
1
Introduction
In December 2019, new cases of severe pneumonia were first detected in Wuhan, China, and the cause was determined to be a novel beta coronavirus related to the severe acute respiratory syndrome (SARS) coronavirus that emerged from a bat reservoir in 2002 (Wu et al., 2020). Within 6 months, this new virus—SARS coronavirus 2 (SARS-CoV-2)—has spread worldwide, infecting at least 10 million people with an estimated 500,000 deaths. Coronavirus disease 2019 (COVID-19), the disease caused by SARS-CoV-2, was declared by the World Health Organization a public health emergency of international concern on January 30, 2020, and a pandemic on March 11, 2020 (WHO, 2020b). To date, there is no approved effective treatment or vaccine for COVID-19, and it continues to spread in many countries. COVID-19 has caused unprecedented global economic and social disruption. In the United States alone, 25 million people have become unemployed and the real gross domestic product contracted 4.8 percent at an annual rate during the first quarter of 2020, with projected losses increasing moving forward (CBO, 2020). Surging numbers of severely ill patients have strained health systems, and population lockdowns to curtail virus transmission have disrupted social interactions, education, and businesses small and large.
Clearly, multiple outbreaks suggest that preparedness and response strategies need modernization. Modern advances in DNA sequencing, genomics, epidemiology, and big data analyses provide new paradigms for tracing symptomatic and asymptomatic transmission networks and identifying sites of spread and at-risk populations, thereby enabling the capacity to break or delay virus transmission to mitigate social and economic disruption and reduce morbidity and mortality. Doing so also allows limited
resources to be targeted to key sites of disease expansion, such as long-term care facilities or specific places of work. Another advantage of these 21st-century pathogen disease-tracing methods is that they provide critical time for the implementation of public health intervention strategies, medical countermeasure development, and disease control. As the 21st century has already seen the emergence of four pandemic viruses (chikungunya virus, Zika virus, 2009 H1N1 influenza, and SARS-CoV-2), several viral epidemics (2003 SARS, 2012 Middle East respiratory syndrome [MERS]-CoV, 2014 Ebola virus in West Africa, and 2018 Ebola virus in the Democratic Republic of the Congo), and intermittent sporadic outbreaks of other viruses such as H7N9 influenza (WHO, 2020a), global health dictates a critical need for modernization and integration of public, private, and federal public health response efforts designed for rapid deployment to protect the health of populations and the economy.
CORONAVIRUS EVOLUTION AND SARS-CoV-2
Coronaviruses demonstrate the capacity for continuous emergence to cause significant and potentially pandemic disease in humans and animals. In the 21st century, three new human coronaviruses have emerged to cause epidemic or pandemic disease outbreaks including SARS-CoV in 2003, MERS-CoV in 2012, and SARS-CoV-2 in 2019 (WHO, 2020a). Concomitantly, three novel coronaviruses have emerged in the 21st century to cause major pandemics in swine, including porcine epidemic diarrhea virus, porcine delta coronavirus, and severe acute diarrhea disease virus in China (Vlasova et al., 2020). The continual emergence of coronavirus epidemics and pandemics underscores the critical importance of developing robust metrics of genome evolution, which could be used to inform the medical and public health communities of high-impact mutations and changes in evolutionary trajectories that might impact spread to human or domesticated animals.
Coronaviruses have large (28–32 kb), message-sense RNA genomes. The replicative machinery of the virus is encoded in the first 20 kb of the genome as two large open reading frames. Downstream of this machinery, all coronaviruses encode essential structural proteins, membrane (M), envelope (E), spike (S), and nucleocapsid (N). For SARS-CoV-2, the best characterized human epithelial cell receptor is angiotensin-converting enzyme 2, which is bound by the virus’s spike protein. This S protein contains the receptor-binding domain that is the target for neutralizing antibody and the focus of vaccine development efforts. RNA viruses depend on an RNA-dependent polymerase (RdRp) to replicate the viral genome. Much of the reason for the high mutation rates in RNA viruses is the error-prone nature of RdRp enzymes. To compensate for their large genome size, coronaviruses have
adapted to mutate less frequently by encoding a novel proofreading 3′-to-5′ exoribonuclease that associates with the polymerase complex to improve genome replication fidelity (Smith et al., 2015).
Although coronavirus mutation rates are slower than for many other RNA viruses, recombination, insertion, and deletion events also produce changes in the viral genome. Genetic recombination drives the creation of viral diversity in many positive-strand RNA viruses by the formation of novel chimeric genomes. During controlled mixed infections in vitro, rates of coronavirus genome RNA recombinations approach 25 percent or more and can be accompanied by deletions and insertions. Intergenic and intragenic recombination allow for rapid acquisition of novel functions and modular exchange of functional components between viruses. Under natural conditions, recombination-based processes have resulted in viruses with altered host range as well as altered immunogenicity and virulence, and thus provide a rapid approach to escape antibody neutralization (Ballesteros et al., 1997; Gallagher et al., 1990; Sánchez et al., 1992).
These processes provide extensive opportunities to overcome reductions due to population bottlenecks caused by antiviral drugs, host immunity, or human-to-human and animal-to-human transmission events. Monitoring RNA virus genomic change is an important step for anticipating viral emergence, predicting disease severity, evaluating drug and vaccine performance, and tracing symptomatic and asymptomatic transmission networks throughout a host population. An important consideration for any genetic epidemiology study is an understanding of the basic factors and selective pressures influencing viral evolution. Monitoring the evolution of genetic diversity in SARS-CoV-2 has the potential to inform targets for countermeasures, such as antiviral drugs and vaccines, and to improve diagnostics (van Dorp et al., 2020).
Phylogenetic studies estimate that SARS-CoV-2 spilled over to humans in late 2019 (Dawood, 2020; van Dorp et al., 2020; Wu et al., 2020). One study of more than 7,000 sequences found 198 sites at which the SARS-CoV-2 genome appeared to have already undergone recurrent independent mutations (van Dorp et al., 2020). Analysis of genomic sequences from the Global Initiative on Sharing All Influenza Data database found eight novel recurrent mutations and potentially coexistent European, North American, and Asian strains characterized by different mutation patterns (Pachetti et al., 2020).
THE POWER OF GENOMICS IN UNDERSTANDING SARS-CoV-2
Viral genome sequence data are an increasingly important tool for detecting and understanding the spread of infectious diseases in real time and for mounting effective responses. Advances in the speed, granularity,
affordability, and portability of genomic sequencing technologies have created transformative potential for widespread rapid genomic surveillance during infectious disease outbreaks, particularly when data from genomic sequencing are integrated with and analyzed alongside patient-based clinical and population-based epidemiological data (Ladner et al., 2019). The rapidly advancing field of phylodynamics uses Bayesian statistical frameworks to obtain both epidemiological and evolutionary information from pathogens to trace their history, infer transmission dynamics, and construct phylogenetic trees (Baele et al., 2016; Grenfell et al., 2004). Prior to the advent of viral genome sequencing and phylodynamics, estimates of critical epidemic parameters to inform the public health response to an outbreak, such as the basic reproductive number (R0), relied exclusively on epidemiological incidence data (Grubaugh et al., 2019). Today, the ability to collectively harness genomic, epidemiological, and clinical data contributes to enhanced, multidimensional understanding of an outbreak and enables molecularly precise and targeted responses that were not previously possible. Sequencing of viral genomes can help to answer—and in some cases, may provide the only way to answer—questions that are foundational to understanding, mitigating, and controlling a virus outbreak: the identity and novelty of the causal virus; its origin in terms of reservoir host and geography; its introduction in humans; its linkages with other outbreaks; and its potential to evolve and locally adapt (Grubaugh et al., 2019).
When combined with epidemiological information, genomic sequencing can be particularly useful for investigating outbreaks of RNA viruses, such as SARS-CoV-2. Calls have already been made to enhance the response to the COVID-19 pandemic by integrating genomic, epidemiological, and clinical data (Koks et al., 2020). Rapidly developing and deploying precise and targeted interventions and countermeasures will require a more granular understanding of exposure and infection by virus variants within and across populations, as well as the genetic, comorbidity, social, and environmental cofactors that modulate disease severity.
Virus genome sequencing is a cornerstone of the emerging field of “genomic epidemiology,” which leverages phylodynamic approaches to clarify pathogen transmission patterns and events in greater detail than is possible with traditional epidemic investigations (Gardy and Loman, 2018; Grubaugh et al., 2019) (see Box 1-1).
Increasing evidence supports the value of viral genome sequence data across all stages of an infectious disease outbreak. During the initial stages of an outbreak, unbiased DNA sequencing of infected tissue can help to genetically identify the causative novel pathogen from which rapid screening tests can be developed. The data can also contribute to identifying the reservoir host and geographic location of the virus’s origin. Compared to traditional approaches, such as interview-based contact tracing, approaches
that integrate genomics offer quicker and more comprehensive methods to build an understanding of a virus’s transmission chain and dynamics (e.g., human-to-human or zoonotic) (Grubaugh et al., 2019; Houldcroft et al., 2017). When integrated with other sets of contextual metadata, genomic epidemiology has transformed the ability to map the spatiotemporal patterns, social drivers, and transmission chains through which cases are emerging as an outbreak continues to unfold (Grubaugh et al., 2019). For example, genomic data and location can serve as proxies for estimating epidemiological connections (Wohl et al., 2020). Understanding the spatiotemporal characteristics of virus transmission—within and across different populations—as well as the virus’s genetic changes over time can inform the design of more effective, targeted interventions and countermeasures (Ladner et al., 2019). During periods between outbreaks, genomic epidemiology also contributes to tracking the evolution and transmission dynamics of viruses in both humans and reservoir species (Grubaugh et al., 2019).
The use of genomic data has substantial practical implications for public health practice around infectious disease control by improving the capacities for ongoing surveillance, rapid diagnosis, and real-time disease tracking (Gardy and Loman, 2018). In situations where there is prior knowledge of mutations that affect specific characteristics of the virus such as virulence, drug susceptibility and antigenicity, whole genome sequencing (WGS) of pathogens—which can now be conducted in (near) real time directly from clinical samples—can provide this information during an outbreak (Koks et al., 2020; Ladner et al., 2019). This information can also enable point-of-care molecular diagnostics and inform individualized treatment regimens akin to the use of human genetic data in precision medicine (Gardy and Loman, 2018; Houldcroft et al., 2017) (see Figure 1-1). At the population level, WGS combined with epidemiological data can use pathogen mutations as markers of transmission events to “reveal patterns of epidemic transmission at a fine-scale resolution” to inform more precise and targeted large-scale public health interventions than traditional approaches (Ladner et al., 2019). WGS of pathogens fits within the broader
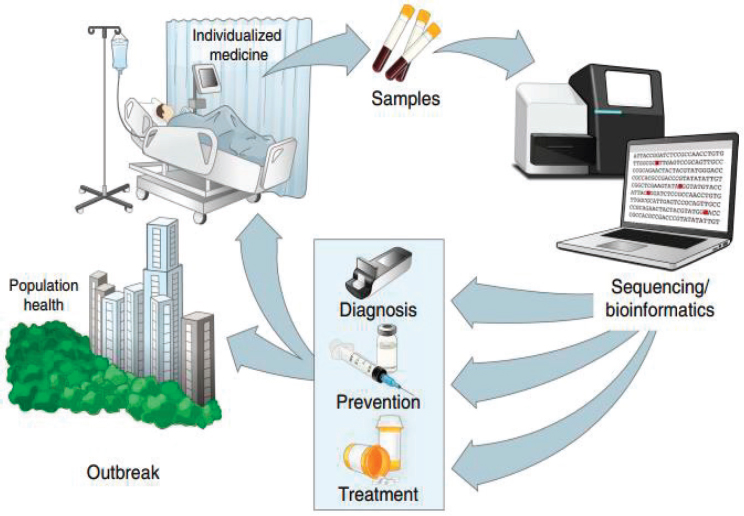
SOURCES: Reprinted by permission from Springer Nature: Springer Nature, Nature Medicine, Precision epidemiology for infectious disease control; Ladner et al., 2019.
paradigm of the One Health approach, which considers human, animal, and environmental health as a whole. Given that most emerging infectious diseases have zoonotic origins and they often spillover to humans in settings of high biological diversity, the application of genomic epidemiology across all three domains could bolster the One Health approach to surveillance, prevention, and control of those diseases (Gardy and Loman, 2018).
STUDY CHARGE
After a rapid telephonic consultation on May 7, 2020,1 with the U.S. Department of Health and Human Services’ Office of the Assistant Secretary for Preparedness and Response and Office of Science and Technology Policy, the National Academies of Sciences, Engineering, and Medicine convened an ad hoc committee to lay out a framework to define and
___________________
1 See https://www.nationalacademies.org/event/05-07-2020/standing-committee-on-emerging-infectious-diseases-and-21st-century-health-threats-expert-call-on-genotypic-drift-and-potential-phenotypic-manifestations-of-sars-cov-2 (accessed June 25, 2020).
describe the data needs for a system to track and correlate viral genome sequences with clinical and epidemiological data. Such a system would help ensure the integration of data on viral evolution with detection, diagnostic, and countermeasure efforts. The full charge to the committee is presented in Box 1-2. The committee comprises experts in the fields of infectious disease and epidemiology; clinical care; immunology, evolutionary biology, microbiology, and molecular genetics; data sharing and genomic surveillance; legal and regulatory issues; and therapeutic and diagnostic development. The biographies of the committee members are presented in Appendix A.
ABOUT THIS REPORT
Study Approach
As the nation is in the midst of the pandemic, the committee deliberated and developed this report and the recommendations presented herein on a compressed timeline. The committee held three virtual meetings in June 2020, two of which included public information-gathering sessions that allowed the committee to hear from the study sponsors and other experts and stakeholders. At the first meeting, a representative of the U.S. Centers for Disease Control and Prevention’s SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology, and Surveillance initiative spoke about the program. At the second meeting, the public session included several speaker panels covering scientific principles of viral evolution (including genomic epidemiology and phylodynamics), policy and ethical concerns, and examples from prior and ongoing initiatives. The public meeting agendas can be found in Appendix B. Staff and committee members conducted targeted searches of literature to ensure adequate background knowledge of the issue at the time of this writing. Given the rapidly evolving nature of the work around SARS-CoV-2, the committee closely monitored ongoing initiatives and concurrent, complementary work throughout the study process.
Organization of the Report
The report is organized into five chapters. Chapter 2 discusses applications of genomic epidemiology in previous infectious disease outbreaks and Chapter 3 highlights current efforts related to SARS-CoV-2. Together, these chapters explore the evolution of genomic epidemiology to present day. Chapter 4 presents a framework to track and correlate viral genome sequences with clinical and epidemiological data and Chapter 5 discusses regulatory and governance considerations.
REFERENCES
Baele, G., M. A. Suchard, A. Rambaut, and P. Lemey. 2016. Emerging concepts of data integration in pathogen phylodynamics. Systematic Biology 66(1):e47–e65.
Ballesteros, M. L., C. M. Sánchez, and L. Enjuanes. 1997. Two amino acid changes at the n-terminus of transmissible gastroenteritis coronavirus spike protein result in the loss of enteric tropism. Virology 227(2):378–388.
CBO (Congressional Budget Office). 2020. Interim economic projections for 2020 and 2021. https://www.cbo.gov/system/files/2020-05/56351-CBO-interim-projections.pdf (accessed June 25, 2020).
Dawood, A. A. 2020. Mutated COVID-19 may foretell a great risk for mankind in the future. New Microbes and New Infections 35:100673.
Forster, P., L. Forster, C. Renfrew, and M. Forster. 2020. Phylogenetic network analysis of SARS-CoV-2 genomes. Proceedings of the National Academy of Sciences 117(17):9241–9243.
Gallagher, T. M., S. E. Parker, and M. J. Buchmeier. 1990. Neutralization-resistant variants of a neurotropic coronavirus are generated by deletions within the amino-terminal half of the spike glycoprotein. Journal of Virology 64(2):731–741.
Gardy, J. L., and N. J. Loman. 2018. Towards a genomics-informed, real-time, global pathogen surveillance system. Nature Reviews Genetics 19(1):9–20.
Grenfell, B. T., O. G. Pybus, J. R. Gog, J. L. Wood, J. M. Daly, J. A. Mumford, and E. C. Holmes. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303(5656):327–332.
Grubaugh, N. D., J. T. Ladner, P. Lemey, O. G. Pybus, A. Rambaut, E. C. Holmes, and K. G. Andersen. 2019. Tracking virus outbreaks in the twenty-first century. Nature Microbiology 4(1):10–19.
Houldcroft, C. J., M. A. Beale, and J. Breuer. 2017. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology 15(3):183–192.
Koks, S., R. W. Williams, J. Quinn, F. Farzaneh, N. Conran, S. J. Tsai, G. Awandare, and S. R. Goodman. 2020. COVID-19: Time for precision epidemiology. Experimental Biology and Medicine (Maywood) 245(8):677–679.
Ladner, J. T., N. D. Grubaugh, O. G. Pybus, and K. G. Andersen. 2019. Precision epidemiology for infectious disease control. Nature Medicine 25(2):206–211.
Pachetti, M., B. Marini, F. Benedetti, F. Giudici, E. Mauro, P. Storici, C. Masciovecchio, S. Angeletti, M. Ciccozzi, R. C. Gallo, D. Zella, and R. Ippodrino. 2020. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. Journal of Translational Medicine 18(1):179.
Sánchez, C. M., F. Gebauer, C. Suñé, A. Mendez, J. Dopazo, and L. Enjuanes. 1992. Genetic evolution and tropism of transmissible gastroenteritis coronaviruses. Virology 190(1):9–105.
Smith, E. C., J. B. Case, H. Blanc, O. Isakov, N. Shomron, M. Vignuzzi, and M. R. Denison. 2015. Mutations in coronavirus nonstructural protein 10 decrease virus replication fidelity. Journal of Virology 89(12):6418–6426.
van Dorp, L., M. Acman, D. Richard, L. P. Shaw, C. E. Ford, L. Ormond, C. J. Owen, J. Pang, C. C. S. Tan, F. A. T. Boshier, A. T. Ortiz, and F. Balloux. 2020. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infection, Genetics and Evolution 83:104351.
Vlasova, A. N., Q. Wang, K. Jung, S. N. Langel, Y. S. Malik, and L. J. Saif. 2020. Porcine coronaviruses. Emerging and Transboundary Animal Viruses 79–110.
WHO (World Health Organization). 2020a. Disease outbreaks by year. https://www.who.int/csr/don/archive/year/en (accessed July 2, 2020).
WHO. 2020b. Timeline of WHO’s response to COVID-19. https://www.who.int/news-room/detail/29-06-2020-covidtimeline (accessed July 7, 2020).
Wohl, S., H. C. Metsky, S. F. Schaffner, A. Piantadosi, M. Burns, J. A. Lewnard, B. Chak, L. A. Krasilnikova, K. J. Siddle, C. B. Matranga, B. Bankamp, S. Hennigan, B. Sabina, E. H. Byrne, R. J. McNall, R. R. Shah, J. Qu, D. J. Park, S. Gharib, S. Fitzgerald, P. Barreira, S. Fleming, S. Lett, P. A. Rota, L. C. Madoff, N. L. Yozwiak, B. L. MacInnis, S. Smole, Y. H. Grad, and P. C. Sabeti. 2020. Combining genomics and epidemiology to track mumps virus transmission in the United States. PLOS Biology 18(2):e3000611.
Wu, F., S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes, and Y.-Z. Zhang. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269.
This page intentionally left blank.










