
5
National and International Governance of Heritable Human Genome Editing
A responsible translational pathway toward potential clinical uses of heritable human genome editing (HHGE) requires that national and international governance foundations be in place prior to any clinical use. Chapter 5 discusses the elements that would need to be part of such systems. This chapter begins by discussing how HHGE intersects with, and poses challenges for, current oversight systems for medical technologies. The chapter then describes the mechanisms that a country would need to establish to ensure responsible oversight of any future clinical uses of HHGE. Finally, the chapter emphasizes the need for international coordination around developments that affect HHGE. The chapter does not delve into detail on how national and international governance systems for HHGE would ultimately be implemented by countries and by the international community—ongoing dialogues including the work of the World Health Organization’s (WHO’s) Expert Advisory Committee are exploring this area in greater depth. However, this chapter concludes with recommendations for core components of these efforts.
A RESPONSIBLE GOVERNANCE SYSTEM FOR HERITABLE HUMAN GENOME EDITING
HHGE would entail a form of assisted reproductive technology (ART) used to generate an embryo with an altered genome with a view to establishing a pregnancy. A governance system for the use of HHGE would need to include the ability to oversee all stages of the translational pathway
described in Chapter 4. These stages include basic and preclinical research to develop methodologies for HHGE that can sufficiently control and characterize the effects of genome editing; national legislative, advisory, and regulatory decision making charged with determining whether a clinical use of HHGE could be considered; and evaluation of outcomes resulting from any clinical use of a genome-edited embryo to establish a pregnancy.
Considerations for Societal and Stakeholder Engagement on Heritable Human Genome Editing
Prior to the clinical use of HHGE in any country, one important requirement is for public engagement on whether it would be acceptable to use HHGE in that country and, if so, for what purposes and with what governance mechanisms. Genome editing in human embryos should not proceed past preclinical laboratory research unless it is deemed acceptable by a country and unless there are approvals by the relevant bodies to consider it for potential clinical use. The question of precisely how such discussions should proceed was beyond this Commission’s charge; however, presentations and submissions to the Commission’s call for evidence emphasized a number of additional points to inform future deliberations (see Box 5-1).
HERITABLE EDITING IN THE CONTEXT OF CURRENT REGULATORY SYSTEMS
A governance system for HHGE would share similarities with the oversight structures that currently guide appropriate conduct in other areas of biomedical research and clinical practice. Because HHGE entails the use of genome editing technologies as a form of assisted reproduction to enable prospective parents to have a child with an altered genome, it shares some characteristics with existing oversight systems in both somatic gene therapies and ARTs. However, the clinical use of HHGE would also pose challenges to current systems.
How Heritable Human Genome Editing Would Relate to the Regulation of Gene Therapies
Many somatic cell gene therapies currently undergoing clinical development rely on using genome editing technologies. Somatic cell gene therapies have a history of highly regulated oversight in the countries in which they have been carried out, including the United States, Japan, China, India, and countries in Europe. In the United States, the European Union, and China,
for example, somatic genome editing is regulated primarily using the frameworks established for prior generations of gene therapies (NASEM, 2017).
A number of clinical trials based on somatic genome editing have been initiated in the United States. The regulatory process involves the institutional reviews required for human clinical trials as well as additional institutional oversight by biosafety committees and federal review. Federal oversight includes requirements for prospective approval from the national
regulatory authority, the U.S. Food and Drug Administration (FDA), via an Investigational New Drug license or its equivalent. Once clinical trials commence, centralized reporting of adverse events and longitudinal data collected during the clinical trial phases are required for submission of an application to the FDA for approval as a therapeutic to be marketed in a clinical context.
Other countries have similar regulatory systems intended to ensure the safety and efficacy of somatic gene therapies tested or approved for use in humans. Regional organizations, such as the European Medicines Agency, promote the development of scientific guidelines in areas such as gene and cell therapy products, and there are other ongoing international dialogues aimed at improving the consistency of somatic gene therapy regulations.1 In addition, countries including Chile, Colombia, Mexico, and Panama have incorporated explicit prohibitions on the use of somatic genome editing for purposes that might be perceived as enhancement (Abou-El-Enein et al., 2017; NASEM, 2017). While somatic genome editing shares a similarity with heritable genome editing in that both types of uses rely on genome-editing technologies, there are important differences that challenge the applicability of its regulatory frameworks to HHGE.
Somatic therapies fit within an oversight paradigm in which medical interventions are developed and deployed to treat an existing patient with a genetic condition. The effects of the editing are limited to that individual’s cells and tissues and are not inheritable, and the largely individual-level harms and benefits can be assessed and explained as part of gaining informed consent. HHGE, on the other hand, provides a reproductive option for prospective parents to have a potential future child without transmitting a disease-causing genotype. The heritable genomic alteration and the potential harms, benefits, and uncertainties that arise may affect not only that child but also any offspring of that child, raising societal concerns and leading to effects that may not be apparent until subsequent generations.
How Heritable Human Genome Editing Would Relate to the Existing Regulation of Assisted Reproductive Technologies
As noted above, HHGE would constitute a form of ART, and ARTs have a very different history of regulatory oversight from that of somatic gene therapies. Laws regarding the use of ART vary substantially among countries. While there are important lessons to be gleaned from such regulatory experiences (Cohen et al., 2020), this variation will make it difficult to achieve coordinated oversight of HHGE using current ART regulatory systems.
___________________
A survey conducted in 2018 by the International Federation of Fertility Societies (IFFS), that spanned 89 of the 132 countries believed to be offering ART, found that 64 percent of countries that responded to the survey had legislation regulating the use of these technologies, with a focus on licensing clinics, physicians, and laboratories. Penalties for violating regulations ranged from admonishment to imprisonment, with the most frequently used sanctions reportedly being financial penalties, loss of license, and threat of criminal prosecution (IFFS, 2019).
One of the ARTs most relevant to the discussion of HHGE is preimplantation genetic testing (PGT), in which cells removed from an early embryo created through in vitro fertilization (IVF) are genetically analyzed and only embryos having specified genotypes are transferred to establish a pregnancy. The majority of countries responding to the IFFS survey reported that they allow PGT for prevention of monogenic disease. These countries were split almost equally with respect to whether laws or regulations permitting the use of PGT were accompanied by further guidelines restricting how it could be used.
Research analyzing national approaches to the use of PGT found that they were typically based on the seriousness of a condition (Isasi et al., 2016). For example, Mexican legislation prohibits PGT for any purpose other than “the elimination or reduction of serious diseases or defects,” while other countries require a “substantial risk” of the disease occurring or that the disease is “untreatable” or “incurable” (Isasi et al., 2016). The United Kingdom is an example of a country that utilizes “seriousness” in its evaluation of PGT applications and where the use of IVF with PGT is permitted only to prevent specific genetic conditions that have been approved by the Human Fertilisation and Embryology Authority. Its list of permitted conditions now totals more than 600 and includes the use of PGT to select an embryo that is immunologically matched to a sibling with a disease (savior sibling PGT).2 In France, only couples at high risk of having a child affected by a “particularly serious and not curable” genetic disease are allowed to use PGT under the national public health code, with oversight by the country’s biomedical agency (Agence de la Biomédecine). Requests for PGT are evaluated on a case-by-case basis rather than against a list of allowed uses. A review committee at each major reproductive medicine center evaluates the requested use and reports annually to the Agence de la Biomédecine on its decisions. This enables retrospective analysis of the criteria by which such requests are judged, which take account of factors such as risk of disease, anticipated disease manifestation, and family medical history. In China, regulations on ARTs were published by the
___________________
2 See https://www.hfea.gov.uk/treatments/embryo-testing-and-treatments-for-disease/approved-pgd-and-ptt-conditions/.
Ministry of Health in 2001 and were amended in 2003 into a document titled “Technical Standards, Basic Requirements, and Ethical Principles on Human ARTs and Related Technologies and Human Sperm Bank.” Any medical institutions permitted to carry out human ARTs are required to meet these regulations and standards and to obtain an approval certificate from the Ministry of Health. Medical institutions offering these technologies are required by law to have ethics committees, which review certain proposed methods or some specific cases. PGT has been used for those couples that are at high risk of having a child with single-gene disease, chromosome disorders, or sex-linked genetic disease, but it is not allowed for sex selection. In the United States, ARTs are offered in the context of the practice of clinical medicine without a requirement for regulatory approval. There are no federal restrictions on the conditions for which PGT can be used. Instead, PGT use is guided by any state laws that may restrict uses, professional guidelines, and the choices of clinicians and prospective parents (Bayefsky, 2016, 2018).
Mitochondrial replacement techniques (MRT) represent a novel form of ART, which are currently permitted for clinical use in the United Kingdom. The approach taken by the United Kingdom to develop a translational pathway and oversight regime for this technology can provide an informative model to help guide the development of national oversight systems relevant to HHGE. As described in Chapter 1, the characteristics include a controlled step-wise process under the auspices of appropriate national regulators; limitation to cases involving parents wishing to have a genetically-related child unaffected by serious disease; limited licensure to use in single cases rather than blanket approval, with ongoing review before subsequent licenses are issued; a comprehensive informed consent process; long-term follow-up of offspring; and prohibition of uses beyond the permitted indication.
Lessons Applicable to the Creation of an Oversight System for Heritable Human Genome Editing
As with other medical technologies, an oversight system for HHGE would need to address all stages of a research and clinical translation pathway. Because multiple actors contribute to any translational pathway, responsibilities at individual, institutional, national, and international levels will be required. Investigators and clinicians will need to adhere to relevant norms, guidelines, standards, and policies. For example, these may include or draw on policies developed for governance of gene therapies and for governance of ARTs. Well-specified processes will need to be established for institutional boards to review clinical use protocols, including appropriate protections for participants. Prior to the initiation of any clinical
use, approvals will be required from relevant national advisory bodies and national regulatory authorities that assess the context of proposed use, preclinical evidence, clinical protocols, and plans for follow-up. Processes will need to be implemented for national and international discussion, coordination, and sharing of results on relevant scientific, ethical, and societal developments impacting the assessment of HHGE’s safety, efficacy, and societal acceptability (see Box 5-2).
Legal and Regulatory Frameworks
The legal and regulatory status of HHGE varies considerably among countries. HHGE is currently prohibited by law in dozens of countries
including many in Europe as well as the United States, where federal budget provisions currently prevent the FDA from considering any application for clinical use of HHGE (Kaiser, 2019). Any clinical use of HHGE in these countries would require changes to the relevant legislation.
All countries in which HHGE research and clinical applications may be pursued will need regulatory mechanisms to oversee use of HHGE and impose sanctions where appropriate, as well as a clearly communicated way for concerns about possible violations of regulations to be reported. Because HHGE would be deployed within an existing culture of IVF and ART clinics, it will be important to engage with this community on the issues posed by HHGE prior to any clinical uses. However, relying on professional conduct guidelines and self-regulation for an emerging and con-
troversial technology such as HHGE may be insufficient. At a minimum, laws or regulations incorporating penalties for any unauthorized use of HHGE should be considered in countries that do not currently have them.
Each country that considers the development of HHGE will end up drawing on the regulatory infrastructure and oversight authorities available under its laws and regulations. For a country such as the United Kingdom, the Human Fertilisation and Embryology Act could be further amended to permit HHGE, as it was in 2008 to enable the Authority to evaluate applications for MRT. If the U.S. government were to decide to permit clinical use of HHGE, it would also need to consider whether the FDA or other state and federal regulatory bodies need additional authorities to oversee the practice of assisted reproductive medicine, since HHGE would take place in ART clinics. Other countries may similarly need to wrestle with how HHGE could fit within or challenge national medical oversight systems and determine whether they need to create new oversight paradigms or whether existing oversight mechanisms could be modified to sufficiently address the oversight needs for HHGE.
REQUIREMENTS FOR NATIONAL OVERSIGHT SYSTEMS FOR HERITABLE HUMAN GENOME EDITING
Regardless of the details of the regulatory systems that a country may design for HHGE, national regulatory authorities or their equivalents would need to establish the specific criteria that must be met for any translational application of HHGE to proceed in their jurisdictions. To address the characteristics for responsible governance of HHGE identified above, all countries in which it is being considered would need to have mechanisms in place to oversee translational progress toward potential clinical use of HHGE, to prevent unpermitted uses and to sanction any misconduct. The issues that will need to be addressed through national systems wherever HHGE is proposed to be undertaken include:
- giving clear and unambiguous direction to researchers and clinicians about the legality of HHGE;
- ensuring that researchers and clinicians adhere to norms of responsible science, including relevant human rights and bioethics principles (see Box 5-3) and applicable guidelines, standards, and policies;
- providing transparency on any applications for HHGE under consideration;
- providing transparency to the world community of any intention to allow an approved clinical use of HHGE;
- creating clear processes and mechanisms for review, approval, and oversight of any initial human clinical uses of HHGE;
- establishing mechanisms to circumscribe the clinical use of HHGE, including to limit and control any uses beyond the scope of a permitted indication; and
- being responsive to international scientific consensus regarding the current state of HHGE technologies, especially in areas of safety and proposed uses, with the goal of coordinating protocols and sharing data to the maximum extent possible.
THE NEED FOR A SYSTEM OF GLOBAL COORDINATION AND COLLABORATION
While countries have decision-making authority concerning the research toward, or clinical use of HHGE, it is critical to also have international scientific and ethical cooperation on HHGE. A translational pathway for HHGE therefore requires governance systems that extend beyond those of individual countries to enable transparent discussion about any approved clinical uses of HHGE and the resulting outcomes. This is because:
- There is a collective interest of humanity in the use of a novel technology that can result in heritable changes to the human genome.
- The research and clinical communities developing these technologies are global, and the technologies have implications beyond national borders.
- Citizens from different countries seeking access to HHGE will travel to countries where it becomes available.
- Any initial uses of HHGE following the pathways described in this report would involve a small number of people, and it would be important to collect and compare information across national boundaries in order to more fully understand first-in-human safety and efficacy data and to promote common approaches.
With respect to both biomedical research and clinical practice, in general, countries have framed licensing powers and accompanying professional duties within their legislation and regulations on health and their health systems, through the creation of statutory oversight bodies, or, more rarely, via legislation on specific sectors or technologies. The approach varies from country to country or it is defined in regional alliances. Any proposed mechanism for international governance of HHGE will need to provide for at least three functions:
- An international scientific advisory panel to provide ongoing technical assessment and evaluation of developments in the science and technologies on which HHGE depends and to make recommendations about their suitability and readiness for particular clinical uses.
- An international body for evaluating and making recommendations on crossing major thresholds associated with the clinical use of HHGE, based on consideration of a wide range of societal and scientific perspectives. In the current context, a threshold represents a boundary that distinguishes a currently accepted use from another that is not currently accepted. Before crossing any threshold, it will be important for the global community to assess not only progress in scientific research but also what additional ethical and societal concerns the circumstances of particular uses could raise, as well as any results, successes, or concerns that had been observed from any human uses of HHGE that had been conducted thus far.
- An international mechanism by which individuals or organizations in one country can bring forward technical or ethical concerns arising from HHGE work conducted in their own country or in another country.
These necessary functions are explored below.
An International Scientific Advisory Panel to Monitor and Assess Relevant Scientific and Clinical Developments
As emphasized throughout the report, before any country should consider approving the clinical use of HHGE, further technical developments are essential. There is therefore a need for the ongoing technical assessment and evaluation of developments in the science and technologies on which HHGE depends, as well as making recommendations about their suitability and readiness for particular clinical uses. Multiple gaps in the ability to fully characterize such genome editing or assess its effects make it premature to use any HHGE approaches at the time of this writing, and articulating the essential characteristics of a translational pathway does not mean that a country should necessarily permit even initial clinical uses.
There is, therefore, a need for an international advisory body to regularly review the latest scientific evidence and to evaluate its potential impact on the feasibility of HHGE. The necessary functions of such scientific review include:
- assessing or making recommendations on further research developments that would be required to reach technical or translational milestones as research on HHGE progresses;
- providing information to national regulatory authorities or their equivalents to inform their own assessment and oversight efforts;
- facilitating coordination or standardization of study designs to promote the ability to compare and pool data across studies and trans-nationally;
- advising on specific measures to be used as part of the long-term follow-up of any children born following HHGE; and
- reviewing data on clinical outcomes from any regulated uses of HHGE and advising on the risks, benefits, and uncertainties of possible further applications.
There are existing international activities that play a valuable role in contributing to the technical assessment of the science and technologies underlying HHGE. The two international summits on human genome editing convened by various scientific academies (including the U.S. National Academies of Sciences and Medicine, the U.K.’s Royal Society, the Chinese Academy and the Hong Kong Academy, and others) have brought together the scientific community for scientific presentations relevant to HHGE. A third summit is planned for 2021.
Professional societies in science or medicine can also play a role in scientific review and standards development. In stem cell research, the International Society for Stem Cell Research has an ongoing mechanism for the
creation and revision of guidelines (ISSCR, 2016) for research and clinical practice in the stem cell field. In the ART field, the U.S. Society for Assisted Reproductive Technology (SART) provides access to data from IVF clinics for research and comparison and is developing a standardized document for informed consent in collaboration with the American Association of Law Schools.3
However, these activities are largely informal and ad hoc. The examples demonstrate that although existing structures and processes can fulfill some of the functions necessary, none do or can perform the collection of functions recommended for ongoing technical assessment and evaluation of the technologies foundational to HHGE.
For this reason, the Commission recommends the creation of an International Scientific Advisory Panel (ISAP) that would provide regular scientific and technical assessments as part of the international governance efforts for HHGE described above (see Figure 5-1). An ISAP would need the endorsement of national governments to have the standing and influence required to perform these functions. It would also need to be flexible given the potential for rapid advances in areas of science that contribute to the feasibility of HHGE. The panel would need to convene regularly in person or virtually, likely at least once per year, with additional meetings and discussions as needed. To be most effective, such a panel would need to have diverse, multidisciplinary membership and include independent experts who can assess scientific evidence of safety and efficacy of both genome editing and associated ARTs. It should include international experts from multiple disciplines including genetics, genome editing, reproductive medicine, pediatric and adult medicine, bioethics, law, and other fields. This combination is similar to that for Data Safety and Monitoring Boards or Data Monitoring Committees for large, often multi-site, clinical trials, which seek to ensure relevant expertise in clinical specialty areas, clinical trial methodologies and analysis, biostatistics, and often in the ethics of design, conduct, and interpretation of clinical trials.4 Because the panel would be assessing evidence that could be used to support progress toward initial use of HHGE for serious monogenic diseases, the panel would also greatly benefit from including representatives of the public, such as members of genetic disease and disability communities.
___________________
3 See www.sart.org. Recent SART clinic reports are available at https://www.sartcorsonline.com/rptCSR_PublicMultYear.aspx?reportingYear=2017 for 2017, and preliminary 2018 data at https://www.sartcorsonline.com/rptCSR_PublicMultYear.aspx?reportingYear=2018.
4 See FDA guidance at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishment-and-operation-clinical-trial-data-monitoring-committees; and NIH guidelines at https://www.nidcr.nih.gov/research/human-subjects-research/toolkit-and-education-materials/interventional-studies/data-and-safety-monitoring-board-guidelines.
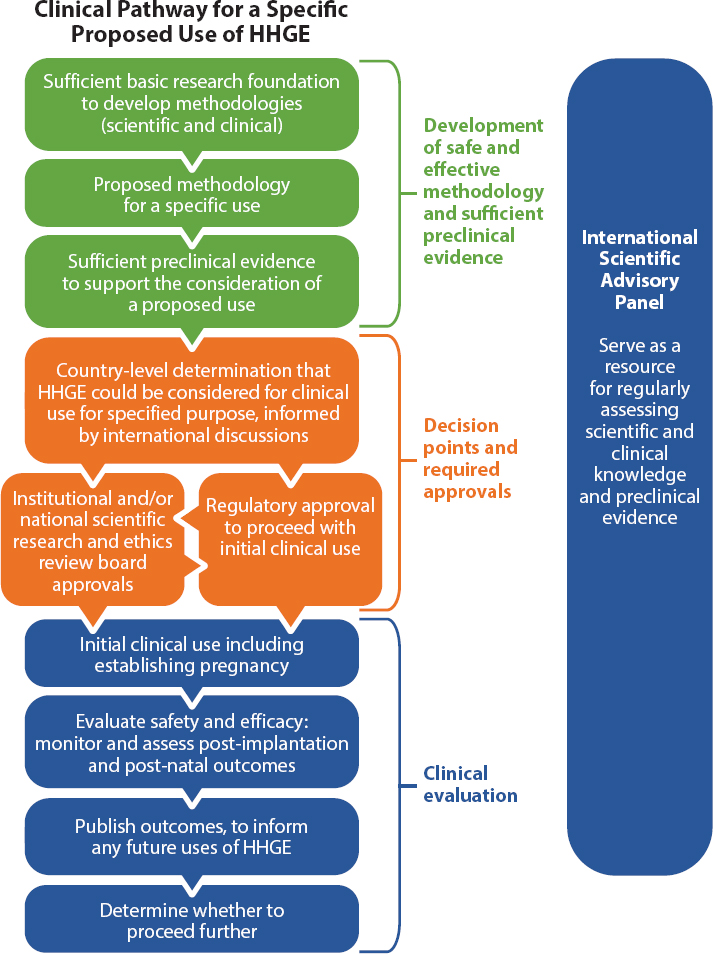
Existing national and international networks could be drawn on to identify members who could be nominated to such a panel. For example, national academies of sciences and medicine, the global network of science academies (the InterAcademy Partnership), national and international professional societies in relevant areas, genetic disease and disability communities, and scientific, medical, and technical experts in relevant government ministries might all serve to identify colleagues who are leaders in their disciplines and could bring the expertise and cooperative spirit required to this task. National and international discussions would be needed to agree on the panel’s terms of reference, its convener, and how its activities would be funded.
The Commission is not wed to any particular body or organization for establishing an ISAP but emphasizes its recommendation that any translational pathway toward HHGE requires establishing a systematic and rigorous way to fulfill the five functions described above in order to enable independent expert review of scientific and clinical evidence to inform national and international governance.
Advances in areas of science relevant to HHGE, as well as in the practice of IVF and PGT, will have implications for whether the translational pathway criteria specified in Chapter 4 can be met. As this pathway was developed for the very first possible uses of HHGE considering the current state of science, it will be important to be open to scientific developments that could alter the methodologies employed to meet the requirements. It will also be important to assess evidence gained from further basic research and preclinical testing and from any future initial human uses.
The Commission strongly believes that successfully carrying out these functions requires more than the current informal and ad hoc systems.
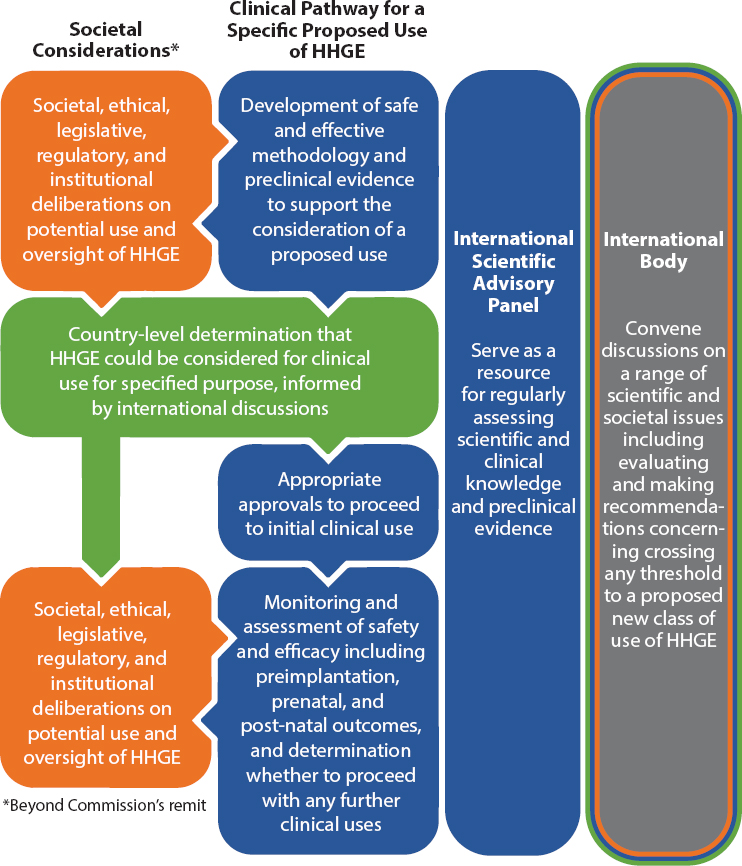
International Body for Evaluating and Making Recommendations before Crossing Heritable Human Genome Editing Thresholds
This report has categorized possible clinical uses of HHGE according to the assessment of the potential harms and benefits they present, with a focus on initial clinical uses. However, decisions about whether to allow HHGE, and, if so, for what purposes, should be based on a wider set of considerations than just scientific assessments. Initial human use of HHGE beyond preclinical development represents a decision that should be based on science, ethics, and societal implications. It will be important for countries to engage in discussions about when, if ever, it is acceptable to move forward with HHGE within their countries and, if so, where to set thresholds on allowable uses. Subsequent decisions about whether to cross additional thresholds to allow further uses of HHGE will similarly require transparent international discussions convened by an institution responsible
for ensuring that these discussions are held regularly and that they engage a diversity of viewpoints (see Figure 5-2).
There is already a range of international bodies whose responsibilities include convening international discussions on the development and regulation of medical technologies. Organizations such as WHO, the Organisation for Economic Cooperation and Development (OECD), and
the United Nations Educational, Scientific and Cultural Organization (UNESCO), for example, all have the requisite experience that could enable an inclusive and transparent debate about whether and how to proceed with HHGE. Other organizations could also be selected for this purpose.
Regardless of where housed or how structured, this international body would need a wide range of perspectives, including from (i) stakeholder communities that could be affected by future uses of HHGE, such as members of disability and disease communities; (ii) scientific fields, including medicine and social sciences; and (iii) law, ethics, and regulation. This should include experts from countries where there are communities that have increased incidence of genetic disease due to factors such as founder mutations or high rates of consanguineous union. As with the current Commission, assessments from this process would inform and be advisory to national and international decision making.
If initial clinical uses of HHGE were ever permitted, those uses would only be considered in a carefully prescribed set of circumstances and would likely entail only a small number (on the order of 10–20) of cases. Assuming analysis of the outcomes of any initial uses did not raise further concerns about the safety and efficacy of HHGE, it might be deemed appropriate to consider uses in circumstances beyond those initially envisaged by this Commission. Before progressing beyond those initial cases toward any further clinical uses of HHGE, it would be important for the global community to pause and reassess not only the state of the science but also what additional ethical and societal concerns new circumstances of uses could raise. New classes of use may or may not precisely align with the six categories defined in Chapter 3. Making recommendations on whether it is appropriate to cross subsequent thresholds in the use of HHGE would be a key role for an international body with responsibility for convening the international debate on HHGE.
Potential uses of HHGE beyond the circumstances set out by this Commission open the door to impacting reproductive options for a significantly larger group of people. Making HHGE available to couples in Category B beyond the narrow circumstances described in Chapter 3 would represent a significant expansion in the possible scope of this technology. As a result, a respected body would be needed that can assess whether it is feasible to envision new responsible translational pathways and what these pathways should entail.
This process should be complemented by other efforts by civil society to promote international cooperation on approaches to responsible development of medical technologies. For example, the Global Observatory on Genome Editing is being set up to foster international, interdisciplinary discussions on genome editing (Hurlbut et al., 2018). Similarly, the Association
for Responsible Research and Innovation in Genome Editing (ARRIGE) was launched in 2018 to promote global governance of genome editing.5 Both ARRIGE and the Global Observatory promote cross-sector discussions of whether genome editing technologies should be used and, if so, for what purposes.
There are also international processes that focus more on promoting responsible scientific conduct, for example, the Good Clinical Practice guidance for clinical trials developed by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), whose members and observers include national regulatory agencies, industry, and international organizations.6 The ICH develops its guidelines through a process that includes formation of an expert working group to draft a technical document on an issue, followed by development by regulatory members of a draft guideline. The draft guideline undergoes a process of consultation and revision before being adopted by ICH. Although governance of HHGE would require engaging a broader and more diverse community than encompassed by ICH, this step-wise process encourages input and buy-in from the represented stakeholders. The launch by WHO in 2019 of initial phases of a global registry for genome editing clinical trials also represents an important step in the ability to monitor advances in HHGE and to maintain awareness of actions being taken within national jurisdictions.7
A Mechanism to Bring Forward Concerns about Research or Clinical Use of Heritable Human Genome Editing
After the announcement in 2018 that children had been born in China following the use of HHGE, an important question posed was how individuals who may have known about the work being conducted could have raised concerns, particularly if they were in one country and the investigator and the research being undertaken were in another. The Commission is not aware of a precise precedent for such an international mechanism that is accessible to anyone who would like to raise a concern.
Future governance of HHGE requires an international mechanism for individuals and groups to raise concerns about possible violations of regulations or activities related to any clinical practice of HHGE in jurisdictions without regulations. There should be a highly visible, easily discoverable
___________________
5 See https://www.arrige.org.
6 See https://www.ich.org.
7 Information on the registry is available at https://www.who.int/health-topics/ethics/human-genome-editing-registry/. The registry collects information on clinical trials using somatic genome editing as well as any clinical trials that would be conducted using HHGE.
entity to which people everywhere may direct their concerns about activity in any country. In developing this mechanism, it will be important to keep in mind that raising concerns about scientific or clinical practices can have personal and professional ramifications for the person making the complaint. It is therefore important to maintain anonymity for anyone using this service. Similarly, details of a complaint should not be made public without prior investigation to protect individuals, institutions, and businesses from false accusations. Such investigations would be the responsibility of national regulatory authorities where available. These authorities would be informed by the international mechanism that a complaint had been made against someone within their jurisdiction.
Although there is no exact precedent, there are relevant examples that can inform the design of such a mechanism. The World Anti-Doping Agency (WADA) has a means by which anyone can report an “Alleged Anti-Doping Rule Violation or any act or omission that could undermine the fight against doping.”8 Some research funders have also developed mechanisms to facilitate the investigation of complaints made against researchers they fund.
CONCLUSION AND RECOMMENDATIONS
The pursuit of a translational pathway toward the clinical use of HHGE would represent the controlled alteration of a human embryonic genome using genome-editing tools, offered as part of an assisted reproduction intervention. All countries pursuing research on or considering the use of HHGE will need to establish oversight systems for this technology, even though national regulatory frameworks for HHGE will differ in their structures and approaches. The governance structures needed for HHGE will also require new models of international coordination. Complex scientific and clinical information will need to be assessed to identify whether the criteria for clinically evaluating a proposed use of HHGE can be met and to incorporate any resulting outcomes into future discussions and decision making. Achieving national and international coordination will pose challenges. But this is exactly why it will be critical to create robust processes by which there can be appropriate and transparent shared responsibility for moving HHGE forward thoughtfully and cautiously, only if there is clear scientific consensus to continue and only if a given country decides to permit its use.
The Commission recommends the following actions as part of this process.
___________________
Essential Elements of Oversight Systems for Heritable Human Genome Editing
Important national and international governance mechanisms should be established before any clinical use of HHGE.
Recommendation 8: Any country in which the clinical use of heritable human genome editing (HHGE) is being considered should have mechanisms and competent regulatory bodies to ensure that all of the following conditions are met:
- individuals conducting HHGE-related activities, and their oversight bodies, adhere to established principles of human rights, bioethics, and global governance;
- the clinical pathway for HHGE incorporates best practices from related technologies such as mitochondrial replacement techniques, preimplantation genetic testing, and somatic genome editing;
- decision making is informed by findings from independent international assessments of progress in scientific research and the safety and efficacy of HHGE, which indicate that the technologies are advanced to a point that they could be considered for clinical use;
- prospective review of the science and ethics of any application to use HHGE is diligently performed by an appropriate body or process, with decisions made on a case-by-case basis;
- notice of proposed applications of HHGE being considered is provided by an appropriate body;
- details of approved applications (including genetic condition, laboratory procedures, laboratory or clinic where this will be done, and national bodies providing oversight) are made publicly accessible, while protecting family identities;
- detailed procedures and outcomes are published in peer-reviewed journals to provide dissemination of knowledge that will advance the field;
- the norms of responsible scientific conduct by individual investigators and laboratories are enforced;
- researchers and clinicians show leadership by organizing and participating in open international discussions on the coordination and sharing of results of relevant scientific, clinical, ethical, and societal developments impacting the assessment of HHGE’s safety, efficacy, long-term monitoring, and societal acceptability;
- practice guidelines, standards, and policies for clinical uses of HHGE are created and adopted prior to offering clinical use of HHGE; and
- reports of deviation from established guidelines are received and reviewed, and sanctions are imposed where appropriate.
Recommendation 9: An International Scientific Advisory Panel (ISAP) should be established with clear roles and responsibilities before any clinical use of heritable human genome editing (HHGE). The ISAP should have a diverse, multidisciplinary membership and should include independent experts who can assess scientific evidence of safety and efficacy of both genome editing and associated assisted reproductive technologies. The ISAP should:
- provide regular updates on advances in, and the evaluation of, the technologies that HHGE would depend on and recommend further research developments that would be required to reach technical or translational milestones;
- assess whether preclinical requirements have been met for any circumstances in which HHGE may be considered for clinical use;
- review data on clinical outcomes from any regulated uses of HHGE and advise on the scientific and clinical risks and potential benefits of possible further applications; and
- provide input and advice on any responsible translational pathway to the international body described in Recommendation 10, as well as at the request of national regulators.
Recommendation 10: In order to proceed with applications of heritable human genome editing (HHGE) that go beyond the translational pathway defined for initial classes of use of HHGE, an international body with appropriate standing and diverse expertise and experience should evaluate and make recommendations concerning any proposed new class of use. This international body should:
- clearly define each proposed new class of use and its limitations;
- enable and convene ongoing transparent discussions on the societal issues surrounding the new class of use;
- make recommendations concerning whether it could be appropriate to cross the threshold of permitting the new class of use; and
- provide a responsible translational pathway for the new class of use.
Recommendation 11: An international mechanism should be established by which concerns about research or conduct of heritable human genome editing that deviates from established guidelines or recommended standards can be received, transmitted to relevant national authorities, and publicly disclosed.
























