
The workshop’s second session explored the ethical issues surrounding patient selection, enrollment, and consent for gene-based therapies and how those differ from conventional clinical trials. This session also identified resources that can help patients and providers accurately understand the potential risks and benefits of participating in a gene-based clinical trial and explored communication strategies aimed at helping patients make informed decisions about participating in trials for gene-based therapies. The discussion was moderated by Mildred Cho, a research professor of pediatrics and the associate director of the Center for Biomedical Ethics at Stanford University. Courtney Fitzhugh, a Lasker clinical research scholar
in the Laboratory of Early Sickle Mortality Prevention at NHLBI, discussed the complexities of patient selection, enrollment, and consent in the context of hematopoietic stem cell transplantation to treat sickle cell disease, while John Tisdale, a senior investigator and the director of the Cellular and Molecular Therapeutics Laboratory at NHLBI, did the same for gene therapies for sickle cell disease. Jennifer Puck, a professor in the pediatrics department at the University of California, San Francisco, addressed these issues in the context of gene therapy for severe combined immunodeficiency (SCID) in the Navajo population. Pat Furlong, the founding president and the chief executive officer of Parent Project Muscular Dystrophy, provided a patient perspective on informed consent, enrollment, and other ethical issues surrounding gene therapy clinical trials. Following these four presentations, there were an additional three speakers who provided patient and family perspectives: Ronald Bartek, a co-founder and the president of the Friedreich’s Ataxia Research Alliance; María José Contreras, a mother of two sons with DMD; and Tesha Samuels, who participated in a gene-based sickle cell disease clinical trial run by Tisdale in 2018. An open discussion with the panelists followed these presentations.
THE COMPLEXITIES OF PATIENT SELECTION, ENROLLMENT, AND CONSENT IN THE CONTEXT OF HEMATOPOIETIC STEM CELL TRANSPLANTATION TO TREAT SICKLE CELL DISEASE
Sickle cell disease, Fitzhugh explained, is caused by a point mutation that causes the hemoglobin protein to polymerize upon deoxygenation, which in turn triggers the transformation of red blood cells from flexible, biconcave disks to rigid, sickle-shaped cells that can block capillaries and small veins. Sickling episodes can occur at any time, and they cause debilitating pain, strokes, liver disease, retinopathy that can lead to blindness, painful leg ulcers, avascular necrosis, and organ damage (Thein and Howard, 2018). Patients with sickle cell disease tend to need hip replacements at a young age, are more prone to develop infections, and often suffer from kidney failure requiring dialysis. Fitzhugh said that the survival rate of children with sickle cell disease has improved substantially since the late 1970s, largely thanks to newborn screening, penicillin prophylaxis, and pneumococcal vaccination. However, the median age at death for adults with sickle cell disease (age 46 in a recent cohort) has changed little over the past 40 years (Fitzhugh et al., 2015; Hassell, 2010).
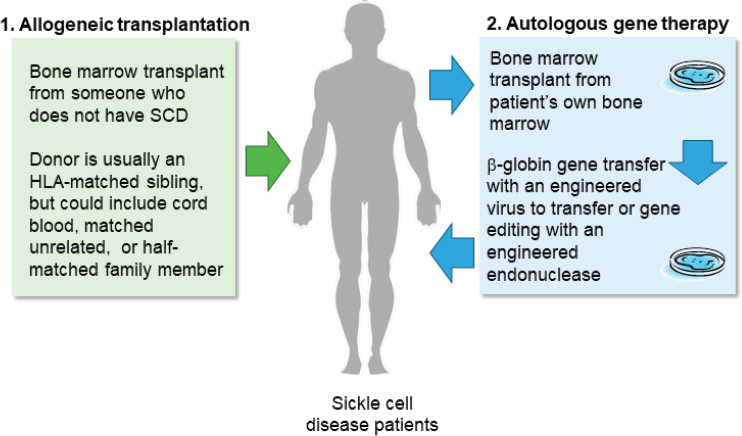
For individuals with sickle cell disease, hematopoietic stem cell transplantation offers a curative option. The most common type of transplant, Fitzhugh said, uses a sibling who is a complete tissue match as a donor to completely replace the patient’s bone marrow with that of the donor. One
study of 1,000 patients with sickle cell disease who underwent myeloablative chemotherapy pretreatment and matched sibling transplants found that in adults the 5-year overall survival rate was 92.9 percent and event-free survival was 91.4 percent, while for patients younger than age 16 the corresponding rates were 95 percent and 93 percent (Gluckman et al., 2017). However, the cumulative incidence of grades II–IV acute graft-versus-host disease (GVHD) was 14.8 percent and the rate of chronic GVHD was 14.3 percent—an unacceptably high incidence of a condition that can cause hardening of the skin, lung scarring, and death. In treating sickle cell disease, the goal is to avoid GVHD because it can potentially be worse for the patients, Fitzhugh said. In addition, many adults who already have organ damage cannot tolerate the myeloablative process.
To overcome this problem, Fitzhugh worked with Tisdale to develop a regimen that uses alemtuzumab, a medication for treating chronic lymphocytic leukemia, to suppress the immune system and deplete it of lymphocytes for 1 month, combined with low-dose total body irradiation to make space in the bone marrow and provide additional immunosuppression. The regimen also includes the drug sirolimus, a compound with immunosuppressive, antitumor, and antiviral properties, in an attempt to mitigate the risk of GVHD. A key difference between this regimen and the standard one with myeloablative chemotherapy is that this one does not completely replace the patient’s bone marrow with that of the donor, and, indeed, Fitzhugh said, it is not necessary to replace all of the bone marrow to cure sickle cell disease. The key, she said, is to have at least 20 percent of the red blood cells coming from the donor. “That is because of the vast differences in half-lives between a normal red cell, which lasts about 3 months, and a sickled red cell, that survives for 5 to 20 days,” said Fitzhugh.
In a study examining the new regimen, none of the 55 patients who received the transplanted hematopoietic stem cells experienced GVHD, though one patient became dependent on blood transfusions for 1.5 years following the transplant, Fitzhugh said (unpublished results). In addition, seven patients rejected the graft (due to graft failure), and six of those individuals had their sickle cell disease return, with the seventh patient dying from an intracranial hemorrhage caused by her sickle cell disease. The overall survival rate was 93 percent, and event-free survival was 87 percent. Unlike the situation with the myeloablative regimen, where most patients are expected to not be able to have children on their own, 8 of the patients using this milder regimen with lower doses of irradiation have had 13 healthy babies post-transplant.
The major problem with this approach is that only about 15 percent of individuals with sickle cell disease will have a sibling who is a complete tissue match. Fitzhugh and her colleagues therefore offer haploidentical transplantation that allows parents, children, and half-matched siblings
to serve as donors. The downside with haploidentical transplantation is that there is a higher risk of graft rejection and GVHD. One study of this approach found that in a cohort of 12 individuals who received two doses of cyclophosphamide post-transplant, only 6 remained free of sickle cell disease because of a relatively high rate of graft rejection (Fitzhugh et al., 2017b). However, more recent results in the haploidentical setting are much more encouraging.
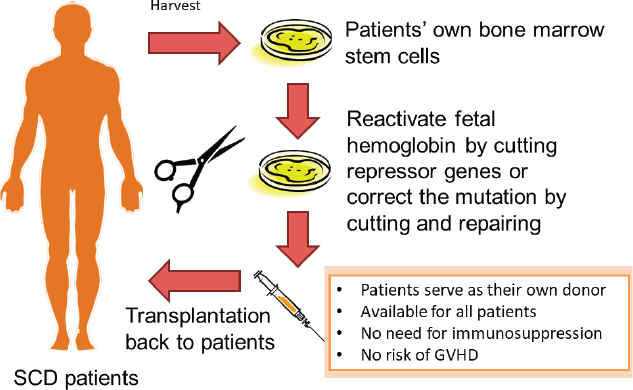
Although Fitzhugh did not discuss gene therapy approaches to treat sickle cell disease—a topic that was covered by Tisdale, the following speaker—Fitzhugh did describe what she and her colleagues tell patients about gene therapy. To start, she tells them, patients can serve as their own donors, which means that it should be available to all patients. There is no need for immunosuppression and no risk of GVHD when the patient is the donor. Myeloablative conditioning is still necessary, and the short- and long-term success of gene therapies is not yet known. In addition, patients with significant organ damage are currently excluded from receiving gene therapy due to the need for high dose chemotherapy. After receiving this information, as well as being briefed on the pros and cons of donor bone marrow transplantation and being assessed for the severity of their disease, patients can decide whether to move forward with a transplant, and if so, what option to choose, Fitzhugh said. “We have to ensure that in each individual patient, the potential benefits outweigh the risks,” she said. A common reason that patients choose gene therapy is to not inconvenience family members or put them at risk from donating bone marrow, she added.
More recent protocols at NHLBI do not include children, given that 98 percent of children with sickle cell disease will survive to age 18, making it hard to justify any procedure with a significant risk of mortality associated with it. It is difficult to explain this to patients and advise them to wait because their disease is not severe enough at that time, Fitzhugh said. In her experience, she said, even after the risks associated with transplantation have been explained to them, some patients are surprised when they reject the graft.
As a result of those experiences, Fitzhugh said, she and her colleagues worked with an ethics team to study the process of decision making by sickle cell disease patients who decide to participate in high-risk clinical research. The ethics team conducted interviews with 26 patients to evaluate motivations, the decision-making process, the patients’ understanding of research, and retrospective reflections. Two-thirds of the patients were capable of clearly describing the purpose of research, and all patients were aware that transplant and gene therapy studies carry side effects and risks, including death, cancer, and GVHD. Of the 26 patients surveyed, 22 acknowledged that the treatment might not work, and the main concerns of the patients included worries that they would have an unsuccessful response
and might die, that they might experience pain, and that they might suffer from long-term side effects.
Most of the patients described performing a personal risk–benefit calculation when deciding about participation, and all patients who decided to enroll cited the intolerability of their sickle cell disease or the hope for a better future without the disease. Those who declined enrollment felt that their current status was not bad enough to justify the risks of the trial, and half of the patients who did enroll cited altruistic motivations, although none reported altruism as their primary motive for participating in the clinical trial. When asked what role family, faith, and other patients played in their decision making, most patients reported that family provided moral support and reassurance. Eleven of the patients had spoken to patients who had positive outcomes, five had spoken with patients who had negative outcomes, and seven of the patients had not spoken to other patients.
THE COMPLEXITIES OF PATIENT SELECTION, ENROLLMENT, AND CONSENT IN THE CONTEXT OF GENE THERAPIES TO TREAT SICKLE CELL DISEASE
Continuing on the theme of sickle cell disease, Tisdale said that both allogeneic bone marrow transplantation and autologous gene therapy work by either replacing or repairing bone marrow stem cells so that the body produces hemoglobin that will not polymerize and cause sickling (see Figure 3-1). As Fitzhugh mentioned, it was recently found that only 20 percent of white blood cells need to come from either repaired or replaced bone marrow in order for the disease to be reversed (Fitzhugh et al., 2017a).
Tisdale described one challenging issue related to the fact that the vector his team uses to deliver the correcting gene to bone marrow cells is derived from HIV. Sickle cell disease has a higher incidence in individuals of African American descent, and when some patients with the disease learn that the vector is based on the virus that causes AIDS, he said, they may think back to the Tuskegee experiment1 and become reluctant to participate in the trial. “The bottom line is that it takes a lot of education in the patient population and long-term follow-up of patients in a setting where they are getting care from physicians that they trust,” he said.
After conducting a series of mouse and large animal studies to determine the potentially therapeutic dose of the vector–gene construct to admin-
___________________
1 The Tuskegee experiment refers to a clinical study (Tuskegee Study of Untreated Syphilis in the Negro Male) conducted between 1932 and 1972 by the U.S. Public Health Service, which violated many bioethical research standards. The aim of the study was to understand the natural history of untreated syphilis; however, the African American men involved in the study did not receive complete and clear information about the study and its associated risks. Researchers also did not give study participants penicillin (a known cure for syphilis).
NOTE: HLA = human leukocyte antigen; SCD = sickle cell disease.
SOURCE: John Tisdale workshop presentation, November 13, 2019.
ister to humans, Tisdale and his colleagues designed a clinical trial to first study the construct’s safety profile, with efficacy as a secondary endpoint. As the trial proceeded, he and his colleagues made several adjustments to the stem cell source and manufacturing method, which resulted in more patients experiencing improvements.
Addressing how to include pre-symptomatic individuals in clinical trials for gene therapy, Tisdale said that the field has not yet reached a place where it can consider relaxing the stringent inclusion criteria that much. “We need to know it is working, and we need to quantitate the benefit in patients for whom the risk–benefit ratio favors the intervention,” he explained. “Once we have de-risked the procedure itself, or have the success rate known, then we can begin to apply that in pre-symptomatic patients.”
Getting the results of the trial back to the patients who participated in it is one part of the clinical trials process that needs improving, Tisdale said. His team now makes a point of going to patient advocacy meetings and holding meetings in their clinic to update patients on the results of ongoing clinical trials. “I think it is helpful to engage the patient population as real team members in this effort,” he said.
Regarding what to do if the initial application of gene therapy does not work, Tisdale said that for individuals who are in good shape, one possibility would be to follow-up with a matched sibling transplant after gene therapy fails. It would be beneficial to see long-term follow-up data
comparing the success of gene therapy with the success of cell-based therapy to know if one approach is better than the other, he said.
Another approach to treating sickle cell disease involves the use of new gene-editing technologies, such as those involving CRISPR/Cas9. One gene-editing method aims to boost levels of fetal hemoglobin in individuals with sickle cell disease. In the months following birth, babies stop making fetal hemoglobin and begin to produce adult hemoglobin. Certain individuals carry genetic mutations that lead to the persistent production of fetal hemoglobin, said Tisdale, and in someone with sickle cell disease these mutations are protective and result in a very mild form of the disease because fetal hemoglobin does not polymerize and cause sickling (NIH Director’s Blog, 2019). Although it is more difficult than turning the production of fetal hemoglobin back on, another gene-editing approach aims to correct the mutation that causes sickle cell disease (see Figure 3-2), Tisdale said, and his group has shown that it can correct approximately 30 percent of the hemoglobin genes and deactivate another 60 percent of the faulty hemoglobin genes, leaving only 10 percent of the faulty hemoglobin. “The majority of the hemoglobin in these red cells are now the correctly spelled beta globin protein,” he said, “and this is far in excess of the 20 percent we need to fix this disease.”
To explore how gene editing would be received by stakeholders, one group of investigators convened 15 focus groups in seven U.S. cities to
NOTE: GVHD = graft-versus-host disease; SCD = sickle cell disease.
SOURCE: John Tisdale workshop presentation, November 13, 2019.
explore attitudes and beliefs toward gene editing within the sickle cell disease community (Hollister et al., 2019; Persaud et al., 2019). Focus groups were shown a short educational video on somatic genome editing and its potential use for sickle cell disease and then given a survey related to genome editing and participation in future clinical trials. The survey was followed up by open discussion periods with the researchers. According to these studies, the factors that motivated people to participate in a gene-editing trial included hope in technology, altruism, the shortcomings of current treatment, and increased awareness of the importance of clinical trials. Deterrents included uncertainty about the consequences of gene editing, the permanence of the change to the genome, trial burden, patients’ mistrust of the medical community, reproductive risk, cost, and a lack of access. Mediating factors included religiosity and the capacity to manage disease and life. Patients reported that they wanted specific details about the trial, the expected interpatient variability, optimal timing, and the track record of the treatment. On a final note, Tisdale stressed that he had been talking about somatic cell–based gene therapy and gene editing, not working with germ-line cells, and that the National Institutes of Health (NIH) has been clear that none of this work will use human embryos.
THE COMPLEXITIES OF PATIENT SELECTION, ENROLLMENT, AND CONSENT IN THE CONTEXT OF GENE THERAPY FOR SEVERE COMBINED IMMUNODEFICIENCY
SCID, a condition in which the body cannot fight infections because it cannot mount an immune response, can result from defects in many genes, Puck told the workshop audience. Babies born with this disease start to lose weight from age 2 to 4 months, and they will not survive unless given a working immune system. This was first accomplished in 1968, when an infant boy with SCID received the first successful bone marrow transplant, using his sister as the donor. This boy is now a 52-year-old man who is healthy and was able to father a child, said Puck.
Because individuals with SCID do not have a working immune system, donors can be parents, matched unrelated donors, or cord blood, Puck said. One form of SCID, characterized by adenosine deaminase (ADA) deficiency, can be treated with the missing ADA enzyme. The first gene therapies used in humans were developed for ADA and X-linked forms of SCID, Puck said, and they are considered curative. Because most SCID cases are sporadic, newborn screening would be needed to identify infants prior to them developing infectious complications, Puck said, given that survival is compromised by infections at the time of the gene therapy treatment (Pai et al., 2014).
A specific form of SCID caused by the recessive mutation DCLRE-1CY192X in the Artemis gene2 occurs at a higher rate in the Navajo and closely related Apache tribes of Native Americans. This genetic variant was present in the survivors of the 1864 “Long Walk,” which killed 90 percent of the people who started the forced resettlement journey from Arizona to New Mexico, thus creating a genetic bottleneck that is thought to have increased the population incidence of this recessive gene. It is estimated that 1 in 2,000 Navajo and Apache infants have this form of SCID, Puck said.
In an unpublished study conducted at the University of California, San Francisco, by Morton Cowan, a small group of Navajo SCID patients were treated with a bone marrow transplant and followed for 24 years. Those individuals, diagnosed very early in life because they had an affected sibling, had a higher survival rate than individuals who were diagnosed because of infections, Puck said. Researchers have since discovered a blood-borne biomarker for SCID called T-cell receptor excision circles, which serves as a marker of T-cell maturation. Today, screening for SCID is part of the Recommended Uniform Screening Panel, a list of disorders that the Secretary of the Department of Health and Human Services encourages states to test for as part of newborn screening programs. However, when Puck and her colleagues undertook a SCID screening study in Navajos in 2009, this method was relatively new, and there was quite a bit of distrust among the Navajo based on past exploitation by population genetics researchers, Puck said. In fact, the Navajo institutional review board initially prohibited genetic testing on study samples and required written, face-to-face consent. Nonetheless, in 2011, after 1,800 research samples were collected and analyzed, the test was adopted reservation-wide as the standard of care.
Recent studies conducted by the Primary Immune Deficiency Treatment Consortium demonstrated that the survival rate after receiving a donor transplant depends on the exact SCID genotype, with the Artemis genotype having the worst survival rate, likely because the genetic defect results in impaired DNA repair, which is not isolated to the immune system (Haddad et al., 2018). This form of SCID is the most difficult to treat with allogeneic bone marrow cell transplant, Puck said, and autologous gene therapy may be a better approach. In collaboration with Scott McIvor at the University of Minnesota, Puck and her colleagues created a self-inactivating lentiviral vector containing the human DCLRE1C promoter, and as of October 2019 her group used that vector to treat four newly diagnosed infants and three older children with SCID who had been previously transplanted but developed insufficient immunity. Two of the infants and two of the older children were Navajo. Newborn screening will be a critical element of this
___________________
2 The Artemis protein is involved in V(D)J recombination, a process important for the development and maturation of T and B cells.
trial going forward, Puck said, because it provides an unbiased population-level ability to make an early diagnosis, which in turn promotes fair access to treatment.
In closing, Puck said that the Navajos now embrace SCID screening and early treatment, though achieving optimal outcomes is challenged by distance from medical facilities, poverty, and social difficulties. Having a network of trusted local physicians has been critical to this effort, Puck said, and she and her team travel to the reservation annually to hold SCID clinics. It is important to note that the entire therapeutic process for SCID can be extremely stressful for Navajo families who have to remain with their child for upward of 3 months as the child develops his or her own T cells.
THE CHALLENGES OF USING GENE THERAPY TO TREAT DUCHENNE MUSCULAR DYSTROPHY
As Richard Finkel noted in the first panel session, DMD is an X chromosome–linked childhood genetic disease. DMD most often affects boys, although in rare cases girls can develop a milder form of the condition (MDA, 2020). Pat Furlong explained that about 30 percent of cases of DMD are due to random spontaneous mutations in which there is no family history of DMD. Other families find out they are carriers of a DMD causative genetic variant after they have had children. The standard of care for individuals with DMD has evolved over the years, with corticosteroid treatment now enabling them to walk until they are 10 to 13 years old, but most affected children still lose mobility in their arms by age 16 or 17, require noninvasive ventilation in their late teens, and die at an average age of 28.
Dystrophin, the gene responsible for DMD, is one of the largest genes in the human genome and was first cloned in 1986 (Monaco et al., 1986). Researchers have identified more than 1,000 mutations in the dystrophin gene that result in DMD. When the protein product of dystrophin was identified in 1987, Furlong said, many investigators were confident that gene therapy for DMD would be straightforward. However, she continued, more than 30 years later clinical trials for a gene-based therapy for DMD are just beginning. The trials are using a viral vector that delivers a small piece of the dystrophin gene because the entire gene is too large to deliver successfully with existing vectors.
Parents who have been recruited to put their children in trials often recall being shown an image of a 65-year-old man with DMD playing tennis, Furlong said. This individual is asymptomatic and provided the rationale for researchers to develop a synthetic, shortened version of dystrophin. The near-universal response when parents see this man’s image, she said,
is a desire to access this treatment for their children. However, she added, what they may not be told is that this man was not given the gene therapy construct, known as a microdystrophin, but instead has had this microdystrophin existing naturally in his genome since birth. Nonetheless, the clinical trial has started to recruit 4- to 7-year-olds. These early trials require the child and a parent to stay at the clinical site for approximately 30 days after treatment, Furlong said. Parents are worried about the age restrictions, especially if their children are slightly older or younger than the target age range, Furlong said. Parents also worry about protecting their children from environmental exposures that might trigger an immune response to the viral vector if and when they are accepted into a trial, she explained.
The requirement to stay at the clinical site means that families without resources or support at home or who have employers who may not be enthusiastic about granting 30 days of leave are unlikely to participate in the trial. Furlong said she has seen families move to be near a clinical site or take out second mortgages on their homes to pay for the indirect costs associated with participating in a gene therapy clinical trial. The current informed consent process is not very informative, Furlong said, and families may not even realize that by agreeing to have their child participate in a gene therapy trial, they are opting out of every one of the other 27 active clinical trials taking place in the DMD space.
Aside from worries about the muscle biopsies that are done under anesthesia and the doubling of the steroid doses that comes with getting the therapy, another common parental concern is whether the child is receiving the experimental treatment or is part of the control group that will eventually receive the active therapy once the trial is over. Furlong also said that in some cases the elevated steroid dose results in negative side effects and behavioral issues in the children. All of these factors, she said, create a large burden on families while also giving them hope.
One concern brought up by Furlong involves muscle turnover in those children treated with gene therapy. Because it is unclear if children can be re-dosed with gene therapy, she asked, will cases of teenage-onset DMD appear? She said that it is important to think about when to deliver subsequent doses of the gene therapy, given that waiting until older boys start showing signs means that there will be parts of their body subject to irreparable damage from which their bodies cannot recover.
A final issue Furlong mentioned was the question of what to tell the children who participate in these clinical trials. She recalled one boy who was in the trial but not showing improvement, and he started acting out likely because of his disappointment that the “magic medicine” was not working as well as everyone had hoped. Information that is communicated to children and families involved in pediatric gene therapy trials is impor-
tant because they look forward to these types of therapeutic opportunities, Furlong said, but the trials come with many complicated issues.
PATIENT AND FAMILY PERSPECTIVES
Contreras, whose family lives in Chile, has two sons with DMD and her older son, 5-year-old Franco, is enrolled in the microdystrophin gene therapy clinical trial. She and her husband learned about gene therapy and clinical trials for DMD after months of intensive research, which led them to contact the organizers of three clinical trials. One researcher got back in touch with the Contreras family and told them that the trials were not accepting international patients. The result was that the family secured visas that allowed them to move to the United States and find work. “We are so grateful, and at the same time very aware of all of the families around the world, but also within the United States, that just cannot afford the direct and indirect costs of participating in a gene therapy clinical trial,” she said.
One concern of Contreras and her husband was that their son would receive a placebo, a risk they were willing to take. What was not acceptable to them was the possibility that their son would receive a suboptimal dose of the therapy, so they chose to prioritize participating in the clinical trial that did not include a dose escalation component. After Franco was accepted into a trial, the family was told to keep their younger son Julián away from Franco for the next 30 days so that he would not be exposed to the viral vector, develop immunity to it, and then be ineligible for accessing gene therapy in the future. As a result, her husband Pablo took a 24-hour, round-trip journey from Columbus, Ohio, to Santiago, Chile, to take Julián back and made it to Ohio in time for Franco’s infusion. A few days later, Pablo returned to Chile while María stayed in Columbus with Franco.
Contreras said that for her family, one of the most sensitive ethical issues is a lack of a sibling protocol. “We are giving Franco an opportunity that at this point we are not sure we will be able to provide to Julián,” she said. For pediatric incurable diseases such as DMD, the need for a sibling protocol is urgent, she said. Other needs, Contreras said, include an expedited regulatory process and improved trial accessibility for patients in early and advanced phases of the disease, for international populations, for boys and girls, and for families that cannot afford to participate in clinical trials. Trial design could be improved by the development of innovative protocols that minimize placebo arms and use natural history studies, master protocols that may combine different therapeutic approaches, and the increased involvement of patients and families in protocol design. “Our voices should be heard not only when we say yes and sign the consent form, but throughout the process,” she said in closing.
Another perspective on these issues came from Samuels, a participant in a gene therapy clinical trial for sickle cell disease. Diagnosed at age 2, her symptoms grew progressively worse, and by the time she was 7 she experienced her first aplastic anemia crisis, which occurs when bone marrow does not produce enough red blood cells. At 13 years old, Samuels suffered a transient ischemic attack, which for months presented as a mild stroke that affected the left side of her face and arms. Soon after, the pain associated with her disease became severe enough that by the time she was a sophomore in high school, she was being home schooled so that she could continue to get an education while being treated with high doses of opioids, monthly blood transfusions, and nightly 10-hour infusions of deferoxamine mesylate to prevent the iron toxicity related to those blood infusions.
Samuels detailed more of the medical emergencies she suffered over the years and noted that she got married in 2008 and had an ectopic pregnancy that triggered another crisis in 2011. In 2015, she was told that liver dialysis would be the next step, which set her off on a persistent search for a clinical trial to join. That persistence paid off, and she enrolled in the NHLBI trial using an autologous gene therapy transplant rather than requiring a matched donor bone marrow transplant.
The gene therapy process that Tisdale and his colleagues laid out for her in great detail, both in writing and verbally, was almost too much to take in for her and her family, Samuels said. “I was contemplating signing up for something that would put my body through high doses of chemo, make me menopausal before the age of 40, take months in a hospital, and agree to at least 3 years of monitoring, and when I considered those things and what I had already gone through, this to me was a godsend,” she explained. “It took a year for me to convince my family of that fact, but I got them to see that as well.”
When Samuels went to the clinical center at NIH in March 2018 for the gene therapy, she did not know what to expect, she said, and it was difficult to keep her emotions in check. However, she said, being surrounded daily by doctors, nurses, nutritionists, and other members of the care team made it a bit easier to trust the process, despite all of the ongoing challenges. It also helped to be able to call people who had gone through the debilitating chemotherapy process before, Samuels said. On the day of the workshop, which took place almost 2 years after the gene therapy administration, Samuels reported that she no longer experiences daily pain, no longer needs narcotics to get out of bed, and has not needed a blood transfusion since August 2018. Today, she said, she makes it a point to “live out loud,” because she was unable to do so for such a long time. “I came here to maintain my hope in the health and rebirth of what science is doing today,” she said in closing.
Bartek began his short presentation by explaining that there is no current gene therapy clinical trial for Friedreich’s ataxia, a genetic neuromuscular disorder, although there are groups currently advancing gene therapy research programs for this disease. The issue of developing gene therapies for rare diseases was the focus of a 2-day workshop held in 20183 that was sponsored by the National Center for Advancing Translational Sciences (NCATS) and FDA’s Office of Tissues and Advanced Therapies. “For 2 days, we heard academic and industry investigators talk about the issues that were confounding their gene therapy trials,” Bartek said.
From those presentations, he said, it was clear that while the successes achieved differed from trial to trial, there were several shared challenges. He compiled a list of five to six issues that were confounding all of these investigations, and after sharing this list with the NCATS leadership, a new program was created under the auspices of the Cures Acceleration Network Review Board to develop advanced technology platform solutions to address the issues he had identified.
Turning to the family perspective on gene therapy clinical trials, Bartek said that safety is a concern, but given that there are no approved treatments for many of these diseases, families are often willing to take substantial risks. Perhaps the gravest concern of families is whether their loved ones will receive a therapeutic dose of the investigational agent. In his opinion, Bartek said, the paradigm of starting trials in adults or only symptomatic patients to prove safety has to change, especially because time is of the essence for pediatric patients; similarly, he said, the assumption that gene therapy is to be a “one-and-done” treatment also must change. “Can we either use different vectors or different routes of administration so that over time we can get a second dose?” he asked. He also posed several other questions, including
- Are there alternatives to a placebo in a clinical trial?
- Is it possible to work around exclusion from a trial based on a previous exposure to these viral vectors?
- Is it possible to know in advance how therapeutic a gene therapy will be?
- Can we educate the patient and the family about expectations?
- How long will it take the field to get to these particular rare diseases?
___________________
3 The August 2018 workshop, The Growing Promise of Gene Therapy Approaches to Rare Diseases, was jointly sponsored by the National Center for Advancing Translational Sciences and the Food and Drug Administration’s Center for Biologics Evaluation and Research. More information about the workshop and the agenda can be found at https://events-support.com/events/NCATS_Gene_Therapy_2018 (accessed February 19, 2020).
Regarding the last question, Bartek said that there are some 7,000 rare genetic diseases and that the current pace of development is far too slow to treat a meaningful number of these diseases in the foreseeable future.
One recommendation Bartek made was for the field to support the creation of a common manufacturing facility that would have capacity to provide sufficient material for a small Phase 1 clinical trial led by academic investigators rather than leaving very early investigations to a pharmaceutical company that would have to invest $200 million in a manufacturing facility before even starting a clinical development program. Another recommendation from Bartek was for NCATS and FDA to collaborate on developing a standardized clinical trial design that would be widely applicable to rare diseases. In conclusion, Bartek said, it is important that novel gene therapies be fairly priced in order to promote equitable access for patients and sustainable reimbursement for payers.
DISCUSSION
Following the presentations, all of the speakers participated in a moderated panel discussion that included questions from the audience. Points raised in this discussion period primarily centered on the development of educational materials for patients and families and issues with the informed consent process.
Patient- and Family-Centered Educational Materials and Access to Statisticians
Educational materials about new technologies such as CRISPR/Cas-9, zinc finger nucleases, and other gene-editing techniques would be useful in helping patients and families understand the clinical process, one workshop participant said. NIH is developing such materials to make the process more understandable for patients and their families, Tisdale responded. The reason that his team uses CRISPR/Cas-9 instead of some of the other gene-editing techniques has to do with patents and ready access to the technology, he said. The participant went on to ask the two NIH speakers why their gene therapy trial stopped using the hydroxyurea treatment, which can reduce the number of pain episodes substantially. Tisdale answered that hydroxyurea also reduces the number of bone marrow stem cells that can be harvested from patients, which would reduce the likelihood of reaching the therapeutic target dose. Thus, in order to harvest sufficient quantities of bone marrow stem cells from patients, hydroxyurea treatments stop and exchange transfusions are used instead, Tisdale said, as a way to yield a better product that is less prone to downstream complications.
A workshop participant asked if any of the patients or family members on the panel had a chance to discuss concerns with statisticians involved with a clinical trial before or during enrollment. Samuels said no, but said she thought that might be a good option in the future. Bartek said that the Friedreich’s Ataxia Research Alliance has had tremendous access to the statistical community, including enlisting statisticians to help fortify and analyze the organization’s natural history database. Recently, in collaboration with FDA and the National Organization for Rare Disorders, his organization has populated its natural history database by adding placebo arm data from clinical trials with help from statisticians. Furlong added that Parent Project Muscular Dystrophy has been working with the Critical Path Institute to develop a disease progression model.
The Informed Consent Process
Regarding informed consent, Cho said that many patients and families are experts in their own diseases and understand the risks and benefits of participating in a clinical trial. At the same time, she said, empirical studies have shown that while people may understand that there are risks of death and severe morbidity from participating in clinical trials, the issue is the extent to which individuals believe that they are in the population that is going to be subject to those risks. Given the difficulty that people have of reconciling the existence of risks in clinical trials with their beliefs about their chances for successful outcomes in those trials, she asked if the panelists had any suggestions for overcoming that cognitive dissonance. A gene therapy consent form can be more than 20 pages of tiny, single-spaced type, making it nearly impossible to explain everything in it in one sitting, Puck said, let alone allow the patient to take in and process all of that information. She suggested that it would be a good idea to have many conversations about participating in gene therapy clinical trials and to let people know that they maintain control over stopping their participation right up to the moment they are infused with the treatment, at which point they need to be partners with the research team going forward. “I think communication, trust, and partnership do not develop over one consent form,” Puck said. Instead, it takes days or weeks, at least, to develop this sort of relationship, she said.
Fitzhugh agreed with Puck and said she asks patients and families to take the consent form home, read it on their own schedule, and write down any questions they have. She also counsels them to talk to their own family physicians and to reach out to other patients. Samuels said that she received the consent form 1 year before she decided to sign it, giving her time to pore over the document and come to her own conclusions. In addition, she
said, she had had several conversations with the research team nurses and a meeting with family and friends before she signed the form.
The process should shift from a consent form as a timepoint event to a consent process, Contreras said, and the research teams should always suggest reaching out to other patients who have gone through the procedure. In the case of her son Franco, she said, the family was asked to re-consent to a change in the protocol that would double the steroid dose after receiving the therapeutic agent. “I would have appreciated if I had received more information from the sponsors and the medical team as to why these changes were made and what the rationale was for making them,” she said. Furlong suggested that it might be helpful if there were a summary page preceding the actual consent form that would refer to the most important pieces of information. Similarly, for re-consent, the front page could point to the exact spot in the original consent form to which proposed changes apply.
Panelists were asked if they had ever encountered cases where parents and children were at odds over signing the consent form. Furlong said that typically young boys with DMD do what their parents say, and Puck said that the children she has worked with and who are old enough cognitively to understand the process have been extremely enthusiastic about participating in a clinical trial, even after learning that the procedure may not work for them.


















