
3
Current Genomic Epidemiology Efforts Related to SARS-CoV-2
A collection of loosely affiliated and competing networks are acquiring and sharing genomic, clinical, and epidemiological data related to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Data are being generated at an escalating pace around the world, while networks for rapidly sharing those data are being established at the regional, national, and global levels. This chapter provides an overview of select SARS-CoV-2 data sources, identifies limitations of those sources, and highlights breakthrough efforts to combine and analyze genomic sequence data with clinical and epidemiological data for SARS-CoV-2. The proceeding chapter sets out the key considerations for such a framework to bring these data sources together.
CURRENT SARS-CoV-2 DATA SOURCES
U.S. Centers for Disease Control and Prevention SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology, and Surveillance
The U.S. Centers for Disease Control and Prevention’s (CDC’s) SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology, and Surveillance1 (SPHERES) consortium was initiated in May 2020 to improve public health by coordinating a large-scale nationwide genomic sequencing effort across the United States. SPHERES aims to accelerate the generation and sharing of high-quality viral sequencing data from clinical and
___________________
1 See https://www.cdc.gov/coronavirus/2019-ncov/covid-data/spheres.html (accessed June 25, 2020).
public health laboratories as well as to set standards to streamline the collection of consistent metadata for integrated analyses. All 50 states now have next-generation sequencing capacity, but the scale up of genomic research efforts has been hampered by lack of workforce capacity and limited coordination of stakeholders across academia, nonprofit, private, and public entities. Using a crowdsourced model, SPHERES hopes to gather sequencing data in a high-quality, representative, and consistent manner that can be used to establish a national baseline needed to monitor trends and inform evidence-based public health responses to the public health emergency; current efforts are built with the architecture designed for influenza surveillance. SPHERES aims specifically to (1) maintain high-level monitoring, (2) use sequencing data to set national and regional baselines, (3) conduct sustainable, longitudinal data collection, (4) use sequencing data to support contact tracing efforts, (5) monitor viral genetic diversity over time, and (6) foster new collaborations and innovation through the public–private partnership model.
SPHERES currently has a patchwork funding environment, with sources including federal funding from CDC and the National Institutes of Health (NIH), academic laboratories, philanthropy, and other private partners, raising concerns of sustainability. In the context of uneven testing and sequencing practices across the country, SPHERES consortium sampling has been patchy and largely passive, with 21 states having submitted no sequencing data as of early June 2020. Consequently, the sampling is non-representative in terms of geography and likely also viral genomic diversity. A further limitation is that SPHERES is not focused on collection and linkage of clinical data, in part because many laboratories do not provide detailed patient-level clinical metadata beyond a minimum set of identifiers.
National Center for Biotechnology Information
The National Center for Biotechnology Information (NCBI)2 at NIH serves as a primary repository for all genomic sequencing. In accordance with NIH Data Sharing policies, genomic scientists rapidly deposit and release assembled SARS-CoV-2 genomes and raw metagenomic reads in NCBI’s GenBank and SRA databases, linked under BioProjects. NCBI provides a resource that includes links to SARS-CoV-2 reference sequences and several methods to explore the data. While different experimental methods have been established to obtain viral genomes, standards for completeness are in place. Standardization could be bolstered with SARS-CoV-2 samples distributed by the National Institute of Standards and Technology for sequencing and submission, thus benchmarking across centers. NCBI also runs a Pathogen Detection program that integrates bacterial pathogen
___________________
2 See https://www.ncbi.nlm.nih.gov/sars-cov-2 (accessed June 25, 2020).
genomic sequences from patients, food, and environmental sources to track foodborne disease outbreaks, assess virulence, and detect antimicrobial resistance. This program might help to inform a similar integrated model for detecting and tracking viruses using genomic sequencing.
Global Initiative on Sharing All Influenza Data
As described in Chapter 2, the Global Initiative on Sharing All Influenza Data3 (GISAID) is a major global platform for rapidly and openly sharing data on all influenza viruses, and has been adapted to include SARS-CoV-2. GISAID collects geographical and species-specific genomic, clinical, and epidemiological data to contribute to understanding how viruses evolve and spread during outbreaks. The initial SARS-CoV-2 genome sequences from China were posted to GISAID; nearly every major SARS-CoV-2 study utilizes GISAID.
Nextstrain
Nextstrain4 is an open-source platform designed to harness the potential of genomic data from a variety of infectious disease pathogens to support epidemiological research and outbreak response. It hosts a GISAID-enabled interface of publicly available sequence data from every continent5 and provides powerful analytic and visualization tools that can be used to explore data at various scales (e.g., global, continent, country, region). Nextstrain’s work on SARS-CoV-2 has largely focused on identifying major clades—defined as clades that have reached 20 percent global frequency—as well as emerging clades. The Nextstrain tool can address questions about which regions of the genome are most variable, estimate the rate of infection, and apply some use of metadata (e.g., coloring sequences on the phylogenetic tree based on age of the person infected).
National COVID Cohort Collaborative
The National COVID Cohort Collaborative (N3C)6 is a centralized and secure portal for patient-level COVID-19 clinical data and a platform
___________________
3 See https://www.gisaid.org (accessed June 25, 2020).
4 See https://nextstrain.org (accessed June 25, 2020).
5 See https://nextstrain.org/ncov/global (accessed June 25, 2020).
6 The National COVID Cohort Collaborative is a collaboration among National Center for Advancing Translational Sciences–supported Clinical and Translational Science Awards Program hubs, the National Center for Data to Health, and distributed clinical data networks, with overall stewardship by NIH’s National Center for Advancing Translational Sciences. More information is available at https://covid.cd2h.org/N3C (accessed June 25, 2020).
for deploying and evaluating methods and tools for clinicians, researchers, and health care professionals. N3C is a collaboration under the auspices of NIH’s National Center for Advancing Translational Sciences7 in response to COVID-19, joining resources of the National Center for Data to Health8 and the Clinical and Translational Science Awards.9 It also serves as a resource for clinicians and researchers with granular and complex clinical questions. N3C was developed to improve efficiency and accessibility of analyses with COVID-19 clinical data, to expand the ability to analyze coronavirus diseases, and to demonstrate novel approaches for collaborative data sharing. It is governed by a single, central Institutional Review Board (IRB) and has five workstreams: data partnership and governance; phenotype and data acquisition; data ingestion and harmonization; collaborative analytics; and synthetic data. The collaborative is currently building a dataset of patients with COVID-19 that can be securely linked to external datasets using coded identifiers in an enclave model that enables linking without the sharing of overtly identifiable health information.
Examples of Regional Initiatives
Broad Institute
The Broad Institute10 in Boston, Massachusetts, has been a lead innovator in genomic research for 15 years through its Genomics Platform,11 which aims to create foundational genomics resources and to facilitate large-scale, pioneering projects to understand the genomic basis of diseases. Currently, the viral genomics group at the Broad Institute is collaborating with Massachusetts General Hospital and the Massachusetts Department of Public Health to investigate the introduction and spread of COVID-19 in the Boston area. Their work suggests that more than 30 introduction events from both domestic and international sources occurred in the area (see Figure 3-1), with subsequent super-spreader events that involved rapid and initially asymptomatic transmission in a congregate living facility as well as widespread transmission at a pharmaceutical conference that underpinned the outbreak in the state (Virological, 2020).
Since 2016, the Broad Institute has partnered with the Massachusetts State Public Health Laboratory and CDC to build distributed capacity for genomic sequencing through a train-the-trainer program for regional- and
___________________
7 See https://ncats.nih.gov (accessed June 25, 2020).
8 See https://cd2h.org (accessed June 25, 2020).
9 See https://ncats.nih.gov/ctsa (accessed June 25, 2020).
10 See https://www.broadinstitute.org/coronavirus/epidemiology-surveillance#top (accessed June 25, 2020).
11 See https://www.broadinstitute.org/genomics (accessed June 25, 2020).
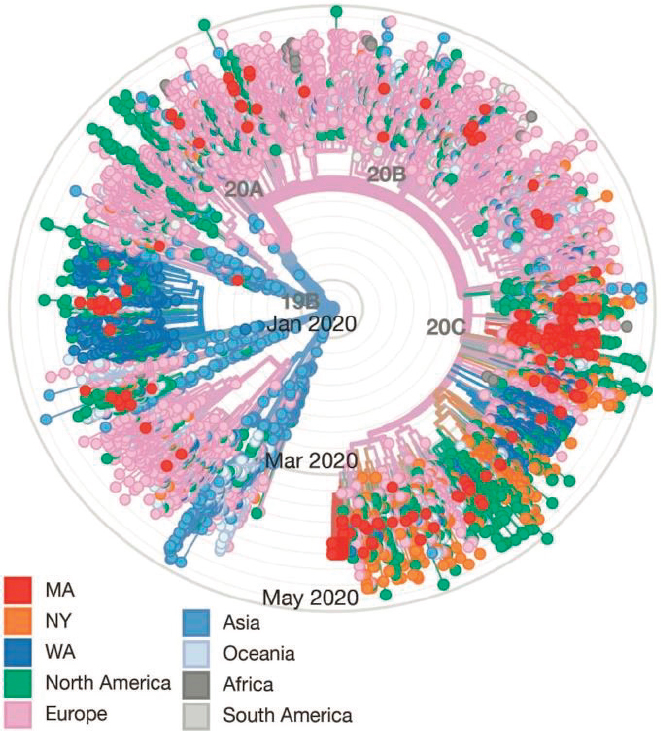
NOTES: 331 complete SARS-CoV-2 genomes from Massachusetts (red) were sequenced at the Broad Institute and are spread throughout the tree. At least 30 putative introductions to the Boston area are posited based on the phylogenetic distribution of the earliest (most central) samples. The earliest documented case in Massachusetts was a subject who returned from Wuhan on January 29, 2020, with symptoms (single red dot located in Western inner circle of tree) with no detected transmissions (no descendent viruses observed). By contrast, clusters of SARS-CoV-2 genomic sequences seen in Southern and Eastern quadrants in February/March, together with epidemiologic data, support multiple local transmissions derived from single sources (super-spreading event).
SOURCES: Virological, 2020 (graphic), through adaptation from Nextstrain and data from GISAID.
state-level laboratory personnel. These programs could serve as a model for developing national coordination among state public health laboratories.
Chan Zuckerberg Biohub
The Chan Zuckerberg Biohub12 has three ongoing COVID-19 efforts being carried out in partnerships with the University of California, San Francisco; the California Department of Public Health; and local- and county-level departments of public health (DPHs). CLIAHUB is a Clinical Laboratory Improvement Amendments–certified laboratory that provides COVID-19 testing to counties and clinics across California. Samples from positive tests are routed directly for full genomic sequencing. COVID-Tracker is a minimum viable product built on Nextstrain by the Biohub for the visualization of data and importation of epidemiological metadata for use by county DPH officials. Last, COVIDNet is a collaboration with the state and county DPHs, academic partners, and commercial laboratories through which partners send positive samples to the Biohub, commercial laboratories, and other academic laboratories in California for the viral genomes to be sequenced. As of July 7, 2020, these efforts have yielded more than 650 genomes from 10 California counties deposited in GISAID, with an estimated 40,000 genomes expected to be completed over the next year.
Wadsworth Center
The Wadsworth Center,13 the public health laboratory for the New York State Department of Health, was the first laboratory in New York to receive Emergency Use Authorization for SARS-CoV-2 testing. While rapidly expanding its own capacity, Wadsworth supported other commercial and hospital laboratories in the state to develop and ramp up testing capacities for SARS-CoV-2. Building on its investment in statewide surveillance of antibiotic-resistant bacteria, food- and water-borne pathogens, and influenza virus, Wadsworth deployed a genomic epidemiological model to track the SARS-CoV-2 caseload in New York State. Thus far, more than 300 genomes from different time periods during the pandemic and covering many regions of the state—including New York City (NYC), the initial epicenter—have been identified. The Wadsworth Center is working closely with the NYC public health laboratory and regional academic and hospital laboratories to collect and integrate clinical metadata. The Governor of New York Andrew Cuomo utilized this genomic epidemiological data, along with daily analysis of the regional
___________________
12 See https://www.czbiohub.org (accessed June 25, 2020).
13 See https://www.wadsworth.org (accessed June 25, 2020).
test positivity rate, as part of the approach to manage New York State’s response to SARS-CoV-2.
Other Initiatives
COVID-19 Genomics UK Consortium
The COVID-19 Genomics UK (COG-UK) Consortium14 was established with £20 million ($25 million) of funding to increase the capacity to collect, sequence, and analyze whole genomes of SARS-CoV-2 virus samples collected in the United Kingdom with an explicit commitment to open science and data sharing. COG-UK was created to deliver clinically actionable data to local medical centers and the UK government to guide health interventions and policies. Collaborating partners include public health agencies, university laboratories, regional university hubs and health organizations, and sequencing centers, including the Wellcome Sanger Institute. More than 20,000 sequences have been published thus far on the COG-UK website, which includes links to all samples and metadata as well as protocols developed by consortium members for preparing, conducting, and analyzing sequences. COG-UK also regularly submits data to GISAID and can be viewed in Nextstrain. Among several workstreams, the project includes a working group on metadata, patient linkage, epidemiology, and health informatics. The UK Academy of Medical Sciences also launched an open-access database15 to map and share UK preclinical research on COVID-19 therapies, as well as informing strategic decision making by policy makers and funders.
Global Alliance for Genomics and Health
The Global Alliance for Genomics and Health16 is a policy-framing and technical standards-setting organization seeking to enable responsible genomic data sharing within a human rights framework. Its actions to facilitate rapid sharing of high-quality data by the human genetics community can support timely and effective responses during global disease outbreaks. Composed of more than 500 organization members, including COG-UK, other members of the alliance include
- Canadian COVID Genomics Network,17 a national collaboration to coordinate data sharing and analyses across the country and to
___________________
14 See https://www.cogconsortium.uk (accessed June 25, 2020).
15 See https://covidpipeline.acmedsci.ac.uk (accessed June 25, 2020).
16 See https://www.ga4gh.org/covid-19 (accessed June 25, 2020).
17 See https://www.genomecanada.ca/en/news/genome-canada-leads-40-million-genomics-initiative-address-covid-19-pandemic (accessed June 25, 2020).
- accelerate genome sequencing to inform clinical and public health approaches;
- COVID-19 Beacon,18 a tool to integrate sequencing data, identify specific genetic mutations, and create visualizations of the geographic and evolutionary origins of pathogens;
- COVID-19 Portal,19 a European portal dedicated to the rapid collection and sharing of genomic data from the global research community;
- Galaxy COVID-19,20 a resource that compiles best practices, infrastructure, and workflows to support genomic analyses of SARS-CoV-2 data; and
- Public Health Alliance for Genomic Epidemiology,21 a global coalition working to develop consensus standards, share best practices, and advocate for open science and data sharing in public health microbial bioinformatics.
Partnerships Among the Private Sector, Academia, and Public Health Agencies
New partnerships among the private sector, academia, and public health agencies are also collaborating to support data collection and genomic analysis of SARS-CoV-2. In Washington State, Microsoft’s data science team is working with the Washington State Department of Health to build more efficient systems for collecting and managing large volumes of data about disease incidence and hospitalization (Edmond, 2020). This partnership was catalyzed when the Washington State Electronic Laboratory Reporting System was overwhelmed by the influx of negative SARS-CoV-2 test results reported early in the pandemic. In California, Amazon Web Services is partnering with the University of California, San Francisco, to perform genomic sequencing on samples from people with COVID-19 in the Bay area (Kent, 2020).
___________________
18 See https://covid-19.dnastack.com/app/workspace/eyJrIjoiY292aWQtcHVibGljIiwiciI6ImFwcCIsImMiOiIwIn0/resource/sequences--eyJrIjoiY292aWQtcHVibGljIiwiciI6ImFwcCIsImMiOiIwIiwibiI6IjEifQ?filter=eyJmaWx0ZXJzIjp7fSwib3JkZXIiOlt7ImZpZWxkIjoiYWNjZXNzaW9uIiwiZGlyZWN0aW9uIjoiQVNDIn1dfQ%3D%3D (accessed June 25, 2020).
19 See https://www.covid19dataportal.org (accessed June 25, 2020).
20 See https://covid19.galaxyproject.org (accessed June 25, 2020).
21 See https://pha4ge.github.io (accessed June 25, 2020).
CURRENT EFFORTS TO INTEGRATE SARS-CoV-2 GENOME SEQUENCE DATA WITH CLINICAL AND EPIDEMIOLOGICAL DATA
The committee identified several breakthrough efforts to combine and analyze SARS-CoV-2 genome sequence data with clinical and epidemiological data that were conducted in Wuhan (China), NYC (United States), and Iceland. These efforts demonstrate the value of integrating data to support transmission tracking in real time as an outbreak unfolds.
In late 2019, a critically ill 41-year-old male presented at the Central Hospital of Wuhan, Hubei province, central China, with lung infiltrates and tested negative for all common respiratory pathogens. To identify the etiologic agent, direct metatranscriptomics were performed on bronchoalveolar lavage fluid with de novo assembly, which yielded a 30-kilobase genome with 89 percent sequence identity to a group of SARS-like coronaviruses previously identified in bats (Wu et al., 2020). Concurrently, critically ill patients with unidentified severe pneumonia disease were admitted to the intensive care unit of another hospital in Wuhan; it was noted that many worked at the same local seafood market. Because the outbreak was occurring with similar epidemiological features to SARS infections (e.g., occurring in winter, patients having links to a food market), researchers first tested and identified a positive signal for a coronavirus. Next, they used direct metagenomic sequencing of bronchoalveolar lavage fluid to identify non-human microbial DNA. With some additional targeted sequencing and analysis, they independently identified the complete SARS-CoV-2 genome (Zhou et al., 2020). Foreshadowing zoonotic investigations, both scientific research teams noted the genomic similarity of SARS-CoV-2 to the 2003 SARS-CoV and to published bat coronaviruses and epidemiological links of early cases to the indoor market selling seafood and other live wild animals. Importantly, both groups submitted SARS-CoV-2 genomes to NCBI/National Library of Medicine GenBank and GISAID, which immediately became the reference for an international effort to screen for new cases (Wu et al., 2020; Zhou et al., 2020).
In early 2020, increasing numbers of patients with clinical conditions consistent with a diagnosis of SARS-CoV-2 presented globally, including in the United States; as screening capacity increased, so too did the number of reported cases. NYC became an epicenter of SARS-CoV-2 infections—with 172,000 cases and 13,000 deaths reported in March and April 2020—prompting the Icahn School of Medicine at Mount Sinai in NYC to activate its extant Pathogen Surveillance Program,22 which had been established the previous year to generate real-time genetic information on pathogens
___________________
22 See https://icahn.mssm.edu/research/genomics/research/pathogen-surveillance (accessed June 25, 2020).
found to cause disease in its patients (Gonzalez-Reiche et al., 2020). With IRB approval, patient consent, and biospecimen handling already in place, investigators acquired clinical samples from 84 patients who tested positive for SARS-CoV-2 in the first weeks of March 2020. Its genome scientists accessed the reference genome and rapidly acquired full genome sequences of these clinical cases. Phylogenetic analyses were performed using the 2,363 other SARS-CoV-2 genomic sequences deposited in GISAID in March 2020. The NYC isolates were distributed throughout the phylogenetic tree, suggesting multiple independent introductions (Gonzalez-Reiche et al., 2020). By standardizing to Nextstrain’s clade nomenclature, 87 percent of these NYC isolates were assigned to a clade that was the dominant clade in Europe at the time and suggested that travel from Europe accounted for the majority of those cases. Machine learning and Bayesian phylodynamic analyses generated an estimated period of untracked global transmission from late January to mid-February 2020. A few of the genomes closely matched strains from Washington State, which supported independent domestic introductions. These 84 patients were NYC residents from 21 neighborhoods across 4 boroughs in NYC and 2 towns in Westchester County. Based on zip code information, two monophyletic clusters—of 17 and 4 cases, respectively—were distributed across the NYC region, which suggested extensive, local, undetected transmission (Gonzalez-Reiche et al., 2020). As described previously in this chapter, many other academic centers, including the Broad Institute in Cambridge, Massachusetts, and the Chan Zuckerburg Biohub in San Francisco, California, established similar genomic tracking programs to identify the initial seeding and the subsequent spread of SARS-CoV-2 in their communities.
Many countries mounted a national response to control the spread of SARS-CoV-2, but Iceland’s response was notable in that it immediately and actively leveraged genomic–epidemiological technology to develop innovative solutions. Although Iceland is geographically isolated and has a relatively small population of 360,000, the country welcomes 2 million tourists per year (Stofa, 2019). Importantly, deCODE Genetics-Amgen23 of Iceland has been performing human population-level genomic sequencing to discover genetic risk factors for disease for 25 years. Iceland was able to conduct targeted testing on 9,199 persons at high risk for infection based on symptoms or recent travel history as well as population-based screening of 13,000 residents (Gudbjartsson et al., 2020). In total, 6 percent of the population was screened, with 13.3 percent of the targeted patients testing positive and 0.6–0.8 percent of the random-population screening testing positive. GenBank and GISAID received 581 genomes that were sequenced from these clinical samples. By leveraging the sequences in
___________________
23 See https://www.decode.com (accessed June 25, 2020).
the GISAID repository, these genomes were assigned to 42 distinct clades, which provides a lower bound on the number of introductions to Iceland (Gudbjartsson et al., 2020). Genomic sequences revealed early virus importations followed by community and family spread of distinct viruses indicating other sources. Genomic sequencing also revealed some unanticipated links, such as a cluster of 14 people who were subsequently found to be linked through missing intermediates, which helped to explain community spread. Although the United States is 1,000 times more populous than Iceland and has 10 cities with populations greater than 1 million, Iceland provides an example of integration across sectors and entities, leading to improved public health surveillance and disease control.
CONCLUDING REMARKS
The committee identified multiple limitations to current sources of genomic, clinical, and epidemiological data on SARS-CoV-2. Key limitations include insufficient funding, poor coordination, limited capacity for data integration, unrepresentative data, and lack of an adequately trained workforce with the multifaceted expertise needed to conduct this work. Funding to support these platforms and databases is inadequate both during and between outbreaks, and the funding that is available is not distributed uniformly across efforts. A passive and non-strategic system like SPHERES is inadequate. Fundamental governance and collaboration issues extending from the top down have led to the fragmentation of approaches and varying capacities at local and national levels. Many state public health laboratories are siloed, with disparate methods and widely varying levels of expertise in genomic sequencing for disease surveillance. Local jurisdictions are disproportionately resourced and have wide variability in their capacities to use data during an outbreak. Data architecture is similarly limited and fragmented at the local, state, and national levels, which is a barrier to rapid and open data sharing. Public health personnel often have no formal degree training in public health and thereby lack sufficient training in data science; more trainers in genomic epidemiology are needed to develop actionable interventions.
Robustly integrating genomic data with clinical and epidemiological data would require a health care and public health system with sufficient infrastructure, coordination, and capacity to integrate and analyze the data. However, integrating genomics with medical records, diagnostic outputs, and public health data is difficult in the United States due to fragmented record keeping as well as institutional and regulatory hurdles. Improving the representativeness of sampling will require greater local-level capacity and improving baseline surveillance, as the current reliance on cluster investigations is inadequate to identify viral genetic diversity. Data requests
often pose further challenges to already overburdened clinical and reference laboratories, which conduct a majority of sample testing. Building national capacity to integrate genomic, clinical, and epidemiological data would have immediate impact for the nation’s response to SARS-CoV-2 and position the United States to respond to any future microbial threat.
Conclusion: Current sources of SARS-CoV-2 genome sequence data, and current efforts to integrate these data with relevant epidemiological and clinical data, are patchy, typically passive, reactive, uncoordinated, and underfunded in the United States. As a result, currently available data are unrepresentative of many important population features, biased, and inadequate to answer many of the pressing questions about the evolution and transmission of the virus, and the relationships of genome sequence variants with virulence, pathogenesis, clinical outcomes, and the effectiveness of countermeasures. Thus, the viral sequence data and associated data needed are not being collected.
RECOMMENDATION 1. The U.S. Department of Health and Human Services should ensure the generation of representative, high-quality full genome sequences of SARS-CoV-2 across the United States, and in the future, from emerging epidemic or pandemic pathogens, in order that these data can be used to meet key needs for genomic surveillance.
- Pathogen samples must be obtained from individuals who represent a broad diversity of factors such as race and ethnicity, gender, age, geography, and other demographic features such as housing type, clinical manifestations and outcomes, and transmissibility.
- Capacity for genomic sequencing should be developed and supported at many geographically distributed sites performing testing, including public health laboratories and academic and medical centers.
- Representative SARS-CoV-2 clinical samples from across the United States should be collected and sequenced on an ongoing basis to provide baseline data and facilitate near-real-time transmission tracking.
- Genome sequences should be shared openly on publicly accessible databases, such as the National Center for Biotechnology Information linked to the Global Initiative on Sharing All Influenza Data.
REFERENCES
Edmond, C. 2020. How Microsoft is responding to the COVID-19 pandemic in Washington State. https://news.microsoft.com/on-the-issues/2020/04/17/microsoft-covid-19-washington-state (accessed July 6, 2020).
Gonzalez-Reiche, A. S., M. M. Hernandez, M. J. Sullivan, B. Ciferri, H. Alshammary, A. Obla, S. Fabre, G. Kleiner, J. Polanco, Z. Khan, B. Alburquerque, A. van de Guchte, J. Dutta, N. Francoeur, B. S. Melo, I. Oussenko, G. Deikus, J. Soto, S. H. Sridhar, Y.-C. Wang, K. Twyman, A. Kasarskis, D. R. Altman, M. Smith, R. Sebra, J. Aberg, F. Krammer, A. García-Sastre, M. Luksza, G. Patel, A. Paniz-Mondolfi, M. Gitman, E. M. Sordillo, V. Simon, and H. van Bakel. 2020. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 369(6501):279–301.
Gudbjartsson, D. F., A. Helgason, H. Jonsson, O. T. Magnusson, P. Melsted, G. L. Norddahl, J. Saemundsdottir, A. Sigurdsson, P. Sulem, A. B. Agustsdottir, B. Eiriksdottir, R. Fridriksdottir, E. E. Gardarsdottir, G. Georgsson, O. S. Gretarsdottir, K. R. Gudmundsson, T. R. Gunnarsdottir, A. Gylfason, H. Holm, B. O. Jensson, A. Jonasdottir, F. Jonsson, K. S. Josefsdottir, T. Kristjansson, D. N. Magnusdottir, L. le Roux, G. Sigmundsdottir, G. Sveinbjornsson, K. E. Sveinsdottir, M. Sveinsdottir, E. A. Thorarensen, B. Thorbjornsson, A. Löve, G. Masson, I. Jonsdottir, A. D. Möller, T. Gudnason, K. G. Kristinsson, U. Thorsteinsdottir, and K. Stefansson. 2020. Spread of SARS-CoV-2 in the Icelandic population. New England Journal of Medicine 382(24):2302–2315.
Kent, J. 2020. Amazon, UCSF partner for COVID-19 genome sequencing projects. https://healthitanalytics.com/news/amazon-ucsf-partner-for-covid-19-genome-sequencing-projects (accessed July 7, 2020).
Stofa, F. M. 2019. Tourism in Iceland in figures: 2018. https://www.ferdamalastofa.is/static/files/ferdamalastofa/talnaefni/tourism-in-iceland-2018_2.pdf (accessed July 6, 2020).
Virological. 2020. Introduction and spread of SARS-CoV-2 in the greater Boston area. https://virological.org/t/introduction-and-spread-of-sars-cov-2-in-the-greater-boston-area/503 (accessed June 24, 2020).
Wu, F., S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Y. Hu, Z.-W. Tao, J.-H. Tian, Y.-Y. Pei, M.-L. Yuan, Y.-L. Zhang, F.-H. Dai, Y. Liu, Q.-M. Wang, J.-J. Zheng, L. Xu, E. C. Holmes, and Y.-Z. Zhang. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269.
Zhou, P., X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, H.-D. Chen, J. Chen, Y. Luo, H. Guo, R.-D. Jiang, M.-Q. Liu, Y. Chen, X.-R. Shen, X. Wang, X.-S. Zheng, K. Zhao, Q.-J. Chen, F. Deng, L.-L. Liu, B. Yan, F.-X. Zhan, Y.-Y. Wang, G.-F. Xiao, and Z.-L. Shi. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273.
This page intentionally left blank.














