
7
Interpreting and Validating Results from High-Throughput Screening Approaches
Today’s sequencing and “-omics” technologies are so powerful that scientists can collect huge amounts of data in relatively little time. This is one of the reasons for the tremendous promise of functional genomics. However, to have confidence in these data, it is important to be able to validate the results, which can be challenging.
The speakers in this session spoke of their experiences in validating different types of high-throughput screening (HTS) approaches, but one commonality emerged: validation generally requires carefully thought-out hard work. The session was moderated by Trudy Mackay of Clemson University. The presenters were Emma Farley of the University of California, San Diego; Philip Benfey of Duke University; Grace Anderson of Octant; and Gary Churchill of The Jackson Laboratory. A panel discussion period followed the presentations.
LESSONS ON DESIGN AND VALIDATION FROM A CRISPR LOSS-OF-FUNCTION SCREEN ON KRAS-MUTANT CANCERS
Hindsight, when used correctly, can offer some valuable lessons for the future, said Grace Anderson of Octant as they described their own experience on a project from their first year of graduate school that aimed to study acquired resistance in KRAS-mutant cancers. They mentioned that “blind spots” of the past can help identify “blind spots” in current technological approaches.
Cancer, Anderson explained, is an umbrella term used to describe more than 200 unique diseases that are highly diverse. In fact, the only thing
cancers have in common is the hallmark of uncontrolled cell growth. Over the past 15 years or so, researchers have come to appreciate the diversity of mutations that occur in cancer, and they have identified commonly mutated driver oncogenes that are similar in many of these diverse cancer types. Researchers have done a good job in identifying targets and developing inhibitor molecules to block the protein products of these commonly mutated oncogenes. However, these molecular targeted therapies have a huge problem. Virtually every one of them faces intrinsic and acquired resistance from the cancer cells it is attacking.
The best example of an acquired resistance, Anderson said, is what happens when BRAF-mutant melanoma is treated with vemurafenib. BRAF is an activating mutation in about 50 percent of all melanomas. Testing has shown that not all patients with a BRAF mutation will respond in the same way to vemurafenib. Some exhibit little to no response; this is referred to as having intrinsic resistance to the drug. Others, even those with metastatic melanoma lesions all over their body, seem to respond well at first, with the lesions completely disappearing. However, every patient that exhibits this complete response eventually relapses, with the drug no longer working; this is acquired resistance (Chapman et al., 2011; Wagle et al., 2011; Sosman et al., 2012).
As a first-year graduate student, Anderson set out with a colleague, Peter Winter, to study this phenomenon in cancers driven by mutations in another gene, KRAS. It is challenging to target KRAS directly due to the exceptionally high binding affinity KRAS has to its endogenous ligand, GTP. With this in mind, Anderson said, they chose to look into downstream methods of targeting the associated pathway. Looking back, Anderson recognizes that they would design the study differently, knowing what they know now. These lessons come in the form of changing library design and validation.
Anderson was interested in KRAS mutations in cancer for a couple of reasons. The most obvious was that KRAS is mutated in about one-fifth of all cancers, and nearly ubiquitously in some, such as pancreatic cancer (Cox et al., 2014). Furthermore, KRAS mutations lead to constitutive signaling through its associated mitogen-activated protein kinase (MAPK) pathway, which is involved in cell cycle progression, among other functions. However, monotherapies blocking downstream signaling proteins, such as MEK or ERK, seem to be insufficient to stop the cancer, which is likely due to the complexity of the pathway as well as its built-in compensatory mechanisms. To overcome this, it would be necessary to look toward combination treatment strategies.
There were some open questions that they thought a CRISPR loss-of-function screening approach would help to answer, Anderson said: What are the pathways that, when inhibited, can sensitize a patient to ERK inhibition
in KRAS-driven disease? How do these pathways vary with respect to the specific tissue in which the KRAS mutation is present? And, what accounts for the diversity of responses to seemingly appropriate therapies?
To start, they selected about 400 interesting cancer-related genes, which, Anderson noted, was a heavily biased list. They screened 12 cell lines and 4 tissue types, for a total of about 94 screens. The CRISPR loss-of-function screens used drug treatment to inhibit either MEK or ERK. Genes that “dropped out” in drug-treated samples were candidate “sensitizers” to that drug (Anderson et al., 2017).
They first noted that very few of the hits spanned more than two tissue types. In fact, only one hit spanned all of them, which was a known pathway reactivation gene, REF. Thus, Anderson noted, most of the boundaries of sensitizers are at the tissue level, an important finding for clinical trial design because most clinical trials were being defined in terms of genotype. This type of testing was, “setting the trial up for failure,” Anderson said, if the drug was not going to work in several tissue types.
More generally, Anderson said, even though they were able to answer some questions, there was not a lot of novel basic science that came out of the screens. Reflecting on this, Anderson noted that they may have made more discoveries by not biasing their library so much.
Moving to the issue of validation, Anderson said that they validated 44 of their 46 hits with two separate assays. The two gene hits that did not validate were damaged genes, with the damage being an artifact of the CRISPR cutting. There were a variety of problems with the validation, however. For one, they did not know the true effect sizes of their phenotypes because they did not include any true lethal genes, such as ribosomal genes, a standard practice today.
Perhaps the biggest issue was that, because all of the hits worked well in both validation assays, they had no way of prioritizing which of the 44 hits to test in vivo.
Later they developed a long-term in vitro assay that allowed them to differentiate among the hits. “What we found,” Anderson said, “was for these 22 combinations in colorectal cancer, all of which looked amazing on short-term validation, there was crazy diversity in how well they could actually suppress resistance long term.” This is now the standard for how researchers validate their lethality screens in the targeted therapy space. In conclusion, Anderson said, heavily biasing the library was probably a pretty big mistake.
In designing a library, one has to find the proper balance between genome-scale and a small-scale targeted library. In retrospect, Anderson said, they should have come up with some way of scaling down from the entire genome without handpicking genes that are important in cancer.
Last, when validating, it is important to choose the proper assays for the phenotype. In their case, they were interested in long-term durability, and so any shorter assays were not appropriate.
USING FUNCTIONAL GENOMICS TO UNDERSTAND DEVELOPMENT
In the next talk Philip Benfey described applying functional genomics tools to study development in a plant—in particular, the development of the Arabidopsis root. “When I say development, our primary focus is on how you go from an undifferentiated stem cell to a fully differentiated cell and how that cell can function to make an organ that has some real purpose in the world. And the organ we focus on is the root.”
Studying Arabidopsis roots simplifies that problem of development because of the way the root grows in one direction along one dimension and because it has radial symmetry—that is, the different cell types essentially grow in concentric tubes, one inside the other, surrounding an inner cylinder of vascular tissue. Thus, the position of a cell can be specified by only two variables: how far from the tip it is and how far from the centerline it is (see Figure 7-1). The stem cells, which are the source of all the other cells, are found at the tip of the root, and because the cells do not move in relation to one another during development, the youngest cells will always be found at the root tip and cell age increases with distance from the tip, meaning that one sees a developmental timeline as you move up the root. Cell development resembles an assembly line where one starts with fairly undifferentiated materials and ends with specialized materials. These two variables of the Arabidopsis root, distance from centerline and distance from the tip, are sufficient to specify cell type and development stage.
About 20 years ago, Benfey said, he and his team studied expression patterns in the different cell types at different times in development. He used 19 different markers of cell type and sliced individual roots into 12 sections, each a certain distance from the tip and thus at a particular developmental stage. When he examined the data from each of the 19 markers at the 12 developmental stages, he found clear expression patterns related to both cell type and developmental stage (Brady et al., 2007; Dinneny et al., 2008). What was surprising at the time—although it would not be surprising today, he said—was how much cell-specific expression they found.
“We went on to show that that cell-type-specific expression was also responsive to different environmental stresses,” he said, explaining that this was an unexpected result. It seemed unusual that there would be cell-type specific responses to the same stressor.
There were, however, a couple of weaknesses with this approach, Benfey said. First, it required the marker lines, and developing them for a new
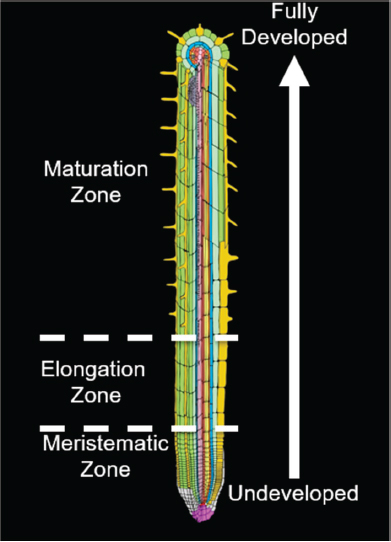
SOURCE: Philip Benfey presentation, slide 2.
species would have required a lot of work. Second, it was difficult to monitor responses over time, again because of the amount of work involved. “As a high-throughput approach, it left a lot to be desired.”
A newer alternative approach is to use single-cell RNA-sequencing (RNA-seq), which generates essentially the same data but from individual cells rather than from cell collections all of a single type. In the past year, Benfey said, there have been five different publications of single-cell RNA-seq studies done on the Arabidopsis root, all of which relied heavily on the annotations that his group had previously developed (Denyer et al., 2019; Jean-Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019; Zhang et al., 2019). However, these studies were missing a lot of the fine detail about changing expression patterns as the root developed. His team decided to approach this by combining some of the published datasets with their own datasets, ultimately using data from 80,000 cells to create a “reference transcriptome” that included information on transcription separated both by cell type and age. Ultimately, the goal with this reference transcriptome would be to use it in a way that is similar to how a reference genome is used.
At that point, Benfey turned to a separate, if related, topic—his team’s efforts to understand what is happening in the initial divisions of a stem cell in the Arabidopsis root. To do that, he said, they take a reductionist approach and examine what happens in a single stem cell (see Figure 7-2).
The cell first divides in the direction of growth to regenerate itself. Then the upper of the two resulting cells—the one farther from the root tip—divides along the longitudinal axis to produce the first two cells of what will become two very different lineages. “This is an asymmetric cell division in the sense that the two cells will have different fates,” Benfey said.
In particular, in wild-type (WT) Arabidopsis, the result of that asymmetric division is the creation of two inner layers, the cortex and the endodermis, which are situated between the outer layer (the epidermis) and the inner cylinder of vascular tissue. But in a shortroot (SHR) mutant variety of Arabidopsis, named “shortroot,” there is only a single layer between the epidermis and the vascular tissue that only has attributes of the cortex, the outer of the two asymmetric layers (Benfey et al., 1993).
A second variety called “scarecrow” also has just a single layer there, but that single layer has attributes of both the endodermis and the cortex (Benfey et al., 1993). In other words, it never makes that initial asymmetric division, but instead the resulting cells have characteristics of both cell types.
After cloning the genes responsible for SHR and scarecrow, Benfey’s team was surprised to find that SHR was not expressed in either of the two lineages from the asymmetric cell division. The SHR protein, a transcription factor, is made internally and then moves out to the adjacent cell layer
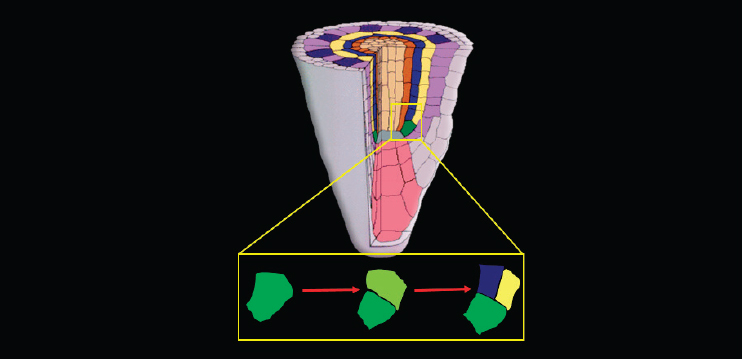
SOURCE: Philip Benfey presentation, slide 19.
(Nakajima et al., 2001). As discussed in Bergmann’s talk (see Chapter 3), plants can signal by moving proteins from one cell to another. As it moves, SHR comes into contact and interacts with the SCARECROW protein, which is another member of this plant-specific transcription factor family, and SCARECROW prevents SHR from continuing to move to the outer layers (Di Laurenzio et al., 1996; Helariutta et al., 2000; Nakajima et al., 2001; Cui et al., 2007).
When SHR interacts with SCARECROW, it binds to the SCARECROW promoter, producing a positive feedback loop (Cruz-Ramírez et al., 2012). The protein complex also binds to the promoters of a highly specialized member of the cell cycle machinery that is critical to the asymmetric cell division. To understand the dynamics of the various interactions, Benfey’s group, in collaboration with the Scheres group at Wageningen University, created a mathematical model that predicted a bi-stable behavior in which SCARECROW is at either a low or high level. “The naïve prediction from that,” he said, “is that if it’s a low level, the switch is off, and if it’s a high level, the switch is on. And when the switch is on, you get the cell division.”
Next they studied the dynamics of the process by studying the behavior of SHR, SCARECROW, and the subsequent asymmetric cell division. To do this, they used light sheet microscopy, which allowed them to examine the levels and kinetics of both SHR and SCARECROW. They could perform live imaging to watch the levels of both proteins in the different cells of the growing roots and even see the asymmetric cell divisions. The behavior they observed did not completely match up with the prediction of bi-stability from the mathematical model, Benfey said, but perhaps the more important fact is that they were actually able to test the predictions of their mathematical model by watching the expression levels in real time.
Finally, Benfey mentioned two other techniques that his team is working with. One is using single-cell RNA-seq to follow spatial and temporal expression in the root. The second is building synthetic circuits with transcription factors and other pieces, embedding them in a larger network, and then modifying the circuits to see what effect it has on that larger network.
In conclusion, he spoke briefly about the challenges his team faced. Time courses are critical, not only for what happens after the induction of SHR and other key regulators, but also for what happens in response to environmental perturbations. The challenge, Benfey said, is how to analyze those time courses. It is not obvious how one can use standard time-course algorithms to study something where there are 10,000 cells, each with sparse data and a different time course. Synthetic networks offer a way of validating one’s models in the deepest way, Benfey said. As engineers will say, “You don’t really understand it until you can make it yourself.” Finally, he said, integrating functional genomic approaches with time-lapse imaging is also a great challenge, but one with a significant potential payoff.
VALIDATING RESULTS FROM HIGH-THROUGHPUT ENHANCER SCREENS
Enhancers are short stretches of DNA that, when bound to by proteins called transcription factors, control the temporal and spatial expression of a gene. These enhancers provide instructions for tissue-specific gene expression. Emma Farley of the University of California, San Diego, spoke about issues related to the HTS of enhancers.
“I think of [enhancers] as switches in the genome that control when and where genes are expressed,” she said. The major shortcoming of screens for enhancers is the lack of functional validation. She mentioned that many studies use correlative methods to predict putative enhancers in the genome, or test how sequence changes in enhancers affect phenotype. These studies often identify tens of thousands of putative enhancers and yet only a handful are validated functionally with experimental assays. This is typical and points to a weakness in the current use of HTS—the lack of true functional validation of the large datasets. Such validation is imperative to help researchers learn how much of what they are inferring is actually true.
To provide further insight, Farley described some of her own work and the issues that arise concerning validating results from HTS. Referring to Figure 7-3, she offered a simple example of the sort of approach she takes to understand how the sequence of an enhancer encodes tissue-specific gene expression and what changes in an enhancer affect gene expression and phenotype.
The simple enhancer illustrated in the figure contains two types of binding sites, one shown in orange and one in blue. As Farley explained, the ball-shaped object is a gastrula-stage embryo, and the cells on the visible side of that embryo could give rise to either skin or nervous system.
The embryo in the top left shows the signaling of fibroblast growth factor (FGF), which activates the transcription factor ETS, and the embryo on the top right shows the cells on the visible side of the embryo expressing the transcription factor GATA. The binding of ETS to the blue sites in the enhancer and of GATA to the orange sites in the enhancer act to turn on the gene otx, which is one of the first genes to specify nervous tissue in an embryo. The bottom right of the figure shows a later-stage embryo, where the head, tail, and nervous system can be seen.
A major issue in functional genomics is understanding which regions of the genome encode enhancers, where and when these enhancers are active, what genes they control, and which single-nucleotide polymorphisms (SNPs) impact enhancer function. To illustrate this problem, Farley said there are more than 100,000 clusters of ETS and GATA binding sites in the genome; some are non-functional, some act as enhancers that turn on in the nervous system, and some turn on in the gut and the heart. Farley
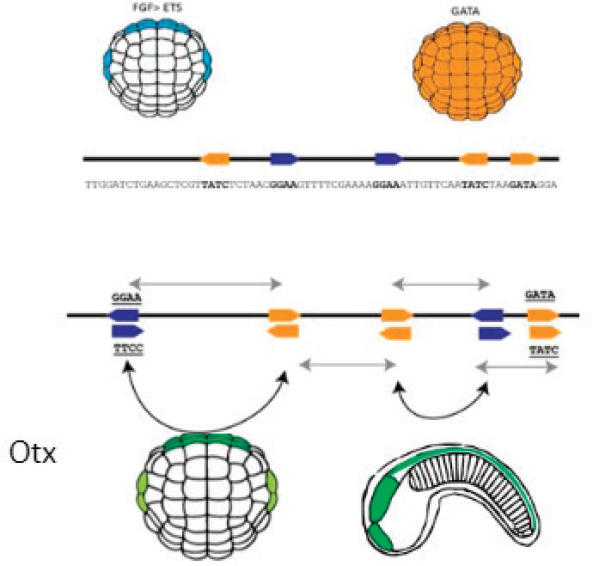
SOURCE: Emma Farley presentation, slide 3.
went on to ask why this particular (Otx-a enhancer) sequence is turning on in the nervous system.
In an ideal world, one would take the sequence and then change various nucleotides and see how that affects gene expression, while keeping constant those nucleotides known to be important in order to make it more likely that the resulting variants are actually functional.
However, the complexity of the required analysis increases rapidly with the number of nucleotides, which, Farley said, is one of the reasons she chose to work with this enhancer—because it is very small. But even in a small enhancer, making every sequence combination would still produce 1,030 different synthetic enhancer variants, an amount that is currently impossible to test and analyze. The other reason for choosing this enhancer is that it has a common logic, as the enhancer is activated by a signaling pathway (FGF>ETS; see Figure 7-3) in combination with a tissue-specific determinant (GATA). Farley noted that she did not just want to understand
how this enhancer works but how enhancers regulated by ETS and GATA, and indeed how enhancers regulated by this common logic of signaling factor and tissue determinant, generally work. She mentioned that her lab not only looks at how sequence encodes gene expression, but also how the role of organization of bindings sites (the order, orientation, and spacing of these sites) encode gene expression.
She works with embryos of the sea squirt Ciona intestinalis. In doing her experiments she takes an enhancer, attaches it to a promoter and a green fluorescent protein (GFP) reporter, and electroporates the construct into fertilized eggs. Then, wherever the enhancer turns on in the resulting embryo, it can be detected using GFP.
“We can electroporate hundreds of thousands or millions of fertilized embryos in a single hour-long experiment,” Farley said, “and the idea is to make all of these different variants and then test them in whole embryos to see how changes in sequence … impact gene expression.” The core of the binding site is kept constant, and the rest of the sequence is randomized. Each enhancer sequence is attached to a promoter, GFP, and a unique barcode. If an enhancer turns on transcription, it will make a messenger RNA (mRNA) of the barcode, which can be detected, measured, and associated with the appropriate enhancer sequence.
After testing millions of enhancer sequences, Farley identified 20,000 that were active at or above WT levels of Otx gene expression. To partially validate the data, she re-tested 100 of the enhancers, some of which were active and some inactive according to their data, using the same general assay, but looking for GFP expression under a microscope instead of at pooled sequencing data. Those results lined up with her original findings, and this gave her faith in the sequencing data.
Next, she examined her set of 20,000 functional enhancers for common features and found two motifs that were high-affinity sites for ETS and GATA. Although the high-throughput data suggested that high-affinity sites were enriched in the functional enhancers from the HTS, when she measured the affinity of the sites within the WT Otx enhancer, she found that there were some high- but also some very-low-affinity sites. To test the hypothesis that only the high-affinity sites were needed, she took enhancer variants from the library that were not functional and sought to make them functional.
“I think that it’s key when you’re doing these high-throughput screens,” she said, “that you test your hypotheses in the most challenging way you can think of. We thought trying to make inert DNA into a functional enhancer was a real challenge because it’s incredibly hard to build tissue-specific enhancers.”
When they tried this by just mutating the sequence flanking the core of the binding site to make high-affinity binding sites, they found that they could indeed turn an inert piece of DNA into a functional enhancer, but they had lost tissue specificity. With further experimentation, she said, they discovered that the lower-affinity sites seen in the WT enhancer were needed for tissue specificity.
Exploring further, the next step was to examine how the organization of the binding sites affects gene expression. They tried manipulating the spacing between the binding sites and found that changing the spacing between the binding sites affected the levels of gene expression.
Interestingly, the spacing seen in the WT enhancer is not optimized to give the highest level of expression. Because the WT enhancer has both binding sites that are of suboptimal affinity and a spacing of the binding sites that is suboptimal for transcriptional output, she tested what would happen if both the affinity and the organization of the binding sites were optimized. The answer was that tissue specificity completely disappeared with optimization (Farley et al., 2015).
To test whether this was a general principle, Farley was successful with work on similar enhancers and other tissues. Such validation, Farley said, is important if one wants to “translate principles and make claims about the rules of life.”
After that, she said, she ran into a roadblock working with the sea squirt because it is difficult in that species to replace one enhancer with another, so she switched to mice. It was a blessing in disguise, she said, because to test if the principles she discovered are truly generalizable, it was important to test them in a completely different organism.
She worked with the well-known enhancer ZRS, which drives expression of the sonic hedgehog gene in developing limb buds. As she had seen with the sea squirt, the ETS binding sites on the enhancer for sonic hedgehog were low affinity. As it happens, she said, there is a human mutation that causes polydactyly, and no one had understood why that mutation has this particular impact, but she noticed that the mutation tripled the affinity of a binding site for ETS (Albuisson et al., 2011). Her work in the sea squirts led to the hypothesis that this mutation could lead to a less precise expression of the sonic hedgehog gene and an extra digit. They tested this in mice and found that optimizing the affinity of the binding site did indeed produce extra digits, demonstrating that this principle of enhancers translates across species.
Wrapping up, Farley said that the key to using HTS is reducing complexity as much as possible. “When we first started,” she said, “all we did was change the sequence outside the core of the binding sites, and, by doing
that, we were able to understand what was necessary and sufficient to drive expression.” This led to work in which they explored changing order, orientation, and spacing of the binding sites to investigate further principles behind enhancer sequences.
Farley noted that the key aspects of HTSs are
- Experimental design:
- Biological context
- Experimental tractability
- Reducing variability and confounding factors
- Validating at multiple levels:
- Validating the primary data
- Rigorous testing of any hypotheses obtained from the HTS analysis
- Making and testing predictions to further challenge your hypothesis
Validating one’s findings in many contexts shows the generalizability of any principles identified. The major roadblocks facing people in this field, she said, include finding a way to reduce the complexity of a problem, the fact that validation of high-throughput screens is very slow, and the fact that no organism provides all the tools needed to answer these questions, so one must decide whether to develop the necessary tools or change organisms.
Looking to the future, Farley said, “I think we need a culture shift. I think functional genomics used to be about correlation, but now I think it should be about causality at scale.” The field needs to develop a framework for validation that relies heavily on testing of predictions, she said. Funding initiatives are needed that focus on innovations to validate large-scale screens more efficiently, whether it is through automated imaging, the integration of other -omics, transgenic tools, or phenotyping at scale.
HARNESSING GENETIC DIVERSITY TO UNDERSTAND MAINTENANCE OF PLURIPOTENCY IN EMBRYONIC STEM CELLS
The session’s final speaker, Gary Churchill of The Jackson Laboratory, began his presentation by announcing that he was going to lead off with his conclusion. Touching on some of the themes from the previous talks, he said that if functional genomics is to make the transition from a correlational approach to a causal approach, genetic variation holds great promise as an experimental perturbation. Under certain circumstances one
can assume that genotypes precede phenotypes. However, he continued, this is an assertion, and it requires some really strong assumptions that can be somewhat alleviated by the construction of artificially structured genetic populations. Mediation analysis is often used to explore the mechanisms of how independent variables, such as a genotypes, influence dependent variables, such as phenotypes, but there are some problems with this approach that cannot be entirely eliminated. Furthermore, functional genomics researchers are faced with the challenging situation of having an overwhelming number of results coming from these analyses but a limited bandwidth with which to do validation.
“So my question is,” Churchill said, “when is validation needed and when are correlational results sufficient to just move on with our lives?”
With that, he began his presentation by describing the mouse populations that are bred at The Jackson Laboratory. There are eight inbred founder strains that are genetically very different from one another, and there are both inbred and outbred populations. The diversity outbreds have a balanced population structure, with more than 400 recombinations per animal, and a high heterogeneity, with the disadvantage that each mouse is unique. The inbreds, or collaborative crosses, have reproducible genomes and a high genetic diversity, but fewer recombinations per line.
Next, Churchill introduced expressional quantitative trait locus (eQTL) and protein quantitative trait locus (pQTL) mapping. These are locations in the genome that are linked to variation in either gene expression levels, that is, mRNA levels (eQTLs) or protein levels (pQTLs). Churchill’s group carried out a study comparing eQTLs and pQTLs in liver tissue in mice, and the overlap between the two “was so close to random that it was shocking.” But a closer look revealed that the overlap between the local eQTLs and pQTLs—that is, those near the corresponding genes—was almost perfect, while the overlap between distal eQTLs and pQTLs was almost entirely absent. “That tells us a lot about how proteins and RNAs are regulated,” he said (Chick et al., 2016).
With those data the team applied mediation analysis in an effort to reveal some of the details of the relationship between the RNAs and the proteins. Such an analysis requires starting with a hypothesis, so the team hypothesized that variation in the DNA causes variation in the RNA, which in turn causes variation in the protein. They use a standard strategy called the causal steps method, which involves four logical statements (see Figure 7-4). “We asked that the protein have a QTL, that the RNA have a QTL, and that these both co-localize”—(i) and (ii) in the figure. “We asked that if we calculate the partial correlation by regressing out the protein, the RNA should still have a QTL” (iii). “And we asked that if we regress out the RNA, the protein no longer has the QTL” (iv).
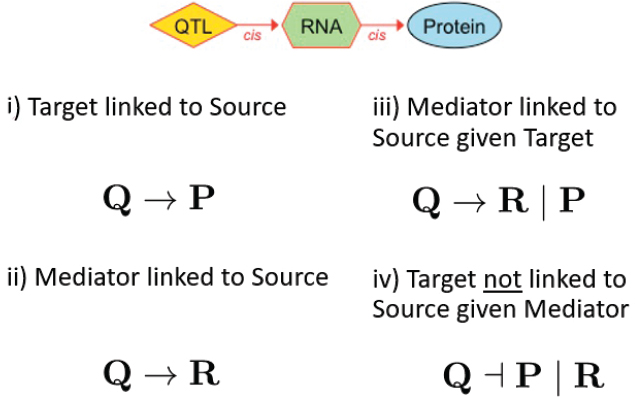
SOURCE: Gary Churchill presentation, slide 4.
The fourth statement is the stumbling block, Churchill said. “That’s the one that we can’t prove. And it’s the one that causes us all the headaches and all the focus.”
With that background, Churchill moved to the main subject of his talk—how he and his team used this approach to understand the maintenance of ground-state pluripotency in mouse embryonic stem cell lines. Ground-state pluripotency is the ability of cells to make unlimited copies of themselves and also, under the right conditions, to differentiate into any sort of cell in the body, which is, in a sense, the most primitive and unlimited form of pluripotency. To study how this pluripotency is maintained, Churchill’s group derived a large set of embryonic stem cells from individual DO mice and performed RNA-seq and assay for transposase-accessible chromatin using sequencing (ATAC-seq) to get data on RNA levels and on chromatin accessibility across the genome, respectively.
When they mapped out their data, they found characteristic patterns where there were strong signals of local regulation with a significant amount of distal variation. And that distal variation sometimes seemed to cluster in bands where a single genetic locus appeared to affect the expression of many genes or a single genetic locus seemed to affect the openness of many ATAC peaks in the genome, corresponding to regions in the genome where the chromatin was accessible (see Figure 7-5).
Interpreting this according to their prior hypotheses, the data indicated that there was a genetic variant that affected the chromatin in multiple
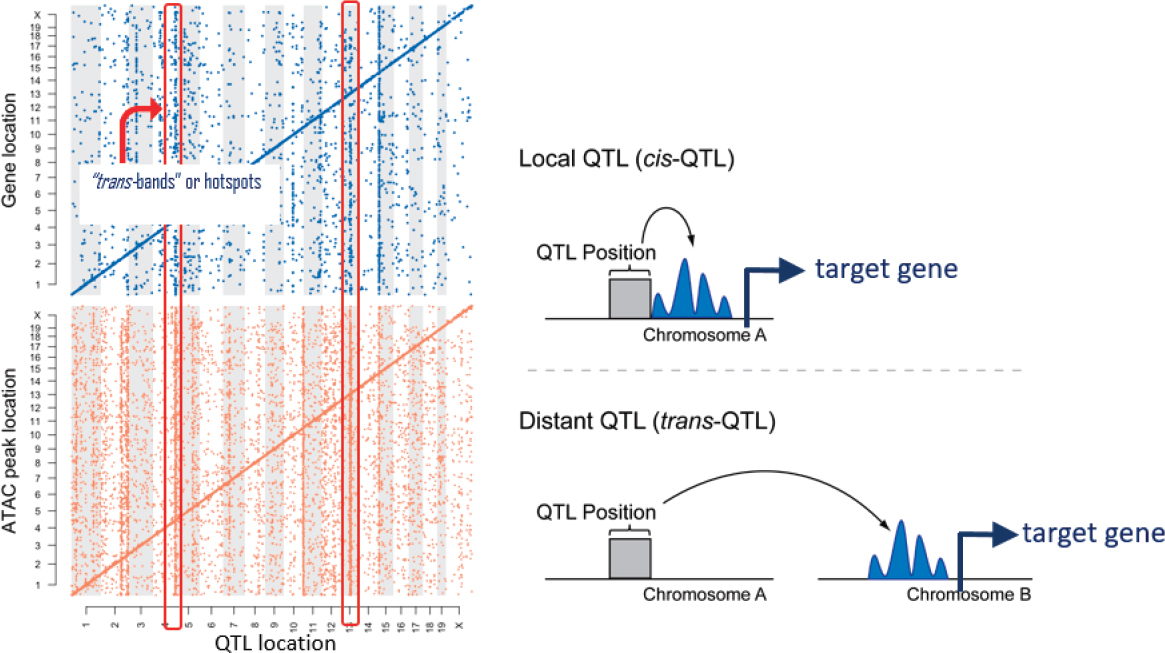
SOURCE: Gary Churchill presentation, slide 13.
places and that, in turn, affected the expression of multiple genes. They found that there were many more ATAC peaks than there were genes, Churchill said, but “for virtually every gene where we saw variation and expression, there was one or more ATAC peaks which were clear mediators, and they followed the right allele effects and everything was cool.”
The more challenging part of the study was to understand what was going on with the “hotspots,” the collections of distal regulatory elements that appeared as bands in the maps. One hypothesis was that there is a genetic variant that by some mechanism affects a distal ATAC-seq defined open chromtin region, which then regulates the expression of the gene.
There were many hotspots throughout the genome that Churchill identified in this work, but for the purposes of the presentation he focused on a hotspot on chromosome 15. His team found a particular gene in the region that seemed to mediate a lot of the downstream target genes, and that was the gene lifr for the leukemia inhibitory factor (LIF) receptor. This gene is involved in regulating the differentiation of embryonic stem cells and pluripotency (Graf et al., 2011).
When Churchill examined the situation more closely, he found greater complexity than was apparent at first. Looking at the genetic pattern at that locus among the eight founder mouse strains at The Jackson Laboratory, he found that of the downstream target genes, many of which are involved in pluripotency, there was high expression in four of the strains and low expression in the other four. Furthermore, the four strains with low expression were “recalcitrant” strains for which the researchers found it difficult to maintain embryonic stem cell lines. By contrast, the four with high expression were “permissive” strains for which the embryonic stem cell lines were easy to maintain. “The downstream targets do the right thing,” Churchill said. “The LIF receptor is doing the right thing.”
Furthermore, they were able to identify an SNP correlated with the difference in lifr expression among the four recalcitrant strains and the four permissive strains. It is about 10,000 base pairs upstream of the gene, and so it is likely to be some sort of enhancer.
Finally, the SNP is just seven base pairs upstream of a known binding side for the transcription factor Nr5a2, and the evidence suggests that Nr5a2 regulates lifr. “Indeed, in the case of the high-expression allele, Nr5a2 is probably the key transcription factor,” he said. “It just binds.” He continued, “if you change the context a little bit, all of a sudden the effect of Nr5a2 becomes quantitative.”
To validate the role of the SNP, Churchill swapped out the two versions of the SNP between a high-lifr strain of mice and a low-lifr strain, so that the high-lifr strain now had the SNP from the low-lifr strain, and vice versa. When he did this, the response to LIF of embryonic stem cells from the two different strains of mice flipped, confirming the key role of the SNP.
Summing up, Churchill said, “we mapped an important regulator of pluripotency maintenance in these embryonic stem cells. We identified a causal gene, and we identified a single causal SNP, and we went to great lengths to validate it.”
DISCUSSION
Mackay, the moderator, opened the discussion with her own question. Any functional genomic screen will produce statistically significant results, depending on what has been screened for, she noted, and many of the implicated genes will have unknown functions. Also, most validation strategies are one hit at a time. So, first, how should these results be interpreted in terms of networks, “bearing in mind that most known networks are based on loss-of-function mutations”? Second, how could one approach validating entire networks rather than one node at a time?
Churchill responded that it is important to keep in mind that when one performs such a screen and has thousands of hits, there will be an error rate—something that many people do not recognize. To determine what that error rate is, it is necessary to do some sort of validation experiment, perhaps with a complementary experiment that does not share the same biases and pitfalls as the original experiment.
Next, Farley suggested, “If you have thousands of regions that you think are having an effect on some phenotype, [you] would test them.” For example, if a study produces a set of ATAC-seq peaks, test as many as possible, if not all. The difficulty will vary depending on the type of data, she said, but with transcriptional regulation studies, for instance, “you could go about asking those questions if you design your experiments right and you design massively parallel reporter assays in the correct way.”
Benfey had a different take on the question. Much of what researchers do is find something interesting and then go deep into it, learning everything they want to learn, and that is a valuable approach. “I’m not that convinced that having totally comprehensive analysis validation is the way we need to go,” he said.
Nathan Springer of the University of Minnesota pushed back on a theme that he had heard in a couple of the presentations—that researchers need to move beyond correlation to causation. There is value in that, he acknowledged, but “I want to be careful placing all of the value there.” As an example, he pointed to the dairy cattle industry, which has made tremendous progress in genomic prediction without spending much time on mechanisms. These researchers care about predicting a trait, not in exploring the genes underlying that trait.
Farley responded that she did not doubt that there is great value to many of the studies that do not touch on causation, but she believes that
“we should learn which assumptions we’re making are accurate and which ones are not.” She was not arguing that researchers should functionally validate everything they can, but she does believe that researchers should start doing some studies that examine causality because such studies have been underrepresented in the work on how genomes encode phenotypes to date. Understanding causation could end up making correlative studies even more powerful.
Churchill suggested that whether a study should look at correlation or causation depends on its goal. Understanding causation can be particularly important in biomedical research where the ultimate goal is to learn enough to develop effective therapies.
Joanna Kelley of Washington State University, playing devil’s advocate, asked why, if validation is so important, shouldn’t the field put all its effort into “studying the role of transcription factors, ATAC-seq peaks, all of these things, in one organism, to really understand deeply how all of these different functional genomic things that we’re measuring work”? In short, why not focus on one organism in depth rather than researchers each trying to validate in their own systems, which presents so many challenges?
“We were asked to talk about how to validate high-throughput screens,” Farley said, “and so I was using my research as an example of how I think we should at the moment validate high-throughput screens to understand the underlying patterns in the information that we’re trying to understand, to get to how changes in our genome actually impact phenotype.” Everyone is asking different questions, and whatever the question is, it is important to test one’s assumptions.
Marc Halfon of the University at Buffalo suggested that how one validates often depends on the question one is trying to answer and what one hopes to accomplish. Also, researchers need to be aware of “validation creep,” which Halfon described as “when you forget that the stuff that you haven’t validated may still not be right or a certain percentage of that may not be right.” If research succumbs to this validation creep and builds on data that have not been validated, it can be dangerous, he said. On the other hand, digging into the few findings that have been validated can be valuable. “You can do a lot with that,” he said. “The risk is forgetting that you haven’t validated all the rest.” Or if one wishes to build on the remaining 19,990, it is important to have more extensive validation.
Farley agreed. “I think the issue is just what you said. We do these genomic-scale studies, and then often there are general statements made based on those genome-wide studies” rather than on the 10 things that have been validated. The other issue is how researchers should approach this issue over the long term. “We can’t possibly measure every parameter in every cell in every time point,” she said, “so what are the ways that we can find the structure or the patterns embedded in DNA sequence?”


















