
2
Application of Genomic Epidemiology in Previous Infectious Disease Outbreaks
When genomic, clinical, and epidemiological data analyses are integrated, they can provide a real-time picture of an outbreak that is more nuanced, precise, and rich than any of the data types considered in isolation. Genomic epidemiology can inform rapid deployment of targeted interventions to protect the public as an outbreak unfolds. Previous efforts to combine and analyze viral genome sequence data with clinical and epidemiological data have demonstrated the value of this approach and have indicated its potential to transform the response to infectious disease outbreaks in the future.
PREVIOUS EFFORTS TO INTEGRATE ANALYSES OF GENOMIC, CLINICAL, AND EPIDEMIOLOGICAL DATA
SARS-CoV
The severe acute respiratory syndrome coronavirus (SARS-CoV) outbreak was among the first epidemics characterized by extensive genome sequencing and was defined by a chronological set of sequence changes, providing a unique opportunity to identify the genetic basis for zoonotic virus transmission and pathogenesis across species during a growing epidemic (Chinese SARS Molecular Epidemiology Consortium, 2004; Kan et al., 2005; Lu et al., 2004; Ruan et al., 2003). First, direct sequencing of clinical samples from the index patients identified a novel coronavirus as the microbial origin of disease, which led to rapid development of diagnostic tests. Sequence analyses and epidemiological data demonstrated that
several independent zoonotic strain introductions caused human cases and this predated the expanding outbreak in early fall 2002. Second, hospitals were epicenters for disease expansion events characterized by sequential mutation and super-spreading events, leading to travelers seeding cases and hospitals in Hong Kong, Taiwan, and Vietnam and then globally. Sequence comparisons with SARS-like CoV in open markets rapidly identified civets and raccoon dogs as potential reservoirs for human infections, leading to the rapid closure of open markets and suppression of the expanding outbreak (Guan et al., 2003). In general by comparing the early, middle-, and late-phase human isolates (GZ02, CHUK-W1, and Urbani, respectively) to the civet isolates SZ16 or HZ/SZ/61/03, sequencing revealed 9–12 amino acid changes in ORF1a and ORF1b, 6–17 in the S glycoprotein, 3–4 in ORF3a, 1 in the M glycoprotein, and the ORF8 29 nucleotide deletion. Insufficient patient metadata usually prevented definitive association of any particular mutation with cross-species transmission, person-to-person transmission, and increased pathogenesis and virulence. In the S glycoprotein, changes at K479N and to a lesser extent S487T were shown to be essential for efficient hACE2 recognition and human infection, while the change G5S in the M glycoprotein appears to enhance virus yields per cell (Li et al., 2005; Pacciarini et al., 2008). The functions of the other mutations that evolved during the expanded SARS-CoV 2003 epidemic remain unknown. Thus, genomic sequencing led to identification of the source of human infection and mutations that facilitated human infection.
Ebola Virus
The power of genomic epidemiology to shape the response to an infectious disease outbreak became increasingly evident during the Ebola virus epidemic in West Africa (Gardy and Loman, 2018; Ladner et al., 2019). In studies published around the outbreak’s peak, large-scale whole genome sequencing efforts using portable sequencing platforms and real-time analyses were able to elucidate transmission dynamics and trends. For instance, viral genomic sequencing was used to establish the outbreak’s origin in a single spillover event—rather than multiple zoonotic transmissions—followed by sustained human-to-human transmission (Holmes et al., 2016).1 Crucially, analyses of genomic and epidemiological data later established that there was transmission through breast milk (Holmes, 2017) and that sexual transmission was possible for asymptomatic survivors long after they had recovered (Christie et al., 2015; Diallo et al., 2016; Mate et al., 2015),
___________________
1 In contrast to the transmission dynamics for Ebola virus, genomic epidemiology efforts were able to establish that human cases of Lassa fever, which is endemic in areas of West Africa, have been caused by multiple distinct spillover events from rodents rather than extensive human-to-human transmission (Andersen et al., 2015; Ladner et al., 2019; Siddle et al., 2018).
a previously unknown factor that contributed to both the initial epidemic and subsequent flare-ups (Whitmer et al., 2018). This led to immediate changes in policy and guidance for male survivors to include the recommendation that they have repeated semen tests for the presence of Ebola virus RNA after recovery (Ladner et al., 2019), as well as the implementation of behavior modifications to reduce transmission. Genome sequencing studies during the epidemic were able to identify mutations that were later determined to be likely instances of novel adaptation to human hosts (Diehl et al., 2016; Dietzel et al., 2017; Urbanowicz et al., 2016). More exhaustive post hoc genomic sequencing reconstructed the geographic spread of the virus across the region, suggesting possible missed interventions that could be incorporated for future preparedness planning (Dudas et al., 2017). Ebola virus demonstrated the critical role for genomic–epidemiological analyses to span local, regional, and national levels, integrated as a real-time response during an epidemic that included transmission from asymptomatic carriers (Gardy and Loman, 2018).
Zika Virus
During the 2015–2016 epidemic of the Zika virus in the Americas, whole genome sequencing, phylogenetic molecular clock analysis, and epidemiological data for this mosquito-borne virus revealed that viral strains in the Americas share a common ancestor with strains in French Polynesia and that the virus was likely circulating in Brazil more than 1 year earlier than initially believed (Faria et al., 2016, 2017; Metsky et al., 2017). Genomic surveillance also revealed a previously unreported 2017 outbreak of Zika in Cuba based on viral sequencing of samples collected in the United States and Europe from travelers to Cuba. Genomics indicated viral introduction from neighboring islands in 2016, 1 year after peak transmission elsewhere in the Caribbean, indicating that aggressive anticipatory mosquito control measures by Cuban public health authorities likely delayed the outbreak (Grubaugh et al., 2019). Sequencing of Zika virus genomes from humans and mosquitoes in Florida suggested that hundreds of introductions of the virus may have been necessary to drive that outbreak (Grubaugh et al., 2017, 2018). Therefore, genomic data can inform outbreak response and mitigation efforts through traveler education and surveillance (Ladner et al., 2019).
Seasonal Influenza
Existing domestic and international networks for tracking the epidemiology of seasonal influenza have leveraged genomic epidemiology to track strain variants and understand vaccine responses in ways that are directly
applicable to SARS-CoV-2. Influenza viruses evolve constantly, making surveillance crucial for vaccine development and modification over time. Because annual seasonal influenza vaccines are developed and produced ahead of flu season based on expert predictions, ongoing data collection is critical for rapidly identifying the major strain(s) circulating in humans during influenza season and assessing the antigenic properties of new strains. The U.S. Centers for Disease Control and Prevention’s (CDC’s) FluView2 is a robust surveillance tool for tracking the prevalence of influenza viruses over time and in different population groups. The system collects data from state, local, tribal, and territorial health departments about the location and timing of influenza activity, viruses that are circulating and how they are changing, and clinical outcomes such as outpatient illness, hospitalization, and death (CDC, 2020). The Global Initiative on Sharing All Influenza Data (GISAID) is a network launched by scientists to enable broader sharing of the genetic sequences of influenza viruses (GISAID, 2020). GISAID plays a key role in facilitating data sharing among World Health Organization Collaborating Centers and National Influenza Centers, as well as contributing to vaccine recommendations (GISAID, 2020). In the context of the coronavirus disease 2019 (COVID-19) pandemic, similar collection of viral genetic sequence data, clinical data, epidemiological data, geographic data, and animal virus data could be harnessed to help researchers understand how SARS-CoV-2 is evolving and spreading (GISAID, 2020).
Mumps
Mumps, which is an infectious disease caused by a paramyxovirus, has been well controlled in the United States with a 99 percent decline in incidence since a vaccine was introduced in 1967 (CDC, 2019). However, from 2016–2017, hundreds of cases were reported to the Massachusetts Department of Public Health by 18 colleges and universities, as well as other close-contact settings (Wohl et al., 2020). Viral genomic sequencing of 158 cases from Massachusetts and 43 cases from other states, collected contemporaneously, revealed that a single viral lineage has been circulating since 2006 mostly within the United States, rather than globally (Wohl et al., 2020). Finer scale genomic analyses demonstrated that multiple introductions of individuals (some with travel history to other regions) onto college campuses started smaller clusters of transmission. Importantly, this study demonstrated how clinical metadata such as “contact links” (e.g., dormitories) or “activity links” (e.g., sports teams or clubs) could be aggregated and combined with genomic data to identify risk factors, untangle, and stem a local–national outbreak of mumps (Wohl et al., 2020).
___________________
2 See https://www.cdc.gov/flu/weekly/fluviewinteractive.htm (accessed June 25, 2020).
Antibiotic-Resistant Bacteria
Genomic epidemiology studies of multidrug-resistant bacteria have demonstrated the capacity for sequencing within hospitals and public health labs to generate clinically actionable data with which to optimize strategies to prevent transmission within hospitals. As the evolution of resistance continues to outpace the development of new antibiotics, preventing transmission of multidrug-resistant bacteria is critical to the mitigation of this threat. Controlling the spread of multidrug-resistant bacteria has required regions and localities to develop systems to integrate patient movement and genomic data. For example, as carbapenem-resistant Klebsiella pneumoniae was expanding its hold in U.S. hospitals, one center undertook genomic sequencing combined with epidemiology to reconstruct its earliest transmission events and articulate specific surveillance and clinical practices, which helped to stem an outbreak (Snitkin et al., 2012). Similarly, expanded genomic–epidemiological analyses demonstrated how the transmission of carbapenem-resistant Klebsiella pneumoniae across a four-county area of Illinois was linked by a patient-sharing network of hospitals, nursing homes, and long-term acute care facilities (Snitkin et al., 2012, 2017). As part of the National Action Plan for Combating Antibiotic-Resistant Bacteria, a national strategy was established that included the Antibiotic Resistance Laboratory Network to perform and coordinate genomic–epidemiological studies.3 Genomic sequence data have made it easier for facilities to communicate about possible undetected transmissions.
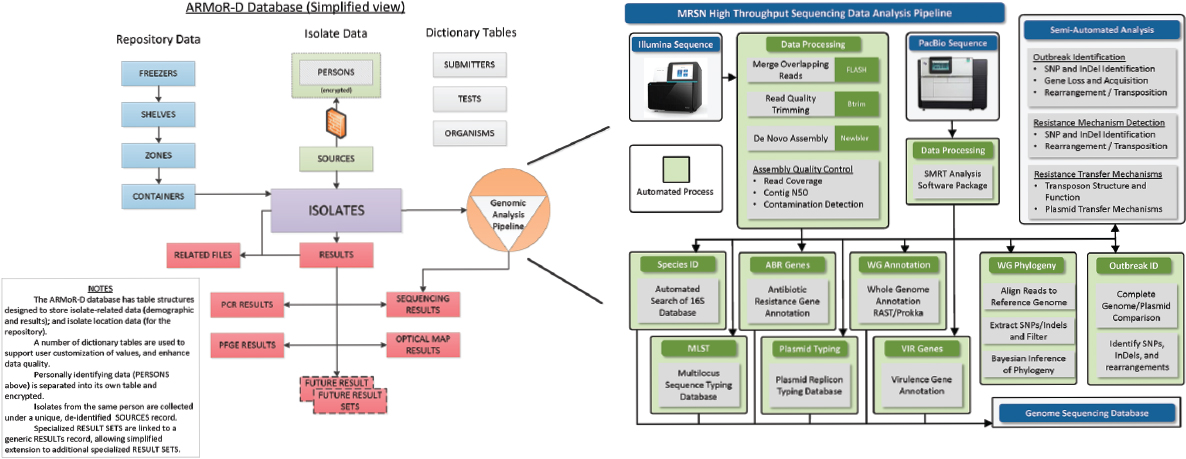
Also in response to the escalating threat from multidrug-resistant bacteria, the U.S. Department of Defense (DoD) launched in 2009 the Antimicrobial Resistance Monitoring and Research (ARMoR) Program, its own global network to track and characterize multidrug-resistant bacteria (Lesho et al., 2014, 2016). The ARMoR Program is an enterprise-wide initiative, consisting of epidemiologists, bioinformaticists, microbiology researchers, policy makers, hospital-based infection preventionists, and health care providers, that implemented next-generation sequencing across the DoD health care system and surveillance network to gain insight into the molecular epidemiology of carbapenemase-producing bacteria to elicit more accurate and actionable data for infection control (see Figure 2-1). Through this program, outbreaks and emerging pathogens were detected earlier, adjustments were made in clinical standard operating procedures and patient care policies, and advances were made in diagnostic assay and software development.
___________________
3 See https://aspe.hhs.gov/pdf-report/national-action-plan-combating-antibiotic-resistant-bacteria-progress-report-year-3 (accessed June 25, 2020).
Outbreaks of Foodborne Pathogenic Bacteria
The value of genomic epidemiology is most evident for outbreaks of foodborne pathogenic bacteria. Genomic epidemiology is helpful for food safety regulators and is integrated into many surveillance systems such as the U.S. Food and Drug Administration’s (FDA’s) GenomeTrakr Network4 (Ladner et al., 2019). This is demonstrated by one of many examples: a year-long investigation of 56 patients from 24 states who were identified by a national surveillance network as infected with shiga-toxin producing E. coli (Crowe et al., 2017). A lengthy diet questionnaire revealed an enrichment of patients who baked during the week before illness onset, eating or tasting homemade batter or dough. Across multiple states, patients provided the dry ingredients used in the recipe. It was found that the flour came from the same large domestic producer, E. coli was cultivated from this flour, and genome sequencing confirmed the match between patient and ingredient (Crowe et al., 2017). The ease of sharing and the richness and precision of genomic sequence data has catalyzed greater coordination across local, state, and national levels.
BEST PRACTICES AND KEYS TO FUTURE SUCCESS
Efforts to date to apply genomic epidemiology to infectious disease outbreaks demonstrate the value of a multipronged, overlapping approach in which data from multiple sources—genomic, clinical, and epidemiological—are overlaid upon each other (Grubaugh et al., 2019; Houldcroft et al., 2017; Ladner et al., 2019). Collective use of this type of approach can enhance communication between different entities (e.g., public health laboratories, regional medical centers, academic hospitals, national organizations, and federal agencies) and provide a richer and more nuanced picture of the epidemiological landscape of an infectious disease outbreak, enabling the investigation and potential confirmation of hypotheses that would be difficult to pursue without the integration of genomic information into the approach. An instructive example is the surveillance framework developed to evaluate the circumstances of mosquito-borne transmission of yellow fever virus (Faria et al., 2018). By integrating rapid viral genomic surveillance with epidemiological and spatial data—from both humans and animals—it was shown that the outbreak originated from multiple sylvatic (forest-dwelling mosquito) transmission events from animals to humans, rather than urban human-to-human transmission by mosquitoes (Faria et al., 2018). Therefore, this type of framework can help to estimate the risk
___________________
4 See https://www.fda.gov/food/whole-genome-sequencing-wgs-program/genometrakr-network (accessed June 25, 2020).
of human viral exposure across space and time while also monitoring the likelihood of different routes of transmission.
Collaboration among stakeholders across multiple sectors is one of the keys for future success of these types of multipronged approaches. One established effort in this vein is FDA’s GenomeTrakr, a distributed network of public, private, and academic laboratories that utilizes precision epidemiology, including whole genome sequencing for pathogen identification (Ladner et al., 2019). The network supports several real-time collaborative projects currently under way, including for analyzing samples of Listeria monocytogenes bacteria and Salmonella enteritidis bacteria (FDA, 2020). In total, GenomeTrakr has sequenced more than 462,000 isolates across a network spanning 15 federal laboratories and 25 state and university laboratories (FDA, 2020). As noted in the E. coli example described above, foodborne disease outbreaks can benefit from genomic analysis to allow the linking of cases to a point source of infection. The genomic data associated with the outbreaks, however, must be combined with relevant epidemiological data to identify risk that allow for specific interventions to be meaningful (Hill et al., 2017). As one 2016 review noted, despite the progress made around the use of whole genome sequencing in foodborne disease outbreaks and public health responses, there remains significant work to be done to ensure methodological consistency and standardization of sequencing approaches used by public health laboratories (Deng et al., 2016). In addition, even as collaboration across stakeholders in foodborne disease detection has advanced, the continued improvement of real-time, data-gathering efforts and incorporation of machine learning to inform predictive efforts represent the next frontier (Hill et al., 2017). By bringing together key stakeholders across the government and private sectors, the network is able to expedite real-time investigation of foodborne illness outbreaks and mitigate their effects on the public.
Expanding the global scope of genomic epidemiology as a practical method for timely and effective outbreak response will require building the technical capacities for genome sequencing and analysis in public agencies and private facilities. In particular, this will require collaborating with and supporting lower-resource settings that are disproportionately impacted by outbreaks of infectious diseases. Initiatives currently under way to support such efforts include the Association of Public Health Laboratories–CDC bioinformatics fellowship program,5 the H3Africa initiative,6 and public–private partnerships such as EcoHealth Alliance7 and Metabiota8 (Ladner et al., 2019).
___________________
5 See https://www.aphl.org/fellowships/Pages/Bioinformatics.aspx (accessed June 25, 2020).
6 See https://h3africa.org (accessed June 25, 2020).
7 See https://www.ecohealthalliance.org/program/wab-net (accessed June 25, 2020).
8 See https://metabiota.com (accessed June 25, 2020).
REFERENCES
Andersen, K. G., B. J. Shapiro, C. B. Matranga, R. Sealfon, A. E. Lin, L. M. Moses, O. A. Folarin, A. Goba, I. Odia, P. E. Ehiane, M. Momoh, E. M. England, S. Winnicki, L. M. Branco, S. K. Gire, E. Phelan, R. Tariyal, R. Tewhey, O. Omoniwa, M. Fullah, R. Fonnie, M. Fonnie, L. Kanneh, S. Jalloh, M. Gbakie, S. Saffa, K. Karbo, A. D. Gladden, J. Qu, M. Stremlau, M. Nekoui, H. K. Finucane, S. Tabrizi, J. J. Vitti, B. Birren, M. Fitzgerald, C. McCowan, A. Ireland, A. M. Berlin, J. Bochicchio, B. Tazon-Vega, N. J. Lennon, E. M. Ryan, Z. Bjornson, D. A. Milner, Jr., A. K. Lukens, N. Broodie, M. Rowland, M. Heinrich, M. Akdag, J. S. Schieffelin, D. Levy, H. Akpan, D. G. Bausch, K. Rubins, J. B. McCormick, E. S. Lander, S. Günther, L. Hensley, S. Okogbenin, C. Viral Hemorrhagic Fever, S. F. Schaffner, P. O. Okokhere, S. H. Khan, D. S. Grant, G. O. Akpede, D. A. Asogun, A. Gnirke, J. Z. Levin, C. T. Happi, R. F. Garry, and P. C. Sabeti. 2015. Clinical sequencing uncovers origins and evolution of Lassa virus. Cell 162(4):738–750.
CDC (U.S. Centers for Disease Control and Prevention). 2019. Mumps. https://www.cdc.gov/vaccines/pubs/pinkbook/mumps.html (accessed July 7, 2020).
CDC. 2020. U.S. influenza surveillance system: Purpose and methods. https://www.cdc.gov/flu/weekly/overview.htm (accessed June 24, 2020).
Chinese SARS Molecular Epidemiology Consortium. 2004. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303(5664):1666–1669.
Christie, A., G. J. Davies-Wayne, T. Cordier-Lassalle, D. J. Blackley, A. S. Laney, D. E. Williams, S. A. Shinde, M. Badio, T. Lo, S. E. Mate, J. T. Ladner, M. R. Wiley, J. R. Kugelman, G. Palacios, M. R. Holbrook, K. B. Janosko, E. de Wit, N. van Doremalen, V. J. Munster, J. Pettitt, R. J. Schoepp, L. Verhenne, I. Evlampidou, K. K. Kollie, S. B. Sieh, A. Gasasira, F. Bolay, F. N. Kateh, T. G. Nyenswah, and K. M. De Cock. 2015. Possible sexual transmission of Ebola virus—Liberia, 2015. Morbidity and Mortality Weekly Report 64(17):479–481.
Crowe, S. J., L. Bottichio, L. N. Shade, B. M. Whitney, N. Corral, B. Melius, K. D. Arends, D. Donovan, J. Stone, K. Allen, J. Rosner, J. Beal, L. Whitlock, A. Blackstock, J. Wetherington, L. A. Newberry, M. N. Schroeder, D. Wagner, E. Trees, S. Viazis, M. E. Wise, and K. P. Neil. 2017. Shiga toxin–producing E. coli infections associated with flour. New England Journal of Medicine 377(21):2036–2043.
Deng, X., H. C. d. Bakker, and R. S. Hendriksen. 2016. Genomic epidemiology: Whole-genome-sequencing–powered surveillance and outbreak investigation of foodborne bacterial pathogens. Annual Review of Food Science and Technology 7(1):353–374.
Diallo, B., D. Sissoko, N. J. Loman, H. A. Bah, H. Bah, M. C. Worrell, L. S. Conde, R. Sacko, S. Mesfin, A. Loua, J. K. Kalonda, N. A. Erondu, B. A. Dahl, S. Handrick, I. Goodfellow, L. W. Meredith, M. Cotten, U. Jah, R. E. Guetiya Wadoum, P. Rollin, N. Magassouba, D. Malvy, X. Anglaret, M. W. Carroll, R. B. Aylward, M. H. Djingarey, A. Diarra, P. Formenty, S. Keïta, S. Günther, A. Rambaut, and S. Duraffour. 2016. Resurgence of Ebola virus disease in Guinea linked to a survivor with virus persistence in seminal fluid for more than 500 days. Clinical Infectious Diseases 63(10):1353–1356.
Diehl, W. E., A. E. Lin, N. D. Grubaugh, L. M. Carvalho, K. Kim, P. P. Kyawe, S. M. McCauley, E. Donnard, A. Kucukural, P. McDonel, S. F. Schaffner, M. Garber, A. Rambaut, K. G. Andersen, P. C. Sabeti, and J. Luban. 2016. Ebola virus glycoprotein with increased infectivity dominated the 2013–2016 epidemic. Cell 167(4):1088–1098.
Dietzel, E., G. Schudt, V. Krähling, M. Matrosovich, and S. Becker. 2017. Functional characterization of adaptive mutations during the West African Ebola virus outbreak. Journal of Virology 91(2).
Dudas, G., L. M. Carvalho, T. Bedford, A. J. Tatem, G. Baele, N. R. Faria, D. J. Park, J. T. Ladner, A. Arias, D. Asogun, F. Bielejec, S. L. Caddy, M. Cotten, J. D’Ambrozio, S. Dellicour, A. Di Caro, J. W. Diclaro, S. Duraffour, M. J. Elmore, L. S. Fakoli, O. Faye, M. L. Gilbert, S. M. Gevao, S. Gire, A. Gladden-Young, A. Gnirke, A. Goba, D. S. Grant, B. L. Haagmans, J. A. Hiscox, U. Jah, J. R. Kugelman, D. Liu, J. Lu, C. M. Malboeuf, S. Mate, D. A. Matthews, C. B. Matranga, L. W. Meredith, J. Qu, J. Quick, S. D. Pas, M. V. T. Phan, G. Pollakis, C. B. Reusken, M. Sanchez-Lockhart, S. F. Schaffner, J. S. Schieffelin, R. S. Sealfon, E. Simon-Loriere, S. L. Smits, K. Stoecker, L. Thorne, E. A. Tobin, M. A. Vandi, S. J. Watson, K. West, S. Whitmer, M. R. Wiley, S. M. Winnicki, S. Wohl, R. Wölfel, N. L. Yozwiak, K. G. Andersen, S. O. Blyden, F. Bolay, M. W. Carroll, B. Dahn, B. Diallo, P. Formenty, C. Fraser, G. F. Gao, R. F. Garry, I. Goodfellow, S. Günther, C. T. Happi, E. C. Holmes, B. Kargbo, S. Keïta, P. Kellam, M. P. G. Koopmans, J. H. Kuhn, N. J. Loman, N. Magassouba, D. Naidoo, S. T. Nichol, T. Nyenswah, G. Palacios, O. G. Pybus, P. C. Sabeti, A. Sall, U. Ströher, I. Wurie, M. A. Suchard, P. Lemey, and A. Rambaut. 2017. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544(7650):309–315.
Faria, N. R., R. Azevedo, M. U. G. Kraemer, R. Souza, M. S. Cunha, S. C. Hill, J. Thézé, M. B. Bonsall, T. A. Bowden, I. Rissanen, I. M. Rocco, J. S. Nogueira, A. Y. Maeda, F. Vasami, F. L. L. Macedo, A. Suzuki, S. G. Rodrigues, A. C. R. Cruz, B. T. Nunes, D. B. A. Medeiros, D. S. G. Rodrigues, A. L. N. Queiroz, E. V. P. da Silva, D. F. Henriques, E. S. T. da Rosa, C. S. de Oliveira, L. C. Martins, H. B. Vasconcelos, L. M. N. Casseb, D. B. Simith, J. P. Messina, L. Abade, J. Lourenço, L. C. J. Alcantara, M. M. de Lima, M. Giovanetti, S. I. Hay, R. S. de Oliveira, P. D. S. Lemos, L. F. de Oliveira, C. P. S. de Lima, S. P. da Silva, J. M. de Vasconcelos, L. Franco, J. F. Cardoso, J. Vianez-Júnior, D. Mir, G. Bello, E. Delatorre, K. Khan, M. Creatore, G. E. Coelho, W. K. de Oliveira, R. Tesh, O. G. Pybus, M. R. T. Nunes, and P. F. C. Vasconcelos. 2016. Zika virus in the Americas: Early epidemiological and genetic findings. Science 352(6283):345–349.
Faria, N. R., J. Quick, I. M. Claro, J. Thézé, J. G. de Jesus, M. Giovanetti, M. U. G. Kraemer, S. C. Hill, A. Black, A. C. da Costa, L. C. Franco, S. P. Silva, C. H. Wu, J. Raghwani, S. Cauchemez, L. du Plessis, M. P. Verotti, W. K. de Oliveira, E. H. Carmo, G. E. Coelho, A. C. F. S. Santelli, L. C. Vinhal, C. M. Henriques, J. T. Simpson, M. Loose, K. G. Andersen, N. D. Grubaugh, S. Somasekar, C. Y. Chiu, J. E. Muñoz-Medina, C. R. Gonzalez-Bonilla, C. F. Arias, L. L. Lewis-Ximenez, S. A. Baylis, A. O. Chieppe, S. F. Aguiar, C. A. Fernandes, P. S. Lemos, B. L. S. Nascimento, H. A. O. Monteiro, I. C. Siqueira, M. G. de Queiroz, T. R. de Souza, J. F. Bezerra, M. R. Lemos, G. F. Pereira, D. Loudal, L. C. Moura, R. Dhalia, R. F. França, T. Magalhães, E. T. Marques, Jr., T. Jaenisch, G. L. Wallau, M. C. de Lima, V. Nascimento, E. M. de Cerqueira, M. M. de Lima, D. L. Mascarenhas, J. P. M. Neto, A. S. Levin, T. R. Tozetto-Mendoza, S. N. Fonseca, M. C. Mendes-Correa, F. P. Milagres, A. Segurado, E. C. Holmes, A. Rambaut, T. Bedford, M. R. T. Nunes, E. C. Sabino, L. C. J. Alcantara, N. J. Loman, and O. G. Pybus. 2017. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546(7658):406–410.
Faria, N. R., M. U. G. Kraemer, S. C. Hill, J. Goes de Jesus, R. S. Aguiar, F. C. M. Iani, J. Xavier, J. Quick, L. du Plessis, S. Dellicour, J. Thézé, R. D. O. Carvalho, G. Baele, C. H. Wu, P. P. Silveira, M. B. Arruda, M. A. Pereira, G. C. Pereira, J. Lourenço, U. Obolski, L. Abade, T. I. Vasylyeva, M. Giovanetti, D. Yi, D. J. Weiss, G. R. W. Wint, F. M. Shearer, S. Funk, B. Nikolay, V. Fonseca, T. E. R. Adelino, M. A. A. Oliveira, M. V. F. Silva, L. Sacchetto, P. O. Figueiredo, I. M. Rezende, E. M. Mello, R. F. C. Said, D. A. Santos, M. L. Ferraz, M. G. Brito, L. F. Santana, M. T. Menezes, R. M. Brindeiro, A. Tanuri, F. C. P. Dos Santos, M. S. Cunha, J. S. Nogueira, I. M. Rocco, A. C. da Costa, S. C. V. Komninakis, V. Azevedo, A. O. Chieppe, E. S. M. Araujo, M. C. L. Mendonça, C. C. Dos Santos, C. D. Dos Santos, A. M. Mares-Guia, R. M. R. Nogueira, P. C. Sequeira,
R. G. Abreu, M. H. O. Garcia, A. L. Abreu, O. Okumoto, E. G. Kroon, C. F. C. de Albuquerque, K. Lewandowski, S. T. Pullan, M. Carroll, T. de Oliveira, E. C. Sabino, R. P. Souza, M. A. Suchard, P. Lemey, G. S. Trindade, B. P. Drumond, A. M. B. Filippis, N. J. Loman, S. Cauchemez, L. C. J. Alcantara, and O. G. Pybus. 2018. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 361(6405):894–899.
FDA (U.S. Food and Drug Administration). 2020. GenomeTrakr network. https://www.fda.gov/food/whole-genome-sequencing-wgs-program/genometrakr-network (accessed June 24, 2020).
Gardy, J. L., and N. J. Loman. 2018. Towards a genomics-informed, real-time, global pathogen surveillance system. Nature Reviews Genetics 19(1):9–20.
GISAID (Global Initiative on Sharing All Influenza Data). 2020. GISAID mission. https://www.gisaid.org/about-us/mission (accessed June 24, 2020).
Grubaugh, N. D., J. T. Ladner, M. U. G. Kraemer, G. Dudas, A. L. Tan, K. Gangavarapu, M. R. Wiley, S. White, J. Thézé, D. M. Magnani, K. Prieto, D. Reyes, A. M. Bingham, L. M. Paul, R. Robles-Sikisaka, G. Oliveira, D. Pronty, C. M. Barcellona, H. C. Metsky, M. L. Baniecki, K. G. Barnes, B. Chak, C. A. Freije, A. Gladden-Young, A. Gnirke, C. Luo, B. MacInnis, C. B. Matranga, D. J. Park, J. Qu, S. F. Schaffner, C. Tomkins-Tinch, K. L. West, S. M. Winnicki, S. Wohl, N. L. Yozwiak, J. Quick, J. R. Fauver, K. Khan, S. E. Brent, R. C. Reiner, P. N. Lichtenberger, M. J. Ricciardi, V. K. Bailey, D. I. Watkins, M. R. Cone, E. W. Kopp, K. N. Hogan, A. C. Cannons, R. Jean, A. J. Monaghan, R. F. Garry, N. J. Loman, N. R. Faria, M. C. Porcelli, C. Vasquez, E. R. Nagle, D. A. T. Cummings, D. Stanek, A. Rambaut, M. Sanchez-Lockhart, P. C. Sabeti, L. D. Gillis, S. F. Michael, T. Bedford, O. G. Pybus, S. Isern, G. Palacios, and K. G. Andersen. 2017. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 546(7658):401–405.
Grubaugh, N. D., N. R. Faria, K. G. Andersen, and O. G. Pybus. 2018. Genomic insights into Zika virus emergence and spread. Cell 172(6):1160–1162.
Grubaugh, N. D., J. T. Ladner, P. Lemey, O. G. Pybus, A. Rambaut, E. C. Holmes, and K. G. Andersen. 2019. Tracking virus outbreaks in the twenty-first century. Nature Microbiology 4(1):10–19.
Guan, Y., B. J. Zheng, Y. Q. He, X. L. Liu, Z. X. Zhuang, C. L. Cheung, S. W. Luo, P. H. Li, L. J. Zhang, Y. J. Guan, K. M. Butt, K. L. Wong, K. W. Chan, W. Lim, K. F. Shortridge, K. Y. Yuen, J. S. Peiris, and L. L. Poon. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302(5643):276–278.
Hill, A. A., M. Crotta, B. Wall, L. Good, S. J. O’Brien, and J. Guitian. 2017. Towards an integrated food safety surveillance system: A simulation study to explore the potential of combining genomic and epidemiological metadata. Royal Society Open Science 4(3):160721.
Holmes, E. C., G. Dudas, A. Rambaut, and K. G. Andersen. 2016. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature 538(7624):193–200.
Houldcroft, C. J., M. A. Beale, and J. Breuer. 2017. Clinical and biological insights from viral genome sequencing. Nature Reviews Microbiology 15(3):183–192.
Kan, B., M. Wang, H. Jing, H. Xu, X. Jiang, M. Yan, W. Liang, H. Zheng, K. Wan, Q. Liu, B. Cui, Y. Xu, E. Zhang, H. Wang, J. Ye, G. Li, M. Li, Z. Cui, X. Qi, K. Chen, L. Du, K. Gao, Y.-T. Zhao, X.-Z. Zou, Y.-J. Feng, Y.-F. Gao, R. Hai, D. Yu, Y. Guan, and J. Xu. 2005. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. Journal of Virology 79(18):11892–11900.
Ladner, J. T., N. D. Grubaugh, O. G. Pybus, and K. G. Andersen. 2019. Precision epidemiology for infectious disease control. Nature Medicine 25(2):206–211.
Lesho, E. P., P. E. Waterman, U. Chukwuma, K. McAuliffe, C. Neumann, M. D. Julius, H. Crouch, R. Chandrasekera, J. F. English, R. J. Clifford, and K. E. Kester. 2014. The Antimicrobial Resistance Monitoring and Research (ARMoR) Program: The U.S. Department of Defense response to escalating antimicrobial resistance. Clinical Infectious Diseases 59(3):390–397.
Lesho, E., R. Clifford, F. Onmus-Leone, L. Appalla, E. Snesrud, Y. Kwak, A. Ong, R. Maybank, P. Waterman, P. Rohrbeck, M. Julius, A. Roth, J. Martinez, L. Nielsen, E. Steele, P. McGann, and M. Hinkle. 2016. The challenges of implementing next generation sequencing across a large healthcare system, and the molecular epidemiology and antibiotic susceptibilities of carbapenemase-producing bacteria in the healthcare system of the U.S. Department of Defense. PLOS ONE 11(5):e0155770.
Li, F., W. Li, M. Farzan, and S. C. Harrison. 2005. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309(5742):1864–1868.
Lu, H., Y. Zhao, J. Zhang, Y. Wang, W. Li, X. Zhu, S. Sun, J. Xu, L. Ling, L. Cai, D. Bu, and R. Chen. 2004. Date of origin of the SARS coronavirus strains. BMC Infectious Diseases 4:3.
Mate, S. E., J. R. Kugelman, T. G. Nyenswah, J. T. Ladner, M. R. Wiley, T. Cordier-Lassalle, A. Christie, G. P. Schroth, S. M. Gross, G. J. Davies-Wayne, S. A. Shinde, R. Murugan, S. B. Sieh, M. Badio, L. Fakoli, F. Taweh, E. de Wit, N. van Doremalen, V. J. Munster, J. Pettitt, K. Prieto, B. W. Humrighouse, U. Ströher, J. W. DiClaro, L. E. Hensley, R. J. Schoepp, D. Safronetz, J. Fair, J. H. Kuhn, D. J. Blackley, A. S. Laney, D. E. Williams, T. Lo, A. Gasasira, S. T. Nichol, P. Formenty, F. N. Kateh, K. M. De Cock, F. Bolay, M. Sanchez-Lockhart, and G. Palacios. 2015. Molecular evidence of sexual transmission of Ebola virus. New England Journal of Medicine 373(25):2448–2454.
Metsky, H. C., C. B. Matranga, S. Wohl, S. F. Schaffner, C. A. Freije, S. M. Winnicki, K. West, J. Qu, M. L. Baniecki, A. Gladden-Young, A. E. Lin, C. H. Tomkins-Tinch, S. H. Ye, D. J. Park, C. Y. Luo, K. G. Barnes, R. R. Shah, B. Chak, G. Barbosa-Lima, E. Delatorre, Y. R. Vieira, L. M. Paul, A. L. Tan, C. M. Barcellona, M. C. Porcelli, C. Vasquez, A. C. Cannons, M. R. Cone, K. N. Hogan, E. W. Kopp, J. J. Anzinger, K. F. Garcia, L. A. Parham, R. M. G. Ramírez, M. C. M. Montoya, D. P. Rojas, C. M. Brown, S. Hennigan, B. Sabina, S. Scotland, K. Gangavarapu, N. D. Grubaugh, G. Oliveira, R. Robles-Sikisaka, A. Rambaut, L. Gehrke, S. Smole, M. E. Halloran, L. Villar, S. Mattar, I. Lorenzana, J. Cerbino-Neto, C. Valim, W. Degrave, P. T. Bozza, A. Gnirke, K. G. Andersen, S. Isern, S. F. Michael, F. A. Bozza, T. M. L. Souza, I. Bosch, N. L. Yozwiak, B. L. MacInnis, and P. C. Sabeti. 2017. Zika virus evolution and spread in the Americas. Nature 546(7658):411–415.
Pacciarini, F., S. Ghezzi, F. Canducci, A. Sims, M. Sampaolo, E. Ferioli, M. Clementi, G. Poli, P. G. Conaldi, R. Baric, and E. Vicenzi. 2008. Persistent replication of severe acute respiratory syndrome coronavirus in human tubular kidney cells selects for adaptive mutations in the membrane protein. Journal of Virology 82(11):5137–5144.
Ruan, Y. J., C. L. Wei, A. L. Ee, V. B. Vega, H. Thoreau, S. T. Y. Su, J.-M. Chia, P. Ng, K. P. Chiu, L. Lim, T. Zhang, C. K. Peng, E. O. L. Lin, N. M. Lee, S. L. Yee, L. F. P. Ng, R. E. Chee, L. W. Stanton, P. M. Long, and E. T. Liu. 2003. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet (London, England) 361(9371):1779–1785.
Siddle, K. J., P. Eromon, K. G. Barnes, S. Mehta, J. U. Oguzie, I. Odia, S. F. Schaffner, S. M. Winnicki, R. R. Shah, J. Qu, S. Wohl, P. Brehio, C. Iruolagbe, J. Aiyepada, E. Uyigue, P. Akhilomen, G. Okonofua, S. Ye, T. Kayode, F. Ajogbasile, J. Uwanibe, A. Gaye, M. Momoh, B. Chak, D. Kotliar, A. Carter, A. Gladden-Young, C. A. Freije, O. Omoregie, B. Osiemi, E. B. Muoebonam, M. Airende, R. Enigbe, B. Ebo, I. Nosamiefan, P. Oluniyi, M. Nekoui, E. Ogbaini-Emovon, R. F. Garry, K. G. Andersen, D. J. Park, N. L. Yozwiak, G. Akpede, C. Ihekweazu, O. Tomori, S. Okogbenin, O. A. Folarin, P. O. Okokhere,
B. L. MacInnis, P. C. Sabeti, and C. T. Happi. 2018. Genomic analysis of Lassa virus during an increase in cases in Nigeria in 2018. New England Journal of Medicine 379(18):1745–1753.
Snitkin, E. S., A. M. Zelazny, P. J. Thomas, F. Stock, D. K. Henderson, T. N. Palmore, and J. A. Segre. 2012. Tracking a hospital outbreak of carbapenem-resistant klebsiella pneumoniae with whole-genome sequencing. Science Translational Medicine 4(148):148ra116.
Snitkin, E. S., S. Won, A. Pirani, Z. Lapp, R. A. Weinstein, K. Lolans, and M. K. Hayden. 2017. Integrated genomic and interfacility patient-transfer data reveal the transmission pathways of multidrug-resistant klebsiella pneumoniae in a regional outbreak. Science Translational Medicine 9(417).
Urbanowicz, R. A., C. P. McClure, A. Sakuntabhai, A. A. Sall, G. Kobinger, M. A. Müller, E. C. Holmes, F. A. Rey, E. Simon-Loriere, and J. K. Ball. 2016. Human adaptation of Ebola virus during the West African outbreak. Cell 167(4):1079–1087.
Whitmer, S. L. M., J. T. Ladner, M. R. Wiley, K. Patel, G. Dudas, A. Rambaut, F. Sahr, K. Prieto, S. S. Shepard, E. Carmody, B. Knust, D. Naidoo, G. Deen, P. Formenty, S. T. Nichol, G. Palacios, and U. Ströher. 2018. Active Ebola virus replication and heterogeneous evolutionary rates in EVD survivors. Cell Reports 22(5):1159–1168.
Wohl, S., H. C. Metsky, S. F. Schaffner, A. Piantadosi, M. Burns, J. A. Lewnard, B. Chak, L. A. Krasilnikova, K. J. Siddle, C. B. Matranga, B. Bankamp, S. Hennigan, B. Sabina, E. H. Byrne, R. J. McNall, R. R. Shah, J. Qu, D. J. Park, S. Gharib, S. Fitzgerald, P. Barreira, S. Fleming, S. Lett, P. A. Rota, L. C. Madoff, N. L. Yozwiak, B. L. MacInnis, S. Smole, Y. H. Grad, and P. C. Sabeti. 2020. Combining genomics and epidemiology to track mumps virus transmission in the United States. PLOS Biology 18(2):e3000611.
This page intentionally left blank.














