
4
Genomic Analyses of Extant Canids
The examination of evolutionary relationships among ancient red wolves, extant red wolf populations (captive and managed), and unidentified Gulf Coast canid populations (GCC populations) will require placing these populations in the context of genomic data from a broader collection of modern canid populations. Fortunately, a solid foundational collection of genomes from various canid species is already publicly available (Appendix E). This chapter draws upon the knowledge about complex speciation clines in other organisms to describe possible ways to analyze how extant red wolf populations and GCC populations might be related to each other and to historical red wolves described in Chapter 3 as well as to the wild populations of coyotes and gray wolves that may have hybridized (i.e., interbred) with red wolves at various times in the course of their history.
The evidence of such hybridization would not affect the possibility that either ancient red wolves or extant GCC populations may still constitute a distinct taxonomic unit that requires intensive conservation measures. Moreover, if gene flow did take place between red wolves and sympatric canids such as coyotes, the significance of this gene flow for the identity of the red wolf as a distinct species might be very different depending on whether the gene flow was recent and massive versus ongoing at low levels for a long period of time. Although many types of genetic data are capable of detecting whether coyote gene flow occurred at all, fewer data types can answer critical questions about the temporal and geographical extent of interbreeding.
Chapter 3 presented a research strategy to study whether the red wolf was an evolutionary lineage that was distinct from coyotes and gray wolves before the anthropologically driven expansion of coyotes into the eastern United States. This chapter builds up on that strategy and presents an approach to determine the continuity of the current GCC populations and extant red wolf populations with the historical (and, if possible, ancient) red wolf populations. Specifically, this chapter describes what the committee considers to be the best approaches to collecting genomic data along with a strong sampling strategy and a collection of current analytical tools that can be used to address the following questions:
- Are the red wolves from the extant captive and managed populations a continuous lineage with those in the eastern United States before the coyote expansion in the 20th century?
- Are the Gulf Coast canid populations part of the same lineage as the canids in the eastern United States before the coyote expansion?
- Is the genetic diversity of extant red wolf populations a subset of the diversity of the Gulf Coast canid populations?
This chapter also examines the strengths and limitations of some types of genomic data and analytical approaches that have been used to address similar questions in canids and other mammals and discuss whether and how they can be applied to resolve questions regarding the continuity between ancient red wolves and the extant red wolf populations as well as about the taxonomic identity of the unidentified canids.
COLLECTION OF GENOMIC DATA
Strengths and Limitations of Approaches Currently Available to Generate Genomic Data to Determine the Taxonomic Identities of the Extant Red Wolf Populations and the Gulf Coast Canids
Different types of genomic markers provide different levels of power to address particular research questions, and the feasibility of using each depends on the sample types available (Box 3-1, Chapter 3). Specifically, to answer questions about lineage continuity among the extant red wolves, the GCC populations, and the historical red wolf populations in the southeastern United States, whole-genome sequence data provide several advantages over reduced representation sequencing approaches (RAD-seq; see Box 3-1), which are the approaches most recently used in North American canid studies (e.g., vonHoldt et al., 2011, 2016; Heppenheimer et al., 2018a, 2020; but see vonHoldt et al., 2016 for use of whole-genome data). Furthermore, whole-genome sequence data offers a wider range of possible analytical options given their high-resolution capabilities, and the fact that whole-genome sequencing can be performed even on some degraded sample types (see Chapter 3). Below, the main limitations of two other commonly used approaches, RAD-seq and single nucleotide polymorphism (SNP) arrays (see Box 3-1), are discussed and the advantages of using whole-genome sequence data are explained.
Recently, double-digest restriction-site-associated DNA sequencing (ddRAD-seq; see Box 3-1) was used to assess the diversity of unidentified canids in Galveston Island, Texas, and Louisiana (Heppenheimer et al., 2020), which provided sufficient information to identify an unusual abundance of red wolf alleles that flagged these populations as potential conservation targets. However, there are several disadvantages to relying exclusively on ddRAD data (Arnold et al., 2013) (see Box 3-1). Even if ddRAD is used to process high-quality tissue samples from living individuals, this restriction enzyme digestion procedure can bias the genetic information extracted from a sample, altering the patterns of genetic diversity enough to confound simple downstream demographic inferences (Shafer et al., 2017; see also Chapter 3 for drawbacks of applying this approach to ancient samples). Moreover, RAD loci are spaced far apart in the genome and, given that they only represent a fraction of the genome, they may miss key pieces of genetic information that are localized in particular genomic regions not represented in the RAD data set. For example, the red wolf genome might maintain a distinct genetic profile because of unique adaptations in key genes that act as permeable barriers to introgression (Harrison and Larson, 2014), but there is no guarantee that the red wolf RAD-seq profile will capture any loci linked to such genes. RAD-seq is equally likely to miss even long introgressed haplotypes that are diagnostic of recent gene flow between species.
One older technology that could be used as an alternative to ddRAD is the SNP array (see Box 3-1), which is comparably inexpensive on a per-sample basis. However, SNP arrays have their own inherent biases that potentially limit their utility for addressing key questions regarding red wolf speciation and lineage continuity. Specifically, most canid SNP arrays are designed for domestic dog or other common canid sequences (i.e., they only consider the variation present in those species). As such, they may miss genomic variation endemic to other canid populations such as the red wolf. Indeed, existing arrays with more than 650,000 SNPs that are designed to analyze DNA isolated from blood (Coates et al., 2009; Garvin et al., 2010), and smaller arrays of less than 1,000 SNPs designed to test fecal samples (Fitak et al., 2015) both lack resolution and random marker selection, thus limiting their applicability to studies of ancestral and hybrid lineages. Most SNP arrays are inappropriate for analyses of gene flow or demography due to biases in how the SNPs are ascertained, though it is possible to construct SNP arrays in a way that makes them usable for quantifying gene flow between populations. The most widely used such array is the AxiomTM Human Origins 1 array (Patterson et al., 2012), and, to the committee’s knowledge, there is no comparable cleanly ascertained array that is commercially available for use in canids.
The most promising option for achieving enough genomic representation and statistical power to address questions related to the evolutionary relationships between historical red wolves, the extant red wolf populations, and the GCC populations is whole-genome sequencing (Box 3-1), a less biased alternative to ddRAD-seq. Although the greater per-sample cost of this approach could limit the ability of project funding to allow sampling many individuals, the committee believes that the tradeoff is warranted by the potential of whole-genome data to provide a more comprehensive picture regarding canid species diversity and fine-scale resolution. Moreover, although high-coverage genomic data will undoubtedly be important, sample sizes could be increased by assaying a larger number of individuals with low-coverage sequencing that could still provide sufficient resolution, but at lower cost per sample. It is also feasible to save libraries of DNA that are initially extracted for low-coverage sequencing and sequence them at higher coverage in the future when more funds become available, without collecting or processing additional tissue samples. Technology even exists for obtaining ultra-low-coverage shotgun sequencing data from fecal samples (Snyder-Mackler et al., 2016), although this methodology is still in its infancy. Resources are likely best directed toward safely capturing even a small number of live animals and generating whole-genome shotgun sequence (Box 3-1) data from peripheral blood, as these data should provide the best chance to ascertain the presence and timing of gene flow among canid lineages as well as any loci that might be driving or maintaining population differentiation of the red wolf lineage.
Finding 4-1: Three main types of genomic data have been used to analyze the taxonomic identity of the red wolf. While all of these data types can provide useful information, whole-genome sequence data constitute the least biased data type that can provide maximum genomic representation and strong inferential power to address questions related to the taxonomic identity and continuity among extant and historical canid populations.
SAMPLING
Samples Required to Address the Genetic Continuity of the Extant Captive and Managed Populations, the Gulf Coast Canid Populations, and the Historical Red Wolf Populations in the Southeastern United States
The acquisition and analysis of ancient and historical samples of canids throughout North America (as presented in Chapter 3) will be critical to properly analyzing the genetic lineage continuity of the extant captive and managed populations and the GCC populations with the reference
red wolf population before the expansion of coyotes into the eastern United States. To do this, it will be important to acquire high-quality biological samples from current captive and managed red wolf and GCC populations to produce high-coverage whole-genome sequence data. Opportunistic sampling of tissue from roadkill and feces is not recommended, as it adds another layer of confounding to the data. Degradation patterns characteristic of ancient DNA begin to accumulate immediately after death, especially in the hot, humid climates where these animals live (McDonough et al., 2018). In general, acceptable sample types that will produce high-quality and high-coverage whole-genome sequence data (30x; see Box 3-1) include fresh tissues of all types (blood, skin biopsy, muscle, organ, etc.). If only samples such as buccal swab are available, then low-coverage genomes (4x-8x; see Box 3-1) should be expected.
Besides collecting data from putative red wolf populations, there is a need for obtaining representative whole-genome sequence data from extant populations of gray wolves and coyotes that can serve as reference data for assessing levels of admixture in the captive and managed red wolf and GCC populations, which is important in determining the extent of red wolf ancestry in each population. Preliminary genomic analyses have been performed and published for modern North American canids and will be useful to guide sampling priorities. Analyses performed to date have distinguished unique lineages within the coyote (Heppenheimer et al., 2018b), gray wolf (vonHoldt et al., 2016), and red wolf (Heppenheimer et al., 2018a; Murphy et al., 2019). Sampling guidelines for extant canids should follow those outlined in Chapter 3 with respect to sampling all North American canids and including representatives of the complete geographic range for each species (or putative species). In addition, sampling should represent all known lineages of the North American canids so far reported in the literature.
To obtain sufficient representation for the coyote population, sampling should include at least all three coyote lineages defined in Heppenheimer et al. (2018b, 2020). This will make it possible to analyze the genetic diversity of the captive and managed red wolves and the GCC populations to determine the extent of admixture with the geographically overlapping coyote population, which will likely include specimens with some degree of red wolf admixture. An additional goal is to sample coyotes throughout their North American geographic range. A broad representation would be important for addressing better the different hypotheses linking coyotes to red wolf both historically and recently. Because of the huge geographic disparity between the two eastern coyote lineages and the much larger western lineage, a minimum of three samples for the two eastern coyote lineages and six samples for the much larger western coyote lineage (ideally, with evenly distributed geographical distribution of each) will be needed for high-coverage whole-genome data analyses. Currently, 21 coyote genomes produced using whole-genome sequencing are available for individuals across their distribution, including 10 from the western (United States), four from the northeastern (United States and Canada), and three from the southeastern (United States and Mexico) regions. Fortunately, these genomes meet the criteria for sampling numbers and approximate geography outlined above, indicating no critical geographic need for sampling or genome sequencing. However, given the wide distribution and relatively large population sizes, the statistical power to detect coyote admixture could be increased if additional samples are collected from throughout the coyote geographic range. Not all existing coyote genomes are sequenced at high enough coverage to enable reliable SNP calling. An efficient approach to account for the most common coyote variation and to increase power to identify recent coyote gene flow would be to sequence tens of additional coyote genomes at low coverage. Such an approach is not as likely to impact estimates of the extent of ancient coyote gene flow. A larger sample size is also necessary to phase genomes and utilize methods that require phased data.
Gray wolf sampling should include three genomes produced using whole-genome sequencing from each of the four North American lineages of gray wolf, as identified in
vonHoldt et al. (2016)—the Great Lakes region (3 genomes available), the Eastern wolf (3 genomes available), the Mexican wolf (1 genome available), and the North American region (e.g., Canada, Alaska, and Yellowstone; 19 genomes available)—and should also include a gray wolf genome as an outgroup (e.g., the Chinese wolf). The geographic representation of existing gray wolf genomes is sufficient to address the questions outlined in this chapter, with the only need being for additional sequencing of the Mexican wolf (1–2 genomes).
A more extensive sampling of the target extant canid populations (i.e., the captive and managed red wolf populations as well as the GCC populations) will be critical to addressing questions about the extent of similarity between them as well as the continuity of each of these extant populations with the historical red wolf population. The prior ddRAD analyses conducted by Heppenheimer et al. (2018a, 2020) can help prioritize individuals for high-coverage whole-genome sequencing in the GCC populations. Heppenheimer et al. (2018a, 2020) found that this population is spatially structured and that in some locations individuals have more coyote ancestry than in others, which implies that more samples are needed to ensure that all distinctive subpopulations are adequately represented in the data.
To enable analysis of spectra of shared common alleles, the sampling of approximately 20 individuals from each of the managed population of red wolves in North Carolina and the GCC populations (in both Louisiana and Texas) is recommended. If available, it would also be important to sample all individuals that founded the original captive population to determine the level of admixture that was already present in the founder population—or at least a sample of the current captive population representing all founder lineages. Currently, three red wolf genomes are available from the captive population (vonHoldt et al., 2016), and one genome is available for the GCC populations (from Texas) (Heppenheimer et al., 2018a), indicating a critical need to sample more red wolf individuals for genome sequencing (founder captive n = 9, managed n = 20, and GCC n = 19).
A large number of domestic dog genomes are available and could provide additional and useful information to the canid reference panel, given the known hybridization between dogs and wild canids. Samples from domestic dogs will be needed if hybridization took place between red wolf and domestic dogs at any point in time. The recently much improved de novo assembly of the original boxer dog genome and recent high-quality de novo genome assemblies of multiple dog breeds will be key for this analysis because de novo assemblies will not propagate the errors found in the first dog genome assembly. For the domestic dog reference panel, incorporating genomes from 5 to 10 domestic dog lineages is recommended; these should include as many of the most ancestral dog lineages as possible because they are not subject to recent bottlenecks associated with intense artificial selection to recent breed standards. Whole-genome sequencing data with de novo assemblies are now available for two German Shepherds (2020), a Labrador retriever (2019), a Great Dane (2019), and a Basenji (2019) (Appendix E), and the production of several other dog breed de novo assemblies is ongoing and expected to be released shortly. Of these de novo genomes currently available the Basenji represents one of the oldest known dog lineages, and this genome will be useful in many of the phylogenetic and population genomic analyses. Additional de novo genome assemblies under consideration include the Akita, Afghan hound, and the northern breeds of Samoyed and Alaskan Malamute. In addition, whole-genome sequences of historical samples collected in Greenland and Russia (Sinding et al., 2020) are available for comparison and may be valuable to include (Appendix E). Finally, researchers have sequences from more than 50 genomes from village dogs originating in Africa, East Asia, and South America as well as Carolina dogs in North America, providing a resource of domestic canines that have not been under selection for particular traits, for inclusion in analysis. Given the depth of domestic dog genome resources available, no additional dog genome sequencing is suggested for comparison to the red wolf.
Finding 4-2: A large number of canid genomes are available that can be used to determine the level of genetic continuity among the extant red wolf populations, GCC populations, and historical red wolves, but further whole-genome sequencing will be required to achieve a more complete representation of the genetic variation across space for some taxa. In particular, additional whole-genome data from one Mexican wolf and one Eastern wolf are required as well as whole-genome data from a larger number of individuals from each of the extant red wolf populations and the GCC populations.
DATA ANALYSES AND INTERPRETATION
Analytical Approaches to Determine Relationships among Extant U.S. Canids and Historical Red Wolf Samples
The red wolf captive breeding population originated from the Gulf Coast area (Texas and Louisiana) about 40 years ago. After animals were captured for the captive program, it was believed that all red wolf individuals had been removed from the area. Today, researchers have found canids in the GCC populations with putative red wolf alleles (i.e., those unique to the captive population) and believe they have also found putative red wolf alleles in current day GCC populations that may not have been represented in the captive breeding population (i.e., “ghost alleles”). These ghost alleles represent unique mutations (Heppenheimer et al., 2018a) in GCC populations from Louisiana and Texas that also lie outside the known genetic profiles of coyote, gray wolf, and dog ancestry. Modeling canid relationships over time can distinguish among possible scenarios regarding the origin of these mutations (e.g., if they were inherited by GCC individuals within the past 40 years from red wolf ancestors who occupied the same geographic territory). If the GCC individuals show signs of prolonged isolation from coyotes, the question remains whether their distinctiveness aligns with that of historical red wolves. Presented here are some analytical approaches that could use whole-genome data to resolve whether there is continuity between the extant red wolf populations, the GCC populations, and the historical red wolves (i.e., those that lived in the southeastern United States before the anthropogenically driven expansion of coyotes).
To answer this question, the same analytical strategy presented in Chapter 3 (section on Data Analysis and Interpretation) to address the question regarding continuity between ancient and historical populations can be applied. Briefly, the reference sample of historical canid specimens from the eastern United States could be used to estimate the allele frequency spectrum of the variation that existed in that population, and the statistical test of Schraiber (2018) (Figure 3-2) could then be employed to infer whether the genetic profile of each of the current populations is consistent with a continuous line of descent from the historical population. While this technique can test for genetic continuity, given the complex history of hybridization among canids in the United States, it would be important to also consider the different approaches that would allow an understanding of the extent of admixture in each population, which will be more extensively covered in the next section. Table 4-1 presents a selection of suitable analytical methods, and summarizes their strengths and weaknesses relative to the questions at hand. Table 4-1 does not preclude the use of other relevant, novel methods, but instead provides a means to judge the appropriateness of other methods based upon their similarities to and differences from the listed methods.
It should be acknowledged that the extant red wolf populations may represent only a small and incomplete ancestry component (a subset) of the historical red wolf due to its strong founder effect (Phillips et al., 2003). This leads to the question of whether the genetic diversity of the extant red wolf populations could have been generated by taking a sample of the GCC gene pool (from the 1970s) and expanding it without any subsequent admixture from coyotes (or another canid population). This question poses a special case of the genetic continuity question presented in the previous paragraph (and in Chapter 3). It asks whether the genetic makeup of the captive and managed red
TABLE 4-1 A Selection of Potential Analytical Methods to Model Lineage Continuity and Admixture over Time among North American Canids
| Analytical Method | Strengths | Weaknesses | |
|---|---|---|---|
| Chapter 3 – determine whether red wolves are a distinct lineage | |||
| badMIXTURE Infer population structure from goodness of fit to the model |
|
|
|
| Chapter 3 – examine historical and ancient red wolf as continuous lineage | |||
| Chapter 4 – examine relationships among extant and historical canids | |||
| Chapter 4 – examine relationship between extant extant red wolf and GCC populations | |||
| Schraiber’s Test Uses historical or ancient allele-frequency spectrum to infer continuity into future time based on expected genetic drift |
|
|
|
| Chapter 3 – examine historical and ancient red wolf as continuous and distinct lineage | |||
| Chapter 4 – distinguish between ancient and recent gene flow | |||
| Patterson’s D statistic or ABBA–BABA test Estimates hybridization using only four genomes at a time (two ancestral, one putative hybrid, and one outgroup) tested by counting sites with ABBA and BABA patterns |
|
|
|
| Analytical Method | Strengths | Weaknesses |
|---|---|---|
| ∂a∂i Infer demographic history of ancient red wolves that did not give rise to modern red wolves or coyotes |
|
|
| gPhoCS (pronounced “g-fox”) Demographic inference using allele sharing within and between populations |
|
|
| ABLE (Approximate Blockwise Likelihood Estimation) Estimates population sizes, divergence times, admixture fractions, and admixture times |
|
|
| Local Ancestry HMM (Hidden Markov Model) Infer extent and time since hybridization based on the ancestry and size of haplotypes in the genome of individuals and admixed populations |
|
|
wolf populations appears to be a subsample of the diversity of the GCC populations, without even allowing for genetic drift or mutation in the short time that has elapsed since the ancestors of the captive and managed red wolf populations were putatively part of the GCC populations.
In practice, even though the extant red wolf populations were established only a short time ago, they have potentially experienced strong genetic drift during the past few generations because these populations were initiated with a small number of individuals and have remained small. When a population is founded by a small number of individuals sampled from a larger population, it often leads to the loss of many genetic variants that were not present in the genomes of the few founders as well as an increase in the frequency of variants that were present in their genomes. This makes it appropriate to apply the same tests described to answer the question on the continuity between
current and historical populations; specifically, to test the hypothesis that the extant red wolves are derived from the same population as the GCC populations found today (see Table 4-1). It is also appropriate to test whether the managed population has interbred with local canids present in its habitat. The next section describes the approaches to assessing the occurrence and time scale of hybridization between putative red wolves and other canids in more detail. If these analyses reveal extensive recent admixture from coyotes into the managed red wolf population, this will underscore the importance of using historical DNA data as a less contaminated red wolf reference for future studies. Additionally, if there has been extensive recent coyote admixture into the GCC populations (or admixture from another canid population), this may imply that although red wolves can be a valid taxonomic group, these particular GCC populations do not derive much of their DNA from the red wolves.
Finding 4-3: The allele frequency spectrum of variation of the reference historical red wolf populations and the statistical test of Schraiber can be used to analyze whole-genome data from the extant red wolf populations and the GCC populations to determine whether these populations represent a continuous line of descent from the historical population. Similarly, this analytical approach could be used to determine whether extant red wolves represent a subset of the GCC populations.
Distinguishing Between Ancient and Recent Gene Flow in Red Wolves
Why Existing Data (mostly RAD-seq) are Insufficient for Accurately Distinguishing between Ancient and Recent Gene Flow in Red Wolves
The timing of introgression can, in principle, be estimated using either RAD or whole-genome sequence data. However, these two data types would require distinct analytical approaches. Whole-genome data sets would provide the most reliable assessment, but methods using RAD-seq data could also be used (although with less power) in those cases when whole-genome sequencing is not feasible given the quality of available samples or the availability of funding to produce whole-genome data for numerous individuals. SNP arrays lack resolution and random marker selection, thus limiting their applicability to studies to resolve ancestral and hybrid lineages.
There has been some success analyzing currently available RAD-seq data and whole-genome data with the help of statistical demographic inference programs such as ∂a∂i (Gutenkunst et al., 2009), gPhoCS (Gronau et al., 2011), and ABLE (Beeravolu et al., 2018) (see Table 4-1). These programs can all infer parameters of population histories that specify times of population divergence, subsequent admixture, and changes in effective population size over time. gPhoCS and ABLE both look at patterns of allele sharing within short RAD-ascertainable regions (i.e., those that can be traced back with high probability to one of the two populations without intervening recombination) and numerically search for the demographic history that maximizes the likelihood of the observed patterns of allele sharing within and between populations. ∂a∂i also searches numerically for the history that maximizes the likelihood of the observed data, but instead of summarizing the data as a series of non-recombining blocks, it calculates the likelihood of the joint allele frequency spectrum of individual SNP variants shared within and between populations, gaining computational tractability by neglecting information about the spatial arrangement of these variants. Although the numerical population sizes, gene flow rates, and divergence times are automatically ascertained by the program, the number and sequence of these demographic events must be programmed by an experienced analyst using their prior knowledge about the biological system and their intuition about what is needed to improve a model that appears too simple to fit the data. Although it is possible to use RAD-seq data in each of these analyses, the use of whole-genome data can provide greater accuracy and precision due to its unbiased nature.
A simpler method, called the ABBA–BABA test or Patterson’s D statistic (Green et al., 2010; Patterson et al., 2012) (Figure 4-1) requires data from only four genomes: a putative hybrid genome H, two genomes P and Q that are related to the ancestral populations that putatively hybridized together, and an outgroup genome O. Patterson’s D statistic does not estimate population sizes, divergence times, or other demographic parameters, but simply gives a binary answer as to whether or not the data provide statistical support for a hybrid origin of the putatively admixed genome. The method assumes that H is more closely related to one of the genomes P and Q, and if the assumption is that the more closely related genome is P, then the null hypothesis under no admixture is that H and P are sister lineages with Q as an outgroup. In that case, H and P should share the same number of derived alleles with Q. This hypothesis can be tested by counting the number of ABBA sites (sites where a derived allele, B, is shared by P and Q but not H or O) and testing whether that number is significantly different from the number of BABA sites (sites where the derived allele B is shared by H and Q but not P or O). If the numbers of these sites are in fact statistically different, with H and Q sharing more derived alleles than P and Q, the implication is that the null hypothesis should be rejected and H is a hybrid with ancestors related to both P and Q. This test can be performed using low-coverage whole-genome data or RAD-seq data, but it cannot be performed using most SNP arrays except for SNP arrays explicitly designed for clean population genetic inference such as the AxiomTM Human Origins array (Patterson et al., 2012). In the context of the canid system under consideration here, the ABBA–BABA test can measure differences between wolf populations
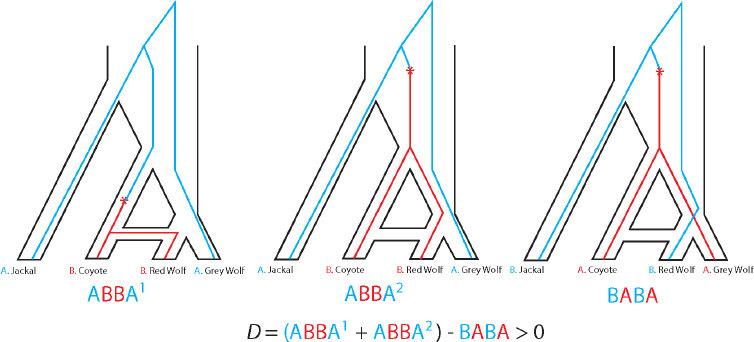
in the proportion of coyote admixture, but it cannot distinguish between long periods of low-level gene flow and recent interbreeding with coyotes brought on by human environmental disruptions.
Since RAD loci are sparsely distributed throughout the genome, they yield little to no information about the existence of any long genomic “tracts” of coyote-like or gray wolf-like ancestry within the extant red wolf populations or the population of unidentified canids. Ancestry tracts are contiguous segments of DNA that can be traced to just one of the ancestral populations (or lineages) that contributed genetic material to a hybrid population (or lineage); in many cases a tract of coyote ancestry within a mostly wolf genome is a genetic excerpt from the genome of a single distant coyote ancestor. Assuming that a hybrid offspring of a coyote and a wolf lived within a population of wolves, and its descendants backcrossed into the wolf population for generations, genetic recombination events would randomly break up the inherited coyote genome into smaller and smaller segments over time, interspersing the broken-up coyote genome with segments of wolf genome. The decay of the introgressed coyote genome into smaller and smaller pieces proceeds at a slow, predictable rate over thousands of years, meaning that if a piece of this genome is identified in a hybrid individual, its length can provide high-resolution information about how long ago the hybrid’s last wolf ancestor likely lived (Pool and Nielsen, 2009; Harris and Nielsen, 2013; Loh et al., 2013). Corbett-Detig and Nielsen (2017) recently developed a hidden Markov model that can infer the lengths and locations of admixture tracts jointly with their times to common ancestry with the population from which they originated.
If red wolf genomic data sets contain long haplotypes with high sequence similarity to coyotes, it is quite likely that these pieces of DNA were inherited from recent coyote ancestors. In contrast, if the haplotypes that show significant similarity to coyotes are short and interspersed with haplotypes that have greater similarity to wolves or dogs, they may not represent recent introgression at all, but simply reflect the genetic structure of the ancestral population from which wolves, dogs, and coyotes all originated. This ancestral structure would exist even if coyotes and wolves had been reproductively isolated from each other for thousands of years and had never interbred after they began diversifying into genetically distinct populations. One important consideration regarding this approach is that its reliability heavily depends on the use of proper reference populations. In this case, the use of coyote genomes from individuals that are not admixed with wolves would be critical. Similarly, red wolf genomes from the historical and ancient specimens discussed in Chapter 3 would be important to use as a reference population for the red wolf.
If many long coyote haplotypes recently entered the gene pool of the unidentified canids and swapped out much of the existing red wolf–related ancestry, this might imply that these populations already have limited conservation potential as a stable reservoir of red wolf genetic distinctiveness. However, if a similar total amount of coyote ancestry entered the red wolf gene pool via low levels of introgression over a longer period of time, it is more likely that weak selection against hybrid individuals has maintained a stable coyote/red wolf speciation cline and purged coyote ancestry away from the most functionally distinctive regions of the red wolf genome. In this scenario, the remaining red wolf ancestry would be more likely to be enriched for a distinctive genetic profile that encodes this species’ distinctive traits.
There are abundant examples of species pairs that hybridize for long periods of time yet maintain genetic distinctiveness, possibly due to weak reproductive incompatibilities or ecological niche specialization. Examples include brown bears, which persist as a distinct species despite hundreds of thousands of years of gene flow from polar bears (Liu et al., 2014; Cahill et al., 2015), as well as the extinct Neandertals and Denisovans who interbred many times with humans and with each other without merging into an indistinct hominin gene pool (Sankararaman et al., 2014; Slon et al., 2018). Although none of these taxa meets the classical definition of biologically isolated species (Mayr, 1942), in each case there exists abundant evidence of long-term isolation and genetic distinctiveness. If the unidentified canids exhibit a similar profile of persistent genetic distinctive-
ness in the face of gene flow, this should be considered when making conservation decisions. In other words, even if red wolves are found to have a long history of intermittent hybridization with coyotes, this would not necessarily make them a less valid taxonomic species than polar bears are considered to be.
Finding 4-4: A number of analytical approaches can provide evidence of hybridization using either RAD-seq or whole-genome data. However, analyses to determine whether hybridization is recent or ancient among North American canid populations will require unbiased whole-genome data sets in order to make strong demographic inferences throughout the genome as well as to identify and determine the size of ancestry tracts.
CONCLUSIONS AND RECOMMENDATIONS
Conclusion 4-1: To trace the genetic history of extant red wolf populations (captive and managed) and the Gulf Coast canid populations, using whole-genome sequencing methods on modern fresh tissue samples will provide the most informative data with which to assess both genetic continuity between populations over space and time and historical admixture with other North American canids.
Conclusion 4-2: Although expensive to produce and computationally intensive to analyze, whole-genome data will provide the best chance to identify introgressed genomic fragments (i.e., ancestry haplotypes) and to estimate the time scale at which gene flow occurred.
Recommendation 4-1: The committee recommends that new whole-genome sequence data be built upon the existing data with a carefully constructed set of contemporary samples including all North American canid genetic lineages and the full geographic range of all North American canids (red wolf, gray wolf, coyote, and domestic dog).
REFERENCES
Arnold, B., R. B. Corbett-Detig, D. Hartl, and K. Bomblies. 2013. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology 22:3179–3190.
Beeravolu, C. R., M. J. Hickerson, L. A. F. Frantz, and K. Lohse. 2018. ABLE: Blockwise site frequency spectra for inferring complex population histories and recombination. Genome Biology 19:145.
Cahill, J. A., I. Stirling, L. Kistler, R. Salamzade, E. Ersmark, T. L. Fulton, M. Stiller, R. E. Green, and B. Shapiro. 2015. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Molecular Ecology 24(6):1205–1217.
Coates, B. S., D. V. Sumerford, N. J. Miller, K. S. Kim, T. W. Sappington, B. D. Siegfried, and L. C. Lewis. 2009. Comparative performance of single nucleotide polymorphism and microsatellite markers for population genetic analysis. Journal of Heredity 100:556–564.
Corbett-Detig, R., and R. Nielsen. 2017. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy. PLOS Genetics 13:e1006529.
Fitak, R. R., A. Naidu, R. W. Thompson, and M. Culver. 2015. PumaPlex 1.0: A new panel of SNP markers for the genetic management of North American pumas. Journal of Fish and Wildlife Management 7(1):13–27.
Garvin, M. R., K. Saitoh, and A. J. Gharrett. 2010. Application of single nucleotide polymorphisms to non-model species: A technical review. Molecular Ecology Resources 10:915–934.
Green, R. E., J. Krause, A. W. Briggs, T. Maricic, U. Stenzel, M. Kircher, N. Patterson, H. Li, W. Zhai, M. H. Fritz, N. F. Hansen, E. Y. Durand, A. S. Malaspinas, J. D. Jensen, T. Marques-Bonet, C. Alkan, K. Prüfer, M. Meyer, H. A. Burbano, J. M. Good, R. Schultz, A. Aximu-Petri, A. Butthof, B. Höber, B. Höffner, M. Siegemund, A. Weihmann, C. Nusbaum, E. S. Lander, C. Russ, N. Novod, J. Affourtit, M. Egholm, C. Verna, P. Rudan, D. Brajkovic, D., Ž. Kucan, I. Gušic, V. B. Doronichev, L. V. Golovanova, C. Lalueza-Fox, M. de la Rasilla, J. Fortea, A. Rosas, R. W. Schmitz, P. L. F. Johnson, E. E. Eichler, D. Falush, E. Birney, J. C. Mullikin, M. Slatkin, R. Nielsen, J. Kelso, M. Lachmann, D. Reich, and S. Pääbo. 2010. A draft sequence of the Neandertal genome. Science 328(5979):710–722.
Gronau, I., M. J. Hubisz, B. Gulko, C. G. Danko, and A. Siepel. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nature Genetics 43:1031–1034.
Gutenkunst, R. N., R. D. Hernandez, S. H. Williamson, and C. D. Bustamante. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLOS Genetics 5(10):e1000695.
Harris, K., and R. Nielsen. 2013. Inferring demographic history from a spectrum of shared haplotype lengths. PLOS Genetics 9(6):e1003521.
Harrison, R. G., and E. L. Larson. 2014. Hybridization, introgression, and the nature of species boundaries. Journal of Heredity 105(Suppl):795–809.
Heppenheimer, E., K. E. Brzeski, R. Wooten, W. Waddell, L. Y. Rutledge, M. J. Chamberlain, D. R. Stahler, J. W. Hinton, and B. M. vonHoldt. 2018a. Rediscovery of red wolf ghost alleles in a canid population along the American Gulf Coast. Genes 9(12):618.
Heppenheimer, E., K. E. Brzeski, J. W. Hinton, B. R. Patterson, L. Y. Rutledge, A. L. DeCandia, T. Wheeldon, S. R. Fain, P. A. Hohenlohe, R. Kays, B. N. White, M. J. Chamberlain, and B. M. vonHoldt. 2018b. High genomic diversity and candidate genes under selection associated with range expansion in eastern coyote (Canis latrans) populations. Ecology and Evolution 8(24):12641–12655.
Heppenheimer, E., K. E. Brzeski, J. W. Hinton, M. J. Chamberlain, J. Robinson, R. K. Wayne, and B. M. vonHoldt. 2020. A genome-wide perspective on the persistence of red wolf ancestry in southeastern canids. Journal of Heredity 11(3):277–286.
Liu, S., E. D. Lorenzen, M. Fumagalli, B. Li, K. Harris, Z. Xiong, L. Zhou, T. S. Korneliussen, M. Somel, C. Babbitt, G. Wray, J. Li, W. He, Z. Wang, W. Fu, X. Xiang, C. C. Morgan, A. Doherty, M. J. O’Connell, J. O. McInerney, E. W. Born, L. Dalén, R. Dietz, L. Orlando, C. Sonne, G. Zhang, R. Nielsen, E. Willerslev, and J. Wang. 2014. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157(4):785–794.
Loh, P. R., M. Lipson, N. Patterson, P. Moorjani, J. K. Pickrell, D. Reich, and B. Berger. 2013. Inferring admixture histories of human populations using linkage disequilibrium. Genetics 193(4):1233–1254.
Mayr, E. 1942. Systematics and the origin of species. New York: Columbia University Press.
McDonough, M. M., L. D. Parker, N. Rotzel McInerney, M. G. Campana, and J. E. Maldonado. 2018. Performance of commonly requested destructive museum samples for mammalian genomic studies. Journal of Mammalogy 99(4):789–802.
Murphy, S. M., J. R. Adams, J. J. Cox, and L. P. Waits. 2019. Substantial red wolf genetic ancestry persists in wild canids of southwestern Louisiana. Conservation Letters 12:e12621.
Patterson, N., P. Moorjani, Y. Luo, S. Mallick, N. Rohland, Y. Zhan, T. Genschoreck, T. Webster, and D. Reich. 2012. Ancient admixture in human history. Genetics 192(3):1065–1093.
Phillips, M. K., V. G. Henry, and B. T. Kelly. 2003. Restoration of the red wolf. In D. L. Mech and L. Boitani (eds.), Wolves: Behavior, ecology, and conservation. Chicago: University of Chicago Press. Pp. 272–288.
Pool, J. E., and R. Nielsen. 2009. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181(2):711–719.
Sankararaman, S., S. Mallick, M. Dannemann, K. Prüfer, J. Kelso, S. Pääbo, N. Patterson, and D. Reich. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507(7492):354–357.
Schraiber, J. G. 2018. Assessing the relationship of ancient and modern populations. Genetics 208(1):383–398.
Shafer, A. B., C. R. Peart, S. Tusso, I. Maayan, A. Brelsford, C. W. Wheat, and J. B. Wolf. 2017. Bioinformatic processing of RAD seq data dramatically impacts downstream population genetic inference. Methods in Ecology and Evolution 8(8):907–917.
Sinding, M. H. S., S. Gopalakrishnan, J. Ramos-Madrigal, M. de Manuel, V. V. Pitulko, L. Kuderna, T. R. Feuerborn, L. A. Frantz, F. G. Vieira, J. Niemann, and J. A. S. Castruita. 2020. Arctic-adapted dogs emerged at the Pleistocene–Holocene transition. Science 368(6498):1495–1499.
Slon, V., F. Mafessoni, B. Vernot, C. de Filippo, S. Grote, B. Viola, M. Hajdinjak, S. Peyrégne, S. Nagel, S. Brown, and K. Douka. 2018. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature 561(7721):113–116.
Snyder-Mackler, N., W. H. Majoros, M. L. Yuan, A. O. Shaver, J. B. Gordon, G. H. Kopp, S. A. Schlebusch, J. D. Wall, S. C. Alberts, S. Mukherjee, and X. Zhou. 2016. Efficient genome-wide sequencing and low-coverage pedigree analysis from noninvasively collected samples. Genetics 203(2):699–714.
vonHoldt, B. M., J. P. Pollinger, D. A. Earl, J. C. Knowles, A. R. Boyko, H. Parker, E. Geffen, M. Pilot, W. Jedrzejewski, B. Jedrzejewska, V. Sidorovich, C. Greco, E. Randi, M. Musiani, R. Kays, C. D. Bustamante, E. A. Ostrander, J. Novembre, and R. K. Wayne. 2011. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Research 21:1294–1305.
vonHoldt, B. M., J. A. Cahill, Z. Fan, I. Gronau, J. Robinson, J. P. Pollinger, B. Shapiro, J. Wall, and R. K. Wayne. 2016. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Science Advances 2(7):e1501714.
This page intentionally left blank.














