
The workshop’s next session explored the implications of the long-term clinical management of patients who participate in gene-based clinical trials and discussed how data from a limited number of patients can be used effectively to determine if a gene-based therapy is safe and effective. The session was moderated by Michael DeBaun, a professor of pediatrics and medicine, the vice chair for clinical and translational research, and the J.C. Peterson Endowed Chair in Pediatric Pulmonology at Vanderbilt University and director of the Vanderbilt-Meharry Center for Excellence in Sickle Cell Disease. Tejashri Purohit-Sheth, the director of the Division of Clinical Evaluation, Pharmacology, and Toxicology in the Office of Tissues and Advanced Therapies, Center for Biologics Evaluation and Research at FDA, discussed the agency’s recommendations regarding long-term follow-up of gene-based therapies. Leslie Robison, the chair of the Department of Epidemiology and Cancer Control and a co-leader of the Cancer Control and Survivorship Program at St. Jude Children’s Research Hospital, spoke about his experience with the long-term surveillance of pediatric and adolescent cancer survivors. David Chonzi, the vice president for pharmacovigilance and epidemiology at Allogene, addressed long-term follow-up for gene
and cellular therapies. Bruce Marshall, the senior vice president of clinical affairs at the Cystic Fibrosis Foundation, discussed the foundation’s role in addressing post-approval regulatory obligations. Bob Levis, one of the early recipients of experimental chimeric antigen receptor T cell (CAR T) therapy for chronic lymphocytic leukemia (CLL), provided a patient’s perspective on long-term follow-up studies.
PERSPECTIVES FROM THE FOOD AND DRUG ADMINISTRATION ON LONG-TERM FOLLOW-UP STUDIES
In her presentation, Purohit-Sheth summarized FDA’s July 2018 draft guidance, Long-Term Follow-Up After Administration of Human Gene Therapy Products.1 Long-term follow-up, she explained, refers to monitoring for adverse events for an extended period of time, which is specified in the clinical studies protocol. The follow-up is meant for individuals who were in clinical studies following the completion of the study as well as for patients receiving gene therapy products after FDA approval. Depending on the risk characteristics of a specific product and what the agency understands about that product, it may not require long-term follow-up, she added.
FDA may require long-term follow-up for gene therapy products because they are intended to achieve a prolonged or permanent therapeutic effect and, as such, long-term exposure may produce unpredictable or unexpected delayed risks for a patient receiving that therapy. Delayed risks can include malignancy, impaired gene function, autoimmune-like reactions, a reactivation after latency and infection, and resistant infections. All of these potential delayed risks depend on the type of vector used in the gene therapy product, Purohit-Sheth said. FDA takes the following characteristics into account when determining which gene therapy products have an increased risk for adverse events:
- the integration activity of the product,
- whether it is a gene-editing construct,
- if the transgene is expressed for a prolonged time,
- if the potential for latency exists, such as with replication-competent herpes virus vectors, and
- if the vector establishes a persistent infection, as occurs with listeria vectors.
___________________
1 The draft guidance is available at https://www.fda.gov/regulatory-information/search-fdaguidance-documents/long-term-follow-after-administration-human-gene-therapy-products (accessed December 23, 2019).
In general, Purohit-Sheth said, the products that FDA considers to have a greater risk of delayed adverse events include those that use integrating viruses, viruses capable of latency reactivation, and genome-editing products. Products considered to have a lower risk of delayed adverse events include those using plasmids, poxvirus, adenovirus, and AAVs. However, if a plasmid has been modified to have the ability to transfer or modify genetic elements, it may be considered to be a higher-risk product, she said.
As part of its draft guidance, FDA has developed a framework to assess the risk of delayed adverse events associated with a gene therapy product (see Figure 5-1). As part of its risk assessment, FDA looks at preclinical data to provide information about the localization, distribution, and persistence of the gene therapy product and to understand possible on-target and off-target effects. When designing preclinical studies, it is important to take into account the gene therapy formulation and the route of administration intended for human use, Purohit-Sheth said. Such studies should also evaluate the product in both male and female animals and evaluate whether product persistence and localization correlate with any adverse effects that may have been muted. Biodistribution studies should use the maximum feasible or clinical dose, and preclinical work should also include kinetic studies. FDA recommends that animal sacrifice should occur at the peak of
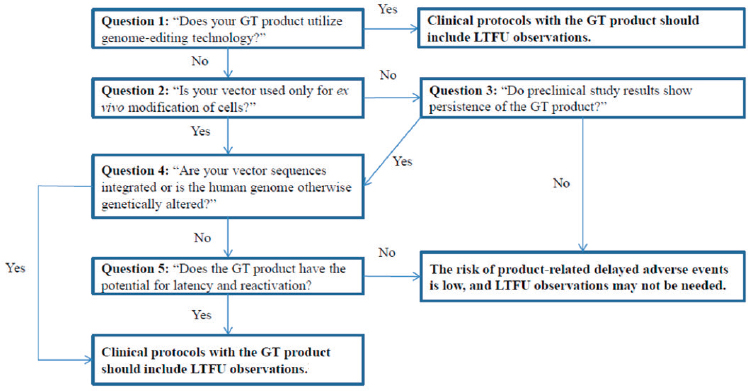
NOTE: GT = gene therapy; LTFU = long-term follow-up.
SOURCES: Tejashri Purohit-Sheth workshop presentation, November 13, 2019. Originally from the FDA draft guidance document Long-Term Follow-Up After Administration of Human Gene Therapy Products (p. 6).
gene therapy product detection and at later timepoints to provide information on product clearance.
Preclinical studies should contain a minimum tissue panel analysis that includes blood and tissue from the injection sites, gonads, brain, liver, kidneys, lung, heart, and spleen as well as additional tissues dependent on the product, vector type and tropism, and the route of administration, Purohit-Sheth said. It is critical when assessing vector persistence and distribution that the assay methodology be both quantitative and sensitive, she said. Assays should be able to detect vector sequences in both animals and humans.
Turning to the subject of clinical considerations for long-term followup studies, Purohit-Sheth said that the agency considers the goals of long-term follow-up, choice of subjects, and study duration. Informed consent for trials that include long-term follow-up will have to include provisions for post-trial consent. The goals of long-term follow-up, she said, are to identify delayed risks associated with exposure to the gene therapy product and to gain insights into the persistence of the gene therapy in the body. The long-term follow-up population, she added, will include all subjects who received the gene therapy in a clinical trial. When designing the long-term follow-up protocol, it is important to consider life expectancy based on the underlying disease, the possibility of multiple comorbidities, and exposure to other agents such as radiation and chemotherapy, which can have their own long-term adverse effects. The duration of a follow-up study should be sufficient to assess any possible adverse events, taking into account the product’s characteristics, such as the observed duration of in vivo product persistence, the observed duration of transgene expression, and other in vivo product characteristics observed during preclinical and clinical studies.
Other considerations include the nature of the exposure to the product, its target organ or cell, and expected survival rates and known background rates of survival in the study population. For integrating vectors and genome-editing products, FDA recommends that long-term follow-up studies should last for 15 years, while for AAV vectors, FDA recommends a 5-year follow-up period. All follow-up studies should proceed using a dedicated clinical protocol with prespecified patient visit schedules, a prespecified sampling plan, the methodology that will be used to assess the persistence of vector sequences, the clinical events that will be monitored, a means of collecting accurate case histories, and a health care provider template for non-investigator caregivers. The protocol should also specify how adverse events will be reported to FDA, how they will be discussed in annual reports, and the procedure for submitting any necessary protocol amendments, such as the need to assess a new risk.
For the first 5 years of a long-term follow-up study, FDA recommends having a detailed plan for scheduled visits and the information to be col-
lected at each visit. Case histories, Purohit-Sheth said, should contain information about any exposure to mutagenic agents and the emergence of new medical conditions of interest. Over the subsequent 10 years, FDA recommends contacting subjects at least yearly by phone, office visit, or questionnaire.
Informed consent should explain the purpose of the long-term followup study, the expected participation and procedures, foreseeable risks, scheduled study visits, and tissue and data collection procedures as well as the basic elements required for any clinical study. It is important to explain the possible adverse events so that the patient can understand the risks and know what to look for over time, Purohit-Sheth said. Informed consent should also include a request, in the event of a patient’s death, for consent for autopsy. On a final note, she said that FDA discusses with applicants at the time they submit their biologics licensure application that they will need to have a post-marketing pharmacovigilance plan that includes routine surveillance. Depending on a particular product’s risks, a risk evaluation and mitigation strategy may also be required.
LONG-TERM SURVEILLANCE OF EXPOSED PEDIATRIC AND ADOLESCENT CANCER SURVIVORS
Survival rates for childhood cancer in the United States are exceptional, Robison said, with 5-year survival rates now exceeding 83 percent. Approximately 1 in 750 U.S. residents is a childhood cancer survivor, with the number of survivors expected to approach 500,000 by 2020 (Robison and Hudson, 2014). Robison noted that this is a small, heterogeneous population for which there are recognized long-term consequences related to the treatment these individuals received as children. To better understand those consequences, the National Cancer Institute (NCI) has funded two large childhood cancer cohorts: the Childhood Cancer Survivor Study (CCSS) cohort of nearly 36,000 survivors from 31 centers across the United States and the St. Jude Lifetime (SJLIFE) cohort of more than 8,200 survivors (see Table 5-1).
The CCSS cohort, now in its 24th year, was originally assembled in response to the realization that many pediatric cancer survivors were not being actively surveilled, Robison said. The majority of the cohort participants had been involved in clinical trials through NCI’s Cooperative Clinical Trials groups, but two-thirds of the patients who had been diagnosed and treated between 1970 and 1985 had not been seen by a pediatric oncologist for more than 10 years. CCSS is currently following more than 24,000 survivors who are distributed geographically across the nation, which Robison said creates some significant challenges in terms of monitoring, contacting, and overall follow-up. The SJLIFE cohort includes
TABLE 5-1 Pediatric Cancer Survivor Cohort Characteristics
| Characteristic | CCSS (Dx 1970–1999) | SJLIFE (Dx 1962–2012) |
|---|---|---|
| Cohort size | 35,937 (24,000+ active participants) | 8,245 (4,688 participants to date) |
| Entry criteria | ≥5 years from diagnosis | ≥5 years from diagnosis |
| Age at cancer diagnosis | <21 years | <25 years |
| Cancers | Leukemia, CNS, HL, NHL, neuroblastoma, soft tissue sarcoma, Wilms, bone tumors | All diagnoses |
| Study design | Retrospective cohort with prospective follow-up, hospital-based | Retrospective cohort with prospective follow-up, hospital-based |
| Methods of contact | Surveys | Clinic visits and surveys |
| Comparison population | Siblings, general population | Frequency-matched community controls, general population |
| Therapeutic exposures | >90% | 100% |
| Ascertainment methods | Self-report, pathology reports, NDI | Med. assessment, self-report, med. record, NDI |
| Collection of germline DNA | >60% | >95% |
NOTE: CCSS = Childhood Cancer Survivor Study; CNS = central nervous system; Dx = diagnosis; HL = Hodgkin lymphoma; NDI = National Death Index; NHL = non-Hodgkin lymphoma; SJLIFE = St. Jude Lifetime.
SOURCE: Leslie Robison workshop presentation, November 13, 2019.
only patients diagnosed and treated at St. Jude Children’s Research Hospital since the institution opened its doors in 1962. All of the participants in both cohorts are at least 5-year cancer survivors.
The CCSS cohort, Robison said, is completely survey-based, with most of the data self-reported by the participants. The SJLIFE cohort is clinically based, with participants returning to the hospital for 3 to 4 days of evaluation. Both cohorts have comparison populations to provide an idea of what events might be expected to occur normally over time and at what rate in an appropriate age-, sex-, and race-matched population. The CCSS cohort uses siblings as its control group, which Robison said is very good for evaluating certain aspects while not as good for evaluating psychosocial or sociodemographic outcomes. SJLIFE, on the other hand, relies on a community control group. For both cohorts, Robison and his colleagues collect detailed therapeutic exposure information and tissue samples that are banked for future study.
Addressing the challenges of assembling a cohort retrospectively, Robison said that 18 percent of the potential CCSS participants either never responded to attempts to reach them or actively refused to participate. Of the approximately 14,000 survivors who initially agreed to join the cohort, 1,300 provided extensive self-reported health information but declined to sign a medical release allowing the researchers to obtain their complete medical records. Ultimately, Robison said, only 72 percent of the eligible population was successfully recruited to join the study, a rate of success that he believes would have been higher had those individuals not been lost in the first place. “We are advocating very strongly that, going forward, this should be done on a prospective basis, consenting at the completion of therapy and starting to collect information on a periodic basis going forward,” Robison said.
One of the strengths of assembling a cohort of survivors, Robison said, is that it is possible to look at multiple outcomes, which is important, given that pediatric cancer survivors are at risk for many different types of adverse outcomes. Data from the SJLIFE cohort revealed that the prevalence of a variety of adverse events, such as the occurrence of abnormal pulmonary function, hearing loss, heart valve disorder, and breast cancer in female survivors, increased over time and that, when assessed in the clinic, survivors were found to be experiencing multiple health issues (Bhakta et al., 2017). “By 45 years of age, on average, a survivor will experience approximately four severe, disabling, or life-threatening conditions,” Robison said. For measuring long-term outcomes, he added, the control group is very important for understanding how much of an increased risk of morbidity there may be for pediatric cancer patients.
Bringing patients back to the clinic for follow-up studies would probably not have been realistic, Robison said, without the very large philanthropic support his team has received for studying the SJLIFE cohort. It is possible to link the cohort to the National Death Index to identify individuals who may be lost to follow-up, and NCI is in the process of creating a virtual national cancer registry that can be used to identify whether any cohort member is subsequently diagnosed with cancer anywhere in the United States. His team has also linked to organ transplant and assisted reproduction registries, but, he said, long-term follow-up in the United States is limited compared with other countries where there is high-quality record linkage.
Various mobile health applications combined with machine learning and artificial intelligence may also provide long-term insights. Robison’s team, for example, is starting to rely on a mobile health application for self-reporting symptoms on a regular basis. This approach has enabled the team to achieve high participation rates for self-reporting of symptoms on a daily basis and of the symptoms’ impact on quality of life on a monthly basis.
In closing, Robison said that his team is very interested in looking at the lifetime outcomes in order to better understand whether pediatric and adolescent cancer survivors, as a result of their cancer and treatment exposures, experience an earlier onset of disease and morbidity than the general population. Concerning gene therapy follow-up, he said that it will be important to have long-term and constant surveillance because there will likely be emerging and late-occurring events within those populations.
LONG-TERM FOLLOW-UP FOR GENE AND CELLULAR THERAPIES
Long-term follow-up for cellular therapies, Chonzi said, should include monitoring for adverse events such as secondary malignancies, autoimmune disorders, new persistent hematological disorders, and other issues such as hypogammaglobulinemia and infections. Following persistence is also important because it is not yet known if all cellular products have the same persistence characteristics in humans.
Before deciding whether to conduct a long-term follow-up study, Chonzi said it is important to ask several key questions:
- Does the product use genome-editing technology?
- Are vector sequences integrated, or is the human genome otherwise genetically altered?
- Does the product have the potential for latency and reactivation?
- Have any specific issues been raised during preclinical studies?
- How long will the study have to be run in order to detect the possible adverse events of interest and concern, particularly if secondary malignancies are a concern?
Because his organization is conducting studies globally, Chonzi said, it also has to pay attention to guidances issued by non-U.S. regulatory agencies. While those are largely consistent with FDA guidances, there are differences that create challenges for sponsors (e.g., guidance on replication-competent retrovirus testing after the first year).2 “We are hoping that as time goes on there is going to be uniformity between the regulators as to how we monitor and follow our patients long-term,” Chonzi said. Regarding CAR T therapy, most of the therapies use gamma retroviruses
___________________
2 For more information, see the 2018 FDA guidance at https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/testing-retroviral-vector-based-human-gene-therapyproducts-replication-competent-retrovirus-during (accessed January 24, 2020) and the 2009 European Medicines Agency guidance at https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-follow-patients-administered-gene-therapy-medicinal-products_en.pdf (accessed January 24, 2020).
or lentiviruses to deliver CAR-encoding sequences into T cells, and these viruses can integrate or have the potential for latency followed by reactivation. Both CAR T products that are on the market have post-license, 15-year long-term follow-up studies ongoing, he noted.
Allogeneic CAR T therapies, which use genome-editing technology, are also under development and have entered clinical testing. At this point, Chonzi said, it is unclear if there will be differences in how autologous and allogeneic cellular therapies will be followed over the long term. Also unclear, he said, is how sponsors will follow integration and genotoxicity over the long term. It would be helpful if regulatory agencies provided more clarity on monitoring for the off-target effects of genome editing and insertional mutagenesis, Chonzi said. He also wondered if there is a way of combining long-term follow-up data from studies with data from the post-marketing experience. Doing so, he said, would allow for harmonizing the two collection methods and two approaches to following patients. Patients in studies could be enrolled in the same registries used for commercial purposes, Chonzi suggested, especially when patients finish the active follow-up of a study.
In closing, Chonzi emphasized the importance of having all stakeholders share their experiences and lessons from long-term follow-up studies. Some groups have done so, he said, but more data are needed on secondary malignancies, autoimmune disorders, and persistent hematological disorders. The field of cellular therapy is growing considerably, and more studies will be coming, he said, adding that “it is even more important for us at this moment in time to try and harmonize how we are collecting the data so that it becomes easier and easier.”
ROLE OF THE CYSTIC FIBROSIS FOUNDATION IN ADDRESSING POST-APPROVAL REGULATORY OBLIGATIONS
Tracking patient outcomes after a gene therapy clinical trial may be improved through access to a patient registry. While there is currently no FDA-approved gene therapy for cystic fibrosis on the market, there are potentially important lessons to learn from the experience of the Cystic Fibrosis Foundation in building their patient registry and using it to follow clinical outcomes in the long term. The Cystic Fibrosis Foundation’s patient registry, which was started in the 1960s, is one of the organization’s crown jewels, Marshall told the workshop. Each night the 133 participating clinical centers download their data to the registry, which then provides analysis back to the centers through a Web-based application called CFSmartReports. The registry also allows for the generation of patient summary reports, population management reports, and clinical trial eligibil-
ity reports, all of which are helpful to clinicians because they bring registry data back to the point of care, Marshall said.
Another important way that the patient registry has been used is in supporting the pharmaceutical industry with post-marketing analyses, Marshall said. He added that FDA approvals of drugs to treat cystic fibrosis have come with post-approval requirements and commitments, which have included
- a 10-year prospective observational study to assess the risk of fibrosing colonopathy3 for reformulated pancreatic enzymes;
- a 5-year prospective observational study to assess the risk of antibiotic resistance to a new inhaled antibiotic; and
- a 5-year prospective observational study to assess the safety of a new modulator of the cystic fibrosis transmembrane conductance regulator (CFTR).
For the first study, in which the goal was to determine the incidence of fibrosing colonopathy, one major challenge was getting all of the different sponsors to harmonize their protocols for identifying fibrosing colonopathy, Marshall said. Anonymized registry patients at participating sites served as the denominator in calculating the incidence rate, and those patients did not require separate consent, he said. The numerator was derived from an IRB-approved, patient-consented study in which suspected cases of fibrosing colonopathy were adjudicated by an expert review panel.
The inhaled antibiotic study, Marshall said, was also IRB-approved and patient consented. The sponsor of this trial established a central laboratory to collect annual respiratory cultures and a standardized approach to collecting and analyzing the specimens; the results of the trial were linked to clinical outcomes in the Cystic Fibrosis Foundation’s registry. For this trial, FDA mandated testing for antibiotic susceptibility, but the sponsor and the Cystic Fibrosis Foundation both agreed it was also important to track clinical outcomes. The data collected in this study, which ended in 2019, did indicate that resistance patterns increased, but with no effect on clinical outcomes, Marshall said.
For the first CFTR modulator, which was approved in 2012, the sponsor conducted an observational study to evaluate the long-term safety of the product in patients with cystic fibrosis. This study used existing anonymized registry data to compare those on the drug to a propensity-matched com-
___________________
3 Fibrosing colonopathy is a potential side effect of high doses of pancreatic enzymes (used to manage pancreatic insufficiency in cystic fibrosis patients) and is characterized by abdominal pain, vomiting, bloody or persistent diarrhea, and insufficient weight gain or weight loss (Atlas and Rosh, 2011).
parator group. The outcomes measured included lung function, pulmonary exacerbation and hospitalization rates, mortality, and the number of lung transplants. An interim analysis of the study data confirmed the effectiveness of this drug at reducing hospitalizations, pulmonary exacerbations, mortality, and the need for organ transplantation and for stabilizing lung function over time (Bessonova et al., 2018).
Looking to the future, Marshall said that over the next 5 years new CFTR modulators should increase the percentage of cystic fibrosis patients who benefit from therapy from about 6 percent to 91 percent. As these therapies are used in infants and life expectancy normalizes for cystic fibrosis patients, there will need to be new approaches to following these individuals over the long term, Marshall said.
In closing, Marshall credited the foundation’s strong infrastructure, which has been in place since the 1960s, as a key factor for success in developing a registry that can facilitate a robust post-marketing research program. He also noted the importance of the ongoing relationships the foundation has with pharmaceutical sponsors. “They knew us. They knew of our Care Center Network, and they knew of our registry, so, it was easy to develop a business relationship with them,” he said. “We also had credibility with the FDA in terms of our registry.”
A PATIENT’S PERSPECTIVE ON LONG-TERM FOLLOW-UP STUDIES
As the fourth patient to receive CAR T therapy for CLL at Penn Medicine in 2013 and one of the first to receive a second round of CAR T therapy in 2017, Levis provided a patient’s perspective of what it is like to participate in a clinical trial—and as he put it, be genetically modified.
First diagnosed with CLL in 2002, Levis was treated initially with the then-standard chemotherapy regimen, which kept his disease in remission for more than 3 years. When he relapsed to a very aggressive form of the disease, he joined a clinical trial testing a promising new antibody-based drug, but he was randomized to the comparison drug, which did not work for him. With 3 to 4 pounds of tumor burden, his platelet and hemoglobin levels falling dangerously low, and little time left to live, Levis joined the first trial for CLL CAR T therapy at the University of Pennsylvania.
On March 12, 2013, he received an infusion of his genetically modified T cells and was told to prepare for challenges ahead. “Sure enough, 8 days later I developed these fevers, was hospitalized, heart rate elevated,” Levis recalled. “It was horrible.” Eight days later, the fevers broke, and the 30 palpable lymph nodes had returned to normal. His hemoglobin and platelet levels had started rising, and a bone marrow biopsy conducted 1 month
later showed no trace of the disease. “It is like magic,” Levis said. “You cannot believe it. You have got your life back.”
Unfortunately, the disease returned 3.5 years later as a slowly progressing form that responded to antibody-based therapy. However, there was still disease present in his bone marrow, so Levis opted to enroll in a second clinical CAR T trial, this time using a humanized construct that the team at Penn Medicine wanted to try along with the antibody-based drug he was taking. Today, he has no tumor burden, the percentage of CLL cells in his marrow has dropped from 40 percent to less than 5 percent, and he feels good on low-dose antibody therapy.
Thinking about long-term follow-up, Levis said he hopes that by continuing to be an “experimental patient at Penn Medicine,” researchers will be able to start answering questions around resistance mechanisms, the need for memory cells for long-term persistence, the role of booster doses sometime after the initial infusion, and reactivation of the protective cells. He closed by repeating something his oncologist told him, which was that this is not even the end of the beginning, and there are new approaches on the horizon.
DISCUSSION
The presentations in the session were followed by a moderated panel discussion, which served as an opportunity for workshop participants to ask questions of the speakers. The panel discussion explored the length of time patients are followed after a gene therapy clinical trial, motivating factors for participation in follow-up studies, and costs for tracking patients over several years.
Determining the Ideal Length for Follow-Up
A workshop participant asked Purohit-Sheth how FDA came up with 15 years as the length of long-term follow-up for gene-based therapies and if it would be possible with more study to identify those patients at the highest risk of developing long-term adverse events rather than following every patient for 15 years. There was a case of secondary malignancy that occurred 14 years after exposure, Purohit-Sheth explained, which is how the 15-year number came about. Regarding learning about long-term risks and delayed adverse events, she said that the field is not yet at a place—as it is with small-molecule drugs—where there are solid expectations as to how these gene-based therapies will work over the long term. “As we procure more information, gain additional understanding,” she said, “certainly there is a possibility that these recommendations may be updated
in the future based on our understanding and our experience with these products.”
A key question involves understanding what level of elevated risk is acceptable, Robison said. Childhood cancer survivors, for example, can have up to a 20-fold increased risk of experiencing long-term adverse events associated with their treatments. What, he asked, is the level of risk that is important for a given population? Is a two-fold increased risk for a serious adverse outcome acceptable? Is it 50 percent increase? Is it five-fold? It will be important to determine what is considered significant and important to monitor, he said. The ultimate goal, he added, will be to understand which particular patients are at high risk because of their individual characteristics and their treatment exposure.
Participation and Payment Challenges Associated with Long-Term Follow-Up
A workshop participant asked Robison what he thought are some of the important steps to take to motivate patients and families to stay involved in long-term follow-up studies. Robison answered that his team uses a number of approaches, but the most important is to educate patients and families and provide feedback so that they understand the importance of continuing to participate over the long term. He noted that the National Academies have recommended that every cancer patient, once they complete treatment, should have a care program that includes a summary of all of their treatments, and he suggested that this field should consider doing the same thing in the name of educating patients and their families.
One concern of Robison’s that had not been addressed, he continued, is how to pay for long-term follow-up studies. One possibility would be to identify risks, demonstrate that those risks are real and targeted to a specific population, and include that information in treatment guidelines. Once that occurs, he said, insurers will have a harder time justifying not covering the cost of long-term surveillance for those outcomes. DeBaun, the session moderator, noted that a large percentage of children and adults in the United States receive health insurance from the government through Medicare or Medicaid. The likelihood of an adult or child with sickle cell disease participating in long-term follow-up with a knowledgeable provider after receiving a curative therapy is less than 1 percent, he said. “Unless FDA acknowledges these non-funded mandates with strategies to follow this vulnerable patient population,” DeBaun said, “it is unlikely that we are going to capture these data even a year to 2 years after curative therapy.”














