10, 2019).
2 For more information, see https://www.ndcn.ox.ac.uk/divisions/fmrib (accessed November 10, 2019).
3 For more information, see https://www.dementiasplatform.uk (accessed November 10, 2019).
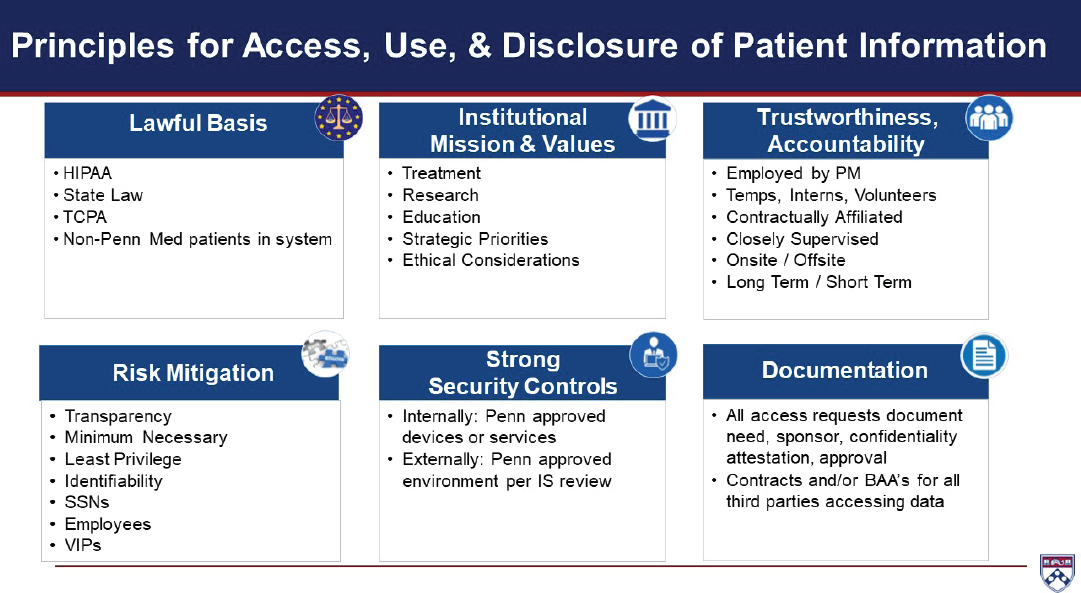
NOTE: BAA = business associate agreement; HIPAA = Health Insurance Portability and Accountability Act; IS = information services; PM = Perelman School of Medicine at the University of Pennsylvania; SSN = Social Security number; TCPA = Telephone Consumer Protection Act.
SOURCE: Presented by William Hanson, September 24, 2019.
and pay an administrative fee for access, said Mackay. She added that an important aspect of the project is that there is no disclosure of any health information to the participants, which limits the ability to recruit potential participants for future studies or for participants to self-identify for future studies, since the health information that would make them eligible for the study cannot be disclosed.
WIN also takes an open science approach and is building a centralized infrastructure to facilitate data sharing, in which responsibility for privacy remains with the study’s principal investigator, according to Mackay. A similar model is being used for the Field platform, which has 35 cohorts and more than 3 million participants, she said. The Field platform infrastructure has been developed not only to facilitate data sharing, but data aggregation and cross-cohort analyses as well. It uses a single point of entry for requesting access to data but, like WIN, assigns responsibility for granting access to the individual data access committees.
DPUK uses another model, where individual cohort owners share their data, but users are unable to download the data, said Mackay. In this model, all computation happens on the platform that holds the dataset—a sort of data playground—said Rosati. The global data platform Vivli also uses this approach as just one of its pillars of data security, said Rebecca Li, executive director of Vivli. They also require anonymized data and manage access to the platform, requiring users to have specialized expertise.
Rosati also mentioned the FDA Sentinel Initiative.4 In developing their privacy rules, she said they decided to pursue a federated model where all participating institutions, both health systems and health insurance companies, put data into a common data model and locally retain them rather than depositing them in a central databank. In addition to protecting data, this system allows investigators to bring analytical tools to multiple datasets, enabling powerful research across different databases, she said.
Mackay noted that every research cohort has a participant panel that plays an important role in determining rules regarding data access. While an individual investigator may argue that participants in his or her study did not consent to data sharing, Mackay pointed out that neither did they consent to the use of those data to further the career of one individual investigator. Moreover, if a cohort sets up the governance in a way that prohibits re-identification, it may remove a participant from the opportunity to take part in a future drug trial of a potentially beneficial treatment. Indeed, said Mackay, while researchers are rightly concerned about security and privacy, the perspective of participants is equally important. As a way of combat-ting the risk aversion that prevents many researchers and institutions from
___________________
4 For more information, see https://www.fda.gov/safety/fdas-sentinel-initiative (accessed November 10, 2019).
sharing data, she suggested discussing case studies of where open science provided positive benefits that could not have been realized had data access been restricted.
KEY PRIVACY-RELATED ISSUES TO BE RESOLVED
No federal laws in the United States prohibit re-identification of individuals, said Haas, adding that this needs to be addressed at the policy and legislative levels. Rosati noted that this is a particular concern in countries like the United States, which does not have a national health system and people worry about losing health insurance, disability insurance, etc. Individual privacy also needs to be addressed with regard to employment laws, said Haas.
Informed consent is a critical component in data sharing via cloud-based platforms, but there are multiple issues still to be addressed. Many participants in the privacy breakout session, including Mackay and Deanna Barch, advocated development of best practices to guide the development of consent forms for clinical trials and participation in repositories and bio-banks. Ideal consent forms enable individuals to share their data with large data-sharing platforms and other collaborative efforts. Haas added that templates for consent should use universally accepted definitions regarding what types of data are particularly sensitive in this changing landscape of regulation and what qualifications should researchers have to gain access to data. Rosati said NIH has developed template language for clinical trials and research repositories, for example, to enable sharing of genome-wide association study (GWAS) data. Barch added that consent forms are also needed that distinguish between data already collected versus data to be collected in the future. She advocated optimizing consent forms so that participants understand their rights of privacy, but consent to as many uses with which they are comfortable.
To implement the use of informed consent language that would both enable data sharing and protect participants, Michael Milham, vice president of research and founding director of the Center for the Developing Brain at the Child Mind Institute, suggested including a range of use cases about how someone’s data might be used in the future. Granting agencies could require such language unless reasons are given for why it should not be required. Milham added that beyond developing best practices, mechanisms are needed to implement the use of consent forms that enable future use of data.
Other unresolved issues discussed in the privacy breakout session overlap with those discussed in the data management and platform governance sessions, discussed further in Chapters 4 and 6. For example, Haas suggested that better definitions need to be developed for what constitutes
sensitive data and what qualifications should be required for an investigator to access shared data. Barch added that there should be some consequence for investigators who violate data use agreements designed to protect individuals’ privacy. Rosati noted that most data use agreements fail to specify whether an individual researcher or the institution is responsible if data are misused. Barch and Hanson discussed the utility of having institution-oriented resources of common practices for data sharing and use in agreements with commercial entities, including details such as secondary relationship governance in the case of the original company being acquired.








