
3
Collecting Genomic Data from Historical and Ancient Canids
Reliable information on ancient and historical canid genetic diversity across North America will be needed to understand the relationship of contemporary Gulf Coast canid (GCC) populations, both wild and managed, with historical and contemporary coyote and wolf populations. Many steps in the collection and analysis of genetic data can introduce biases that affect or constrain the results. This risk is heightened in work with older samples, which are often few in number and contain only limited amounts of often-fragmented DNA. Bias can be mitigated through care in selecting samples, assaying informative genetic markers, and selecting methods to isolate DNA, filter data, and analyze data. This chapter describes the best practices for sample selection and DNA extraction as well as data collection, filtering, analysis, and interpretation for the purposes of addressing two specific questions:
- Are the ancient red wolves (i.e., specimens dated prior to 1800) a distinct lineage from the lineages of gray wolves and coyotes?
- Are the historical red wolves (i.e., those in the 19th century, before the eastward expansion of coyotes) a continuous lineage with ancient wolves in the eastern United States?
SAMPLE SELECTION
To assess whether ancient wolves in all or parts of the eastern United States are a distinct lineage that is also ancestral to historical red wolves, it is essential that sampling for genetic analyses include individuals from a broad geographic range and across different time points. Rather than aiming to collect data exclusively from specimens categorized either by previous studies or by convention—for example, by relying on an existing designation of samples as “red wolf,” “gray wolf,” or “coyote”—sample selection must emphasize diversity in geographic provenance. Animals’ ranges can shift over time, and collection of samples from diverse time-points can capture these changes. In the case of North American canids, specimens should come from two different categories: (1) individuals from across North America prior to 1800 (“ancient” specimens), and
(2) individuals from post-1800 to 1920 (“historical” specimens), after which the coyote expanded eastward, resulting in opportunities for hybridization with other canid populations.
Implemented comprehensively, this sample-selection strategy approach will yield a latitudinal and longitudinal survey of genetic diversity in canids across North America. The resulting data will be foundational in distinguishing among three hypotheses: (1) that the red wolf is a distinct taxonomic lineage that shares a most recent common ancestor with either coyotes or gray wolves, (2) that the red wolf is a lineage that resulted from ancient hybridization between two canid species (e.g., coyote, gray wolf, or other now extinct canid lineages), and (3) that animals termed “red wolves” are possibly the result of recent (i.e., occurring within historical and not ancient time) hybridization between coyotes and gray wolves.
Useful samples for addressing questions regarding red wolf population history exist in museum and archaeological collections. For example, if one is focused primarily on “wolf” specimens in the broadest sense from the historical period (ca. 1800-1940), there are more than 400 skull specimens in existence, and of these, approximately 110 are putative red wolves from the “core” range (see Appendix B). In addition, historical pelts offer another source of DNA from this time period. To sample genomes that date to prior to European contact, wolf bones and teeth from archaeological contexts (e.g., middens or as canine pendants recovered from a living space) and from paleontological contexts may be the most productive sources of DNA. Ideally, genetic data would be obtained from multiple prehistoric and historical specimens from individuals across the known or hypothesized range of the red wolf, as well as east of the Rocky Mountains). These could then serve as a reference set for historical and prehistoric (“ancient”) red wolves.
Whenever possible, it is optimal to collect both genetic and morphological data from each sample selected for study. A morphological assessment of ancient and historical samples can result in ambiguous findings (see Chapter 2), and pairing morphological data with genomic data from the same samples could provide otherwise inaccessible insights. This will require particular care in the study design because only a subset of DNA extraction methods preserve the external morphology (Reich et al., 2010; Bolnick et al., 2012) of bones and teeth. DNA extraction procedures can preclude the subsequent collection of morphological data. Therefore, samples should be fully documented at the outset of work. The documentation should include photography and high-resolution three-dimensional scanning or casting for skeletal tissues.
Sample quality and tissue type can also be important considerations in selecting specimens for study. DNA from historical or prehistoric sources is more difficult to analyze than modern DNA because poor environmental conditions and the passage of time can result in molecular damage and degradation to small fragments (Lawlor et al., 1991; Höss and Pääbo, 1993; Dabney et al., 2013). These DNA fragments may be too small to map to reference genome sequences, even using advanced bioinformatic techniques (Schubert et al., 2012; Prüfer et al., 2014; Meyer et al., 2016). The preservation of DNA in biological materials depends on the characteristics of the sample itself, the environment in which it was found, and the conditions in which it is stored thereafter.
In general, DNA yield is highest from tooth roots, dental calculus, and dense bone, and considerably less from mummified soft tissue, coprolites, and specimens stored in formaldehyde (Kurosaki et al., 1993; O’Rourke et al., 1996; Pruvost et al., 2007; Ozga et al., 2016). Some research suggests that the petrous portion of the temporal bone and dental calculus may harbor the most well-preserved DNA, though the majority of the DNA present in dental calculus is microbial (Pinhasi et al., 2015; Ozga et al., 2016; Nieves-Colón et al., 2018). The environments that are best for DNA recovery characteristically have cool mean annual temperatures and low humidity, and soils with neutral or slightly alkaline pH, or wet anoxic conditions (also with neutral pH) (Hagelberg and Clegg, 1991; Lawlor et al., 1991; Campos et al., 2012; Prüfer et al., 2014). For particularly rare specimens, other taxa from the same site can serve as a proxy for assessing DNA preservation prior to destructive sampling, although different microenvironments within the same
site can affect the success of DNA recovery (Hagelberg and Clegg, 1991; Stone and Stoneking, 1999; Miloš et al., 2007; Douka et al., 2019). The oldest DNA sequences and the samples with the highest endogenous DNA content are from permafrost contexts (Höss and Pääbo, 1993; Lister and Stuart, 2008; Schwarz et al., 2009; Rasmussen et al., 2010; Orlando et al., 2015), which are lacking in the southeastern United States; however, cave sites also provide good preservation environments (e.g., Noonan et al., 2005; Dabney et al., 2013; Delsuc et al., 2018), and ancient wolf samples from these contexts may offer the best chances of successful DNA recovery.
Finding 3-1: To collect a data set maximally useful for discerning the taxonomic status of the red wolf, it will be necessary to include canid samples of broad geographic and temporal provenance and to prioritize individual samples that can yield high-quality morphological and genomic data.
PARTIAL VERSUS WHOLE-GENOME DATA COLLECTION
To assess the relationships among North American canid lineages, identify significant populations, and examine patterns of hybridization among lineages over time with sufficient statistical power, data from whole-genome sequence rather than just a subset of genes or sites is essential. The earliest canine genome assessment methods relied on evaluation of multi-allele microsatellites (Neff et al., 1999). Only a few hundred of these were ever characterized, leaving most of the genome uninterrogated. Methods such as RAD-seq (restriction-site associated DNA markers) and genotyping using large numbers of single nucleotide polymorphisms (SNPs) in array formats that span the genome (Box 3-1) are targeted approaches that sample more, but still only a fraction of the genome. Therefore, none is ideal for addressing questions where the set of potentially informative genomic regions is not well established or may differ between the samples to be compared. In the case of North American canids, for example, a SNP-array effective for assaying varying sites between dog breeds (e.g., Lindblad-Toh et al., 2005; Dreger et al., 2016) could fail to provide data from loci informative for identifying presence of ancient red wolf ancestry. Indeed, commercially available canine SNP chips were designed using whole-genome sequence from domestic dog breeds that are largely of western European descent, and they include few specific variants for testing wild canids. It is worth noting, however, that SNP chip analysis requires considerably less DNA than whole-genome sequencing, and the SNPs which make up the chip (on the order of 500,000) are randomly spaced. With appropriate controls, some information about composition and relatedness can be gleaned from resulting haplotype analysis. RAD-seq is also problematic for comparing samples taken across large expanses of space and time, particularly ancient samples, as it requires a comparable larger amount of high molecular weight DNA. To ensure the optimal use of existing samples, more robust approaches should be employed.
Whole-genome sequencing methods escape the limitations of the above and other methods that target only a fraction of the genome or require comparably larger amounts of DNA. Because whole-genome sequencing assays all sites in the genome, starting assumptions as to which sites will be most informative are not required. In addition, whole-genome data are better suited to address key questions about local variation and the extent and timing of hybridization among populations (Dreger et al., 2016; Payseur and Rieseberg, 2016; Lowry et al., 2017).
To ensure successful whole-genome analyses, sufficient read depth providing statistical confidence in the sequence generated is required. Read coverage describes the average number of times a given base pair is assayed in a sequencing run (Sims et al., 2014). The reference genome used to represent a given species is typically sequenced at 30x coverage or higher, meaning that each genomic region is represented, on average, 30 times in resulting sequence reads. Given that sequencing errors occur using all sequencing approaches, if read coverage is too low, error rates are unacceptably high and it is difficult to identify a true variant at a specific site. This creates the
potential for ongoing uncertainty about genetic variation between a set of samples. The total number of samples to be assayed must be considered in deciding on optimal read coverage for a given study (Kishikawa et al., 2019). High read coverage increases sequencing cost per sample, ultimately limiting the number of samples that can be examined. In some cases, especially when resources are limited, a reference genome exists already, and there is a need to sequence and compare DNA from a large number of additional individuals, researchers apply coverage as low as 1x. Analysis of the resulting low-coverage data sometimes includes likelihood-based inference of genotypes (Korneliussen et al., 2014) rather than high-confidence determination of the nucleotide present at each site in each genome.
In many ancient DNA studies, genome data with low read coverage of <1x are obtained, often limiting the analyses that can be successfully performed (Kousathanas et al., 2017). A mixed strategy that collects some higher coverage genomes (20x or more) to complement lower coverage data (<5x) in many cases can be sufficient to address many questions about admixture and demographic history.
Finding 3-2: Many steps in the collection and analysis of genetic data can introduce bias. Methods that sequence the entire genome, rather than individual sites, will be maximally informative for addressing questions of lineage identity and history.
ANCIENT DNA ANALYSES
Ancient DNA recovery can be challenging, particularly from samples that originated from warm, humid environments such as found in the southeastern United States; thus, the use of current methods that optimize DNA extraction, minimize contamination, and apply next-generation sequencing (NGS) technology is paramount (Figure 3-1). Techniques for the analysis of ancient DNA have evolved rapidly over the past 20 years as NGS methods have become standard. These techniques include new methods of DNA extraction, library construction, and DNA capture. Box 3-2 provides an overview, and a number of reviews discuss these methods in detail (e.g., Heintzman et al., 2015, Orlando et al., 2015). Older methods of ancient DNA analyses were usually based on polymerase chain reaction (PCR) and typically recovered short segments of mitochondrial genome
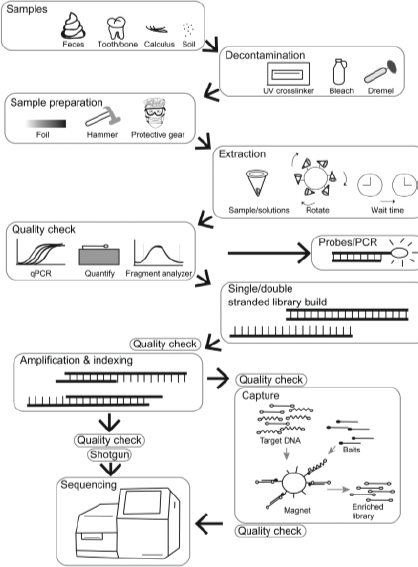
(Higuchi et al., 1984; Pääbo, 1985; Hagelberg et al., 1989). PCR is still applied in contemporary techniques for ancient DNA sequencing, but is typically used for or immediately after application of sequencing indexes and adapters and only for a few cycles.
Regardless of the methods used, because of low quantity and quality of endogenous DNA in ancient samples, contamination from modern sources (and the surrounding environment) is a concern. Samples should be collected in a way that avoids introducing exogenous modern genetic material. They should be stored in a constant-temperature environment (preferably with low humidity, cool temperatures, and little light) because DNA degrades over time under poor storage conditions (Pruvost et al., 2007; Rubio et al., 2009). Once in an ancient DNA laboratory, samples typically undergo additional decontamination procedures that include removing the outer layer of the sample or bleaching and ultraviolet (UV) irradiation of the surface. The ancient DNA laboratory should be separate (both physically and via a positive filtered airflow system) from modern DNA laboratories analyzing similar species. Ideally, the ancient DNA laboratory will have separate areas for sample preparation and subsequent laboratory protocols, and all research will take place using procedures recommended for ancient DNA research (Cooper and Poinar, 2000; Malmström et al., 2007; Pruvost et al., 2007; Rubio et al., 2009; Pilli et al., 2013). Of special concern for this study would be contaminating DNA that could be transferred from dog owners (Leonard et al., 2007; Rohland and Hofreiter, 2007; Pedersen et al., 2015).
Finding 3-3: In work with ancient DNA samples, specialized methods are necessary to minimize the risk of contamination and preserve the integrity of DNA molecules.
DATA FILTERING
Ancient DNA data provide access to different snapshots of time, which is particularly important for inferring evolutionary processes in such taxa as the red wolf that have been heavily affected by human anthropogenic activity. Prior to the development of new NGS-based methods, ancient DNA analyses were PCR-based and focused on small fragments of mitochondrial DNA. Today, sequencing methods are widely applied and are preferable in that they provide genome-wide information. However, for these sequencing data to be robust, filtering is essential. Filtering ensures the removal of low-quality reads, assesses and accounts for damage, and identifies and removes contaminating sequence reads. In addition, sufficient read depth across the genome is needed. There is also a need for analytic methods that can incorporate both ancient and modern samples, taking advantage of the time dimension (Leonardi et al., 2017).
Once sequencing is complete, the filtering of the resulting data removes low-quality sequences, duplicates that were generated during library amplification, and any contamination from modern DNA sources. With the advent of NGS methods, the earlier practice of requiring that each ancient sample be independently analyzed in two different research labs has largely been replaced by application of bioinformatic methods to screen for contamination (Renaud et al., 2019). Because ancient DNA is commonly degraded (both in size and with chemical damage), these bioinformatic methods are used to remove the damaged sequence (typically cytosine to thymine changes due to deamination of cytosines at the ends of the molecule) and to identify and remove modern contamination from the sequence data. These methods examine whether only one individual is represented in the sequence data, quantify the ratio of contaminating to endogenous molecules, and assess the extent of ancient DNA damage (Skoglund et al., 2012, 2014; Fu et al., 2013; Jónsson et al., 2013; Renaud et al., 2019).
The data are then mapped to reference sequences—or de novo assembled if no reference is available—to produce a consensus sequence with identifiable variants needed for subsequent phylogenetic and population genetic analyses. These bioinformatic analyses typically use a mixture of
computer programs and scripts as well as high-performance computing resources, and some of these are specifically designed to address common issues associated with ancient DNA data (Ginolhac et al., 2011; Schubert et al., 2012; Jónsson et al., 2013; Louvel et al., 2016; Peltzer et al., 2016; Günter and Nettleblad, 2019). The majority of NGS analysis pipelines use command line scripting and pre-packaged programs, though researchers can also create custom scripts for data analyses. To avoid potential issues arising from reference bias, it is recommended that data sets are aligned to more than one reference genome (e.g., to dog (ex: Canfam3.1)) and another canid such as the African Golden wolf and the subsequent analyses compared.
Samples may require multiple rounds of extraction, library construction, capture, and sequencing to obtain sufficient read depth and read coverage for subsequent genomic analyses. However, after the ancient DNA data have been appropriately filtered and mapped to a reference genome, subsequent analyses usually incorporate previously published genomic data (both modern and ancient) and proceed using standard population genomic methods of analyses. NGS, along with methods for targeted capture, has transformed ancient DNA research, allowing host genomic analyses as well as the investigation of symbiotic and pathogenic microbes found in hosts (Jónsson et al., 2013).
Finding 3-4: Filtering genomic data collected from ancient samples is essential to eliminate contaminating sequences and biases arising from damage accrued over time.
DATA ANALYSIS AND INTERPRETATION
Assessing whether ancient red wolves constitute a distinct lineage and whether 19th century specimens derive from a lineage continuous with them will require applying methods that can yield reliable results despite the complexities of introgression from other lineages and a possibly limited set of genomic data.
Determining Whether the Ancient Red Wolves (i.e., specimens dated prior to 1800) Are a Distinct Lineage from Those of Gray Wolves and Coyotes
To address the distinctiveness of the ancient red wolf, gray wolf, and coyote lineages, it is essential to first define what is meant by a “distinct lineage.” One reasonable approach would be to ask whether the ancient red wolf gene pool can—or cannot—be adequately modeled as a mixture of the modern gray wolf gene pool and the modern coyote gene pool. Though focused on the problem of inferring population structure, the popular program STRUCTURE can sometimes give the misleading impression that one species is a simple mixture of the gene pools of two other species. This is because the assumption that gene pools mixing together are not related by descent from a common ancestor is built into the statistical framework of that program. The recently developed software badMIXTURE is designed to escape this problem by testing goodness of fit to the assumption that one gene pool is simply the mixture of the other two (Lawson et al., 2018).
If ancient red wolves cannot be adequately modeled as a mixture of gray wolf and coyote gene pools, then demographic inference programs such as ∂a∂i (Diffusion Approximation for Demographic Inference, Gutenkunst et al., 2010) or gPhoCs (Generalized Phylogenetic Coalescent Sampler, Gronau et al., 2011) could be used to infer the most likely demographic history that did give rise to this gene pool. For example, this approach could be used to assess the possibility that the ancestors of red wolves diverged from gray wolves and experienced gene flow with coyotes until a certain time in the past.
Geographically isolated populations can have distinct genetic profiles even when they do not rise to meet the definition of species, as is the case with human ancestry groups. If assessing whether ancient red wolves constituted a “distinct lineage” means asking specifically about the
possibility of biological distinctiveness beyond isolation and genetic drift, a more appropriate approach might be to examine the landscape of coyote-related and gray-wolf-related ancestry along the red wolf genome. One possible finding would be that the extent of red wolf genetic ancestry that can be traced to the coyote varies as a function of recombination rate or distance to the nearest coding sequence. This finding could indicate the existence of red wolf alleles that are at least weakly incompatible with gene flow from the coyote (Sankararaman et al., 2014; Schumer et al., 2018), placing the red wolf somewhere on the continuum of evolution into a distinct species. Such a finding—especially if corroborated by morphological evidence (see Chapter 2)—would give the U.S. Fish and Wildlife Service the certainty to call red wolf a distinct species that is not the product of a recent hybridization between gray wolves and coyotes.
Determining Whether the Historical Red Wolves (i.e., those in the 19th century, before the eastward expansion of coyotes) Are a Continuous Lineage to Ancient Wolves in the Eastern United States
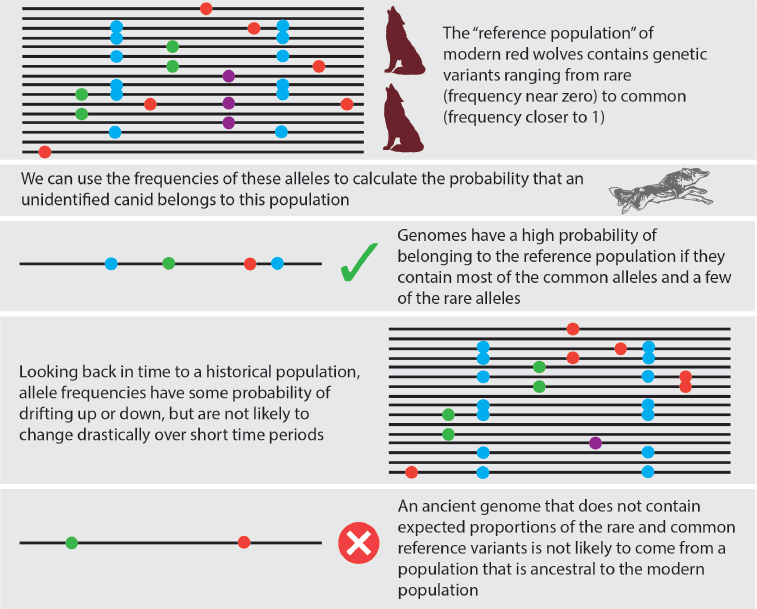
Evidence of the genetic continuity of “historical” 19th century red wolf specimens with ancient samples would imply that the red wolves still existed as a distinct lineage before the coyote expansion to the eastern United States. To assess this possibility, it will be useful to first estimate the allele frequency spectrum—that is, to identify genetic variants and calculate their frequencies—in ancient canid specimens from the eastern United States for which genomic data can be collected using the methods outlined above.
Using such data, it will be possible to infer whether the genetic profile of historical red wolves is consistent with a continuous line of descent from the ancient population (Schraiber, 2018). Specifically, Schraiber’s test (2018) (Figure 3-2) rests on the observation that genetic changes arising through interbreeding with a different lineage will occur quickly relative to other sources of genetic novelty. In particular, it is expected that some genetic drift has occurred during the time between ancient and historical period, meaning that some ancient wolf genetic variants will have become more common by chance and others will be lost. However, allele frequency changes arising through genetic drift and through introduction of new genetic mutations will be slow relative to the rapid genetic changes that would occur through interbreeding and introgression of novel genetic variants through interbreeding with a different population. Notably, using this approach it would be possible to test for genetic continuity between ancient and historical red wolves without reference to any other populations that might have interbred with red wolves during the intervening period, potentially lessening the need to collect data from a very large set of diverse canids.
Other analyses of genomic data, such as the ABBA–BABA test (explained in detail in Chapter 4; see also Figure 4-2), could provide us with complementary information on whether specific populations such as coyotes or gray wolves interbred with red wolves during this period. If coyotes, for example, did not interbreed with red wolves in the interval separating ancient from historical samples, then ABBA–BABA analysis would be expected to support the hypothesis that coyotes form a clean outgroup to the clade consisting of ancient and historical red wolves. Moreover, coyotes should share more genetic variants with ancient red wolves than with historical red wolves since historical red wolves would then be separated from their last common ancestor with coyotes by the period of time stretching from ancient to historical red wolves. This technique was applied by Rasmussen et al. (2014) to show that an ancient human is from a population ancestral to the native North and South American populations but contributed no genetic material to either population after their split.
Another possibility is that the findings might lead to rejection of the hypothesis of interbreeding between red wolves with coyotes or gray wolves during the period between the ancient sampling and historical sampling but also imply the rejection of the hypothesis that the ancient red wolves are
directly ancestral to the historical red wolves. This might imply that red wolves recently interbred with a “ghost” lineage of unknown canids that are either presently extinct or are not represented in the panel of ancient and modern canids. This could be interpreted as additional evidence that modern red wolves are genetically distinct, even though it would make the red wolf more complex to define as a genetic entity.
Finding 3-5: To assess questions of lineage origin and continuity for the red wolf, analytic approaches will need to account for the possibility of previous introgression and uncertainties in its timing.
RECENT RESEARCH ON ANCIENT NORTH AMERICAN CANIDS
As noted in the NASEM (2019) consensus report on the red wolf, genetic research that includes ancient and historical red wolves has been based solely on mitochondrial DNA data obtained using older PCR-based methods and has found that the red wolf haplotypes cluster with the coyote clade
(Roy et al., 1996; Wilson et al., 2003; Rutledge et al., 2010; Brzeski et al., 2016). The only new research on ancient North American canids that has appeared since the publication of the National Academies’ consensus report was a paper published by Loog et al. (2019) that does not include any putative red wolves, though it does use newer methods and NGS to obtain mitochondrial genome data. This study suggests that there was a rapid expansion of wolves out of Beringia about 25,000 years ago with a population bottleneck occurring between 15,000 and 40,000 years ago and that North American wolves predating the last glacial maximum were either replaced by or similar to the Beringian wolves.
CONCLUSIONS AND RECOMMENDATIONS
To understand and define red wolf populations today and from the late 20th century, it is necessary to have genomic data from red wolves that lived prior to the eastward expansion of coyotes and to compare these data with genomic data from other canids along the proposed range boundaries and from further afield. Care in sample selection and coordination across researchers are recommended so that a given sample can be used just once to generate a high-quality data set useful for multiple researchers (Austin et al., 2019). For example, researchers can first collect morphology data or create high-quality images, then extract DNA using best practices (as recommended in Box 3-2) and use that DNA to construct libraries that can be amplified and shared among research groups in order to address complementary questions.
In sum, genome data from ancient and historical canids would make it feasible to investigate the ancestry and population history of red wolves. To be useful, the genome data must be sufficiently fine-grained to make it possible to discern whether the red wolf lineage is distinct from other wolf lineages in North American and to detect the occurrence and timing of historical hybridization events among lineages. For such analyses, a well-planned selection of samples and careful data collection and filtering will be essential.
Conclusion 3-1: Biases in collection and analysis of genetic data can be mitigated through care in selecting samples, application of appropriate methods to isolate DNA, and analysis of data from the entire genome rather than from small subsets of the genome.
Conclusion 3-2: Genome data from ancient and historical canids would make it possible to investigate the ancestry and population history of red wolves. In particular, high-resolution whole-genome data will be needed to determine with high confidence whether the historical red wolf lineage is distinct from other wolf lineages in North America and to screen for the occurrence and timing of possible hybridization among lineages.
Conclusion 3-3: To discern the taxonomic identity and relationships of contemporary and late 20th century red wolf populations with each other and with earlier canids, it will be necessary to have genomic data from individuals that lived prior to the coyote expansion into the eastern United States. In addition, genomic data from other canids that lived along the proposed range boundaries and from farther afield, across a range of time periods, will be needed to provide a data set for comparison.
REFERENCES
Austin, R. M., S. B. Sholts, L. Williams, L. Kistler, and C. A. Hofman. 2019. Opinion: To curate the molecular past, museums need a carefully considered set of best practices. Proceedings of the National Academy of Sciences 116(5):1471–1474.
Baird, N. A., P. D. Etter, T. S. Atwood, M. C. Currey, A. L. Shiver, Z. A. Lewis, E. U. Selker, W. A. Cresko, and E. A. Johnson. 2008. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLOS ONE 3(10):e3376.
Bennett, E. A., D. Massilani, G. Lizzo, J. Daligault, E. M. Geigl, and T. Grange. 2018. Library construction for ancient genomics: Single strand or double strand? Biotechniques 5(6):289–300.
Bolnick, D. A., H. M. Bonine, J. Mata-Míguez, B. M. Kemp, M. H. Snow, and S. A. LeBlanc. 2012. Nondestructive sampling of human skeletal remains yields ancient nuclear and mitochondrial DNA. American Journal of Physical Anthropology 147(2):293–300.
Brandhagen, M. D., O. Loreille, and J. A. Irwin. 2018. Fragmented nuclear DNA is the predominant genetic material in human hair shafts. Genes (Basel) 9(12):640.
Briggs, A. W., J. M. Good, R. E. Green, J. Krause, T. Maricic, U. Stenzel, and S. Pääbo 2009. Primer extension capture: Targeted sequence retrieval from heavily degraded DNA sources. Journal of Visualized Experiments (31):1573.
Brzeski, K. E., M. B. DeBiasse, D. R. Rabon, Jr., M. J. Chamberlain, and S. S. Taylor. 2016. Mitochondrial DNA variation in Southeastern pre-Columbian canids. Journal of Heredity 107(3):287–293.
Burbano, H. A., E. Hodges, R. E. Green, A. W. Briggs, J. Krause, M. Meyer, J. M. Good, T. Maricic, P. L. F. Johnson, Z. Xuan, M. Rooks, A. Bhattacharjee, L. Brizuela, F. W. Albert, M. de la Rasilla, J. Fortea, A. Rosas, M. Lachmann, G. J. Hannon, S. Pääbo. 2010. Targeted investigation of the Neandertal genome by array-based sequence capture. Science 328(5979):723–725.
Campos, P. F., O. E. Craig, G. Turner-Walker, E. E. Peacock, E. Willerslev, M. T. P Gilbert. 2012. DNA in ancient bone—Where is it located and how should we extract it? Annals of Anatomy 194(1):7–16.
Carpenter, M. L., J. D. Buenrostro, C. Valdiosera, H. Schroeder, M. E. Allentoft, M. Sikora, M. Rasmussen, S. Gravel, S. Guillén, G. Nekhrizov, and K. Leshtakov. 2013. Pulling out the 1%: Whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. American Journal of Human Genetics 93(5):852–864.
Chiou, K. L., and C. M. Bergey. 2018. Methylation-based enrichment facilitates low-cost, noninvasive genomic scale sequencing of populations from feces. Scientific Reports 8(1):1975.
Cooper, A., and H. N. Poinar. 2000. Ancient DNA: Do it right or not at all. Science 289(5482):1139.
Dabney, J., M. Knapp, I. Glocke, M. T. Gansauge, A. Weihmann, B. Nickel, C. Valdiosera, N. García, S. Pääbo, J. L. Arsuaga, and M. Meyer. 2013. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proceedings of the National Academy of Sciences 110(39):15758–15763.
Dabney, J., M. Meyer, and S. Pääbo. 2013. Ancient DNA damage. Cold Spring Harbor Perspectives in Biology 5(7):a012567.
Davey, J. W., and M. L. Blaxter. 2010. RADSeq: Next-generation population genetics. Briefings in Functional Genomics 9(5–6):416–423.
Delsuc, F., M. Kuch, G. C. Gibb, J. Hughes, P. Szpak, J. Southon, J. Enk, A. T. Duggan, and H. N. Poinar. 2018. Resolving the phylogenetic position of Darwin’s extinct ground sloth (Mylodon darwinii) using mitogenomic and nuclear exon data. Proceedings of the Royal Society, B: Biological Sciences 285(1878):20180214.
Douka, K., V. Slon, Z. Jacobs, C. B. Ramsey, M. V. Shunkov, A. P. Derevianko, F. Mafessoni, M. B. Kozlikin, B. Li, R. Grün, and D. Comeskey. 2019. Age estimates for hominin fossils and the onset of the Upper Palaeolithic at Denisova Cave. Nature 565(7741):640–644.
Dreger, D. L., M. Rimbault, B. W. Davis, A. Bhatnagar, H. G. Parker, and E. A. Ostrander. 2016. Whole-genome sequence, SNP chips and pedigree structure: Building demographic profiles in domestic dog breeds to optimize genetic-trait mapping. Disease Models & Mechanisms 9(12):1445–1460.
Fu, Q., A. Mittnik, P. L. Johnson, K. Bos, M. Lari, R. Bollongino, C. Sun, L. Giemsch, R. Schmitz, J. Burger, and A. M. Ronchitelli. 2013. A revised timescale for human evolution based on ancient mitochondrial genomes. Current Biology 23(7):553–559.
Gamba, C., K. Hanghøj, C. Gaunitz, A. H. Alfarhan, S. A. Alquraishi, K. A. Al Rasheid, D. G. Bradley, and L. Orlando. 2016. Comparing the performance of three ancient DNA extraction methods for high throughput sequencing. Molecular Ecology Resources 16(2):459–469.
Gansauge, M. T., and M. Meyer. 2013. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nature Protocols 8(4):737–748.
Gansauge, M. T., T. Gerber, I. Glocke, P. Korlević, L. Lippik, S. Nagel, L. M. Riehl, A. Schmidt, and M. Meyer. 2017. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Research 45(10):e79.
Ginolhac, A., M. Rasmussen, M. T. P. Gilbert, E. Willerslev, and L. Orlando. 2011. mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27(15):2153–2155.
Gronau, I., M. J. Hubisz, B. Gulko, C. G. Danko, and A. Siepel. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nature Genetics 43(10):1031.
Günther, T., and Nettelblad, C. 2019. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLOS Genetics 15(7):e1008302. https://doi.org/10.1371/journal.pgen.1008302.
Gutenkunst, R., R. Hernandez, S. Williamson, and C. Bustamante. 2010. Diffusion approximations for demographic inference: DaDi. Nature Precedings, June 30. https://doi.org/10.1038/npre.2010.4594.1.
Hagelberg, E., and Clegg, J. B. 1991. Isolation and characterization of DNA from archaeological bone. Proceedings of the Royal Society, B: Biological Sciences 244(1309):45–50.
Hagelberg, E., B. Sykes, and R. Hedges. 1989. Ancient bone DNA amplified. Nature:485-485.
Higuchi, R., B. Bowman, M. Freiberger, O. A. Ryder, and A. C. Wilson. 1984. DNA sequences from the quagga, an extinct member of the horse family. Nature 312(5991):282–284.
Höss, M., and S. Pääbo. 1993. DNA extraction from Pleistocene bones by a silica-based purification method. Nucleic Acids Research 21(16):3913–3914.
Jónsson, H., A. Ginolhac, M. Schubert, P. L. Johnson, and L. Orlando. 2013. mapDamage2. 0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29(13):1682–1684.
Kircher, M., S. Sawyer, and M. Meyer. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Research 40(1):e3.
Kishikawa, T., Y. Momozawa, T. Ozeki, T. Mushiroda, H. Inohara, Y. Kamatani, M. Kubo, and Y. Okada. 2019. Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Scientific Reports 9(1):1–10.
Knapp, M., and M. Hofreiter. 2010. Next generation sequencing of ancient DNA: Requirements, strategies and perspectives. Genes 1(2):227–243.
Korneliussen, T. S., Albrechtsen, A. and Nielsen, R. 2014. ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics 15(1):356.
Kousathanas A., C. Leuenberger, V. Link, C. Sell, J. Burger, D. Wegmann. 2017. Inferring heterozygosity from ancient and low coverage genomes. Genetics 205(1): 317-332.
Kurosaki, K., T. Matsushita, and S. Ueda. 1993. Individual DNA identification from ancient human remains. American Journal of Human Genetics 53(3):638–643.
Lawlor, D. A., C. D. Dickel, W. W. Hauswirth, and P. Parham. 1991. Ancient HLA genes from 7,500-year-old archaeological remains. Nature 349(6312):785–788.
Lawson, D. J., L. van Dorp, and D. Falush. 2018. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nature Communications 9(1):3258.
Leonard, J. A., O. Shanks, M. Hofreiter, E. Kreuz, L. Hodges, W. Ream, R. K. Wayne, and R. C. Fleischer. 2007. Animal DNA in PCR reagents plagues ancient DNA research. Journal of Archaeological Science 34(9):1361–1366.
Leonardi, M., P. Librado, C. D. Sarkissian, M. Schubert, A. H. Alfarhan, S. A. Alquraishi, K. A. S. Al-Rasheid, C. Gamba, E. Willerslev, and L. Orlando. 2017. Evolutionary patterns and processes: Lessons from ancient DNA. Systematic Biology 66(1):e1–e29.
Lindblad-Toh, K., C. M. Wade, T. S. Mikkelsen, E. K. Karlsson, D. B. Jaffe, M. Kamal, M. Clamp, J. L. Chang, E. J. Kulbokas, M. C. Zody, and E. Mauceli. 2005. Genome sequence, comparative analysis, and haplotype structure of the domestic dog. Nature 438(7069):803–819.
Lister, A. M., and A. J. Stuart. 2008. The impact of climate change on large mammal distribution and extinction: Evidence from the last glacial/interglacial transition. Comptes Rendus Geoscience 340(9–10):615–620.
Loog, L., O. Thalmann, M. H. S. Sinding, V. J. Schuenemann, A. Perri, M. Germonpré, H. Bocherens, H., K. E. Witt, J. A. Samaniego Castruita, M. S. Velasco, I. K. Lundstrøm, N. Wales, G. Sonet, L. Frantz, H. Schroeder, J. Budd, E.-L. Jimenez, S. Fedorov, B. Gasparyan, A. W. Kandel, M. Lázničková-Galetová, H. Napierala, H.-P. Uerpmann, P. A. Nikolskiy, E. Y. Pavlova, V. V. Pitulko, K.-H. Herzig, R. S. Malhi, E. Willerslev, A. J. Hansen, K. Dobney, M. T. P. Gilbert, J. Krause, G. Larson, A. Eriksson, and A. Manica. 2020. Ancient DNA suggests modern wolves trace their origin to a Late Pleistocene expansion from Beringia. Molecular Ecology 29:1596–1610.
Louvel, G., C. Der Sarkissian, K. Hanghøj, and L. Orlando. 2016. meta BIT, an integrative and automated metagenomic pipeline for analysing microbial profiles from high throughput sequencing shotgun data. Molecular Ecology Resources 16(6):1415–1427.
Lowry, D. B., S. Hoban, J. L. Kelley, K. E. Lotterhos, L. K. Reed, M. F. Antolin, and A. Storfer. 2017. Breaking RAD: An evaluation of the utility of restriction site associated DNA sequencing for genome scans of adaptation. Molecular Ecology Resources 17(2):142–152.
Malmström, H., E. M. Svensson, M. T. P. Gilbert, E. Willerslev, A. Götherström, and G. Holmlund. 2007. More on contamination: The use of asymmetric molecular behavior to identify authentic ancient human DNA. Molecular Biology and Evolution 24(4):998–1004.
Mann, A. E., S. Sabin, K. Ziesemer, A. J. Vågene, H. Schroeder, A. T. Ozga, K. Sankaranarayanan, C. A. Hofman, J. A. Fellows Yates, D. C. Salazar-García, B. Frohlich, M. Aldenderfer, M. Hoogland, C. Read, G. R. Milner, A. C. Stone, C. M. Lewis, Jr., J. Krause, C. Hofman, K. I. Bos, and C. Warinner. 2018. Differential preservation of endogenous human and microbial DNA in dental calculus and dentin. Scientific Reports 8:9822.
Marciniak, S., J. Klunk, A. Devault, J. Enk, and H. N. Poinar. 2015. Ancient human genomics: The methodology behind reconstructing evolutionary pathways. Journal of Human Evolution 79:21–34.
Meyer, M., J. L. Arsuaga, C. de Filippo, S. Nagel, A. Aximu-Petri, B. Nickel, I. Martínez, A. Gracia, J. M. B. de Castro, E. Carbonell, and B. Viola. 2016. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531(7595):504–507.
Miloš, A., A. Selmanović, L. Smajlović, R. L. Huel, C. Katzmarzyk, A. Rizvić, and T. J. Parsons. 2007. Success rates of nuclear short tandem repeat typing from different skeletal elements. Croatian Medical Journal 48(4):486.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2019. Evaluating the taxonomic status of the Mexican gray wolf and the red wolf. Washington, DC: The National Academies Press. Available online at https://www.nap.edu/catalog/25351.
Neff M. W., Broman K. W., Mellersch C. S., Ray K., Acland G. M., Aguirre G. D., Ziegle J. S., Ostrander E. A., and Rine J. 1999. A second-generation genetic linkage map of the domestic dog, Canis familiaris. Genetics 151(2): 803-820.
Nieves-Colón, M. A., A. T. Ozga, W. J. Pestle, A. Cucina, V. Tiesler, T. W. Stanton, and A. C. Stone. 2018. Comparison of two ancient DNA extraction protocols for skeletal remains from tropical environments. American Journal of Physical Anthropology 166(4):824–836.
Noonan, J. P., M. Hofreiter, D. Smith, J. R. Priest, N. Rohland, G. Rabeder, J. Krause, J. C. Detter, S. Pääbo, and E. M. Rubin. 2005. Genomic sequencing of Pleistocene cave bears. Science 309(5734):597–599.
O’Rourke, D. H., S. W. Carlyle, and R. L. Parr. 1996. Ancient DNA: Methods, progress, and perspectives. American Journal of Human Biology 8(5):557–571.
Orlando, L., M. T. P. Gilbert, and E. Willerslev. 2015. Reconstructing ancient genomes and epigenomes. Nature Reviews Genetics 16(7):395–408.
Ozga, A. T., M. A. Nieves Colón, T. P. Honap, K. Sankaranarayanan, C. A. Hofman, G. R. Milner, C. M. Lewis Jr, A. C. Stone, and C. Warinner. 2016. Successful enrichment and recovery of whole mitochondrial genomes from ancient human dental calculus. American Journal of Physical Anthropology 160(2):220–228.
Pääbo, S. 1985. Molecular cloning of Ancient Egyptian mummy DNA. Nature 314(6012):644–645.
Payseur, B. A., and L. H. Rieseberg. 2016. A genomic perspective on hybridization and speciation. Molecular Ecology 25(11):2337–2360.
Pedersen, M. W., S. Overballe-Petersen, L. Ermini, C. D. Sarkissian, J. Haile, M. Hellstrom, J. Spens, P. F. Thomsen, K. Bohmann, E. Cappellini, and I. B. Schnell. 2015. Ancient and modern environmental DNA. Philosophical Transactions of the Royal Society, B: Biological Sciences 370(1660):20130383.
Peltzer, A., G. Jäger, A. Herbig, A. Seitz, C. Kniep, J. Krause, and K. Nieselt. 2016. EAGER: Efficient ancient genome reconstruction. Genome Biology 17(1):60.
Pilli, E., A. Modi, C. Serpico, A. Achilli, H. Lancioni, B. Lippi, F. Bertoldi, S. Gelichi, M. Lari, and D. Caramelli, D. 2013. Monitoring DNA contamination in handled vs. directly excavated ancient human skeletal remains. PLOS ONE 8(1):e52524.
Pinhasi, R., D. Fernandes, K. Sirak, M. Novak, S. Connell, S. Alpaslan-Roodenberg, F. Gerritsen, V. Moiseyev, A. Gromov, P. Raczky, and A. Anders. 2015. Optimal ancient DNA yields from the inner ear part of the human petrous bone. PLOS ONE 10(6):e0129102.
Poinar, H. N., M. Hofreiter, W. G. Spaulding, P. S. Martin, B. A. Stankiewicz, H. Bland, R. P. Evershed, G. Possnert, and S. Pääbo. 1998. Molecular coproscopy: Dung and diet of the extinct ground sloth Nothrotheriops shastensis. Science 281(5375):402–406.
Prüfer, K., F. Racimo, N. Patterson, F. Jay, S. Sankararaman, S. Sawyer, A. Heinze, G. Renaud, P. H. Sudmant, C. De Filippo, and H. Li. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505(7481):43–49.
Pruvost, M., R. Schwarz, V. B. Correia, S. Champlot, S. Braguier, N. Morel, Y. Fernandez-Jalvo, T. Grange, and E. M. Geigl. 2007. Freshly excavated fossil bones are best for amplification of ancient DNA. Proceedings of the National Academy of Sciences 104(3):739–744.
Rasmussen, M., Y. Li, S. Lindgreen, J. S. Pedersen, A. Albrechtsen, I. Moltke, M. Metspalu, E. Metspalu, T. Kivisild, R. Gupta, and M. Bertalan. 2010. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463(7282):757–762.
Rasmussen, M., S. L. Anzick, M. R. Waters, P. Skoglund, M. DeGiorgio, T. W. Stafford Jr., S. Rasmussen, I. Moltke, A. Albrechtsen, S. M. Doyle, and G. D. Poznik. 2014. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature 506(7487):225–229.
Reich, D., R. E. Green, M. Kircher, J. Krause, N. Patterson, E. Y. Durand, B. Viola, A. W. Briggs, U. Stenzel, P. L. Johnson, and T. Maricic. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468(7327):1053–1060.
Renaud, G., M. Schubert, S. Sawyer, and L. Orlando. 2019. Authentication and assessment of contamination in ancient DNA. In B. Shapiro, A. Barlow, P. D. Heintzman, M. Hofreiter, J. L. A. Paijmans, and A. E. R. Soares (eds.), Ancient DNA: Methods and Protocols. New York: Humana Press. Pp. 163–194.
Rohland, N., and Hofreiter, M. 2007. Ancient DNA extraction from bones and teeth. Nature Protocols 2(7):1756–1762.
Rohland, N., and D. Reich. 2012. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Research 22(5):939–946.
Rohland, N., E. Harney, S. Mallick, S. Nordenfelt, and D. Reich. 2015. Partial uracil–DNA–glycosylase treatment for screening of ancient DNA. Philosophical Transactions of the Royal Society, B: Biological Sciences 370(1660):20130624.
Roy, M. S., E. Geffen, D. Smith, and R. K. Wayne. 1996. Molecular genetics of pre-1940 red wolves. Conservation Biology 10(5):1413–1424.
Rubio, L., L. J. Martinez, E. Martinez, and S. M. de las Heras. 2009. Study of short- and long-term storage of teeth and its influence on DNA. Journal of Forensic Sciences 54(6):1411–1413.
Rutledge, L. Y., K. I. Bos, R. J. Pearce, and B. N. White. 2010. Genetic and morphometric analysis of sixteenth century Canis skull fragments: Implications for historic eastern and gray wolf distribution in North America. Conservation Genetics 11(4):1273–1281.
Sankararaman, S., S. Mallick, M. Dannemann, K. Prüfer, J. Kelso, S. Pääbo, N. Patterson, and D. Reich. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507(7492):354–357.
Schraiber, J. G. 2018. Assessing the relationship of ancient and modern populations. Genetics 208(1):383–398.
Schubert, M., A. Ginolhac, S. Lindgreen, J. F. Thompson, K. A. Al-Rasheid, E. Willerslev, A. Krogh, and L. Orlando. 2012. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13(1):178.
Schumer, M., C. Xu, D. L. Powell, A. Durvasula, L. Skov, C. Holland, J. C. Blazier, S. Sankararaman, P. Andolfatto, G. G. Rosenthal, and M. Przeworski. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360(6389):656–660.
Schwarz, C., R. Debruyne, M. Kuch, E. McNally, H. Schwarcz, A. D. Aubrey, J. Bada, and H. Poinar. 2009. New insights from old bones: DNA preservation and degradation in permafrost preserved mammoth remains. Nucleic Acids Research 37(10):3215–3229.
Sims, D., I. Sudbery, N. E. Ilott, A. Heger, and C. P. Ponting. 2014. Sequencing depth and coverage: Key considerations in genomic analyses. Nature Reviews Genetics 15(2):121–132.
Skoglund, P., B. H. Northoff, M. V. Shunkov, A. P. Derevianko, S. Pääbo, J. Krause, and M. Jakobsson. 2014. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences 111(6):2229–2234.
Skoglund, P., H. Malmström, M. Raghavan, J. Storå, P. Hall, E. Willerslev, M. T. P. Gilbert, A. Götherström, and M. Jakobsson. 2012. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science 336(6080):466–469.
Stone, A. C., and J. T. Ozga. 2019. Ancient DNA in the study of ancient disease. In J. E. Buikstra (ed.), Ortner’s Identification of Pathological Conditions’ in Human Skeletal Remains. Academic Press. Pp. 183–210.
Stone, A. C., and M. Stoneking. 1999. Analysis of ancient DNA from a prehistoric Amerindian cemetery. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 354(1379):153–159.
Wade, C. M., E. J. Kulbokas, A. W. Kirby, M. C. Zody, J. C. Mullikin, E. S. Lander, K. Lindblad-Toh, and M. J. Daly. 2002. The mosaic structure of variation in the laboratory mouse genome. Nature 420(6915):574–578.
Wales, N., C. Carøe, M. Sandoval-Velasco, C. Gamba, R. Barnett, J. A. Samaniego, J. Ramos Madrigal, L. Orlando, and M. T. P. Gilbert. 2015. New insights on single-stranded versus double-stranded DNA library preparation for ancient DNA. Biotechniques 59(6):368–371.
Wilson, P. J., S. Grewal, T. McFadden, R. C. Chambers, and B. N. White. 2003. Mitochondrial DNA extracted from eastern North American wolves killed in the 1800s is not of gray wolf origin. Canadian Journal of Zoology 81(5):936–940.
Yang, D.Y., B. Eng, J. S. Waye, J. C. Dudar, and S. R. Saunders. 1998. Technical note: Improved DNA extraction from ancient bones using silica-based spin columns. American Journal of Physical Anthropology 105(4):539–543.
This page intentionally left blank.
















