
5
Advancing Research on the Environmental Regulation of Gene Function
After the session devoted to the contributions of non-coding DNA to phenotype, described in Chapter 4, the workshop moved on to the closely related topic of the environmental regulation of gene function. Because the environment exerts its effects on genes in large part through the non-coding elements of the genome, there was inevitably a great deal of overlap between the presentations in the two sessions. Research into how environmental factors affect gene function and, ultimately, an organism’s phenotype requires studying how the genome’s non-coding regions shape gene expression, and vice versa. Still, the two sessions were clearly differentiated by differing emphases. The presentations in Chapter 4 focused on non-coding elements and their interactions with the rest of the genome, whereas the talks described in this chapter were more focused on environmental factors and how they affect the regulation of gene function—and, ultimately, the organism’s phenotype.
The main session described in this chapter was moderated by Philip Benfey of Duke University, and there were four speakers. Sarah Kocher of Princeton University described how the environment shapes social behavior in sweat bees, some of whom create highly organized societies like those of honeybees, while others are solitary. Joanna Kelley of Washington State University described how certain small fish have adapted to live in water that is highly sulfidic. Nathan Springer of the University of Minnesota discussed research aimed at understanding the interaction between genome and environment in corn plants, which has significant economic implications because of the importance of corn as a crop. And Trudy Mackay of Clemson University, who works with Drosophila, described how genotype,
environment, and sex interact to determine an animal’s phenotype. This chapter also includes a talk from David Page of the Whitehead Institute for Biomedical Research, whose talk on the phenotypic effects of sex differences fits the theme of this chapter, while his talk was given during a different session of the workshop.
FACTORS SHAPING VARIATION IN SOCIAL BEHAVIOR
Sarah Kocher’s group at Princeton University studies social insects to gain insight into the factors that shape variation in social behavior. In particular, she said, they are interested in understanding what facilitates the transition between individuals that live and reproduce independently and individuals that come together to reproduce as a group.
Social insects, such as ants and honeybees, are excellent organisms in which to study social behavior because they are extreme examples of social living and have been helpful in understanding the molecular mechanisms behind social behaviors, such as caste differentiation. Using social insects, researchers who are interested in understanding what facilitates the transition between solitary and social living must work in systems that include closely related species of both highly social animals and animals that are solitary or only weakly social.
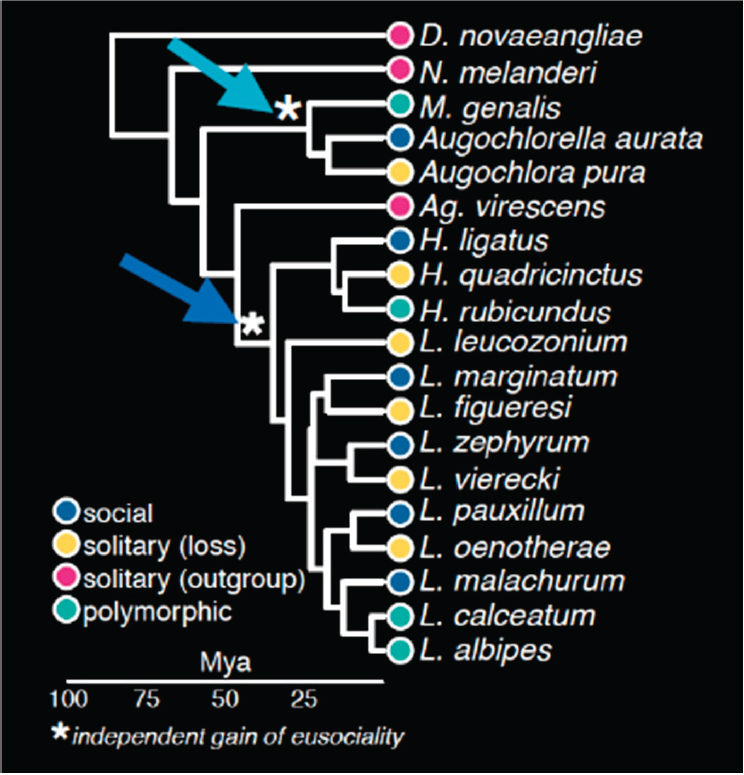
With that in mind, Kocher and her group chose to work on a family of bees called the halictid bees, or the “sweat” bees. This group encompasses the full range of social behavior, she said. Some species are solitary and live and reproduce independently. Others are eusocial and have overlapping generations, cooperative brood care, and a reproductive queen or queens with the other members of a colony not reproducing. Still other species are socially polymorphic, so that in a single species there are some females that establish solitary nests and others that found eusocial nests. It is thought that eusociality evolved twice in the halictids independently and that it has been lost independently about a dozen different times. The result is a family of organisms that exhibits the entire spectrum of social behavior occurring both within species and between species.
When she started working on this system, she began with one of the socially polymorphic species, Lasioglossum albipes. Previous research had found that populations of this bee in the west of France are eusocial, while populations in the east of France are solitary (Plateaux-Quénu et al., 2000). Whether a particular population of these bees is eusocial or solitary appears to depend largely on environmental factors, Kocher said. In the solitary bees, females found nests independently and produce a single brood consisting of reproductives, both males and females. After the bees have overwintered, the fertilized females leave the nest to establish their own nests and start the cycle all over again. In a social nest, by contrast,
the females produce workers first, followed by a reproductive brood. The queens make roughly twice as many reproductives in a social colony as in a solitary nest, and there is a huge fitness payoff to that, Kocher said, but because the queens in the social nests have to produce two broods, first workers and then reproductives, it takes them twice as long to reach reproductive maturity.
That difference in timing is related to the mean temperature of the area in which a nest is located, Kocher said. In those parts of France that are colder, on average, bees have fewer days when it is warm enough to forage during the day, and the populations are solitary. In the warmer areas where there are more days when it is warm enough to forage, the populations are eusocial. In short, she said, it seems that local adaptation is shaping the social/solitary divide.
Earlier research on these bees had shown that if eusocial and solitary populations are brought into the lab and reared under the same conditions, there is no plasticity in social behaviors. This showed that the social behavior of the populations is largely genetic (Plateaux-Quénu et al., 2000).
When she started working on this system, Kocher and her team had to build the genetic resources for the system from the ground up, so they started by generating a reference genome. Having done that and having figured out how to catch the bees in the wild, she brought 25 individuals back to the lab from each of six populations scattered across France, populations that were at different points on the eusocial/solitary spectrum. Her team then generated whole-genome sequences from the bees with the goal of identifying some of the genetic differences associated with the various social patterns.
The first thing they showed was that the populations were not simply incipient species (Kocher et al., 2018). Using a principal component analysis of the genomes showed that the populations did not cluster in the way that would be expected if they were speciating. Instead, Kocher said, “it seems like there have been repeated shifts in social behavior within the species, and this is a signature of local adaptation.”
Next, she carried out a genome-wide association study (GWAS) looking for associations between genotype and social polymorphisms. Several regions across the genome show strong associations with social behavior, which is not surprising given that it is a complex behavior. But one of the most striking observations was a single window where there were seven single nucleotide polymorphisms (SNPs) located in non-coding regions of the gene syntaxin 1A (Munson, 2015). Because the SNPs were all noncoding variants, Kocher’s first question was whether there were differences in the expression of syntaxin 1A in the natural populations that could be related to those SNPs. A quantitative polymerase chain reaction (qPCR) showed that social bees have higher expression levels of syntaxin 1A in their brains than solitary bees.
The next question was whether any of the seven SNPs could help to explain the observed variation in expression. She chose two SNPs that showed the greatest degree of differentiation between social and solitary populations and used luciferase assays to test for enhancer function. One of the SNPs lies within the first intron of syntaxin 1A, a sequence having enhancer activity, and that activity varies according to the allele that an individual carries. In particular, she found that the social allele drives higher levels of gene expression in the same direction seen in social populations of the bees.
Syntaxin is an interesting gene for a number of different reasons, Kocher said. It has been implicated in social behaviors in many different species, including other insect species (Chen et al., 2015) as well as in mice (Fujiwara et al., 2016), where decreasing its expression disrupts normal maternal care. Surprisingly, it has also been linked to autism in humans, in GWASs as well as gene expression studies.
“And so,” Kocher said, “our conclusion from this was that it seems like selection could be acting across these deep evolutionary timescales to shape variation in social behavior in both insects and vertebrate species like ourselves.” For the past several years, she and her group have been trying to identify some of the core mechanisms that might shape variation in social behavior. In particular, working with postdocs Ben Rubin and Beryl Jones, Kocher’s group has been carrying out comparative genomics on a collection of 19 different species of sweat bees, including L. albipes, in an effort to identify the genetic mechanisms shaping variation in their social behavior.
Among the genomic resources they have built are de novo assemblies for the 19 different sweat bee species with a combination of 10× sequencing and high-throughput chromosome conformation capture. The genome sizes average 414 megabases. “To improve annotation for each of these genomes,” Kocher said, “we also did some tissue-specific RNA sequencing for as many species as we could get our hands on.” They now have calculated a mean complete BUSCO, a metric assessing the completeness of a genome assembly, of 94 percent for the 19 different genomes, she said, adding that this is similar to what is seen in many of the other well-developed reference genomes in existence now.
With that dataset they began to investigate the natural selection that led to the different types of social behavior. In particular, in the evolutionary tree of the 19 species (see Figure 5-1), eusociality appeared twice, in two separate branches, and it also disappeared on a number of branches. In the two branches where eusociality developed, Kocher’s team did tests for signatures of positive selection on each and then looked at the genes that intersected both of those branches. They found nine genes that showed positive selection on both branches, and because there were only nine, they did not see a strong signature of gene ontology (GO) enrichment at this level.
NOTE: Mya = million years ago.
SOURCE: Sarah Kocher presentation, slide 26.
However, she said, most of the analytical power in the system is through the multiple losses of sociality. They tested for relaxed selection on each of the branches that represent losses of social behavior and compared those results with species that are eusocial and have maintained that behavior. This comparison identified 443 genes with relaxed selection associated with the loss of eusociality, Kocher said, and those genes were enriched for things such as chromosome condensation and DNA packaging, indicating an important role in chromatin accessibility.
One of her team’s most exciting findings is that four of the nine genes that show the signatures of positive selection with the origins of social
behavior also showed relaxation when social behavior was lost, thus identifying them as genes that play an important role in the evolution of social behavior. “I think that this represents a really powerful way … to identify some of the convergent mechanisms that shape the evolution of social behavior in the system,” she said.
In the last part of her talk, Kocher turned to the role of gene regulation in the development of social behavior. A few studies over the past several years have suggested that changes in gene regulation are important in mediating social behavior and social interactions in many different species (Kapheim et al., 2015). When her team examined the proportion of transcription factor binding sites associated with either solitary or social genomes across the 19 different species, they found three times as many binding sites positively correlated with social genomes as binding sites associated with solitary genomes. This suggests, she said, that there has been some sort of expansion in the binding capacity of the social genomes—which in turn would point to this particular type of change in gene regulation as playing a role in mediating social behavior.
They also looked for signatures of positive selection on putative regulatory elements in the 19 genomes and found about 600 non-coding regions that could be aligned across all 19 different species and that show signatures of positive selection on the social lineages. These non-coding regions are enriched for “exactly the kinds of things that you would expect to find,” she said.
There is enrichment for neural functions, for things that are differentially expressed between social and solitary species, and for the genes linked to autism risk in humans. “So I think this is giving us some hint that maybe the continued development and maintenance of social behaviors may be fine-tuned by changes in gene regulation,” she said.
The bigger take-home point, she said, is that both coding and noncoding sequences shape the evolution of social behavior. “If you’re studying behavior, you have to acknowledge that there’s a genetic component, but there are also environmental components that shape that variation.”
Looking to the future, Kocher said that her team has been focusing on developing ways to generate comparable functional genomic datasets across the different species in order to examine how gene regulation might change with the gains and losses of social behavior. Instead of bringing 19 different species into the lab and doing a common garden—which seems really hard, Kocher said—“we’ve decided that we’re going to take the lab to the bees.” They have carried out pop-up common garden experiments where they use cages to observe nesting behaviors of bees in their natural environment. By comparing social and solitary bees in the same environment, they are able to look at how gene expression changes and how it varies across different species in the same environmental context.
In conclusion, Kocher listed some of her successes: using techniques from population and comparative genomics to provide insights into social evolution and building genomic tools that have allowed them to unmask some of the convergent mechanisms shaping social behavior. One of the challenges they face is generating comparative genomic datasets for 20 different species in a comparable way.
Finally, she said, it is important to have a strong community that can help build some of the necessary functional genomic tools. Because her lab is not large and there are only half a dozen labs in the entire country that work on sweat bees, they have turned to other systems, such as Drosophila, where genomes and genetic tools are readily available, and have adapted those tools to their own purposes.
HOW ENVIRONMENTAL FACTORS INFLUENCE A COMPLEX PHENOTYPE
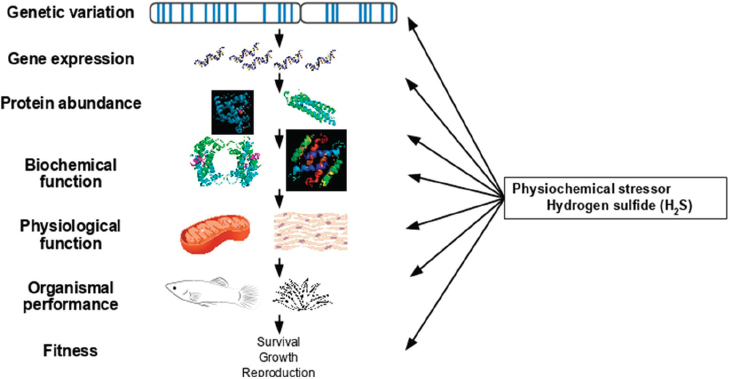
In the next presentation, Joanna Kelley discussed how variation in natural systems can be used to understand complex phenotypes. She began by sketching out the usual frame for understanding the gene–phenotype connection: genetic variation leads to differences in gene expression and in protein abundance, which affect biochemical and physiological function, and, ultimately, the function of an organism and its fitness.
Environmental factors can affect every level of this hierarchy, she said, and the particular environmental factor she studies is the presence of a stressor, hydrogen sulfide, in aquatic environments. This stressor acts on organisms not only directly, but also through its effects on both the abiotic and biotic environment. These in turn influence all of the different hierarchical levels (Tobler et al., 2018; see Figure 5-2). “While we like to think about our beautiful linear path from genetic variation to phenotype,” Kelley said, “in reality it’s much, much more complex.”
Hydrogen sulfide is both a toxicant and a signaling molecule (Tobler et al., 2006). In the regions in Mexico where Kelley carries out her research—and in various volcanic regions throughout the world—the concentrations of hydrogen sulfide (H2S) in springs and other water bodies can be as high as 1 millimolar (Tobler et al., 2006). For organisms that are not adapted to these conditions, H2S at 5 to 40 micromolar can be acutely toxic. It inhibits oxygen transport and cellular respiration and causes and aggravates hypoxia in aquatic environments.
There are, however, fish in many places that have adapted to high levels of H2S, Kelley said. In particular, there are several different species in Mexico. Kelley studies those species adapted for living in a single location to better understand the connection between phenotype and genetic variation.
SOURCES: Joanna Kelley presentation, slide 5; Tobler et al., 2018.
In particular, she studies three different sympatric lineages of poeciliids that live in divergent stream environments. Each of the three species has two populations, one that lives in a sulfidic environment and another that lives in a freshwater environment. The two environments are very close to one another—within about 200 meters. “So,” Kelley said, “we’re going to leverage this comparative contrast with having multiple different species in exactly the same environment to see how that phenotype of survival in hydrogen sulfide arises and connects to underlying genetic variation.”
The three species that she studied are widely distributed across the large phylogeny of Poeciliidae. Because they are not closely related, their separate adaptations to H2S are probably not due to adaptive genes moving from one species into another one. The three species, therefore, allow them to examine convergence and the origins of this phenotype.
Testing the tolerance to H2S in the three species shows that all three of the freshwater populations die relatively quickly once the H2S concentration reaches a certain level, while fish in the sulfidic populations can survive much longer. There are also consistent morphological differences between the sulfidic and freshwater populations, Kelley said, with similar changes in head and body shape across the three species, so there have been convergent shifts in body form as well.
Because much is known about how H2S acts in organisms, she said, it is possible to make predictions about both the genetic and transcriptional changes as well as the physiological and biochemical changes that may be happening in the three species. For example, as H2S comes into a system, it
blocks cytochrome c oxidase (COX), which is a major target of hydrogen sulfide toxicity, so one prediction would be that COX would be modified somehow. There should also be up-regulation of the enzymes detoxifying hydrogen sulfide, down-regulation of enzymes producing hydrogen sulfide within the body, and differential regulation of other molecular targets.
Testing bears out these predictions, Kelley said. For instance, one of the major enzymes involved in the oxidation of hydrogen sulfide is sulfide–quinone reductase (SQR). Lab experiments with two populations of Poecilia mexicana showed that SQR activity in fishes from the sulfidic population increased as the H2S concentration increased, whereas SQR activity in the freshwater population actually decreased with increasing hydrogen sulfide (Greenway et al., 2020).
Similarly, there was greater expression of H2S-related genes in the sulfidic populations than in the freshwater populations, and this was true in all three species, showing convergent shifts in gene expression (Kelley et al., 2016). On the other hand, Kelley’s team saw no evidence of down-regulation related to H2S production.
Kelley pointed out that she was only discussing genes that she knew were related to H2S somehow and that there is actually a huge list of genes that are expressed differently between the populations, but whose role in adapting to H2S is unclear. “They don’t fit into this nice framework of what I understand about their biology,” she said, and are candidates for discovering other ways that these fish are adapting to their environments.
Because most of her group’s experiments had been conducted on wild-caught individuals, it was impossible to tell whether the increased expression of certain genes in the sulfidic populations was because of evolved changes or whether these were responses to the fish being in water with high levels of H2S. To determine which differences in gene expression between ecotypes were due to adaptation and which were due to plasticity, Kelley and her collaborators carried out a common garden experiment in which one of the species was brought into the lab where several generations were raised and then exposed to H2S. This was done with the descendants of both sulfidic and freshwater populations, and there were also control groups where the fish were kept in water without H2S. They found that many, but not all, of the genes whose expression differed between the two populations in the wild actually had evolved changes in gene expression rather than having expression plasticity (Passow et al., 2017).
Kelley noted that these evolved changes between the two populations, freshwater and sulfidic, in a single species are possible. Even though the two populations live only about 200 meters apart, there is little gene flow between the populations. There is some migration of individuals from sulfidic into non-sulfidic populations, so there are low levels of gene flow, but an ancestry analysis found that most of the non-sulfidic individuals had
100 percent non-sulfidic ancestry and all of the sulfidic individuals had 100 percent sulfidic ancestry.
Next Kelley described looking for highly differentiated regions in the genomes of these fish that might underlie the traits related to survival in a hydrogen sulfide environment. This was done by looking primarily for regions that differed in the same ways across all three poeciliids.
They found basically nothing. While the two populations from the three species have phenotypes that diverge in similar ways, the underlying genetic bases for those differences vary among the species. Kelley concluded that the populations took different genetic paths to get to the same place.
In closing, Kelley brought up two issues with which researchers in the field must grapple. First, with which phenotypes should they be working? Are the phenotypes of gene expression or even protein abundance really representative of organismal function? Perhaps, she said, but researchers must be careful because while some phenotypes are easier to measure than others, those are not always the best choice. “We may be most interested in physiological function, but those are hard phenotypes to assay,” she said. “I think it is really important to think about which phenotypes we are really interested in and how much we can learn about organismal performance from gene expression or protein abundance.”
The other issue, she said, is whether researchers should be limiting themselves to organisms that are tractable in the lab. “How tractable does an organism need to be? Are cells or tissues or cell culture sufficient to answer some of our questions?” The answers are not obvious, she said, but the question should be asked.
ENVIRONMENTAL REGULATION OF GENE FUNCTION IN AGRICULTURE
Moving from the first two presentations, which focused on insects and fish living in natural environments, Nathan Springer of the University of Minnesota spoke about crop plants, which have been carefully bred for optimized performance in farm fields. In particular, he focused on maize, which he described as “a species with wonderful diversity at many levels.”
As a geneticist, Springer said, he is interested in learning about the molecular variants that underlie that diversity. Furthermore, he added, he is interested not just in how the variants directly influence crop traits, but also in how they combine to influence them. In corn, he explained, one frequently observes heterosis, or the combining of two different lines to generate a much different, generally superior line. That phenomenon requires an interaction between the variants present in the different parents.
Plants such as corn also offer a particularly good system in which to assess genotype-by-environment (GxE) effects, Springer said. There is
a variety of highly inbred lines of corn that can be planted in different environments and monitored with ease because they stay in one place. Furthermore, he said, there is an economic incentive to learning more about corn. “We’re trying to create GxE effects that would provide higher yield for these genotypes.”
Introducing the key messages of his presentation, Springer said he would be focusing on three critical questions:
- What factors influence variation for gene expression patterns?
- How do genes acquire environmental responsiveness?
- How do we effectively link genomic variation to GxE effects?
Concerning differential gene expression, Springer said that like many of the previous speakers, he had studied how gene expression varies between closely related species or even between different individuals in a single species, and that he agreed with most of what had been said at the workshop. There are many genes that are expressed differently in different individuals, he said, and often variation in cis-regulation is the important factor. “One of the things that I think is important that I didn’t appreciate for a long time,” he said, “is that there are not stronger and weaker alleles.”
To illustrate, he showed a dataset on allelic expression patterns in 23 different tissues taken from 3 different maize genotypes. There were more than 22,000 genes that were differentially expressed in at least one tissue, which, he said, was not surprising. It is similar to the findings that other speakers had reported. What was surprising, however, was the consistency of patterns across tissues and developmental stages. Nearly 1 in 10 of the genes identified as being differentially expressed in at least one tissue were differentially expressed in more than 80 percent of the tissues in which they were expressed. More than 6 in 10 of the identified genes were differentially expressed in 20 to 80 percent of the tissues in which they were expressed, and about 3 in 10 of the genes were differentially expressed in less than 20 percent of the tissues in which they were expressed. Furthermore, Springer added, it was not just that there was tissue specificity for differential expression, but the alleles often varied. Of all differentially expressed genes, 45 percent exhibit these so-called mixed effects. He explained that, “it’s not that one allele has a stronger promoter and just outcompetes the other; it’s that they have different inputs into their tissue-specific expression levels.”
One facet Springer is curious about is how plants generate regulatory novelty. In particular, he asks, how does a plant go from an ancestral state in which a particular allele does not respond to the environment, where it does not have cis-regulatory inputs, to a state where there is some difference in the cis-regulatory elements and the plant now responds to that environmental input? What happens to create this shift?
There may be some ways in which SNPs can do this, he said, but his suspicion is that it will be hard for single-base-pair changes to do that. So he brought up an idea that was put forward years ago by Barbara McClintock—that transposons, as they move throughout the genome, could shuffle or create novel regulatory elements (McClintock, 1950). As an example, he mentioned the Naiba family of elements, of which there are about 500 in the maize genome. More than 80 percent of genes that are near Naiba elements turn on in response to cold stress. Furthermore, alleles that have such an insertion often respond to cold. “So there’s an association between the presence or absence of this transposon near a gene and the expression responsiveness,” he said.
What he really wanted was to do this on a much larger scale. “I’m going to tell you about why this has been difficult, and why I think we’re getting closer,” he said.
For a decade or so, Springer said, corn researchers had a single reference genome, but within the past couple of years that has grown to about 40 fairly high-quality reference genomes, and that has revealed a great deal of additional complexity that must be taken into account. For instance, he described a study of the transposons in the genomes of two corn hybrids that revealed great transposon variation. The maize genome is about 2.5 Gb, and transposons accounted for about 1.35 Gb of that in each of the hybrid genomes. Of that 1.35 Gb, about 500 Mb of transposons—or about 20 percent of the entire genome—were not shared between the two genomes, or at least not at the same position, Springer said. Furthermore, when four hybrid genomes were analyzed, while a subset of transposons were shared across all four lines, more than 78 percent of the transposons varied across the four hybrids, and many of the transposons were present in only one, two, or three of the lines (Anderson et al., 2019).
This is important, he said, because in the four corn genomes they examined, more than 50 percent of the genes were located within 5 kb of a polymorphic transposon. So even if the transposons do not do anything themselves, the three-dimensional architecture, the local neighborhoods, and distances between genetic elements are radically different among these genotypes. That makes it difficult to align various maize genomes, he said. So the 40 reference genomes offer a wonderful opportunity to study and compare multiple genomes, but at the same time they bring new challenges. “How do we even do an alignment of genomes that have highly variable content?”
Switching to the topic of chromatin, Springer said that it had been really heartening to hear all the various talks about using chromatic marks. “This gives us new insights into finding potential elements and functions in genomes,” he said (Ricci et al., 2019). In maize, open chromatin makes up less than 1 percent of the genome, but nucleotide variation in such regions
accounts for more than 40 percent of phenotypic variations (Rodgers-Melnick et al., 2016).
Furthermore, more than 20 percent of all those open chromatin regions are found in transposons, which offers support for the idea that transposons might be carrying regulatory elements and influencing genes.
In the future, Springer said, generating genotypes will no longer be the expensive or difficult part of plant research. “Genotypes are relatively cheap,” he said. “The hard part is measuring phenotype. What is the phenotypic outcome of a genome?” Plant researchers working for private-sector companies sometimes have access to large datasets with phenotypic data, but that is not common in the public sector. To address this issue, the Genomes to Fields initiative, a consortium of plant researchers, have been growing a set of 500 maize genotypes in about 20 to 30 locations over the past 5 summers. “We collect environmental data with the same brand of weather station in every field, monitoring core data on agronomic traits at this point,” Springer said. “This is certainly not enough.” Other phenotype-measuring technologies are needed, he said. For example, his lab flies drones over their field every 2 days gathering data on such things as how fast the plants are growing and canopy closure. They probably need additional sensors, he added, and they also need progress on how they store and share phenotypic data.
Summing up, Springer said, “We really need a better understanding of the phenotypic outcomes associated with our environmental data.” As he pointed out, “in nature environments happen only once. You’ll never get exactly the same environment the next year,” he said. “So we’re throwing away data right now that we should be gathering and keeping forever.”
ENVIRONMENTAL FACTORS AFFECTING QUANTITATIVE TRAITS IN DROSOPHILA
Next, Trudy Mackay of Clemson University spoke about how environmental factors affect quantitative traits in Drosophila. Quantitative traits, she explained, are continuously distributed in natural populations and include such things as height, weight, and blood pressure. A century ago, Sir Ronald Fisher, a British statistician and geneticist, attributed this continuous variation to the presence of multiple genes that each contributed to the overall trait as well as non-genetic variation, which he referred to as “environmental variation” (Fisher, 1918). In Fisher’s model, the genetic and environmental variation were separate factors that, when added together, produced the total phenotypic variation.
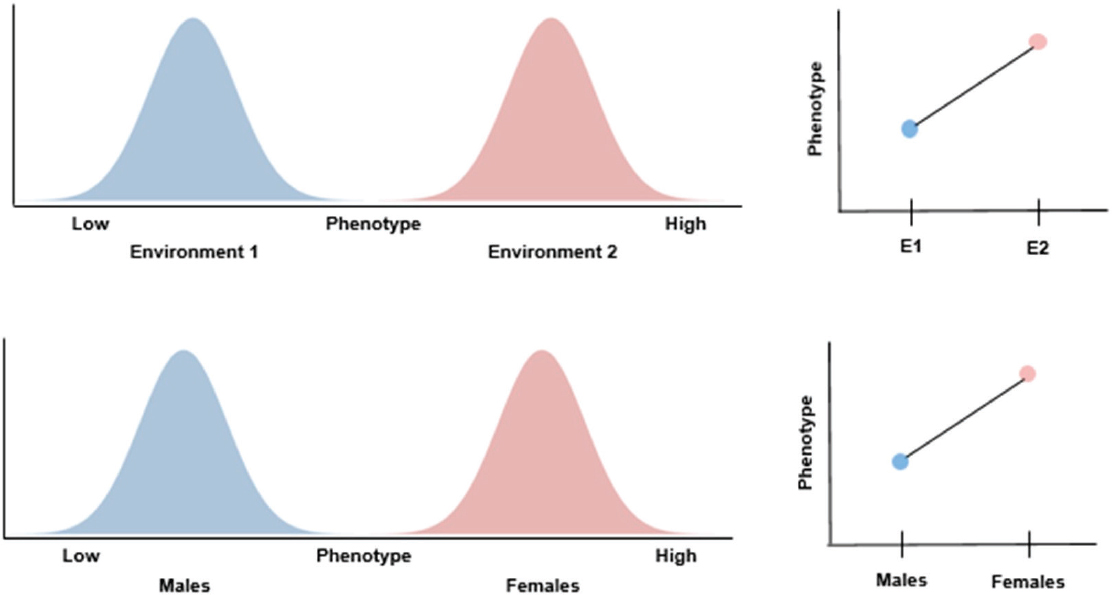
To illustrate, Mackay posited two genetically identical populations, one of which was in a hot environment and the other in a cold environment. The different temperature regimes caused the two populations to differ, on
average, in a particular phenotype. This is phenotypic plasticity, Mackay said, and in her illustration the plasticity was such that the phenotype did not overlap between the two populations (see Figure 5-3).
Organisms that have two sexes, male and female, can also exhibit sexual dimorphism, Mackay noted, and this can add extra variation to the phenotype (see Figure 5-3). In this situation, sex can be considered a genotype as well as an environment, she said. (See David Page’s talk below for more discussion of the role of sex in shaping gene expression.)
Thus, in this model three different factors can lead to phenotypic variation: genetic and environmental variation and sexual dimorphism. In Fisher’s view, these three factors would be additive—the total phenotypic variation would simply be the sum of the genetic and environmental variation plus the sexual dimorphism considered separately. However, the real world is more complicated and includes interactions among all three of these factors. For example, males and females might respond differently to increasing temperatures.
To illustrate how these varying interactions can contribute to phenotypic variation, Mackay discussed work that has been done in her lab with the fruit fly, Drosophila melanogaster. She began by describing the D. melanogaster Genetic Reference Panel (DGRP) that was developed in her lab (Mackay et al., 2012; Huang et al., 2014). The initial version had 205 inbred lines created by 20 generations of full brother–sister matings. The collection of different lines is valuable because the members of each line are largely genetically identical, but they are different from individuals from other lines, and they can be raised in multiple environments in order to study the interaction between genetics and the environment on phenotypes.
In one such experiment, Mackay exposed 72 males and 72 females from each of 186 lines to each of three different thermal environments—18°C, 25°C, and 28°C—and observed how long they lived (Huang et al., 2020). The ones kept at 18°C had the longest lifespans, followed by those at 25°C and 28°C. After collecting these data, Mackay and her team analyzed them to uncover the effects of genotype, environment, sex, and interactions among the three factors on the lifespans of the flies.
They found clear genetic variation within each temperature—some lines survived significantly longer than other lines—as well as sexual dimorphism and phenotypic plasticity (Huang et al., 2020). Importantly the interactions among the three factors were also significant.
There was, for example, genetic variation in sexual dimorphism, so that the differences between the sexes varied in different environments. There was also genetic variation in phenotypic plasticity, and the three-way interaction was highly significant. “What this means,” Mackay said, “is that if I had information on the lifespan of a particular genotype female
SOURCE: Trudy Mackay presentation, slide 3.
at 25 degrees, that tells me very little about that same genotype in other environments.”
An additional complication is how variable the lifespans of the flies are within different lines. “This is obvious when you’re measuring the flies,” she said. “Sometimes the first one dies when it’s 3 days old, and the last one lives to 100 days, whereas for some lines you come in one day, they’re all alive, and the next day they’re all dead.” This characteristic is clearly genetic, she noted, but it also varies according to the environment—this “micro-environmental plasticity,” as it is called, is different for the same line of flies in different environments (Huang et al., 2020).
And while there is no overall sexual dimorphism, Mackay said, the amount of lifespan variation in each sex differs according to the environment. “So life is a little bit more complicated at the level of quantitative traits than one might think,” she said.
Next Mackay described an experiment that examined how much of the genome exhibits variation in gene expression in different environments (Zhou et al., 2012). Her team worked with an outbred Drosophila population that was a mix of 40 DGRP lines—“all sorts of different genotypes, all mixed up,” she said—rather than working with single inbred lines. The flies were exposed to 20 different environments: a control and 19 that involved distinct environmental stressors ranging from heat shock to social crowding. The researchers used microarrays to measure gene expression across the genome. Surprisingly, only about 10 percent of the transcripts show environmental plasticity. By contrast, she said, nearly half of the transcripts exhibited sexual dimorphism, and a few of them had sexual dimorphism that differed across environments. Their conclusion was that the majority of the transcriptome was robust to environmental fluctuations, at least in this outbred population where the effects were averaged over all the different genotypes that they observed.
To close, Mackay offered brief descriptions of two other studies of variation in transcription. In one, her group examined the response of fly brains to cocaine using single-cell expression techniques. They found 691 unique genes expressed differentially in males and 322 in females. The response was highly sexually dimorphic and cell-type specific.
Noting that the first study was done with just one genotype, Mackay described a second with young adult flies from 200 different DGRP lines. RNA sequencing was used to examine gene expression in both sexes of all these lines (Everett et al., 2020). They found that much of the transcriptome was highly sexually dimorphic and that there was enormous genetic variation and sexual dimorphism at the level of transcription—echoing what they had seen at the level of quantitative traits.
In concluding her presentation, Mackay offered her thoughts on the roadblocks facing this sort of work. Generally speaking, she agreed with
what the previous speakers had said. “I think the roadblocks are not intellectual,” she said, “but they’re matters of scale and the money needed to do these kinds of experiments.” The factorial experimental design needed to estimate and map the genetic base of environmental interactions across multiple environments, sexes, genotypes, tissues, developmental stages, exposure time courses, and multiple -omic levels is expensive to carry out.
“For example,” she said, “we would want to look at bulk RNA, single cells, micro RNAs, epigenetic marks, metabolites, proteins, nuclear architecture. That would give us a very full picture of the effect of environmental variation on transcription and how that would relate to quantitative trait.” That list of items mirrors a list of items from the Drosophila Encyclopedia of DNA Elements (ENCODE) project, she said. “I think there would be great value in actually doing a similar large community-based project with genetic variation overlaid.”
Sex Differences in Comparative Functional Genomics
Sex differences should be front and center in comparative functional genomics research. That was the thesis that David Page set forth at the beginning of his talk, and he spent the rest of his presentation expanding on and supporting that suggestion.
Although the X and Y chromosomes are considered to be the sex chromosomes, Page said, “I want to argue … that every chromosome is a ‘sex chromosome’ and that the sex chromosomes have no monopoly, and not even any statistically discernible specialization, in the matters of sex.” In particular, he suggested that the study of sex differences should be moved to the entirety of the organism and should be at the center of functional genomics research.
The first challenge, Page said, “is to decide what we even mean by sex differences.” According to the standard textbook account of mammalian sex determination—which can be generalized to other organisms—during the first 6 weeks of human development the XX and XY fetuses are anatomically indistinguishable. Then, around week 7, the bipotential gonad begins to take on the microscopic appearance of the testis or ovary. The remainder of the classic strict binary of sex determination follows this first appearance of sex organs.
The problem is, however, that not all sex differences are strictly binary, although the textbooks tend to focus on those traits that are. To illustrate, he spoke about height and size. “Males tend to be a little larger than females,” he said. “This is actually true of most mammals, not all, but most,” with the males tending to be 4 to 8 percent larger (Crow, 1997).
There are many other such traits in humans that show sex differences. “For every man who has lupus, there are six women,” Page said. “Or if we
flip it around to autism spectrum disorders, for every girl who’s diagnosed with autism, there are four boys.” However, he added, models of sex determination and sex differentiation, at least in mammals, do not generally accommodate these sorts of non-binary differences.
He has been working with others to develop a new model of sex determination and differentiation. The model will be fluid and dynamic, instead of binary. Furthermore, he said, the model will not be restricted to a few cell types in the reproductive tract but rather will encompass all cell types across the entire body and focus on the entire genome.
The model is based on three key conjectures. The first is that autosomes are read differently in males and females. That is, it is not just the X and Y chromosomes that have gene expression that differs between males and females, but in fact all of the chromosomes are read out a bit differently in the two sexes.
The second conjecture is that this has been true throughout human evolutionary history, going back 600 million years, or since before there were sex chromosomes. It was the appearance of structurally distinct gametes—eggs and sperm—that was the defining feature of the origin of males and females, Page said, and this happened about 600 million years ago.
The final conjecture is that in mammals the autosomes are read differently in every tissue, in every cell type, and at all developmental stages. “I don’t have all the evidence to support that, of course,” Page said, “but it’s a sort of a guiding hypothesis, a speculative form.”
To illustrate the sorts of research that can be motivated by these conjectures, Page described some work that was motivated by the general question of how autosomal gene expression differs between males and females. His team sought to answer three specific questions: How does autosomal gene expression differ in organs that are outside the reproductive tract, that is, in organs that are “the same” in male and females? Are sex biases in gene expression conserved between humans and other mammals used in basic research and in pharmaceutical trials? And do sex differences in gene expression across the genome contribute to sex differences in a trait?
To address the second question, the team surveyed sex differences in gene expression across humans and four other animals—rhesus macaque, mouse, rat, and dog—which all had a common ancestor around 100 million years ago (Naqvi et al., 2019). They looked at 12 tissues present in both sexes that would typically be thought of as being the same in males and females: tissue from the brain, pituitary gland, thyroid gland, heart, lung, liver, spleen, adrenal gland, colon, skin, skeletal muscle, and adipose tissue. For humans they reanalyzed data from the Genotype-Tissue Expression (GTEx) Consortium, while they generated new RNA-sequencing (RNA-seq) data for the other four species.
The team clustered 72 human and 277 non-human mammalian RNA-seq libraries based on nearly 13,000 genes that were one-to-one orthologs across the species. Unsurprisingly, the samples clustered first by tissue and then by species. But after that they clustered—most of the time—by sex. Page notes “that sex is a subtle third-order determinant of each tissue’s transcriptome. We can identify just about any tissue in any of these species as being male or female based on its transcriptome, but it is subtle.” He noted the major contrast between the subtle sex differences one sees in gene expression and the sometimes large sex differences that appear in phenotypes.
The next question, he said, is whether such sex differences in autosomal gene expression actually contribute to sex differences in a trait. As an answer, he described a study that his lab did on sex differences in height. This is an exhaustively studied trait in humans, Page noted. There are at least 700 genes that have been implicated as making minuscule contributions to height variation in both men and women, and it is the same 700 genes for both sexes (Rawlik et al., 2016; Sanjak et al., 2018; Sidorenko et al., 2019). In theory, there could have been somewhat different sets of genes responsible for height differences in males and females, but that is not what has been found. Despite the enormous literature on this topic, no one has come up with an explanation for the average 5-inch height difference between men and women.
His lab has made some progress in that area, Page said, and they started by describing work done on a single gene—the gene for a transcription factor called LCORL. This transcription factor has been genetically associated with increased body size in humans, cattle, and horses, with increased expression of this gene being associated with a decrease in size. What Page’s lab noticed is that LCORL is expressed at slightly higher levels in females than in males—a difference that would tend to result in a slightly smaller body size.
Furthermore, they found that this was generally true among genes that influence height. Other autosomal genes that are expressed at higher levels in females tend to decrease height, while, conversely, autosomal genes with a conserved male bias in expression tend to increase height. “It’s not invariably true,” Page said, “but there was a tendency.” When his team summed the effects on height across the hundreds of implicated genes, they found that the result of the differences in expression explained 12 percent of the observed difference in the average heights between females and males.
The next steps, Page said, will be to collect data from more tissues, more cell types, more developmental stages, and more species, and to explore whether sex biases in gene expression explain sex differences in traits other than height. Furthermore, he would like to understand the roots of
this sex bias in gene expression. Is it a consequence of sex hormones, of sex chromosomes, of both, or is it perhaps something else?
DISCUSSION
The session moderator, Philip Benfey, opened the discussion by referring to the keynote talk by Aviv Regev, who had suggested that researchers in functional genomics think “in terms of a random sampling across a large data population versus a more defined factorial approach,” as Benfey phrased it. He asked the panelists which approach would be most effective in their experimental systems. Mackay and Kelley responded that it would be difficult for them to use random design in large natural systems to answer the questions they are interested in, while Springer said he could see value in both approaches and which one best depended on what he wanted to learn. Kocher agreed with Springer. “I think a lot of it really depends on the type of question that we’re asking,” she said.
Scott Edwards asked about measuring gene expression in the wild versus in the lab. What is the value of studying gene expression in the wild, he asked, and is it only valuable if one can also do a common garden experiment? Kelley said that there is great value to doing it in the wild, but the limitation is that it can be impossible to tell if patterns of expression in the wild are due to adaptation or plasticity. Springer agreed about the value of doing experiments in the wild but added that the greatest value comes when he can also do controlled experiments to get additional information that cannot be gleaned in the wild. Kocher added that it is important to go into the wild and sample individuals there because it provides a baseline for things that are studied in the lab.
Gene Robinson of the University of Illinois at Urbana-Champaign, following up on a comment by Paul Katz of the University of Massachusetts Amherst, spoke about Kocher’s finding that regulation of the levels of syntaxin 1A affected sociality in the sweat bees. This finding illustrates, Robinson said, that phenotypic differences in natural populations are often due to differences in expression levels rather than to the presence or absence of a gene. Therefore, functional genomics tools need to be able to dial up or dial down expression rather than just knocking out or adding genes to the genome. Kelley agreed, saying that the ability to turn expression up or down is hugely important and that many of the genes that she examines are essential for survival.
Benfey made an observation and asked a question. “I’ve been struck over the last 2 days that most of the discussion has been around transcriptional responses,” he said. Is the current focus on transcriptional responses truly a reflection of where the most valuable investigations lie? Mackay
suggested that the current popularity of studying transcriptional responses is due to the fact that this is what is easiest to measure at this point in time, but that in the future more attention should be paid to networks. Kocher added that one can learn a lot from studying sequence variation rather than just transcriptional variation. If researchers can start to identify transcription factors that have been under selection or changes in binding motifs across genome-wide scales, then that can give us something to grab onto when thinking about how these networks might change over time.
Gary Churchill from The Jackson Laboratory described observations he has made about GxE interactions seen with experiments in mice. Noting that, as Mackay had pointed out, sex can be thought of as an environmental variable, mice fed different diets were experiencing another environmental variable, and mice that had been allowed to age were experiencing a third variable. What he and his colleagues have seen is that GxE interactions for sex are largely local and are mediated through RNA to protein; GxE effects of diet are a mix of local interactions mediated through RNA and distal, direct protein-to-protein interactions. The age effects are all distal. “My interpretation of this,” he said, “is that our genomes are exquisitely programmed to be one sex or the other. They’re pretty well programmed to respond to diets. But they’re not designed to age at all.” Then he asked the panel what their thoughts were.
Mackay responded that the sort of complexity that Churchill and others were describing is only going to be understood “when we can stop going from one variant to one transcript and to one trait, and start using our data to infer transcriptional genetic networks based on naturally occurring genetic variation and how those entire regulatory networks translate to organismal phenotypes.” With enough data, she said, it should be possible to map expression quantitative trait loci (eQTLs) that have cis effects, eQTLs that have trans effects, figure out which eQTLs have both, and then start to infer a cis–trans-regulatory network. “We need to start to embrace network theory in order to understand how complex gene regulatory networks are going to affect complex traits,” she said.
Kelley took a different angle in discussing how to deal with complexity. “To answer some of these complex questions or get at these complex phenotypes,” she said, “we really need integrative biology. We need to get mathematicians in the room who are going to develop these network models, we need physiologists, we need behavioral biologists.” In short, she said, it will be important to get all sorts of different disciplines into the same room to address the challenge of complexity.
Katz added that another problem is simply identifying what the bigger questions are. “In other words,” he said, “you have to be exposed to a genome researcher to even understand what your questions are.”
This page intentionally left blank.






















